

Abstract
Methylmercury (MeHg) is a pervasive environmental toxicant, with known detrimental effects on neurodevelopment. Despite a longstanding paradigm of neurotoxicity, where motor deficits are prevalent among those developmentally exposed, consideration of muscle as a MeHg target has received minimal investigation. Recent evidence has identified muscle-specific gene networks that modulate developmental sensitivity to MeHg toxicity. One such network is muscle cell differentiation. Muscle cell differentiation is a coordinated process regulated by the myogenic regulatory factors (MRFs): Myf5, MyoD, MyoG, and MRF4. A previous study demonstrated that MeHg inhibits muscle cell differentiation in vitro, concurrent with reduced MyoG expression. The potential for MeHg to modify the temporal expression of the MRFs to alter differentiation, however, has yet to be fully explored. Using the C2C12 mouse myoblast model, we examined MRF expression profiles at various stages subsequent to MeHg exposure to proliferating myoblasts. MeHg was seen to persistently alter myoblast differentiation capacity, as myod, myog, and mrf4 gene expression were all affected. Myog exhibited the most robust changes in expression across the various culture conditions, while myf5 was unaffected. Following MeHg exposure to myoblasts, where elevated p21 expression indicated departure from proliferation, cells failed to subsequently differentiate, even in the absence of MeHg, as reflected by a concurrent reduction in the MRF4 and myosin heavy chain (MHC) markers of terminal differentiation. Our results indicate that within a brief window of exposure MeHg can disrupt the intrinsic myogenic differentiation program of proliferative myoblasts.
Keywords: methylmercury, muscle, myogenic regulatory factors, cell differentiation
Introduction
Methylmercury (MeHg) is a persistent environmental toxicant. Exposure to humans occurs primarily via consumption of MeHg contaminated seafood. Accidental high-dose exposure incidents have underscored the detrimental effects of MeHg in humans, particularly on neurodevelopment (Bakir, Damluji et al. 1973, Harada 1978). As such, MeHg research has predominantly focused on the nervous system as the primary target of toxicity. Some epidemiological studies in populations that subsist on seafood have found that in utero exposed children exhibit neurodevelopmental deficits (Debes, Weihe et al. 2016, Engstrom, Love et al. 2016, Tatsuta, Murata et al. 2017). Among these deficits, poorer motor skills (Grandjean, Weihe et al. 1998) were observed, indicating potential neuromuscular targets for MeHg. The potential for MeHg to disrupt muscle development has received little attention. A previous study in rats demonstrated that MeHg could accumulate in muscle tissue consequent to in utero exposure (Cambier, Fujimura et al. 2018). Furthermore, developmental MeHg exposure in Drosophila resulted in abnormal muscle morphology (Engel and Rand 2014, Prince and Rand 2017). The precise mechanism by which MeHg elicits toxicity on muscle development, however, remains unexplored.
A genome-wide association study in Drosophila revealed several muscle-specific gene networks that associate with tolerance or susceptibility to developmental MeHg exposure. One such network identified in this study was muscle cell differentiation (Montgomery, Vorojeikina et al. 2014). Muscle cell differentiation is a coordinated process, regulated by the myogenic regulatory factors (MRFs): Myf5, MyoD, MyoG, and MRF4 (Zammit 2017). Whereas each of these factor have a unique influence on myogenesis, Myf5 and MyoD are generally thought of as determinants of myogenic potential while MyoG and MRF4 are known to mediate steps toward terminal differentiation of myocytes into myotubes and fibers (Singh and Dilworth 2013). Accordingly, the MRF transcription factors are temporally expressed as differentiation proceeds from myoblasts to myotubes. Myf5 is expressed in myoblasts, and is typically downregulated in response to differentiation cues. MyoD is persistently expressed in myoblasts as well as myocytes (Ishibashi, Perry et al. 2005). As differentiation is induced MyoG is upregulated (Andres and Walsh 1996) under the control of MyoD (Faralli and Dilworth 2012). Finally, MRF4 is expressed as myotubes form, which also propagates expression of myosin heavy chain (MHC) (Hinterberger, Sassoon et al. 1991). The extent to which MeHg can influence MRFs has yet to be fully investigated. Nonetheless, the highly coordinated temporal regulation of these MRFs highlights the possibility that a discrete window of susceptibility for MeHg toxicity may exist where one of these factors is preferentially targeted.
In a prior study, using differentiating mouse C2C12 myoblasts in vitro, we demonstrated a robust effect of MeHg in reducing MyoG transcripts that accompanied a strong inhibition of differentiation to myocytes and fusion into myotubes (Prince and Rand 2018). Moreover, this effect correlated with decreased nuclear MyoG (Prince and Rand 2018). Notably, MyoD expression was seen to be little affected by MeHg (Prince and Rand 2018). These findings are consistent with limited studies of MeHg effects on muscle in vivo that have demonstrated a decrease muscle fiber size in rats (Usuki, Yasutake et al. 1998) and zebrafish (de Oliveira Ribeiro, Nathalie et al. 2008). While these results support the idea that MeHg might inhibit myoblast differentiation, potentially via select action on MyoG, the influence of MeHg on other MRFs remains unexplored. In addition to the MRFs, the cyclin-dependent kinase inhibitor p21 demonstrates marked change in expression with an increase in myoblasts that are induced to differentiate (Guo, Wang et al. 1995). MeHg effects on p21 in muscle cells has also not been fully examined.
The C2C12 mouse myoblast model offers exceptional control over the study of muscle cell differentiation mechanisms. Proliferative myoblasts can be maintained in high serum (e.g. 20% fetal bovine serum) culture conditions and synchronously induced to differentiate by a shift to low serum containing media (e.g. 2% horse serum). In a prior study we investigated the consequences of the MeHg insult occurring contemporaneously with onset of differentiation, e.g. in a low serum condition (Prince and Rand 2018). In the present work, we aimed to determine if the potent effects of MeHg on myogenic differentiation can be implemented at an earlier stage and in optimal growth conditions (e.g. high serum) that are more representative of myogenic cells in vivo (e.g. in the fetal myotome or in the adult satellite cell niche). Here we show that MeHg exposure to undifferentiated myoblasts uniquely modifies MRF expression and significantly impairs their capacity to subsequently differentiate.
Materials and Methods
Cell culture
C2C12 cells were purchased from ATCC (CRL-1772, Lot # 61633507). Cells were cultured in growth media, Dulbecco’s Modified Eagle Medium (DMEM, Gibco #11995-065) supplemented with 20% fetal bovine serum (FBS, Gibco #10082147) and 1% Penicillin/Streptomycin (PenStrep, Gibco #15140-122). Initially, cells were thawed and seeded in T75 cm2 flasks (Falcon #353136). After three days, cells reached approximately 60% confluency, and were passaged into appropriate tissue culture ware (~1.25×104 cells/ml) for experiments. Once again, cells reached approximately 60% confluency, and were treated as detailed below. To promote differentiation, cells were cultured in differentiation media, DMEM supplemented with 2% horse serum (HS, ATCC #30-2040), 1 µg/ml insulin (Sigma #I6634), and 1% PenStrep.
Treatment
Methylmercury Chloride (MeHg, Sigma #215465) was prepared as a 50 mM stock in dimethyl sulfoxide (DMSO, Fisher #BP231), and stored at −20°C. Stocks were then diluted in growth media prior to treatment, so that the final DMSO concentration was < 0.01%. Cells were treated in growth media ± MeHg for 24 hr, then either collected (myoblasts) or switched to differentiation media without MeHg for an additional 24 (myocytes) or 48 hr (myotubes) (Figure 1).
Figure 1. Experimental paradigm.
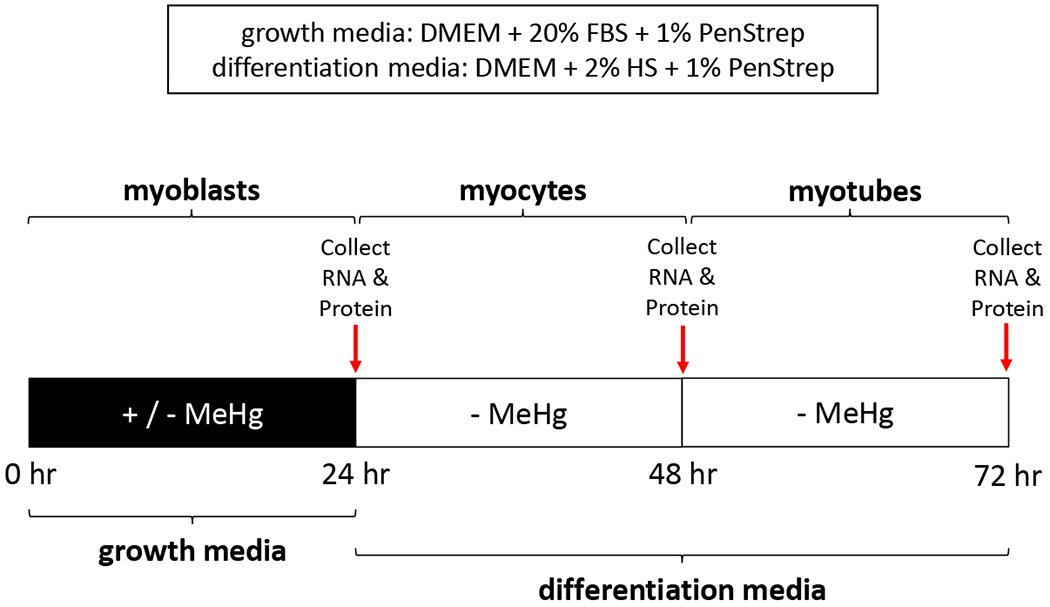
Culture conditions were designed to evaluate MeHg effects implemented in the growth phase on subsequent C2C12 differentiation stages. In all conditions, cell were exposed in growth media ± MeHg for 24 hr. Myoblast RNA and protein was immediately collected after this 24 hr exposure. Subsequent to a switch to differentiation media without MeHg for 24 or 48 hours, RNA and protein was collected at the myocyte and myotube stage, respectively .
Cell viability
Cells were seeded in 6-well plates (Corning #3516) for MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, Sigma # M2128) assay. A 5 mg/ml MTT stock was made in Hank’s Balanced Salt Solution (HBSS, Gibco #14175-095), aliquoted in light sensitive Eppendorf tubes, and stored at −20°C. Stocks were thawed at room temperature prior to experiments. Treatment media was removed, and cells washed with warm HBSS. The appropriate media (growth or differentiation) without MeHg was added back, as well as MTT at a final concentration of 0.25 mg/ml. Cells were incubated for 2 hr at 37°C, the media + MTT removed, and Sorensen’s Buffer (0.1 M glycine, 0.1 M NaOH; pH 10.5) + DMSO added. Absorbance was measured at 570 nm (corrected for background absorbance at 690 nm). Cell viability is reported as percent (%) of control (0 µM MeHg).
Real-time PCR
Cells were seeded in 60 mm petri dishes (Falcon #353004) for RNA isolation. Total RNA was isolated using the Qiagen RNeasy® Mini Kit (Qiagen #74104), and converted to cDNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems #4368813). Real-time PCR was done using iTaq Universal SYBR® Green Supermix (Bio-Rad #1725121) and appropriate forward/reverse primers (Supplemental Table 1). Bio-Rad CFX96™ Real-Time PCR System was programmed according to manufacturer instructions, and quantified cycle number (Cq) for genes of interest (myf5, myod, myog, mrf4) and a housekeeping gene (gapdh) were acquired for subsequent analysis.
Western Blot
Cells were plated in 60 mm petri dishes for whole-cell lysate preparation. Briefly, cells were lysed in Pierce™ RIPA buffer (Thermo #89900) + Halt Protease Inhibitor Cocktail EDTA-free (Thermo #87785), and frozen at −80°C prior to sample preparation. Western blot samples were prepared in 2X Laemmli sample buffer (Bio-Rad #161-0737) + 2-mercaptoethanol (Fisher #BP176) for a final protein concentration of 1 µg/µl, and boiled at 100°C. Samples and the Precision Plus Protein™ Kaleidoscope™ (Bio-Rad #161-0375) ladder were run on 12% polyacrylamide gels (30% acrylamide, 1.5 M Tris HCl, 10% SDS, ddH2O, 10% APS, TEMED). Gels were transferred overnight at 4°C to polyvinylidene difluoride (PVDF, Millipore #IPVH00010) membranes, and stained with Ponceau S (Fisher #BP103-10) to confirm protein transfer. Membranes were blocked in 5% bovine serum albumin (Fisher #BP1600-100), then incubated overnight at 4°C with appropriate primary antibodies. p21 (#556431) and MyoD (#554130) were purchased from BD Pharmigen; MyoG (#F5D) and MHC (#MF-20) were purchased from Developmental Studies Hybridoma Bank; and actin (#A5441) was purchased from Sigma. Membranes were washed, then incubated for 1 hr at room temperature with peroxidase-conjugated goat-anti mouse secondary (Jackson ImmunoResearch #115-035-146). Protein bands were visualized with Clarity™ Western ECL Substrate (Bio-Rad #170-5060), and imaged on the Bio-Rad Chemi-Doc™ MP Imaging System. ImageJ (NIH) was used for band intensity analysis.
Data Analysis
Data are presented as mean ± standard deviation (SD). For all experiments, at least three biological replicates were completed. Each biological replicate represents a newly thawed from frozen vial of C2C12 cells and one passage prior to treatment and experimentation. To calculate delta Ct (ΔCt), the Cq value for gapdh was subtracted from the Cq value for myf5, myod, myog, or mrf4. The 2-ΔΔCt method was used to determine fold change (Livak and Schmittgen 2001). p21, MyoD, MyoG or MHC band intensity was normalized to actin prior to comparison across MeHg concentrations. Shapiro-Wilk Test for Normality and Levene’s Test for Equal Variances were applied to all data. One-way analysis of variance (ANOVA) with Tukey’s Honestly Significant Difference (HSD) or Kruskal-Wallis with Dunn’s Multiple Comparisons, Bonferroni correction were determined in R with significance at p < 0.05. For delta Ct, untreated myocytes or myotubes were compared to untreated myoblasts only, while for all other analysis 0.5 µM or 2.5 µM MeHg were compared to untreated (0 µM MeHg) controls.
Results
Temporal profile of myogenic regulatory factor gene expression in C2C12 cells
First, we sought to establish the baseline profile of MRF expression spanning the transition from myoblasts to myocytes to myotubes during differentiation in the absence of MeHg. MRF gene expression patterns were examined by qPCR at time-points according to our culture conditions (Figure 1) and the results are expressed as delta-Ct values in Table 1. Expression of myf5 in the myocytes was moderately reduced compared to the myoblasts, although not quite significantly (p = 0.06). Unexpectedly, myf5 expression in the myotubes was seen to return to levels near that of myoblasts. Myod expression was moderately enhanced in the myocytes (p < 0.001) as well as the myotubes (p < 0.01) compared to myoblasts. In contrast, Myog expression was substantially increased in the myocytes (p < 0.01) and in the myotubes, however, a skewed distribution in the myoblasts ΔCt values resulted in a statistically insignificant change (p = 0.15) in the latter. Mrf4 expression was increased in the myotubes (p < 0.01), while showing a moderate insignificant decrease in the myocytes.
Table 1.
MRFs delta Ct (ΔCt) values
| Gene | Myoblasts | Myocytes | Myotubes |
|---|---|---|---|
| myf5 | 5.14 ± 0.66 | 6.07 ± 0.17 | 5.27 ± 0.45 |
| myod | 3.96 ± 0.26 | 3.05 ± 0.37a | 3.20 ± 0.16b |
| myog | 5.53 ± 2.32 | 1.59 ± 0.54c | 2.10 ± 0.81 |
| mrf4 | 11.35 ± 0.59 | 12.36 ± 0.45 | 9.36 ± 0.96b |
mean ± SD; n ≥ 3
one-way ANOVA, Tukey’s HSD; p < 0.001
one-way AVOVA, Tukey’s HSD; p < 0.01
Kruskal-Wallis, Dunn’s Multiple Comparisons; p < 0.01
C2C12 mouse myoblast cell viability
Next, we assessed cell viability of the C2C12 cells by MTT assay to identify sub-toxic and toxic MeHg concentrations for our experiments. No significant decrease in cell viability at 0.5 µM MeHg in myoblasts (100.6 ± 10.5%, n = 3; p = 1.00 (Figure 2A)), myocytes (102.5 ± 26.3%, n = 3; p = 0.98 (Figure 2B)), or myotubes (102.3 ± 8.6%, n = 3; p = 0.98 (Figure 2C)) was observed. However, 2.5 µM MeHg produced a significant decrease in cell viability in myoblasts (67.0 ± 11.7%, n = 3; p < 0.05 (Figure 2A)), myocytes (43.6 ± 4.4%, n = 3; p < 0.05 (Figure 2B)), as well as myotubes (50.5 ± 19.6%, n = 3; p < 0.01 (Figure 2C)). Therefore, we evaluated 0.5 µM and 2.5 µM MeHg in all our subsequent experiments.
Figure 2. Cell viability.
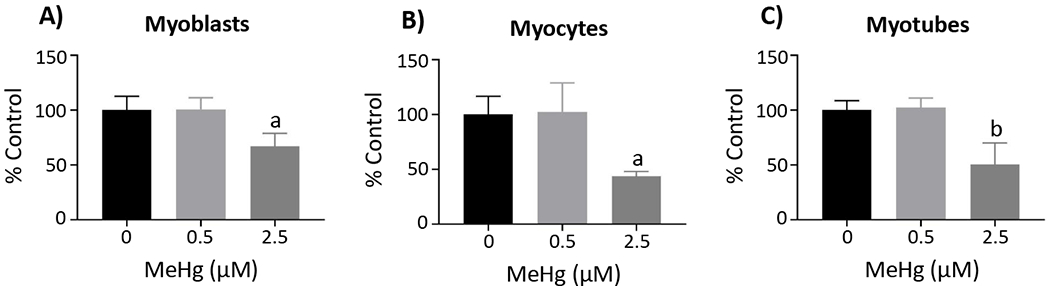
Cell viability was assessed by MTT assay in the myoblasts (A), myocytes (B), and myotubes (C). Data are reported as percent (%) control, and represent the mean ± SD; aone-way ANOVA, Tukey’s HSD, p < 0.05; bone-way ANOVA, Tukey’s HSD, p < 0.01
MeHg effects on myogenic regulatory factor gene expression
We then examined MeHg effects on MRF gene expression (Figure 3 and Supplemental Table 2). Relative to the untreated controls, there were no significant effects on MRF gene expression with 0.5 µM MeHg. Further, myf5 was not significantly altered at 2.5 µM MeHg in the myoblasts, myocytes, or myotubes (Supplemental Table 2). With 2.5µM MeHg, myod (0.67 ± 0.16, n = 4; p <0.05 (Figure 3A)) and myog (0.16 ± 0.07, n = 4; p < 0.001 (Figure 3B)) expression was significantly decreased in the myoblasts. myog expression was also significantly decreased in the myocytes with 2.5µM MeHg, although to a lesser extent (0.48 ± 0.31, n = 5; p < 0.01 (Figure 3D)); whereas myod expression was not significantly lower (0.68 ± 0.19, n = 4; p = 0.26 (Figure 3C)). With 2.5µM MeHg Mrf4 expression was significantly decreased in the myotubes (0.44 ± 0.13, n = 3; p < 0.01 (Figure 3E)). Curiously, with 2.5µM MeHg expression of myod and myog were not significantly altered in the myotubes (Supplemental Table 2).
Figure 3. MeHg effects on MRF gene expression.
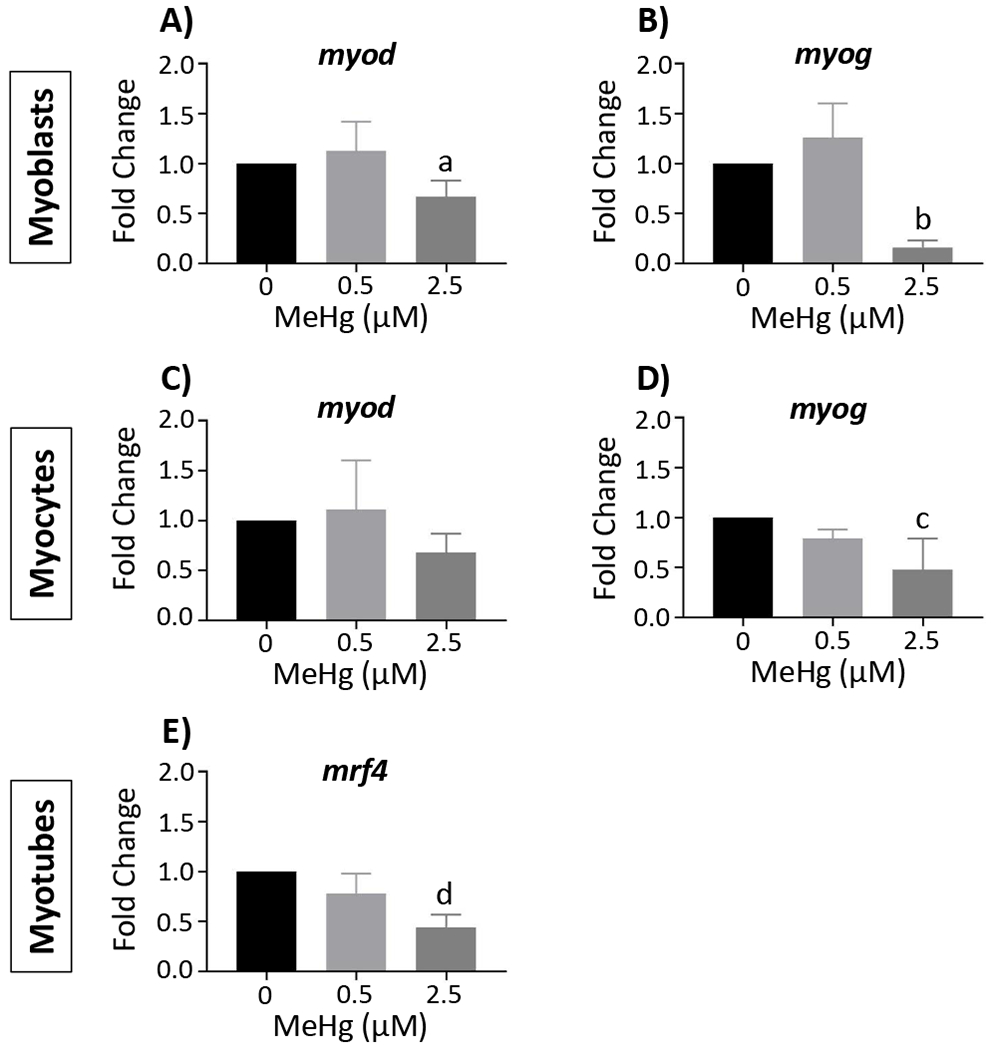
Gene expression was determined by real-time qPCR. Myod and myog expression are presented in the myoblasts (A and B) and myocytes (C and D), respectively, while mrf4 expression is presented in the myotubes (E). Data are reported as fold change, and represent the mean ± SD; aone-way ANOVA, Tukey’s HSD, p < 0.05; bone-way ANOVA, Tukey’s HSD, p < 0.001; cKruskal-Wallis, Dunn’s Multiple Comparisons, p < 0.01; done-way ANOVA, Tukey’s HSD, p < 0.01 (See also suppl. Table 2)
Relative protein level of differentiation factors in C2C12 mouse myoblasts
Before assessing MeHg effects on proteins involved in myoblast differentiation, we measured the relative protein level of these factors in our culture conditions without MeHg and the results are seen in Table 2. First, we included examination of the p21 protein as marker of proliferation status. p21 was significantly increased in the myocytes (p < 0.05) relative to myoblasts; however, levels in myotubes were nearly equivalent to those in the myoblasts . MyoD protein level was unchanged in the myocytes, yet was decreased in the myotubes compared to the myoblasts, albeit without significance (p = 0.07). However, relative to the myocytes, MyoD was significantly decreased in the myotubes (p < 0.01). MyoG protein was significantly increased in the myocytes (p < 0.001) and myotubes (p < 0.05) compared to the myoblasts; however, relative to the myocytes, MyoG was increased to a lesser extent in the myotubes (p < 0.05). MHC levels could be determined in the myotubes, but were absent in the myoblasts or the myocytes.
Table 2.
Relative total protein level (normalized to β-actin)
| Protein | Myoblasts | Myocytes | Myotubes |
|---|---|---|---|
| p21 | 0.93 ± 0.06 | 1.72 ± 0.05a | 1.03 ± 0.09 |
| MyoD | 1.24 ± 0.29 | 1.83 ± 0.67 | 0.28 ± 0.15 |
| MyoG | 0.57 ± 0.17 | 1.94 ± 0.37b | 1.19 ± 0.04c |
| MHC | ND* | ND* | 2.03 ± 0.28 |
mean ± SD; n = 3
ND, not detected
Kruskal-Wallis, Dunn’s Multiple Comparisons; p < 0.05
one-way ANOVA, Tukey’s HSD; p < 0.001
one-way ANOVA, Tukey’s HSD; p <0.05
MeHg effects on protein levels of differentiation factors
To compliment transcript expression analyses, we evaluated MeHg effects on corresponding MRF protein levels, together with p21 protein, in myoblasts, myocytes and myotubes (Figures 4, 5 and 6, respectively). In the myoblasts, there were no significant effects on protein levels for any of the factors at 0.5 µM MeHg (Figure 4). With 2.5µM MeHg, p21 was significantly increased (1.94 ± 0.35, n = 3; p < 0.01 (Figs. 4A and B)) in myoblasts, while MyoG was significantly decreased (0.21 ± 0.11, n = 3; p < 0.05 (Figs. 4A and D)) compared to untreated myoblasts. MyoD levels were unaltered by MeHg in the myoblasts (Figs. 4A and C). In the myocytes, MyoG was significantly decreased with 0.5 µM (0.66 ± 0.02, n = 3; p < 0.05) and 2.5 µM MeHg (0.20 ± 0.12, n = 3; p < 0.001) (Figs. 5A and D). p21 was also decreased with 0.5 µM (0.62 ± 0.10, n = 3; p = 0.22) and 2.5 µM MeHg (0.46 ± 0.30, n = 3; p = 0.08), although not quite significantly (Figs. 5A and B). Again, MyoD levels were unaffected by MeHg in the myocytes (Figs. 5A and C). Interestingly, in the myotubes, p21 (Figs. 6A and B), MyoD (Figs. 6A and C), and MyoG (Figs. 6A and D) were not altered at either MeHg concentration. In contrast, MHC was significantly decreased with 0.5 µM (0.56 ± 0.11, n = 3; p < 0.01) and drastically decreased at 2.5 µM MeHg (0.02 ± 0.02, n = 3; p < 0.001) (Figs. 6A and E).
Figure 4. MeHg effects on protein levels of differentiation factors in myoblasts.
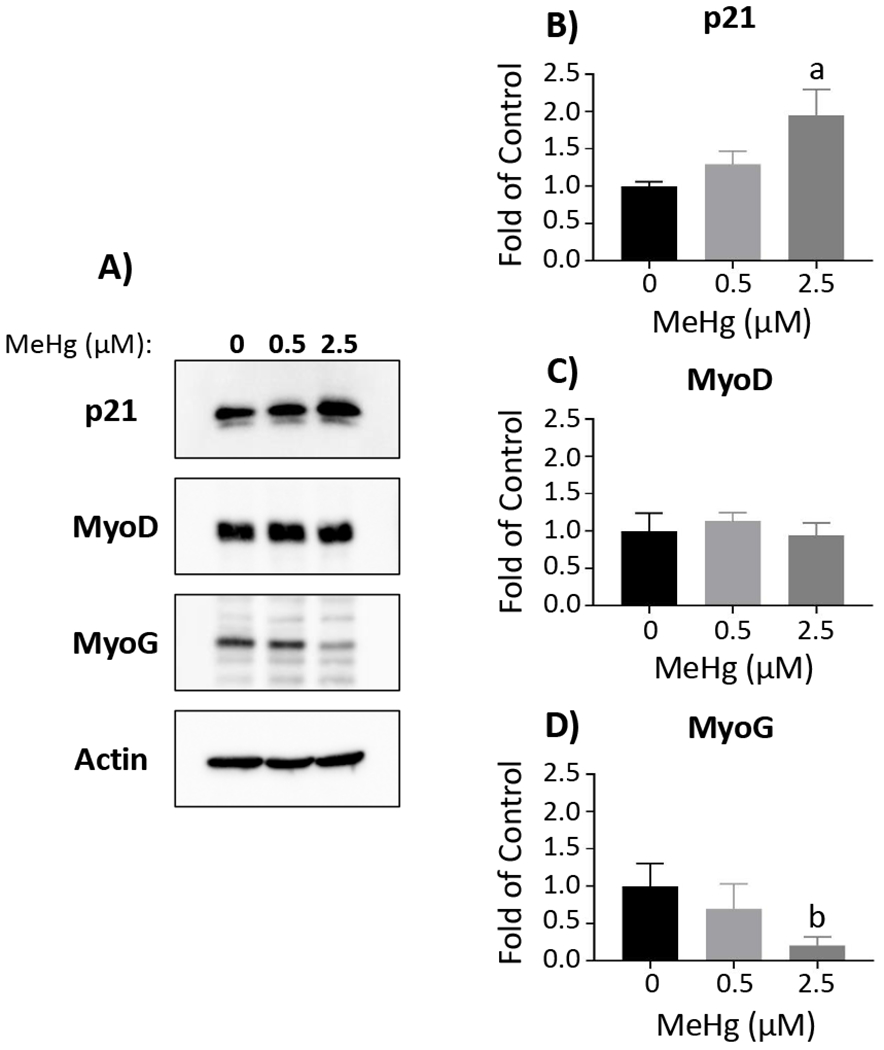
Representative Western blots for p21, MyoD, MyoG, and actin are displayed (A). Band intensity was quantified using ImageJ and normalized to actin, prior to comparison between MeHg concentrations. Total protein was quantified for p21 (B), MyoD (C), and MyoG (D). Data are reported as fold of control, and represent mean ± SD; aone-way ANOVA, Tukey’s HSD, p < 0.01; bone-way ANOVA, Tukey’s HSD, p < 0.05
Figure 5. MeHg effects on protein levels of differentiation factors in myocytes.
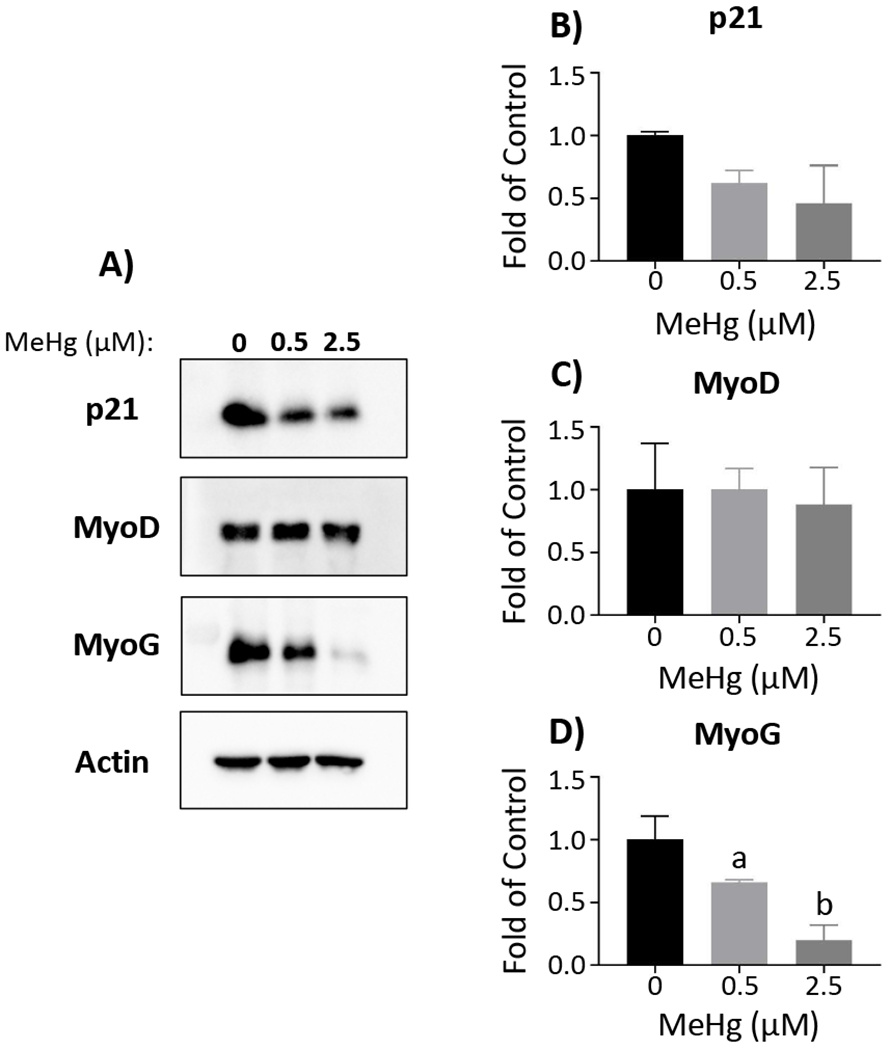
Representative Western blots for p21, MyoD, MyoG, and actin are displayed (A). Band intensity was quantified using ImageJ and normalized to actin, prior to comparison between MeHg concentrations. Total protein was quantified for p21 (B), MyoD (C), and MyoG (D). Data are reported as fold of control, and represent mean ± SD; aone-way ANOVA, Tukey’s HSD, p < 0.05; bone-way ANOVA, Tukey’s HSD, p < 0.001
Figure 6. MeHg effects on protein levels of differentiation factors in myotubes.
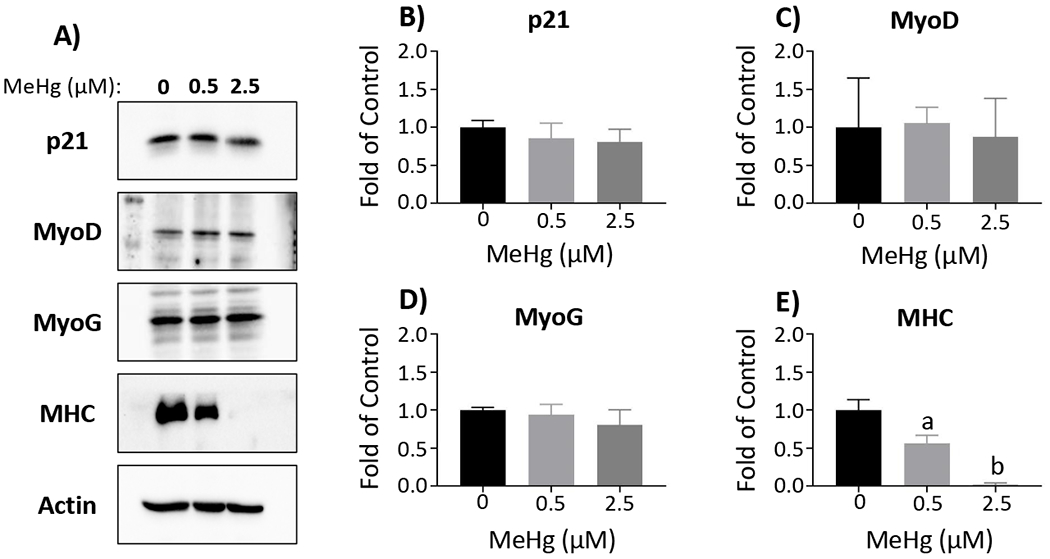
Representative Western blots for p21, MyoD, MyoG, MHC, and actin are displayed (A). Band intensity was quantified using ImageJ and normalized to actin, prior to comparison between MeHg concentrations. Total protein was quantified for p21 (B), MyoD (C), MyoG (D), and MHC (E). Data are reported as fold of control, and represent mean ± SD; aone-way ANOVA, Tukey’s HSD, p < 0.01; bone-way ANOVA, Tukey’s HSD, p < 0.001
Discussion
Despite that motor coordination deficits are common clinical symptoms associated with developmental MeHg exposure, consideration of muscle as a developmental MeHg target has not been extensively explored. Here, we expanded upon emerging evidence that MeHg disrupts normal muscle development, with specific emphasis on dysregulation of a fundamental myogenic differentiation program. First, we established the temporal pattern of MRF expression exhibited in the C2C12 cells under our culture conditions, intended to distinguish myoblasts, myocytes, and myotubes. We confirmed that MeHg has a pointed inhibitory effect on myoblast differentiation. Most striking was that MeHg exposure to the myoblast, while residing in a high-serum growth condition and prior to any differentiation initiating cue, persistently derailed the subsequent ability to differentiate. These results predict that MeHg could have profound effects on muscle development in the fetal myotome, and alternatively, on the adult satellite cells needed for muscle repair and homeostasis.
The unique MRF expression profiles we observed provide insight into how MeHg may elicits its effects predominantly via MyoG. Curiously, MeHg induced little to no changes in expression levels of the master myogenic regulatory factors Myf5 or MyoD. In contrast, effects on MyoG were the most robust, with substantial reduction in gene expression and protein levels in both myoblasts and myocytes subsequent to MeHg exposure. The MyoG protein level was particularly sensitive showing reduction at the lower (0.5µM) MeHg concentration. Equally remarkable is the observation that MyoG transcript and protein levels at the myotube stage were nearly equivalent across all MeHg exposures. This latter profile indicates MyoG expression, while substantially reduced upon MeHg exposure (i.e. in the myoblast), and in the 24 hours after withdrawal of MeHg (i.e. the myocyte), can return to the appropriate level of expression at 48 hours, equal to that seen with the myotubes resulting from untreated myoblasts. Despite this apparent “equilibration” of MyoG levels at the myotube stage, at the higher MeHg exposure MRF4 remains decreased and MHC nearly absent, indicating terminal differentiation has been augmented. Altered expression of MyoG occurs despite the fact that levels of MyoD, a foremost transcriptional regulator of myog (Tapscott 2005), are relatively unaffected. One plausible explanation would be that MeHg might act on MyoD cellular distribution, potentially disrupting its nuclear localization. Alternatively, MeHg might act more directly on myog expression, possibly altering its transcription by epigenetic mechanisms. For example, H3K27 trimethylation represses myog gene expression, and maintains myoblast proliferation (Asp, Blum et al. 2011). MeHg has been demonstrated to enhance H3K27 trimethylation at the brain-derived neurotrophic factor promotor, and repress its expression in the brain (Onishchenko, Karpova et al. 2008). The potential for MeHg to enhance this repressive mark at the myog promotor, however, remains to be explored. Regardless of mechanism, the dynamic profile of MyoG in response to MeHg highlights a potential window of susceptibility for MeHg toxicity in myogenic cells.
One limitation to interpreting the MeHg effects in this system is the apparent toxicity profile revealed by the MTT assay. In a prior study, we showed that MeHg treatment up to 3µM yielded no decrease in viability by MTT assay (Prince and Rand 2018). Here we demonstrate significant reduction in MTT signal by 2.5µM MeHg at all stages. Some discrepancy may be due to the prior study conditions that employed cells at higher density (90% confluence) and differentiation in higher serum levels (i.e. 10% horse serum) than used here (Prince and Rand 2018). Alternatively, it is possible that MTT is simply reflecting an effect on proliferation and not cytotoxicity. In support of this notion, we observe that MeHg at 2.5µM induces elevated p21 in myoblasts, which can be interpreted as a direct reflection of cells moving out of the proliferative state. Furthermore, the fact that MyoG protein and transcript levels at the myotube stage resume equivalent levels between MeHg treated and untreated cells indicates the transcription and translation machinery is intact and suggests the MTT values are reflecting a reduced number of cells due to proliferation effects at the myoblast stage. It remains to be determined directly whether or not the C2C12 myoblasts continue to proliferate with MeHg in our culture conditions.
The increase in p21 protein observed in the present work is consistent with MeHg effects in other studies, which demonstrated enhanced p21 in mouse brain (Ou, Thompson et al. 1999), as well as in neural stem cells (Bose, Onishchenko et al. 2012). Interestingly, we observed that MeHg exposure to myoblasts results in decreased p21 protein at the myocyte stage. This latter profile might be consistent with the overall reduction of MyoG at this stage, as a prior report demonstrated MyoG expression precedes that of p21 in C2C12 cells induced to differentiate. Moreover, myocytes were also found to co-express MyoG and p21 (Andres and Walsh 1996). Thus, the significant decrease in MyoG concurrent with increased p21 that occurs in the myoblasts, likely indicates a more complex relationship between these differentiation factors. It remains to be determined whether p21 function in our culture conditions could be independent of its role in myoblast differentiation. Future studies will aim to characterize the proliferative capacity of the C2C12 cells to further illuminate the role of p21 in MeHg-induced myotoxicity.
By comparison, arsenic (As), another ubiquitous environmental toxicant, has also been demonstrated to inhibit cell differentiation and reduce MyoG expression in C2C12 cells (Yen, Tsai et al. 2010, Steffens, Hong et al. 2011). Similar to the present work, As exposure was initiated in the myoblasts; however, in contrast, exposure continued throughout differentiation. Interestingly, C2C12 cells appeared able to recover from As, as MyoG expression and myotube formation were not different from controls after four days of differentiation (Steffens, Hong et al. 2011). In the present study, as well as our previous work (Prince and Rand 2018), C2C12 cells prove to be unable to recover from acute MeHg exposure. It remains to be determined whether chronic MeHg exposure would yield similar results. Intriguingly, zebrafish exposed to cadmium exhibited myotoxicity, but the MyoD and MyoG patterning in the embryos was unaltered; however, comparable to our results, MHC, myhz in zebrafish, was reduced (Hen Chow and Cheng 2003). Curiously, MHC expression was not altered in adult zebrafish exposed to MeHg (Cambier, Gonzalez et al. 2010), highlighting that observed effects in the present report need further investigation to distinguish whether MeHg exerts a similar profile of toxicity in developing muscle.
In conclusion, MeHg inhibits C2C12 mouse myoblast differentiation. Based on the present report, as well as our previous work, this effect appears independent of whether cells are acutely exposed prior to differentiation, as myoblasts, or in the earliest phase of differentiation, as myocytes (Prince and Rand 2018). We did not, however, make comparisons of initiating MeHg exposures at later post-differentiation time points. Thus, the relative sensitivity of myoblasts, myocytes, or myotubes to MeHg remains to be explored further. As MyoG was most strikingly impacted by MeHg, compared to the other MRFs, future research should focus on whether MeHg directly or indirectly modifies this pivotal myogenic transcription factor. Importantly, this study sets the stage to determine whether these effects are recapitulated in vivo.
Supplementary Material
Acknowledgements
The authors would like to thank Piero Sestili for generously providing the primer sequences for myf5 and mrf4. In addition, the authors would also like to acknowledge Daria Vorojeikina for her technical support.
Funding
This research was supported by NIH R01ES025721 (M.R., PI), the University of Rochester Environmental Health Science Center P30ES001247, and NIH T32ES007026 awarded to Megan Culbreth.
References
- Andres V and Walsh K (1996). “Myogenin expression, cell cycle withdrawal, and phenotypic differentiation are temporally separable events that precede cell fusion upon myogenesis.” J Cell Biol 132(4): 657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asp P, Blum R, Vethantham V, Parisi F, Micsinai M, Cheng J, Bowman C, Kluger Y and Dynlacht BD (2011). “Genome-wide remodeling of the epigenetic landscape during myogenic differentiation.” Proc Natl Acad Sci U S A 108(22): E149–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakir F, Damluji SF, Amin-Zaki L, Murtadha M, Khalidi A, al-Rawi NY, Tikriti S, Dahahir HI, Clarkson TW, Smith JC and Doherty RA (1973). “Methylmercury poisoning in Iraq.” Science 181(4096): 230–241. [DOI] [PubMed] [Google Scholar]
- Bose R, Onishchenko N, Edoff K, Janson Lang AM and Ceccatelli S (2012). “Inherited effects of low-dose exposure to methylmercury in neural stem cells.” Toxicol Sci 130(2): 383–390. [DOI] [PubMed] [Google Scholar]
- Cambier S, Fujimura M and Bourdineaud JP (2018). “A likely placental barrier against methylmercury in pregnant rats exposed to fish-containing diets.” Food Chem Toxicol 122: 11–20. [DOI] [PubMed] [Google Scholar]
- Cambier S, Gonzalez P, Durrieu G, Maury-Brachet R, Boudou A and Bourdineaud JP (2010). “Serial analysis of gene expression in the skeletal muscles of zebrafish fed with a methylmercury-contaminated diet.” Environ Sci Technol 44(1): 469–475. [DOI] [PubMed] [Google Scholar]
- Conerly ML, Yao Z, Zhong JW, Groudine M and Tapscott SJ (2016). “Distinct Activities of Myf5 and MyoD Indicate Separate Roles in Skeletal Muscle Lineage Specification and Differentiation.” Dev Cell 36(4): 375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira Ribeiro CA, Nathalie MD, Gonzalez P, Yannick D, Jean-Paul B, Boudou A and Massabuau JC (2008). “Effects of dietary methylmercury on zebrafish skeletal muscle fibres.” Environ Toxicol Pharmacol 25(3): 304–309. [DOI] [PubMed] [Google Scholar]
- Debes F, Weihe P and Grandjean P (2016). “Cognitive deficits at age 22 years associated with prenatal exposure to methylmercury.” Cortex 74: 358–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel GL and Rand MD (2014). “The Notch target E(spl)mdelta is a muscle-specific gene involved in methylmercury toxicity in motor neuron development.” Neurotoxicol Teratol 43: 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engstrom K, Love TM, Watson GE, Zareba G, Yeates A, Wahlberg K, Alhamdow A, Thurston SW, Mulhern M, McSorley EM, Strain JJ, Davidson PW, Shamlaye CF, Myers GJ, Rand MD, van Wijngaarden E and Broberg K (2016). “Polymorphisms in ATP-binding cassette transporters associated with maternal methylmercury disposition and infant neurodevelopment in mother-infant pairs in the Seychelles Child Development Study.” Environ Int 94: 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faralli H and Dilworth FJ (2012). “Turning on myogenin in muscle: a paradigm for understanding mechanisms of tissue-specific gene expression.” Comp Funct Genomics 2012: 836374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandjean P, Weihe P, White RF and Debes F (1998). “Cognitive performance of children prenatally exposed to “safe” levels of methylmercury.” Environ Res 77(2): 165–172. [DOI] [PubMed] [Google Scholar]
- Guo K, Wang J, Andres V, Smith RC and Walsh K (1995). “MyoD-induced expression of p21 inhibits cyclin-dependent kinase activity upon myocyte terminal differentiation.” Mol Cell Biol 15(7): 3823–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada M (1978). “Congenital Minamata disease: intrauterine methylmercury poisoning.” Teratology 18(2): 285–288. [DOI] [PubMed] [Google Scholar]
- Hen Chow ES and Cheng SH (2003). “Cadmium affects muscle type development and axon growth in zebrafish embryonic somitogenesis.” Toxicol Sci 73(1): 149–159. [DOI] [PubMed] [Google Scholar]
- Hinterberger TJ, Sassoon DA, Rhodes SJ and Konieczny SF (1991). “Expression of the muscle regulatory factor MRF4 during somite and skeletal myofiber development.” Dev Biol 147(1): 144–156. [DOI] [PubMed] [Google Scholar]
- Ishibashi J, Perry RL, Asakura A and Rudnicki MA (2005). “MyoD induces myogenic differentiation through cooperation of its NH2- and COOH-terminal regions.” J Cell Biol 171(3): 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ and Schmittgen TD (2001). “Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method.” Methods 25(4): 402–408. [DOI] [PubMed] [Google Scholar]
- Montgomery SL, Vorojeikina D, Huang W, Mackay TF, Anholt RR and Rand MD (2014). “Genome-wide association analysis of tolerance to methylmercury toxicity in Drosophila implicates myogenic and neuromuscular developmental pathways.” PLoS One 9(10): e110375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onishchenko N, Karpova N, Sabri F, Castren E and Ceccatelli S (2008). “Long-lasting depression-like behavior and epigenetic changes of BDNF gene expression induced by perinatal exposure to methylmercury.” J Neurochem 106(3): 1378–1387. [DOI] [PubMed] [Google Scholar]
- Ou YC, Thompson SA, Ponce RA, Schroeder J, Kavanagh TJ and Faustman EM (1999). “Induction of the cell cycle regulatory gene p21 (Waf1, Cip1) following methylmercury exposure in vitro and in vivo.” Toxicol Appl Pharmacol 157(3): 203–212. [DOI] [PubMed] [Google Scholar]
- Prince LM and Rand MD (2017). “Notch Target Gene E(spl)mdelta Is a Mediator of Methylmercury-Induced Myotoxicity in Drosophila.” Front Genet 8: 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince LM and Rand MD (2018). “Methylmercury exposure causes a persistent inhibition of myogenin expression and C2C12 myoblast differentiation.” Toxicology 393: 113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K and Dilworth FJ (2013). “Differential modulation of cell cycle progression distinguishes members of the myogenic regulatory factor family of transcription factors.” FEBS J 280(17): 3991–4003. [DOI] [PubMed] [Google Scholar]
- Steffens AA, Hong GM and Bain LJ (2011). “Sodium arsenite delays the differentiation of C2C12 mouse myoblast cells and alters methylation patterns on the transcription factor myogenin.” Toxicol Appl Pharmacol 250(2): 154–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapscott SJ (2005). “The circuitry of a master switch: Myod and the regulation of skeletal muscle gene transcription.” Development 132(12): 2685–2695. [DOI] [PubMed] [Google Scholar]
- Tatsuta N, Murata K, Iwai-Shimada M, Yaginuma-Sakurai K, Satoh H and Nakai K (2017). “Psychomotor Ability in Children Prenatally Exposed to Methylmercury: The 18-Month Follow-Up of Tohoku Study of Child Development.” Tohoku J Exp Med 242(1): 1–8. [DOI] [PubMed] [Google Scholar]
- Usuki F, Yasutake A, Matsumoto M, Umehara F and Higuchi I (1998). “The effect of methylmercury on skeletal muscle in the rat: a histopathological study.” Toxicol Lett 94(3): 227–232. [DOI] [PubMed] [Google Scholar]
- Yen YP, Tsai KS, Chen YW, Huang CF, Yang RS and Liu SH (2010). “Arsenic inhibits myogenic differentiation and muscle regeneration.” Environ Health Perspect 118(7): 949–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zammit PS (2017). “Function of the myogenic regulatory factors Myf5, MyoD, Myogenin and MRF4 in skeletal muscle, satellite cells and regenerative myogenesis.” Semin Cell Dev Biol 72: 19–32. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.