

Phosphorylation of Rad51/Rad52 by CDK1 regulates cell cycle–specific activation of homologous recombination.
Abstract
Homologous recombination is exquisitely activated only during specific cell phases. In the G1 phase, homologous recombination activity is completely suppressed. According to previous reports, the activation of homologous recombination during specific cell phases depends on the kinase activity of cyclin-dependent kinase 1 (CDK1). However, the precise regulatory mechanism and target substrates of CDK1 for this regulation have not been completely determined. Here, we report that the budding yeast CDK1, Cdc28, phosphorylates the major homologous recombination regulators Rad51 and Rad52. This phosphorylation occurs in the G2/M phase by Cdc28 in combination with G2/M phase cyclins. Nonphosphorylatable mutations in Rad51 and Rad52 impair the DNA binding affinity of Rad51 and the affinity between Rad52 rings that leads to their interaction. Collectively, our data provide detailed insights into the regulatory mechanism of cell cycle–dependent homologous recombination activation in eukaryotic cells.
INTRODUCTION
DNA double-strand breaks (DSBs) spontaneously occur during cell proliferation. Because these chromosomal breaks can lead to genetic mutations, cell death, and tumor generation, cells have evolved diverse repair pathways. Homologous recombination is the major error-free pathway for repair of DSBs. When homologous sequences in the homologous chromosome are used as a template, the homologous recombination mechanism repairs the DNA lesions without altering the genetic information. DNA damage repair by homologous recombination progresses through the following steps: (i) When a DSB occurs, the end resection process resects the broken ends of the DNA; (ii) the replication protein A (RPA) complex recognizes exposed single-stranded DNA (ssDNA) at the DNA damage site and recruits the major homologous recombination regulator, Rad52, to the site; (iii) the DNA-bound Rad52 sequentially recruits Rad51 to the homologous DNA region to activate strand invasion; and (iv) in the course of DNA synthesis, the damage is repaired on the basis of the homologous sequence (1). Termination of homologous recombination is mainly mediated by synthesis-dependent strand annealing (SDSA) and DSB repair (DSBR). SDSA, which does not produce crossover products, is predominantly activated in mitotic cells. In contrast, DSBR, which generates crossover products by double Holliday junction resolution, is induced mostly in meiotic cells (1, 2).
Cell cycle progression is precisely regulated by cyclin-dependent protein kinases (CDKs). Saccharomyces cerevisiae has five encoded CDKs: Cdc28, Pho85, Kin28, Ssn3, and Ctk1. Among these, Cdc28 (CDK1) functions as a major regulator of cell cycle progression (3). Nine cyclins of S. cerevisiae are generally classified by cell cycle phase as follows: the G1 phase cyclins (Cln1, Cln2, and Cln3), the S phase cyclins (Clb5 and Clb6), and the G2/M phase cyclins (Clb1, Clb2, Clb3, and Clb4) (4). Each cyclin is expressed during a specific cell phase and forms a complex with CDK to phosphorylate target proteins that are related to cell cycle–dependent functions (3, 4). Cell cycle–dependent regulation of homologous recombination has been studied in yeast and mammals over the past decade. For human cells, it has been proposed that CDK1 may affect the function of the major mediators in this cell cycle–dependent regulation of homologous recombination (5). However, the precise regulatory mechanism and target substrates of CDK1 for this regulation have not been fully elucidated. In S. cerevisiae, the process of homologous recombination is highly suppressed in the G1 phase and is induced in the G2/M phase. It has also been reported that this regulation depends on the kinase activity of Cdc28 (6, 7). DNA resection proteins Sae2 and Dna2 have been identified as substrates of Cdc28 (8, 9). End resection, which is the process immediately preceding homologous recombination pathway initiation, comprises two sequential steps: initial resection and extensive resection. Both resection steps are induced by the kinase activity of Cdc28. However, the proteins that mediate homologous recombination directly through cell cycle–dependent regulation remain to be discovered.
Yeast has many advantages for the study of homologous recombination. Chromosome III of S. cerevisiae harbors the mating-type locus and two mating-type alleles known as a and α. HO endonuclease recognizes a short sequence in the mating-type locus and makes a site-specific single DSB. Through the homologous recombination pathway, this damage is repaired on the basis of the genetic information on the opposite mating-type allele, and consequently, the genetic information of the mating-type locus is changed to that of the opposite mating-type allele (10). Thus, in S. cerevisiae, the efficiency of homologous recombination during mitotic growth can be monitored by checking the efficiency of the mating-type switching (6).
In this study, we investigated the unknown targets of Cdc28 for cell cycle–dependent regulation of homologous recombination in S. cerevisiae. We found that both Rad51 and Rad52 are substrates of Cdc28. In addition, the functions of Rad51 and Rad52 for activating homologous recombination are regulated by the G2/M-phase CDK1-dependent phosphorylation. In total, our results suggest a previously unknown mechanism for cell cycle–dependent regulation of homologous recombination activity.
RESULTS
Rad51 and Rad52 are substrates of Cdc28
Cell cycle–dependent regulation of the homologous recombination process has been reported in previous studies (6, 7). The activity of homologous recombination is highly suppressed in the G1 phase and enhanced in the G2/M phase. To check the activation of homologous recombination, we performed a polymerase chain reaction (PCR)–based homologous recombination efficiency test (fig. S1A) (11). If a DSB is generated by the expression of HO endonuclease, then PCR cannot be performed using primers pI and pJ. Confirmation of strand invasion and primer extension can be confirmed by PCR using primers pG and pH. PCR can be completed using primers pC and pD only after the newly synthesized DNA is connected to the opposite end of damaged DNA. Using this test, we confirmed that homologous recombination is not activated in G1-arrested yeast cells even after DSB formation (fig. S1B). In contrast, G2/M-arrested yeast cells progressed through the strand invasion, primer extension, and ligation processes of homologous recombination after DSB formation. These results are consistent with previous reports of other techniques used to check the efficiency of homologous recombination (6, 7).
Although it has been known that cell cycle–dependent regulation of homologous recombination depends on the kinase activity of Cdc28 (in yeast, CDK1) (6, 7), the mechanism for this regulation is still undetermined. To address this issue, we focused on two major homologous recombination regulators: Rad51 and Rad52. As previously reported, rad51Δ and rad52Δ cells could not repair DNA damage that was generated by the DNA-damaging agent methyl methanesulfonate (MMS) (fig. S1C). This result indicated that deletion of either RAD51 or RAD52 completely impairs homologous recombination activity. Furthermore, neither strand invasion nor primer extension processes were completed in the rad51Δ and rad52Δ cells (fig. S1D), suggesting that homologous recombination in these mutants is arrested at an early step. To examine the possibility that Rad51 and Rad52 may be the target of Cdc28 for cell cycle–dependent regulation, we performed an in vitro kinase assay using an adenosine 5′-triphosphate (ATP) analog–sensitive mutant of Cdc28, Cdc28-as1 (12). In the absence of the ATP analog 1NM-PP1, both Rad51 and Rad52 were clearly phosphorylated by Cdc28-as1 (Fig. 1A). When the kinase activity of Cdc28-as1 was suppressed by 1NM-PP1, the phosphorylation of Rad51 and Rad52 was completely abolished. This result suggested that both Rad51 and Rad52 are substrates of Cdc28.
Fig. 1. Rad51 and Rad52 are phosphorylated by Cdc28 in the G2/M phase.
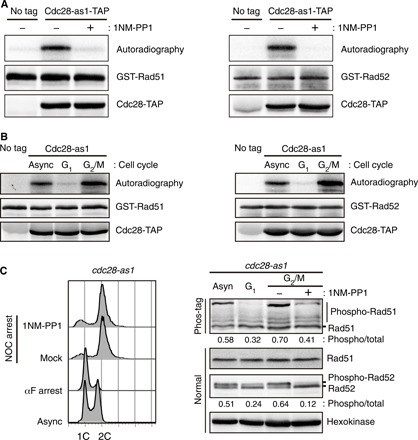
(A) Results from the kinase assay for the phosphorylation of Rad51 (left) and Rad52 (right) using the Cdc28-as1 mutant in vitro. Cdc28-as1-TAP was purified by anti-TAP immunoprecipitation from asynchronous cells. GST-purified Rad51 and Rad52 were used as substrates, and 1NM-PP1 (5 μM) was added to cells to suppress the kinase activity of Cdc28-as1. (B) Results from the kinase assay for the phosphorylation of Rad51 (left) and Rad52 (right) using Cdc28 extracted from cell cycle–arrested cells in vitro. The cell cycle was arrested for 3 hours with α-factor (150 μM) to arrest G1 and nocodazole (15 μg ml−1) to arrest G2/M. Async, asynchronous cells. (C) Analysis of the in vivo phosphorylation level of Rad51 and Rad52 through the cell cycle. The cell cycle was arrested for 3 hours with α-factor (150 μM) to arrest G1. Synchronized cells were released to fresh YPD media with nocodazole (15 μg ml−1) for 45 min. After 45 min of incubation, 5 μM 1NM-PP1 or DMSO was directly added to cell cultures for 1 hour. The DNA content data were analyzed by flow cytometry (left). 1C and 2C indicate single and double DNA haploid content, respectively. Phos-tag (50 μM) and 100 μM MnCl2 were mixed with 6% separating gel for analysis of phospho-Rad51 (right panel). The relative ratio of phosphorylated proteins to total proteins is shown below each lane.
Rad51 and Rad52 are phosphorylated by G2/M-phase CDK1
Given that Rad51 and Rad52 are phosphorylated by the CDK Cdc28, we speculated that Cdc28-dependent phosphorylation of Rad51 and Rad52 might be regulated in a cell cycle–dependent manner. To obtain cell cycle–specific CDK1 proteins, we immunoprecipitated Cdc28 from extracts of either G1-arrested or G2/M-arrested cells. Cdc28-dependent phosphorylation of Rad51 and Rad52 was observed only with the CDK1 from the G2/M-phase cells but not with the CDK1 from the G1-phase cells (Fig. 1B and fig. S2A). We next examined whether cell cycle–dependent phosphorylation of Rad51 and Rad52 occurs in cells by tracking their level of phosphorylation through the cell cycle. Whereas the phosphorylated Rad52 migrated slower than the nonphosphorylated Rad52, the phosphorylated Rad51 was not separated from the nonphosphorylated Rad51 in normal SDS–polyacrylamide gel electrophoresis (PAGE). To analyze the phosphorylation status of Rad51, we used Phos-tag SDS-PAGE, which enables a slower migration of phosphorylated proteins. The phosphorylation of Rad51 and Rad52 was decreased in the G1 phase and substantially increased in the G2/M phase (Fig. 1C). When the kinase activity of Cdc28-as1 was suppressed by 1NM-PP1 treatment, the phosphorylation of Rad51 and Rad52 was decreased even in the G2/M phase. We further confirmed that Rad51 was substantially less phosphorylated in the G1 phase than in the G2/M phase by using the phospho-CDK substrate antibody (fig. S2B). These results suggested that Rad51 and Rad52 are highly phosphorylated by CDK1 in the G2/M phase, not in the G1 phase.
To identify the specific cyclin-CDK1 complexes that are responsible for the phosphorylation of Rad51 and Rad52, we performed a kinase assay on all the cyclins of budding yeast in vitro. Each cyclin was collected by immunoprecipitation and mixed with purified Cdc28 to form a cyclin-CDK1 complex. Both Rad51 and Rad52 were phosphorylated by Cdc28, combined with either Clb2 or Clb3, which are activated in the G2/M phase, but not by Cdc28 combined with the G1 cyclins such as Cln1, Cln2, and Cln3 (Fig. 2, A and B, and fig. S2C). These results suggested that Cdc28 combined with Clb2 or Clb3 phosphorylates Rad51 and Rad52 in the G2/M phase. We also checked the deletion effect of either CLB2 or CLB3 on the in vivo phosphorylation of Rad51 and Rad52. Because Clb2 and Clb3 were redundantly expressed in the S and G2/M phases (fig. S2D), the single deletion of either CLB2 or CLB3 did not markedly affect the phosphorylation of Rad51 and Rad52 in the S and G2/M phases (fig. S2E). However, we observed a moderate reduction in the phosphorylation of Rad52 and Rad51 in clb2∆ and clb3∆ cells, respectively. This result suggested that Rad51 and Rad52 have a kinase preference for Clb3-Cdc28 and Clb2-Cdc28 complexes, respectively.
Fig. 2. Cdc28 combined with Clb2 or Clb3 phosphorylates Ser125 and Ser375 of Rad51 and Thr412 of Rad52.
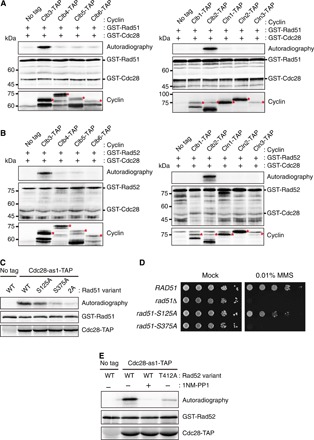
(A) Results from the kinase assay for the phosphorylation of Rad51 using purified Cdc28 and cyclins in vitro. Each cyclin was purified by anti-TAP immunoprecipitation from asynchronous cells. Red asterisks indicate bands of corresponding cyclins. (B) Results from the kinase assay for the phosphorylation of Rad52 using purified Cdc28 and cyclins in vitro. (C) Results from the kinase assay using alanine substitution mutants of Rad51 in vitro. Cdc28-as1-TAP was purified by anti-TAP immunoprecipitation from asynchronous cells. GST-tagged Rad51 variants were expressed in E. coli and purified by GST pull down. S125A, S375A, and 2A indicate Rad51 mutants with alanine substitutions at Ser125, at Ser375, and at both Ser125 and Ser375, respectively. WT, wild type. (D) Results from the serial dilution assay used to assess MMS sensitivity of rad51Δ cells expressing Rad51 variants. Cells were spotted in 10-fold serial dilutions on SC medium in the absence or presence of 0.01% MMS. rad51-S125A and rad51-S375A indicate rad51Δ cells expressing the Rad51 mutant with alanine substitutions at Ser125 and Ser375, respectively. (E) Results from the kinase assay using an alanine substitution mutant of Rad52 in vitro. Cdc28-as1-TAP and GST-tagged Rad52 were purified as described in (C). T412A indicates a Rad52 mutant with an alanine substitution at Thr412.
Ser125 and Ser375 of Rad51 and Thr412 of Rad52 are the target residues of Cdc28
It is known that Cdc28 is a proline-directed kinase (13). However, target residues that are not part of the conventional consensus motif (Ser/Thr-Pro) have also been reported in diverse eukaryotic cells, including those of budding yeast (14, 15). To find the target residues of Cdc28 on Rad51 and Rad52, we analyzed the amino acid sequences of Rad51 and Rad52 by using the Group-based Prediction System (GPS) 3.0 algorithm (16), which has been successfully adopted for predicting protein kinase–specific phosphorylation sites. GPS 3.0 identified Ser125 and Ser375 on Rad51 and Thr412 on Rad52 as putative Cdc28-specific phosphorylation sites (fig. S3A). Ser375 on Rad51 and Thr412 on Rad52 coincide with the consensus Ser/Thr-Pro motif of CDK1, and Ser125 of Rad51 is located within a motif that matches the non-Ser/Thr-Pro motif (Ser/Thr-X-X-Lys/Arg) of CDK1 (15). To examine whether both Ser125 and Ser375 on Rad51 are the target residues of Cdc28, we generated two mutants that each have a single nonphosphorylatable site (Rad51-S125A and Rad51-S375A) and one mutant with two nonphosphorylatable sites (Rad51-2A) by substituting these residues with alanine. In a kinase assay conducted in vitro, Cdc28-dependent phosphorylation was slightly decreased at Rad51-S125A and greatly reduced at Rad51-S375A compared with that of wild-type Rad51 (Fig. 2C). Furthermore, the phosphorylation of Rad51-2A was decreased to a much greater degree than that of Rad51-S375A. We next examined the DNA repair efficiency of the Rad51 nonphosphorylatable mutants by serial dilution assay. Consistent with the results of the kinase assay conducted in vitro, while hypersensitivity to MMS was predominantly induced by the alanine substitution at Ser375, the rad51-S125A cells also exhibited low-level sensitivity to MMS (Fig. 2D). Therefore, we concluded that Cdc28 phosphorylates both Ser125 and Ser375 of Rad51. We also observed that a nonphosphorylatable mutant of Rad52 (Rad52-T412A) showed decreased phosphorylation compared with wild-type Rad52 (Fig. 2E), suggesting that Thr412 of Rad52 is phosphorylated by Cdc28. Corroborating these results, in vivo phosphorylation of Rad51-2A, which was detected by the phospho-CDK substrate antibody, was greatly decreased compared with that of wild-type Rad51 (fig. S3B), and mass spectrometry analysis revealed that Thr412 of Rad52 is phosphorylated in vivo (fig. S3C).
The G2/M-phase CDK1-dependent phosphorylation of Rad51 promotes its DNA binding affinity
To determine the effect of the CDK1-dependent phosphorylation of Rad51 on its homologous recombination activity, we used a serial dilution assay to examine the DNA repair efficiency of cells expressing Rad51-2A. Similar to the rad51Δ cells, the rad51-2A cells showed hypersensitivity to the DNA damage generated by MMS (Fig. 3A). Next, we performed a PCR-based homologous recombination efficiency test using the rad51-2A cells. Although a DSB was induced at the MAT locus by HO endonuclease expression, strand invasion and primer extension did not occur in the rad51-2A cells (Fig. 3B). Consequently, the ligation process, which is the subsequent step of primer extension, also did not occur in the rad51-2A cells. Even in the G2/M phase, during which homologous recombination is highly activated, the rad51-2A cells did not progress through strand invasion and primer extension (fig. S4). These results suggested that the G2/M phase CDK1-dependent phosphorylation of Rad51 has an important role in the process that precedes the strand invasion step. We also tested the DNA repair efficiency and PCR-based homologous recombination efficiency of cells expressing Rad51-2E, a Cdc28-dependent phosphomimicking form of Rad51. The rad51-2E cells failed to recover from the DNA damage (Fig. 3, A and B), suggesting that Rad51-2E does not mimic the effect of Cdc28-dependent phosphorylation. It has been shown that the negative charge and ionic shell produced by aspartate or glutamate substitution often differ from those produced by the phosphorylated residue under physiological conditions (17). Given this finding, we assume that the differences in negative charge and ionic shell between Rad51-2E and Cdc28-dependent phosphorylated Rad51 are sufficiently large such that they have different functions.
Fig. 3. The G2/M-phase CDK1-dependent phosphorylation promotes the DNA binding affinity of Rad51.
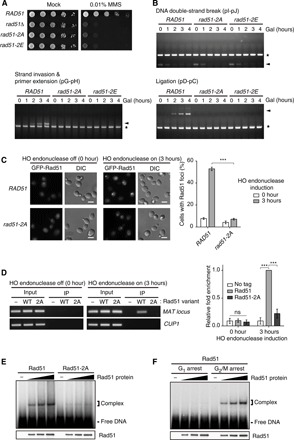
(A) Results from the serial dilution assay used to assess MMS sensitivity of rad51Δ cells expressing Rad51 variants. Cells were spotted in 10-fold serial dilutions on SC medium in the absence or presence of 0.01% MMS. rad51-2A indicates rad51Δ cells expressing the Rad51 mutant with alanine substitutions at Ser125 and Ser375. rad51-2E indicates rad51Δ cells expressing the Rad51 mutant with glutamate substitutions at Ser125 and Ser375. (B) Homologous recombination efficiency test of rad51Δ cells expressing Rad51 variants. Genomic DNA was extracted every 1 hour after 2% galactose addition and analyzed by PCR. Arrowheads indicate the PCR products of the homologous recombination intermediates. Asterisks indicate the PCR products of the control region (ARG5,6). (C) Accumulated GFP-tagged Rad51 and Rad51-2A at the DNA damage sites. To make DSB, HO endonuclease was induced during incubation with 2% galactose for 3 hours. DIC, differential interference contrast. Scale bars, 4 μm. The percentages of cells with Rad51-GFP foci are shown on the right panel. Data are presented as the means ± SD of triplicate experiments. P values were determined by Student’s t test (***P < 0.005). (D) ChIP assay was used to assess the DNA binding affinity of Rad51 and Rad51-2A. HO endonuclease was induced during incubation with 2% galactose for 3 hours. Immunoprecipitated DNA with Rad51 variants was analyzed for the MAT locus by PCR (left panel) and by quantitative real-time PCR (right panel). CUP1 was used as a negative control. Data are presented as the means ± SD of triplicate experiments. P values were determined by a one-sample t test (***P < 0.005). ns, not significant. (E) Results from the EMSA used to assess the ssDNA binding affinity of Rad51 and Rad51-2A. EMSA was performed using a binding buffer that includes 35 mM tris-Cl (pH 7.5), 5 mM ATP, 5 mM MgCl2, 50 mM KCl, bovine serum albumin (100 μg/ml), and 1 mM dithiothreitol. (F) Results from the EMSA used to assess the DNA binding affinity of the G1- and the G2/M-phase Rad51. α-Factor (150 μM) and nocodazole (15 μg ml−1) were treated to synchronize cell cycle to the G1 and G2/M phases, respectively.
ssDNA at the DNA damage site is covered with RPA-Rad52 complexes. This nucleoprotein filament recruits Rad51, and, subsequently, ssDNA-bound Rad51 induces strand invasion into the undamaged homologous DNA region (1, 18). We investigated the effect of the G2/M-phase CDK1-dependent phosphorylation of Rad51 on its recruitment to the DNA damage site. The fluorescence signal of the green fluorescent protein (GFP)–tagged Rad51 was diffusely localized in the nucleus and cytoplasm under normal conditions, while a pattern of small bright foci was detected in the nucleus upon DSB induction by HO endonuclease (Fig. 3C, left). The fluorescence signal of the GFP-tagged Rad51-2A did not readily accumulate in the foci upon the initial DSB induction; that is, after 3 hours of HO endonuclease expression, only 7% of cells showed GFP–Rad51-2A in the nuclear foci, whereas more than 50% of cells showed GFP-Rad51 in the nuclear foci (Fig. 3C, right).
Rad51 foci formation at the DNA damage site requires two prior steps: Rad51 recruitment by the physical interaction with Rad52 and ssDNA binding of Rad51 (1). Using a coimmunoprecipitation assay, we examined the effect of the nonphosphorylatable mutation of Rad51 on the physical interaction between Rad51 and Rad52. As shown in fig. S5A, the protein-protein interaction between Rad51 and Rad52 was not perturbed by the loss of phosphorylation caused by the mutation of Rad51. Next, we checked the effect of the G2/M-phase CDK1-dependent phosphorylation of Rad51 on its DNA binding affinity by chromatin immunoprecipitation (ChIP) assay. In the absence of DSB formation at the MAT locus, the Z1 region in the MAT locus was not precipitated with either the wild-type Rad51 or Rad51-2A (Fig. 3D). After 3 hours of HO endonuclease expression, Rad51 was bound to a high degree to the Z1 region in the MAT locus. The degree of binding of Rad51-2A to the MAT locus was considerably lower compared with that of wild-type Rad51. This result suggested that the G2/M-phase CDK1-dependent phosphorylation of Rad51 promotes its DNA binding affinity upon DSB induction.
In addition, we also examined whether the nonphosphorylatable mutation of Rad51 affects ssDNA binding in vitro by electrophoresis mobility shift assay (EMSA) using Rad51 proteins purified from vegetative yeast cells. Compared with wild-type Rad51, Rad51-2A failed to form the ssDNA-protein complex (Fig. 3E). To further verify that the DNA binding defect of Rad51-2A is caused by the absence of phosphorylation, we removed the phosphorylation of wild-type Rad51 through λ phosphatase treatment. The DNA-protein complex formation of λ phosphatase–treated Rad51 was substantially decreased compared with that of untreated Rad51 (fig. S5B). We also examined whether the G2/M-phase CDK1-dependent phosphorylation of Rad51 is required to enhance its binding affinity to the ssDNA. To test this role of phosphorylation, wild-type Rad51 was purified from either the G1- or G2/M-arrested yeast cells. The Rad51 from the G1-phase cells failed to form the DNA-protein complex, according to EMSA, whereas the G2/M-phase Rad51 was efficiently bound to the ssDNA (Fig. 3F). These results suggested that the ssDNA binding defect of Rad51-2A is caused by the deficiency in CDK1-dependent phosphorylation. Furthermore, Rad51-2A also showed substantially decreased double-stranded DNA (dsDNA) binding affinity compared with wild-type Rad51 (fig. S5C). Overall, it seems that the G2/M-phase CDK1-dependent phosphorylation of Rad51 facilitates its DNA binding during the initial process of homologous recombination.
The G2/M-phase CDK1-dependent phosphorylation of Rad52 does not affect its function early in the process of homologous recombination
To determine whether the G2/M-phase CDK1-dependent phosphorylation of Rad52 is also required to promote homologous recombination activity, we examined the DNA repair efficiency of rad52-T412A cells by serial dilution assay. As shown in Fig. 4A, similar to the rad52Δ cells, the rad52-T412A cells could not repair the MMS-induced DSBs. We found a critical difference between the rad51-2A and rad52-T412A cells in the results of the PCR-based homologous recombination efficiency test. Although the rad51-2A cells could not progress through the strand invasion and primer extension processes, the rad52-T412A cells exhibited normal activation of strand invasion and primer extension processes (Fig. 4B). However, the ligation process did not occur in the rad52-T412A cells. We observed that, even in the G2/M phase–arrested rad52-T412A cells, the ligation process was completely suppressed, whereas the strand invasion and primer extension processes occurred normally (fig. S6A). Together, these results suggested that the G2/M-phase CDK1-dependent phosphorylation of Rad52 is required for its function at a step between the primer extension and ligation processes. In corroboration of these observations, cells expressing a Cdc28-dependent phosphomimicking mutant Rad52-T412E repaired the DNA damage induced by MMS treatment and progressed through all processes of homologous recombination (Fig. 4, A and B, and fig. S6A).
Fig. 4. The G2/M-phase CDK1-dependent phosphorylation does not affect Rad52 functions in the initial step of homologous recombination.
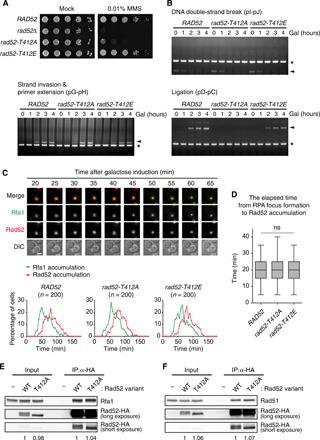
(A) Results from the serial dilution assay used to assess MMS sensitivity of rad52Δ cells expressing Rad52 variants. Cells were spotted in 10-fold serial dilutions on SC medium in the absence or presence of 0.01% MMS. rad52-T412A indicates rad52Δ cells expressing the Rad52 mutant with an alanine substitution at Thr412. rad52-T412E indicates rad52Δ cells expressing the Rad52 mutant with a glutamate substitution at Thr412. (B) Results from the homologous recombination efficiency test for rad52Δ cells expressing Rad52 variants. Genomic DNA was extracted every 1 hour after 2% galactose addition and analyzed by PCR. Arrowheads indicate the PCR products of the homologous recombination intermediates. Asterisks indicate the PCR products of the control region (ARG5,6). (C) Results from the analysis of RPA focus formation and Rad52 accumulation. Images of GFP-tagged Rfa1 and RFP-tagged Rad52 were taken every 5 min after the addition of 2% galactose (top). Scale bar, 2 μm. The percentages of cells with RPA foci (green) and Rad52 foci (red) are shown in the bottom. (D) The elapsed time between RPA foci formation and Rad52 accumulation. A box plot is shown with whiskers from the 5th to the 95th percentile, and the data were normalized to the median of measures from the RAD52 cells (n = 200). P values were determined by the Mann-Whitney U test. (E) Coimmunoprecipitation assay used to assess the binding affinity between Rad52 and Rfa1. Protein complexes with Rad52-HA were precipitated using anti-HA agarose beads. Rfa1 was detected by anti-Rfa1 antibody. The relative ratio of Rfa1 to Rad52, normalized against that of cells with WT Rad52, is shown below each lane. (F) Coimmunoprecipitation assay used to assess the binding affinity between Rad51 and Rad52. Rad51 was detected by anti-Rad51 antibody. The relative ratio of Rad51 to Rad52, normalized against that of cells with WT Rad52, is shown below each lane.
In previous studies, most Rad52 functions were studied during the initiation steps of the homologous recombination pathway (2, 18). Rad52 recognizes RPA-coated ssDNA at the DNA damage site and physically interacts with RPA to form the nucleoprotein filament needed for initiation of homologous recombination (19). Subsequently, Rad51 is recruited to the nucleoprotein filament, which comprises RPA, Rad52, and ssDNA, by the physical interaction with Rad52 (20, 21). We examined the effect of the G2/M-phase CDK1-dependent phosphorylation of Rad52 on its functions in the initiation steps of homologous recombination. To examine the accumulation efficiency of Rad52 at the DNA damage site, we measured the elapsed time between RPA binding and Rad52 accumulation after DSB induction. Similar to a previous study (22), RPA binding and Rad52 accumulation were assessed by nuclear focus formation of GFP-tagged Rfa1, the largest subunit of the RPA complex, and red fluorescent protein (RFP)–tagged Rad52 under fluorescence microscopy. The RPA focus was usually formed approximately 45 min after HO endonuclease induction, and Rad52 started to accumulate another 20 min after RPA focus formation (Fig. 4C, top). This pattern was almost identical among all strains that expressed each of the three Rad52 variants (Fig. 4C, bottom). Statistically, the elapsed time was not notably different among the three strains (Fig. 4D), suggesting that the efficiency of Rad52 accumulation is not affected by the G2/M-phase CDK1-dependent phosphorylation.
We next checked the physical interaction between Rad52 and other homologous recombination-related proteins, such as RPA and Rad51, because these interactions are essential for the activation of homologous recombination (1). Because Rad52 exhibited different migration patterns in SDS-PAGE depending on its phosphorylation status (fig. S6B), we measured and compared the total band intensity of each Rad52 variant. The amount of Rfa1 that coprecipitated with nonphosphorylatable Rad52-T412A was similar to the amount that coprecipitated with wild-type Rad52 (Fig. 4E). The protein-protein interaction between Rad51 and Rad52 was also not perturbed by the nonphosphorylatable mutation of Rad52 (Fig. 4F). This result suggested that the G2/M-phase CDK1-dependent phosphorylation of Rad52 does not regulate its physical interaction with other homologous recombination-related proteins. Together, our findings indicated that the G2/M-phase CDK1-dependent phosphorylation of Rad52 is not required for its function in the initial steps of homologous recombination.
The G2/M-phase CDK1-dependent phosphorylation of Rad52 facilitates the interactions between Rad52 rings that form the superstructure late in the homologous recombination process
Previous reports indicated that Rad52 forms ring structures (23, 24) and that these rings are the bases of superstructures, which are formed dependent on the C-terminal region of Rad52 (25, 26). The ring formation of Rad52 is crucial for its nuclear localization (27) and ssDNA binding in the initial steps of homologous recombination (28). The interaction between Rad52 rings promotes strand annealing of DSB ends (26, 29, 30), and this process is required for the termination steps of homologous recombination, such as the DNA strand annealing in SDSA and the second end capture in DSBR (31–33). Because the functions of Rad52 in the initial steps of homologous recombination were not affected by the alanine substitution of the CDK1 target residue (Fig. 4), we investigated the effect of the CDK1-dependent phosphorylation of Rad52 on its functions in the late steps of homologous recombination. To check the possibility that the G2/M-phase CDK1-dependent phosphorylation may regulate the functions of the Rad52 ring for strand annealing of DSB ends, we measured the efficiency of DNA ligation in vitro. By the use of linear DNA molecules generated through treatment with a restriction enzyme, the efficiency of the DNA ligation can be evaluated by checking the amount of multimeric ligation products. Consistent with previous studies using human Rad52 (26, 29), we observed that yeast Rad52 also stimulated the DNA ligation that was catalyzed by T4 DNA ligase (Fig. 5A). The amount of multimers produced by Rad52-T412A was decreased to ~40% of that produced by wild-type Rad52. Given that DNA ligation is stimulated by the interaction between Rad52 rings (26, 29), we hypothesized that the Rad52 ring interaction for superstructure formation may be regulated by Cdc28-dependent phosphorylation.
Fig. 5. The G2/M-phase CDK1-dependent phosphorylation facilitates the physical interaction between Rad52 proteins.
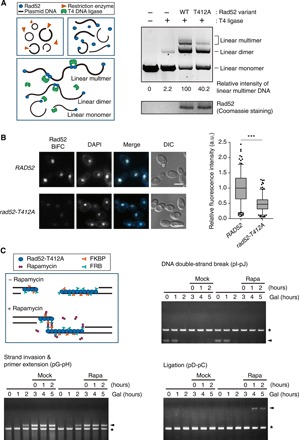
(A) Stimulation of DNA ligation by Rad52 variants in vitro. The left panel presents the schematic of the in vitro DNA ligation test. The efficiency of multimeric product formation was evaluated by measuring the band intensity of the linear multimer (right panel). The relative amount of multimeric product, normalized against that of WT Rad52, is shown below each lane. (B) Results from the BiFC assay used to assess the effect of CDK1-dependent phosphorylation on the physical interaction between Rad52 proteins. Representative BiFC images for the interaction between Rad52 proteins are shown in the left panel. RAD52 indicates cells expressing both VN-Rad52 and VC-Rad52, and rad52-T412A indicates cells expressing both VN-Rad52-T412A and VC-Rad52-T412A. Nuclei were visualized by DAPI (1 μg ml−1) staining. Scale bars, 4 μm. The relative BiFC signal intensity in the RAD52 and rad52-T412A cells is shown in the right panel. A box plot is shown with whiskers from the 5th to the 95th percentile, and the data were normalized to the median of the measures from RAD52 cells (n > 200). P values were determined by the Mann-Whitney U test (***P < 0.005). a.u., arbitrary units. (C) Recovery of the physical interactions between Rad52-T412A proteins by the FKBP-FRB system. The illustration represents a schematic diagram of the artificial recovery of the physical interaction between Rad52-T412A proteins by the FKBP-FRB system. Genomic DNA was extracted every 1 hour after 2% galactose addition and analyzed by PCR. Rapamycin (1 μM) or DMSO was added to cell cultures 3 hours after galactose addition. Arrowheads indicate the PCR products of the homologous recombination intermediates. Asterisks indicate the PCR products of the control region (ARG5,6).
To confirm this hypothesis, we examined whether the T412A mutation disturbs the interaction between Rad52 proteins. For this test, we used the bimolecular fluorescence complementation (BiFC) assay with two fragments of a yellow fluorescence protein variant, Venus. The BiFC assay is a powerful tool to detect protein-protein interactions in the cellular context with minimal perturbation of the structures and functions of the target proteins (34). The BiFC signal of Rad52-T412A was substantially decreased compared with that of wild-type Rad52 (Fig. 5B). Regardless of which fragment of Venus was tagged, the expression levels of Rad52 and Rad52-T412A were similar to each other (fig. S7, A and B), suggesting that the decreased BiFC signal of Rad52-T412A is not due to lower expression. In addition, Rad52-T412A was properly localized to the nucleus similarly to Rad52 (fig. S7C), indicating that the nuclear localization of Rad52 is not disturbed by the alanine substitution at Thr412. Given that the nuclear localization of Rad52 requires ring formation (27), we used transmission electron microscopy to further examine whether Rad52-T412A properly forms ring structures. As shown in fig. S7D, both wild-type Rad52 and Rad52-T412A formed ring structures that were consistent with electron microscopic images of Rad52 presented in previous studies (23, 26). Furthermore, similar to the human Rad52 heptameric ring (24), the yeast Rad52 ring seemed to comprise seven Rad52 proteins. This observation also indicated that the low BiFC signal for Rad52-T412A is not caused by the disassembly of the Rad52 heptameric rings. Together, our data suggested that the interaction between Rad52 rings, which is necessary for superstructure formation, is facilitated by the G2/M-phase CDK1-dependent phosphorylation.
Given the negative effect of the T412A mutation on the interaction between Rad52 rings, we next examined whether enhanced interactions between Rad52-T412 proteins restores the defect such that homologous recombination proceeds. To enhance the interaction between Rad52-T412A proteins, we adopted a chemically induced dimerization system using FKBP (the FK506 binding protein) and FRB (FKBP-rapamycin binding) proteins. When rapamycin is present, FKBP forms a complex with rapamycin, and the FKBP-rapamycin complex subsequently binds to FRB (35). We generated a strain that expressed both Rad52-T412A proteins attached to either FKBP or FRB. Without rapamycin treatment, no PCR products of the ligation process were observed, even after 5 hours of DSB induction, while strand invasion and primer extension processes occurred successfully after 2 hours of DSB induction (Fig. 5C). When rapamycin was added 3 hours after DSB induction, the ligation process recovered to levels similar to those of wild-type cells (Fig. 5C). Cells that expressed both wild-type Rad52 proteins attached to either FKBP or FRB showed normal progression through all processes of homologous recombination, whether rapamycin was present or not (fig. S8A). However, the ligation process that was defective in cells that expressed Rad52-T412A protein without FKBP or FRB attachment was not restored by rapamycin treatment (fig. S8B). Together, these results suggested that the G2/M-phase CDK1-dependent phosphorylation of Rad52 facilitates the interaction between Rad52 rings to induce the end-to-end annealing of damaged DNA in the late process of homologous recombination.
DISCUSSION
In this study, we show that both Rad51 and Rad52 are substrates of the G2/M-phase CDK1 and that their phosphorylation is required for proper activation of homologous recombination. On the basis of our findings, we propose a model for cell cycle–dependent regulation of homologous recombination by the G2/M-phase CDK1 (Fig. 6). In the G1 phase, Cdc28 that have combined with G1 cyclins cannot phosphorylate Rad51 and Rad52, which are the major regulators for the activation of homologous recombination. Consequently, homologous recombination is suppressed in the G1 phase. On the other hand, in the G2/M phase, Cdc28 that have combined with some G2/M cyclins, such as Clb2 and Clb3, phosphorylates Rad51 and Rad52. The phosphorylation of Rad51 enhances its DNA binding affinity such that phosphorylated Rad51 binds to ssDNA that is coated by the RPA complex and Rad52. Rad51 binding enables strand invasion into the homologous DNA region for the initiation of homologous recombination-dependent DNA damage repair. The phosphorylation of Rad52 facilitates interactions between Rad52 rings, which are required for end-to-end annealing, a process for connecting newly synthesized DNA ends to the opposite end of the damaged DNA. After end-to-end annealing, gap-filling synthesis and ligation processes terminate homologous recombination.
Fig. 6. Summary diagram for cell cycle–dependent regulation of homologous recombination by the G2/M-phase CDK1.
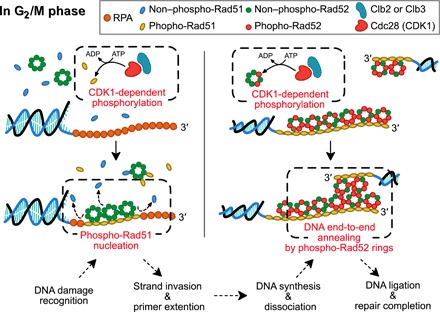
In the G1 phase, Rad51 and Rad52 are not phosphorylated by Cdc28 combined with the G1 phase cyclins. In the G2/M phase, Cdc28 combined with either Clb2 or Clb3 phosphorylates both Rad51 and Rad52. When a DSB occurs, it is recognized by the RPA complex and Rad52. Subsequently, because Cdc28-dependent phosphorylation promotes DNA binding of Rad51, phosphorylated Rad51 proteins form a nucleoprotein complex with ssDNA at the DNA damage site. Rad51 binding allows strand invasion to the homologous DNA region. After DNA synthesis for repair of the DSB, the newly synthesized DNA strand dissociates from its template strand. Because Cdc28-dependent phosphorylation facilitates protein-protein interaction between Rad52 rings, the dissociated strand is annealed to the other recessed end of the DSB by superstructure formation of Rad52 rings that are associated with each strand of broken DNA. Subsequently, the DSBR pathway is completed by the gap filling and ligation process.
Similar to that described in previous studies (6, 7), our examination into cell cycle–dependent regulation of homologous recombination was undertaken by α-factor treatment to induce G1 arrest and nocodazole treatment to induce G2/M arrest. It has been reported that activation of homologous recombination is also observed in the S phase in S. cerevisiae (1, 20). Because replication stress is a major source of DSBs, DNA damage repair by homologous recombination is critically required to maintain genome stability in the S phase. Thus, it is reasonable to assume that a mechanism exists whereby homologous recombination is also activated in the S phase. In this study, we found that the phosphorylation of Rad51 and Rad52 for the progression of homologous recombination is catalyzed by Cdc28 combined with either Clb2 or Clb3. Given that Clb2 and Clb3 are redundantly expressed in the S and G2/M phases (fig. S2D), it is presumable that Cdc28 combined with either Clb2 or Clb3 induces Rad51 and Rad52 to function for the precise regulation of homologous recombination in the S phase.
To activate homologous recombination, the broken ends of DNA should be resected before the recombination factors are assembled. End resection comprises two sequential steps: initial resection and extensive resection (2). It has been reported that Sae2 and Dna2, which are involved in initial resection and extensive resection, respectively, are substrates of CDK1 that are activated precisely for end resection in a cell cycle–dependent manner (8, 9). In addition, in this study, we found cooperative mechanisms for cell cycle–dependent regulation of homologous recombination. Thus, it is likely that homologous recombination is elaborately regulated by stepwise procedures and that timing for this activation depends on the particular cell phases. Although it is still unclear why CDK1 regulates multiple targets for homologous recombination, the following possibilities can be considered: (i) Because partial activation is not sufficient to drive homologous recombination in systems with multiple targets that must be regulated, homologous recombination is not unexpectedly activated in the G1 phase; (ii) although ssDNA is not exposed during end resection suppression, continuous activation of homologous recombination would be harmful to genome stability; and (iii) CDK1 phosphorylates some of the Rad51 and Rad52 pool to maintain a level of active forms appropriate for the precise regulation of homologous recombination. To expand the understanding of the regulatory mechanisms of homologous recombination, it would be interesting to investigate the physiological hierarchy or the interrelation of the CDK1 targets involved in cell cycle–dependent regulation of homologous recombination.
Our results suggest that the G2/M-phase CDK1 stimulates the functions of Rad51 and Rad52 in the initial step and the late step of homologous recombination, respectively. Rad51 binding is a decisive factor for inducing the strand invasion step of the homologous recombination pathway (36). Without Rad51 binding, a DSB is repaired by the Rad51-independent single-strand annealing pathway (36). Given this process, the CDK1-dependent phosphorylation of Rad51 seems to be a key determinant for the selection of an appropriate DSBR pathway. If that presumption is accurate, additional upstream regulators might also be required for fine-tuning the phosphorylation by CDK1. In addition, it is reasonable to think that suppression of Rad52 before it binds to damaged DNA may be the best way to suppress homologous recombination. However, Rad52 binding to DNA is not affected by CDK1-dependent phosphorylation. Previously, we demonstrated that Rad52 has unknown functions for the proper regulation of chromosome segregation, and they are not related to the functions for homologous recombination (37). Given that Rad52 binds to centromeric DNA to regulate chromosome segregation, it is possible that suppressing the DNA binding affinity of Rad52 affects not only its homologous recombination activity but also its other functions. Hence, we speculate that regulation of the interaction between Rad52 rings in the late step of homologous recombination is more advantageous than regulation of the DNA binding affinity of Rad52 during the initial step of homologous recombination for minimizing undesirable effects.
It has been reported that diploid yeast cells are able to progress homologous recombination in the G1 phase (38, 39). The study by Luchnik et al. (38) has demonstrated the different response between haploid and diploid cells against DNA damage under the same conditions; in nonnutrient medium, haploid cells could not repair DNA damage in the G1 phase, whereas diploid cells did. It is presumable that diploid cells, which have two homologous DNA duplexes in the G1 phase, can activate homologous recombination in the G1 phase by another regulatory mechanism. Identifying the different mechanisms between haploid and diploid yeast cells would be helpful to expand the knowledge about regulation of homologous recombination activity.
Although cell cycle–dependent regulation of homologous recombination has been discovered during the past decade, the details of its mechanisms are still mostly unknown. Therefore, our identification of Rad51 and Rad52 as targets of the G2/M-phase CDK1 for the fine-tuned regulation of homologous recombination advances the understanding of the mechanisms needed for maintaining genome stability in eukaryotic cells. It would be interesting to investigate whether the present findings extend to other eukaryotic organisms, including humans.
MATERIALS AND METHODS
Yeast strains and culture conditions
The genotypes of all strains used in this study are listed in table S1. Strains were constructed by PCR-mediated epitope tagging method or PCR-mediated gene deletion, or gene integration using yeast integrative vectors. The desired mutants were selected by prototrophy and subsequently verified by PCR. Yeast cells were grown at 30°C in yeast extract/peptone/glucose (YPD) or synthetic complete (SC) medium lacking amino acids as required. α-Factor (150 μM), hydroxyurea (200 mM), and nocodazole (15 μg ml−1) were treated to synchronize cell cycle to the G1, S, and G2/M phases, respectively. For all chemical treatments, cells were grown to midlog phase [optical density (OD600) = 0.7 to 1.0] and diluted to OD600 = 0.5. Chemical-treated cells were incubated at 30°C for 3 hours. To activate the GAL promoter, cells were grown to midlog phase (OD600 = 0.7 to 1.0) in YP or SC media containing 2% raffinose and diluted to OD600 = 0.5 with raffinose-containing media. Two percent of galactose was directly added to cell cultures. For 1NM-PP1 treatment, cells were grown to midlog phase (OD600 = 0.7 to 1.0) and diluted to OD600 = 0.5. α-Factor (150 μM) was treated for 3 hours to synchronize cell cycle to the G1 phase and washed twice with YPD. Washed cells were resuspended in fresh YPD including nocodazole (15 μg ml−1) and incubated for 45 min. After 45 min of incubation, 5 μM 1NM-PP1 was directly added to cell cultures. Cells were further incubated for 1 hour and harvested for Western blot analysis.
PCR-based homologous recombination test
This assay was performed as previously described (11). Cells were grown in YP media containing 2% raffinose to an OD600 = 1.0 at 30°C. To induce HO endonuclease expression, cell cultures were adjusted to OD600 = 0.5 with YP media containing 2% raffinose, and 2% galactose was directly added. Genomic DNA of each specimen was extracted every 1 hour. PCR analysis of genomic DNA was performed using the following primers: for the detection of DSB generation, 5′-CTTTTAGTTTCAGCTTTCCG-3′ (pI)/5′-ACTCTATAAGGCCAAATGTACAAAC-3′ (pJ); for the detection of strand invasion and primer extension process, 5′-GCAGCACGGAATATGGGACT-3′ (pG)/5′-ATGTGAACCGCATGGGCAGT-3′ (pH); for the detection of ligation process, 5′-AGATGAGTTTAAATCCAGCATACTAG-3′ (pC)/5′-TGTTGTCTCACTATCTTGCCAATAAG-3′ (pD); for the detection of ARG5, 6 as a control DNA region, 5′-CAAGGATCCAGCAAAGTTGGGTGAAGTATGGTA-3′/5′-GAAGGATCCAAATTTGTCTAGTGTGGGAACG-3′.
Measurement of DNA content
Measurement of DNA content was performed as described previously (37). Approximately 1 × 107 cells were harvested and fixed with 5 ml of 70% ethanol for 1 hour at room temperature. The fixed cells were collected and washed once with 1 ml of distilled water and transferred to a microfuge tube. In total, 0.5 ml of 2 mM ribonuclease (RNase) solution [RNase A (2 mg ml−1) in 50 mM tris-Cl (pH 8.0), 15 mM NaCl, boiled for 15 min] was used to treat the harvested cells for 2 to 3 hours at 37°C. After removing the RNase solution, 0.2 ml of pepsin solution [pepsin (5 mg ml−1) in 1 ml of distilled water, 4.5 μl HCl) was directly added to the collected cells for 0.5 to 1 hour at 37°C. Cells were collected by centrifugation and resuspended in 0.5 ml of 50 mM tris-Cl (pH 7.5). This sample was analyzed immediately or stored at 4°C for a few days. To analyze the DNA content, 50 μl of sample solution was added to 1 ml of SYTOX Green solution [1 mM SYTOX Green (Invitrogen) in 50 mM tris-Cl (pH 7.5)], and this mixture was sonicated for a few minutes at a low power. The DNA content was then measured by the FACSCanto flow cytometer (BD Biosciences) using a 488-nm excitation filter and a 510-nm emission filter.
Serial dilution assay
Serial dilution assays for checking DNA damage sensitivity were performed as described in a previous study (37). Cells were grown to OD600 = 1.0 to 2.0 in YPD media and adjusted to OD600 = 0.75 with distilled water for the first dilution. Four 10-fold serial dilutions were prepared by using the first dilution sample. For MMS sensitivity test, 3 μl of five serial dilution samples was subsequently spotted on media plates in the absence or presence of 0.01% MMS and incubated at 30°C for 3 days.
Live-cell imaging and image analysis
Fluorescence microscopy was performed on a Nikon Eclipse Ti inverted microscope and a DeltaVision microscope (Applied Precision) using 100× objective lens with immersion oil. For sample fixation, 1 × 107 cells were harvested and treated with 1 ml of paraformaldehyde solution (4% paraformaldehyde, 3.4% sucrose in distilled water) for 15 min at room temperature. Fixed cells were collected by centrifugation and washed twice with phosphate-buffered saline (PBS). Washed cells were resuspended in 0.5 ml of PBS. Fixed samples were analyzed immediately or stored at 4°C for a few days. To visualize nuclear DNA, the fixed samples were directly treated with 4′,6-diamidino-2-phenylindole (DAPI) (1 μg ml−1) (Invitrogen). A 96-well glass bottom plate (Metrical Bioscience) for the Nikon Eclipse Ti inverted microscope and a coverglass bottom dish (SPL Life Sciences) for the DeltaVision microscope were pretreated with concanavalin A (5 μg ml−1) (Sigma-Aldrich) to allow cells to adhere to the plates. For fluorescence quantification of the BiFC signal and RFP-tagged Rad52, the acquired images were analyzed with ImageJ software (National Institutes of Health). The fluorescence intensity was quantified by manual selection of Rad52 signals that colocalized with DAPI signals, and the background intensity was obtained from a random position that was not colocalized with DAPI signals. The relative fluorescence intensity was calculated by subtracting the background intensity from the fluorescence intensity of Rad52 BiFC or RFP signals.
Time-lapse microscopy of living cells was performed using a DeltaVision microscope. Cells were grown in SC media containing 2% raffinose to an OD600 = 1.0 at 30°C, and 2% galactose was directly added to cell cultures. Cells were adjusted to an OD600 = 0.2 with SC media containing 2% galactose, and 150 μl of cell culture was placed on concanavalin A–treated coverglass bottom dish. Fluorescence images were acquired every 5 min in an environmental chamber of the DeltaVision microscope. To maintain cell growing condition, the environmental chamber was heated to 30°C while time-lapse images were acquired.
Antibodies and immunoblotting
To extract proteins from S. cerevisiae, harvested cells were washed twice with PBS and resuspended in 100 μl of lysis buffer with protease inhibitors and phosphatase inhibitors [150 mM NaCl, 50 mM tris (pH 7.5), 0.15% NP-40, 1 mM phenylmethylsulfonyl fluoride, 1 mM benzamidine, leupeptin (1 μg ml−1), pepstatin (1 μg ml−1), 10 mM sodium fluoride, 20 mM β-glycerolphosphate, 10 mM sodium orthovanadate, and 10 mM sodium pyrophosphate]. Cells were disrupted by bead beating using 0.5-mm glass beads. Lysates were clarified by centrifugation at 14,000 revolutions per minute (RPM) for 10 min on a 4°C precooled microcentrifuge. Protein concentration was determined by Bradford assay. Protein samples were mixed with 6× SDS sample buffer and boiled for 5 min at 95°C. SDS-PAGE was performed with 8% separating gel. For Phos-tag SDS-PAGE, 50 μM Phos-tag and 100 μM MnCl2 were mixed with 6% separating gel. For λ phosphatase assay, lysate was incubated with 600 U of λ phosphatase (New England Biolabs), 1× NEBuffer for protein metallophosphatases, and 1 mM MnCl2 at 30°C for 2 hours.
Immunoblotting was performed using the following antibodies: an anti–phospho–mitogen-activated protein kinase (MAPK)/CDK substrates (Cell Signaling Technology, 23255) for phospho-Rad51, an anti-Rfa1 antibody (Abcam, ab221198) for intact Rfa1, an anti-Rad51 antibody (Abcam, ab63798) for intact Rad51, a horseradish peroxidase (HRP)–conjugated anti–influenza hemagglutinin (HA) antibody (Santa Cruz Biotechnology, SC-7392 HRP) for Rad52-HA, an anti-myc antibody (Santa Cruz Biotechnology, SC-40) for Rad52-myc, an HRP-conjugated anti-GFP antibody (Santa Cruz Biotechnology, SC-9996 HRP) for GFP-tagged proteins, an anti-mouse immunoglobulin G (IgG) antibody (Sigma-Aldrich, A9044) for Cdc28-TAP (tandem affinity purification), an anti–glutathione S-transferase (GST) antibody (Santa Cruz Biotechnology, SC-138) for GST-tagged proteins, an polyclonal anti-GFP antibody (Rockland, 600-103-215) for VN (the N-terminal fragment of Venus)/VC (the C-terminal fragment of Venus)–tagged Rad52, and an anti-hexokinase antibody (Rockland, 100-4159) for endogenous hexokinases.
Coimmunoprecipitation assay
Cells were grown in 100 ml of YPD to OD600 = 1.0. Cells were harvested and washed twice with precooled PBS. Harvested cells were divided into two microfuge tubes and resuspended in 800 μl of lysis buffer with protease inhibitors and phosphatase inhibitors [150 mM NaCl, 50 mM tris (pH 7.5), 0.15% NP-40, 1 mM phenylmethylsulfonyl fluoride, 1 mM benzamidine, leupeptin (1 μg ml−1), pepstatin (1 μg ml−1), 10 mM sodium fluoride, 20 mM β-glycerolphosphate, 10 mM sodium orthovanadate, and 10 mM sodium pyrophosphate] for each tube. Cells were disrupted by bead beating using 0.5-mm glass beads. After clearance of cell debris by centrifugation at 14,000 RPM, 50 μl of aliquot was set aside to make an INPUT sample. For the immunoprecipitation of HA-tagged Rad52, 1.5 ml of aliquot was incubated with 30 μl of anti-HA agarose beads (Sigma-Aldrich) for 4 hours at 4°C. For the immunoprecipitation of myc-tagged Rad52, 1.5 ml of aliquot was incubated with 7 μl of anti-myc antibody for 2 hours at 4°C, and, subsequently, 30 μl of protein A–Sepharose beads (GE Healthcare) was directly added. After an additional 2 hours of incubation at 4°C, the beads in the microfuge tube were collected by centrifugation at 2000 RPM. The beads were washed five times with lysis buffer. Washed beads were mixed with 30 μl of 2× sample buffer and boiled for 5 min at 95°C. SDS-PAGE was performed with 8% separating gel.
ChIP assay
ChIP assay was performed as described previously (37). Cells were grown in 100 ml of YPD to OD600 = 1.0, and 3 ml of paraformaldehyde solution (37.5% paraformaldehyde, 20 mM NaOH in PBS) was treated to cell culture for 15 min. Six milliliters of 2.5 M glycine was treated to cell culture to stop paraformaldehyde fixation, followed by incubation at 30°C for 5 min. Cells were harvested and washed twice with precooled PBS. Harvested cells were divided into two microfuge tubes and resuspended in 400 μl of ChIP lysis buffer [50 mM Hepes/KOH (pH 7.5), 500 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 1 mM phenylmethylsulfonyl fluoride, 1 mM benzamidine, leupeptin (1 μg/ml), and pepstatin (1 μg/ml)] for each tube. Cells were disrupted by bead beating using 0.5-mm glass beads. Lysates were collected in a microfuge tube, and sonication was performed with Sonicator-XL2020 (Misonix) to get sheared chromatin fragments of an average size of 500 to 1000 base pairs. After clearance of cell debris by centrifugation at 14,000 RPM, 50 μl of aliquot was set aside to make an INPUT sample. A total of 750 μl of aliquot was incubated with 7 μl of anti-Rad51 antibody (Abcam) at 4°C for 3 hours, and, subsequently, 30 μl of protein A–Sepharose beads (GE Healthcare) was directly added. After 3 hours of incubation at 4°C, the beads in the microfuge tube were collected by centrifugation at 2000 RPM. The beads were washed twice with 1 ml of ChIP lysis buffer, once with 1 ml of deoxycholate buffer [10 mM tris-Cl (pH 8.0), 250 mM LiCl, 0.5% NP-40, 0.5% sodium deoxycholate, and 1 mM EDTA] and once with 1 ml of TE buffer [10 mM tris-Cl (pH 7.5), 1 mM EDTA]. Washed beads were mixed with 50 μl of TES buffer [50 mM tris-Cl (pH 8.0), 10 mM EDTA, and 1% SDS] and incubated at 65°C for 10 min. By centrifugation at 14,000 RPM, supernatant was collected and transferred to a microfuge tube. Precipitated beads were mixed again with 150 μl of TES buffer and centrifuged. A total of 150 μl of supernatant was transferred to the microfuge tube with first 50 μl of supernatant. Cell lysate for an INPUT sample was also mixed with 150 μl of TES buffer. An INPUT sample and immunoprecipitation sample were incubated at 65°C overnight. Overnight incubated samples were mixed with 200 μl of TE buffer and 12.5 μl of proteinase K (20 mg ml−1) and incubated at 37°C for 2 hours. After proteinase K incubation, 400 μl of phenol/chloroform/isoamyl alcohol (25:24:1) was added to each microfuge tube, and mixtures were centrifuged at 14,000 RPM. A total of 350 μl of supernatant was transferred to a new microfuge tube and mixed with 44 μl of 3 M sodium acetate and 20 μl of glycogen (1 mg ml−1). This mixture was mixed with 1 ml of −20°C precooled ethanol and incubated at −20°C for 10 min. To collect DNA, the mixture was centrifuged at 14,000 RPM for 5 min, and precipitated DNA was washed with −20°C precooled 70% ethanol. Precipitated DNA was resolved to 40 μl of TE buffer including RNase A (200 μg/ml), and DNA solution was incubated at 37°C for 1 hour. Quantitative PCR was performed on QuantStudio 3 (Applied Biosystems) with SensiFAST SYBR Lo-ROX Mix (Bioline). The primers used in the ChIP assay for the MAT locus were 5′-CCTGGTTTTGGTTTTGTAGAGTGG-3′ and 5′-GAGCAAGACGATGGGGAGTTTC-3′ (40), and those for noncentromeric DNA (CUP1) were 5′-TCTTTTCCGCTGAACCGTTCCAGC-3′ and 5′-GGCATTGGCACTCATGACCTTCAT-3′.
In vitro kinase assay
For in vitro kinase assay using Cdc28-as1-TAP, Rad51 and Rad52 variants were purified from Escherichia coli by GST affinity purification, and Cdc28-as1-TAP was purified from S. cerevisiae by immunoprecipitation. Histone H1 (Sigma-Aldrich, H5505) was used as a control substrate. In total, 100 ml of cells expressing either untagged Cdc28 or Cdc28-as1-TAP was cultured in YPD media to an OD600 = 1.0 at 30°C. Cell lysates were incubated with 50 μl of IgG-Sepharose beads (GE Healthcare) at 4°C for 3 hours. Ten microliters of washed IgG-Sepharose beads was incubated with 5 μg of Rad51, Rad52 variants, histone H1, kinase buffer [25 mM Hepes (pH 7.6), 10 mM MgCl2, 5 mM β-glycerolphosphate, and 50 μM ATP in distilled water], and 4 μCi [γ-32P]-ATP at 30°C for 60 min. For 1NM-PP1 treatment, 5 μM of 1NM-PP1 in dimethyl sulfoxide (DMSO) or the same volume of DMSO was added to the kinase reaction mixture. Incorporation of 32P was visualized using BAS-2500 (Bio-Rad).
For in vitro kinase assay using cyclins, cyclins were purified from S. cerevisiae by immunoprecipitation, and Cdc28 was purified from E. coli by GST affinity purification. Histone H1 (Sigma-Aldrich, H5505) was used as a control substrate. In total, 50 ml of cells expressing each cyclin was cultured in YPD media to an OD600 = 1.0 at 30°C. Cell lysates were incubated with 25 μl of IgG-Sepharose beads (GE Healthcare) at 4°C for 3 hours. Ten microliters of washed IgG-Sepharose beads was incubated with 3.5 μg of Cdc28; 5 μg of Rad51, Rad52 variants, histone H1, and kinase buffer; and 4 μCi [γ-32P]-ATP at 30°C for 60 min. Incorporation of 32P was visualized using BAS-2500 (Bio-Rad).
Mass spectrometry
Protein samples were prepared as described in the ChIP assay. Cells were grown in 100 ml of YPD to OD600 = 1.0. Cells were harvested and washed twice with precooled PBS. Harvested cells were divided into two microfuge tubes and resuspended in 800 μl of lysis buffer with protease inhibitors and phosphatase inhibitors [150 mM NaCl, 50 mM tris-Cl (pH 7.5), 0.15% NP-40, 1 mM phenylmethylsulfonyl fluoride, 1 mM benzamidine, leupeptin (1 μg ml−1), pepstatin (1 μg ml−1), 10 mM sodium fluoride, 20 mM β-glycerolphosphate, 10 mM sodium orthovanadate, and 10 mM sodium pyrophosphate] for each tube. Cells were disrupted by bead beating using 0.5-mm glass beads. After clearance of cell debris by centrifugation at 14,000 RPM, 1.5 ml of aliquot was incubated with 30 μl of IgG-Sepharose beads (GE Healthcare) for Rad52-TAP. After incubation at 4°C for 4 hours, the beads in the microfuge tube were collected by centrifugation at 2000 RPM. The beads were washed five times with lysis buffer. Washed beads were mixed with 30 μl of 2× sample buffer and boiled for 5 min at 95°C. SDS-PAGE was performed with 8% separating gel. Precipitated proteins were visualized by Coomassie blue staining. Mass spectrometry analysis was performed by the Proteomics Core Facility at the School of Biological Sciences in Seoul National University, which is supported by the Center for RNA Research, Institute for Basic Science.
Stimulation of DNA ligation
DNA ligation test was performed as described previously with some modifications (26, 29). TAP-tagged Rad52 variants were immunoprecipitated by incubation with 25 μl of IgG-Sepharose beads for 2 hours at 4°C. Rad52 proteins were purified by tobacco etch virus (TEV) protease (Invitrogen) treatment for 2 hours at 30°C. Protein concentration was determined by Bradford assay. Rad52 (1 μM) was incubated for 30 min at 30°C with Sma I–digested, linearized pTZ18R (10 mM) in 20 mM triethanolamine-HCl (pH 7.5). Subsequently, 2 μl of 10× T4 ligation buffer and 1 Weiss unit of T4 DNA ligase (Fermentas) were added directly and incubated at 16°C overnight. Reactions were deproteinized by the addition of 2 ml of stop buffer (2% SDS, 0.6 mg ml−1 proteinase K) and incubation at 37°C for 15 min. Products were analyzed by 0.7% agarose gel electrophoresis using tris-acetate-EDTA buffer containing 1× GelRed Nucleic Acid Gel Stain (Biotium).
Electrophoresis mobility shift assay
Rad51 variants fused to HA through TEV cleavage sequence were immunoprecipitated by incubation with 25 μl of HA-agarose beads for 2 hours at 4°C. Rad51 proteins were purified by TEV protease (Invitrogen) treatment for 2 hours at 30°C. Protein concentration was determined by Bradford assay. To make the ssDNA probe for EMSA, 325 mM ssDNA was incubated with 1× T4 polynucleotide kinase buffer, 20 U of T4 polynucleotide kinase, and 40 μCi [γ-32P]-ATP at 37°C for 30 min (5′-CGGGTGTCGGGGCTGGCTTAACTATGCGGCATCAGAGCAGATTGTACTGAGAGTGCACCATATGCGGTGTGAAATACCGCACAGATGCGT-3′). To make the dsDNA probe for EMSA, the above ssDNA probe was annealed with complementary ssDNA, and isotope labeling was performed as described above. T4 polynucleotide kinase reaction was stopped by boiling at 65°C for 20 min, and the radioactive probe was purified by Prober DNA purification column (iNtRON Biotechnology). Rad51 proteins were mixed with 2 μl of probe and 2 μl of 5× binding buffer [175 mM tris-Cl (pH 7.5), 25 mM ATP, 250 mM KCl, 4 mM MgCl2, 5 mM dithiothreitol, and bovine serum albumin (500 μg ml−1) in distilled water] and incubated at 30°C for 60 min. Products were analyzed by 5% polyacrylamide–tris-borate EDTA (TBE) gel in 0.5× TBE buffer. Incorporation of 32P was visualized using BAS-2500 (Bio-Rad) and Typhoon FLA 7000 (GE Healthcare). For λ phosphatase treatment, the Rad51 protein sample was incubated with 800 U of λ phosphatase (New England Biolabs) at 30°C for 3 hours. For cell cycle phase-dependent EMSA, cultures were treated with 150 μM α-factor and nocodazole (15 μg ml−1) for the G1 and G2/M phase arrest, respectively.
Electron microscopy
Electron microscopy was performed as described previously with some modifications (26). Rad52 variants were purified from E. coli by GST affinity purification. Purified proteins were diluted with a buffer containing 20 mM tris-Cl (pH 7.5), 100 mM KCl, 5% glycerol, 5 mM β-mercaptoethanol, and 0.1 mM EDTA. Three microliters of aliquot of the sample was applied to a 400-mesh grid coated with carbon film that had been freshly glow discharged. The grid was stained with 2% uranyl acetate for 1 min. Transmission electron microscopy images of Rad52 complex were obtained by using a Talos L120C transmission electron microscope (Thermo Fisher Scientific).
Statistics
On the basis of previous literature, we assumed that the data such as Fig. 3 (C, right panel, and D, right panel) have a normal distribution, and the data such as Figs. 4D, 5B (right panel), and fig. S7C (right panel) have a non-normal distribution. Two-sided Student’s t test was used for Fig. 3C (right panel). Mann-Whitney U test was used for non-normal distribution data such as Figs. 4D, 5B (right panel), and fig. S7C (right panel). One-sample t test was used for normalized data by specified column such as Fig. 3D (right panel). Statistical tests were also indicated in individual figure legends and supplementary figure legends.
Figure 3C (right panel)
For RAD51, n = 203, 191, and 201 (at 0 hour) and n = 149, 130, and 105 (at 3 hours) cells were analyzed in each independent experiment. For rad51-2A, n = 166, 160, and 189 (at 0 hour) and n = 158, 126, and 125 (at 3 hours) cells were analyzed in each independent experiment. The data represent the means ± SD of triplicate experiments.
Figure 3D (right panel)
Extracts from three independent experiments were analyzed for each strain. The data represent the means ± SD of triplicate experiments. P values were determined by the one-sample t test. Immunoprecipitated MAT locus DNA in Rad51 strain column was used as a hypothetical mean.
Figure 4D
For the RAD52 strain, a total of 200 cells were analyzed. For the rad52-T412A strain, a total of 200 cells were analyzed. For the rad52-T412E strain, a total of 200 cells were analyzed. Box plot was normalized by the median of Rad52 cell with whiskers from the 5th to the 95th percentile. P values were determined by the Mann-Whitney U test.
Figure 5B (right panel)
For the RAD52 strain, a total of 207 cells were analyzed. For the rad52-T412A strain, a total of 204 cells were analyzed. Box plot was normalized by the median of the RAD52 strain with whiskers from the 5th to the 95th percentile. P values were determined by the Mann-Whitney U test.
Figure S7C (right panel)
For the RAD52 strain, a total of 218 cells were analyzed. For the rad52-T412A strain, a total of 207 cells were analyzed. Box plot was normalized by the median of the RAD52 strain with whiskers from the 5th to the 95th percentile. P values were determined by the Mann-Whitney U test.
Supplementary Material
Acknowledgments
We thank Chaok Seok for technical assistance in the interpretation of Rad51 structure and the members of our laboratory for the helpful discussions. LC-MS/MS analysis was performed by the Proteomics Core Facility at the School of Biological Sciences in Seoul National University, which is supported by the Center for RNA Research, Institute for Basic Science. Funding: This work was supported by the National Research Foundation of Korea (grant nos. 2015R1A2A1A01007871 and 2018R1A2B2009169), funded by the Ministry of Education, Science and Technology, Republic of Korea. Author contributions: G.L. and W.-K.H. designed the research; G.L. and Y.C. performed the research; G.L. and W.-K.H. analyzed the data; and G.L. and W.K.H. wrote the manuscript. Competing Interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/6/eaay2669/DC1
Fig. S1. Deletion of either RAD51 or RAD52 impairs DNA damage repair process.
Fig. S2. Deletion of either CLB2 or CLB3 affects the phosphorylation of Rad51 and Rad52 in cells.
Fig. S3. CDK1 phosphorylates S125 and S375 of Rad51 and T412 of Rad52 in cells.
Fig. S4. Nonphosphorylatable mutation of Rad51 impairs the strand invasion process even in the G2/M phase–arrested cells.
Fig. S5. CDK1-dependent phosphorylation regulates the DNA binding affinity of Rad51.
Fig. S6. Nonphosphorylatable mutation of Rad52 impairs ligation process even in the G2/M phase–arrested cells.
Fig. S7. Both wild-type Rad52 and Rad52-T412A form ring structures.
Fig. S8. Rapamycin treatment does not restore the defect in the ligation process in cells that express Rad52-T412A protein without FKBP or FRB attachment.
Fig. S9. Full images of Western blots.
Table S1. Yeast strains used in this study.
REFERENCES AND NOTES
- 1.Symington L. S., Rothstein R., Lisby M., Mechanisms and regulation of mitotic recombination in Saccharomyces cerevisiae. Genetics 198, 795–835 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mathiasen D. P., Lisby M., Cell cycle regulation of homologous recombination in Saccharomyces cerevisiae. FEMS Microbiol. Rev. 38, 172–184 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Michael A. E. H., Mendenhall D., Regulation of Cdc28 cyclin-dependent protein kinase activity during the cell cycle of the yeast Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 62, 1191–1243 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bloom J., Cross F. R., Multiple levels of cyclin specificity in cell-cycle control. Nat. Rev. Mol. Cell Biol. 8, 149–160 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Hanamshet K., Mazina O. M., Mazin A. V., Reappearance from obscurity: Mammalian Rad52 in homologous recombination. Genes 7, E63 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ira G., Pellicioli A., Balijja A., Wang X., Fiorani S., Carotenuto W., Liberi G., Bressan D., Wan L., Hollingsworth N. M., Haber J. E., Foiani M., DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 431, 1011–1017 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aylon Y., Liefshitz B., Kupiec M., The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. EMBO J. 23, 4868–4875 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huertas P., Cortés-Ledesma F., Sartori A. A., Aguilera A., Jackson S. P., CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature 455, 689–692 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen X., Niu H., Chung W.-H., Zhu Z., Papusha A., Shim E. Y., Lee S. E., Sung P., Ira G., Cell cycle regulation of DNA double-strand break end resection by Cdk1-dependent Dna2 phosphorylation. Nat. Struct. Mol. Biol. 18, 1015–1019 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee C.-S., Haber J. E., Mating-type gene switching in Saccharomyces cerevisiae. Microbiol. Spectr. 3, MDNA3-0013-2014 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Ohuchi T., Seki M., Branzei D., Maeda D., Ui A., Ogiwara H., Tada S., Enomoto T., Rad52 sumoylation and its involvement in the efficient induction of homologous recombination. DNA Repair 7, 879–889 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Bishop A. C., Ubersax J. A., Petsch D. T., Matheos D. P., Gray N. S., Blethrow J., Shimizu E., Tsien J. Z., Schultz P. G., Rose M. D., Wood J. L., Morgan D. O., Shokat K. M., A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature 407, 395–401 (2000). [DOI] [PubMed] [Google Scholar]
- 13.Nguyen Ba A. N., Moses A. M., Evolution of characterized phosphorylation sites in budding yeast. Mol. Biol. Evol. 27, 2027–2037 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Harvey S. L., Charlet A., Haas W., Gygi S. P., Kellogg D. R., Cdk1-dependent regulation of the mitotic inhibitor Wee1. Cell 122, 407–420 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Suzuki K., Sako K., Akiyama K., Isoda M., Senoo C., Nakajo N., Sagata N., Identification of non-Ser/Thr-Pro consensus motifs for Cdk1 and their roles in mitotic regulation of C2H2 zinc finger proteins and Ect2. Sci. Rep. 5, 7929 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xue Y., Ren J., Gao X., Jin C., Wen L., Yao X., GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol. Cell. Proteomics 7, 1598–1608 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hunter T., Why nature chose phosphate to modify proteins. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367, 2513–2516 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sung P., Krejci L., Van Komen S., Sehorn M. G., Rad51 recombinase and recombination mediators. J. Biol. Chem. 278, 42729–42732 (2003). [DOI] [PubMed] [Google Scholar]
- 19.Sugiyama T., Kowalczykowski S. C., Rad52 protein associates with replication protein A (RPA)-single-stranded DNA to accelerate Rad51-mediated displacement of RPA and presynaptic complex formation. J. Biol. Chem. 277, 31663–31672 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Lisby M., Rothstein R., Mortensen U. H., Rad52 forms DNA repair and recombination centers during S phase. Proc. Natl. Acad. Sci. U.S.A. 98, 8276–8282 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krejci L., Song B., Bussen W., Rothstein R., Mortensen U. H., Sung P., Interaction with Rad51 is indispensable for recombination mediator function of Rad52. J. Biol. Chem. 277, 40132–40141 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Lisby M., Barlow J. H., Burgess R. C., Rothstein R., Choreography of the DNA damage response: Spatiotemporal relationships among checkpoint and repair proteins. Cell 118, 699–713 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Shinohara A., Shinohara M., Ohta T., Matsuda S., Ogawa T., Rad52 forms ring structures and co-operates with RPA in single-strand DNA annealing. Genes Cells 3, 145–156 (1998). [DOI] [PubMed] [Google Scholar]
- 24.Stasiak A. Z., Larquet E., Stasiak A., Müller S., Engel A., Van Dyck E., West S. C., Egelman E. H., The human Rad52 protein exists as a heptameric ring. Curr. Biol. 10, 337–340 (2000). [DOI] [PubMed] [Google Scholar]
- 25.Ranatunga W., Jackson D., Lloyd J. A., Forget A. L., Knight K. L., Borgstahl G. E. O., Human RAD52 exhibits two modes of self-association. J. Biol. Chem. 276, 15876–15880 (2001). [DOI] [PubMed] [Google Scholar]
- 26.Lloyd J. A., Forget A. L., Knight K. L., Correlation of biochemical properties with the oligomeric state of human rad52 protein. J. Biol. Chem. 277, 46172–46178 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Plate I., Albertsen L., Lisby M., Hallwyl S. C. L., Feng Q., Seong C., Rothstein R., Sung P., Mortensen U. H., Rad52 multimerization is important for its nuclear localization in Saccharomyces cerevisiae. DNA Repair 7, 57–66 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saotome M., Saito K., Yasuda T., Ohtomo H., Sugiyama S., Nishimura Y., Kurumizaka H., Kagawa W., Structural basis of homology-directed DNA repair mediated by RAD52. iScience 3, 50–62 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van Dyck E., Stasiak A. Z., Stasiak A., West S. C., Binding of double-strand breaks in DNA by human Rad52 protein. Nature 398, 728–731 (1999). [DOI] [PubMed] [Google Scholar]
- 30.Wu Y., Kantake N., Sugiyama T., Kowalczykowski S. C., Rad51 protein controls Rad52-mediated DNA annealing. J. Biol. Chem. 283, 14883–14892 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nassif N., Penney J., Pal S., Engels W. R., Gloor G. B., Efficient copying of nonhomologous sequences from ectopic sites via P-element-induced gap repair. Mol. Cell. Biol. 14, 1613–1625 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davis A. P., Symington L. S., The yeast recombinational repair protein Rad59 interacts with Rad52 and stimulates single-strand annealing. Genetics 159, 515–525 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McIlwraith M. J., West S. C., DNA repair synthesis facilitates RAD52-mediated second-end capture during DSB repair. Mol. Cell 29, 510–516 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Sung M.-K., Huh W.-K., Bimolecular fluorescence complementation analysis system for in vivo detection of protein-protein interaction in Saccharomyces cerevisiae. Yeast 24, 767–775 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Gallego O., Specht T., Brach T., Kumar A., Gavin A.-C., Kaksonen M., Detection and characterization of protein interactions in vivo by a simple live-cell imaging method. PLOS ONE 8, e62195 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bhargava R., Onyango D. O., Stark J. M., Regulation of single-strand annealing and its role in genome maintenance. Trends Genet. 32, 566–575 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lim G., Huh W.-K., Rad52 phosphorylation by Ipl1 and Mps1 contributes to Mps1 kinetochore localization and spindle assembly checkpoint regulation. Proc. Natl. Acad. Sci. U.S.A. 114, E9261–E9270 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luchnik A. N., Glaser V. M., Shestakov S. V., Repair of DNA double-strand breaks requires two homologous DNA duplexes. Mol. Biol. Rep. 3, 437–442 (1977). [DOI] [PubMed] [Google Scholar]
- 39.Lee P. S., Petes T. D., Mitotic gene conversion events induced in G1-synchronized yeast cells by gamma rays are similar to spontaneous conversion events. Proc. Natl. Acad. Sci. U.S.A. 107, 7383–7388 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saponaro M., Callahan D., Zheng X., Krejci L., Haber J. E., Klein H. L., Liberi G., Cdk1 targets Srs2 to complete synthesis-dependent strand annealing and to promote recombinational repair. PLOS Genet. 6, e1000858 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/6/eaay2669/DC1
Fig. S1. Deletion of either RAD51 or RAD52 impairs DNA damage repair process.
Fig. S2. Deletion of either CLB2 or CLB3 affects the phosphorylation of Rad51 and Rad52 in cells.
Fig. S3. CDK1 phosphorylates S125 and S375 of Rad51 and T412 of Rad52 in cells.
Fig. S4. Nonphosphorylatable mutation of Rad51 impairs the strand invasion process even in the G2/M phase–arrested cells.
Fig. S5. CDK1-dependent phosphorylation regulates the DNA binding affinity of Rad51.
Fig. S6. Nonphosphorylatable mutation of Rad52 impairs ligation process even in the G2/M phase–arrested cells.
Fig. S7. Both wild-type Rad52 and Rad52-T412A form ring structures.
Fig. S8. Rapamycin treatment does not restore the defect in the ligation process in cells that express Rad52-T412A protein without FKBP or FRB attachment.
Fig. S9. Full images of Western blots.
Table S1. Yeast strains used in this study.