

Abstract
Introduced species can impose profound impacts on the evolution of receiving communities with which they interact. If native and introduced taxa remain reproductively semi‐isolated, human‐mediated secondary contact may promote genetic exchange across newly created hybrid zones, potentially impacting native genetic diversity and invasive species spread. Here, we investigate the contributions of recent divergence histories and ongoing (post‐introduction) gene flow between the invasive marine mussel, Mytilus galloprovincialis, and a morphologically indistinguishable and taxonomically contentious native Australian taxon, Mytilus planulatus. Using transcriptome‐wide markers, we demonstrate that two contemporary M. galloprovincialis introductions into south‐eastern Australia originate from genetically divergent lineages from its native range in the Mediterranean Sea and Atlantic Europe, where both introductions have led to repeated instances of admixture between introduced and endemic populations. Through increased genome‐wide resolution of species relationships, combined with demographic modelling, we validate that mussels sampled in Tasmania are representative of the endemic Australian taxon (M. planulatus), but share strong genetic affinities to M. galloprovincialis. Demographic inferences indicate late‐Pleistocene divergence times and historical gene flow between the Tasmanian endemic lineage and northern M. galloprovincialis, suggesting that native and introduced taxa have experienced a period of historical isolation of at least 100,000 years. Our results demonstrate that many genomic loci and sufficient sampling of closely related lineages in both sympatric (e.g. Australian populations) and allopatric (e.g. northern hemisphere Mytilus taxa) ranges are necessary to accurately (a) interpret patterns of intraspecific differentiation and to (b) distinguish contemporary invasive introgression from signatures left by recent divergence histories in high dispersal marine species. More broadly, our study fills a significant gap in systematic knowledge of native Australian biodiversity and sheds light on the intrinsic challenges for invasive species research when native and introduced species boundaries are not well defined.
Keywords: demographic history, hybrid, introgression, marine invasions, mussels, Mytilus, non‐native species, transcriptome
1. INTRODUCTION
The ability of introduced species to alter the ecology and evolution of native communities is a fundamental issue for understanding the long‐term impacts of biological invasions (Colautti & Lau, 2015; Rius, Turon, Bernardi, Volckaert, & Viard, 2015). When introduced species are distinct in morphology, life history or ecology from native residents, studies have documented profound effects on receiving communities at multiple levels of biological organization. Successful invaders may directly or indirectly displace native species though predation or competition (e.g. Arcella, Perry, Lodge, & Feder, 2014; Branch & Steffani, 2004), inflict damage to local habitats (e.g. Robinson, Griffiths, Branch, & Govender, 2007) and prompt cascading community‐level impacts that can transform entire ecosystems (e.g. Griffiths, Hockey, Erkom Schurink, & Roux, 1992; Shine, 2010). From a molecular perspective, native and introduced species that have been isolated for short periods of time or have experienced historical contact throughout their evolutionary histories may retain genomes semi‐permeable to gene flow (Roux et al., 2016). If native and introduced taxa remain reproductively semi‐isolated, secondary contact can promote ongoing genetic exchange across hybrid zones, imposing less discernible, but potentially severe genetic impacts that can result in complex evolutionary outcomes for endemic populations (Ellstrand & Schierenbeck, 2000).
In the absence of complete reproductive barriers, introgression may promote successful introductions through the spread of locally favoured variants into introduced genomic backgrounds (Hovick & Whitney, 2014; Schierenbeck & Ellstrand, 2009). Hybridization may also impede invasions by trapping species barriers at environmental boundaries (Bierne, Welch, Loire, Bonhomme, & David, 2011; El Ayari, Menif, Hamer, Cahill, & Bierne, 2019; Kovach et al., 2016). Conversely, introgression into native genomic backgrounds may alter native genetic diversity (Blum, Walters, Burkhead, Freeman, & Porter, 2010; Fitzpatrick et al., 2010; Todesco et al., 2016) or eliminate parental genotypes entirely through introgression swamping (Arcella et al., 2014; Glotzbecker, Walters, & Blum, 2016; Riley, Bradley Shaffer, Randal Voss, & Fitzpatrick, 2003). Despite potentially significant consequences for endemic diversity and invasive species spread, hybrid invasions are likely to go undetected if native and introduced species boundaries are not well defined (Geller, Darling, & Carlton, 2010). Indeed, comparative genomic studies have revealed a high occurrence of weakly differentiated and semi‐reproductively isolated species within the “grey zone” of the speciation continuum naturally occurring in both terrestrial and marine systems (De Queiroz, 2007; i.e. 0.075–2% average transcriptome‐wide molecular divergence; Roux et al., 2016), highlighting taxonomic issues pertinent for delineating closely related lineages (Galtier, 2019). For invasive species research, however, the “grey zone” raises additional challenges for detecting species introductions and understanding the outcomes of secondary contact. Furthermore, when hybridization is possible between native and introduced taxa, genetic tools and multilocus genotyping become essential for resolving the consequences of hybridization for endemic populations (Viard, David, & Darling, 2016).
In the marine environment, semi‐reproductively isolated species complexes are a common and persistent issue for detecting marine invasions (Bouchemousse, Liautard Haag, Bierne, & Viard, 2016; Viard et al., 2016). Many marine species exhibit high fecundity and dispersal potential (through planktonic larvae) that support elevated rates of gene flow and low genetic differentiation between populations (Gagnaire et al., 2015). Weak differentiation is also sustained by large effective population sizes that slow down genetic drift, such that high levels of ancestral polymorphisms are common features of many diverging marine taxa (e.g. Fraïsse, Belkhir, Welch, & Bierne, 2016; Gagnaire, Normandeau, & Bernatchez, 2012). Genomic methods based on differentiation (i.e. F‐statistics) alone may fail to distinguish between recently diverged native and introduced species or identify sources of introduced populations (Tepolt, 2015; Viard et al., 2016). Furthermore, because lineages with large effective populations sizes are not expected to reach genome‐wide reciprocal monophyly for many generations (up to 10Ne generations; Keightley & Eyre‐Walker, 2012), both incomplete lineage sorting and recent (post‐introduction) gene flow may lead to shared polymorphisms between semi‐isolated species (Marko & Hart, 2011). Ongoing introgression is therefore difficult to recognize and quantify when native and introduced taxa show either weak divergence or genomes shaped by complex speciation histories of intermittent historical contact (Fraïsse et al., 2016). Additionally, because both F ST and linkage disequilibrium‐based population clustering approaches assume a mutation‐drift equilibrium and a single demographic model (i.e. Wright's island model; Wright, 1951), such methods cannot provide explicit tests of migration or demographic history underlining patterns of genetic ancestry (Patterson et al., 2012; Pickrell & Pritchard, 2012). In turn, neglecting complex demographic scenarios that have shaped the genetic backgrounds of closely related taxa may mislead interpretations of population relationships and introgression between endemic and introduced marine populations (Rougemont & Bernatchez, 2018).
Methods that model demographic histories across many loci offer powerful approaches for resolving the contributions of ancestral polymorphism and recent introgression to shared variation between species (Fagundes et al., 2007; Fu & Li, 1997; Pritchard, Seielstad, Perez‐Lezaun, & Feldman, 1999). Coalescent genealogy samplers, for example, allow explicit inferences of divergence and migration rate parameters (e.g. isolation‐with‐migration models; Hey & Nielsen, 2004, 2007; Kuhner, 2009; Marko & Hart, 2012; Sousa, Carneiro, Ferrand, & Hey, 2013), but rely on full‐likelihood calculations that are computationally intractable for large genomic data sets or complex demographic histories experienced by marine taxa (Roux et al., 2016). Approximate Bayesian computations (ABC) allow tests of alternative divergence models and rely on few samples per taxon for robust inferences of divergence histories while avoiding full‐likelihood computations (Beaumont, 2010; Beaumont, Zhang, & Balding, 2002; Bertorelle, Benazzo, & Mona, 2010; Pritchard et al., 1999; Roux et al., 2016). ABC approaches have been highly informative for reconstructing invasion routes (Barker, Andonian, Swope, Luster, & Dlugosch, 2017; Estoup & Guillemaud, 2010; Lombaert et al., 2011) and identifying introgression in contact zones (Estoup, Beaumont, Sennedot, Moritz, & Cornuet, 2004; Estoup, Wilson, Sullivan, Cornuet, & Moritz, 2001; Guillemaud, Beaumont, Ciosi, Cornuet, & Estoup, 2010; Pascual et al., 2007; Roux, Tsagkogeorga, Bierne, & Galtier, 2013). Coalescent approximations can also strengthen comparative inferences of historical relationships between weakly differentiated introduced and native taxa when taxonomic boundaries are also challenged by invasive introgression.
Marine mussels in the genus Mytilus are a compelling example of a morphologically cryptic and reproductively semi‐isolated group of species that have also experienced complex evolutionary histories of past hybridization and contemporary human‐mediated secondary contact. The Mediterranean native, M. galloprovincialis, is recognized as one of the world's most widespread invasive species and is surprisingly the only Mytilus congener known to pose invasion threats globally (Lowe, Browne, Boudjelas, De, & Poorter, 2000; McDonald, Seed, & Koehn, 1991). Despite several pre‐ and postzygotic reproductive isolating mechanisms between Mytilus species (e.g. Bierne, Bonhomme, Boudry, Szulkin, & David, 2006; Bierne, Bonhomme, & David, 2003; Bierne, Borsa, et al., 2003; Skibinski, Beardmore, & Cross, 1983), M. galloprovincialis has a well‐documented history of hybridizing with native congeners where their ranges overlap throughout its introduced distribution in the northern hemisphere (e.g. Japan, Brannock, Wethey, & Hilbish, 2009; California, Rawson, Agrawal, & Hilbish, 1999; Saarman & Pogson, 2015). There is also strong evidence for differential introgression with sister species, Mytilus edulis, in some parts of its present day native range across mosaic hybrid zones in Europe (Bierne, Borsa, et al., 2003; Fraïsse et al., 2016; Fraïsse, Roux, et al., 2018; Gosset & Bierne, 2013; Rawson & Hilbish, 1998; Roux et al., 2014). Interspecific admixture with M. edulis has subsequently led to pronounced genetic differentiation between M. galloprovincialis lineages from the Mediterranean Sea and Atlantic Europe (Fraïsse et al., 2016; Quesada, Wenne, & Skibinski, 1995). These divergent M. galloprovincialis lineages display partial reproductive isolation (El Ayari et al., 2019) and have both been implicated in independent invasions into California (Daguin & Borsa, 2000; McDonald & Koehn, 1988) and South Africa (Branch & Steffani, 2004), respectively.
Despite a number of genetic investigations, less is known about the invasive distribution of M. galloprovincialis in other parts of the southern hemisphere (Daguin & Borsa, 2000; Gardner, Zbawicka, Westfall, & Wenne, 2016; Gérard, Bierne, Borsa, Chenuil, & Féral, 2008; Hilbish et al., 2000; Larraín, Zbawicka, Araneda, Gardner, & Wenne, 2018; McDonald et al., 1991; Oyarzún, Toro, Cañete, & Gardner, 2016). The widespread occurrence of northern M. galloprovincialis haplotypes along temperate coastlines in Chile, New Zealand and Australia suggests that introduced populations are established in coastal regions (e.g. Gardner et al., 2016; Larraín et al., 2018; Westfall & Gardner, 2010). However, the existence of morphologically cryptic Mytilus lineages endemic to the southern hemisphere has sustained ongoing confusion regarding M. galloprovincialis introductions in these regions (Ab Rahim et al., 2016; Colgan & Middlefart, 2011; Dias, Fotedar, & Snow, 2014; Larraín et al., 2018; Westfall & Gardner, 2010). In Australia, fossil Mytilus shells predating European contact (New South Wales, Donner & Jungner, 1981; South Australia, Hope, Lampert, Edmondson, Smith, & Tets, 1977; Tasmania, Colhoun, Turner, & Van de Geer, 1982; reviewed in McDonald et al., 1991; Hilbish et al., 2000) support an endemic taxon. Yet, resolving the taxonomic affinity of the native Australian species, originally named Mytilus planulatus Lamarck 1819, has been complex: initial genetic studies using size polymorphic nuclear markers suggested high genetic similarity to northern M. galloprovincialis and described the native taxon as an endemic southern hemisphere lineage of M. galloprovincialis (Borsa, Daguin, & Bierne, 2007; Daguin & Borsa, 2000; McDonald et al., 1991). Later phylogenetic comparisons of the mitochondrial marker COI, however, dated the origins of southern Mytilus to the late Pleistocene approximately 0.84 (0.5–1.3 mya, Gérard et al., 2008) and 1.2 million years ago, implicating deeper historical isolation between northern and southern taxa (Hilbish et al., 2000).
To date, taxonomic delineation of the Australian endemic taxon has been hampered by a limited number of reliable loci that has also precluded detailed investigations of its recent evolutionary history. In particular, high levels of ancient incomplete lineage sorting among Mytilus species (resulting in various gene topologies) are likely to obscure signals of present day introgression and further amplify discordant species relationships when few loci are examined (Fraïsse, Haguenauer, et al., 2018). Thus, it remains unresolved (a) whether Australian native Mytilus comprise a lineage sufficiently divergent from northern M. galloprovincialis to warrant species status as M. planulatus, and (b) whether the history of Australian mussels reflects recent divergence only, or includes ongoing hybridization with introduced congeners. Here, we use transcriptome‐wide population genomic analyses with ABC inferences to test alternative hypotheses regarding the origins of Australian Mytilus mussels (hereafter referred to as its current nomenclature, M. planulatus) and resolve the contributions of past and ongoing (post‐introduction) gene flow with introduced M. galloprovincialis in south‐eastern Australia. This study represents the first transcriptome‐wide investigation of demographic history and introgression between introduced and Australian endemic Mytilus species.
2. METHODS
2.1. Sample collection, RNA extraction and sequencing
Mussels were collected from wild populations from rocky intertidal or subtidal environments (Table 1). Outgroup specimens (Mytilus californianus, n = 3; Mytilus trossulus, n = 3; M. edulis n = 3) were collected from known contemporary allopatric ranges to minimize the possibility of sampling hybrid individuals. We collected Atlantic (n = 5) and Mediterranean (n = 10) M. galloprovincialis belonging to two genetically divergent lineages separated by the Almeria‐Oran front, including a population east of the Siculo–Tunisian Strait dividing the eastern and western Mediterranean (Fraïsse et al., 2016; Table 1). In Australia, we targeted previously unsampled locations to include populations where introductions would be likely, such as large shipping harbours (i.e. Sydney Harbour, n = 9) and a second location (i.e. Batemans Bay, n = 9), to extend previous sampling efforts across the eastern coast of Australia. We additionally included samples from Tasmania (n = 5), where high frequencies of the divergent southern mitochondrial haplotypes have been reported (Colgan & Middlefart, 2011). Individuals were genotyped for the species diagnostic marker Glu‐5’ (Rawson, Joyner, Meetze, & Hilbish, 1996) to obtain a first clue about species identity. Preliminary assignment of Australian samples (total samples = 23; Table 1) as M. planulatus was based on the F‐type (female) mitochondrial marker COIII using primers from Riginos, Hickerson, Henzler, and Cunningham (2004) and phylogenetic analyses using neighbour‐joining statistics implemented in Geneious 8.1. Total RNA was extracted from 10 to 20 mg of mantle tissue (preserved in RNAlater) using the RNeasy Plant Mini Kit and following the animal tissue protocol with an additional DNAse treatment to remove genomic DNA. Individual cDNA libraries were constructed and barcoded using the TruSeq stranded mRNA kit (Illumina), with average insert sizes of 250–300 bp. Paired‐end (125 bp fragments) libraries were sequenced across three lanes of an Illumina Hiseq2000 or across a single lane of an Illumina Hiseq4000.
Table 1.
Details of samples and collection locations
| Taxon | Sampling location | Range | Individuals sequenced |
|---|---|---|---|
| Mytilus californianus | Scripps Institute of Oceanography, California, USA | Native | 3 |
| Mytilus trossulus | Lighthouse Park, British Columbia, Canada | Native | 3 |
| Mytilus edulis | Darling Marine Station, Maine, USA | Native | 3 |
| Mytilus galloprovincialis | Primel, France (Atlantic)* | Native | 5 |
| Crique, Les Issambres, France (Mediterranean‐West)* | Native | 5 | |
| Herceg Novi, Montenegro (Mediterranean‐East)* | Native | 5 | |
| Mytilus galloprovincialis | Sydney Harbour, New South Wales, Australia a | Introduced a | 9 |
| Batemans Bay, New South Wales, Australia a | Introduced a | 9 | |
| Mytilus planulatus | Spring Bay, Tasmania, Australia | Native | 5 |
Samples in bold indicate species identity (based on genome‐wide analyses) and range. Populations marked with an asterisk were used to construct a de novo reference transcriptome assembly for M. galloprovincialis.
M. galloprovincialis samples are introgressed with M. planulatus.
RNA‐seq data sets were trimmed and filtered to select for the highest quality reads. Three native range M. galloprovincialis populations (Table 1) were used to construct a de novo reference transcriptome assembly free of contaminant sequences. Details regarding RNA‐seq read filtering, processing and de novo transcriptome assembly are outlined in the Appendix S1. The resulting 159,985 nuclear sequences were used as a reference assembly for variant discovery and as input for all downstream analyses.
2.2. Genomic data filtering
RNA‐seq reads from Australian samples and four northern Mytilus taxa were mapped to the M. galloprovincialis de novo reference transcriptome (Appendix S1) using Bowtie2 (Langmead & Salzberg, 2012), and PCR duplicates were removed using Picard MarkDuplicates (http://picard.sourceforage.net). Different subsequent analyses required different filtering schemes (Figure S1): for genomic analyses (with the exception of ABC inference), single nucleotide polymorphisms (SNPs) were called using Freebayes (https://github.com/ekg/freebayes) and filtered in VCFtools (Danecek et al., 2011). Variant sites below a minimum genotype quality of 30 and a minimum mean depth coverage of 10 reads were excluded. For principal component analyses and genomic analyses of species relationships (network analysis and topology weighting), we removed singletons, indel variants and positions with missing data. Genotypes were statistically phased using beagle v4.1 (Browning & Browning, 2007). We generated consensus sequences for individual haplotypes using the corresponding VCF file and reference assembly in BCFtools v1.3.1. For analyses investigating population structure and admixture (ADMIXTURE and TreeMix), we additionally removed SNPs with a minor allele frequency of less than 5%, but retained positions with up to 20% missing data. For variant calling and filtering pertaining to ABC inference, refer to Approximate Bayesian Computations (ABC) of demographic history.
2.3. Analyses of population structure and admixture
We first established whether individuals sampled in Australia belonged to introduced or endemic genomic backgrounds by comparing Australia mussel genotypes against northern native range M. galloprovincialis. Principal component analysis and genomic clustering analyses using the program ADMIXTURE (Alexander, Novembre, & Lange, 2009) were undertaken to ascertain the presence of the putative endemic M. planulatus and the possibility of admixture between native and introduced populations. In ADMIXTURE, we estimated individual ancestry proportions with M. edulis as an outgroup taxon. VCFtools and PLINK v1.90 (Purcell et al., 2007) were used to convert the filtered VCF output to BED format files as input, which reduced the original data set to 34,097 biallelic SNPs across 3,945 contigs. We ran ADMIXTURE with 100 iterations and used the cross‐validation procedure with 50 replicates for K = 1 to K = 10 genetic clusters.
Second, to test for introgression and to validate potential sources of gene flow, we performed joint analyses of migration and historical population relationships in TreeMix v1.12 (Pickrell & Pritchard, 2012). TreeMix uses allele frequency correlations between populations to infer a maximum‐likelihood population tree representative of the phylogenetic relationships between groups. Migration edges are subsequently added between branches with varying strength (branch weight = w) and directions to determine whether incorporating admixture events improves the likelihood of the tree given the genetic data. Mytilus edulis was used as an outgroup to focus inferences on recent admixture events and we accounted for linkage disequilibrium by performing analyses on windows of 100 variants. We examined the residual plot of pairwise population genetic covariances to infer the possibility of gene flow, where negative residual standard error values suggest closer relationships between populations than compared to the population tree with no migration events. We then modelled 1–10 migration events sequentially to see whether adding migration edges to the phylogeny improved the likelihood fit to the data. We calculated the standard error of migration events with the –se option without sample size correction (option –noss). We used a stepwise comparison Akaike information criterion (AIC) between sequential migration models to determine whether adding a migration edge significantly improved the likelihood of the population tree. We calculated AIC values as (−2(ln(likelihood)) + 2K), where K is the number of free parameters in the model. We did not consider additional migration events when the difference between nested models was less than two (∆AIC < 2).
Third, the three‐population (f3) test of admixture (Keinan, Mullikin, Patterson, & Reich, 2007; Reich, Thangaraj, Patterson, Price, & Singh, 2009) was used to verify evidence of migration inferred by TreeMix. We estimated the f3 statistic using the threepop function. The f3 statistic estimates whether allele frequency differences between each population combination deviate more than expected due to incomplete lineage sorting, thereby suggesting recent admixture. Significant migration is inferred if the f3 statistic is negative and has a z‐score of ≤−3.8 (equivalent to a p‐value < .0001), which is determined through a jackknifing procedure over 100‐SNP windows.
2.4. Genomic analysis of species relationships
To visualize genomic relationships between Australian lineages against four northern Mytilus outgroup taxa (Table 1), we performed a genomic network analysis of individual haplotype sequences using the neighbour‐net method in SplitsTree4 v4.14.6 (Huson & Bryant, 2006). The phylogenetic network was generated from a concatenated nucleotide sequence (constructed in R) consisting of 27,343 SNPS from 2,620 nuclear contigs, using default settings. We also estimated a phylogenetic network based on 144 SNPs from 12 protein‐coding female mitochondrial genes. The distinctive Mytilus male mitotypes were not recovered due to low coverage in the transcriptome data.
To quantify how species relationships between M. planulatus and northern hemisphere Mytilus species vary across the nuclear genome, we estimated the relative contributions of three possible topologies (i.e. grouping M. planulatus with one of three outgroup species: M. galloprovincialis (Mediterranean), M. edulis or M. trossulus) to the nuclear species tree using a heuristic topology weighting analysis in TWISST (topology weighting by iterative sampling of subtrees; Martin & Van Belleghem, 2017). TWISST estimates the weight or contribution of all possible unrooted topologies for each locus by resampling a single haplotype per taxon to generate all possible subtrees for that locus. To minimize unresolved topologies due to present day introgression, we included only putative non‐introgressed M. planulatus samples from Tasmania showing no evidence of admixture in initial analyses (discussed in section above). We also excluded the most distant outgroup, M. californianus, to limit comparisons to three possible topologies. Consensus haplotypes for each locus were analysed individually; we inferred locus‐specific genealogies with the R package ‘Ape’ using the neighbour‐joining method and F84 distances (Felsenstein & Churchill, 1996). To exclude poorly resolved phylogenies (e.g. Martin & Van Belleghem, 2017), we performed the analysis on a subset of 343 genealogies with a minimum tree length of 0.025, which is equivalent to 5 SNPs every 200 bp.
2.5. Approximate Bayesian Computations (ABC) of demographic history
We used an ABC framework to test the hypothesis that M. planulatus from Tasmania (putative endemic lineage) have experienced an independent evolutionary history from northern M. galloprovincialis. We evaluated six alternative models of Australian Mytilus origins representing a spectrum of divergence histories between northern and southern taxa: panmixia (pan), divergence in isolation (div), isolation with migration (im), divergence with ancient gene flow (divAGF), divergence with recent (invasive) secondary contact (divSC) and divergence with ancient gene flow and recent secondary contact (divAGFSC) (Refer to Figure 1).
Figure 1.
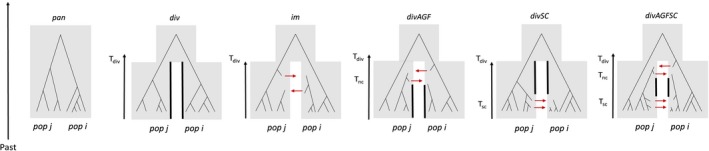
Competing divergence models between northern M. galloprovincialis (pop j) and native M. planulatus (pop i). Models assumed constant effective population sizes and divergence from an ancestral panmictic population at time Td iv. The pan model assumes populations belong to the same gene pool. The div model assumes populations evolve independently with no gene flow since their divergence. In the im model, populations diverge with ongoing gene flow to the present day. The divAGF model assumes bidirectional migration is restricted to the early stages of speciation from T div to a more recent time (T nc) up to the last glacial maximum (20,000 years ago), after which populations evolve independently with no migration. This scenario is consistent with transequatorial migration between hemispheres facilitated by cyclical glacial cooling of the oceans during the late Pleistocene. In the divSC model, populations evolve in allopatry until recent human‐mediated secondary contact (T sc), when populations begin to exchange genes. This scenario tests explicitly for the presence of post‐introduction gene flow from northern M. galloprovincialis into Australian populations assuming that the onset of migration occurs after the earliest record of European contact (<600 years ago). Finally, the divAGFSC model assumes that populations diverged with ancient migration for a period of time, after which they evolve in allopatry; genetic exchange is re‐established at T sc following recent secondary contact via human‐mediated introductions
We compared the genomic backgrounds of M. planulatus sampled in Tasmania against two divergent M. galloprovincialis lineages from the Mediterranean and Atlantic. Because of the power afforded by analysing large numbers of loci across the genome (i.e. thousands of independent genealogies), model‐based inferences of isolation and migration are robust to small sample sizes (i.e. n = 5–10) (Fraïsse, Roux, et al., 2018; Robinson, Bunnefeld, Hearn, Stone, & Hickerson, 2014). Details relating to (a) the filtering of empirical genetic data sets, (b) parameterizing and generating coalescent simulations of genetic data under separate demographic models, (c) demographic model selection, (d) model validation, (e) incorporating parameter heterogeneity and (f) demographic parameter estimation are outlined in the Appendix S1.
Briefly, as input for ABC analyses, we mapped reads against a reduced protein‐coding M. galloprovincialis transcriptome assembly and called variants using the reads2snps program. Subsequent filtering and analyses were conducted using custom R scripts (https://github.com/dinmatias) implementing an existing ABC pipeline (Roux et al., 2016; https://github.com/popgenomics/popPhylABC). The resulting empirical data sets consisted of 1,362 loci (Mediterranean‐Tasmania) and 1,539 loci (Atlantic‐Tasmania). For the simulated data, we used msnsam to generate one million multilocus simulations under each demographic model, for each population pair (Figure 1; Ross‐Ibarra et al., 2008). Initial models assumed equal (homogeneous) effective population size (N e) among loci and homogeneous migration rates (m) every generation. Each simulation was parameterized by model‐specific demographic parameters (Table S1) that are described in the Appendix S1. A standard set of 39 summary statistics (e.g. Fraïsse, Roux, Welch, & Bierne, 2014) of divergence and polymorphism were calculated for each simulation and for the empirical genetic data using mscalc (Ross‐Ibarra et al., 2008).
We evaluated the posterior support for alternative demographic models by performing a categorical regression (neural network method) on the model identity and summary statistics of the posterior samples (Beaumont, 2010) using the packages ‘abc’ (Csilléry, François, & Blum, 2012) and ‘nnet’ (Ripley, Venables, & Ripley, 2016) in R. We validated the power of our approach by performing the same analyses on 1,000 pseudo‐observed data sets (PODS) for each model simulated from the prior distribution. From this cross‐validation, we determined the overall precision (rate of correctly supporting a true model) and misclassification (type I error: rate by which incorrect models are supported) of our approach.
For initial ABC comparisons, simulated demographic models assumed genome‐wide homogeneous N e and m. However, modelling the effects of linked selection on genome‐wide variation has been shown to improve the accuracy of demographic inferences in Mytilus species (Roux et al., 2014) and other semi‐isolated marine taxa (e.g. Ciona sp., Roux et al., 2013; sea bass, Tine et al., 2014; Salmo salar, Rougemont & Bernatchez, 2018). To account for the combined effects of variable among‐locus rates of genetic drift and differential migration (Roux et al., 2016), we re‐simulated a series of nested models incorporating heterogeneous N e and/or heterogeneous m under the best demographic scenario (inferred from initial homogeneous model comparisons) to estimate demographic parameters. We varied the initial N e and m values for a certain proportion of loci by either (a) decreasing the initial parameter value (hetero1) or (b) allowing loci to have a lower or higher parameter values than the initial draw (hetero2). Finally, demographic parameters were estimated for each population pair using the posterior distribution approximated by accepted simulations under the best inferred demographic model.
3. RESULTS
3.1. Population structure and evidence for genome‐wide admixture
Principal component analysis of 20,509 SNPs revealed separation between Australian samples and northern M. galloprovincialis, explaining 7.31% (PC1) and 6.55% (PC2) of variance among individual genotypes (Figure 2a). Populations from Sydney Harbour and Batemans Bay showed intermediate placement between samples from Tasmania and northern M. galloprovincialis populations. Divergence of these populations across the second PC axis points to likely admixture with divergent lineages of M. galloprovincialis from the Mediterranean and Atlantic. ADMIXTURE analyses for K = 4 clusters discriminated between genetic groups identified in the PCA (Figure 2b). Individuals sampled in Batemans Bay and Sydney Harbour showed greater than 50% shared ancestry proportions with northern M. galloprovincialis, suggesting at least two independent introductions into Australia of divergent source lineages: Atlantic M. galloprovincialis into Batemans Bay and Mediterranean M. galloprovincialis into Sydney Harbour, with subsequent admixture with these native populations. Analyses for all K clusters did not provide evidence of mixed ancestry proportions in Tasmania. Admixture proportions were consistent when analyses were performed using one SNP per contig to account for linkage effects, as nearby SNPs are not independent (Figure S2).
Figure 2.
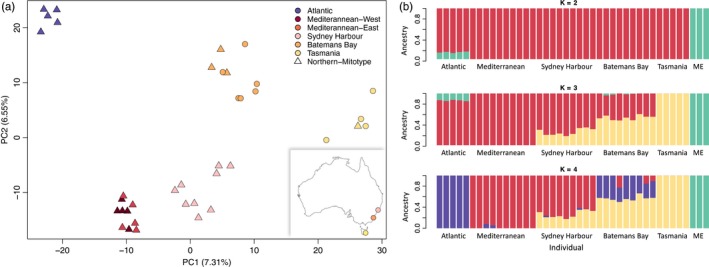
(a) Principal component analysis of three populations sampled in Australia (geographic location shown in inset map) and northern M. galloprovincialis from its native range in the Atlantic and Mediterranean Sea. Colours correspond to populations, and individuals marked with a triangle indicate samples carrying northern clade (M. galloprovincialis) mitochondrion (refer to Figure 4). (b) ADMIXTURE analyses for K = 2–4 genetic clusters, including M. edulis (ME) as an outgroup taxon. Each bar represents an individual with genetic elements belonging to one or more ancestral clusters, corresponding to different colours
For TreeMix analyses, the population tree without migration explained 95.88% of variation in the allele frequency covariance matrix based on 34,097 SNPs (Figure S3). However, we observed high residual covariance between both Sydney Harbour and Batemans Bay with northern populations. The addition of four admixture events explained more than 99% of the genotypic variance and provided the highest likelihood fit (based on stepwise AIC comparisons) compared to models with fewer migration edges (Figure 3a); however, only two out of the four migration edges (those into Australian populations) accounted for most of the explained genotypic variance. We found significant p‐values (p « .001) for individual migration edges, although the direction of the migration edges should not be interpreted at face value (Figure 3a).
Figure 3.
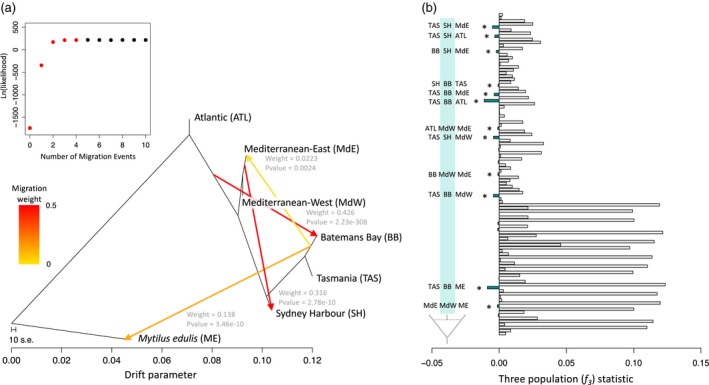
Tests of migration inferred by TreeMix. (a) Maximum‐likelihood population tree with four migration events including Australian M. planulatus, M. galloprovincialis and M. edulis as an outgroup. The addition of four admixture events significantly improved the fit of the population tree to the genetic data compared to a model with no migration. (b) The right panel shows f 3 statistics for all population combinations including M. edulis as an outgroup. Significant and negative f 3 values indicated with an asterisk and the corresponding three‐population combination is shown to the left of the asterisk, where the middle population marked with green shows evidence of admixture with putative ancestral populations indicated on either side
Results from TreeMix confirmed evidence for introgression from eastern Mediterranean M. galloprovincialis into Sydney Harbour (w = 33%) and migration from the Atlantic M. galloprovincialis population into Batemans Bay (w = 40%), suggesting contemporary admixture between native and introduced populations. Results did not provide evidence for migration into Tasmanian mussels. The slight signal of admixture between Batemans Bay and M. edulis suggests shared genetic elements are likely a result of secondary contact with Atlantic M. galloprovincialis populations that share ancestry with M. edulis through past and ongoing introgression (Fraïsse et al., 2016). The weak strength of this admixture event (w = 12%) is consistent with small proportions of M. edulis ancestry observed among Atlantic M. galloprovincialis individuals in clustering analyses (Figure 2b; K = 3). Similarly, slight evidence of admixture between the eastern Mediterranean and Batemans Bay likely indicates allele sharing with Mediterranean M. galloprovincialis through introgression with Atlantic populations prior to introduction; however, this migration edge was not strongly supported (p > .001; Figure 3a).
For f3 statistics, we found significantly negative values (p < .0001) for almost all population combinations involving either Sydney Harbour or Batemans Bay as the admixed population, with M. planulatus (Tasmania) and northern M. galloprovincialis as putative ancestral populations (Figure 3b). Additionally, we detected signatures of M. edulis genetic elements in both Australian (e.g. Batemans Bay) and Mediterranean M. galloprovincialis (Mediterranean‐West) genetic backgrounds. Three‐population tests involving Tasmania as an admixed population did not yield significant values for any population combinations, supporting the hypothesis that samples in this region are representative of the endemic lineage.
3.2. Genomic analysis of species relationships
Consensus haplotype genetic networks of Australian samples and four Mytilus outgroup taxa constructed from 12 mitochondrial genes and 2,620 nuclear contigs revealed discordance between the mitochondrial tree and the average nuclear tree. Individuals carrying the Australian (female) mitotype formed a distinct divergent clade (Figure 4). In contrast, the same individuals clustered together alongside M. galloprovincialis when nuclear loci were analysed. TWISST analyses of species relationships corroborated that gene trees grouping M. planulatus with M. galloprovincialis dominated the nuclear genome (Figure S4). The mean weighting for topologies placing M. planulatus as a sister species to the invasive taxon was 54%, supporting a close relationship between these two species (Figure S4). Only 39/343 loci (11%) had fully resolved topologies (topology weight = 1.0), all of which grouped M. planulatus with M. galloprovincialis. Visual inspection of these topologies revealed that all loci are paraphyletic (do not form species‐specific clades) with M. galloprovincialis, suggesting high levels of ongoing incomplete lineage sorting in Tasmanian samples or genetic exchange, although we did not recover evidence supporting introgression in this population in any analyses. Topologies grouping M. planulatus with M. edulis and M. trossulus showed mean weightings across contigs of 21% and 23%, respectively (Figure S4), corroborating ancient incomplete lineage sorting resolved into multifarious gene tree topologies in the Mytilus species tree phylogeny.
Figure 4.
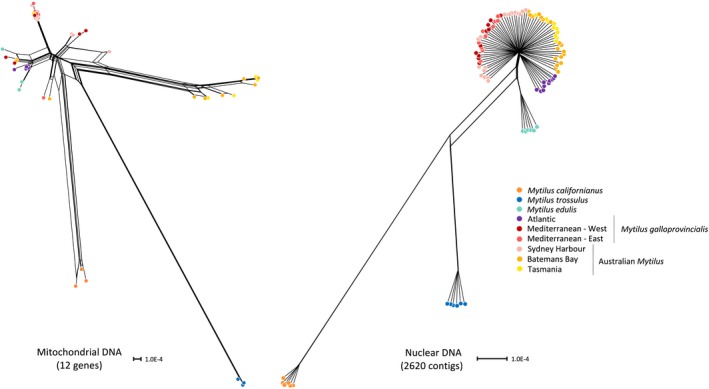
Consensus haplotype genetic network of Australian samples (M. planulatus samples from Tasmania are shown in yellow) and four outgroup taxa. Network phylogenies are constructed from 12 mitochondrial genes (left) and 2,620 nuclear contigs (right)
3.3. Historical demographic inference using ABC
3.3.1. Levels of endemic genetic diversity and divergence at synonymous sites
The majority of biallelic polymorphic sites were shared between M. planulatus and M. galloprovincialis (average shared polymorphic sites across loci = 5.36–5.83), compared to private polymorphisms in M. planulatus (average private polymorphic sites = 2.87–4.07). Levels of nucleotide diversity at synonymous sites (averaged across loci) were similar for all populations based on Tajima's pi (Tajima, 1983) ranging between 0.023 and 0.024 for Tasmanian M. planulatus and 0.021 (Mediterranean) and 0.026 (Atlantic) for northern M. galloprovincialis. Pairwise comparisons indicated low population differentiation evidenced by few fixed variants (0–0.02 averaged across contigs) and low F ST or absolute (d xy) and net (D a) divergence values between Tasmania and Mediterranean (mean F ST = 0.052; d xy = 0.25; D a = 0.003) or Atlantic (mean F ST = 0.087; d xy = 0.30; D a = 0.005) M. galloprovincialis, indicating that population differentiation is largely driven by the presence of private alleles. Departures of the site frequency spectrum measured as mean Tajima's D values (Tajima, 1989) varied between populations; we observed negative values close to neutrality in Tasmania (average D = −0.151) and greater negative values in M. galloprovincialis (Mediterranean D = −0.475; Atlantic D = −0.445), indicating an excess of rare alleles due to population expansions, gene flow from unsampled populations or signatures of directional selective processes.
3.3.2. Historical isolation between northern and southern hemispheres
We compared six models of divergence between Tasmanian M. planulatus and M. galloprovincialis using an ABC framework. We first inferred the best demographic model by comparing models with homogeneous among‐locus parameters. The model of divergence with ancient gene flow (divAGF) received the highest posterior support (>82% posterior probability using neural network inference and acceptance threshold 0.001) for both population pairs (Table 2). The strict divergence model (div) provided the second highest posterior probability in most comparisons. We observed consistent rejection of the isolation‐with‐migration (im) model suggesting that historical gene flow was followed by divergence in isolation. Model comparisons returned no support for panmixia and weak support for all models that included recent gene flow associated with contemporary introductions < 600 years ago.
Table 2.
Summary of demographic model selection under an approximate Bayesian computation (ABC) framework
| Tolerance | Method | Demographic model probability: proportion of accepted simulations | |||||
|---|---|---|---|---|---|---|---|
| pan | Div | im | divSC | divAGF | divAGFSC | ||
| (A) | |||||||
| 0.001 | Rejection | 0.0000 | 0.1320 | 0.0417 | 0.0188 | 0.7835 | 0.0240 |
| 0.01 | Rejection | 0.0000 | 0.1955 | 0.0708 | 0.0546 | 0.6130 | 0.0660 |
| 0.001 | Rejection | – | 0.1588 | 0.0266 | 0.0100 | 0.7894 | 0.0152 |
| 0.01 | Rejection | – | 0.1972 | 0.0664 | 0.0398 | 0.6421 | 0.0545 |
| 0.001 | Neural Net | – | 0.0328 | 0.0375 | 0.0056 | 0.9094 | 0.0142 |
| 0.01 | Neural Net | – | 0.0618 | 0.0051 | 0.0026 | 0.9267 | 0.0038 |
| (B) | |||||||
| 0.001 | Rejection | 0.0000 | 0.3868 | 0.0037 | 0.0015 | 0.6047 | 0.0033 |
| 0.01 | Rejection | 0.0000 | 0.3052 | 0.0135 | 0.0069 | 0.6654 | 0.0090 |
| 0.001 | Rejection | – | 0.4252 | 0.0036 | 0.0028 | 0.5654 | 0.0030 |
| 0.01 | Rejection | – | 0.3129 | 0.0116 | 0.0062 | 0.6611 | 0.0081 |
| 0.001 | Neural Net | – | 0.0527 | 0.0019 | 0.0059 | 0.8273 | 0.1123 |
| 0.01 | Neural Net | – | 0.0408 | 0.0010 | 0.0009 | 0.9557 | 0.0015 |
Model posterior probabilities assuming homogeneous N e and m parameters for A) the Mediterranean‐Tasmania population pair; and B) the Atlantic‐Tasmania population pair. In model comparisons where not all six demographic models had accepted values within the applied threshold, the simple rejection method (i.e. linear regression) was applied. Bold indicates the highest probability model for each comparison.
Model choice validation indicated that we could discriminate between the best inferred model (divAGF) and models including recent genetic exchange (im, divSC, divAGFSC). The divAGF model had the highest posterior probability in 59% (Atlantic) and 61% (Mediterranean) of model comparisons using PODs generated under the same model (i.e. precision; Figure S5, Table S3). However, we found that 41% (Atlantic) and 39% (Mediterranean) of PODs simulated under the divAGF model were misclassified as divergence in isolation (div). Measures of robustness in the accuracy of model discrimination indicated a minimum threshold for model probability ≥ 83% required to yield a robustness of 95% or greater for the divAGF model, corroborating initial model choice inference (Table 2). Overall, there was clear discrimination between models excluding (divAGF, div) and including (im, divSC, divAGFSC) ongoing gene flow since divergence, suggesting that M. planulatus and M. galloprovincialis have experienced a period of historical isolation.
3.3.3. Genome‐wide heterogeneous genetic drift and migration
Comparisons of the heterogeneous models (under the best demographic scenario; divAGF) allowing among‐locus variation in N e and m provided an improved model fit to the observed genetic data when compared to the divAGF model with homogeneous parameters (Table S4). Specifically, we found that models allowing N e to vary both below and above the initial N e parameter value (hetero2) and where m was either homogeneous (Mediterranean‐Tasmania) or heterogeneous (Atlantic‐Tasmania) outperformed other models (N e homo and N e hetero1) with a cumulative posterior probability > 90% for both population comparisons. Substantial support for hetero2 models suggests that heterogeneity in N e is better captured with models incorporating both extremes of variation across loci; processes leading to both low levels of variation (e.g. due to linked selection, Rougemont & Bernatchez, 2018) and high diversity at synonymous loci (e.g. due to polygenic balancing selection, Charlesworth, 2006; introgression from unsampled ghost populations, Butlin et al., 2014; variable levels of ancestral diversity, Guerrero & Hahn, 2017) are therefore important factors to account for when reconstructing population histories and estimating demographic parameters.
3.3.4. Demographic parameter inference from the best models
For the best heterogeneous divAGF models, divergence time parameters were contained within the prior distribution and well differentiated in both population comparisons (Mediterranean‐Tasmania Mode: 116,352 generations, CI 95% [90,858–173,929]; Atlantic‐Tasmania Mode: 134,157 generations, CI 95% [98,371–274,804]; Figure 5). Divergence time estimates suggest that Australian M. planulatus started diverging in allopatry between 100,000 and up to 600,000 years ago and are likely to have experienced low levels of historical gene flow. However, estimates of ancient migration (ma), including the time of onset of ancient gene flow since the present (T nc), were poorly resolved and could not be estimated with accuracy.
Figure 5.
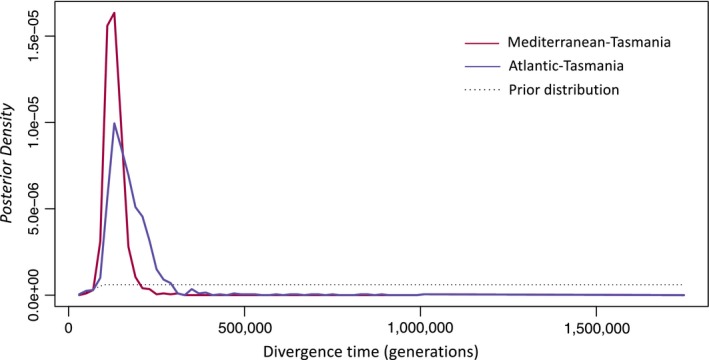
Posterior distributions for divergence time (T div) parameter estimates inferred from ABC analyses for two population pairs based on highest probability demographic model (divAGF) accounting for heterogeneous N e and m parameters. Parameter plots correspond to models with the highest estimated probability in which the N e parameter was hetero2 and m was either homogeneous (Mediterranean‐Tasmania; mode: 116,352 generations) or hetero1 (Atlantic‐Tasmanian; mode: 134,157 generations)
4. DISCUSSION
Detecting species introductions and documenting cases of recent introgression are notoriously challenging when native and introduced species inhabit the “grey zone” of the speciation continuum, where species boundaries are often contentious (Roux et al., 2016). Here, we utilize the power of transcriptome‐wide markers to uncover the complex divergence history of Mytilus mussels in Australia. Unexpectedly, we find that contemporary introductions of M. galloprovincialis into south‐eastern Australia originate from genetically distinct northern hemisphere lineages, implicating at least two independent introductions into this region. Contingent on resolving species relationships, we also provide evidence that both introductions are associated with recent admixture with the native Australian taxon, M. planulatus. Through increased genome‐wide resolution of species relationships, combined with demographic modelling, we validate that M. planulatus sampled in Tasmania are representative of the endemic Australian lineage and have been isolated from northern M. galloprovincialis for at least 100,000 years. Taken together, our study demonstrates the utility of genomic data for detangling the contributions of contemporary invasive hybridization from signatures left by historical gene flow and recent divergence histories in high dispersal marine species.
4.1. Multiple introductions from northern hemisphere source lineages
Our study uncovers two new and important results regarding M. galloprovincialis introductions and its interactions with endemic southern hemisphere taxa, building upon previous genetic studies (e.g. Borsa et al., 2007; Gérard et al., 2008; Westfall & Gardner, 2010). First, we demonstrate that introductions into Australia are derived from genetically distinct source lineages of M. galloprovincialis from the Mediterranean Sea and the Atlantic coast of Europe. Genotypic variance across nuclear SNPs revealed differentiation between populations from Sydney Harbour and Batemans Bay from a more divergent Tasmanian lineage and M. galloprovincialis sampled from its native range (Figure 2a). Within these two populations, all individuals displayed mixed ancestry with high genomic contributions (i.e. 33%–82% ancestry proportions; Figure 2b) from northern M. galloprovincialis, pointing to at least two contemporary introductions of divergent northern genotypes into mainland Australia (i.e. Westfall & Gardner, 2010). The second important finding is that both introductions have led to repeated instances of hybridization and introgression with native M. planulatus. Tests of migration in TreeMix revealed strong evidence for introgression from eastern Mediterranean M. galloprovincialis into Sydney Harbour and from Atlantic M. galloprovincialis into Batemans Bay (Figure 3a). Aside from the Tasmanian population, a striking observation is that we did not identify any nonintrogressed M. planulatus or pure northern M. galloprovincialis individuals among Australian samples. This result suggests that admixture may be widespread and that both introductions are accompanied by introgression, most likely from the native into the introduced genetic backgrounds (Currat, Ruedi, Petit, & Excoffier, 2008).
Multiple introductions are a common feature of biological invasions for many non‐native marine species (Riquet, Daguin Thiébaut, Ballenghien, Bierne, & Viard, 2013; Rius et al., 2015; Viard et al., 2016). Successive introductions of large numbers of larvae are likely to promote secondary contact and subsequent admixture between genetically differentiated lineages (Keller & Taylor, 2010; Rius & Darling, 2014). For example, differentiated lineages of the invasive European green crab, Carcinus maenas, have been independently introduced into eastern Northern America (Darling, Bagley, Roman, Tepolt, & Geller, 2008; Tepolt & Palumbi, 2015), and postintroduction admixture between warm‐adapted and cold‐adapted lineages has been proposed as a factor in the establishment of invasive genotypes beyond their previous range limits (Darling, Tsai, Blakeslee, & Roman, 2014; Jeffery et al., 2018; Tepolt & Somero, 2014). Our findings are in line with general perceptions that successful marine introductions are likely to involve propagules from multiple and potentially diverse sources (Lockwood, Cassey, & Blackburn, 2005; Rius et al., 2015). It is tempting to speculate that variation in thermal physiology has contributed to successful introductions of Atlantic M. galloprovincialis in more southern (cooler) habitats relative to Sydney Harbour, where Mediterranean M. galloprovincialis predominate. However, there is no evidence to date suggesting differences in temperature tolerance among M. edulis‐introgressed Atlantic mussels and those of purely Mediterranean origins. Similarly, whether post‐introduction admixture between divergent M. galloprovincialis lineages has enhanced the success of introduced populations will require additional investigations examining these synergies (Rius et al., 2015).
While genetic studies in marine systems have not explicitly investigated whether introgression directly promotes successful introductions, it is evident that hybridization can influence both invasive and native species in a number of ways (Le Roux & Wieczorek, 2009; Sloop, Ayres, & Strong, 2009). Hybridization may alleviate Allee effects on introduced populations through purely demographic processes (Gagnaire et al., 2018; Johannesson, 1988; Mesgaran et al., 2016). Differential introgression of introduced diversity into native genomic backgrounds may also promote the spread on non‐native alleles and increase opportunities for adaptation in introduced environments (Fitzpatrick et al., 2010). In Australia, we find signatures of divergent M. edulis genetic elements in one admixed population (i.e. Batemans Bay) likely obtained through post‐introduction admixture with introgressed Atlantic M. galloprovincialis (rather than direct gene flow with M. edulis). These findings are consistent with previous investigations implicating asymmetric introgression of M. edulis genes into Atlantic M. galloprovincialis populations in south‐western Europe (Bierne, Borsa, et al., 2003; Fraïsse et al., 2014; Gosset & Bierne, 2013; Rawson & Hilbish, 1998; Roux et al., 2014). Additionally, a recent study (Simon et al., 2019) has shown that Mediterranean‐lineage M. galloprovincialis introduced into five Atlantic shipping ports display mixed M. edulis ancestry and genetic separation from local genomic backgrounds, suggesting that admixture with M. edulis has occurred prior to regional introductions. While it is not evident whether introgressed M. edulis genetic elements are adaptive for introduced populations, similar patterns of introgression in Atlantic ports (Simon et al., 2019) and Australian introduced populations (this study) raise important considerations regarding the possibility of differential introgression of divergent outgroup variants into native genetic diversity, where continued dispersal of admixed genotypes may benefit colonizers (Keller & Taylor, 2010; Rius & Darling, 2014). Given the small numbers of sampled individuals in the present study, however, and the potential for high variance in allele frequencies, we could not test specific hypotheses regarding levels of gene flow at specific loci to explore these conjectures. Nevertheless, our findings reinforce the notion that sufficient sampling of closely related sister lineages in both sympatric (i.e. endemic M. planulatus) and allopatric ranges (i.e. M. edulis outgroup) is imperative for accurate interpretations of intraspecific genomic differentiation and signatures of introgression associated with marine introductions.
4.2. Late‐Pleistocene divergence between native and introduced Australian mussels
Documenting the spread of M. galloprovincialis in Australia has been the subject of a number of previous genetic investigations; however, distinguishing between introduced and native taxa has been hampered by low genetic differentiation between populations and high levels of marker discordance (Westfall & Gardner, 2010). Representing a much larger proportion of genetic variation than previous approaches, we confirm strong species tree discordance between mitochondrial and genome‐wide nuclear loci (Gérard et al., 2008; Hilbish et al., 2000). Network analysis of 12 protein‐coding mitochondrial genes from Australian populations placed southern lineage mitochondrial haplotypes in a clade divergent from northern M. galloprovincialis (Figure 4). Additionally, haplotypes across all genes remained paraphyletic for M. galloprovincialis and M. edulis sister taxa despite 2.5 million years of divergence (Roux et al., 2014), consistent with historical isolation between southern and northern hemisphere lineages that predates the split between M. edulis and M. galloprovincialis (Gérard et al., 2008). In contrast, variation across the nuclear genomic background of Australian mussels alongside northern hemisphere taxa validated strong genetic affinities to M. galloprovincialis, suggesting a closer genetic relationship to the invasive taxon than implicated by mitochondrial loci (e.g. Fraïsse, Haguenauer, et al., 2018; Hilbish et al., 2000).
Discordant species relationships between mitochondrial and nuclear genomes are expected to arise due to the fourfold difference in effective population size between genomes, with stronger genetic drift and faster lineage sorting in mitochondrial genes (Toews & Brelsford, 2012). For example, strong differentiation in the mitochondrial genome of M. planulatus may be explained by historical bottlenecks associated with the first colonizations of Australia by northern Mytilus taxa followed by a period of allopatry (Gérard et al., 2008). Introgression through past or contemporary gene flow may erode differentiation at nuclear loci, further pronouncing mito‐nuclear discordance between northern and southern hemisphere lineages. TreeMix and ABC inferences, however, recovered weak support for contemporary gene flow from northern M. galloprovincialis into Tasmanian mussels (Figure 3), suggesting that samples in this region are largely representative of the endemic Australian taxon. We also found little evidence that Tasmanian mussels are introgressed with M. edulis through past admixture as suggested by previous authors (e.g. Borsa et al., 2007). Instead, high variance in gene tree topologies in TWISST indicated extensive levels of incomplete lineage sorting with northern M. edulis and M. trossulus, which may account for the presence of outgroup alleles in some populations (Figure S4; Westfall & Gardner, 2013). Furthermore, paraphyly among M. planulatus and M. galloprovincialis haplotypes in locus‐specific topologies (analysed in TWISST) suggests ongoing incomplete lineage sorting (i.e. shared ancestral polymorphism) between native and introduced taxa.
Focusing on Tasmanian mussels as representative of endemic M. planulatus, demographic inferences provided the strongest support for a model of divergence with low levels of historical gene flow with northern M. galloprovincialis. Parameter estimations indicated recent divergence times between 100,000–600,000 years ago, suggesting that M. planulatus likely differentiated from proto‐M. galloprovincialis postdating the separation of northern hemisphere species. While divergence time estimates assume a generation time of 2 years, these approximate values are compatible with midden fossils placing endemic Mytilus in Australia since the end of the late glacial retreat (>10,000 years bp). Estimates are also consistent with previous studies proposing separation times between northern and southern hemisphere Mytilus species ~0.5 to 1.3 million years ago based on mitochondrial loci (Gérard et al., 2008; Hilbish et al., 2000). More broadly, our findings support a scenario of parallel transequatorial migrations leading to the origins of southern hemisphere mussels, that is M. planulatus in Australasia which is more related to M. galloprovincialis (this study) and M. platensis in South America and the Kerguelen Islands which is more related to M. edulis in the northern hemisphere (Fraïsse, Haguenauer, et al., 2018). Under this scenario, however, mitochondrial introgression swamping following secondary contact between M. galloprovincialis and M. edulis (Fraïsse, Haguenauer, et al., 2018; Gérard et al., 2008) has been proposed to reconcile incongruent patterns of deep divergence at mitochondrial loci and reciprocal monophyly (with northern haplotypes) for all southern hemisphere lineages (Gérard et al., 2008).
Based on the genomic data presented here, we infer that shared polymorphisms and modest signals of genome‐wide differentiation between M. planulatus (Tasmania) and M. galloprovincialis are mostly due to recent divergence histories possibly associated with historical gene flow (likely facilitated by glacial melting and the formation of cool‐water refugia across the equator; Lindberg, 1991). However, we found no evidence for contemporary introgression from human‐mediated introductions of M. galloprovincialis in this region, suggesting that Tasmanian mussels represent naturally occurring endemic genetic diversity. Mytilus planulatus is currently recognized as valid nomenclature in the World Register of Marine Species (WORMS) database. Strong sequence similarities and permeable barriers to gene flow with M. galloprovincialis, however, support previous proposals to assign the endemic Australian lineage with a regional subspecies status (e.g. Borsa et al., 2007; Daguin & Borsa, 2000; Gérard et al., 2008; Hilbish et al., 2000; Westfall & Gardner, 2010). Further clarification will be required on whether M. planulatus has experienced historical introgression with other southern clades in New Zealand and South America (e.g. Larraín et al., 2018). Genetic investigations to date have used either insufficient loci to resolve species relationships and migration (e.g. Westfall & Gardner, 2010) or have sampled only a single landmass (e.g. Gardner et al., 2016). A recent genetic survey has confirmed genomic differentiation between southern clades (Gérard et al., 2008), including the Kergulen islands; however, introgression with M. planulatus was not investigated (Fraïsse, Haguenauer, et al., 2018). The relationship between two southern Mytilus taxa (i.e. M. planulatus and M. platensis) will therefore require further investigation.
4.3. Monitoring M. galloprovincialis introductions in Australia and implications for invasive species research
Mytilus mussels are well‐known ecosystem engineers, and changes in their abundance and range could impose significant alterations to intertidal communities (Braby & Somero, 2006). Ecological impacts of M. galloprovincialis in other parts of the world, including niche displacement of native taxa (California, Rawson et al., 1999; South Africa, Bownes & McQuaid, 2006) and negative effects on aquaculture populations through parasite hitchhiking (Dias et al., 2014; Jones & Creeper, 2006), warrant caution against M. galloprovincialis introductions into Australian coastlines and industries. For the case of Australian Mytilus, however, it is evident that many markers are required for identification of introduced and endemic populations. Our results confirm that diagnostic methods used to identify hemisphere origins based on mitochondrial loci (e.g. Ab Rahim et al., 2016; Colgan & Middelfart, 2011; Dias et al., 2014) are not a reliable method for differentiating native and introduced Mytilus populations, especially when hybridization is pervasive. We also demonstrate that multiple introduced sources are only detected when significant structure exists in the native range. It is therefore likely that for many marine species, even genomic data will not be sufficiently robust to resolve exact sources of populations in the absence of strong differentiation, despite adequate sampling of native range variation (e.g. eastern versus western Mediterranean M. galloprovincialis). In such cases, surveys for high gene flow marine invasive species using environmental DNA (Kelly, Port, Yamahara, & Crowder, 2014) may not be able to detect intraspecific non‐native diversity or even interspecific variation, despite holding promise for identifying broader taxonomic groups (Bourne, Hudson, Holman, & Rius, 2018).
Knowledge regarding the potential ecological consequences associated with introduced populations will therefore be essential for monitoring and minimizing the spread of introduced lineages (Braby & Somero, 2006). Considering Mytilus as a case study, Saarman and Pogson (2015) recently documented low levels of asymmetric introgression from native M. trossulus into invasive M. galloprovincialis in its introduced range in California, where patterns of introgression were consistent with predictions that populations furthest from the source of introduction should experience stronger gene flow into the invading genomic background (Currat et al., 2008). These findings suggest that the direction of introgression and the potential for introgression swamping towards endemic taxa are likely driven by the relative sizes of native and non‐native populations, rather than by selection processes. Documented cases of introgression between invasive and native taxa, including newly described parallel hybrid zones between M. planulatus and M. galloprovincialis, provide opportunities to evaluate how introgression with native congeners may accelerate or impede the spread of introduced species. We hypothesize that an absence of pure parental individuals in the present study (with the exception of Tasmania) indicates significant contributions of introgression to the genomic composition of Australian mussels, likely during the early stages of the introductions. Future research in Mytilus and other marine invasive species should focus on temporal and spatial sampling of greater numbers of individuals to assess the rate and direction of introgression and the potential impacts of introgression swamping of native genomes on ecologically relevant timescales (Glotzbecker et al., 2016; Riley et al., 2003; Todesco et al., 2016). Such data will be critical for informing our general understanding of the role of hybrid zone expansions in marine invasive spread and the scope and potential of long‐term (evolutionary) impacts of biological introductions on receiving marine communities.
CONFLICT OF INTEREST
None declared.
Supporting information
ACKNOWLEDGEMENTS
We would like to thank all of the colleagues who helped with Australian and northern hemisphere mussel collections, in particular, C. da Silva, J. Thia, G. Rouse, M. Hart, L. Malard and B. Popovic. Thank you to F. Viard and members of the Riginos Laboratory for useful suggestions and comments on the manuscript, and to members of the Palumbi Laboratory and Hacky Hour UQ for bioinformatics help. The QRIScloud computing cluster at the University of Queensland provided computational infrastructure for this project. This work is supported by the Australian Biological Resources Study (ABRS) National Taxonomy Research Grant (grant number RF216‐11), and awards from the National Sciences and Engineering Research Council of Canada (to I.P), the Society for the Study of Evolution (to I.P) and the University of Queensland (to I.P).
Popovic I, Matias AMA, Bierne N, Riginos C. Twin introductions by independent invader mussel lineages are both associated with recent admixture with a native congener in Australia. Evol Appl. 2020;13:515–532. 10.1111/eva.12857
DATA AVAILABILITY STATEMENT
The raw RNA‐seq data are deposited to the NCBI sequence read archive (BioProject ID: PRJNA560413). Genomic data sets that support the findings of this study are openly available in Genomic Observatories MetaDatabase (https://geome-db.org) and the Dryad digital repository at ://doi.org/10.5061/dryad.540cc05 (Popovic, Matias, Bierne, & Riginos, 2019a, 2019b, 2019c).
REFERENCES
- Ab Rahim, E. S. , Nguyen, T. T. , Ingram, B. , Riginos, C. , Weston, K. J. , & Sherman, C. D. (2016). Species composition and hybridisation of mussel species (Bivalvia: Mytilidae) in Australia. Marine and Freshwater Research, 67(12), 1955–1963. 10.1071/MF15307 [DOI] [Google Scholar]
- Alexander, D. H. , Novembre, J. , & Lange, K. (2009). Fast model‐based estimation of ancestry in unrelated individuals. Genome Research, 19, 1655–1664. 10.1101/gr.094052.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcella, T. E. , Perry, W. L. , Lodge, D. M. , & Feder, J. L. (2014). The role of hybridization in a species invasion and extirpation of resident fauna: Hybrid vigor and breakdown in the rusty crayfish, Orconectes rusticus . Journal of Crustacean Biology, 34(2), 157–164. 10.1163/1937240X-00002204 [DOI] [Google Scholar]
- Barker, B. S. , Andonian, K. , Swope, S. M. , Luster, D. G. , & Dlugosch, K. M. (2017). Population genomic analyses reveal a history of range expansion and trait evolution across the native and invaded range of yellow starthistle (Centaurea solstitialis). Molecular Ecology, 26(4), 1131–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont, M. A. (2010). Approximate Bayesian computation in evolution and ecology. Annual Review of Ecology, Evolution, and Systematics, 41, 379–406. 10.1146/annurev-ecolsys-102209-144621 [DOI] [Google Scholar]
- Beaumont, M. A. , Zhang, W. , & Balding, D. J. (2002). Approximate Bayesian computation in population genetics. Genetics, 162(4), 2025–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertorelle, G. , Benazzo, A. , & Mona, S. (2010). ABC as a flexible framework to estimate demography over space and time: Some cons, many pros. Molecular Ecology, 19(13), 2609–2625. 10.1111/j.1365-294X.2010.04690.x [DOI] [PubMed] [Google Scholar]
- Bierne, N. , Bonhomme, F. , Boudry, P. , Szulkin, M. , & David, P. (2006). Fitness landscapes support the dominance theory of post‐zygotic isolation in the mussels Mytilus edulis and M. galloprovincialis . Proceedings of the Royal Society of London B: Biological Sciences, 273(1591), 1253–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierne, N. , Bonhomme, F. , & David, P. (2003). Habitat preference and the marine‐speciation paradox. Proceedings of the Royal Society of London B: Biological Sciences, 270(1522), 1399–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierne, N. , Borsa, P. , Daguin, C. , Jollivet, D. , Viard, F. , Bonhomme, F. , & David, P. (2003). Introgression patterns in the mosaic hybrid zone between Mytilus edulis and M. galloprovincialis . Molecular Ecology, 12(2), 447–461. 10.1046/j.1365-294X.2003.01730.x [DOI] [PubMed] [Google Scholar]
- Bierne, N. , Welch, J. , Loire, E. , Bonhomme, F. , & David, P. (2011). The coupling hypothesis: Why genome scans may fail to map local adaptation genes. Molecular Ecology, 20(10), 2044–2072. 10.1111/j.1365-294X.2011.05080.x [DOI] [PubMed] [Google Scholar]
- Blum, M. J. , Walters, D. M. , Burkhead, N. M. , Freeman, B. J. , & Porter, B. A. (2010). Reproductive isolation and the expansion of an invasive hybrid swarm. Biological Invasions, 12(8), 2825–2836. 10.1007/s10530-010-9688-9 [DOI] [Google Scholar]
- Borsa, P. , Daguin, C. , & Bierne, N. (2007). Genomic reticulation indicates mixed ancestry in Southern‐Hemisphere Mytilus spp. mussels. Biological Journal of the Linnean Society, 92(4), 747–754. [Google Scholar]
- Bouchemousse, S. , Liautard Haag, C. , Bierne, N. , & Viard, F. (2016). Distinguishing contemporary hybridization from past introgression with postgenomic ancestry‐informative SNP s in strongly differentiated Ciona species. Molecular Ecology, 25(21), 5527–5542. [DOI] [PubMed] [Google Scholar]
- Bourne, S. D. , Hudson, J. , Holman, L. E. , & Rius, M. (2018). Marine invasion genomics: Revealing ecological and evolutionary consequences of biological invasions In Oleksiak M. F., & Rajora O. P. (Eds.), Population Genetics: Marine Organisms, Population Genomics (pp. 1–36). Cham: Springer. [Google Scholar]
- Bownes, S. J. , & McQuaid, C. D. (2006). Will the invasive mussel Mytilus galloprovincialis Lamarck replace the indigenous Perna perna L. on the south coast of South Africa? Journal of Experimental Marine Biology and Ecology, 338(1), 140–151. 10.1016/j.jembe.2006.07.006 [DOI] [Google Scholar]
- Braby, C. E. , & Somero, G. N. (2006). Ecological gradients and relative abundance of native (Mytilus trossulus) and invasive (Mytilus galloprovincialis) blue mussels in the California hybrid zone. Marine Biology, 148(6), 1249–1262. 10.1007/s00227-005-0177-0 [DOI] [Google Scholar]
- Branch, G. M. , & Steffani, C. N. (2004). Can we predict the effects of alien species? A case‐history of the invasion of South Africa by Mytilus galloprovincialis (Lamarck). Journal of Experimental Marine Biology and Ecology, 300(1–2), 189–215. 10.1016/j.jembe.2003.12.007 [DOI] [Google Scholar]
- Brannock, P. M. , Wethey, D. S. , & Hilbish, T. J. (2009). Extensive hybridization with minimal introgression in Mytilus galloprovincialis and M. trossulus in Hokkaido. Japan. Marine Ecology Progress Series, 383, 161–171. 10.3354/meps07995 [DOI] [Google Scholar]
- Browning, S. R. , & Browning, B. L. (2007). Rapid and accurate haplotype phasing and missing‐data inference for whole‐genome association studies by use of localized haplotype clustering. The American Journal of Human Genetics, 81(5), 1084–1097. 10.1086/521987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butlin, R. K. , Saura, M. , Charrier, G. , Jackson, B. , André, C. , Caballero, A. , … Rolán‐Alvarez, E. (2014). Parallel evolution of local adaptation and reproductive isolation in the face of gene flow. Evolution, 68(4), 935–949. 10.1111/evo.12329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, D. (2006). Balancing selection and its effects on sequences in nearby genome regions. PLoS Genetics, 2(4), e64 10.1371/journal.pgen.0020064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colautti, R. I. , & Lau, J. A. (2015). Contemporary evolution during invasion: Evidence for differentiation, natural selection, and local adaptation. Molecular Ecology, 24(9), 1999–2017. 10.1111/mec.13162 [DOI] [PubMed] [Google Scholar]
- Colgan, D. J. , & Middelfart, P. (2011). Mytilus mitochondrial DNA haplotypes in southeastern Australia. Aquatic Biology, 12(1), 47–53. 10.3354/ab00323 [DOI] [Google Scholar]
- Colhoun, E. A. , Turner, E. , & Van de Geer, G. (1982). Late Pleistocene marine molluscan faunas from four sites in Tasmania. In: Papers and Proceedings of the Royal Society of Tasmania. (Vol. 116, pp. 91–97). [Google Scholar]
- Csilléry, K. , François, O. , & Blum, M. G. (2012). abc: An R package for approximate Bayesian computation (ABC). Methods in Ecology and Evolution, 3(3), 475–479. 10.1111/j.2041-210X.2011.00179.x [DOI] [PubMed] [Google Scholar]
- Currat, M. , Ruedi, M. , Petit, R. J. , & Excoffier, L. (2008). The hidden side of invasions: Massive introgression by local genes. Evolution, 62(8), 1908–1920. 10.1111/j.1558-5646.2008.00413.x [DOI] [PubMed] [Google Scholar]
- Daguin, C. , & Borsa, P. (2000). Genetic relationships of Mytilus galloprovincialis Lamarck populations worldwide: Evidence from nuclear‐DNA markers. Geological Society, London, Special Publications, 177(1), 389–397. [Google Scholar]
- Danecek, P. , Auton, A. , Abecasis, G. , Albers, C. A. , Banks, E. , DePristo, M. A. , … Durbin, R. (2011). The variant call format and VCFtools. Bioinformatics, 27(15), 2156–2158. 10.1093/bioinformatics/btr330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling, J. A. , Bagley, M. J. , Roman, J. , Tepolt, C. K. , & Geller, J. B. (2008). Genetic patterns across multiple introductions of the globally invasive crab genus Carcinus . Molecular Ecology, 17(23), 4992–5007. [DOI] [PubMed] [Google Scholar]
- Darling, J. A. , Tsai, Y. H. E. , Blakeslee, A. M. , & Roman, J. (2014). Are genes faster than crabs? Mitochondrial introgression exceeds larval dispersal during population expansion of the invasive crab Carcinus maenas . Royal Society Open Science, 1(2), 140202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Queiroz, K. (2007). Species concepts and species delimitation. Systematic Biology, 56(6), 879–886. 10.1080/10635150701701083 [DOI] [PubMed] [Google Scholar]
- Dias, P. J. , Fotedar, S. , & Snow, M. (2014). Characterisation of mussel (Mytilus sp.) populations in Western Australia and evaluation of potential genetic impacts of mussel spat translocation from interstate. Marine and Freshwater Research, 65(6), 486–496. [Google Scholar]
- Donner, J. , & Jungner, H. (1981). Radiocarbon dating of marine shells from southeastern Australia as a means of dating relative sea‐level changes. Annales Academiae Scientiarum Fennicae. Series A. 3, Geologica‐‐Geographica, 131, 1–44. [Google Scholar]
- El Ayari, T. , El Menif, N. T. , Hamer, B. , Cahill, A. E. , & Bierne, N. (2019). The hidden side of a major marine biogeographic boundary: A wide mosaic hybrid zone at the Atlantic‐Mediterranean divide reveals the complex interaction between natural and genetic barriers in mussels. Heredity, 122, 770–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellstrand, N. C. , & Schierenbeck, K. A. (2000). Hybridization as a stimulus for the evolution of invasiveness in plants? Proceedings of the National Academy of Sciences, 97(13), 7043–7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estoup, A. , Beaumont, M. , Sennedot, F. , Moritz, C. , & Cornuet, J. M. (2004). Genetic analysis of complex demographic scenarios: spatially expanding populations of the cane toad, bufo marinus. Evolution, 58(9), 2021–2036. 10.1111/j.0014-3820.2004.tb00487.x [DOI] [PubMed] [Google Scholar]
- Estoup, A. , & Guillemaud, T. (2010). Reconstructing routes of invasion using genetic data: Why, how and so what? Molecular Ecology, 19(19), 4113–4130. 10.1111/j.1365-294X.2010.04773.x [DOI] [PubMed] [Google Scholar]
- Estoup, A. , Wilson, I. J. , Sullivan, C. , Cornuet, J.‐M. , & Moritz, C. (2001). Inferring population history from microsatellite and enzyme data in serially introduced cane toads, Bufo Marinus. Genetics, 159(4), 1671–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagundes, N. J. , Ray, N. , Beaumont, M. , Neuenschwander, S. , Salzano, F. M. , Bonatto, S. L. , & Excoffier, L. (2007). Statistical evaluation of alternative models of human evolution. Proceedings of the National Academy of Sciences, 104(45), 17614–17619. 10.1073/pnas.0708280104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein, J. , & Churchill, G. A. (1996). A Hidden Markov Model approach to variation among sites in rate of evolution. Molecular Biology and Evolution, 13(1), 93–104. 10.1093/oxfordjournals.molbev.a025575 [DOI] [PubMed] [Google Scholar]
- Fitzpatrick, B. M. , Johnson, J. R. , Kump, D. K. , Smith, J. J. , Voss, S. R. , & Shaffer, H. B. (2010). Rapid spread of invasive genes into a threatened native species. Proceedings of the National Academy of Sciences, 107, 3606–3610. 10.1073/pnas.0911802107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraïsse, C. , Belkhir, K. , Welch, J. J. , & Bierne, N. (2016). Local interspecies introgression is the main cause of extreme levels of intraspecific differentiation in mussels. Molecular Ecology, 25(1), 269–286. 10.1111/mec.13299 [DOI] [PubMed] [Google Scholar]
- Fraïsse, C. , Haguenauer, A. , Gerard, K. , Weber, A.‐A.‐T. , Bierne, N. , & Chenuil, A. (2018). Fine‐grained habitat‐associated Genetic Connectivity in an Admixed Population of Mussels in the Small Isolated Kerguelen Islands. Biorxiv, 239244. 10.1101/239244 [DOI] [Google Scholar]
- Fraïsse, C. , Roux, C. , Gagnaire, P.‐A. , Romiguier, J. , Faivre, N. , Welch, J. J. , & Bierne, N. (2018). The divergence history of European blue mussel species reconstructed from Approximate Bayesian Computation: The effects of sequencing techniques and sampling strategies. Peer J, 6, e5198 10.7717/peerj.5198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraïsse, C. , Roux, C. , Welch, J. J. , & Bierne, N. (2014). Gene flow in a mosaic hybrid zone: Is local introgression adaptive? Genetics, 197, 939–951. 10.1534/genetics.114.161380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y.‐X. , & Li, W.‐H. (1997). Estimating the age of the common ancestor of a sample of DNA sequences. Molecular Biology and Evolution, 14(2), 195–199. 10.1093/oxfordjournals.molbev.a025753 [DOI] [PubMed] [Google Scholar]
- Gagnaire, P.‐A. , Broquet, T. , Aurelle, D. , Viard, F. , Souissi, A. , Bonhomme, F. , … Bierne, N. (2015). Using neutral, selected, and hitchhiker loci to assess connectivity of marine populations in the genomic era. Evolutionary Applications, 8(8), 769–786. 10.1111/eva.12288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnaire, P.‐A. , Lamy, J.‐B. , Cornette, F. , Heurtebise, S. , Dégremont, L. , Flahauw, E. , … Lapègue, S. (2018). Analysis of genome‐wide differentiation between native and introduced populations of the cupped oysters Crassostrea gigas and Crassostrea angulata . Genome Biology and Evolution, 10(9), 2518–2534. 10.1093/gbe/evy194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnaire, P.‐A. , Normandeau, E. , & Bernatchez, L. (2012). Comparative genomics reveals adaptive protein evolution and a possible cytonuclear incompatibility between European and American eels. Molecular Biology and Evolution, 29(10), 2909–2919. 10.1093/molbev/mss076 [DOI] [PubMed] [Google Scholar]
- Galtier, N. (2019). Delineating species in the speciation continuum: A proposal. Evolutionary Applications, 12(4), 657–663. 10.1111/eva.12748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner, J. P. , Zbawicka, M. , Westfall, K. M. , & Wenne, R. (2016). Invasive blue mussels threaten regional scale genetic diversity in mainland and remote offshore locations: The need for baseline data and enhanced protection in the Southern Ocean. Global Change Biology, 22(9), 3182–3195. 10.1111/gcb.13332 [DOI] [PubMed] [Google Scholar]
- Geller, J. B. , Darling, J. A. , & Carlton, J. T. (2010). Genetic perspectives on marine biological invasions. Annual Review of Marine Science, 2, 367–393. 10.1146/annurev.marine.010908.163745 [DOI] [PubMed] [Google Scholar]
- Gérard, K. , Bierne, N. , Borsa, P. , Chenuil, A. , & Féral, J.‐P. (2008). Pleistocene separation of mitochondrial lineages of Mytilus spp. mussels from Northern and Southern Hemispheres and strong genetic differentiation among southern populations. Molecular Phylogenetics and Evolution, 49(1), 84–91. [DOI] [PubMed] [Google Scholar]
- Glotzbecker, G. J. , Walters, D. M. , & Blum, M. J. (2016). Rapid movement and instability of an invasive hybrid swarm. Evolutionary Applications, 9(6), 741–755. 10.1111/eva.12371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosset, C. C. , & Bierne, N. (2013). Differential introgression from a sister species explains high FST outlier loci within a mussel species. Journal of Evolutionary Biology, 26(1), 14–26. [DOI] [PubMed] [Google Scholar]
- Griffiths, C. L. , Hockey, P. , Van Erkom Schurink, C. , & Le Roux, P. J. (1992). Marine invasive aliens on South African shores: Implications for community structure and tropillc functioning. South African Journal of Marine Science, 12(1), 713–722. 10.2989/02577619209504736 [DOI] [Google Scholar]
- Guerrero, R. F. , & Hahn, M. W. (2017). Speciation as a sieve for ancestral polymorphism. Molecular Ecology, 26(20), 5362–5368. 10.1111/mec.14290 [DOI] [PubMed] [Google Scholar]
- Guillemaud, T. , Beaumont, M. A. , Ciosi, M. , Cornuet, J.‐M. , & Estoup, A. (2010). Inferring introduction routes of invasive species using approximate Bayesian computation on microsatellite data. Heredity, 104(1), 88 10.1038/hdy.2009.92 [DOI] [PubMed] [Google Scholar]
- Hey, J. , & Nielsen, R. (2004). Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis . Genetics, 167(2), 747–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey, J. , & Nielsen, R. (2007). Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proceedings of the National Academy of Sciences, 104(8), 2785–2790. 10.1073/pnas.0611164104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilbish, T. J. , Mullinax, A. , Dolven, S. I. , Meyer, A. , Koehn, R. K. , & Rawson, P. D. (2000). Origin of the antitropical distribution pattern in marine mussels (Mytilus spp.): Routes and timing of transequatorial migration. Marine Biology, 136(1), 69–77. [Google Scholar]
- Hope, J. H. , Lampert, R. J. , Edmondson, E. , Smith, M. J. , & Van Tets, G. F. (1977). Late Pleistocene faunal remains from Seton rock shelter, Kangaroo Island, South Australia. Journal of Biogeography, 4, 363–385. 10.2307/3038194 [DOI] [Google Scholar]
- Hovick, S. M. , & Whitney, K. D. (2014). Hybridisation is associated with increased fecundity and size in invasive taxa: Meta‐analytic support for the hybridisation‐invasion hypothesis. Ecology Letters, 17(11), 1464–1477. 10.1111/ele.12355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson, D. H. , & Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Molecular Biology and Evolution, 23, 254–267. 10.1093/molbev/msj030 [DOI] [PubMed] [Google Scholar]
- Jeffery, N. W. , Bradbury, I. R. , Stanley, R. R. , Wringe, B. F. , Van Wyngaarden, M. , Lowen, J. B. , et al. (2018). Genome‐wide evidence of environmentally mediated secondary contact of European green crab (Carcinus maenas) lineages in eastern North America. Evolutionary Applications, 11, 869–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannesson, K. (1988). The paradox of Rockall: Why is a brooding gastropod (Littorina saxatilis) more widespread than one having a planktonic larval dispersal stage (L. littorea)? Marine Biology, 99(4), 507–513. 10.1007/BF00392558 [DOI] [Google Scholar]
- Jones, J. B. , & Creeper, J. (2006). Diseases of pearl oysters and other molluscs: A Western Australian perspective. Journal of Shellfish Research, 25(1), 233–238. 10.2983/0730-8000(2006)25[233:DOPOAO]2.0.CO;2 [DOI] [Google Scholar]
- Keightley, P. D. , & Eyre‐Walker, A. (2012). Estimating the rate of adaptive molecular evolution when the evolutionary divergence between species is small. Journal of Molecular Evolution, 74(1–2), 61–68. 10.1007/s00239-012-9488-1 [DOI] [PubMed] [Google Scholar]
- Keinan, A. , Mullikin, J. C. , Patterson, N. , & Reich, D. (2007). Measurement of the human allele frequency spectrum demonstrates greater genetic drift in East Asians than in Europeans. Nature Genetics, 39(10), 1251 10.1038/ng2116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller, S. R. , & Taylor, D. R. (2010). Genomic admixture increases fitness during a biological invasion. Journal of Evolutionary Biology, 23(8), 1720–1731. 10.1111/j.1420-9101.2010.02037.x [DOI] [PubMed] [Google Scholar]
- Kelly, R. P. , Port, J. A. , Yamahara, K. M. , & Crowder, L. B. (2014). Using environmental DNA to census marine fishes in a large mesocosm. PLoS ONE, 9(1), e86175 10.1371/journal.pone.0086175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovach, R. P. , Hand, B. K. , Hohenlohe, P. A. , Cosart, T. F. , Boyer, M. C. , Neville, H. H. , et al. (2016). Vive la résistance: Genome‐wide selection against introduced alleles in invasive hybrid zones. Proceedings of the Royal Society B: Biological Sciences, 283(1843), 20161380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhner, M. K. (2009). Coalescent genealogy samplers: Windows into population history. Trends in Ecology & Evolution, 24(2), 86–93. 10.1016/j.tree.2008.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. , & Salzberg, S. L. (2012). Fast gapped‐read alignment with Bowtie 2. Nature Methods, 9(4), 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larraín, M. A. , Zbawicka, M. , Araneda, C. , Gardner, J. P. , & Wenne, R. (2018). Native and invasive taxa on the Pacific coast of South America: Impacts on aquaculture, traceability and biodiversity of blue mussels (Mytilus spp.). Evolutionary Applications, 11(3), 298–311. [Google Scholar]
- Le Roux, J. , & Wieczorek, A. M. (2009). Molecular systematics and population genetics of biological invasions: Towards a better understanding of invasive species management. Annals of Applied Biology, 154(1), 1–17. 10.1111/j.1744-7348.2008.00280.x [DOI] [Google Scholar]
- Lindberg, D. R. (1991). Marine biotic interchange between the northern and southern hemispheres. Paleobiology, 17(3), 308–324. 10.1017/S0094837300010629 [DOI] [Google Scholar]
- Lockwood, J. L. , Cassey, P. , & Blackburn, T. (2005). The role of propagule pressure in explaining species invasions. Trends in Ecology & Evolution, 20(5), 223–228. 10.1016/j.tree.2005.02.004 [DOI] [PubMed] [Google Scholar]
- Lombaert, E. , Guillemaud, T. , Thomas, C. E. , Lawson handley, L. J. , Li, J. , Wang, S. , … Estoup, A. (2011). Inferring the origin of populations introduced from a genetically structured native range by approximate Bayesian computation: Case study of the invasive ladybird Harmonia axyridis . Molecular Ecology, 20(22), 4654–4670. 10.1111/j.1365-294X.2011.05322.x [DOI] [PubMed] [Google Scholar]
- Lowe, S. , Browne, M. , Boudjelas, S. , & De Poorter, M. (2000). 100 of the world's worst invasive alien species: a selection from the global invasive species database (Vol. 12). Invasive Species Specialist Group Auckland. [Google Scholar]
- Marko, P. B. , & Hart, M. W. (2011). The complex analytical landscape of gene flow inference. Trends in Ecology & Evolution, 26(9), 448–456. 10.1016/j.tree.2011.05.007 [DOI] [PubMed] [Google Scholar]
- Marko, P. B. , & Hart, M. W. (2012). Retrospective coalescent methods and the reconstruction of metapopulation histories in the sea. Evolutionary Ecology, 26(2), 291–315. 10.1007/s10682-011-9467-9 [DOI] [Google Scholar]
- Martin, S. H. , & Van Belleghem, S. M. (2017). Exploring evolutionary relationships across the genome using topology weighting. Genetics, 206, 429–438. 10.1534/genetics.116.194720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald, J. H. , & Koehn, R. K. (1988). The mussels Mytilus galloprovincialis and M. trossulus on the Pacific coast of North America. Marine Biology, 99(1), 111–118. [Google Scholar]
- McDonald, J. H. , Seed, R. , & Koehn, R. K. (1991). Allozymes and morphometric characters of three species of Mytilus in the Northern and Southern Hemispheres. Marine Biology, 111(3), 323–333. 10.1007/BF01319403 [DOI] [Google Scholar]
- Mesgaran, M. B. , Lewis, M. A. , Ades, P. K. , Donohue, K. , Ohadi, S. , Li, C. , & Cousens, R. D. (2016). Hybridization can facilitate species invasions, even without enhancing local adaptation. Proceedings of the National Academy of Sciences, 113(36), 10210–10214. 10.1073/pnas.1605626113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyarzún, P. A. , Toro, J. E. , Cañete, J. I. , & Gardner, J. P. (2016). Bioinvasion threatens the genetic integrity of native diversity and a natural hybrid zone: Smooth‐shelled blue mussels (Mytilus spp.) in the Strait of Magellan. Biological Journal of the Linnean Society, 117(3), 574–585. [Google Scholar]
- Pascual, M. , Chapuis, M. P. , Mestres, F. , Balanya, J. , Huey, R. B. , Gilchrist, G. W. , et al. (2007). Introduction history of Drosophila subobscura in the New World: A microsatellite‐based survey using ABC methods. Molecular Ecology, 16(15), 3069–3083. [DOI] [PubMed] [Google Scholar]
- Patterson, N. , Moorjani, P. , Luo, Y. , Mallick, S. , Rohland, N. , Zhan, Y. , … Reich, D. (2012). Ancient admixture in human history. Genetics, 192, 1065–1093. 10.1534/genetics.112.145037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell, J. K. , & Pritchard, J. K. (2012). Inference of population splits and mixtures from genome‐wide allele frequency data. PLoS Genetics, 8(11), e1002967 10.1371/journal.pgen.1002967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovic, I. , Matias, A. M. A. , Bierne, N. , & Riginos, C. (2019a). ReferenceTranscriptome_Files Dryad, 10.5061/dryad.540cc05 [DOI]
- Popovic, I. , Matias, A. M. A. , Bierne, N. , & Riginos, C. (2019b). FilteredVCF_Files_PopGenomicAnalyses Dryad, 10.5061/dryad.540cc05 [DOI]
- Popovic, I. , Matias, A. M. A. , Bierne, N. , & Riginos, C. (2019c). Reads2snps_VCF_Files Dryad, 10.5061/dryad.540cc05 [DOI]
- Popovic, I. , Matias, A. M. A. , Bierne, N. , & Riginos, C. (2019d). BioProject ID: PRJNA560413; NCBI sequence read archive.
- Pritchard, J. K. , Seielstad, M. T. , Perez‐Lezaun, A. , & Feldman, M. W. (1999). Population growth of human Y chromosomes: A study of Y chromosome microsatellites. Molecular Biology and Evolution, 16(12), 1791–1798. 10.1093/oxfordjournals.molbev.a026091 [DOI] [PubMed] [Google Scholar]
- Purcell, S. , Neale, B. , Todd‐Brown, K. , Thomas, L. , Ferreira, M. A. R. , Bender, D. , … Sham, P. C. (2007). PLINK: A tool set for whole‐genome association and population‐based linkage analyses. The American Journal of Human Genetics, 81(3), 559–575. 10.1086/519795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesada, H. , Wenne, R. , & Skibinski, D. O. (1995). Differential introgression of mitochondrial DNA across species boundaries within the marine mussel genus Mytilus . Proceedings of the Royal Society of London. Series B: Biological Sciences, 262(1363), 51–56. [Google Scholar]
- Rawson, P. D. , Agrawal, V. , & Hilbish, T. J. (1999). Hybridization between the blue mussels Mytilus galloprovincialis and M. trossulus along the Pacific coast of North America: Evidence for limited introgression. Marine Biology, 134(1), 201–211. [Google Scholar]
- Rawson, P. D. , & Hilbish, T. J. (1998). Asymmetric introgression of mitochondrial DNA among European populations of blue mussels (Mytilus spp.). Evolution, 52(1), 100–108. [DOI] [PubMed] [Google Scholar]
- Rawson, P. D. , Joyner, K. L. , Meetze, K. , & Hilbish, T. J. (1996). Evidence for intragenic recombination within a novel genetic marker that distinguishes mussels in the Mytilus edulis species complex. Heredity, 77(6), 599 10.1038/hdy.1996.187 [DOI] [PubMed] [Google Scholar]
- Reich, D. , Thangaraj, K. , Patterson, N. , Price, A. L. , & Singh, L. (2009). Reconstructing Indian population history. Nature, 461(7263), 489 10.1038/nature08365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riginos, C. , Hickerson, M. J. , Henzler, C. M. , & Cunningham, C. W. (2004). Differential patterns of male and female mtDNA exchange across the Atlantic Ocean in the blue mussel, Mytilus edulis . Evolution, 58(11), 2438–2451. 10.1111/j.0014-3820.2004.tb00873.x [DOI] [PubMed] [Google Scholar]
- Riley, S. P. , Bradley Shaffer, H. , Randal Voss, S. , & Fitzpatrick, B. M. (2003). Hybridization between a rare, native tiger salamander (Ambystoma californiense) and its introduced congener. Ecological Applications, 13(5), 1263–1275. 10.1890/02-5023 [DOI] [Google Scholar]
- Ripley, B. , Venables, W. , & Ripley, M. B. (2016). Package “nnet”. R Package Version, 7–3. [Google Scholar]
- Riquet, F. , Daguin Thiébaut, C. , Ballenghien, M. , Bierne, N. , & Viard, F. (2013). Contrasting patterns of genome‐wide polymorphism in the native and invasive range of the marine mollusc Crepidula fornicata . Molecular Ecology, 22(4), 1003–1018. [DOI] [PubMed] [Google Scholar]
- Rius, M. , & Darling, J. A. (2014). How important is intraspecific genetic admixture to the success of colonising populations? Trends in Ecology & Evolution, 29(4), 233–242. 10.1016/j.tree.2014.02.003 [DOI] [PubMed] [Google Scholar]
- Rius, M. , Turon, X. , Bernardi, G. , Volckaert, F. A. , & Viard, F. (2015). Marine invasion genetics: From spatio‐temporal patterns to evolutionary outcomes. Biological Invasions, 17(3), 869–885. 10.1007/s10530-014-0792-0 [DOI] [Google Scholar]
- Robinson, J. D. , Bunnefeld, L. , Hearn, J. , Stone, G. N. , & Hickerson, M. J. (2014). ABC inference of multi‐population divergence with admixture from unphased population genomic data. Molecular Ecology, 23(18), 4458–4471. 10.1111/mec.12881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, T. B. , Griffiths, C. L. , Branch, G. M. , & Govender, A. (2007). The invasion and subsequent die‐off of Mytilus galloprovincialis in Langebaan Lagoon, South Africa: Effects on natural communities. Marine Biology, 152(2), 225–232. 10.1007/s00227-007-0697-x [DOI] [Google Scholar]
- Ross‐Ibarra, J. , Wright, S. I. , Foxe, J. P. , Kawabe, A. , DeRose‐Wilson, L. , Gos, G. , … Gaut, B. S. (2008). Patterns of polymorphism and demographic history in natural populations of Arabidopsis lyrata . PLoS ONE, 3(6), e2411 10.1371/journal.pone.0002411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rougemont, Q. , & Bernatchez, L. (2018). The demographic history of Atlantic Salmon (Salmo salar) across its distribution range reconstructed from Approximate Bayesian Computations. Evolution, 72(6), 1261–1277. [DOI] [PubMed] [Google Scholar]
- Roux, C. , Fraïsse, C. , Castric, V. , Vekemans, X. , Pogson, G. H. , & Bierne, N. (2014). Can we continue to neglect genomic variation in introgression rates when inferring the history of speciation? A case study in a Mytilus hybrid zone. Journal of Evolutionary Biology, 27(8), 1662–1675. [DOI] [PubMed] [Google Scholar]
- Roux, C. , Fraïsse, C. , Romiguier, J. , Anciaux, Y. , Galtier, N. , & Bierne, N. (2016). Shedding light on the grey zone of speciation along a continuum of genomic divergence. PLoS Biology, 14(12), e2000234 10.1371/journal.pbio.2000234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux, C. , Tsagkogeorga, G. , Bierne, N. , & Galtier, N. (2013). Crossing the species barrier: Genomic hotspots of introgression between two highly divergent Ciona intestinalis species. Molecular Biology and Evolution, 30(7), 1574–1587. 10.1093/molbev/mst066 [DOI] [PubMed] [Google Scholar]
- Saarman, N. P. , & Pogson, G. H. (2015). Introgression between invasive and native blue mussels (genus Mytilus) in the central California hybrid zone. Molecular Ecology, 24(18), 4723–4738. [DOI] [PubMed] [Google Scholar]
- Schierenbeck, K. A. , & Ellstrand, N. C. (2009). Hybridization and the evolution of invasiveness in plants and other organisms. Biological Invasions, 11(5), 1093 10.1007/s10530-008-9388-x [DOI] [Google Scholar]
- Shine, R. (2010). The ecological impact of invasive cane toads (Bufo marinus) in Australia. The Quarterly Review of Biology, 85(3), 253–291. [DOI] [PubMed] [Google Scholar]
- Simon, A. , Couteau, J. , Nielsen, E. E. , Sussarellu, R. , Burgeot, T. , … Skazina, M. (2019). Replicated anthropogenic hybridisations reveal parallel patterns of admixture in marine mussels. BioRxiv, p. 590737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibinski, D. , Beardmore, J. A. , & Cross, T. F. (1983). Aspects of the population genetics of Mytilus (Mytilidae; Mollusca) in the British Isles. Biological Journal of the Linnean Society, 19(2), 137–183. 10.1111/j.1095-8312.1983.tb00782.x [DOI] [Google Scholar]
- Sloop, C. M. , Ayres, D. R. , & Strong, D. R. (2009). The rapid evolution of self‐fertility in Spartina hybrids (Spartina alterniflora × foliosa) invading San Francisco Bay, CA. Biological Invasions, 11(5), 1131–1144. 10.1007/s10530-008-9385-0 [DOI] [Google Scholar]
- Sousa, V. M. C. , Carneiro, M. , Ferrand, N. , & Hey, J. (2013). Identifying loci under selection against gene flow in isolation with migration models. Genetics, 194, 211–233. 10.1534/genetics.113.149211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F. (1983). Evolutionary relationship of DNA sequences in finite populations. Genetics, 105, 437–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F. (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics, 123, 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepolt, C. K. (2015). Adaptation in marine invasion: A genetic perspective. Biological Invasions, 17(3), 887–903. 10.1007/s10530-014-0825-8 [DOI] [Google Scholar]
- Tepolt, C. K. , & Palumbi, S. R. (2015). Transcriptome sequencing reveals both neutral and adaptive genome dynamics in a marine invader. Molecular Ecology, 24(16), 4145–4158. 10.1111/mec.13294 [DOI] [PubMed] [Google Scholar]
- Tepolt, C. K. , & Somero, G. N. (2014). Master of all trades: Thermal acclimation and adaptation of cardiac function in a broadly distributed marine invasive species, the European green crab, Carcinus maenas . Journal of Experimental Biology, 217(7), 1129–1138. 10.1242/jeb.093849 [DOI] [PubMed] [Google Scholar]
- Tine, M. , Kuhl, H. , Gagnaire, P.‐A. , Louro, B. , Desmarais, E. , Martins, R. S. T. , … Reinhardt, R. (2014). European sea bass genome and its variation provide insights into adaptation to euryhalinity and speciation. Nature Communications, 5, 5770 10.1038/ncomms6770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todesco, M. , Pascual, M. A. , Owens, G. L. , Ostevik, K. L. , Moyers, B. T. , Hübner, S. , … Rieseberg, L. H. (2016). Hybridization and extinction. Evolutionary Applications, 9(7), 892–908. 10.1111/eva.12367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toews, D. P. , & Brelsford, A. (2012). The biogeography of mitochondrial and nuclear discordance in animals. Molecular Ecology, 21(16), 3907–3930. 10.1111/j.1365-294X.2012.05664.x [DOI] [PubMed] [Google Scholar]
- Viard, F. , David, P. , & Darling, J. A. (2016). Marine invasions enter the genomic era: Three lessons from the past, and the way forward. Current Zoology, 62(6), 629–642. 10.1093/cz/zow053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westfall, K. M. , & Gardner, J. P. (2010). Genetic diversity of Southern hemisphere blue mussels (Bivalvia: Mytilidae) and the identification of non‐indigenous taxa. Biological Journal of the Linnean Society, 101(4), 898–909. 10.1111/j.1095-8312.2010.01549.x [DOI] [Google Scholar]
- Westfall, K. M. , & Gardner, J. P. (2013). Interlineage Mytilus galloprovincialis Lmk. 1819 hybridization yields inconsistent genetic outcomes in the Southern hemisphere. Biological Invasions, 15(7), 1493–1506. [Google Scholar]
- Wright, S. (1951). The genetical structure of populations. Annals of Eugenics, 15(1), 323–354. 10.1111/j.1469-1809.1949.tb02451.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Popovic, I. , Matias, A. M. A. , Bierne, N. , & Riginos, C. (2019a). ReferenceTranscriptome_Files Dryad, 10.5061/dryad.540cc05 [DOI]
- Popovic, I. , Matias, A. M. A. , Bierne, N. , & Riginos, C. (2019b). FilteredVCF_Files_PopGenomicAnalyses Dryad, 10.5061/dryad.540cc05 [DOI]
- Popovic, I. , Matias, A. M. A. , Bierne, N. , & Riginos, C. (2019c). Reads2snps_VCF_Files Dryad, 10.5061/dryad.540cc05 [DOI]
- Popovic, I. , Matias, A. M. A. , Bierne, N. , & Riginos, C. (2019d). BioProject ID: PRJNA560413; NCBI sequence read archive.
Supplementary Materials
Data Availability Statement
The raw RNA‐seq data are deposited to the NCBI sequence read archive (BioProject ID: PRJNA560413). Genomic data sets that support the findings of this study are openly available in Genomic Observatories MetaDatabase (https://geome-db.org) and the Dryad digital repository at ://doi.org/10.5061/dryad.540cc05 (Popovic, Matias, Bierne, & Riginos, 2019a, 2019b, 2019c).