

Abstract
Previously published comparative functional genomic data sets from primates using frozen tissue samples, including many data sets from our own group, were often collected and analyzed using nonoptimal study designs and analysis approaches. In addition, when samples from multiple tissues were studied in a comparative framework, individuals and tissues were confounded. We designed a multitissue comparative study of gene expression and DNA methylation in primates that minimizes confounding effects by using a balanced design with respect to species, tissues, and individuals. We also developed a comparative analysis pipeline that minimizes biases attributable to sequence divergence. Thus, we present the most comprehensive catalog of similarities and differences in gene expression and DNA methylation levels between livers, kidneys, hearts, and lungs, in humans, chimpanzees, and rhesus macaques. We estimate that overall, interspecies and inter-tissue differences in gene expression levels can only modestly be accounted for by corresponding differences in promoter DNA methylation. However, the expression pattern of genes with conserved inter-tissue expression differences can be explained by corresponding interspecies methylation changes more often. Finally, we show that genes whose tissue-specific regulatory patterns are consistent with the action of natural selection are highly connected in both gene regulatory and protein–protein interaction networks.
Gene regulatory differences between humans and other primates are hypothesized to underlie human-specific traits (King and Wilson 1975). Over the last decade, dozens of comparative genomic studies focused on characterizing mRNA expression level differences between primates in a large number of tissues (e.g., Enard et al. 2002; Khaitovich et al. 2005; Blekhman et al. 2008; Brawand et al. 2011; Barbash and Sakmar 2017), typically focusing on differences between humans and other primates. A few studies have also characterized inter-primate differences in regulatory mechanisms and phenotypes other than gene expression levels, such as DNA methylation levels, chromatin modifications and accessibility, and protein expression levels (Cain et al. 2011; Pai et al. 2011; Hernando-Herraez et al. 2013, 2015a,b; Ward et al. 2013; Stergachis et al. 2014; Zhou et al. 2014; Villar et al. 2015). These studies often construct catalogs of gene expression levels and other mechanisms. These catalogs have been useful to better understand the evolutionary processes that led to adaptations in humans (Enard et al. 2002; Cáceres et al. 2003; Karaman et al. 2003; Khaitovich et al. 2004a, 2005; Gilad et al. 2005; Lemos et al. 2005; Blekhman et al. 2008, 2010; Nowick et al. 2009; Babbitt et al. 2010; Pai et al. 2011; Shibata et al. 2012; Capra et al. 2013; Khan et al. 2013) and ancestral or derived phenotypes that may be relevant to human diseases (Cooper and Shendure 2011; Romero et al. 2012).
One caveat that is shared among practically all comparative studies in primates is related to difficulty in obtaining multiple tissue samples from the same individual. To date, there have been no published comparative studies in primates that have analyzed multiple tissues sampled from the same individuals across multiple species in a balanced design (Romero et al. 2012). As a result, regulatory differences between tissues are always confounded with regulatory differences between individuals (Blekhman et al. 2008; Brawand et al. 2011; Pai et al. 2011; Chen et al. 2019). In turn, catalogs from these studies cannot be used to compare tissue-specific regulatory differences between species to inter-tissue differences in regulatory variation within species (see “Discussion” in Pai et al. 2011).
Our group and others often use previously published catalogs of comparative data from primates in the different studies. Although we do not expect previously observed patterns to be erroneous, we are aware that data on gene-specific interspecies regulatory differences, and especially data that pertain to comparisons of divergence across tissues, may be inaccurate for the reasons we discussed above. We thus designed the current study to produce a new comprehensive catalog of comparative gene expression and DNA methylation data from humans, chimpanzees, and rhesus macaques, attempting to minimize possible confounders.
The goal of our study is not to challenge previous conclusions or document specific differences between the current and previous data. Instead, we aim to provide a new and more accurate comparative catalog of inter-tissue and interspecies differences in gene regulation between humans and other primates, with substantial sample and study design documentation. Overall, we believe that this catalog can be useful for many future applications and can serve as a new benchmark for regulatory divergence in primates.
Results
Study design and data collection
To comparatively study gene expression levels and DNA methylation patterns in primates, we collected primary heart, kidney, liver, and lung tissue samples from four human, four chimpanzee, and four rhesus macaque individuals (Fig. 1A; Supplemental Table S1A). From these 48 samples, we harvested RNA and DNA in parallel (Methods). After confirming that the RNA from all samples was of acceptable quality (Supplemental Fig. S1A; Supplemental Table S1B), we performed RNA-sequencing to obtain estimates of gene expression levels. Additional details about the donors, tissue samples, sample processing, and sequencing information can be found in “Methods” and Supplemental Table S1.
Figure 1.
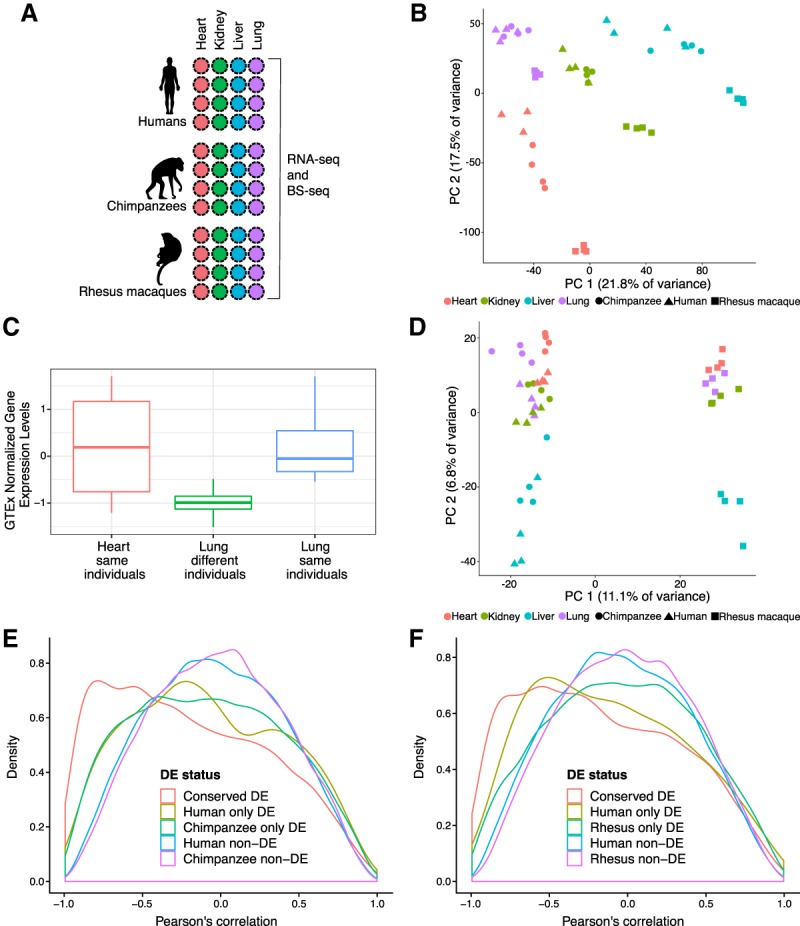
Surveying gene expression and DNA methylation in diverse tissues across primates. (A) Study design. (B) Principal components analysis (PCA) of gene expression levels in 47 samples. (C) Normalized gene expression (quantile-transformed RPKMs) from four donors in the GTEx heart collection (“heart same individuals”) compared to the lungs from different (“lung different individuals”) and the same donors’ lungs (“lung same individuals”) in AC020922.1. (D) PCA of average methylation levels 250 bp upstream and downstream in 47 samples. (E) Density function of the correlation between gene expression and DNA methylation levels in human–chimpanzee orthologous genes. (F) Density function of the correlation between gene expression and DNA methylation levels in genes orthologous across humans and rhesus macaques.
We estimated gene expression levels using an approach designed to prevent biases driven by sequence divergence across species, similar to the approach of Blake et al. (2018b). Briefly, we mapped RNA-sequencing reads to each species’ respective genome. To compare gene expression levels across species, we only calculated the number of reads mapping to exons that can be classified as clear orthologs across all three species (Supplemental Table S1B). We excluded data from genes that were lowly expressed in more than half of the samples as well as data from one human heart sample that was an obvious outlier, probably owing to a sample swap (Supplemental Fig. S2A,B). We normalized the distribution of gene expression levels to remove systematic expression differences between species (maximizing the number of genes with invariant expression levels across species corresponds to our null hypothesis) (Methods). Through this process, we obtained TMM- and cyclic loess-normalized log2 counts per million (CPM) values for 12,184 orthologous genes to be used in downstream analyses (Supplemental Table S2).
Elements of study design, including sample processing, have been shown to impact gene expression data (Gilad and Mizrahi-Man 2015). Consequently, we tested the relationship between a large number of technical factors recorded throughout our experiments and the biological variables of interest in our study, namely tissue and species (Methods; Supplemental Material; Supplemental Table S3A,B). We found that there were no technical confounders with tissue but two technical factors were confounded with species: time postmortem until collection and RNA extraction date (Supplemental Fig. 1B,C). Because of the opportunistic nature of sample collection, these confounders are practically impossible to avoid in comparative studies of primates (especially apes). We discuss possible implications of these confounders throughout the paper.
Gene expression varies more across tissues than across species
We first examined broad patterns in the gene expression data. A principal component analysis (PCA) and a separate clustering analysis indicated that, as expected (Brawand et al. 2011; Barbosa-Morais et al. 2012; Merkin et al. 2012), the primary sources of gene expression variation are tissue (regression of PC1 by tissue = 0.81; P < 10−14; regression of PC2 by tissue = 0.70; P < 10−10) (Fig. 1B; Supplemental Tables S1A,B, S3A,B; Supplemental Fig. S3), followed by species (regression of PC2 by species = 0.27; P < 10−3) (Supplemental Tables S1A,B, S3A,B). We then confirmed that, globally, gene expression levels across tissues from the same individual are more highly correlated than gene expression levels across tissues from different individuals (Supplemental Fig. S2C). This observation supports the intuitive notion that collecting and analyzing multiple tissues from the same individual is highly desirable in functional genomics studies.
We sought further explicit evidence that incorporating balanced collection of multiple tissues from the same individuals is an effective study design. To do so, we used lung and heart data from the GTEx Consortium (The GTEx Consortium 2017). We first identified differentially expressed (DE) genes between lung and heart; we designated these classifications, which are based on hundreds of samples, as the “truth” (Supplemental Material; Supplemental Table S3E). Next, we repeatedly identified DE genes between lung and heart using GTEx data from randomly chosen sets of just four samples from each tissue. We then compared the results to DE genes identified from an equivalent analysis of sets of four samples from each tissue, in which the tissue samples originated from the same donor. Compared to the designated “truth,” DE analyses using data from tissue samples that are matched for donors result in a higher ratio of true positives to false positives than analyses using tissue samples that are unmatched for donors (P = 0.03) (Supplemental Table S3F). Given the small number of false positives in both data sets, study design is unlikely to impact large-scale, highly robust trends across species. However, this study design choice is particularly important if one is interested in individual genes (as shown by an example in Fig. 1C).
Putatively functional tissue-specific gene expression patterns
To analyze the pairwise regulatory differences across tissues and species, we used the framework of a linear model (Methods). We first identified (at FDR <1%) 3695–7027 (depending on the comparison we considered) differences in gene expression levels between tissues, within each species (Table 1; Supplemental Table S4A–C). Overall, the patterns of inter-tissue differences in gene expression levels are similar in the three species, significantly more so than expected by chance alone (P < 10−16, hypergeometric distribution) (Supplemental Material; Supplemental Table S5). A range of 17%–26% of inter-tissue DE genes have conserved inter-tissue expression patterns in all three species (Supplemental Table S5). Regardless of species, we found the fewest inter-tissue DE genes when we contrasted liver and kidney, and the largest number of DE genes between liver and either heart or lung (Table 1; Supplemental Table S4B). However, because our data were produced from bulk RNA-sequencing, we were unable to determine the impact of cell composition on the number of inter-tissue DE genes.
Table 1.
Pairwise DE genes at FDR 1%

We used the same framework of linear modeling to identify gene expression differences between species, within each tissue (Supplemental Table S4A). Depending on the tissue and species we considered, we identified between 799 and 4098 interspecies DE genes (at FDR = 1%) (Table 1). As expected, given the known phylogeny of the three species, within each tissue we classified far fewer DE genes between humans and chimpanzees than between either of these species and rhesus macaques (Supplemental Table S4B).
It is a common notion that genes with tissue-specific expression patterns may underlie tissue-specific functions. Previous catalogs of such patterns in primates were always confounded by the effect of individual variation (because each tissue was sampled from a different individual). To classify tissue-specific genes using our data, we focused on genes that are either up-regulated or down-regulated in a single tissue relative to the other three tissues (within one or more species). We define such genes as having a “tissue-specific” expression pattern, acknowledging that this definition may only be relevant in the context of the four tissues we considered here.
Using this approach and considering the human data across all tissue comparisons, we identified 5284 genes with tissue-specific gene expression patterns (FDR 1%) (Fig. 2A–D). By performing similar analyses using the chimpanzee and rhesus macaque data, we found that the degree of conservation of tissue-specific expression patterns is higher than expected by chance (P < 10−16) (Fig. 2A–D). This observation is robust with respect to the statistical cutoffs we used (Supplemental Table S6), indicating that many of these conserved tissue-specific regulatory patterns are likely of functional significance.
Figure 2.
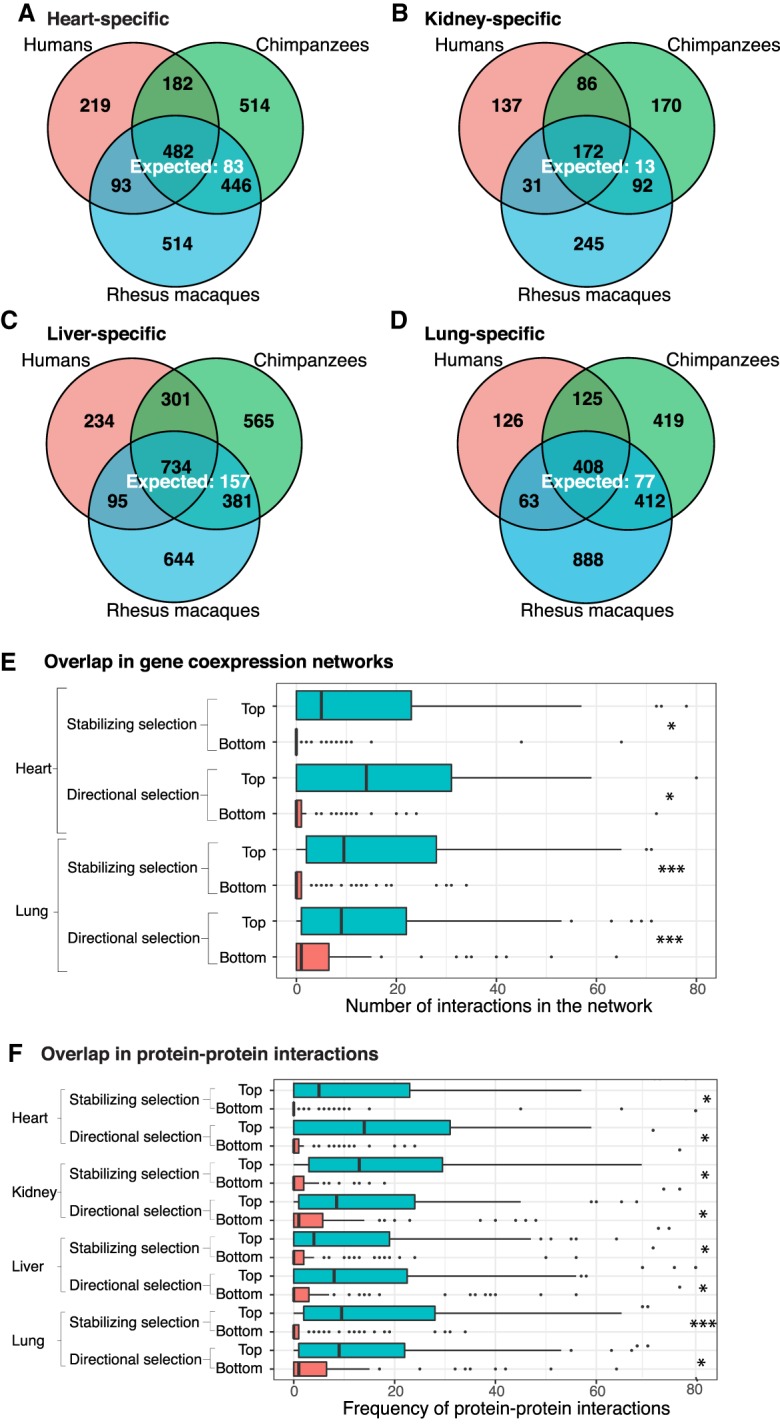
Tissue-specific DE genes (FDR = 0.01). The number of conserved tissue-specific DE genes across all three species is greater than the number expected by chance: (A) heart; (B) kidney; (C) liver; (D) lung. In each tissue, genes with tissue-specific regulatory patterns that are consistent with the action of natural selection (top) are more likely to appear in gene coexpression networks (E) and have an increased number of protein–protein interactions (F) than those that are less consistent with the action of natural selection (bottom). (*) P < 0.05; (***) P < 0.001. The x-axes of E and F are cut off at 80 interactions for readability. In both cases, <5% of the data points are beyond this cutoff.
To broadly analyze the biological function of genes with conserved tissue-specific expression, we performed a Gene Ontology (GO) enrichment analysis (Supplemental Material). We found these genes are indeed highly enriched with functional annotations that are relevant to the corresponding tissue (Supplemental Tables S7A–D, S8). For example, genes with conserved heart-specific expression patterns were enriched in GO categories related to muscle filament sliding (e.g., ACTA1, MYL2) and cardiac muscle contraction (e.g., MYBPC3, TNNI3).
Functional analysis of gene regulatory differences
We sought further evidence that the classification of genes with conserved tissue-specific expression patterns is meaningful. To do so, we considered transcription coexpression networks (Stuart et al. 2003; Zhang and Horvath 2005) based on GTEx data from heart and lung (Pierson et al. 2015). We found that genes with conserved tissue-specific expression patterns are more likely to appear as nodes in the networks than genes without tissue-specific expression patterns or genes whose tissue-specific expression patterns are not conserved (P < 10−5). When we only considered genes that appear as nodes in the network, we found that genes with conserved tissue-specific expression patterns are more likely to be classified as hubs in the networks than genes without tissue-specific expression patterns or genes whose tissue-specific expression patterns are not conserved (P < 0.007).
Motivated by these findings, we focused on gene expression patterns that are consistent with the action of natural selection (as described in Blekhman et al. 2008; Supplemental Material; Supplemental Table S7E). We found that genes whose expression patterns are consistent with the action of either stabilizing or directional selection (top 10%) (Supplemental Table S7F) have more interactions with other genes in the network than genes whose expression patterns are not consistent with the action of natural selection (bottom 10%; P < 0.05 for all comparisons) (Fig. 2E). This observation is fairly robust with respect to percentile cutoff (Supplemental Table 7F).
We repeated a similar analysis using protein–protein interaction data from the Human Protein Atlas (Uhlén et al. 2015; Yu et al. 2015; Lindskog 2016; Thul and Lindskog 2018) in all four tissues. We again found that genes whose expression patterns are consistent with selection have more annotated protein–protein interactions (P < 0.05 in all eight comparisons) (Fig. 2F; Supplemental Table 7G). These interaction results suggest that functionally important genes are carefully regulated. Furthermore, this tight regulation occurs at both the gene expression and protein levels in primates.
Variation in DNA methylation across tissues and species
We used low-coverage whole-genome bisulfite sequencing (BS-seq) to study DNA methylation patterns in each sample. The bisulfite conversion reaction efficiency was higher than 99.4% for all samples (Supplemental Table S1C). Following sequencing, we mapped the high-quality BS-seq reads to in silico bisulfite-converted genomes of the corresponding species. We measured DNA methylation levels in 12.5 million to 22.9 million CpG sites per sample, with a minimum coverage of two sequencing reads per site (Supplemental Table S1C).
We estimated local methylation levels by smoothing the data across nearby CpG sites (Supplemental Material; Supplemental Figs. S4–S6; Hansen et al. 2012). To facilitate a comparison of methylation levels across species, we annotated 10.5 million orthologous CpGs in the human and chimpanzee genomes, as well as a smaller set of 2.4 million orthologous CpGs in all three primate genomes (Supplemental Table S1C–E). To identify differences in DNA methylation levels between tissues and species we again used a linear model framework (Methods; Fig. 1D). Focusing on DNA methylation patterns across tissues within species, we identified between 7026 and 41,280 differentially methylated regions between tissues, within species (T-DMRs) (Table 2; Supplemental Table S9A; Blake et al. 2018a). Pairwise comparisons between hearts and lungs showed the lowest number of T-DMRs, regardless of species (7026 in rhesus macaques, 8524 in chimpanzees, 14,208 in humans), whereas comparisons involving heart and liver showed the largest number of T-DMRs (22,561 in humans, 28,767 in chimpanzee, and 41,280 in rhesus macaques) (Table 2). We found that human T-DMRs overlapped genic and regulatory features significantly more than expected by chance. In particular, there is an enrichment of T-DMRs in intergenic regions, introns, 5′ UTRs, 3′ UTRs, and active enhancers (P < 0.04 for all tests) (as defined by Andersson et al. 2014; Supplemental Table S9B).
Table 2.
Pairwise differentially methylated regions (DMRs) in autosomal chromosomes

We found strong evidence for T-DMR conservation across all three species (P < 10−16 across all comparisons) (Supplemental Table S10A). Although this level of conservation is higher than expected by chance, we recognize that in each tissue comparison we performed, we had incomplete power to identify T-DMRs and so the true conservation of T-DMRs is expected to be even higher. To compare T-DMRs across species more effectively, we considered DNA methylation data from all T-DMR orthologous regions that were classified as such in at least one species. When we performed hierarchical clustering using orthologous DNA methylation data from these T-DMRs, the data clustered first by tissue, then by species (Supplemental Fig. S7). This trend is robust with respect to the species used to initially locate T-DMRs (Supplemental Figs. S8, S9). Thus, our results suggest that in general, inter-tissue DNA methylation differences within a species tend to be conserved, consistent with the observations of previous studies (Martin et al. 2011; Molaro et al. 2011; Pai et al. 2011; Hernando-Herraez et al. 2013, 2015b).
We next focused specifically on tissue-specific DMRs, as these may contribute to tissue-specific function. In contrast to differences in DNA methylation between any pair of tissues, a tissue-specific DMR is defined as having a similar methylation level in three of the tissues we considered, but a significantly different DNA methylation level in the remaining tissue. We found that there were more DMRs specific to liver (3278 to 11,433 DMRs depending on the species) than to kidney (2300 to 3957 DMRs), heart (1597 to 2969 DMRs), or lung (453 to 5018 DMRs) (Fig. 3A–D; Supplemental Table S10B). Tissue-specific DMRs are highly conserved regardless of the comparisons we made (P < 10−13 for all comparisons, at least 25% bp overlap was required to be considered shared).
Figure 3.

Tissue-specific DMRs (FDR = 0.01). The number of conserved tissue-specific DMRs is greater than expected by chance: (A) heart; (B) kidney; (C) liver; (D) lung. Genes with the closest TSSs to conserved tissue-specific DMRs are enriched for relevant functional annotations in hearts (E) and livers (F).
In all four tissues, >59% of conserved DMRs are hypomethylated in a tissue-specific manner. We evaluated the overlap between tissue-specific DMRs and genomic regions marked with H3K27ac, a mark often associated with active gene expression (The ENCODE Project Consortium 2012). We found that conserved hypomethylated tissue-specific DMRs were annotated with H3K27ac more frequently than tissue-specific DMRs identified only in humans (P < 0.001, difference of proportions test) (Supplemental Material; Supplemental Table S10C). We then asked about the potential impact of these conserved hypomethylated tissue-specific DMRs on the expression of nearby genes. We found that genes with the closest TSSs to conserved tissue-specific DMRs are highly enriched with relevant functional annotations in hearts and livers (the tissues with the largest numbers of conserved hypomethylated tissue-specific DMRs) (Fig. 3E,F; Supplemental Table S10D; Supek et al. 2011). For example, conserved heart-specific DMRs are closest to genes in cardiovascular-related pathways, including ventricular cardiac muscle cell development, canonical Wnt signaling pathway, and the MAPK7 cascade. Overall, these observations suggest that conserved tissue-specific DMRs are likely to underlie tissue-specific gene regulation in primates.
Interspecies differences in gene expression and DNA methylation levels
Our comparative catalog can be used to identify DNA methylation differences that could potentially explain gene expression differences across species and tissues. To do so, we first identified 7,725 orthologous genes with expression data and corresponding promoter DNA methylation data in humans and chimpanzees, and 4155 orthologous genes with the same information for all three species. We then determined to what extent divergence in DNA methylation levels could potentially underlie interspecies differences in gene expression by comparing the gene expression effect size associated with “species” before and after accounting for methylation levels. To determine significant effect size differences, we applied adaptive shrinkage (Stephens 2017)—a flexible empirical Bayes approach for estimating false discovery rate (Methods). We note that this mediation approach does not consider the possibility that a third, unobserved event may be causally responsible for both the DNA methylation and expression patterns.
Considering DE genes between humans and chimpanzees (in at least one tissue), we found that between 11% and 25% of genes (depending on tissue) showed a difference in the effect of species on gene expression levels once average promoter methylation levels were accounted for (significant difference in effect size classified at FSR 5%, represented by red in Supplemental Fig. S10; Supplemental Table S11A). As a control analysis, we considered only the genes that were not originally classified as DE between humans and chimpanzees, and we found that the difference in the effect size of species on gene expression levels was reduced in <1% of genes once DNA methylation was accounted for (FSR 5%) (Supplemental Fig. S10; Supplemental Table S11A); thus, our approach is well calibrated.
We applied the same approach to the human and rhesus macaque data and found that the percentage of genes for which gene expression differences could potentially be explained by DNA methylation differences ranges from 21% in the lung to 40% in the liver (Supplemental Fig. S11; Supplemental Table S11B). This observation may reflect the more extreme gene expression differences between humans and rhesus macaques than between humans and chimpanzees (before accounting for DNA methylation levels, P < 0.003 in all tissues, t-test comparing the absolute values of the effect sizes for both groups of DE genes).
Next, we examined the genes in which DNA methylation differences may underlie inter-tissue gene expression differences (example in Fig. 4A–C). Using adaptive shrinkage, we found that 9%–17% of inter-tissue gene expression differences could potentially be explained by DNA methylation differences across tissues (FSR 5%). When we performed the control analysis and considered only data from genes that were not DE between tissues, <1% of effect sizes differed once we accounted for the DNA methylation data (Fig. 4F; Supplemental Table S11C–E).
Figure 4.

Inter-tissue DNA methylation and gene expression levels (FDR = 0.05 and FSR = 0.05). (A–C) A representative example of the PRKACA gene in which the variation of methylation levels (A) may explain the differences in gene expression levels (B) between human heart and kidney. (C) The residuals of normalized gene expression levels after regressing out methylation levels. (D–G) Next, we compared the tissue effect sizes before and after controlling for DNA methylation levels in inter-tissue DE and non-DE genes, separately. Genes in red are significant at S-value <0.05. Effect size differences in conserved DE genes in human heart relative to human kidney (D), nonconserved DE genes in human heart relative to human kidney (E), and non-DE genes in human heart and human kidney (F). (G) The conserved DE genes are enriched for heart-related function. (H) Variation in DNA methylation is more likely to explain variation in conserved DE genes than nonconserved DE genes (DE in the human tissues listed, but not in the same tissues in chimpanzees).
Finally, we focused on regulatory patterns that are most likely to be functional—namely, conserved inter-tissue gene regulatory differences. These differences were more likely to be explained by variation in DNA methylation levels than nonconserved inter-tissue gene expression differences (minimum difference is 7%, P < 0.005 for all comparisons; at FDR <5% and FSR <5%) (Fig. 4D,E,H; Supplemental Table S11C–E). This observation is robust with respect to the FDR and FSR cutoff used (Supplemental Table S11C–E). Indeed, the correlation between DNA methylation and gene expression data is higher for genes with conserved inter-tissue expression patterns compared to genes whose expression patterns are not conserved (Fig. 1E,F).
One way to maintain conserved inter-tissue gene expression differences could be through DNA methylation level differences. We compared the genes whose variation in inter-tissue gene expression can potentially be explained by variation in DNA methylation levels (assuming no independent effect of an unobserved factor) to all genes with conserved inter-tissue expression differences. We found that these genes are enriched for “essential tissue functions” (Supplemental Table S11F). For example, the heart genes are enriched for cardiac and smooth muscle contraction, whereas those in liver are enriched for regulation of cholesterol transport and hormone secretion (Fig. 4G; Supplemental Table S11F). These observations suggest that DNA methylation levels may mark or even drive differences in the expression levels in functionally relevant genes.
Discussion
We designed a comparative study of gene regulation in humans, chimpanzees, and rhesus macaques that minimized confounding effects and bias. Consistent with previous studies, we found a high degree of conservation in gene expression levels when we considered the same tissue across species (Barlow 1993; Brawand et al. 2011; Sharp et al. 2011; Lin et al. 2014; Gallego Romero et al. 2015). We also found evidence for conservation of tissue-specific DMRs. Our observations are qualitatively consistent with those of previous studies that mostly used microarrays to measure DNA methylation levels (Pai et al. 2011; Hernando-Herraez et al. 2015b; Tsankov et al. 2015), however, the high resolution of our BS-seq data allowed us to examine a much larger number of CpG sites. Thus, we were able to show that although DNA methylation can potentially explain a modest proportion of expression differences between tissues (Pai et al. 2011), it is more likely to impact conserved tissue-specific gene expression levels.
We created and made available the most comprehensive, and likely most accurate comparative catalog of gene expression and methylation levels in humans, chimpanzees, and rhesus macaques. Comparative functional genomic studies in primates, including from our own laboratory, often are not designed to test specific hypotheses. Rather, many of these comparative genome-scale studies aim to build catalogs of similarities and differences in gene regulation between humans and other primates. These catalogs have been shown to be quite useful; for example, they can be used to identify interspecies regulatory changes that have likely evolved under natural selection (Enard et al. 2002; Cáceres et al. 2003; Karaman et al. 2003; Khaitovich et al. 2004a, 2005; Gilad et al. 2005; Lemos et al. 2005; Blekhman et al. 2008, 2010; Nowick et al. 2009; Babbitt et al. 2010; Pai et al. 2011; Shibata et al. 2012; Capra et al. 2013; Khan et al. 2013), and thereby help us better understand the evolutionary processes that led to adaptations in humans. These catalogs are also used to establish informed models of the relative importance of changes in different molecular mechanisms to regulatory evolution (Khaitovich et al. 2004b; Warnefors and Eyre-Walker 2012) and to inform us about ancestral or derived phenotypes that may be relevant to human diseases (Cooper and Shendure 2011; Gallego Romero et al. 2015). Ultimately, comparative catalogs of gene regulatory phenotypes are used to develop and test specific hypotheses regarding the connection between interspecies regulatory changes and physiological, anatomical, and cognitive phenotypic differences between species.
In this study, we used a comparative catalog to identify species-specific and, in particular, tissue-specific regulatory patterns, because these genes are often drug targets (Dezső et al. 2008) and are likely important for the evolution of human traits (Blekhman et al. 2008). We showed that genes with conserved tissue-specific regulatory patterns have more regulatory interactions and protein–protein interactions than genes whose regulatory patterns are not conserved or are not tissue-specific. These patterns became even more pronounced when we focused on genes whose expression patterns are consistent with the action of natural selection. Together, these observations consistently support the inference that when genes perform an important function that needs to be carefully regulated, evolution can act on multiple levels of the regulatory cascade in primates.
Focusing on species-specific patterns of tissue-specific gene regulation, our observations can help formulate specific functional hypotheses regarding human-specific adaptations. For example, genes with tissue-specific gene regulation identified only in humans are enriched for GO pathways that may contribute to human-specific features, including sodium ion import across the plasma membrane in kidneys (e.g., SLC9A3 and TRPM4), the glycogen biosynthetic process in livers (e.g., PGM1 and AKT1), and paraxial mesoderm morphogenesis in lungs (e.g., MST1R and MAP9).
Consideration of study design and record keeping
Regardless of the model system used and the types of data that are collected, study design is critical. Perhaps because comparative studies in primates typically rely on opportunistic sample collection, there are not recognized standards for study design that are kept and consistently reported in most existing studies (including many earlier studies from our group). We thus believe that it is worthwhile to explicitly discuss a few important considerations regarding study design and the recording of metadata.
Without a balanced study design, it would have been impossible to independently estimate the effects of individual, tissue, and species on our data. Because the sources of confounding factors are difficult to predict in advance, we strongly recommend that samples are collected using a balanced design with respect to as many parameters as possible. These include the distribution of tissue samples per individual, the number of individuals from each species, sex, age range, cause of death and collection time (in the case of postmortem tissues), or sample collection and cell culturing (in the case of iPSC-based models). All steps of sample processing (RNA extraction, library preparation, and so forth) should be done in batches that are randomized or balanced with respect to species, tissue, and any other variables of interest.
Most important, all sample processing steps should be recorded in a sample history file that includes anything that happened to any sample. We have documented many of these steps in Supplemental Table S1A–E. This documentation can help provide evidence that a phenomenon is driven by biological rather than technical factors. It may also benefit future studies by facilitating effective meta-analysis of multiple data sets, which would help to address the problems of tissue availability and small sample sizes. We believe that, moving forward, it should be a requirement that these metadata are available with every published comparative genomic data set.
Methods
Sample description
We collected heart (left ventricle), kidney (cortex), liver, and lung tissues from four individual donors in human (Homo sapiens, all of reported Caucasian ethnicity), chimpanzee (Pan troglodytes), and Indian rhesus macaque (Macaca mulatta), for a total of 48 samples (3 species × 4 tissues × 4 individuals) (Fig. 1A). The choice of these tissues was guided by their relative homogeneity with respect to cellular composition (e.g., Balashova and Abdulkadyrov 1984), which does not change substantially across primate species. In contrast, other tissues, such as brain subparts, differ substantially in cellular composition across primates (Brodal 1983), which could potentially confound the analyses.
Human samples were obtained from the National Disease Research Interchange (IRB protocol 14378B). Non-human samples were obtained from several sources, including the Yerkes primate center and the Southwest Foundation for Biomedical Research, under IACUC protocol 71619. When possible, samples were collected from adult individuals whose cause of death was unrelated to the tissues studied.
RNA library preparation and sequencing
In total, we prepared 48 unstranded RNA-sequencing libraries as previously described (Marioni et al. 2008; Blekhman et al. 2010). Twenty-four barcoded adapters were used to multiplex different samples on two pools of libraries. RNA-sequencing libraries were sequenced on 26 lanes on four different flow-cells on an Illumina HiSeq 2500 sequencer in either the Gilad laboratory or at the University of Chicago Genomics Facility (50-bp single-end reads) (Supplemental Material; Supplemental Table S1B).
Quantifying the number of RNA-seq reads from orthologous genes
We used FastQC (version 0.10.0; https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) to generate a read quality report and TrimGalore (version 0.2.8; https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), a wrapper based on Cutadapt (version 1.2.1) (Martin 2011), to trim adapter sequences from RNA-seq reads. We trimmed using a stringency of 3. To cut the low-quality ends of reads, we used a quality threshold (Phred score) of 20. Reads shorter than 20 nt after trimming were eliminated before mapping (Supplemental Table S1B).
For each sample, we used TopHat2 (version 2.0.8b) (Kim et al. 2013) to map the reads to the correct species’ genome: human reads to the hg19 genome, chimpanzee reads to the panTro3 genome, and rhesus macaque reads to the rheMac2 genome (Supplemental Material). Expression level estimates may be biased across the species owing to factors such as mRNA transcript size and different genome annotation qualities. To circumvent these issues, we only retained reads that mapped to a set of 30,030 Ensembl gene orthologous meta-exons available for each of the three genomes, as described and used previously (Blekhman et al. 2010; Blekhman 2012; Gallego Romero et al. 2015). We defined the number of reads mapped to orthologous genes as the sum of the reads mapped to the orthologous meta-exons of each gene. We quantified gene expression levels using the program coverageBed from the BEDTools suite (Quinlan and Hall 2010) and then performed TMM and cyclic loess normalization (Supplemental Material).
Because all three releases of GTEx use hg19 and only a fraction of the 1000 Genomes Project data are available in GRCh38 coordinates, we also used hg19. However, to show that the results we report would not change much if we used the GRCh38 build, we leveraged the fact that differential expression analysis compares gene expression levels from groups of samples (e.g., human liver samples to human lung samples). Therefore, we compared the ranks of the normalized gene expression levels in the 15 human samples mapped using hg19 to the same samples mapped to GRCh38. The correlations of these ranks were extremely high (median Pearson's correlation = 0.96). These strong correlations suggest that our general conclusions—and indeed, many genes we identified as DE—would remain if we had used the GRCh38 build.
Analysis of technical variables
To assess whether the study's biological variables of interest—tissue and species—were confounded with the study's recorded sample and technical variables, we used a previously described approach (Blake et al. 2018b).
For the 12 RNA-seq related technical variables that were the most highly correlated with tissue or species, we assessed which technical variables constitute the “best set” of independent variables to be included in a linear model for gene expression levels. Because of the partial correlations between the variables, we applied lasso regression using the package “glmnet” (Friedman et al. 2010). Before performing the analysis, we also protected our variables of interest, tissue and species, in the model for each gene. We summarized each technical variable's influence across the genes by counting the number of times each technical variable was included in the best set of the gene models. We found that none of the technical variables appeared in more than 25% of the best sets (i.e., >25% of the gene models). Therefore, we chose not to include these technical variables in our model for testing differential expression.
Finally, during our analysis of technical factors, we discovered that RNA extraction date was confounded with species. In 2012, we extracted RNA from the chimpanzee samples on March 8, from the human samples on 3 d between March 12 and 29, and from the rhesus samples on March 6. To test the relationship between the date of RNA extraction and gene expression PCs in humans, we performed individual linear models on PCs 1–5 using RNA extraction date as a predictor. None of the models were statistically significant at FDR 10%, suggesting that tissue type is more highly associated with gene expression levels than RNA extraction date.
Differential expression analysis using a linear model-based framework
To perform differential expression analysis, we used the same approach as Blake et al. (2018b). We applied a linear model-based empirical Bayes method (Smyth 2004; Smyth et al. 2005) that accounts for the mean-variance relationship of the RNA-sequencing read counts, using weights specific to both genes and samples (Law et al. 2014).
To be considered a “tissue-specific DE gene” under our stringent definition, the gene must be in the same direction and statistically significant in all pairwise comparisons including the given tissue but not significant in any comparison without that tissue. For example, for a gene to be classified as having heart-specific up-regulation in a given species, the gene needed to be up-regulated (a significant, positive effect size) in heart versus liver, heart versus lung, heart versus kidney, but not significantly different between the liver versus lung, liver versus kidney, or kidney versus lung, in the same species. Under the more lenient definition of tissue-specific DE genes, we compared the gene expression level of one tissue to the mean of the other three tissues. To do so, we grouped the three tissues together and again used the limma + voom framework to identify significant differences in one tissue versus the group of the other tissues (Law et al. 2014; Ritchie et al. 2015).
To identify interspecies differences in gene expression patterns across tissues within species (tissue-by-species interactions), we used the limma + voom framework and looked for the significance of tissue-by-group interactions. In one analysis, the groups were great ape versus rhesus macaque and in another analysis, the groups were human versus non-human primates. To minimize the number of interactions, we compared one tissue relative to a group of the other three tissues (e.g., great ape vs. rhesus macaque, heart vs. non-heart). Significant tissue-specific interactions were detected using the adaptive shrinkage method, ashr (Stephens 2017). Specifically, for each test, we input the regression estimates from limma to ashr: regression coefficients, posterior standard errors, and posterior degrees of freedom. We used the default settings in ashr to calculate the shrunken regression coefficients (called the “posterior mean” in ashr), false discovery rate (FDR, also known as Q-value), and false sign rate (FSR, also known as S-value: the probability that sign of the estimated effect size is wrong in either direction). We assigned directionality based on the sign of the posterior mean and determined significance based on the FSR.
The impact of matched tissue samples on DE results
To determine the impact of matched tissue samples on DE results, we compared inter-tissue DE analysis results when using tissues from the same or different individuals in GTEx v7 data (The GTEx Consortium 2017). We first subset the GTEx raw gene expression count data to only individuals for which there was gene count information in the heart and lung tissues, for genes included in all three tissues. (The most GTEx samples were in heart and lung, so we decided to focus on these samples.) Furthermore, to minimize the number of covariates needed in the linear model, we decided to only analyze individuals of the same sex and whose samples were sequenced on the same platform (sex = 1 and platform = 1 from the GTEx documentation). We then normalized the data and performed DE analysis using a voom + limma pipeline. In the linear model, we included tissue and three GTEx-provided covariates (covariates 1 and 2 and inferred covariate 1 from the covariate file for each tissue) as fixed effects and individual as a random effect. We chose to renormalize the raw counts data rather than use the normalized counts from GTEx because the voom + limma pipeline requires raw counts to assign voom weights. We considered the output of the inter-tissue DE analysis for all individuals (DE vs. non-DE genes at FDR 5%) as the “ground truth.” To evaluate the impact of our study design, we then subset the gene expression information to the individuals for which there is information in all three tissues. We obtained gene expression level information from four randomly selected individuals and used the voom + limma pipeline to identify inter-tissue DE genes. Next, we compared the list of DE genes from this analysis to the ground truth list. We performed this downsampling procedure for tissues from the same four individuals as well as from four different individuals 10 times each and compared the number of true and false positives from the tests. For the analysis with eight different individuals, there were no repeated individuals, so we did not use the “duplicateCorrelation” function in voom.
BS-seq library preparation, sequencing, and mapping
We prepared a total of 48 whole-genome BS-seq libraries from extracted DNA as previously described (Tung et al. 2012; Banovich et al. 2014). We aligned the trimmed reads to the human (hg19, February 2009), chimpanzee (panTro3, October 2010), or rhesus macaque (rheMac2, January 2006) genomes, and to the lambda phage genome using the Bismark aligner (version 0.8.1) (Krueger and Andrews 2011).
We estimated the percentage of methylation at an individual cytosine site by the ratio of the number of cytosines (unconverted) found in mapped reads at this position, to the total number of reads covering this position (sequenced as cytosine or thymine, i.e., converted or unconverted) using the methylation extractor tool from Bismark (version 0.8.1). We additionally collapsed information from both DNA strands (because CpG methylation status is highly symmetrical on opposite strand) (Lister et al. 2009) to achieve better precision in methylation estimates across the genome.
To obtain CpGs that mapped to multiple species, the chimpanzee and rhesus macaque CpG sites were mapped to human coordinates (hg19) using chain files from ftp://hgdownload.cse.ucsc.edu/goldenPath/hg19/liftOver/ and the liftOver tool from the UCSC Genome Browser (Karolchik et al. 2014). These files had previously been filtered for paralogous regions and repeats, but we also removed positions that were not remapped to their original position when we mapped from human back to their original genome. Chimpanzee and rhesus macaque CpG sites were mapped to human, even if their orthologous positions were not a CpG site in human.
Identifying differentially methylated regions (DMRs)
We were interested in identifying regions showing consistent differences between pairs of tissues or pairs of species, taking biological variation into account. To identify DMRs we used the linear model-based framework in the Bioconductor package bsseq (version 0.10.0) (Hansen et al. 2012). For a given pairwise comparison (e.g., human liver vs. human heart), the bsseq package produces a signal-to-noise statistic for each CpG site similar to a t-test statistic, assuming that methylation levels in each condition have equal variance. As recommended by Hansen et al. (2012), we used a low-frequency mean correction to improve the marginal distribution of the t-statistics. Similar to previous studies using this methodology, a t-statistic cutoff of −4.6, 4.6 was used for significance (Hansen et al. 2011, 2014). DMRs were defined as regions containing at least three consecutive significant CpGs, an average methylation difference of 10% between conditions, and at least one CpG every 300 bp (Hansen et al. 2012). We used BEDTools (version 2.26.0) (Quinlan and Hall 2010) to calculate the number of overlapping DMRs across tissues and/or species (Blake et al. 2018a). We required overlapping DMRs to have a minimum base pair overlap of at least 25%, unless otherwise stated.
To be considered a tissue-specific DMR, the region was required to be a significant tissue DMR (T-DMR) in one tissue compared to the other three tissues pairwise (in the same direction) but not a significant T-DMR across any of the other three tissues in pairwise comparisons. We again used BEDTools to ensure a minimum base pair overlap of 25%. Once the tissue-specific DMRs were identified within a species, we then classified them as species-specific or conserved. To be considered conserved (across humans and chimpanzees or all three species), the tissue-specific DMR had to be significant in all species in the comparison and have a minimum base pair overlap of the T-DMR of at least 25%.
Calculating the average methylation levels of conserved promoters
To calculate the DNA methylation levels of orthologous CpGs around the transcription start site (TSS) of orthologous genes, we first had to determine the orthologous TSSs. We began with the 12,184 orthologous genes in our RNA-sequencing analysis. Of these, we found that 11,131 of these orthologous genes had an hg19 RefSeq TSS annotation (https://sourceforge.net/projects/seqminer/files/Reference%20coordinate/refGene_hg19_TSS.bed/download). We used liftOver to find orthologous sites in the chimpanzees and rhesus macaque genomes in 9682 of those 11,131 genes. We then determined which of the hg19 RefSeq TSS annotations were closest to the first hg19 orthologous exon, and we repeated this process with the other two species and their respective genomes. We found that 9604 of 9682 of the closest TSS annotations in humans had the same liftOver coordinates in the other two species. We then calculated the distance between the first orthologous exon to the TSS site in all three species individually. To minimize this difference between the three species, we filtered all genes with a maximum distance difference across the species of larger than 2500 bp (for reference, the 75th percentile of the maximum difference in distance was 2078 bp). After this filtering step, 7263 autosomal genes remained, and 4155 genes had at least two orthologous CpGs 250 bp upstream of and 250 bp downstream from the orthologous TSS. We chose a 250-bp window around the TSS based on DNA methylation levels around the promoter in Banovich et al. (2014) and calculated the average of orthologous CpGs within this window for the 4155 genes. Using the same method but in humans and chimpanzees only, we found and calculated the average of orthologous CpGs within this window for 7725 genes.
Joint analysis of promoter DNA methylation and gene expression levels
To determine whether DNA methylation may underlie interspecies differences in gene expression levels, we used a joint analysis method as described below. For each gene, we analyzed the gene expression levels, along with the accompanying average methylation levels 250 bp upstream of and downstream from the TSS (found above). For a given tissue, we first determined the effect of species on gene expression levels using a linear model, with species and RIN score as fixed effects (Model 1). Next, we parameterized a linear model attempting to predict expression levels exclusively from methylation levels. We refer to these residuals as “methylation-corrected” gene expression values. We then used these values to again determine the effect of species, this time on gene expression levels “corrected” for methylation, using a linear model with species and RIN score as fixed effects (Model 2). To determine the contribution of DNA methylation levels to interspecies differences in gene expression, we computed the difference in the species effect size between Model 1 and Model 2 for each gene, as well as the standard error of the difference. Large effect size differences between Models 1 and 2 for a given gene suggest that methylation status may be a significant driver of DE. To assess the significance of this difference, we used adaptive shrinkage (ashr) (Stephens 2017) to compute the posterior mean of the differences in the effect sizes, using vashr (Lu and Stephens 2016), with the degrees of freedom equal to the number of samples in the linear model minus 2. The shrunken variances from vashr were used in the ashr posterior mean computation. From this procedure, we obtained the number of genes where species has a significant difference in effect sizes before and after regressing out methylation. We assessed significance using the S-value statistic (FS) (Stephens 2017). Using the S-values, rather than the Q-values, not only takes significance into account but also has the added benefit of assessing our confidence in the direction of the effect.
We performed the above analysis separately for interspecies DE genes and non-DE genes, and in each tissue individually. We identified interspecies DE genes in our tissue of interest as those with a significant species term in the model of species and RIN score as fixed effects. We assessed significance of DE genes at FDR 5%, unless otherwise noted.
We also applied the same analysis framework to determine whether DNA methylation may underlie inter-tissue differences in gene expression levels. For the inter-tissue DE genes and non-DE genes, we replaced “species” with “tissue” as a fixed effect in models 1 and 2. We assessed significance with various FDR and FSR thresholds, as specified in the text.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE112356. Data and scripts used in this paper are available at https://github.com/Lauren-Blake/Reg_Evo_Primates and in the Supplemental Code. The results of our scripts can be viewed at https://lauren-blake.github.io/Regulatory_Evol/analysis/.
Competing interest statement
The authors declare no competing interests.
Supplementary Material
Acknowledgments
Members of the Gilad, Marques-Bonet, Robinson-Rechavi, Stephens, and Pritchard laboratories provided helpful discussions and comments on the manuscript. In particular, Matthew Stephens provided guidance on integrating the DNA methylation and gene expression data, and Michelle Ward and Natalia Gonzales provided helpful comments on a draft of the manuscript. We thank Kasper Hansen, Jenny Tung, and Luis Barreiro for discussions regarding multispecies methylation analysis. We acknowledge Athma Pai for sharing a methylation protocol, Bryce van de Geijn for help with the WASP pipeline, and Charlotte Soneson, Jacob Degner, and Unjin Lee. We also thank the Yerkes Primate Center and Southwest Foundation for Biomedical Research, Anne Stone, and Jéssica Hernández Rodríguez for providing and/or helping to ship the tissue samples. L.E.B. was supported by the National Science Foundation Graduate Research Fellowship (DGE-1144082). Additionally, L.E.B. and I.E. were funded by the National Institutes of Health (NIH) Genetics and Gene Regulation Training Grant (T32 GM07197). J.R. was funded by a Swiss National Science Foundation postdoc mobility fellowship (PBLAP3-134342) and the FP7 Marie Curie International Outgoing Fellowship PRIMATE_REG_ EVOL. This project was funded in part by the Office of Research Infrastructure/Office of the Director (ORIP/OD) P51OD011132 grant. T.M.-B. is supported by BFU2017-86471-P (Ministry of Economy and Competitiveness/European Regional Development Fund, European Union), NIH U01 MH106874 grant, Howard Hughes Medical Institute: International Early Career, Obra Social “La Caixa” and Secretaria d'Universitats i Recerca and Centres de Recerca de Catalunya Programme del Departament d'Economia i Coneixement de la Generalitat de Catalunya. The content presented in this article is solely the responsibility of the investigators and does not necessarily reflect the official views of the funders. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.254904.119.
Freely available online through the Genome Research Open Access option.
References
- Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, Chen Y, Zhao X, Schmidl C, Suzuki T, et al. 2014. An atlas of active enhancers across human cell types and tissues. Nature 507: 455–461. 10.1038/nature12787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babbitt CC, Fedrigo O, Pfefferle AD, Boyle AP, Horvath JE, Furey TS, Wray GA. 2010. Both noncoding and protein-coding RNAs contribute to gene expression evolution in the primate brain. Genome Biol Evol 2: 67–79. 10.1093/gbe/evq002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balashova V, Abdulkadyrov K. 1984. Cellular composition of hemopoietic tissue of the liver and spleen in the human fetus. Arkh Anat 86: 80. [PubMed] [Google Scholar]
- Banovich NE, Lan X, McVicker G, van de Geijn B, Degner JF, Blischak JD, Roux J, Pritchard JK, Gilad Y. 2014. Methylation QTLs are associated with coordinated changes in transcription factor binding, histone modifications, and gene expression levels. PLoS Genet 10: e1004663 10.1371/journal/pgen.1004663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbash S, Sakmar TP. 2017. Brain gene expression signature on primate genomic sequence evolution. Sci Rep 7: 17329 10.1038/s41598-017-17462-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa-Morais NL, Irimia M, Pan Q, Xiong HY, Gueroussov S, Lee LJ, Slobodeniuc V, Kutter C, Watt S, Colak R, et al. 2012. The evolutionary landscape of alternative splicing in vertebrate species. Science 338: 1587–1593. 10.1126/science.1230612 [DOI] [PubMed] [Google Scholar]
- Barlow DP. 1993. Methylation and imprinting: from host defense to gene regulation? Science 260: 309–310. 10.1126/science.8469984 [DOI] [PubMed] [Google Scholar]
- Blake LE, Roux J, Gilad Y. 2018a. Pairwise DMRs for, “A comparison of gene expression and DNA methylation patterns across tissues and species” (Version 1). 10.5281/zenodo.1495829 [DOI] [PMC free article] [PubMed]
- Blake LE, Thomas SM, Blischak JD, Hsiao CJ, Chavarria C, Myrthil M, Gilad Y, Pavlovic BJ. 2018b. A comparative study of endoderm differentiation in humans and chimpanzees. Genome Biol 19: 162 10.1186/s13059-018-1490-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blekhman R. 2012. A database of orthologous exons in primates for comparative analysis of RNA-seq data. Nat Preced hdl: 10.101/npre.2012.7054.1 [DOI] [Google Scholar]
- Blekhman R, Oshlack A, Chabot AE, Smyth GK, Gilad Y. 2008. Gene regulation in primates evolves under tissue-specific selection pressures. PLoS Genet 4: e1000271 10.1371/journal.pgen.1000271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blekhman R, Marioni JC, Zumbo P, Stephens M, Gilad Y. 2010. Sex-specific and lineage-specific alternative splicing in primates. Genome Res 20: 180–189. 10.1101/gr.099226.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brawand D, Soumillon M, Necsulea A, Julien P, Csárdi G, Harrigan P, Weier M, Liechti A, Aximu-Petri A, Kircher M, et al. 2011. The evolution of gene expression levels in mammalian organs. Nature 478: 343–348. 10.1038/nature10532 [DOI] [PubMed] [Google Scholar]
- Brodal A. 1983. The perihypoglossal nuclei in the macaque monkey and the chimpanzee. J Comp Neurol 218: 257–269. 10.1002/cne.902180303 [DOI] [PubMed] [Google Scholar]
- Cáceres M, Lachuer J, Zapala MA, Redmond JC, Kudo L, Geschwind DH, Lockhart DJ, Preuss TM, Barlow C. 2003. Elevated gene expression levels distinguish human from non-human primate brains. Proc Natl Acad Sci 100: 13030–13035. 10.1073/pnas.2135499100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain CE, Blekhman R, Marioni JC, Gilad Y. 2011. Gene expression differences among primates are associated with changes in a histone epigenetic modification. Genetics 187: 1225–1234. 10.1534/genetics.110.126177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capra JA, Erwin GD, McKinsey G, Rubenstein JL, Pollard KS. 2013. Many human accelerated regions are developmental enhancers. Philos Trans R Soc Lond B Biol Sci 368: 20130025 10.1098/rstb.2013.0025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Swofford R, Johnson J, Cummings BB, Rogel N, Lindblad-Toh K, Haerty W, Palma FD, Regev A. 2019. A quantitative framework for characterizing the evolutionary history of mammalian gene expression. Genome Res 29: 53–63. 10.1101/gr.237636.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Shendure J. 2011. Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nat Rev Genet 12: 628–640. 10.1038/nrg3046 [DOI] [PubMed] [Google Scholar]
- Dezső Z, Nikolsky Y, Sviridov E, Shi W, Serebriyskaya T, Dosymbekov D, Bugrim A, Rakhmatulin E, Brennan RJ, Guryanov A, et al. 2008. A comprehensive functional analysis of tissue specificity of human gene expression. BMC Biol 6: 49 10.1186/1741-7007-6-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enard W, Khaitovich P, Klose J, Zöllner S, Heissig F, Giavalisco P, Nieselt-Struwe K, Muchmore E, Varki A, Ravid R, et al. 2002. Intra- and interspecific variation in primate gene expression patterns. Science 296: 340–343. 10.1126/science.1068996 [DOI] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J, Hastie T, Tibshirani R. 2010. Regularization paths for generalized linear models via coordinate descent. J Stat Softw 33: 1–22. 10.18637/jss.v033.i01 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego Romero I, Pavlovic BJ, Hernando-Herraez I, Zhou X, Ward MC, Banovich NE, Kagan CL, Burnett JE, Huang CH, Mitrano A, et al. 2015. A panel of induced pluripotent stem cells from chimpanzees: a resource for comparative functional genomics. eLife 4: e07103 10.7554/eLife.07103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad Y, Mizrahi-Man O. 2015. A reanalysis of mouse ENCODE comparative gene expression data. F1000 Res 4: 121 10.12688/f11000research.16536.12681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad Y, Rifkin SA, Bertone P, Gerstein M, White KP. 2005. Multi-species microarrays reveal the effect of sequence divergence on gene expression profiles. Genome Res 15: 674–680. 10.1101/gr.3335705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The GTEx Consortium. 2017. Genetic effects on gene expression across human tissues. Nature 550: 204–213. 10.1038/nature24277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, Wen B, Wu H, Liu Y, Diep D, et al. 2011. Increased methylation variation in epigenetic domains across cancer types. Nat Genet 43: 768–775. 10.1038/ng.865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KD, Langmead B, Irizarry RA. 2012. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol 13: R83 10.1186/gb-2012-13-10-r83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KD, Sabunciyan S, Langmead B, Nagy N, Curley R, Klein G, Klein E, Salamon D, Feinberg AP. 2014. Large-scale hypomethylated blocks associated with Epstein-Barr virus–induced B-cell immortalization. Genome Res 24: 177–184. 10.1101/gr.157743.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernando-Herraez I, Prado-Martinez J, Garg P, Fernandez-Callejo M, Heyn H, Hvilsom C, Navarro A, Esteller M, Sharp AJ, Marques-Bonet T. 2013. Dynamics of DNA methylation in recent human and great ape evolution. PLoS Genet 9: e1003763 10.1371/journal.pgen.1003763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernando-Herraez I, Garcia-Perez R, Sharp AJ, Marques-Bonet T. 2015a. DNA methylation: insights into human evolution. PLoS Genet 11: e1005661 10.1371/journal.pgen.1005661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernando-Herraez I, Heyn H, Fernandez-Callejo M, Vidal E, Fernandez-Bellon H, Prado-Martinez J, Sharp AJ, Esteller M, Marques-Bonet T. 2015b. The interplay between DNA methylation and sequence divergence in recent human evolution. Nucleic Acids Res 43: 8204–8214. 10.1093/nar/gkv693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaman MW, Houck ML, Chemnick LG, Nagpal S, Chawannakul D, Sudano D, Pike BL, Ho VV, Ryder OA, Hacia JG. 2003. Comparative analysis of gene-expression patterns in human and African great ape cultured fibroblasts. Genome Res 13: 1619–1630. 10.1101/gr.1289803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik D, Barber GP, Casper J, Clawson H, Cline MS, Diekhans M, Dreszer TR, Fujita PA, Guruvadoo L, Haeussler M, et al. 2014. The UCSC Genome Browser database: 2014 update. Nucleic Acids Res 42: D764–D770. 10.1093/nar/gkt1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaitovich P, Muetzel B, She X, Lachmann M, Hellmann I, Dietzsch J, Steigele S, Do HH, Weiss G, Enard W, et al. 2004a. Regional patterns of gene expression in human and chimpanzee brains. Genome Res 14: 1462–1473. 10.1101/gr.2538704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaitovich P, Weiss G, Lachmann M, Hellmann I, Enard W, Muetzel B, Wirkner U, Ansorge W, Pääbo S. 2004b. A neutral model of transcriptome evolution. PLoS Biol 2: e132 10.1371/journal.pbio.0020132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaitovich P, Hellmann I, Enard W, Nowick K, Leinweber M, Franz H, Weiss G, Lachmann M, Pääbo S. 2005. Parallel patterns of evolution in the genomes and transcriptomes of humans and chimpanzees. Science 309: 1850–1854. 10.1126/science.1108296 [DOI] [PubMed] [Google Scholar]
- Khan Z, Ford MJ, Cusanovich DA, Mitrano A, Pritchard JK, Gilad Y. 2013. Primate transcript and protein expression levels evolve under compensatory selection pressures. Science 342: 1100–1104. 10.1126/science.1242379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36 10.1186/gb-2013-14-4-r36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MC, Wilson AC. 1975. Evolution at two levels in humans and chimpanzees. Science 188: 107–116. 10.1126/science.1090005 [DOI] [PubMed] [Google Scholar]
- Krueger F, Andrews SR. 2011. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27: 1571–1572. 10.1093/bioinformatics/btr167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, Smyth GK. 2014. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 15: R29 10.1186/gb-2014-15-2-r29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos B, Meiklejohn CD, Cáceres M, Hartl DL. 2005. Rates of divergence in gene expression profiles of primates, mice, and flies: stabilizing selection and variability among functional categories. Evolution 59: 126–137. 10.1554/04-251 [DOI] [PubMed] [Google Scholar]
- Lin S, Lin Y, Nery JR, Urich MA, Breschi A, Davis CA, Dobin A, Zaleski C, Beer MA, Chapman WC, et al. 2014. Comparison of the transcriptional landscapes between human and mouse tissues. Proc Natl Acad Sci 111: 17224–17229. 10.1073/pnas.1413624111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindskog C. 2016. The Human Protein Atlas—an important resource for basic and clinical research. Expert Rev Proteomics 13: 627–629. 10.1080/14789450.2016.1199280 [DOI] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. 2009. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462: 315–322. 10.1038/nature08514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Stephens M. 2016. Variance adaptive shrinkage (vash): flexible empirical Bayes estimation of variances. Bioinformatics 32: 3428–3434. 10.1093/bioinformatics/btw483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. 2008. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res 18: 1509–1517. 10.1101/gr.079558.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17: 10 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- Martin DI, Singer M, Dhahbi J, Mao G, Zhang L, Schroth GP, Pachter L, Boffelli D. 2011. Phyloepigenomic comparison of great apes reveals a correlation between somatic and germline methylation states. Genome Res 21: 2049–2057. 10.1101/gr.122721.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkin J, Russell C, Chen P, Burge CB. 2012. Evolutionary dynamics of gene and isoform regulation in mammalian tissues. Science 338: 1593–1599. 10.1126/science.1228186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molaro A, Hodges E, Fang F, Song Q, McCombie WR, Hannon GJ, Smith AD. 2011. Sperm methylation profiles reveal features of epigenetic inheritance and evolution in primates. Cell 146: 1029–1041. 10.1016/j.cell.2011.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowick K, Gernat T, Almaas E, Stubbs L. 2009. Differences in human and chimpanzee gene expression patterns define an evolving network of transcription factors in brain. Proc Natl Acad Sci 106: 22358–22363. 10.1073/pnas.0911376106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai AA, Bell JT, Marioni JC, Pritchard JK, Gilad Y. 2011. A genome-wide study of DNA methylation patterns and gene expression levels in multiple human and chimpanzee tissues. PLoS Genet 7: e1001316 10.1371/journal.pgen.1001316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson E, GTEx Consortium, Koller D, Battle A, Mostafavi S, Ardlie KG, Getz G, Wright FA, Kellis M, Volpi S, et al. 2015. Sharing and specificity of co-expression networks across 35 human tissues. PLoS Comput Biol 11: e1004220 10.1371/journal.pcbi.1004220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. 2015. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43: e47 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero IG, Ruvinsky I, Gilad Y. 2012. Comparative studies of gene expression and the evolution of gene regulation. Nat Rev Genet 13: 505–516. 10.1038/nrg3229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AJ, Stathaki E, Migliavacca E, Brahmachary M, Montgomery SB, Dupre Y, Antonarakis SE. 2011. DNA methylation profiles of human active and inactive X chromosomes. Genome Res 21: 1592–1600. 10.1101/gr.112680.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata Y, Sheffield NC, Fedrigo O, Babbitt CC, Wortham M, Tewari AK, London D, Song L, Lee BK, Iyer VR, et al. 2012. Extensive evolutionary changes in regulatory element activity during human origins are associated with altered gene expression and positive selection. PLoS Genet 8: e1002789 10.1371/journal.pgen.1002789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK. 2004. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3: Article3 10.2202/1544-6115.1027 [DOI] [PubMed] [Google Scholar]
- Smyth GK, Michaud J, Scott HS. 2005. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics 21: 2067–2075. 10.1093/bioinformatics/bti270 [DOI] [PubMed] [Google Scholar]
- Stephens M. 2017. False discovery rates: a new deal. Biostatistics 18: 275–294. 10.1093/biostatistics/kxw041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stergachis AB, Neph S, Sandstrom R, Haugen E, Reynolds AP, Zhang M, Byron R, Canfield T, Stelhing-Sun S, Lee K, et al. 2014. Conservation of trans-acting circuitry during mammalian regulatory evolution. Nature 515: 365–370. 10.1038/nature13972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart JM, Segal E, Koller D, Kim SK. 2003. A gene-coexpression network for global discovery of conserved genetic modules. Science 302: 249–255. 10.1126/science.1087447 [DOI] [PubMed] [Google Scholar]
- Supek F, Bošnjak M, Škunca N, Šmuc T. 2011. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6: e21800 10.1371/journal.pone.0021800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thul PJ, Lindskog C. 2018. The human protein atlas: a spatial map of the human proteome. Protein Sci 27: 233–244. 10.1002/pro.3307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankov AM, Gu H, Akopian V, Ziller MJ, Donaghey J, Amit I, Gnirke A, Meissner A. 2015. Transcription factor binding dynamics during human ES cell differentiation. Nature 518: 344–349. 10.1038/nature14233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung J, Barreiro LB, Johnson ZP, Hansen KD, Michopoulos V, Toufexis D, Michelini K, Wilson ME, Gilad Y. 2012. Social environment is associated with gene regulatory variation in the rhesus macaque immune system. Proc Natl Acad 109: 6490–6495. 10.1073/pnas.1202734109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, et al. 2015. Proteomics. Tissue-based map of the human proteome. Science 347: 1260419 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
- Villar D, Berthelot C, Aldridge S, Rayner TF, Lukk M, Pignatelli M, Park TJ, Deaville R, Erichsen JT, Jasinska AJ, et al. 2015. Enhancer evolution across 20 mammalian species. Cell 160: 554–566. 10.1016/j.cell.2015.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward MC, Wilson MD, Barbosa-Morais NL, Schmidt D, Stark R, Pan Q, Schwalie PC, Menon S, Lukk M, Watt S, et al. 2013. Latent regulatory potential of human-specific repetitive elements. Mol Cell 49: 262–272. 10.1016/j.molcel.2012.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnefors M, Eyre-Walker A. 2012. A selection index for gene expression evolution and its application to the divergence between humans and chimpanzees. PLoS One 7: e34935 10.1371/journal.pone.0034935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu NY, Hallström BM, Fagerberg L, Ponten F, Kawaji H, Carninci P, Forrest AR, The FANTOM Consortium, Hayashizaki Y, Uhlén M, et al. 2015. Complementing tissue characterization by integrating transcriptome profiling from the Human Protein Atlas and from the FANTOM5 consortium. Nucleic Acids Res 43: 6787–6798. 10.1093/nar/gkv608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Horvath S. 2005. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol 4: Article17 10.2202/1544-6115.1128 [DOI] [PubMed] [Google Scholar]
- Zhou X, Cain CE, Myrthil M, Lewellen N, Michelini K, Davenport ER, Stephens M, Pritchard JK, Gilad Y. 2014. Epigenetic modifications are associated with inter-species gene expression variation in primates. Genome Biol 15: 547 10.1186/s13059-014-0547-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.