


Abstract
The developmental potential of cells, termed pluripotency, is highly dynamic and progresses through a continuum of naive, formative and primed states. Pluripotency progression of mouse embryonic stem cells (ESCs) from naive to formative and primed state is governed by transcription factors (TFs) and their target genes. Genomic techniques have uncovered a multitude of TF binding sites in ESCs, yet a major challenge lies in identifying target genes from functional binding sites and reconstructing dynamic transcriptional networks underlying pluripotency progression. Here, we integrated time-resolved ‘trans-omic’ datasets together with TF binding profiles and chromatin conformation data to identify target genes of a panel of TFs. Our analyses revealed that naive TF target genes are more likely to be TFs themselves than those of formative TFs, suggesting denser hierarchies among naive TFs. We also discovered that formative TF target genes are marked by permissive epigenomic signatures in the naive state, indicating that they are poised for expression prior to the initiation of pluripotency transition to the formative state. Finally, our reconstructed transcriptional networks pinpointed the precise timing from naive to formative pluripotency progression and enabled the spatiotemporal mapping of differentiating ESCs to their in vivo counterparts in developing embryos.
INTRODUCTION
Pluripotency describes the potential of cells to differentiate into derivatives of all three embryonic germ layers: endoderm, mesoderm and ectoderm. It is an intrinsic and highly dynamic cellular property bookended by naive and primed states (1) and underpins the stemness of cells (2). Between the naive and primed states, a maturation phase of pluripotency termed as the formative state is characterized by rewired transcriptional networks and signaling apparatus but remains relatively homogeneous and unspecified (3). Importantly, the formative state is thought to represent an executive phase wherein cells undergo remodelling of transcriptional, epigenetic, signalling and metabolic network to acquire multilineage competence and responsiveness to specification cues (4). This raises the possibility that the exit from the naive state and the progression to the formative state are mandatory en route steps towards the specification of primary germ layers during mammalian development (4,5). Embryo-derived stem cells exist in the spectrum of naive to primed pluripotent states, which may be mirrored by embryonic cells in vivo. In particular, naive mouse embryonic stem cells (ESCs) derived from the inner cell-mass of pre-implantation mouse blastocysts (E3.75–E4.75) are considered to capture the naive pluripotent state, and epiblast-like cells (EpiLCs) induced from ESCs are considered to represent the formative phase of pluripotency and are the in vitro counterparts of post-implantation epiblasts (E5.5–E6.5) (6). In ESCs, this transition between cell states is tightly controlled by core ESC transcription factors (TFs), which bind to cis-regulatory elements located within promoters and distal enhancers of their target genes to regulate the activity of transcriptional networks controlling pluripotency (7,8). However, the target genes that make up the transcriptional networks and their dynamic rewiring as ESCs progress from naive to formative pluripotent states remain poorly characterised.
The advance of chromatin immunoprecipitation followed by ultrafast sequencing (ChIP-seq) techniques has enabled genome-wide profiling of TF binding sites (TFBSs) in a multitude of cell types including ESCs (9,10). These TF chromatin binding profiles have provided a means of identifying candidate genes regulated by TFs in a cell type-specific manner (11,12). TFBSs can generally be categorized as promoter-proximal sites that are located close to an annotated transcription start site (TSS) or distal sites with no nearby TSSs (13,14). One simple approach to identify TF target genes is to assign the gene with the nearest TSS (in terms of nucleotide distance) to each TFBS identified in a ChIP-seq experiment (15). While this approach may work for promoter-proximal sites, it becomes much less reliable for distal sites because the assignment of genes with the nearest TSS to distal sites ignores tertiary chromatin structures such as enhancer-promoter interactions (16–18), and can thereby result in a large number of false positive identifications (19).
Recent knowledge gleaned from genome-wide RNA Polymerase II (Pol2) chromatin interaction analysis with paired-end-tag (Pol2-ChIA-PET) assays (20) and chromosome conformation capture assays such as Hi-C (21) has enabled the quantification of long-range three-dimensional chromatin interactions in ESCs. The information collated from linking distal TFBSs to TSSs that have Pol2-ChIA-PET and/or Hi-C supported interactions has enabled a more accurate identification of genes that are potentially regulated by distal sites. Several previous studies have utilized Pol2-ChIA-PET data to validate long-range regulatory interactions predicted by machine learning-based models in multiple cell-types (22–24). Despite these advances, current approaches are still unable to discriminate functionally relevant TFBSs and precisely identify their proximal and distal target genes out of all putative candidates, as physical contact does not always imply functional regulation by TFs (25). Therefore, to reconstruct dynamic transcriptional networks underpinning the pluripotency progression, a key challenge is to distinguish functionally relevant TFBSs and target genes that are regulated in ESCs during their differentiation.
Using an established system to induce naive mouse ESCs to post-implantation EpiLCs (6,26), we recently profiled the temporal dynamics of trans-omic layers (27) as pluripotency progresses from naive to formative state (4). Here, we first extended a statistical learning framework based on our adaptive sampling and ensemble model (AdaEnsemble) (28,29) to predict a list of high-confidence proximal and distal target genes regulated by ESC TFs controlling the transition from naive to formative state by using the trans-omic data. Using the AdaEnsemble-identified TF target genes, we show that TFs associated with the naive state are more likely to regulate genes that are TFs themselves compared to TFs associated with formative state, suggesting denser TF hierarchies for signal propagation in naive pluripotency. Further analysis revealed that genes regulated by formative TFs are marked by permissive epigenomic signatures in naive ESCs suggesting that they are poised for expression prior to pluripotency transition. Finally, we reconstructed the transcriptional networks that underpin distinct pluripotent states of stem cells in vitro and mapped their counterparts in vivo in mouse epiblasts using the spatiotemporally resolved transcriptomics data we generated recently (30). Together, the dynamic rewiring of these transcriptional networks sheds light on the timing of pluripotency transition, providing new insights into the transcriptional regulation of naive and formative TF target genes.
MATERIALS AND METHODS
Trans-omic data pre-processing
RNA-seq, ChIP-seq, and MS-based proteomics data were processed as previously described (27). Briefly, sequence reads from RNA-seq experiments were aligned to the mouse reference genome (mm9 assembly) using STAR (31) version 2.5.2a, allowing up to three mismatches and acquiring unique reads only. Gene expression across eight time points of the differentiation were quantified using HTSeq (32) version 0.6.1 against Ensembl mouse gene annotation. Sequence reads from ChIP-seq experiments were aligned to mm9 using Bowtie (33) version 0.12.8 allowing at most two mismatches and accepting only reads mapped to unique genomic regions. Histone modifications and Pol2 ChIP-seq signals were quantified for each gene by computing reads per kilobase million (RPKM) of 1Kb region centred around the transcription start site of each gene, and this is repeated for each of the eight time points. For MS-based proteomics data, raw MS files were processed using MaxQuant (34) version 1.5.3.29 against mouse UniProt database with a FDR set at 0.01 for both peptide identification and protein inference. Proteins were quantified using LFQ intensity (log2) at each of the nine profiled time points.
Differentially regulated TFs
Analysis of variance (ANOVA) tests were performed on time-course RNA-seq and MS-based proteomic data to identify differentially regulated genes on mRNA and protein levels, respectively. Benjamini-Hochberg correction that controls for FDR was applied to account for multiple testing. Subsequently, for each gene, an integrated 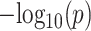 of mRNA and protein was derived as:
of mRNA and protein was derived as:
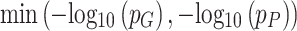 |
where 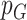 and
and 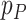 are adjusted p-values from ANOVA analysis of mRNA and protein for each gene, respectively. Similarly, an integrated log2 fold change of mRNA and protein was calculated for each gene as:
are adjusted p-values from ANOVA analysis of mRNA and protein for each gene, respectively. Similarly, an integrated log2 fold change of mRNA and protein was calculated for each gene as:
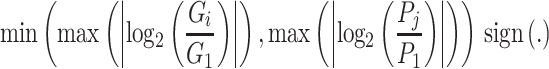 |
where 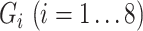 and
and 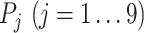 are expression levels of mRNA and protein at a time point. The
are expression levels of mRNA and protein at a time point. The 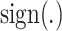 function determine the sign of the selected fold change. Genes with a smaller than 0.05 integrated P-value and a >1 integrated log2 fold change, as well as being annotated as a TF in public databases (35,36) were noted as differentially regulated TFs during the pluripotency progression. Those that have been profiled previously using ChIP-seq in ESCs (i.e. Sox2, Nanog, Esrrb, Klf4, Nr5a2, Otx2 and c-Myc) were selected for analysis.
function determine the sign of the selected fold change. Genes with a smaller than 0.05 integrated P-value and a >1 integrated log2 fold change, as well as being annotated as a TF in public databases (35,36) were noted as differentially regulated TFs during the pluripotency progression. Those that have been profiled previously using ChIP-seq in ESCs (i.e. Sox2, Nanog, Esrrb, Klf4, Nr5a2, Otx2 and c-Myc) were selected for analysis.
TFBS identification and classification
Sequence reads of Sox2, Nanog, Esrrb, Klf4, Nr5a2, Otx2 and c-Myc ChIP-seq experiments, generated from ESCs, were processed uniformly by aligning to the mouse reference genome (mm9) using Bowtie (33) version 0.12.8 allowing at most two mismatches of read mapping to only unique genomic positions. Aligned reads were processed using SISSRs (37) using default settings for genome-wide TFBS identification. For each TF, SISSRs identified binding sites were subsequently classified as proximal binding sites if the mid-point of a binding site is within 1 kb from a Refseq annotated transcription start site or distal binding sites otherwise.
Chromatin interaction and putative TF target genes
Chromatin interaction between distal TF binding sites and promoters were determined based on published Pol2-ChIA-PET (20) dataset in ESCs. A TFBS was considered to interact with a promoter if the 1 kb region centred around the mid-point of the binding site and the 1 kb region centred around a transcription start site (gene promoter) has been observed to interact with Pol2-ChIA-PET interaction data. Genes that are involved in chromatin interaction with the distal binding sites of a TF were considered as putative distal target genes of that TF. Together with proximal target genes (i.e. genes with promoters that are bound by the same TF within 1kb), they were denoted as putative target genes of the TF under consideration.
Application of AdaEnsemble for TF target prediction
AdaEnsemble utilises ESC Pol2-ChIA-PET data together with each TF ChIP-seq profile data from ESCs to create a ‘noisy’ initial class label vector and iteratively optimises this class label vector by evaluating their likelihood with the dynamics of trans-omic data under the assumption that target genes of the same TF would have certain transcriptomic, proteomic and epigenomic profiles that distinguish them from non-target genes. The final prediction result is a matrix of genes with predicted confidence of being targets of a set of core TFs during the ESC to EpiLC transition from which the transcriptional networks could be reconstructed and dynamics resolved.
For each TF, all its putative target genes were treated as positive instances and a set of randomly selected genes that are not included in the positive set are used as negative instances. The size of the negative set is maintained as the same as the positive set to keep the class distribution balanced. AdaEnsemble is an extension of AdaSampling for positive-unlabelled learning as described previously (29) with a modified version of kNN classifier specifically designed in this study to evenly weight the contribution of the transcriptome, proteome and epigenome data. In particular, the modified kNN utilises a weighted Euclidean distance to quantify the dissimilarity 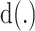 of two genes
of two genes 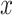 and
and 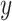 as follows:
as follows:
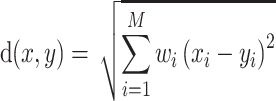 |
where 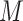 is the number of features and
is the number of features and 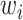 is the weight associated with the
is the weight associated with the 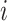 th feature (
th feature (  ).
).
Predictability assessment for epigenomic data
Significantly more features can be extracted from temporal epigenomic data (6 marks times 8 time points = 48 features) compared to the transcriptome (eight features) and proteome (nine features). To account for this, we performed an initial training of AdaEnsemble using only transcriptome and proteome data (z-scores standardised across time) and a subsequent augmented training where the ‘intermediate’ predictions from the initial training were used to assess the predictability of each epigenomic feature. This is quantified by calculating an AUC value for each histone mark and Pol2 at each of the eight profiled time points with respect to the intermediate predictions. The final predictions for each TF were made by combining the transcriptomic, proteomic, and AUC-weighted epigenomic data by using the above modified kNN classifier.
Estimation of false positive predictions
To estimate the percentage of false positive predictions in AdaEnsemble, for each TF, the trained AdaEnsemble model was used to classify a set of randomly selected genes that were not included in the putative target gene list. Note that the size of these randomly selected gene sets matches the size of the putative target gene sets. Also, these randomly selected gene sets were for model testing and were different from those used as negative instances in the model training process. By treating the randomly selected genes as negative (i.e. non-target genes), we next estimated the false positive rate (FPR) as the number of positive predictions from the random set divided by the number of all genes in the random set.
Dynamic network reconstruction and analysis
AdaEnsemble-identified TF target genes that are themselves TFs were included for visualizing the transition of transcriptional networks from naive to formative pluripotency using igraph R package with a layout calculated by using Kamada–Kawai force-directed algorithm (38). The temporal mRNA expression of a gene 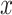 was first standardized across time points using z-score transformation:
was first standardized across time points using z-score transformation:
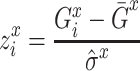 |
where for each gene 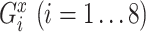 is the expression level of mRNA at a time point
is the expression level of mRNA at a time point 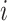 , and
, and 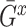 and
and 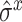 are the average expression and variance across time points. After the standardization, the expression between a pair of genes
are the average expression and variance across time points. After the standardization, the expression between a pair of genes 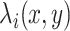 on the mRNA level at a time point of
on the mRNA level at a time point of 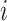 (
(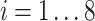 ) was calculated as the average of their standardised temporal mRNA expression values at each time point:
) was calculated as the average of their standardised temporal mRNA expression values at each time point:
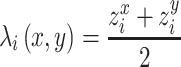 |
To quantify overall expression change, the pairwise expression calculated above was averaged for all target genes of either naive or formative TFs. For calculating the relative change, the overall expression change calculated for the naive TFs was subtracted by that of the formative TFs at each time point. For visualizing dynamic changes in the transcriptional networks, the pairwise expression 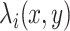 was scaled to 0 and 1.
was scaled to 0 and 1.
Quantification of transcriptional regulation
For each TF, to quantify the transcriptional regulation of its target genes that are themselves TFs, we first identified the maximum and the minimum log2 fold changes at any of all time points and calculated the difference between these two values for all target genes. For a target gene 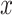 , this value (denoted as
, this value (denoted as 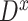 ) is calculated as below:
) is calculated as below:
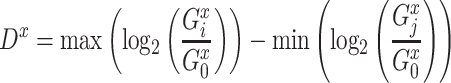 |
where 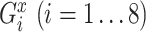 and
and 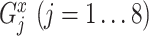 are the expression levels of mRNA at a time point. Next, we took the summation of the difference in the log2 fold changes for target genes that are themselves TFs (denoted as
are the expression levels of mRNA at a time point. Next, we took the summation of the difference in the log2 fold changes for target genes that are themselves TFs (denoted as 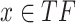 ) and divided this by the summation for all target genes.
) and divided this by the summation for all target genes.
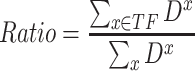 |
The above ratio from this calculation is interpreted as the contribution of target genes that are themselves TFs to the total transcriptional regulation.
Pathway enrichment analysis
Pathway enrichment analysis, in terms of over-representation of genes from a pathway, was performed using Limma R package (39) against the gene ontology (GO) terms from GO database (40). P-values were adjusted using the Benjamini and Hochberg.
Spatiotemporal mapping of differentiating ESCs
To determine the in vivo counterparts of ESCs during pluripotency transition, we mapped cells from each time point to E5.5–E7.5 epiblasts of mouse embryos based on the TF target genes identified in this study. Briefly, Geo-seq samples of embryos were used for mapping (41) and the area under the recovery curve across the ranking of genes was calculated for each Geo-seq sample and ESCs from each time point using the AUCell R package (v1.2.4) (42). The area under the recovery curve was then used as the activity score which measures the enrichment of in vitro ESCs at each time point and in vivo spatial epiblasts.
Finally, the corn plots of differentiating ESCs at each time point were generated based on the enrichment on each Geo-seq sample. The corn plots illustrate gene expression or gene-set enrichment activity on the respective embryonic positions. Spatial coordinates in the 2D plot are as follows: the proximal–distal location in descending numerical order (1 = most distal site) and in the transverse plane of the germ layers: epiblast/ectoderm, anterior (A), posterior (P) containing the primitive streak, right (R)—anterior (R1) and posterior (R2), left (L)—anterior (L1) and posterior (L2).
RESULTS
Selection of dynamically regulated TFs during pluripotency progression
The expression level of TFs is a key determinant of the expression of their downstream target genes (Figure 1A) and thus the transcriptional networks they orchestrate (36,43). To precisely pinpoint the timing of transcriptional network transitions during pluripotency progression, we selected a panel of TFs that are dynamically regulated during this process. First, we assessed the mRNA expression and protein abundance of a list of known TFs compiled from multiple sources (35,36) in our trans-omic data (27) profiling the ESC to EpiLC transition (Figure 1B and C). We then defined the significance threshold as an absolute log2 fold change equal to or greater than 1 (i.e. 2-fold increased or decreased) at one or more time points and a p-value from an FDR corrected ANOVA-test of less than or equal to 0.05 from the integrated transcriptome and proteome data (see more details in ‘Materials and Methods’ section). This threshold captures both the magnitude of change as well as reproducibility within biological replicates across the time points to ensure that selected TFs are significantly altered during the ESC to EpiLC transition. Using this approach, we found 110 TFs that are dynamically regulated during the differentiation process (Figure 1C; Supplementary Table S1). Lastly, we filtered the dynamically regulated TFs, selecting a panel of seven that have been profiled in a compendium of ChIP-seq datasets in ESCs (14). The seven selected TFs are known to promote ESC self-renewal or differentiation through previous genetic and/or functional studies (Figure 1C). Five out of this seven are down-regulated during the pluripotency transition, Sox2 (44), Nanog (45,46), Esrrb (47), Klf4 (48), and Nr5a2 (49) and two are up-regulated, Otx2 (26) and c-Myc (50).
Figure 1.

Identification of dynamically regulated TFs in pluripotency progression. (A) Schematics of TF expression and their binding at promoter-proximal (P1, P2 and P3) and distal (D1, D2 and D3) sites during pluripotency progression from naive to formative states. Dash lines (red) represent interactions between distal TF binding sites and their target genes. (B) Schematic summary of the time-course trans-omic dataset utilised in this study for reconstructing and characterising the transcriptional networks in pluripotency progression from naive ESCs to EpiLCs that represent formative state. (C) Volcano plot of genes profiled on both transcriptome and proteome levels in the trans-omic dataset (27). TFs that are dynamically regulated (DR) during the ESC to EpiLC transition are highlighted in blue and within these DR TFs, those that have been profiled previously using ChIP-seq in ESCs are highlighted in red. (D) Pie charts showing the distribution of promoter-proximal target genes (TSS ± 1 kb) and putative distal target genes (TSS > 1 kb) with chromatin loops (Pol2-ChIA-PET) for each TF according to its ChIP-seq profile in ESCs.
After calling the binding sites for each of these TFs from their respective ChIP-seq datasets using SISSRs (37), we categorized them as promoter-proximal (binds within 1 kb of a TSS) and distal binding sites (binds 1 kb away from TSSs) either without or with chromatin loop support (interacts with one or more TSSs through chromatin looping, as supported by the Pol2-ChIA-PET data) (Supplementary Figure S1A). We next identified putative proximal and distal target genes for each TF (see ‘Materials and Methods’ section) (Figure 1D). Assessing the relative ratio of putative proximal versus distal target genes for each TF, we found that while c-Myc and Otx2 have more putative proximal target genes (72% and 57%, respectively), other TFs have relatively more putative distal target genes (57–78%).
Prediction of TF target genes during pluripotency progression using AdaEnsemble
Identification of genes that are regulated by master TFs is an essential step for reconstructing transcriptional networks during the pluripotency progression. Here, we integrated time-resolved trans-omic data with publicly available Pol2-ChIA-PET (20) and ChIP-seq datasets from ESCs, and formulated the task of identifying TF target genes as learning with the presence of class label noise from all putative proximal and distal target genes. We extended our AdaEnsemble model (28,29) for TF target gene prediction (Supplementary Figure S1B). To achieve highly time-sensitive and context-specific predictions, we processed the trans-omic dataset into time-resolved gene features from the transcriptome, proteome and epigenome that can be utilized by AdaEnsemble (see more details in ‘Materials and Methods’ section). Because the expression level of a TF is a key determinant of the expression of its downstream target genes, we hypothesized that target genes of the same TF would have certain transcriptomic, proteomic and epigenomic profiles that distinguish them from non-target genes. The mRNA expression and protein abundance of the seven TFs (Figure 2A and Supplementary Figure S2A) show that, consistent with Figure 1C, Sox2, Nanog, Esrrb, Klf4 and Nr5a2 are dramatically down-regulated, whereas Otx2 and c-Myc are significantly up-regulated across the 72 h time-course. Figure 2A summarizes the standardized mRNA and protein expression profiles of both the putative proximal and distal target genes of Sox2 and c-Myc supported by chromatin loop (top panel) and those identified by AdaEnsemble (bottom panel). We found that genes identified by AdaEnsemble (P > 0.9) exhibit a much greater dynamics on both mRNA and protein levels during ESC differentiation. Notably, we found that the temporal patterns of the AdaEnsemble-identified target genes clearly resemble the temporal changes in mRNA expression and protein abundance of Sox2 and c-Myc (Figure 2A). Similar results are observed for Nanog, Esrrb, Klf4, Nr5a2 and Otx2 (Supplementary Figure S2A and B), and the full list of prediction scores for all putative target genes of the seven TFs are reported in Supplementary Table S2.
Figure 2.

Prediction and validation of TF target genes in transition from naive to formative pluripotency using AdaEnsemble and trans-omic data. (A) Time-course showing the log2 fold change (compared to time = 0) in expression for mRNA (green) and protein (blue) for Sox2 and c-Myc. Bars represent standard deviation among biological replicates (n = 2 for mRNA and n = 4 for protein). Pearson's correlation coefficient for concordance between protein and mRNA are shown. (B) Time-course expression profiles of putative target genes (i.e. putative candidates) supported by chromatin loops from Pol2-ChIA-PET (top) and AdaEnsemble predicted target genes (bottom) for Sox2 and c-Myc, respectively. Profiles are divided into those from promoter-proximal and distal target genes, and then further divided into those from mRNA and protein levels. (C, D) Cumulative distribution showing degree of correlation (Pearson's) between time-course expression profiles of each TF with its putative target genes (green and black) and those predicted by AdaEnsemble (red and yellow), on mRNA (C, red) and protein (D, yellow) levels. P-values were computed using Wilcoxon Mann-Whitney U test (two-sided). (E) Log2 fold change in mRNA after Sox2, Nr5a2, Nanog, Klf4, Esrrb knockdown, or Otx2 knockout in ESCs compared to WT ESCs. Wilcoxon Mann–Whitney U test (one-sided) are performed on AdaEnsemble-identified target genes (iv) versus all quantified genes (i), proximal and distal target genes by nearest TSS assignment of all TF binding sites identified in ChIP-seq data (ii), and putative target genes by chromatin loop support (Pol2-ChIA-PET) (iii), respectively, and the largest p-value is displayed as an upper bound for all pairwise comparisons for each TF. (F) Similar to (E) but in c-Myc overexpressed ESCs compared to WT ESCs.
We next partitioned the TFBSs for each TF into those that have high probability of target gene prediction (P > 0.9; herein referred to as high-TFBSs) and those with low probability of prediction (P < 0.5; herein referred to as low-TFBSs). For Sox2, Nanog, Esrrb, Nr5a2 and Klf4, we combined their TFBSs and refer to the combined sets as high-TFBSs and low-TFBSs of naive TFs. We found that high-TFBSs are enriched for more H3K27ac mark and have less H3K27me3 mark compared to low-TFBSs (Supplementary Figure S2C), suggesting that high-TFBSs correspond to active genomic regions and low-TFBSs to less active regions. Finally, we quantified the goodness of fit of the expression profiles of the putative target genes and the AdaEnsemble-identified target genes to those of their respective TFs. Compared to putative target genes, we found that the AdaEnsemble-identified target genes generally have a significantly higher correlation with their respective TFs both at the mRNA (Figure 2C) and protein (Figure 2D) levels. Overall, our findings from the AdaEnsemble prediction suggest that the temporal behaviour of the high-confidence target genes are similar to the expression profiles of their respective TFs during the pluripotency transition, in line with the role of TFs in regulating the expression of their target genes.
Expression of AdaEnsemble-identified target gene sets upon perturbation of their respective TFs
To validate the AdaEnsemble predictions for each TF, we evaluated the AdaEnsemble model by using genes that are not included as putative candidates in the model training process. This allowed us to estimate the false positive rate (FPR) because any positive predictions from these genes are likely to be false positive predictions (see ‘Materials and Methods’ section). Using this approach, we estimated FPR of the AdaEnsemble prediction for each of the seven TFs (Supplementary Figure S3A). We found that the prediction threshold (i.e. the probability w.r.t. positive) of the AdaEnsemble is negatively correlated with the FPR. Moreover, estimation at an FPR of 0.1 corresponded closely to the AdaEnsemble prediction probability of 0.9.
To cross-validate the AdaEnsemble predictions with independent data sources, we quantified the log2 fold change of each target gene using previously published gene expression data (see ‘Materials and Methods’) of the transcriptome of wide-type (WT) ESCs and those with knockout, knockdown, or overexpression of Sox2, Nanog, Esrrb, Klf4, Nr5a2, Otx2 or c-Myc. Compared to putative targets (i.e. all proximal genes and distal genes supported by Pol2-ChIA-PET), gene sets identified by AdaEnsemble are significantly more down- or up-regulated (Wilcoxon rank sum test, P < 0.05) after the knockdown/knockout (Figure 2E) or overexpression (Figure 2F), respectively, of the corresponding TFs. In comparison, gene sets selected by using the nearest TSS assignment (Supplementary Table S3) do not show significant expression changes after TF ablation or overexpression (Figure 2E and F). Interestingly, we found that the AdaEnsemble prediction largely maintained the ratio of proximal and distal target genes throughout the probability thresholds (Supplementary Figure S3B and Figure 1D), suggesting that our model is not biased towards selecting either proximal or distal targets.
Predictability of histone marks and Pol2 ChIP-seq for TF target genes
While the temporal changes of the transcriptome and proteome are critical for capturing the dynamic modality of the regulation of TF target genes, the utility of individual histone modifications and Pol2 in TF target prediction remains to be determined. To this end, we implemented an ‘initial training’ phase to create intermediate positive and negative predictions with only the transcriptomic and proteomic data. These predictions were subsequently utilized to assess predictability of each histone mark and Pol2 ChIP-seq occupancies at the TSS of each gene at each time point (see ‘Materials and Methods’ section). Using this procedure implemented in AdaEnsemble (Supplementary Figure S1B), we determined the overall predictive power of each histone mark and Pol2 ChIP-seq data (Figure 3A) and their predictive power with respect to each TF (Supplementary Figure S4A). While these data suggest that not all epigenomic features are equally predictive of TF target genes, we found that Pol2 and H3K27ac are the most predictive marks, in general, consistent with the understanding that these two marks are enriched at active promoters and enhancers (51,52). In contrast, we found that H3K4me1 is more predictive for target genes of naive pluripotency TFs (Sox2, Nanog, Esrrb, Nr5a2 and Klf4; together referred hereon as naive TFs), whereas H3K4me3 is more predictive for target genes of formative pluripotency TFs (Otx2 and c-Myc; together referred hereon as formative TFs) (Figure 3B). This supports a distal enhancer binding preference of naive TFs, and in comparison, a relatively stronger promoter binding preference of formative TFs. The relative predictability of histone marks and Pol2 were similar in naive TFs (r = 0.9 ± 0.08) but it differed significantly from formative TFs (r = 0.75 ± 0.16, Wilcoxon rank sum test P = 0.019) (Supplementary Figure S4B).
Figure 3.

Evaluation of predictability of histone marks and Pol2 ChIP-seq on TF target genes. (A) Overall predictive power (in terms of area under the ROC curve [AUC]) of histone marks and Pol2 ChIP-seq data, averaged across all seven TFs, when assessed against the intermediate predictions from the initial training phase of AdaEnsemble. (B) Bi-clustered heatmap showing relationship among histone modifications and Pol2 ChIP-seq in terms of predictiveness (scaled AUC) in TF target genes. The dashed box highlights H3K4me1 and H3K4me3 marks for their contrast between naive pluripotency TFs (Sox2, Nanog, Esrrb, Nr5a2, Klf4) and formative pluripotency TFs (Otx2 and c-Myc). (C) Levels of H3K4me1 (6 h) and H3K4me3 (48 h) signal at the promoters of the target genes of naive and primed pluripotency TFs.
Because H3K4me1 at 6 h and H3K4me3 at 48 h showed the most divergent predictabilities in terms of their discriminative power for naive and primed TF target genes, we next computed the average ChIP-seq signal of these two marks at the promoters of genes (i) identified by AdaEnsemble, (ii) supported by putative chromatin loops and (iii) assigned by nearest TSS approach (Figure 3C). We found that the AdaEnsemble-identified target genes of naive TFs have higher H3K4me1 signal than those defined using the two other methods and, in contrast, those predicted for primed TFs have higher H3K4me3 signal than their counterparts.
Characterisation of transcriptional networks governed by naive and formative TFs
Using the AdaEnsemble-identified TF target genes, we next quantified the degree of overlap in transcriptional networks regulated by the seven TFs using the Jaccard index (Figure 4A). Consistent with the clustering of epigenomic features (Figure 3B), transcriptional networks controlled by multiple TFs are segregated into the naive module (i.e. Nanog, Sox2, Esrrb, Nr5a2 and Klf4) and the formative module (c-Myc and Otx2) (Figure 4A). Within the naive module, 197 of the 824 (∼24%) target genes are regulated by two or more naive TFs (Supplementary Figure S5A). For the formative module, 539 target genes are shared between Otx2 and c-Myc, and account for ∼65% and ∼35% of all formative TF target genes, respectively (Figure 4B). We next assessed the number of naive TFs that regulate a putative target gene (i.e. all proximal target genes and distal target genes supported by chromatin loop) on the basis of AdaEnsemble prediction (Figure 5B). Our results showed that approximately the same number of genes are regulated by multiple naive TFs (38.4%) compared to those that are regulated by a single naive TF (40.8%). Interestingly, genes regulated by more than one naive TF tend to have more active chromatin features, such as high H3K4me1/3, H3K27ac and Pol2 occupancy and low H3K27me3 and H3K9me2 marks (Supplementary Figure S5C). Therefore, genes bound by multiple naive TFs may have enhanced expression (53).
Figure 4.

Characterisation of TF target genes and transcriptional networks in naive and formative pluripotency. (A) Heatmap showing proportion (as quantified by Jaccard index) of genes regulated by pairs of TFs. (B) Venn diagram showing a three-way overlap of target genes of naive TFs (union), Otx2 and c-Myc. (C) Over-representation of gene ontology (GO) of target genes unique to naive TFs (naive-specific), Otx2 (Otx2-specific) and c-Myc (c-Myc-specific). (D) Contribution of target genes that are themselves TFs toward the total transcriptional regulation. (E) Correlation between (D) and the percentage of target genes that are themselves TFs (y-axis) for each of the seven TFs.
Figure 5.

Formative target genes in naive pluripotency. (A) Boxplot for comparing H3K27me3 and H3K27ac signal at the promoter of AdaEnsemble-identified TF target genes of naive and formative TFs during pluripotency progression. (B) Time-course of average log2 fold change (relative to 0 min) in mRNA expression for the target genes of naive and formative TFs. Shaded areas represent standard deviation among target genes. (C) Chromatin accessibility (ATAC-seq) of c-Myc, Otx2, and naive TF binding sites in ESCs and EpiLCs. (D) Schematic illustration of the poised chromatin associated with formative target genes in naive pluripotency.
For the target genes that are uniquely regulated by the naive TFs, Otx2 or c-Myc (Figure 4B), analysis of the gene ontology (GO) representation showed that naive-specific target genes are enriched for pathways such as negative regulation of FGF signalling and WNT receptor signalling (Figure 4C), in agreement with the known association of these signalling pathways in naive pluripotency (1). Target genes specifically regulated by c-Myc show strong enrichment for GO terms associated with RNA processing (54,55), which are known to be associated with the maintenance of pluripotency and differentiation potential of ESCs (56). Finally, we found target genes specifically regulated by Otx2 are associated with DNA-strand break repair and replication initiation (Figure 4C).
Interestingly, we found that, compared to the formative module, the naive module in general has a higher proportion of transcriptional regulation contributed by target genes that are themselves TFs (Figure 4D). Further investigation shows that, the transcriptional regulation contributed by target genes that are themselves TFs is positively correlated with their percentage in the total target genes (Figure 4E). Moreover, we found that for a given TF the ratio of distal versus proximal binding sites are positively correlated with the percentage of its target genes that are themselves TFs (Supplementary Figure S5D). These results suggest that denser TF hierarchies may exist for signal propagation in naive pluripotency wherein naive TFs rely on multiple layers of transcriptional regulation, whilst Otx2 and c-Myc may regulate genes that have more explicit biological functions that are specifically associated with the ESC to EpiLC transition.
Poised formative transcriptional networks in naive pluripotency
Epigenomic remodelling is critical for pluripotency progression (57). To delineate the rewiring of the epigenomic landscape during pluripotency transition, we analysed histone marks of naive and formative modules across the profiled time points. We found that, from 24 hours onwards, target genes specific to the naive module showed a gradual gain in H3K27me3 (a histone mark associated with the Polycomb repressive complex 2 (58)) at their promoters, an indicator of a transcriptionally repressive state, suggesting the repression of genes in the naive module (Figure 5A). The increase in H3K9me2 level, a marker of heterochromatin, demonstrated a similar specificity towards the naive module (Supplementary Figure S6A). The gradual deposition of the repressive marks is in line with the global transcriptional down-regulation of the target genes in the naive module (Figure 5A). By contrast, despite the apparent transcriptional activation of the target genes in the formative module (Figure 5B), we found that the H3K27me3 and H3K9me2 remains largely unchanged at these gene promoters throughout the ESC to EpiLC transition (Figure 5A and Supplementary Figure S6A). Likewise, we observed minimal increase in H3K27ac and H3K4me3 marks (indicators of active transcription) at the promoters of the formative target genes, whilst decreasing trends were observed for genes associated with the naive state (Figure 5A and Supplementary Figure S6B), consistent with the dissolution of the naive transcriptional networks with the induction of differentiation. The difference in variabilities may reflect the difference in the number of target genes for each TF. To further characterise the end-point of the differentiation, we also assessed the chromatin accessibility of the binding sites of c-Myc, Otx2, and the combined naive TFs using ATAC-seq generated from both initial (ESCs) and final (EpiLCs) time points (59). We found that while chromatin accessibility at naive TF binding sites reduced from the initial to the final time point, the binding sites occupied by c-Myc and Otx2 at the initial time point is open and remains open to a similar degree at the final time point (Figure 5C). Together, these results suggest that genes associated with the formative state are epigenetically poised for transcription in naive pluripotency and a limiting factor for activation of the formative transcriptional networks may be the expression level of formative TFs (Figure 5D).
Precise timing in transcriptional network rewiring during pluripotency progression
To reveal the dynamics of core transcriptional networks during the transition between the two pluripotent states, we filtered the AdaEnsemble-identified target genes for those that have a prediction confidence >0.95 and are themselves TFs (Figure 6A). As expected, the target genes are largely clustered into two modules with respect to their expression patterns across the timepoints (Supplementary Figure S7A). We subsequently reconstructed the dynamics of the time-resolved networks by taking the mean of the expression of each TF–gene pair in the networks (Figure 6B and Supplementary Table S4). We observe that, during the early time points after induction of differentiation, transcriptional regulation mediated by the naive module dominates the transcriptional networks. The collective expression of the naive module decreased concurrently with the increasing expression of the formative module, which became the dominant transcriptional networks at later time points (Supplementary Figure S7B).
Figure 6.

Dynamic rewiring of transcriptional networks from naive to formative pluripotency. (A) TF networks prior to differentiation (naive pluripotent state). AdaEnsemble-identified TF target genes (prediction probability > 0.95) that are themselves TFs are included in the network reconstruction. Edges measure mean expression of each TF–gene pair. (B) Dynamic change of TF networks during the transition from naive to formative pluripotent states. Edge colour across times reflects the change of expression of each TF–gene pair in the transcriptional networks. (C) Relative change in all pairwise expressions from naive to formative states (see ‘Materials and Methods’ section for details). (D) Mapping the differentiating ESCs to the equivalent epiblast cell populations by the activity of reconstructed transcriptional networks at different time points. Colour of the corn plots denotes enrichment from low (green) to high (red). (E) Correlation between the expression of transcription networks from single cells of the E5.5 epiblast and the ESC to EpiLC transcriptional networks at different time points.
To elucidate the precise timing from the reconstructed transcriptional networks, we summarised all pairwise gene expressions to reveal a progressively diminishing influence of the naive TFs across the 72 h of differentiation. The hierarchical clustering of pairwise correlation of the eight time-points across all TFs in the reconstructed transcriptional networks shows a transition point between 12 and 24 h after the initiation of differentiation (Supplementary Figure S7C), suggesting that the transition between naive and formative states may occur between these two time-points. Indeed, the relative changes of all pairwise expressions illustrate the same picture (Figure 6C), where the collective transcriptional network of the formative module overtakes that of the naive module, thereby dominating the transcriptional landscape at later stages of differentiation. We next spatiotemporally mapped differentiating ESCs at each time point to data from mouse epiblasts from E5.5 to E7.5 (41,60) (Supplementary Figure S7D) using our reconstructed transcriptional networks (see ‘Materials and Methods’ section). Specifically, E5.5 to E6.0 cells of the epiblasts are proposed to represent the formative state. In agreement with the timing of emergence of formative pluripotency (Figure 6C), we found the epiblast cell population at E5.5 and E6.0, corresponding to the formative state (4), closely resembles differentiating ESCs at 24 h and onwards but not prior (Figure 6D). We subsequently compared our transcriptional networks of differentiating ESCs at each time point to the E5.5 epiblasts that were profiled using single-cell RNA-seq (61). We found that following the transition to formative dominated stages, the transcriptional networks of the formative module peaked at 48 hours of differentiation and subsided afterwards (Figure 6E), suggesting that the in vivo establishment of formative state in about 48 hours post-induction, consistent with the spatiotemporal mapping results (Figure 6D). Together, these data reveal that the timeline of the rewiring of transcriptional networks during the progression from naive to formative pluripotency is tightly controlled during early embryogenesis.
DISCUSSION
The pluripotency progression of ESCs is controlled by the expression of specific transcriptional networks. Yet our ability to reconstruct these dynamic transcriptional networks underpinning the pluripotency exit is hampered by challenges in discriminating functionally relevant TFBSs and their proximal and distal target genes. Importantly, the rewiring of long-range interactions has been found to be a hallmark in pluripotent state transition in ESCs (62). Therefore, it becomes especially important within the context of the ESC to EpiLC transition to accurately define proximal and distal target genes in order to obtain an accurate depiction of the underlying transcriptional networks and their dynamics.
An array of computational methods exists for identifying TF target genes. These include correlation and machine learning-based approaches that associate expression of candidate genes with DNase I hypersensitivity signals (63), chromatin features (23), and histone marks (11–12,22,64) across multiple cell types (24,65–66), for predicting target genes of proximal and distal TFBSs. Nevertheless, these methods rely on (epi-)genomic features measured across multiple cell types and therefore cannot reveal specific dynamic changes in transcriptional networks during the transition from one cell type to another (24,67). The advent of genome-wide ChIA-PET (68,69) and Hi-C assays (21,70) has enabled the quantification of long-range three-dimensional chromatin interactions. While several recent studies have utilised this structural information as a ‘gold standard’ to validate predicted target genes of distal TFBSs (22–24,71), few methods have utilized chromatin tertiary structure information for the purposes of TF target prediction. Due to the limitation in resolution of and the biological noise from chromatin interaction analysis (a few thousand bps) (72–74) and ChIP-seq techniques (75), not all genes bound in adjacency based on ChIP-seq profile or in distance supported by chromatin interaction are truly regulated by that TF. Moreover, having a physical contact does not always imply a functional regulation of a potential target gene by a TFBS (25).
Here, we incorporated chromatin conformation information and developed an AdaEnsemble model that learns from dynamic trans-omic data, while taking into account experimental and biological data uncertainties. Although epigenomic features such as various histone modification marks have previously been utilized for TF target prediction, few studies have looked at the predictiveness of individual histone marks for target genes of specific TFs. By using the initial training procedure implemented in AdaEnsemble (Supplementary Figure S1B), we found that not all histone marks and Pol2 ChIP-seq are equally predictive at each time point (Figure 3 and Supplementary Figure S4). To account for the difference in predictiveness of each epigenomic mark and Pol2, we subsequently incorporated this information as feature weights in a weighted k-nearest neighbour (kNN) classifier in the augmented learning phase for predicting the final TF target genes. Using public functional datasets, we demonstrated the robustness of our approach by showing that the expression of many predicted target genes is significantly altered with knockdown/knockout or overexpression of their respective TFs (Figure 2E). While these functional datasets provide additional evidence for predicted TF target genes, they may not be useful for precise validation of all target genes because of secondary effects such as compensation by other TFs as well as the difficulty in determining the optimal time for transcriptome profiling following TF knockdown, knockout, or overexpression. Therefore, rather than to comprehensively identify the target genes for each TF, we focused on characterizing the properties of naive and formative transcriptional networks from the high-confidence TF target genes.
Our analyses of the AdaEnsemble-identified TF target genes suggest that naive pluripotency is enriched with pathways such as negative regulation of FGF signalling and Wnt signalling. Consistent with previous studies showing that miRNA depletion and blockade of Dgcr8-dependent miRNA biogenesis is critical to silencing self-renewal (76,77), we found that one of the major functional outputs of the formative transcriptional network is ncRNA processing (Figure 4C). Indeed, we observed that key mediators of RNA processing, including Isy1, Dgcr8 and Drosha, are among the high-confidence targets of c-Myc (Supplementary Table S2) and have been demonstrated to be important for pluripotency exit in ESCs (77–79). Although recent studies have demonstrated an important role for Otx2 for pluripotency transition from naive to formative state (80,81), the target genes of Otx2 have not fully identified and explored. Here, we found that targets of Otx2 are enriched for DNA repair and replication initiation. Together, the functional output of the formative transcriptional networks, geared towards DNA replication, post-transcriptional RNA processing and translational initiation (Figure 4C), may herald cell lineage commitment.
Recent studies propose formative cells to be executors of pluripotency, serving as mandatory intermediates en route to multi-lineage specification (4,82). Our analysis suggests that the pivotal transformation, wherein the formative transcriptional networks override the naive transcriptional networks, occurs at 12–24 h of differentiation induction (Figure 6C). Furthermore, our results showing the proximity of transcriptome between differentiating ESCs and the epiblast have allowed us to pinpoint the developmental equivalence of the EpiLCs and the early post-implantation epiblast (E5.5 and E6.0) (Figure 6D), which are reputed to be at the formative phase of the transition of pluripotency state (3,6). It is hypothesised that the dominant transcriptional networks at the formative pluripotent state will be driven by the combinatorial activity of TFs including Otx2, Sox3 and Oct6, which are also neuroectoderm lineage specifiers (4). Results of our study showed that the Otx2/c-Myc-driven transcriptional networks, which also feature the interaction of Sox2 and Pou5f1, peaked by 48 hours of in vitro differentiation. Our findings have therefore provided the first glimpse of the dynamic architecture of the formative pluripotency networks.
DATA AVAILABILITY
AdaEnsemble is implemented in the AdaSampling R package and is publicly available from [https://CRAN.R-project.org/package=AdaSampling]. DyTN, a shiny application summarising the reconstructed transcriptional networks and visualising their dynamics during the pluripotency progression, can be explored at (http://shiny.maths.usyd.edu.au/DyTN/). Mouse ESC to EpiLC time-course RNA-seq, ChIP-seq, and MS-proteomics data were downloaded from Gene Expression Omnibus (GEO) with accession number GSE117896 and PRIDE with accession number PXD010621 (27). Mouse ESC TF ChIP-seq data were downloaded from GEO with accession number GSE44288 for Nanog and Sox2 (83); GSE11431 for Esrrb, Klf4 and Myc (50); GSE19019 for Nr5a2 (49) and GSE56138 for Otx2 (26). Mouse ESC Pol2-ChIA-PET data were downloaded from GEO with accession number GSE44067 (20). Mouse ESC Microarray data were downloaded from GEO with accession number GSE4679 for Sox2 KD and Esrrb KD (84); GSE26520 for Nanog KD and Nr5a2 KD (85); GSE9775 for Klf2,4,5 KD (86); GSE56138 for Otx2 KD (26) and GSE60344 for Myc overexpression (87). Mouse ESC and EpiLC ATAC-seq data were downloaded from GEO with accession number GSE93147 (59). Mouse embryo spatial epiblast RNA-seq data were downloaded from GSE120963 (30) and mouse embryo epiblast single-cell RNA-seq data from GSE100597 (61).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dinuka Perera for developing the shiny application and our colleagues from School of Mathematics and Statistics, and Charles Perkins Centre for their discussion and feedback.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Discovery Early Career Researcher Award [DE170100759 to P.Y.]; National Health and Medical Research Council (NHMRC) Investigator Grant [1173469 to P.Y.]; Australian Research Council (ARC) Postgraduate Research Scholarship [to H.J.K.]; NHMRC Senior Principal Research Fellowship [1110751 to P.P.L.T.]; University of Sydney Postdoctoral Fellowship [to S.J.H.]; Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences [1Z1AES102625 to R.J.]; Shanghai Natural Science Foundation [18ZR1446200 to G.P.]; Science and Technology Planning Project [2017B030314056 to G.P.]. Funding for open access charge: Australian Research Council [DE170100759]; National Health and Medical Research Council [1173469].
Conflict of interest statement. None declared.
REFERENCES
- 1. Weinberger L., Ayyash M., Novershtern N., Hanna J.H.. Dynamic stem cell states: naive to primed pluripotency in rodents and humans. Nat. Rev. Mol. Cell Biol. 2016; 17:155–169. [DOI] [PubMed] [Google Scholar]
- 2. Kalkan T., Olova N., Roode M., Mulas C., Lee H.J., Nett I., Marks H., Walker R., Stunnenberg H.G., Lilley K.S. et al.. Tracking the embryonic stem cell transition from ground state pluripotency. Development. 2017; 144:1221–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kinoshita M., Smith A.. Pluripotency Deconstructed. Dev. Growth Differ. 2018; 60:44–52. [DOI] [PubMed] [Google Scholar]
- 4. Smith A. Formative pluripotency: the executive phase in a developmental continuum. Development. 2017; 144:365–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kalkan T., Smith A.. Mapping the route from naive pluripotency to lineage specification. Philos. Trans. R. Soc. London B Biol. Sci. 2014; 369:20130540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hayashi K., Ohta H., Kurimoto K., Aramaki S., Saitou M.. Reconstitution of the mouse germ cell specification pathway in culture by pluripotent stem cells. Cell. 2011; 146:519–532. [DOI] [PubMed] [Google Scholar]
- 7. Yeo J.-C., Ng H.-H.. The transcriptional regulation of pluripotency. Cell Res. 2013; 23:20–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ng H.-H., Surani M.A.. The transcriptional and signalling networks of pluripotency. Nat. Cell Biol. 2011; 13:490–496. [DOI] [PubMed] [Google Scholar]
- 9. Consortium ENCODE. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Klein S.A., Seidel D.J., Pearson B.D., Singer S.F., Michaels P.J., Cox D.I., Seidel D.J., Eskridge R.E., Peterson T.C., Vose R.S. et al.. The transcriptional landscape of the mammalian genome. Science. 2005; 309:1559–1563. [DOI] [PubMed] [Google Scholar]
- 11. Rodelsperger C., Guo G., Kolanczyk M., Pletschacher A., Kohler S., Bauer S., Schulz M.H., Robinson P.N.. Integrative analysis of genomic, functional and protein interaction data predicts long-range enhancer-target gene interactions. Nucleic Acids Res. 2011; 39:2492–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. O’Connor T., Bodén M., Bailey T.L.. CisMapper: predicting regulatory interactions from transcription factor ChIP-seq data. Nucleic Acids Res. 2016; 45:e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Oldfield A.J., Yang P., Conway A.E., Cinghu S., Freudenberg J.M., Yellaboina S., Jothi R.. Histone-fold domain protein NF-Y promotes chromatin accessibility for cell type-specific master transcription factors. Mol. Cell. 2014; 55:708–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang P., Oldfield A., Kim T., Yang A., Yang J.Y.H., Ho J.W.K.. Integrative analysis identifies co-dependent gene expression regulation of BRG1 and CHD7 at distal regulatory sites in embryonic stem cells. Bioinformatics. 2017; 33:1916–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M., Bejerano G.. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010; 28:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Andersson R., Gebhard C., Miguel-Escalada I., Hoof I., Bornholdt J., Boyd M., Chen Y., Zhao X., Schmidl C., Suzuki T. et al.. An atlas of active enhancers across human cell types and tissues. Nature. 2014; 507:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van Arensbergen J., van Steensel B., Bussemaker H.J.. In search of the determinants of enhancer–promoter interaction specificity. Trends Cell Biol. 2014; 24:695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pennacchio L.A., Bickmore W., Dean A., Nobrega M.A., Bejerano G.. Enhancers: five essential questions. Nat. Rev. Genet. 2013; 14:288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sanyal A., Lajoie B.R., Jain G., Dekker J.. The long-range interaction landscape of gene promoters. Nature. 2012; 489:109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang Y., Wong C.-H.C.-H., Birnbaum R.Y., Li G., Favaro R., Ngan C.Y., Lim J., Tai E., Poh H.M., Wong E. et al.. Chromatin connectivity maps reveal dynamic promoter-enhancer long-range associations. Nature. 2013; 504:306–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B.. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012; 485:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Roy S., Siahpirani A.F., Chasman D., Knaack S., Ay F., Stewart R., Wilson M., Sridharan R.. A predictive modeling approach for cell line-specific long-range regulatory interactions. Nucleic Acids Res. 2015; 43:8694–8712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Whalen S., Truty R.M., Pollard K.S.. Enhancer-promoter interactions are encoded by complex genomic signatures on looping chromatin. Nat. Genet. 2016; 48:488–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. He B., Chen C., Teng L., Tan K.. Global view of enhancer-promoter interactome in human cells. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:E2191–E2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shlyueva D., Stampfel G., Stark A.. Transcriptional enhancers: from properties to genome-wide predictions. Nat. Rev. Genet. 2014; 15:272–286. [DOI] [PubMed] [Google Scholar]
- 26. Buecker C., Srinivasan R., Wu Z., Calo E., Acampora D., Faial T., Simeone A., Tan M., Swigut T., Wysocka J.. Reorganization of enhancer patterns in transition from naive to primed pluripotency. Cell Stem Cell. 2014; 14:838–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang P., Humphrey S.J., Cinghu S., Pathania R., Oldfield A.J., Kumar D., Perera D., Yang J.Y.H., James D.E., Mann M. et al.. Multi-omic profiling reveals dynamics of the phased progression of pluripotency. Cell Syst. 2019; 8:427–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang P., Liu W., Yang J.. Positive unlabeled learning via wrapper-based adaptive sampling. Proceedings of the Twenty-Sixth International Joint Conference on Artificial Intelligence. 2017; California: International Joint Conferences on Artificial Intelligence Organization; 3273–3279. [Google Scholar]
- 29. Yang P., Ormerod J.T., Liu W., Ma C., Zomaya A.Y., Yang J.Y.H.. AdaSampling for positive-unlabeled and label noise learning with bioinformatics applications. IEEE Trans. Cybern. 2019; 49:1932–1943. [DOI] [PubMed] [Google Scholar]
- 30. Peng G., Suo S., Cui G., Yu F., Wang R., Chen J., Chen S., Liu Z., Chen G., Qian Y. et al.. Molecular architecture of lineage allocation and tissue organization in early mouse embryo. Nature. 2019; 572:528–532. [DOI] [PubMed] [Google Scholar]
- 31. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Anders S., Pyl P.T., Huber W.. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015; 31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Langmead B., Trapnell C., Pop M., Salzberg S.L.. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009; 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cox J., Mann M.. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008; 26:1367–1372. [DOI] [PubMed] [Google Scholar]
- 35. Zhang H.-M., Chen H., Liu W., Liu H., Gong J., Wang H., Guo A.-Y.. AnimalTFDB: a comprehensive animal transcription factor database. Nucleic Acids Res. 2012; 40:D144–D149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vaquerizas J.M., Kummerfeld S.K., Teichmann S.A., Luscombe N.M.. A census of human transcription factors: function, expression and evolution. Nat. Rev. Genet. 2009; 10:252–263. [DOI] [PubMed] [Google Scholar]
- 37. Jothi R., Cuddapah S., Barski A., Cui K., Zhao K.. Genome-wide identification of in vivo protein-DNA binding sites from ChIP-Seq data. Nucleic Acids Res. 2008; 36:5221–5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Csardi G., Nepusz T.. The igraph software package for complex network research. InterJournal Complex Syst. 2006; 1695:1–9. [Google Scholar]
- 39. Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K.. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. The Gene Ontology Consortium Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015; 43:D1049–D1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Peng G., Suo S., Chen J., Chen W., Liu C., Yu F., Wang R., Chen S., Sun N., Cui G. et al.. Spatial transcriptome for the molecular annotation of lineage fates and cell identity in mid-gastrula mouse embryo. Dev. Cell. 2016; 36:681–697. [DOI] [PubMed] [Google Scholar]
- 42. Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.C., Geurts P., Aerts J. et al.. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods. 2017; 14:1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lu P., Vogel C., Wang R., Yao X., Marcotte E.M.. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat. Biotechnol. 2007; 25:117–124. [DOI] [PubMed] [Google Scholar]
- 44. Avilion A.A., Nicolis S.K., Pevny L.H., Perez L., Vivian N., Lovell-Badge R.. Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev. 2003; 17:126–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chambers I., Colby D., Robertson M., Nichols J., Lee S., Tweedie S., Smith A.. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell. 2003; 113:643–655. [DOI] [PubMed] [Google Scholar]
- 46. Mitsui K., Tokuzawa Y., Itoh H., Segawa K., Murakami M., Takahashi K., Maruyama M., Maeda M., Yamanaka S.. The homeoprotein nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell. 2003; 113:631–642. [DOI] [PubMed] [Google Scholar]
- 47. Festuccia N., Osorno R., Halbritter F., Karwacki-Neisius V., Navarro P., Colby D., Wong F., Yates A., Tomlinson S.R., Chambers I.. Esrrb is a direct Nanog target gene that can substitute for Nanog function in pluripotent cells. Cell Stem Cell. 2012; 11:477–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Takahashi K., Yamanaka S.. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006; 126:663–676. [DOI] [PubMed] [Google Scholar]
- 49. Heng J.-C.D., Feng B., Han J., Jiang J., Kraus P., Ng J.-H., Orlov Y.L., Huss M., Yang L., Lufkin T. et al.. The nuclear receptor Nr5a2 Can Replace Oct4 in the reprogramming of murine somatic cells to pluripotent cells. Cell Stem Cell. 2010; 6:167–174. [DOI] [PubMed] [Google Scholar]
- 50. Chen X., Xu H., Yuan P., Fang F., Huss M., Vega V.B., Wong E., Orlov Y.L., Zhang W., Jiang J. et al.. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008; 133:1106–1117. [DOI] [PubMed] [Google Scholar]
- 51. Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J., Hanna J., Lodato M.A., Frampton G.M., Sharp P.A. et al.. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cinghu S., Yang P., Kosak J.P., Conway A.E., Kumar D., Oldfield A.J., Adelman K., Jothi R.. Intragenic enhancers attenuate host gene expression. Mol. Cell. 2017; 68:104–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chronis C., Fiziev P., Papp B., Butz S., Bonora G., Sabri S., Ernst J., Plath K.. Cooperative binding of transcription factors orchestrates reprogramming. Cell. 2017; 168:442–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Van Riggelen J., Yetil A., Felsher D.W.. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer. 2010; 10:301–309. [DOI] [PubMed] [Google Scholar]
- 55. Zheng G.X.Y., Do B.T., Webster D.E., Khavari P.A., Chang H.Y.. Dicer-microRNA-Myc circuit promotes transcription of hundreds of long noncoding RNAs. Nat. Struct. Mol. Biol. 2014; 21:585–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Guttman M., Donaghey J., Carey B.W., Garber M., Grenier J.K., Munson G., Young G., Lucas A.B., Ach R., Bruhn L. et al.. LincRNAs act in the circuitry controlling pluripotency and differentiation. Nature. 2011; 477:295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kurimoto K., Yabuta Y., Hayashi K., Ohta H., Kiyonari H., Mitani T., Moritoki Y., Kohri K., Kimura H., Yamamoto T. et al.. Quantitative dynamics of chromatin remodeling during germ cell specification from mouse embryonic stem cells. Cell Stem Cell. 2015; 16:517–532. [DOI] [PubMed] [Google Scholar]
- 58. Hansen K.H., Bracken A.P., Pasini D., Dietrich N., Gehani S.S., Monrad A., Rappsilber J., Lerdrup M., Helin K.. A model for transmission of the H3K27me3 epigenetic mark. Nat. Cell Biol. 2008; 10:1291–1300. [DOI] [PubMed] [Google Scholar]
- 59. Chen A.F., Liu A.J., Krishnakumar R., Freimer J.W., DeVeale B., Blelloch R.. GRHL2-dependent enhancer switching maintains a pluripotent stem cell transcriptional subnetwork after exit from naive pluripotency. Cell Stem Cell. 2018; 23:226–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cui G., Suo S., Wang R., Qian Y., Han J.D.J., Peng G., Tam P.P.L., Jing N.. Mouse gastrulation: Attributes of transcription factor regulatory network for epiblast patterning. Dev. Growth Differ. 2018; 60:463–472. [DOI] [PubMed] [Google Scholar]
- 61. Mohammed H., Hernando-Herraez I., Savino A., Scialdone A., Macaulay I., Mulas C., Chandra T., Voet T., Dean W., Nichols J. et al.. Single-cell landscape of transcriptional heterogeneity and cell fate decisions during mouse early gastrulation. Cell Rep. 2017; 20:1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Novo C.L., Javierre B.M., Cairns J., Segonds-Pichon A., Wingett S.W., Freire-Pritchett P., Furlan-Magaril M., Schoenfelder S., Fraser P., Rugg-Gunn P.J.. Long-range enhancer interactions are prevalent in mouse embryonic stem cells and are reorganized upon pluripotent state transition. Cell Rep. 2018; 22:2615–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Thurman R.E., Rynes E., Humbert R., Vierstra J., Maurano M.T., Haugen E., Sheffield N.C., Stergachis A.B., Wang H., Vernot B. et al.. The accessible chromatin landscape of the human genome. Nature. 2012; 489:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shen Y., Yue F., McCleary D.F., Ye Z., Edsall L., Kuan S., Wagner U., Dixon J., Lee L., Lobanenkov V. V. et al.. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012; 488:116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ernst J., Kheradpour P., Mikkelsen T.S., Shoresh N., Ward L.D., Epstein C.B., Zhang X., Wang L., Issner R., Coyne M. et al.. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011; 473:43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yip K.Y., Cheng C., Bhardwaj N., Brown J.B., Leng J., Kundaje A., Rozowsky J., Birney E., Bickel P., Snyder M. et al.. Classification of human genomic regions based on experimentally determined binding sites of more than 100 transcription-related factors. Genome Biol. 2012; 13:R48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yip K.Y., Cheng C., Gerstein M.. Machine learning and genome annotation: a match meant to be. Genome Biol. 2013; 14:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Fullwood M.J., Wei C.L., Liu E.T., Ruan Y.. Next-generation DNA sequencing of paired-end tags (PET) for transcriptome and genome analyses. Genome Res. 2009; 19:521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhang J., Poh H.M., Peh S.Q., Sia Y.Y., Li G., Mulawadi F.H., Goh Y., Fullwood M.J., Sung W.K., Ruan X. et al.. ChIA-PET analysis of transcriptional chromatin interactions. Methods. 2012; 58:289–299. [DOI] [PubMed] [Google Scholar]
- 70. Belton J.M., McCord R.P., Gibcus J.H., Naumova N., Zhan Y., Dekker J.. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods. 2012; 58:268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hafez D., Karabacak A., Krueger S., Hwang Y.C., Wang L.S., Zinzen R.P., Ohler U.. McEnhancer: Predicting gene expression via semi-supervised assignment of enhancers to target genes. Genome Biol. 2017; 18:199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rao S.S.P., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T., Sanborn A.L., Machol I., Omer A.D., Lander E.S. et al.. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014; 159:1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jin F., Li Y., Dixon J.R., Selvaraj S., Ye Z., Lee A.Y., Yen C.-A., Schmitt A.D., Espinoza C.A., Ren B.. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. 2013; 503:290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Heidari N., Phanstiel D.H., He C., Grubert F., Jahanbani F., Kasowski M., Zhang M.Q., Snyder M.P.. Genome-wide map of regulatory interactions in the human genome. Genome Res. 2014; 24:1905–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kidder B.L., Hu G., Zhao K.. ChIP-Seq: technical considerations for obtaining high-quality data. Nat. Immunol. 2011; 12:918–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang Y., Medvid R., Melton C., Jaenisch R., Blelloch R.. DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nat. Genet. 2007; 39:380–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Melton C., Judson R.L., Blelloch R.. Opposing microRNA families regulate self-renewal in mouse embryonic stem cells. Nature. 2010; 463:621–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Du P., Pirouz M., Choi J., Huebner A.J., Clement K., Meissner A., Hochedlinger K., Gregory R.I.. An intermediate pluripotent state controlled by MicroRNAs is required for the naive-to-primed stem cell transition. Cell Stem Cell. 2018; 22:851–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Cirera-Salinas D., Yu J., Bodak M., Ngondo R.P., Herbert K.M., Ciaudo C.. Noncanonical function of DGCR8 controls mESC exit from pluripotency. J. Cell Biol. 2017; 216:355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Acampora D., Di Giovannantonio L.G., Simeone A.. Otx2 is an intrinsic determinant of the embryonic stem cell state and is required for transition to a stable epiblast stem cell condition. Development. 2013; 140:43–55. [DOI] [PubMed] [Google Scholar]
- 81. Yang S.H., Kalkan T., Morissroe C., Marks H., Stunnenberg H., Smith A., Sharrocks A.D.. Otx2 and Oct4 drive early enhancer activation during embryonic stem cell transition from naive pluripotency. Cell Rep. 2014; 7:1968–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Rossant J., Tam P.P.L.. New insights into early human development: lessons for stem cell derivation and differentiation. Cell Stem Cell. 2017; 20:18–28. [DOI] [PubMed] [Google Scholar]
- 83. Whyte W.A., Orlando D.A., Hnisz D., Abraham B.J., Lin C.Y., Kagey M.H., Rahl P.B., Lee T.I., Young R.A.. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013; 153:307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ivanova N., Dobrin R., Lu R., Kotenko I., Levorse J., DeCoste C., Schafer X., Lun Y., Lemischka I.R.. Dissecting self-renewal in stem cells with RNA interference. Nature. 2006; 442:533–538. [DOI] [PubMed] [Google Scholar]
- 85. Nishiyama A., Sharov A.A., Piao Y., Amano M., Amano T., Hoang H.G., Binder B.Y., Tapnio R., Bassey U., Malinou J.N. et al.. Systematic repression of transcription factors reveals limited patterns of gene expression changes in ES cells. Sci. Rep. 2013; 3:1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Jiang J., Chan Y.-S., Loh Y.-H., Cai J., Tong G.-Q., Lim C.-A., Robson P., Zhong S., Ng H.-H.. A core Klf circuitry regulates self-renewal of embryonic stem cells. Nat. Cell Biol. 2008; 10:353–360. [DOI] [PubMed] [Google Scholar]
- 87. Hishida T., Nakachi Y., Mizuno Y., Katano M., Okazaki Y., Ema M., Takahashi S., Hirasaki M., Suzuki A., Ueda A. et al.. Functional compensation between Myc and PI3K signaling supports self-renewal of embryonic stem cells. Stem Cells. 2015; 33:713–725. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
AdaEnsemble is implemented in the AdaSampling R package and is publicly available from [https://CRAN.R-project.org/package=AdaSampling]. DyTN, a shiny application summarising the reconstructed transcriptional networks and visualising their dynamics during the pluripotency progression, can be explored at (http://shiny.maths.usyd.edu.au/DyTN/). Mouse ESC to EpiLC time-course RNA-seq, ChIP-seq, and MS-proteomics data were downloaded from Gene Expression Omnibus (GEO) with accession number GSE117896 and PRIDE with accession number PXD010621 (27). Mouse ESC TF ChIP-seq data were downloaded from GEO with accession number GSE44288 for Nanog and Sox2 (83); GSE11431 for Esrrb, Klf4 and Myc (50); GSE19019 for Nr5a2 (49) and GSE56138 for Otx2 (26). Mouse ESC Pol2-ChIA-PET data were downloaded from GEO with accession number GSE44067 (20). Mouse ESC Microarray data were downloaded from GEO with accession number GSE4679 for Sox2 KD and Esrrb KD (84); GSE26520 for Nanog KD and Nr5a2 KD (85); GSE9775 for Klf2,4,5 KD (86); GSE56138 for Otx2 KD (26) and GSE60344 for Myc overexpression (87). Mouse ESC and EpiLC ATAC-seq data were downloaded from GEO with accession number GSE93147 (59). Mouse embryo spatial epiblast RNA-seq data were downloaded from GSE120963 (30) and mouse embryo epiblast single-cell RNA-seq data from GSE100597 (61).