

Abstract
Distal renal tubular acidosis is a rare renal tubular disorder characterized by hyperchloremic metabolic acidosis and impaired urinary acidification. Mutations in three genes (ATP6V0A4, ATP6V1B1 and SLC4A1) constitute a monogenic causation in 58–70% of familial cases of distal renal tubular acidosis. Recently, mutations in FOXI1 have been identified as an additional cause. Therefore, we hypothesized that further monogenic causes of distal renal tubular acidosis remain to be discovered. Panel sequencing and/or whole exome sequencing was performed in a cohort of 17 families with 19 affected individuals with pediatric onset distal renal tubular acidosis. A causative mutation was detected in one of the three “classical” known distal renal tubular acidosis genes in 10 of 17 families. The seven unsolved families were then subjected to candidate whole exome sequencing analysis. Potential disease causing mutations in three genes were detected: ATP6V1C2, which encodes another kidney specific subunit of the V-type proton ATPase (1 family); WDR72 (2 families), previously implicated in V-ATPase trafficking in cells; and SLC4A2 (1 family), a paralog of the known distal renal tubular acidosis gene SLC4A1. Two of these mutations were assessed for deleteriousness through functional studies. Yeast growth assays for ATP6V1C2 revealed loss-of-function for the patient mutation, strongly supporting ATP6V1C2 as a novel distal renal tubular acidosis gene. Thus, we provided a molecular diagnosis in a known distal renal tubular acidosis gene in 10 of 17 families (59%) with this disease, identified mutations in ATP6V1C2 as a novel human candidate gene, and provided further evidence for phenotypic expansion in WDR72 mutations from amelogenesis imperfecta to distal renal tubular acidosis.
Keywords: distal tubule, renal acidification, renal tubular acidosis, pediatric nephrology
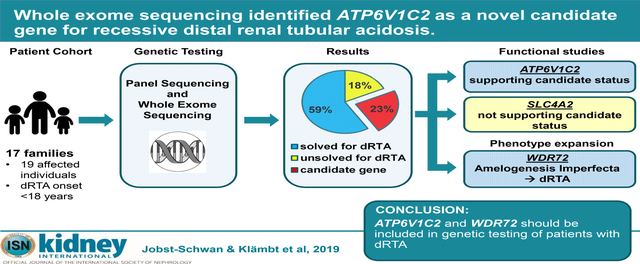
Graphical Abstract
INTRODUCTION
Distal renal tubular acidosis (dRTA) is a rare renal tubular disorder characterized by hyperchloremic, hypokalemic metabolic acidosis accompanied by impaired urinary acidification. In many cases, this metabolic condition leads to growth retardation, osteomalacia, severe muscle weakness, nephrolithiasis, nephrocalcinosis (NC), and, if untreated, progression to renal failure.1, 2 While the major causes of dRTA in adults are autoimmune diseases, childhood onset dRTA is mainly due to monogenic mutations. Mutations in the genes ATP6V0A4 and ATP6V1B1, both encoding subunits of the V-type proton ATPase, are causative in the majority of recessive cases of dRTA and are often accompanied by sensorineural deafness (SD) due to gene co-expression in the inner ear.1, 3, 4 Mutations in SLC4A1 have been identified as causative for autosomal recessive as well as dominant cases of dRTA without SD.5 These 3 “classical genes” account for 58–70% of familial dRTA cases,1, 6 suggesting the existence of yet undefined monogenic causes of dRTA. Accordingly, mutations in the gene FOXI1 have been identified in 2 families with dRTA and functional studies of the mutation show loss-of-function of the variant.7 Furthermore, very recently WDR72 mutations have been proposed to expand the human phenotype of amelogenesis imperfecta (AI) by dRTA,8, 9 underlining the potential for further identification of novel dRTA disease genes.
In order to identify novel monogenic causes for dRTA, we performed panel sequencing and/or whole exome sequencing (WES) in a cohort of 19 affected individuals from 17 families diagnosed with pediatric onset dRTA of unassigned molecular diagnosis. 10 of the 17 families had been prescreened for the 3 classical dRTA genes by gene panel sequencing. The unsolved cases, and all patients recruited subsequently, were subject to WES. This sequential procedure allowed diagnosis of mutations in 1 of the 3 classical dRTA genes in 10/17 families (59%). Furthermore, we identified novel mutations in WDR72 in 2 families with dRTA and AI, and in 2 potential novel dRTA genes in 2 families: ATP6V1C2, and SLC4A2. Functional studies for ATP6V1C2 and SLC4A2 were performed, and were conclusive for loss-of-function for the ATP6V1C2 mutation.
RESULTS
We evaluated 19 affected individuals from 17 families with clinically diagnosed dRTA that presented before the age of 25 years (Fig. 1). Among these 19 individuals, 17 had NC or renal stones and 8 presented with SD (Table 1). 10 families were reported to be consanguineous. Initial panel screening was performed by multiplex PCR in 11 individuals from 10 families for the 3 classical dRTA genes (ATP6V0A4, ATPV1B1, SLC4A1). Genomic DNAs of the 3 families not solved by panel sequencing and 7 additional families subsequently recruited were then subjected to WES (Fig. 1).
Figure 1: Flow Diagram for gene detection by panel sequencing and whole exome sequencing of likely causative monogenic mutations leading to distal renal tubular acidosis (dRTA), and candidate genes in 17 families.

By panel sequencing 10 families were tested for 3 known dRTA genes. Among those 10 families, 3 families were “unsolved” and assessed for whole exome sequencing, together with 7 additional families. A likely causative mutation in a known gene was detected in 10 families (59%) and in 4 families mutations indicating novel candidate dRTA genes were discovered.
TABLE 1.
Clinical and genetic data of 10 families with dRTA and mutations in known dRTA genes.
| Gene Family - Individual | Nucleotide change | Amino acid change | Exon (zygosity, segregation) | PPH2 score | SIFT | Mut taster | Amino acid conservation to species | Gnomad allele frequencies (hom/hemi/het/wt) | Mutation reported in Reference | ACMG classification | Gender | Ethnic origin | Parental consanguinity | Age of onset, yrs | Acidosis | Hypokalemia | Urine pH >5.5 under acidosis | Hypercalciuria | Nephro-Calcinosis | Deafness | T reatment |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ATP6V0A4, ATPase H+ Transporting V0 Subunit A4 (NM_020632.2) | |||||||||||||||||||||
| F589 -22 |
c.2446A>G | p.Lys816Glu | 23 (hom, m,p) | 0.999 | Del | Dis | S. cerevisiae | not reported | novel | likely pathogenic | F | Turkey | Yes | 2 | Yes | Yes | Yes | Yes | Yes | No | NaHCO3, KHCO3 |
| B964 -21 |
c.1346G>A | p.Arg449His | 15 (hom) | 1 | Del | Dis | S. cerevisiae | 0/0/3/246,228 | 62 | likely pathogenic | F | n/a | Yes | <1 | Yes | Yes | Yes | Yes | Yes | No | K-Citrate, NaHCO3 |
| B1673 -21 -22 |
c.1312_1315 del | p.Asp438 Metfs*13 | 14 (hom) | n/a | n/a | n/a | n/a | not reported | 63 | pathogenic | F F |
Serbia Serbia |
No No |
n/d n/d |
Yes Yes |
Yes Yes |
Yes Yes |
Yes Yes |
Stones Stones |
Yes Yes |
K/Na-Citrate, KHCO3 |
| ATP6V1B1, ATPase H+ Transporting V1 Subunit B1 (NM_001692.3) | |||||||||||||||||||||
| F391 -21 |
c.1037C>G | p.Pro346Arg | 10 (hom, m, p) | 1 | Del | Dis | S. cerevisiae | 0/0/4/245,492 | 4 | pathogenic | F | Germany (Turkish) | Yes | <1 | Yes | Yes | Yes | n/a | Yes | Yes | NaHCO3, KHCO3 |
| F608 -21 |
c.497del | p.Thr166 Argfs*9 | 6 (hom) | n/a | n/a | n/a | n/a | 0/0/2/246,220 | 4 | pathogenic | F | Jordan | No | 6 | Yes | Yes | n/d | n/d | Yes | Yes | NaHCO3, KHCO3 |
| F769 -21 |
c.242T>C | p.Leu81Pro | 3 (hom, m, p) | 1 | Del | Dis | S. cerevisiae | 0/0/1/245,488 | 4 | pathogenic | F | Switzerland | No | <1 | Yes | n/d | Yes | n/d | Yes | Yes | NaHCO3, K- Citrate |
| B1459 -21 |
c.1037C>G | p.Pro346Arg | 10 (hom) | 1 | Del | Dis | S. cerevisiae | 0/0/4/245,492 | 4 | pathogenic | F | Yemen | No | <1 | Yes | Yes | n/d | Yes | Yes | Yes | K-Citrate |
| SLC4A1, Solute Carrier Family 4 Member 1 (NM_000342.2) | |||||||||||||||||||||
| F1049 -21 |
c.1766G>A | p.Arg589His | 14 (het) | 0.996 | Del | Dis | C. intestinalis | not reported | 5 | pathogenic | F | Switzerland (Turkish) | n/a | 2 | Yes | n/d | Yes | n/d | Yes | No | K/Na-Citrate |
| B3155 -21 |
c.2573C>A c.1199_1225del |
p.Ala858Asp p.Ala400_Ala408del |
19 (het) 11 (het) |
0.774 n/a |
Del n/a |
Dis n/a |
D. melanogaster | 0/0/17/245,910 0/0/13/282,788 |
64 | pathogenic pathogenic |
F | Malaysia | Yes | 3 | Yes | n/d | n/d | n/d | Yes | No | NaHCO3 |
| B2838 -21 |
c.2573C>A | p.Ala858Asp | 19 (hom) | 0.774 | Del | Dis | D. melanogaster | 0/0/17/245,910 | 64 | pathogenic | F | Pakistani | Yes | n/d | Yes | No | Yes | Yes | Yes | No | K/Na-Citrate |
Del, deleterious; Dis, disease-causing; hemi, hemizygous; het, heterozygous; hom, homozygous; m, maternal; mo., months; n/d, no data; p, paternal; PPH2 score, humvar PolyPhen2 prediction score; SIFT, Sorting intolerant from tolerant; tol, tolerated; VUS, variant of unknown significance; wt, wildtype; yrs, years.
Known genes
Using our established evaluation based on ACMG criteria for deleteriousness for alleles of monogenic disease genes10 we identified likely causative mutations in 10/17 families (59%) in 1 of the 3 classical dRTA genes (Figure 1, Table 1). Specifically, we identified recessive mutations in ATP6V0A4 (NM_020632.2) in 3 families (4 individuals) and in ATP6V1B1 (NM_001692.3) in 4 families (4 individuals), and SLC4A1 (NM_000342.2) mutations in 3 families (3 individuals, 1 recessive homozygous, 1 recessive compound heterozygous, 1 dominant heterozygous mutation) (Table 1). 9 of the 10 identified mutations had been previously reported. The homozygous mutation in ATP6V0A4 in family F589 (c.2446A>G, p.Lys816Glu) is a novel mutation affecting a lysine that has been conserved throughout evolution starting from yeast. The mutation has strong prediction scores and is unreported in either homozygous or heterozygous state in the healthy exome and genome database gnomAD (Table 1).
ATP6V1C2
In the 7 dRTA cases in whom no causative mutation was found, we performed WES analysis and utilized the WES datasets for homozygosity mapping to evaluate consanguineous cases for homozygous recessive mutations within homozygous haplotypes.11 We identified a very likely deleterious recessive mutation in the gene ATP6V1C2 (NM_001039362.1) in family F588 (Fig. 2A, Table 2, Suppl. Fig. S1A). Like ATP6V0A4 and ATP6V1B1, ATP6V1C2 encodes a subunit (subunit C) of the V-type proton ATPase (Fig. 3A).12 In contrast to its paralog ATP6V1C1, ATP6V1C2 is predominantly expressed in the kidney with high expression in renal intercalated cells (IC) (Fig. 2D).13, 14 The gene has been implicated as a human candidate disease gene for dRTA but no mutations have been identified so far.14 The mutation c.503T>C, p.Ile168Thr changes the nonpolar aa residue Ile to the polar Thr. Alignment of the orthologs protein sequences reveals evolutionary conservation of Ile or the biochemically similar Leu from yeast to humans (Fig. 2A). Ile168 is located in a hydrophobic knuckle (Suppl. Fig. S2) that slides against another hydrophobic surface of the C-subunit during the ATPase catalytic cycle.15 We modeled this human mutation in the yeast ortholog VMA5 (YKL080W) that encodes the yeast V-ATPase C subunit and introduced the corresponding mutation (p.Ile178Thr) into the genomic copy of VMA5 to obtain vma5 p.Ile178Thr mutant strains. Thermal protein stability was predicted for the 3 states of Vma5 reported by Zhao et al.15 Protein stability prediction (CUPSAT) suggests stabilization of state 1 (Suppl. Fig. S3A) but destabilization of state 2 (Suppl. Fig. S3B) and state 3 (Suppl. Fig. S3C).16 These results predict stabilization of the protein in state 1, possibly preventing the dynamic conformational changes likely required for stable function of the active ATPase.
Figure 2: Whole exome sequencing in 4 families with dRTA identifies potentially disease-causing recessive mutations in 3 genes, ATP6V1C2, WDR72, and SLC4A2, that are expressed in intercalated cells.

Exon structure and protein domains of human ATP6V1C2 (A), WDR72 (B), and SLC4A2 (C) cDNA. Positions of start codons (ATG) and stop codons (TAA, TGA) are indicated. Exon numbers are marked on a black or white background. Protein domain lengths are indicated by the colored boxes. Mutation positions are indicated by black arrows in relation to the exon and the protein domain (see also Table 1). Chromatograms of recessive mutations identified in dRTA patients are indicated under each protein domain diagram. A-C. Black arrowheads or red highlights denote altered nucleotide. CLUSTAL-generated amino acid sequence alignments of ATP6V1C2 and SLC4A2 orthologs are shown for the regions surrounding sites of missense mutation (A, C). (D) Single cell type specific average expression of dRTA genes. Data was modified from Park et al.24 The heat map is based on z-scores. Each column represents a cell type and each row represents one gene. aa: amino acid; bp: base pairs. Endo Endothelium, Podo Podocytes, PT proximal tubule, LOH loop of Henle, DCT distal convoluted tubule, CD-PC collecting duct - primary cells, CD-IC collecting duct - intercalated cells.
TABLE 2.
Clinical and genetic data of 4 families with dRTA with mutations in the novel candidate genes ATP6V1C2, WDR72, and SLC4A2.
| Gene Family - Individual | Nucleotide change | Amino acid change | Exon (zygosity, segregation) | PPH2 score | SIFT | Mut taster | Amino acid conservation to species | Gnomad allele frequencies (hom/hemi/het/wt) | Mutation reported | ACMG classification | Gender | Ethnic origin | Parental consanguinity | Age of onset, yrs | Acidosis pH HCO3 (mmol/L) | Hypo-Kalemia (mmol/L) | Urine pH >5.5 under acidosis | Hypercalciuiria | Nephrocalcinosis | Deafness | Treatment | Comment |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ATP6V1C2, ATPase H+ Transporting V1 Subunit C2 (NM_001039362.1) | ||||||||||||||||||||||
| B1022 -21 |
c.503T>C | p.Ile168Thr | 7 (hom, m, p) | 0.975 | Del | Dis | C. intestinalis | 0/0/224/277222 | novel | likely pathogenic | F | Egypt | Yes | <1 | Yes ph 7.2 HCO3 10 | Yes 1.8 | 8.6 | No | No | No | Polycitrate Potassium | Death age 9 mo due to renal failure |
| WDR72, WD Repeat Domain 72 (NM_182758.3) | ||||||||||||||||||||||
| F382 -25 |
c.477_485dup | p.(Ile159_Cys161dup) | 5 (hom) | n/a | n/a | n/a | n/a | not reported | novel | likely pathogenic | F | Turkey | Yes | 5 | Yes pH 7.1 HCO3 11.5 | Yes 2.5 | 8 | Yes | Yes | No | NaHCO3 KHCO3 | Enamel Defect |
| B2673 -21 -22 |
c.764_768delGGCAG | p.Gly255Val fs*40 | 8 (hom) | n/a | n/a | n/a | n/a | 0/0/1/246104 | novel | pathogenic | F M |
Indian | No | 3 <1 |
Yes 11 Yes 10 |
Yes 2.4 Yes 2.8 |
7 7.5 |
No No |
No No |
Yes n/d |
NaHCO3 KHCO3 | Enamel Defect Enamel Defect |
| SLC4A2, Solute Carrier Family 4 Member 2 (NM_01199692.2) | ||||||||||||||||||||||
| F588 -21 |
c.2107G>A | p.Ala703Thr | 14 (hom, m, p) | 0.995 | Del | Dis | G. gallus | 0/0/1/250904 | novel | VUS | F | Turkey | Yes | 1 | Yes 7.1 | Yes 2.5 | 7 | Yes | Yes | Yes | NaHCO3, KHCO3 | |
Del, deleterious; Dis, disease-causing; hemi, hemizygous; het, heterozygous; hom, homozygous; m, maternal; mo., months; n/d; no data; p, paternal; PPH2 score, humvar PolyPhen2 prediction score; SIFT, Sorting intolerant from tolerant; tol, tolerated; wt, wildtype; yrs, years.
Figure 3: vma5 (I178T) mutants display a growth defect and reduced V-ATPase activity.

(A) Structure of V-ATPase (modified from12) including a peripheral domain (V1) important for ATP hydrolysis and an integral domain (V0) responsible for proton translocation. The V1 domain includes A and B subunits in a hexameric arrangement connected to the V0 domain via peripheral stalks comprising subunits C, E, G, H and the N-terminal domain of subunit a. The V0 domain includes a ring of proteolipids (c) adjacent to subunits a and e. (B) 3 independent isolates (9, 11, 15) containing the I178T vma5 mutation fail to grow on YEPD, pH 7.5, CaCl2 plates, whereas no growth loss was evident on YEPD, pH 5 plates. (C) Mean concanamycin A-sensitive V-ATPase activity in vacuolar vesicles from WT and vma5 I175T mutants (n=3 for each, error bars represent S.E.) (D) The levels of both membrane-bound (vacuolar V0 subunit a, Vph1) and peripheral V1 subunits (A, B, and C) were visualized by immunoblot and normalized to mature ALP (mALP), a vacuolar membrane protein. mALP runs as 2 bands as in the wild-type sample. If V-ATPase activity and therefore, vacuolar proteolytic activity, is reduced, a third higher molecular weigth pro-ALP band (pALP) appears, which is present in the mutants. The levels of Vph1 were similar in vacuolar vesicles prepared from 3 different mutant strains compared to WT (ns, P=0.94), but the level of the Vma5 protein was reduced (***, P ≤ 0.001), as were the levels of the V1 subunits, A (**, P ≤ 0.01) and B (*, P ≤ 0.05).
(E) V1 subunits (A, B, and C) were measured via immunoblot in yeast whole cell lysates of WT and 3 different mutant strains and normalized to PKC as loading control. Levels of the C subunit are reduced in the mutant strains (**, P ≤ 0.01).
To further assess loss-of-function of the p.Ile168Thr mutation in ATP6V1C2 (p.Ile178Thr in yeast vma5), we tested the growth of 3 independent mutant isolates compared to wild-type (WT) cells grown on YEPD medium buffered to pH 5 and on YEPD medium buffered to pH 7.5 containing 60 mM CaCl2 (Fig. 3B). Vma mutants with no V-ATPase activity are able to grow at pH 5, while an elevated pH or Ca2+ concentration is lethal to yeast mutants lacking the V-ATPase activity.17, 18 Here, we showed that vma5 (Ile178Thr) mutant indeed failed to grow on YEPD, pH 7.5 +CaCl2 plates, suggesting a significant loss of V-ATPase function (Fig. 3B).
To further explore the source of the growth defect, we isolated vacuolar vesicles from WT and mutant yeast strains. Vacuolar vesicles isolated from 3 independent mutant strains had concanamycin A-sensitive V-ATPase activity, but this activity was much lower than V-ATPase activity in WT vacuoles (Figure 3C), We visualized the levels of both membrane-bound (vacuolar V0 subunit a, Vph1) and peripheral V1 subunits (A, B, and C) by immunoblot. Importantly, the V0 and V1 subcomplexes can assemble independently and the V0 subcomplex reaches the vacuole even in the complete absence of V1 subunits. Consistent with this, the levels of V0 subunit Vph1, ortholog of human subunit a (Fig. 3A) were comparable between the WT and mutant strains (Fig. 3D), indicating that the V0 complex is correctly assembled and transported to the vacuole. In contrast, the level of the Vma5 protein in the mutant vacuolar vesicles was reduced, as were the levels of V1 subunits A and B (Fig. 3D). By immunoblot of whole cell lysates, we investigated whether the reduced Vma5 levels reflected impaired V-ATPase assembly or poor stability of the mutant Vma5 polypeptide (Fig. 3E). Vma5 protein was barely detectable in whole cell lysates from mutant vma5 Ile178Thr, suggesting protein instability. However, the low detectable levels of vma5 Ile178Thr in vacuolar vesicles indicate that mutant Vma5 can assemble into V-ATPase at the vacuole (Fig. 3D).
WDR72
In 2 additional dRTA families (F382 and B2673; 3 individuals), we identified homozygous mutations in the gene WDR72 (NM_182758.3), located in a segment of homozygosity by descent (Fig. 2B, Fig. 4, Suppl. Fig. S1B–D, Table 2). WDR72 is a paralog of WDR7 which interacts with human dRTA protein ATP6V1B1 and co-localizes with V-ATPases in IC. WDR7 was shown to promote V-ATPase activity and to mediate intracellular vesicle acidification.19 WDR72 may function similarly. The mutation c.477_485dup, p.Ile159_Cys161dup showed recessive segregation within family F382 and is predicted to cause an in-frame insertion of 3 aa in the third WD40 domain of WDR72 (Fig. 2B, Fig. 4A). In family B2673 with 2 affected siblings with dRTA (Fig. 4C), the homozygous mutation c.764_768delGGCAG, p.Gly255Valfs*40 results in a premature stop codon and is a presumptive loss-of-function variant. Neither mutation was reported in the gnomAD database in a homozygous state. Single cell RNA sequencing databases indicate that WDR72 is highly expressed in IC (Fig. 2D). WDR72 mutations can cause autosomal recessive amelogenesis imperfecta (AI) in humans20 and mice.21 Very recently, 4 families with association of dRTA and AI and a WDR72 mutation have been described.8, 9 Interestingly, upon “reverse phenotyping” by contacting our clinical collaborators, we learned that all of our here described WDR72 mutant subjects have distinct features of AI as well (Fig. 4, Table 2). We thereby confirmed that WDR72 mutations can be associated with a syndromic disease of AI and dRTA.8,9
Figure 4: Patients with WDR72 mutation, dRTA and amelogenesis imperfecta.

Pedigree structure of family F382 (A) and B2673 (C). (B, D) show enamel defects in patient II:5, F382 and patient II:1 and II:2, B2673. het, heterozygous; hom, homozygous.
SLC4A2
In family F588 we identified a homozygous missense mutation in the gene SLC4A2 (NM_01199692.2), located within a segment of homozygosity by descent (Table 2, Fig. 2C, Suppl. Fig. S1E). SLC4A2 encodes the Anion Cl−/HCO3− exchanger 2 (AE2) and is a paralog of the known dRTA gene SLC4A1 encoding a basolateral Cl−/HCO3− exchanger of the collecting duct Type A IC. The SLC4A2 locus was previously proposed as a recessive dRTA candidate gene on the basis of genetic linkage to a 14 Mb region of chromosome 7q, but no disease causing mutation could be confirmed at that time.22 The SLC4A2 mutation c.2107G>A, p.Ala703Thr in family F588 replaces the short hydrophobic side chain of Ala703 with a longer, more hydrophilic side chain which may alter its predicted interaction with hydrophobic residue Ile761 (Suppl. Fig. S4). Indeed, CUPSAT17 predicts a destabilizing effect of the Ala703Thr substitution as modeled on the crystal structure of the human SLC4A1 paralog transmembrane domain (Suppl. Fig. S5).16, 23
To test the deleteriousness of the Ala703Thr missense mutation, we generated the corresponding human (hAE2) and mouse (mAE2) cDNA constructs. Transient expression of WT hAE2 in MDCKII cells showed the expected predominant peripheral membrane localization (Fig. 5A, C) which was unaltered in cells expressing hAE2 Ala703Thr (Fig. 5B, D), suggesting unimpaired biosynthetic trafficking in this context. The trafficking similarities of HA-tagged and untagged hAE2 polypeptides was reflected in comparable 36Cl− uptake activities of cRNA-injected Xenopus laevis oocytes (Suppl. Fig. S6A). Additional assays of functional expression in Xenopus oocytes revealed hAE2 Ala703Thr-mediated Cl−/HCO3− and Cl−/Cl− exchange activities sensitive to the stilbene disulfonate inhibitor, DIDS, that were indistinguishable from those of WT hAE2 (Fig. 5E, F). The extracellular pH-dependence of hAE2-mediated Cl−/Cl− exchange was similar to that of WT hAE2 (Fig. 5G, H). Moreover, WT hAE2 activations by acidic intracellular pH (Suppl. Fig. S7) and by hypertonicity (Suppl. Fig. S8) were indistinguishable in oocytes expressing hAE2 Ala703Thr. The 2.4 mM K1/2 for extracellular Cl− exhibited by WT hAE2 was identical to that of the mutant (Suppl. Fig. S9). NH4+-stimulated hAE2-mediated, DIDS-sensitive Cl−/Cl− exchange was slightly reduced compared to WT hAE2 (Suppl. Fig. S10), but not to a degree that would imply pathological significance.
Figure 5. Indistinguishable peripheral membrane localization in MDCK monolayers and indistinguishable rates and regulation by extracellular pH of 36Cl−/HCO3− and 36Cl−/Cl− exchange in oocytes expressing hAE2 and hAE2 variant A703T.

(A, B) MDCKII cells plated at high density on glass cover slips were transiently transfected with hAE2 or hAE2 A703T, and immunostained with a rabbit anti-AE2 primary antibody and goat anti-rabbit Cy3-conjugated secondary antibody. Nuclei are DAPI-stained. (C, D) Same experiment as in (A, B) showing MDCKII cells transiently expressing hAE2-HA or hAE2 A703T-HA, stained with anti-HA primary antibody. Wildtype and variant polypeptides both exhibited prominent peripheral membrane localization. Shown are representative images from one of 3 identical experiments with similar results; scalebars 20 μm. (E) 36Cl− efflux traces from representative individual oocytes previously injected with 2 ng cRNA encoding hAE2 or hAE2 A703T, or from an uninjected oocyte, during sequential exposure to baths containing nominally impermeant Na cyclamate (96 mM), Na cyclamate (72 mM)/bicarbonate (24 mM), and NaCl (96 mM), followed by final addition of the AE2 inhibitor, DIDS. (F) 36Cl− efflux rate constants for Cl−/HCO3− and Cl−/Cl− exchange by oocytes expressing hAE2 (n=6) or variant hAE2 A703T (n=6), compared to uninjected control oocytes (n=3). Values are means ± S.E.M. (G) 36Cl− efflux traces from representative individual oocytes previously injected with 2ng cRNA encoding hAE2 or hAE2 A703T cRNA, or from an uninjected oocyte, during sequential exposure to baths of the indicated increasing pH values. (H) Normalized 36Cl− efflux rate constants for oocytes expressing hAE2 and for hAE2 A703T, both fit to a single sigmoidal curve yielding pHo(50) values of 7.19±0.04. Means ± S.E.M. for 5 oocytes in each group.
In summary, as variant A703T did not relevantly alter AE2 function in the multiple experimental conditions tested, our data failed to support SLC4A2 as a viable dRTA candidate gene.
DISCUSSION
To identify potential additional monogenic causes of dRTA, we performed panel sequencing and/or WES combined with homozygosity mapping in 17 families with clinically diagnosed childhood onset dRTA. In 10/17 families (59%), we identified causative mutations in 1 of the 3 known, classical dRTA genes. In addition, we discovered potentially disease-causing mutations the 3 genes ATP6V1C2, WDR72 and SLC4A2, and functionally evaluated the mutations in ATP6V1C2 and SLC4A2. These studies confirmed ATP6V1C2 as a novel recessive dRTA candidate gene, confirmed expansion of the phenotypic spectrum of AI due to WDR72 mutations, but failed to detect functional evidence supporting the genetic data that suggested SLC4A2 as a candidate dRTA gene.
Genetic testing of our cohort provided a molecular diagnosis for 59% of all cases with a clinical dRTA phenotype, a percentage comparable to that of Ashton et al (57%)6 among European children, but lower than that of Palazzo et al (70%)1 among Italian children.
WES and homozygosity mapping initially identified ATP6V1C2, WDR72, and SLC4A2 as potential novel dRTA genes. Candidate status of all 3 genes was supported by high expression levels of each in the renal IC cluster in single cell mRNA sequencing data (Fig. 2D).24
As for known dRTA disease genes ATP6V0A4 and ATP6V1B1, the novel candidate gene ATP6V1C2 is a subunit of the V-type H+- ATPase. We showed the yeast Vma5 V-ATPase C subunit mutation p.Ile178Thr corresponding to human ATP6V1C2 mutation Ile168Thr had properties of a loss-of-function mutation (Fig. 3). Yeast strains expressing the mutant C subunit showed reduced V-ATPase activity and exhibited a Vma5 growth phenotype. Although the mutant C subunit was slightly detectable in isolated vacuolar vesicles, it was not detectable in whole cell lysates and V-ATPase complexes were insufficient to support full V-ATPase function (Fig. 3). Therefore, these findings suggest loss-of-function for the corresponding human mutation c.503T>C, p.Ile168Thr, and confirm ATP6V1C2 as a novel dRTA candidate gene.
We also identified 2 novel homozygous WDR72 mutations in 2 families with dRTA and AI (Fig. 2B, Fig. 4). Mutation c.764_768delGGCAG encodes a frameshift leading to an early stop codon, and therefore can be considered a loss-of-function variant. The second mutation encodes a 3 aa insertion within the third WD40 domain of WDR72 (Fig. 2B). In genome-wide association studies, WDR72 has been associated with loss-of kidney function and chronic kidney disease.25, 26 Recessive mutations in WDR72 are a known cause for AI (OMIM 613211), causing vesicle trafficking defects in ameloblasts and thereby impairing enamel formation.20, 27 The association of AI and dRTA has been described very recently in 4 families with WDR72 mutations.8, 9 WDR72 mutations may thus underlie several dRTA cases with accompanying dental abnormalities reported without molecular diagnosis.28–30
The biological function of WDR72 is still largely unknown. Wdr72−/− mice mimic the human enamel defect31 but the renal phenotype has not been investigated to date. WDR72 is an intracellular protein, with a predicted structure including N-terminal WD40 repeats forming 2 β-propellers and a C-terminal α-solenoid tail. WDR7, a human paralog of WDR72, regulates Ca2+-dependent exocytosis of neurotransmitter release at synapsis.32 Furthermore, WDR7 interacts with human dRTA protein ATP6V1B1 and co-localizes with V-ATPases in IC. WDR7 was shown to promote V-ATPase activity and to mediate intracellular vesicle acidification.19 WDR72 may have similar functions in IC with involvement in vesicle trafficking. However, further investigations into the pathogenesis of WDR72 mutations in dRTA are warranted. Our findings have thus confirmed the phenotypic expansion of WDR72 mutations from isolated AI to a syndromic disease that features both AI and dRTA.8
SLC4A2 encodes the AE2 Cl−/HCO3− exchanger and is a paralog of the known dRTA gene SLC4A1 encoding a basolateral Cl−/HCO3− exchanger of the collecting duct Type A IC. Moreover, Slc4a2 was cloned from and immunolocalized in guinea pig cochlea,33 and SLC4A2 mRNA was detected in cochlear epithelial cells differentiated from human iPSC.34 A dRTA disease gene region was mapped by linkage analysis in 9 dRTA families to a 14 cM region of human chromosome 7q33–34 encompassing the SLC4A2 gene.22 We identified a missense mutation in SLC4A2 in an unbiased candidate approach by WES in a patient with dRTA and hearing loss. However, functional expression of human or mouse AE2 protein in Xenopus oocytes revealed no significant functional differences between WT and mutant proteins in baseline Cl−/HCO3− or Cl−/Cl− exchange activities or in Cl−/Cl− exchange regulation by intracellular or extracellular pH, by hypertonicity, or by ammonium (Fig. 5, Suppl. Fig. S6–S10). These experiments did not exclude the possibility that the mutation impairs activity or regulated expression of AE2 in IC in situ or in another model epithelial cell type grown in different conditions.35
Slc4a2 knock-out (KO) mice are toothless, are severely growth-retarded, develop achlorhydria and abnormal gastric epithelium, and die within the first 40 days of life.36 Although the patient lacks these features of the mouse KO phenotype, a hypomorphic missense mutation might not resemble a KO.
SLC4A2 residue 703 corresponds to SLC4A1 residue 400. Heterozygous deletion of residues 400–408 causes the benign hematological condition, Southeast Asian Ovalocytosis (SAO), without renal phenotype. However, the rare homozygous SAO deletion is associated with severe dyserythropoietic hemolytic anemia and dRTA,37 and causes anemia and dRTA in compound heterozygous states as well. SLC4A2 Ala703 is located at the C-terminal end of AE2’s N-terminal cytoplasmic domain, near the start of the first of AE2’s 14 transmembrane helices. The missense mutation p.Ala703Thr is predicted to possibly interfere with protein stability (Suppl. Fig. S4),16 through a conformational change in the flexible linker region connecting cytoplasmic and transmembrane domains, as in AE1,38 or through potentially altered modulation by proteinase CK2 or other regulators.39 In summary, no significant hAE2 loss-of-function resulted from the c.2107G>A substitution allele, as detected by our assays. Identification of additional dRTA patients with SLC4A2 mutations and further experimental studies will be necessary to maintain SLC4A2 as a candidate disease gene for dRTA and hearing loss.
We have presented combined panel sequencing and WES for the rapid and reliable molecular diagnosis of patients with a clinical diagnosis of dRTA. However, panel sequencing had been performed before the publication of both papers that associated WDR72 mutations with dRTA. Thus, this temporal connection underlines the advantages of WES over panel sequencing as unsolved WES datasets may be reevaluated after discovery of novel genetic causes of the disease. As costs for WES are declining rapidly, WES should be considered as a first choice for genetic testing. Furthermore, we have established ATP6V1C2 as a novel recessive dRTA gene in humans, and confirmed the phenotype expansion of recessive WDR72 mutations from isolated AI to syndromic AI with dRTA. However, our functional data failed to reinforce the genetic evidence supporting SLC4A2 as a candidate dRTA gene. Thus, WES provides a powerful tool to identify novel dRTA genes and, by directing functional validation studies, helps elucidate pathogenic mechanisms of dRTA.
METHODS
Study participants and genetic testing
This study was approved by the institutional review board (IRB) of Boston Children’s Hospital (BCH). We obtained informed consent, clinical data, pedigree information, and DNA samples from subjects clinically diagnosed with dRTA manifesting before the age of 25 years.
Panel screening was performed by multiplex PCR for the genes ATP6V1B1, ATP6V0A4 and SLC4A1 in 11 individuals from 10 families as previously described.40, 41 WES was performed as previously described42 in 11 individuals from 10 families including 3 individuals from 3 families unsolved by panel sequencing. A mean depth of coverage of the known dRTA and candidate genes of 79.9X was achieved (Suppl. Table 1).
Homozygosity mapping was calculated based on whole exome sequencing data. In brief, aligned BAM files were processed using Picard and SAMtools as described by other groups.43 Single nucleotide variant calling was performed using Genome Analysis Tool Kit (GATK).44 The resulting VCF files were used to generate homozygosity mapping data and visual outputs using the program Homozygosity Mapper.45
Variants were evaluated for mutations in known dRTA genes. Remaining variants were ranked based on their predicted impact on protein sequence and function considering evolutionary conservation among orthologs across phylogeny, and web-based pathogenicity prediction programs (PolyPhen-246, SIFT47 and MutationTaster48). Remaining variants were evaluated by literature review and by phenotype correlation. Deleteriousness was assessed using on our established criteria based on ACMG.10 Unsolved WES datasets were subjected to candidate gene analysis. Remaining variants were confirmed in original patient DNA by Sanger sequencing as previously described (Suppl. Fig. S11).49 Whenever parental DNA was available, segregation analysis was performed.
Yeast growth assay
The ATP6V1C2 mutation isoleucine 178 to threonine (vma5I178T) that reflects the human mutation Ile168Thr was introduced into the genomic copy of VMA5 in WT yeast strain SF838–5Aα by a variation of the delitto perfetto method.50 AA 150–214 of VMA5 were replaced with URA3, and transformants were selected on fully supplemented minimal medium lacking uracil.51 The I178T mutation was first introduced into a plasmid borne copy of WT VMA5 by Quikchange mutagenesis (Agilent) using oligonucleotides VMA5 I178T forward 5’CTGTCAGATCCTTGCATGATACTGTCAAGCCCGAAGACTTCGTTC-3’ and VMA5 I178T reverse 5’GAACGAAGTCTTCGGGCTTGACAgTATCATGCAAGGATCTGACAGAAAG-3’, introduction of the mutation was confirmed by sequencing. A VMA5 fragment containing the I178T mutation was then PCR-amplified and used to transform the vma5Δ150–214::URA3 strain. Transformants that had replaced URA3 with the mutated fragment were selected on medium containing 5-fluoro-orotic acid (5-FOA), which selects against the URA3 marker. 3 separate transformants that grew on 5-FOA but had lost growth on medium lacking uracil were analyzed further. Growth of WT and vma5 (I175T) strains was compared by growing all strains to log-phase, diluting to 0.1 OD/ml and then doing sequential 10-fold serial dilutions of each strain before pinning onto YEPD plates buffered to pH 5 with 50 mM phosphate and 50 mM succinate or YEPD plates buffered to pH 7.5 containing 60 mM CaCl2.
Isolation and characterization of vacuolar vesicles
Vacuolar vesicles were isolated as described previously.52 ATPase activity was determined at 37°C through a coupled enzyme assay ( V-ATPase activity was activity sensitive to 200 nM concanamycin A). Immunoblots were performed as described previously.53 Vacuolar vesicles were solubilized in cracking buffer, and separated by SDS-PAGE, and transferred to nitrocellulose. Mouse monoclonal antibodies 8B1, 13D11, and 10D7, were used to detect V1 subunits A and B and Vph1, respectively. Vma5 (V1 subunit C) was detected with rabbit polyclonal antiserum against Vma5 (a generous gift from Tom Stevens, University of Oregon). Whole cell lysates were obtained by several rounds glass bead lysis of cells in cracking buffer, followed by heating to 95°C. Lysates from 0.5 OD600 units of cells were probed for V1 A and B as described above. Lysates probed for V1C came from 2.5 oD600 units of cells.
Xenopus oocyte experiments
Construction and mutagenesis of cDNA expression plasmids
Mouse Ae2a (Slc4a2) was subcloned into the Xenopus oocyte expression vector pXT7 and used as previously described.54 Human AE2a (SLC4A2) cDNA was purchased from OriGene and subcloned into pXT7. SLC4A2 mutations mouse Ae2 A699T and human AE2 A703T were generated by four primer polymerase chain reaction (PCR) as described before.55 All constructs were subcloned into pCDNA3 for transient transfection of MDCK cells.
Expression of cRNAs in Xenopus oocytes
Capped cRNA was transcribed from linearized cDNA templates with T7 polymerase (Ambion, Austin, TX). RNA integrity was confirmed by agarose gel electrophoresis in formaldehyde. Mature female Xenopus (Dept. of Systems Biology, Harvard Medical School; or NASCO, Madison, WI) were maintained and subjected to partial ovariectomy under hypothermic tricaine anesthesia following protocols approved by the Institutional Animal Care and Use Committee of Beth Israel Deaconess Medical Center. Stage V-VI oocytes were prepared by overnight incubation of ovarian fragments in MBS with 1.5 mg/ml collagenase B (Alfa Aesar), followed by a 20 min rinse in Ca2+-free MBS with subsequent manual selection and defolliculation as needed. Oocytes were injected on the same day with cRNA (0.5–50 ng) or with water in a volume of 50 nl. Injected and uninjected oocytes were then maintained 2–6 days at 19°C in MBS [(in m M) 85 NaCl, 1 KCl, 2.4 NaHCO3, 0.82 Mg(SO4)2, 0.33 Ca(NO3)2, 0.41 CaCl2, and 10 HEPES (final pH 7.40)] containing gentamicin until used for experiments.
Isotopic influx experiments
36Cl− influx studies were carried out in 96 well plates as previously described56 for periods of 15, 20, or 30 min in ND-96 consisting of (in mM) 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, and 5 HEPES (pH 7.40). In Cl−-free ND-96 or in partial Cl− substitution solutions, NaCl was replaced mole-for-mole with Na cyclamate or, as needed, the Cl− salts of K+, Ca2+, and Mg2+ were substituted on an equimolar basis with the corresponding gluconate salts. Addition to flux media of the weak acid salt sodium butyrate (40 mM) was in equimolar substitution for Na cyclamate. Addition of NH4Cl (20 mM) was in equimolar substitution for NaCl. In pH dependence experiments, 5 mM HEPES was replaced on an equimolar basis with a biological buffer of appropriate pKa: pH 5.0, MES; pH 6.0, MES; pH 7.0, HEPES; pH 8.0, HEPES; pH 9.0, CHES (pH adjusted with NaOH or HCl). Hypertonic solutions were formulated by addition of sodium chloride, sodium glutamate or mannitol as described, and osmotic strength was measured by freezing point depression (Osmette A, Precision Systems Inc., Natick MA).
Bath volume was 150 μl and bath [Cl−] was 103.6 mM, except as indicated in experiments conducted in hyperosmolar conditions. 0.25 μCi 36Cl− (ICN, Irvine, CA) was included in each well. Influx experiments were terminated with 3 washes in cold Cl−-free ND96, followed by oocyte lysis in 150 μl of 2% sodium dodecyl sulfate (SDS). 2 ml scintillation fluid (SX-18 ScintiVerse, Fisher) was added to each lysed oocyte, and radiation uptake was measured in a PerkinElmer Tri-Carb scintillation counter. Triplicate 10 μL aliquots of initial influx solution were used to calculate 36Cl− specific activity. Cl− uptake by oocytes was calculated by comparing oocyte cpm to bath specific activity.
Isotopic efflux experiments
For unidirectional 36Cl− efflux studies, individual oocytes in Cl−-free ND-96 were injected with 50 nl of 260 mM Na36Cl (20,000–24,000 cpm). Following a 5 min recovery period in Cl−-free ND-96, the efflux assay was initiated by transfer of individual oocytes to 6 ml borosilicate glass tubes, each containing 1 ml efflux solution as specified. At intervals of 1 or 3 min, 0.95 ml of this efflux solution was removed for scintillation counting and replaced with an equal volume of fresh efflux solution. Following completion of the assay with a final efflux period either in Cl−-free cyclamate solution or in the presence of the inhibitor DIDS (200 μM), each oocyte was lysed in 150 μl of 2% SDS. Samples were counted for 3–5 min such that the magnitude of 2SD was <5% of the sample mean.
To vary pHi, oocytes were pre-exposed to 40 mM Na butyrate (substituting for Na cyclamate) for 30 min prior to initiation of an efflux experiment, to acidify oocyte pHi to pH ~6.7.57 Upon removal of bath butyrate (with substitution by Na cyclamate) during the course of the efflux experiment, pHi rapidly alkalinized back towards initial pHi while pHo remained constant. Variation of pHo was achieved at near-constant pHi. Some oocyte groups were exposed to 20 mM NH4Cl during the course of efflux experiments, acidifying pHi to pHi 7.1–7.0.58
Efflux data were plotted as ln (% cpm remaining in the oocyte) vs. time. Efflux rate constants for 36Cl− were measured from linear fits to data from the last 3 time points sampled within each experimental period. For each experiment, water-injected or uninjected oocytes from the same frog were subjected to parallel measurements with cRNA-injected oocytes. Each experimental condition was tested in oocytes from at least 2 frogs. The following 2 exclusion criteria were defined for “non-specific” efflux or “leaky” oocytes in efflux experiments. One was <50% reduction in efflux rate constant in the presence of DIDS or in the absence of exchangeable bath anion. The second was loss of >85% of injected isotope prior to completion of the efflux assay.
Immunocytochemistry
MDCKII cells were plated on glass coverslips and transfected (Altogen Biosystems MDCK Transfection Reagent Kit) per manufacturer’s instructions. After 48 h cells on coverslip were fixed with 3% PFA in PBS for 30 min at room temperature (RT), then permeabilized with PBS + 0.1%(v/v) Triton X-100 for 15 min at RT, and blocked with PBS containing 2% BSA. Primary antibodies used were rabbit polyclonal anti-mouse AE2 directed against C-terminal aa 1224–123759–61 and mouse monoclonal anti-HA tag (Cell Signaling), each overnight at 4°C at 1:2000 dilutions, then for 1 h at RT with secondary Cy3-conjugated goat anti-rabbit Ig or goat anti-mouse Ig (each at 1:1000 dilution).
Statistics
Data are reported as mean ± SE. Flux data were compared by Student’s paired or unpaired 2-tailed T tests (Microsoft Excel), or by ANOVA with Tukey post-hoc analysis (SigmaPlot). pH dependence data were fit to a 4-parameter Hill equation in SigmaPlot 8.0.
Supplementary Material
Supplementary Figure S1. Whole exome sequencing (WES) and homozygosity mapping reveals 3 potential candidate genes for recessive distal renal tubular acidosis (dRTA).
Supplementary Figure S2: The hs. ATP6V1C2 mutation c.503T>C, p.Ile168Thr affects a hydrophobic pocket in ATP6V1C2.
Supplementary Figure S3. Protein stability prediction (CUPSAT) suggests a destabilizing effect of the mutation in yeast VMA5.
Supplementary Figure S4. The hs. SLC4A2 mutation c.503T>C, p.Ile168Thr may affect a hydrophobic interaction in SLC4A2.
Supplementary Figure S5. Protein stability prediction (CUPSAT) suggests a destabilizing effect of the p.Ala703Thr mutation in SLC4A2.
Supplementary Figure S6. Similar levels of Cl− uptake by oocytes expressing hAE2 WT and its variant A703T.
Supplementary Figure S7. Indistinguishable inhibition by acidic intracellular pHi exhibited by hAE2 WT and variant A703T.
Supplementary Figure S8. Indistinguishable activation by hyperosmolarity of Cl− uptake mediated by hAE2 WT and by variant A703T.
Supplementary Figure S9. Indistinguishable K1/2 for extracellular Cl− for 36Cl−/Cl− exchange mediated by hAE2 WT and variant A703T.
Supplementary Figure S10. Slightly reduced activation by NH4+ of 36Cl−/Cl− exchange mediated by hAE2 variant A703T compared to hAE2 WT.
Supplementary Figure S11. Sequencing traces and family pedigrees of individuals with dRTA.
Supplementary Table S1. Information on mean coverage of known and candidate dRTA genes per family.
Translational statement.
Distal renal tubular acidosis (dRTA) causes metabolic acidosis, electrolyte imbalance and, if untreated, renal failure by mutations in 3 classical genes. We performed panel sequencing and/or whole exome sequencing in 17 dRTA families to identify novel genetic causes. In 10 families a molecular diagnosis in 1 of the 3 classical genes was established and in 4 families a dRTA candidate mutation (ATP6V1C2, WDR72, SLC4A2) was identified. Functional studies confirmed ATP6V1C2 as a candidate gene, but excluded SLC4A2. Furthermore, we generated further evidence for a phenotypic expansion for WDR72 mutations from amelogenesis imperfecta to dRTA. Thus, in future, APT6V1C2 and WDR72 mutations should be included in genetic testing for dRTA patients.
ACKNOWLEDGEMENTS
We thank the physicians and the participating families for their contribution. We thank Leslie Speanas, Brittany Fisher, and Kassaundra Amann for recruitment of study participants. F.Hildebrandt is the William E. Harmon Professor of Pediatrics. This research was supported by grants from the National Institutes of Health DK1069274, DK1068306, and DK064614 to F.H. and 5U54HG006504 to R.P.L., and GM126020 to P.M.K.. T.J.S. (281319475), V.K. (403877094) and F.B. (404527522) are supported by the Deutsche Forschungsgemeinschaft. A.M. was supported by a NIH Training Grant in Pediatric Nephrology (T32DK007726), by the 2017 Post-doctoral Fellowship Grant from the Harvard Stem Cell Institute Kidney Group, and by the American Society of Nephrology Lipps Research Program 2018 Polycystic Kidney Disease Foundation Jared J. Grantham Research Fellowship. WES performed at Yale Center for Mendelian Genomics and Cologne Center for Genomics.
Footnotes
STATEMENT OF DISCLOSURE
None of other authors have competing financial interests to disclose.
Supplemental Material
Supplementary information is available at Kidney International’s website
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Palazzo V, Provenzano A, Becherucci F, et al. The genetic and clinical spectrum of a large cohort of patients with distal renal tubular acidosis. Kidney Int 2017; 91: 1243–1255. [DOI] [PubMed] [Google Scholar]
- 2.Lopez-Garcia SC, Emma F, Walsh SB, et al. Treatment and long-term outcome in primary distal renal tubular acidosis. Nephrol Dial Transplant 2019. [DOI] [PubMed] [Google Scholar]
- 3.Smith AN, Skaug J, Choate KA, et al. Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nature genetics 2000; 26: 71–75. [DOI] [PubMed] [Google Scholar]
- 4.Karet FE, Finberg KE, Nelson RD, et al. Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nature genetics 1999; 21: 84–90. [DOI] [PubMed] [Google Scholar]
- 5.Bruce LJ, Cope DL, Jones GK, et al. Familial distal renal tubular acidosis is associated with mutations in the red cell anion exchanger (Band 3, AE1) gene. J Clin Invest 1997; 100: 1693–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ashton EJ, Legrand A, Benoit V, et al. Simultaneous sequencing of 37 genes identified causative mutations in the majority of children with renal tubulopathies. Kidney Int 2018; 93: 961–967. [DOI] [PubMed] [Google Scholar]
- 7.Enerback S, Nilsson D, Edwards N, et al. Acidosis and Deafness in Patients with Recessive Mutations in FOXI1. J Am Soc Nephrol 2018; 29: 1041–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rungroj N, Nettuwakul C, Sawasdee N, et al. Distal renal tubular acidosis caused by tryptophan-aspartate repeat domain 72 (WDR72) mutations. Clin Genet 2018; 94: 409–418. [DOI] [PubMed] [Google Scholar]
- 9.Zhang H, Koruyucu M, Seymen F, et al. WDR72 Mutations Associated with Amelogenesis Imperfecta and Acidosis. Journal of dental research 2019: 22034518824571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hildebrandt F, Heeringa SF, Ruschendorf F, et al. A systematic approach to mapping recessive disease genes in individuals from outbred populations. PLoS Genet 2009; 5: e1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun-Wada GH, Wada Y. Role of vacuolar-type proton ATPase in signal transduction. Biochim Biophys Acta 2015; 1847: 1166–1172. [DOI] [PubMed] [Google Scholar]
- 13.Sun-Wada GH, Murata Y, Namba M, et al. Mouse proton pump ATPase C subunit isoforms (C2-a and C2-b) specifically expressed in kidney and lung. The Journal of biological chemistry 2003; 278: 44843–44851. [DOI] [PubMed] [Google Scholar]
- 14.Smith AN, Borthwick KJ, Karet FE. Molecular cloning and characterization of novel tissue-specific isoforms of the human vacuolar H(+)-ATPase C, G and d subunits, and their evaluation in autosomal recessive distal renal tubular acidosis. Gene 2002; 297: 169–177. [DOI] [PubMed] [Google Scholar]
- 15.Zhao J, Benlekbir S, Rubinstein JL. Electron cryomicroscopy observation of rotational states in a eukaryotic V-ATPase. Nature 2015; 521: 241–245. [DOI] [PubMed] [Google Scholar]
- 16.Parthiban V, Gromiha MM, Schomburg D. CUPSAT: prediction of protein stability upon point mutations. Nucleic acids research 2006; 34: W239–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Forster C, Kane PM. Cytosolic Ca2+ homeostasis is a constitutive function of the V-ATPase in Saccharomyces cerevisiae. The Journal of biological chemistry 2000; 275: 38245–38253. [DOI] [PubMed] [Google Scholar]
- 18.Nelson H, Nelson N. Disruption of genes encoding subunits of yeast vacuolar H(+)ATPase causes conditional lethality. Proceedings of the National Academy of Sciences of the United States of America 1990; 87: 3503–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Merkulova M, Paunescu TG, Azroyan A, et al. Mapping the H(+) (V)-ATPase interactome: identification of proteins involved in trafficking, folding, assembly and phosphorylation. Scientific reports 2015; 5: 14827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.El-Sayed W, Parry DA, Shore RC, et al. Mutations in the beta propeller WDR72 cause autosomal-recessive hypomaturation amelogenesis imperfecta. Am J Hum Genet 2009; 85: 699–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Song W, Wang Y, Chu Q, et al. Loss of transforming growth factor-beta1 in epithelium cells affects enamel formation in mice. Archives of oral biology 2018; 96: 146–154. [DOI] [PubMed] [Google Scholar]
- 22.Karet FE, Finberg KE, Nayir A, et al. Localization of a Gene for Autosomal Recessive Distal Renal Tubular Acidosis with Normal Hearing (<strong>rdRTA2</strong>) to 7q33–34. The American Journal of Human Genetics 65: 1656–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arakawa T, Kobayashi-Yurugi T, Alguel Y, et al. Crystal structure of the anion exchanger domain of human erythrocyte band 3. Science 2015; 350: 680–684. [DOI] [PubMed] [Google Scholar]
- 24.Park J, Shrestha R, Qiu C, et al. Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 2018; 360: 758–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Franceschini N, Haack K, Almasy L, et al. Generalization of associations of kidney-related genetic loci to American Indians. Clinical journal of the American Society of Nephrology : CJASN 2014; 9: 150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kottgen A, Glazer NL, Dehghan A, et al. Multiple loci associated with indices of renal function and chronic kidney disease. Nature genetics 2009; 41: 712–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Katsura KA, Horst JA, Chandra D, et al. WDR72 models of structure and function: a stage-specific regulator of enamel mineralization. Matrix Biol 2014; 38: 48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Misgar RA, Hassan Z, Wani AI, et al. Amelogenesis Imperfecta with Distal Renal Tubular Acidosis: A Novel Syndrome? Indian J Nephrol 2017; 27: 225–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ravi P, Ekambaranath TS, Arasi SE, et al. Distal renal tubular acidosis and amelogenesis imperfecta: A rare association. Indian J Nephrol 2013; 23: 452–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jain M, Agarwal MP, Wasir JS, et al. Amelogenesis imperfecta and distal renal tubular acidosis presenting as hypokalemic periodic paralysis. J Assoc Physicians India 1999; 47: 1205–1206. [PubMed] [Google Scholar]
- 31.Wang SK, Hu Y, Yang J, et al. Critical roles for WDR72 in calcium transport and matrix protein removal during enamel maturation. Molecular genetics & genomic medicine 2015; 3: 302–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kawabe H, Sakisaka T, Yasumi M, et al. A novel rabconnectin-3-binding protein that directly binds a GDP/GTP exchange protein for Rab3A small G protein implicated in Ca(2+)-dependent exocytosis of neurotransmitter. Genes to cells : devoted to molecular & cellular mechanisms 2003; 8: 537–546. [DOI] [PubMed] [Google Scholar]
- 33.Stankovic KM, Brown D, Alper SL, et al. Localization of pH regulating proteins H+ATPase and Cl−/HCO3− exchanger in the guinea pig inner ear. Hear Res 1997; 114: 21–34. [DOI] [PubMed] [Google Scholar]
- 34.Hosoya M, Fujioka M, Sone T, et al. Cochlear Cell Modeling Using Disease-Specific iPSCs Unveils a Degenerative Phenotype and Suggests Treatments for Congenital Progressive Hearing Loss. Cell reports 2017; 18: 68–81. [DOI] [PubMed] [Google Scholar]
- 35.Mumtaz R, Trepiccione F, Hennings JC, et al. Intercalated Cell Depletion and Vacuolar H(+)-ATPase Mistargeting in an Ae1 R607H Knockin Model. Journal of the American Society of Nephrology : JASN 2017; 28: 1507–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gawenis LR, Ledoussal C, Judd LM, et al. Mice with a targeted disruption of the AE2 Cl−/HCO3− exchanger are achlorhydric. The Journal of biological chemistry 2004; 279: 30531–30539. [DOI] [PubMed] [Google Scholar]
- 37.Picard V, Proust A, Eveillard M, et al. Homozygous Southeast Asian ovalocytosis is a severe dyserythropoietic anemia associated with distal renal tubular acidosis. Blood 2014; 123: 1963–1965. [DOI] [PubMed] [Google Scholar]
- 38.Jiang J, Magilnick N, Tsirulnikov K, et al. Single particle electron microscopy analysis of the bovine anion exchanger 1 reveals a flexible linker connecting the cytoplasmic and membrane domains. PloS one 2013; 8: e55408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ibrahim SH, Turner MJ, Saint-Criq V, et al. CK2 is a key regulator of SLC4A2-mediated Cl(−)/HCO3(−) exchange in human airway epithelia. Pflugers Archiv : European journal of physiology 2017; 469: 1073–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Braun DA, Lawson JA, Gee HY, et al. Prevalence of Monogenic Causes in Pediatric Patients with Nephrolithiasis or Nephrocalcinosis. Clin J Am Soc Nephrol 2016; 11: 664–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Halbritter J, Diaz K, Chaki M, et al. High-throughput mutation analysis in patients with a nephronophthisis-associated ciliopathy applying multiplexed barcoded array-based PCR amplification and next-generation sequencing. Journal of Medical Genetics 2012; 49: 756–767. [DOI] [PubMed] [Google Scholar]
- 42.Daga A, Majmundar AJ, Braun DA, et al. Whole exome sequencing frequently detects a monogenic cause in early onset nephrolithiasis and nephrocalcinosis. Kidney Int 2018; 93: 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van der Auwera GA, Carneiro MO, Hartl C, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Current protocols in bioinformatics 2013; 43: 11 10 11–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seelow D, Schuelke M, Hildebrandt F, et al. HomozygosityMapper--an interactive approach to homozygosity mapping. Nucleic acids research 2009; 37: W593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nature methods 2010; 7: 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature protocols 2009; 4: 1073–1081. [DOI] [PubMed] [Google Scholar]
- 48.Schwarz JM, Cooper DN, Schuelke M, et al. MutationTaster2: mutation prediction for the deep-sequencing age. Nature methods 2014; 11: 361–362. [DOI] [PubMed] [Google Scholar]
- 49.Otto EA, Ramaswami G, Janssen S, et al. Mutation analysis of 18 nephronophthisis associated ciliopathy disease genes using a DNA pooling and next generation sequencing strategy. J Med Genet 2011; 48: 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Storici F, Resnick MA. The delitto perfetto approach to in vivo site-directed mutagenesis and chromosome rearrangements with synthetic oligonucleotides in yeast. Methods Enzymol 2006; 409: 329–345. [DOI] [PubMed] [Google Scholar]
- 51.Amberg DC, Botstein D, Beasley EM. Precise gene disruption in Saccharomyces cerevisiae by double fusion polymerase chain reaction. Yeast 1995; 11: 1275–1280. [DOI] [PubMed] [Google Scholar]
- 52.Roberts CJ, Raymond CK, Yamashiro CT, et al. Methods for studying the yeast vacuole. Methods Enzymol 1991; 194: 644–661. [DOI] [PubMed] [Google Scholar]
- 53.Kane PM, Kuehn MC, Howald-Stevenson I, et al. Assembly and targeting of peripheral and integral membrane subunits of the yeast vacuolar H(+)-ATPase. The Journal of biological chemistry 1992; 267: 447–454. [PubMed] [Google Scholar]
- 54.Stewart AK, Kurschat CE, Burns D, et al. Transmembrane domain histidines contribute to regulation of AE2-mediated anion exchange by pH. American journal of physiology Cell physiology 2007; 292: C909–918. [DOI] [PubMed] [Google Scholar]
- 55.Clark JS, Vandorpe DH, Chernova MN, et al. Species differences in Cl− affinity and in electrogenicity of SLC26A6-mediated oxalate/Cl− exchange correlate with the distinct human and mouse susceptibilities to nephrolithiasis. The Journal of physiology 2008; 586: 1291–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chernova MN, Jiang L, Shmukler BE, et al. Acute regulation of the SLC26A3 congenital chloride diarrhoea anion exchanger (DRA) expressed in Xenopus oocytes. The Journal of physiology 2003; 549: 3–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stewart AK, Chernova MN, Kunes YZ, et al. Regulation of AE2 anion exchanger by intracellular pH: critical regions of the NH(2)-terminal cytoplasmic domain. American journal of physiology Cell physiology 2001; 281: C1344–1354. [DOI] [PubMed] [Google Scholar]
- 58.Nakhoul NL, Hering-Smith KS, Abdulnour-Nakhoul SM, et al. Transport of NH(3)/NH in oocytes expressing aquaporin-1. American journal of physiology Renal physiology 2001; 281: F255–263. [DOI] [PubMed] [Google Scholar]
- 59.Gawenis LR, Bradford EM, Alper SL, et al. AE2 Cl−/HCO3− exchanger is required for normal cAMP-stimulated anion secretion in murine proximal colon. Am J Physiol Gastrointest Liver Physiol 2010; 298: G493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stehberger PA, Shmukler BE, Stuart-Tilley AK, et al. Distal renal tubular acidosis in mice lacking the AE1 (band3) Cl−/HCO3− exchanger (slc4a1). J Am Soc Nephrol 2007; 18: 1408–1418. [DOI] [PubMed] [Google Scholar]
- 61.Alper SL, Stuart-Tilley AK, Biemesderfer D, et al. Immunolocalization of AE2 anion exchanger in rat kidney. Am J Physiol 1997; 273: F601–614. [DOI] [PubMed] [Google Scholar]
- 62.Stover EH, Borthwick KJ, Bavalia C, et al. Novel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. J Med Genet 2002; 39: 796–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Besouw MTP, Bienias M, Walsh P, et al. Clinical and molecular aspects of distal renal tubular acidosis in children. Pediatr Nephrol 2017; 32: 987–996. [DOI] [PubMed] [Google Scholar]
- 64.Bruce LJ, Wrong O, Toye AM, et al. Band 3 mutations, renal tubular acidosis and South-East Asian ovalocytosis in Malaysia and Papua New Guinea: loss of up to 95% band 3 transport in red cells. Biochem J 2000; 350 Pt 1: 41–51. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1. Whole exome sequencing (WES) and homozygosity mapping reveals 3 potential candidate genes for recessive distal renal tubular acidosis (dRTA).
Supplementary Figure S2: The hs. ATP6V1C2 mutation c.503T>C, p.Ile168Thr affects a hydrophobic pocket in ATP6V1C2.
Supplementary Figure S3. Protein stability prediction (CUPSAT) suggests a destabilizing effect of the mutation in yeast VMA5.
Supplementary Figure S4. The hs. SLC4A2 mutation c.503T>C, p.Ile168Thr may affect a hydrophobic interaction in SLC4A2.
Supplementary Figure S5. Protein stability prediction (CUPSAT) suggests a destabilizing effect of the p.Ala703Thr mutation in SLC4A2.
Supplementary Figure S6. Similar levels of Cl− uptake by oocytes expressing hAE2 WT and its variant A703T.
Supplementary Figure S7. Indistinguishable inhibition by acidic intracellular pHi exhibited by hAE2 WT and variant A703T.
Supplementary Figure S8. Indistinguishable activation by hyperosmolarity of Cl− uptake mediated by hAE2 WT and by variant A703T.
Supplementary Figure S9. Indistinguishable K1/2 for extracellular Cl− for 36Cl−/Cl− exchange mediated by hAE2 WT and variant A703T.
Supplementary Figure S10. Slightly reduced activation by NH4+ of 36Cl−/Cl− exchange mediated by hAE2 variant A703T compared to hAE2 WT.
Supplementary Figure S11. Sequencing traces and family pedigrees of individuals with dRTA.
Supplementary Table S1. Information on mean coverage of known and candidate dRTA genes per family.