

Abstract
Glutathione is a major redox buffer, reaching millimolar concentrations within cells and high micromolar concentrations in airways. While glutathione has been traditionally known as an antioxidant defense mechanism that protects the lung tissue from oxidative stress, glutathione more recently has become recognized for its ability to become covalently conjugated to reactive cysteines within proteins, a modification known as S-glutathionylation (or S-glutathiolation or protein mixed disulfide). S-glutathionylation has the potential to change the structure and function of the target protein, owing to its size (the addition of three amino acids) and charge (glutamic acid). S-glutathionylation also protects proteins from irreversible oxidation, allowing them to be enzymatically regenerated. Numerous enzymes have been identified to catalyze the glutathionylation/deglutathionylation reactions, including glutathione S-transferases and glutaredoxins. Although protein S-glutathionylation has been implicated in numerous biological processes, S-glutathionylated proteomes have largely remained unknown. In this paper, we focus on the pathways that regulate GSH homeostasis, S-glutathionylated proteins, and glutaredoxins, and we review methods required toward identification of glutathionylated proteomes. Finally, we present the latest findings on the role of glutathionylation/glutaredoxins in various lung diseases: idiopathic pulmonary fibrosis, asthma, and chronic obstructive pulmonary disease.
Keywords: allergic airway disease, asthma, chronic obstructive pulmonary disease, glutaredoxin, glutathiolation, glutathione, glutathione S-transferase, glutathionylation, fibrosis
GSH METABOLISM AND UTILIZATION
Glutathione is considered a major redox buffer that can exist in the thiol-reduced (GSH) and thiol-oxidized (GSSG, glutathione disulfide) forms (219). The three amino acids that constitute GSH are either derived through dietary sources or can be synthesized de novo in cases of glycine and glutamate (259, 274) and converting methionine through the transsulfuration pathway in the case of cysteine (232). GSH is metabolized through the γ-glutamyl cycle that involves six enzymes: glutamate cysteine ligase (GCL, also known as γ-glutamylcysteine synthetase), glutathione synthase (GS), γ-glutamyltranspeptidase (GGT), γ-glutamylcyclotransferase (GGCT), 5-oxoprolinase (OPLAH), and dipeptidase (DP) (Fig. 1A) (146, 151). The synthesis of GSH from the individual amino acids requires two ATP-dependent steps and two enzymes: the formation of γ-glutamylcysteine through the enzymatic reaction of GCL followed by the synthesis of GSH from γ-glutamylcysteine and glycine through the enzymatic reaction of GS. In mammals, GCL consists of two subunits, the catalytic subunit GCL-C, which is subject to feedback inhibition by GSH, and the modulatory subunit GCL-M. GGTs break down GSH to cysteinylglycine (Cys-Gly) and transfer the γ-glutamyl group to an amino acid or a peptide. GGCT then releases glutamate from the glutamyl amino acid/peptide and converts it to 5-oxoproline. Finally, OPLAH regenerates glutamate from 5-oxoproline, whereas DP breaks down Cys-Gly into individual amino acids. In addition, a separate class of cytosolic enzymes, glutathione-specific γ-glutamylcyclotransferase (ChaC)1 and ChaC2, found in the yeast Saccharomyces cerevisiae, mouse, and human, discovered more recently, can metabolize GSH directly into 5-oxoproline and Cys-Gly (118, 129). Collectively, the existence of multiple and redundant enzyme systems points to an evolutionarily conserved need for a precise glutathione level and its precursor amino acids. GSSG can be reduced to two molecules of GSH by glutathione reductase (GR) oxidizing NADPH to NADP+ (247). The pentose phosphate pathway, an offshoot of the glycolysis cascade, is a major pathway that in turn reduces NADP+ to NADPH, thereby maintaining the glutathione redox buffer (231). The reported GSH concentrations in cells are in the range of 1–11 mM (219). The GSH/GSSG ratio in the cytosol ranges from 100:1 to >500:1 in different cell types (71), while in the endoplasmic reticulum (ER), the reported GSH/GSSG ratio ranges from 1:1 to 7:1 by use of different methods of measurements (8, 169). Lack of adequate blocking, in addition to autooxidation, may result in an overestimation of GSSG values, leading to aforementioned variations in reported GSH/GSSG ratios, and these concerns continue to plague the field (73). GSH is involved in multiple cellular reactions. GSH is consumed by glutathione peroxidases to reduce oxidized lipids (27). GSH is also involved in phase II metabolism where glutathione S-transferases (GSTs) conjugate GSH to electrophiles or other compounds in detoxification processes (281). GSH is also utilized by leukotriene C4 synthase to form leukotriene C4, the parent compound of the cysteinyl leukotrienes (132). Lastly, GSH is conjugated to proteins in a process called protein S-glutathionylation (PSSG) as a protective mechanism that prevents cysteines from overoxidation and also changes the protein structure and function, reviewed in detail below (Fig. 1B).
Fig. 1.

A: reduced glutathione (GSH) metabolism. Glutamate cysteine ligase (GCL), composed of a catalytic subunit GCLC and a regulatory unit GCLM, catalyzes the formation of γ-glutamylcysteine (1). Glutathione synthetase (GS) then completes the synthesis of GSH by adding glycine to γ-glutamylcysteine (2). GSH is broken down to cysteinylglycine by γ-glutamyl transpeptidase (GGT) (3). The γ-glutamyl group can be recycled through multiple steps where it is converted to 5-oxoproline by γ-glutamyl cyclotransferase (GGCT) (4) before it is converted back to glutamate by 5-oxoprolinase (OPLAH) (5). Cysteinylglycine is broken down into cysteine and glycine by dipeptidase (DP), where they are recycled or are shunted into other metabolic pathways (6). Alternatively, glutathione-specific γ-glutamylcyclotransferase (ChaC)1 and ChaC2 break down GSH into cysteinylglycine and 5-oxoproline (7). B: GSH utilization. GSH is conjugated to 1) oxidized lipid (LOOH) and through the catalytic reaction of glutathione peroxidase (GPXs) reduces oxidized lipid to its reduced form (LOH); 2) electrophiles and other xenobiotics through the catalytic reaction of glutathione S-transferases (GSTs) in phase II drug metabolism; 3) leukotriene A4 through the action of leukotriene C4 synthase (LTC4S), forming leukotriene C4; and 4) proteins to prevent protein cysteines from undergoing irreversible oxidation. GSH is also used in the regeneration of the sulfhydryl group to its reduced state through the catalytic activity of glutaredoxins.
PSSG: CHEMISTRY, REDOX REGULATION, DETECTION, AND PREDICTION OF PSSG SITES
One mechanism whereby GSH affects cellular physiology that has emerged within the past 10–15 years is through its covalent reaction with reactive cysteines within proteins, a process known as protein S-glutathionylation (PSSG, also known as glutathiolation or protein mixed-sulfide.) Protein S-glutathionylation represents a special type of protein disulfide, wherein the disulfide bond is formed between a protein thiol and glutathione. PSSG can occur in multiple ways. Invariably, some sort of oxidative stress precedes the formation of the disulfide bond between a protein thiol group and GSH. Proteins can be directly S-glutathionylated in vitro in the presence of compounds such as GSSG, S-nitrosoglutathione (GSNO), and glutathione disulfide S-oxide (GS(O)SG) by using supraphysiological concentrations (17, 136, 168, 208). Within cells, PSSG can be induced through the inhibition of GR to artificially increase GSSG concentration (260, 267) or by directly exposing cells to oxidants such as H2O2, diamide, peroxynitrite (ONOO−), or GSSG ethyl ester (1, 124, 260). These oxidants can lead to diverse oxidations of protein thiols including the formation of sulfenic acids or nitros(yl)ated species that can be a gateway for subsequent PSSG (72, 82, 160). More relevant to normal cellular physiology are studies demonstrating that cellular redox perturbations that occur in physiological settings also lead to S-glutathionylation of target proteins. For example, activation of NADPH oxidases (NOXs), or oxidants derived from mitochondria, have been shown to promote S-glutathionylation, and a number of PSSG targets have been identified (116, 145, 255). These more recent observations demonstrate that PSSG is not merely representative of cell stress but is instead a feature of normal cellular redox homeostasis.
Early methods to detect PSSG include the use of anti-GSH antibody and 35S-labeled glutathione (37). Other avenues to detect PSSG include treatment of cells with biotinylated GSH (Bio-GSH), biotinylated GSSG (Bio-GSSG), biotinylated glutathione ethyl ester (Bio-GEE), and fluorescein isothiocyanate-labeled glutathione (FITC-GSH) (26, 133, 238). However, these modified forms of glutathione suffer the drawback of having a bulky molecule conjugated to GSH, which might reduce its accessibility to reactive protein cysteines that might be S-glutathionylated (61). Using click chemistry, another iteration of glutathione has been developed where the side chain of the glycine residue is replaced with an azide, an alkynyl, or an allyl group (61, 119, 217, 218). The subsequent S-glutathionylated proteins detected with this modified GSH are biotin labeled through click chemistry using a corresponding modified biotin such as biotin alkyne or azide-PEG3-biotin. The biotin-labeled S-glutathionylated proteins can then be enriched using streptavidin/avidin bead pulldown. These modified forms of glutathione with glycine derivatives (GSH with azido-Ala and GSH with allyl-Gly) avoid the concern of bulky molecule conjugation mentioned above. All of the aforementioned methods to detect PSSG with GSH analogs suffer from the limitation that they are added to cell culture media or are incubated with cell lysates, where they compete with endogenous glutathione and/or interfere with normal redox homeostasis.
Since the administration of modified versions of GSH cannot be used in the studies of PSSG in human tissues, alternative methods have been developed to identify protein S-glutathionylation based on specific derivatization of PSSG followed by resin-based affinity capture of the target proteins (81, 162) (Fig. 2). These resin-assisted enrichment methods take advantage of the specificity of the deglutathionylating activity of glutaredoxins (discussed below) to derivatize free thiol groups from S-glutathionylated proteins. The first step in this procedure includes cell or tissue lysis and permeabilization in the presence of a thiol-blocking agent to exclude capturing already reduced protein cysteines. Samples are thereafter incubated with GLRX to convert PSSG to proteins with newly derivatized free thiol groups. Next, these newly formed free thiol groups can be enriched directly using sulfhydryl-reactive beads such as thiopropyl sepharose beads or after incubation with modified biotins that are sulfhydryl reactive such as HPDP-biotin, which can then be enriched with streptavidin/avidin beads (70, 235). The enriched proteins are then trypsin digested and labeled with isobaric reagents such as tandem mass tag (TMT) or iTRAQ for mass spectrometry-based identification and quantification. The specific cysteine residues of a protein that are S-glutathionylated can also be identified if labeled with sulfhydryl-reactive biotins through a specific mass shift of the cysteine-containing fragment ion. In addition to these methods, our laboratory has developed an in situ method for visualizing PSSG in intact tissues. This method is applicable to paraffin-embedded sections and can reveal regional and cell-specific differences in PSSG in healthy and diseased conditions (2). Lastly, in addition to the qualitative detection of PSSG, or identification of specific PSSG targets, total PSSG can also be quantified biochemically. The quantification of total PSSG involves steps to remove free GSH/GSSG and precipitate proteins, followed by reduction to release the GSH moiety from S-glutathionylated proteins, and finally quantify GSH released (200, 256, 279). Several bioinformatics tools have been developed to predict S-glutathionylation of specific cysteine residues from a given protein that can be glutathionylated, with specificity and sensitivity ranging from 50 to 75% (180, 241, 284, 285). A web-based database, dbGSH, has also been created, where all experimentally verified S-glutathionylated proteins are manually curated (43).
Fig. 2.
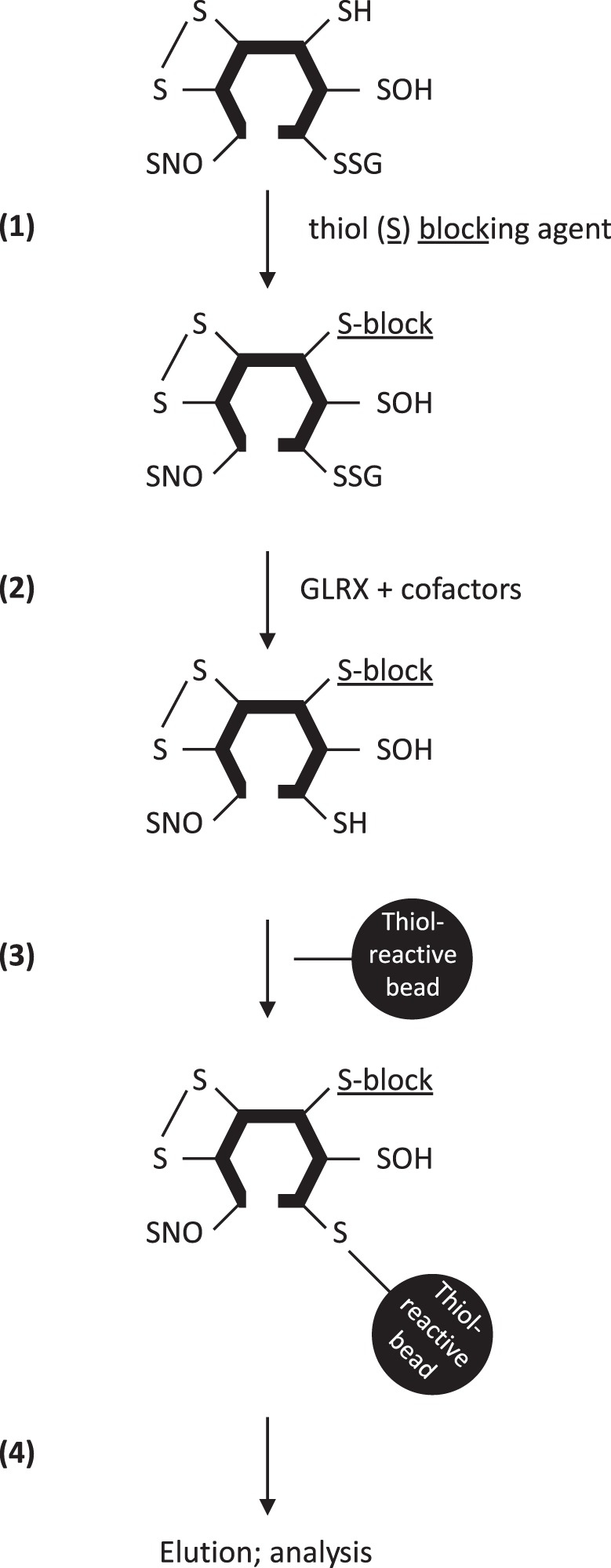
Schema summarizing the steps required to identify S-glutathionylated proteins using glutaredoxin-1 (GLRX)-based derivatization and thiol affinity capture. Existing free thiols in proteins are first blocked by thiol-blocking agents (1). S-glutathionylated cysteines are thereafter derivatized by adding GLRX and cofactors (2), resulting in the emergence of new free thiols. New thiols are enriched using thiol-reactive beads (3), and enriched proteins are eluted thereafter for further analysis (4), using mass spectrometry or Western blotting as examples. We refer the reader to the text for detailed descriptions about these procedures.
GLUTATHIONYLATION/DEGLUTATHIONYLATION CHEMISTRY AND RELEVANCE TO BIOLOGICAL SIGNALING PATHWAYS
Protein S-glutathionylation, glutaredoxins, and GSH have increasingly garnered more attention in recent years as reflected by the increasing amount of publications with glutathionylation, glutaredoxin, or glutathione as key terms. Protein S-glutathionylation/deglutathionylation has been implicated in the regulation of a multitude of biological functions and diseases analogous to other reversible posttranslational modifications of amino acids. This can be attributed to the fact that the conjugation of GSH, a three-amino acid peptide, changes a protein’s tertiary structure (166, 280). The S-glutathionylation of a protein could also affect the formation of protein complexes. For example, S-glutathionylation of histone H3 induces structural changes and leads to nucleosomal instability (69, 186, 206). S-glutathionylation of cytoskeletal proteins such as actin, tubulin, and L-plastin inhibits their polymerization and leads to altered cellular functions, such as arrested mitosis and neutrophil and monocyte polarization, chemotaxis, adhesion, and phagocytosis (42, 54, 216, 255, 258). Lastly, S-glutathionylation may serve as a signal for protein degradation. For example, the caspase-dependent 14-3-3ζ degradation under low-density lipoprotein plus high-glucose-induced metabolic stress in monocytes is increased following 14-3-3ζ glutathionylation (120). S-glutathionylation of the 20S proteasome core has been shown to increase its proteolytic activity during oxidative stress, perhaps as a coping mechanism of cells under stress (227).
PSSG regulates cellular functions in multiple ways. For example, various enzymes involved in cellular bioenergetics, such as aldolase, uncoupling proteins UCP2 and UCP3, and mitochondrial complex I have been shown to be S-glutathionylated and to inhibit their respective enzymatic activities (100, 154, 187). Multiple subunits (a1 and b1) of the Na+-K+ pump have been shown to be S-glutathionylated where the addition of the GSH adduct is at least partially modulated by its associative protein, the FXYD domain-containing ion transport regulator 3 (FXYD), resulting in decreased ATPase activity (19, 144, 145). Endothelial nitric oxide synthase (eNOS), the major isoform of NOS that regulates vascular function through the production of nitric oxide (NO), is a highly studied PSSG-regulated protein (286). eNOS produces NO in its dimer form (65), and S-glutathionylation of eNOS at Cys689 and Cys908 uncouples eNOS, switching the production of NO to O2·−, thereby disrupting the NO signaling pathway, with implications for vascular biology (41). PSSG also indirectly regulates cellular signaling pathways through the modification of protein kinases and phosphatases, thus altering the phosphorylation/dephosphorylation of target proteins (30, 31, 52, 115, 121, 161, 245, 269). For example, glutathionylation of ribosomal S6 kinase-1 inhibits its kinase activity and leads to downstream inhibition of NOS activation due to the inability of NOS to be phosphorylated (245). Another protein kinase, Ca2+/calmodulin-dependent protein kinase I (CaMKI), is shown to be inhibited by S-glutathionylation at its active-site cysteine (115). On the other hand, adenosine monophosphate-activated protein kinase (AMPK) is activated by S-glutathionylation at its AMPKα catalytic subunit (52). S-glutathionylation also modulates MAPK phosphatase-1 activity through increased proteasomal degradation (121). S-glutathionylation of Signal transducer and activator of transcription 3 (STAT3) has been demonstrated to interfere with its phosphorylation, resulting in its inactivation (30, 31, 161, 269). For example, S-glutathionylation of STAT3 at Cys328/542 inhibits STAT3 phosphorylation at Tyr705 (31) and has been correlated to increased apoptosis and decreased cell viability in alantolactone- or cynaropicrin-induced oxidative stress (30, 161).
The PSSG/glutaredoxin system also can affect other oxidative posttranslational modifications indirectly, through the control of thioredoxin (TXN) and peroxiredoxins (PRDX) redox systems, which are interconnected to control the cellular redox environment. For example, TXN has been shown to be glutathionylated at Cys72 and lead to the inhibition of its insulin disulfide reductase activity (37). TXN can be inactivated following oxidation of noncatalytic site cysteines, leading to the formation of a two-disulfide form of TXN that is inactive. The GLRX system allows regeneration of active TXN via a monothiol mechanism, described below (53). PRDX can be inactivated following their overoxidation, involving the formation of sulfinic (-SO2H) and sulfonic acid (-SO3H) residues (57). S-glutathionylation has been shown to protect PRDX from overoxidation by preventing the formation of sulfinic acid species, allowing rapid regeneration of reduced active PRDX following GLRX-mediated restoration of the SH group (192). Of special note is the importance of S-glutathionylation of PRDX6, a 1-Cys peroxiredoxin. Glutathione S-transferase pi (GSTP) has been shown to be critical in the catalysis of S-glutathionylation of PRDX6, increasing the accessibility of the otherwise buried sulfenic acid or sulfenylamide forms of PRDX6, allowing subsequent regeneration of its catalytic activity through the GLRX/GSH system (156, 202).
DISCOVERY AND BIOCHEMISTRY OF GLUTAREDOXINS
Glutaredoxin (GLRX) was first described in the 1950s–60s (175, 199). The enzyme was called “transhydrogenase”, as it was thought that hydrogen was transferred from two GSH molecules to reduce the disulfide bond. It was later demonstrated that this enzyme catalyzes deglutathionylation in a two-step reaction where a thiol-disulfide exchange between GSH and the protein disulfide bond occurs in the first step (9) (Fig. 3A). The name “thioltransferase” was thus suggested to more accurately describe this protein. Independently, an enzyme was discovered in Escherichia coli that acted as an alternate hydrogen donor to the thioredoxin system for ribonucleoside diphosphate reductase (97, 99). The author, Arne Holmgren, suggested the name “glutaredoxin”, as glutathione supplied the reducing power in the reaction. Glutaredoxins/thioltransferases were subsequently discovered in other mammals and other kingdoms (11, 89, 102, 134, 214). The name “thioltransferase” fell out of preference, and the enzyme has been known as glutaredoxin since.
Fig. 3.

Human glutaredoxin-1 (GLRX) primary sequence, 3D structure, and deglutathionylation mechanism. A: GLRX catalyzes deglutathionylation. GLRX deglutathionylates proteins and is glutathionylated in the process (1). GLRX is regenerated by consuming GSH, which is in turn regenerated through the reduction of oxidized glutathione (GSSG) by glutathione reductase (GR) with NADPH as the final electron donor (2). B: the 5 cysteines that comprise human GLRX are indicated. Note that only Cys23 is required for deglutathionylation activity. Cys8, Cys79, and Cys83 have been linked to oxidative inactivation of GLRX.
GLRXs are generally known as enzymes that catalyze deglutathionylation activity (to be reviewed in a later section.) However, GLRXs have also been demonstrated to be promoting protein glutathionylation on rare occasions (17, 138, 195, 211, 230). GLRXs belong to the thioredoxin protein superfamily that catalyzes disulfide bond formation and share a distinct “thioredoxin fold” structural motif that consists of three α-helices flanking a four-stranded β-sheet (150, 158). GLRXs can be divided into two classes, the monothiol glutaredoxins such as human GLRX3 (PICOT) and GLRX5 with an active site amino acid sequence of CGFS (111, 263), and the dithiol glutaredoxins such as human GLRX1–2 with an active site sequence of C(P/S)(Y/F/D)C (152, 183). The dithiol glutaredoxins were originally classified as disulfide reductases that catalyze the disulfide reduction of certain proteins and other small molecule oxidation such as hydroxyethyl disulfide (HED) and S-sulfocysteine (Cys-SO3) (12, 98, 141, 153, 157, 194, 262). GLRXs catalyze deglutathionylation in a ping-pong mechanism whereby the first step involves the nucleophilic displacement of the GSH moiety by the active site cysteine, followed by the rate-limiting step where the thiolate ion of the active site cysteine is regenerated by consuming one molecule of GSH (228) (Fig. 3A). GLRX’s specificity toward the γ-linkage-glutamyl moiety in both the γ-linkage-glutamyl moiety of glutathionylated proteins and free GSH is conferred by the GSH binding groove not found in other members of the thioredoxin superfamily (29, 239, 276). GLRXs contain a varying number of cysteines, ranging from two cysteines in E. coli GLRX (101), to five cysteines in human GLRX (183) (Fig. 3B). The NH2-terminal active site cysteine of the active site motif (Cys23 of human GLRX) is required for both disulfide bond reduction and deglutathionylation (67, 68, 270, 271). This cysteine differs from the COOH-terminal active site cysteine (Cys26) in that Cys23 is exposed to the exterior of the protein and is solvent accessible, whereas Cys26 is buried and is inaccessible to solvent (117, 266). The NH2-terminal active site cysteine has an unusually low pKa value of ∼3.5, contributing to the formation of thiolate ion (67, 165, 271). Several factors contribute to the low pKa, including the ion-pair interactions between the conserved positively charged residue and the thiolate ion (108, 266, 271), the interaction of the thiolate ion and the electrostatic field of the α-helix by the helix dipole (123) and the proline residue immediately after the NH2-terminal active site cysteine (66, 110).
The functional relevance of other cysteines in deglutathionylation catalysis varies among species. In E. coli GLRX, the COOH-terminal active site cysteine contributes to the deglutathionylation reaction, as cysteine-to-serine mutation significantly reduces deglutathionylation activity (28, 215). In contrast, mammalian GLRX requires only the active site NH2-terminal cysteine for the deglutathionylation, as the mutation of individual or all four cysteines other than active site Cys23 does not change the deglutathionylating activity (117, 268, 270, 271). Mutation of the COOH-terminal active site cysteine to serine (Cys26Ser in human GLRX) actually increases the enzymatic activity of GLRX (268, 271).
Several methods exist to measure the enzymatic activities of glutaredoxins. In the ribonucleotide reductase-coupled assay, the disulfide reduction activity is measured by the formation of [3H]dCDP from [3H]CDP, as glutaredoxin is required for GSH-dependent reduction of oxidized ribonucleotide reductase to catalyze the reduction of ribonucleotides to deoxyribonucleotides (96–99). The more commonly used assay to measure deglutathionylating activity is the NADPH-coupled assay using substrates such as HED or S-sulfocysteine, where the glutaredoxin activity is measured by the consumption of NADPH (11, 66–68, 96, 134, 165, 228). Other methods to more directly measure the deglutathionylation activity also exist, for example, peptides with glutathionylated cysteine adjacent to a tryptophan residue (189, 215), fluorescent eosin-glutathione or eosin-glutathionylated BSA (48, 111), or 35S-glutathionylated proteins (114). Each assay has its limitations. The ribonucleotide reductase-coupled assay is not suitable to measure the deglutathionylation activity selectively, as the assay relies on the disulfide bond reduction of the oxidized ribonucleotide reductase and, as such, measuring general disulfide bond reduction instead of deglutathionylation. Most substrates used in the NADPH-coupled assay are small molecules that do not represent GSH bound to bulky biological protein cysteines. Furthermore, monothiol glutaredoxins do not have any measurable enzymatic activity using the HED/NADPH-coupled assay, whereas the deglutathionylation activity of monothiol glutaredoxins has been documented in vivo (246, 278). Using 35S-glutathionylated proteins from whole cells, GLRX was demonstrated to preferentially deglutathionylate proteins of lower molecular weights (114). Thus, it is important to consider carefully which activity assay to utilize when investigating the biological relevance of glutaredoxin activity.
HUMAN GLUTAREDOXINS
Glutaredoxin-1
Human glutaredoxin-1 (GLRX) is primarily localized to cytosol and is encoded by 321 bases that translate to 106 amino acid residues, with the active site CPYC motif. The pKa of the active site cysteine (Cys23) is ∼3.5 (165), which is similar to the active site cysteine of GLRX in other mammals. The human gene encoding GLRX was mapped onto the chromosome arm 5q (178). The TATA box and the CCAAT sequence are located at 30 and 160 bp, respectively, upstream of the transcription start point (184). The promoter region contains 69 predicted transcription factor binding sites, including transcription factors implicated in apoptosis (c-Jun), cytokine production (NF-κB1, RelA), cytokine response (STAT1, STAT5), and redox sensing (AP-1) (Table 1) (60, 164, 184). Consequently, a number of growth factors and cytokines have been shown to affect GLRX mRNA expression. Notably, exposure to the profibrogenic growth factor, transforming growth factor-β1 (TGF-β1) led to decreased GLRX mRNA in mammary, tracheal and alveolar epithelial cells. Conversely, GLRX mRNA was increased in tracheal epithelial cells exposed to interferon-γ (IFN-γ) (135, 188, 209) or activation of the NF-κB pathway (4).
Table 1.
Transcription factors predicted to bind to HsGLRX promoter region
| AP-1 | GRα | POU2F1 |
| AP-2α | Grβ | POU2F2 |
| AR | HNF-1A | PPARα:RXRα |
| ATF-1 | HNF-1B | PRα |
| ATF3 | HNF-1C | PRβ |
| c-Ets-1 | HNF-3α | PU.1 |
| c-Ets-2 | HNF-4α | PXR-1:RXRα |
| c-Jun | HOXD10 | RARβ |
| c-Myb | HOXD9 | RBP-Jκ |
| C/EBPα | IRF-1 | RelA |
| C/EBPβ | IRF-2 | RXRα |
| CTF | LEF-1 | SRY |
| E2F-1 | MEF-2A | STAT1β |
| EBF | NF-1 | STAT4 |
| Egr-3 | NF-AT1 | STAT5A |
| ELF-1 | NF-AT1 | T3Rβ1 |
| Elk-1 | NF-AT2 | TCF4 |
| ENKTF-1 | NF-κB1 | TCF4E |
| ERα | NF-Y | TFII-I |
| FOXP3 | NFI/CTF | TFIID |
| GATA-1 | p53 | VDR |
| GATA-2 | Pax-5 | XBP-1 |
| GR | PEA3 | YY1 |
Prediction was done using PROMO, a web-based program for transcription factor binding site prediction (http://alggen.lsi.upc.es). GLRX, glutaredoxin-1; Hs, Homo sapiens; AP, activator protein 1; AR, androgen receptor; ATF, activating transcription factor; C/EBP, CCAAT box enhancer-binding protein; CTF, CCAAT box-binding transcription factor; E2F, E2 transcription factor; EBF, early β-cell factor; Egr, early growth response protein; ELF, E74-like factor; Elk-1, ETS like-1 protein; ENKTF, enkephalin transcription factor; ER, estrogen receptor; FOXP, forkhead box P3; GATA, GATA-binding protein; GR, glutathione reductase; Gr, glucocorticoid receptor; HNF, hepatocyte nuclear factor; HOXD, homeobox D; IRF, IFN regulatory factor; LEF, lymphoid enhancer-binding factor; MEF, mouse embryo fibroblast; NF, nuclear factor; NF-AT, nuclear factor of activated T cells; Pax, paired-box protein; PEA, polyoma enhancer activator; POU2F2, Pit-Oct-Unc family transcription factor 2F2; PR, progesterone receptor; PU.1, PU box transcription factor; PXR, pregnane X receptor; RXR, retinoid X receptor; RAR, retinoic acid receptor; RBP, retinol-binding protein; RelA, Rel-like domain-containing; SRY, sex-determining region Y; STAT, signal transducer and activator of transcription; T3R, thyroid hormone receptor; TCF4, transcription factor 4; TF, transcription factor; VDR, vitamin D receptor; XBP, X-box binding protein; YY1, yin yang 1.
Human GLRX is sensitive to oxidative stress. Incubation of GLRX with GSSG, H2O2, or ONOO− was shown to significantly decrease its enzymatic activity (88, 196, 229). Incubation of GLRX with GSSG also resulted in a mixture of oligomers, glutathionylation at noncatalytic cysteines, and disulfide formation between noncatalytic cysteines (88, 240). Lastly, Cys8 is solvent exposed, and Cys8Ser mutation prevents its aggregation (179). These results suggest a mechanism whereby structural cysteines regulate GLRX through oxidation processes. The physiological relevance of GLRX regulation through these cysteines remains to be fully understood. As will be described below, S-glutathionylation of GLRX, at presumably noncatalytic cysteines, has been linked to its oxidative inactivation in settings of fibrotic lung disease (6).
Glutaredoxin-2
Human glutaredoxin-2 (GLRX2) has three isoforms, GLRX2a (mitochondrial) and the nuclear and cytosolic GLRX2b and GLRX2c (147, 152). GLRX2 is composed of either 164 (mitochondrial isoform; GLRX2a) or 165 amino acids (nuclear isoform; GLRX2b), with a molecular mass of 18–19 kDa. The gene encoding GLRX2 was mapped on to chromosome arm 1q and contains five exons (exons 1a, 1b, 2, 3, and 4) (147, 152). Both isoforms contain exons 2, 3, and 4 where the nuclear translocation signal is located. Alternative splicing of exon 1b gives rise to isoforms 2b and 2c (147), whereas the mitochondrial form includes an additional exon 1a, which contains the mitochondrial translocation signal (74, 152). GLRX2 contains the conserved active site motif, with an amino acid sequence of CSYC and the thioredoxin fold, and shares ~36% sequence identity with GLRX (74). GLRX2 contains four cysteines, the two cysteines at the active site, and two others (Cys28 and Cys113) that form a disulfide bond (109). GLRX2 is a Fe/S cluster-binding protein. The dimeric holo GLRX2 consists of two GLRX2 monomers, a 2Fe/2S cluster and two GSH molecules and is coordinated through the active site Cys37 (18, 109). GLRX2 has also been demonstrated to catalyze deglutathionylation, although its specific activity of deglutathionylation is 10-fold less than that of GLRX (152). Interestingly, holo GLRX2 is unstable in an aerobic environment or in the presence of oxidants and can regain the “lost” enzymatic activity, suggesting a redox-sensing property of GLRX in addition to its role as an additional pool of deglutathionylating enzymes in response to oxidative stress (139, 140, 265).
Glutaredoxin-3 (PICOT)
Glutaredoxin-3 (GLRX3) was originally discovered as a cytosolic, protein kinase Cθ (PKCθ)-interacting protein, and the original name is PICOT (PKC-interacting cousin of thioredoxin) (263). Unlike GLRX and GLRX2, GLRX3 is a multidomain, monothiol glutaredoxin. GLRX3 contains a thioredoxin domain but not the conserved thioredoxin active site motif. Mammalian GLRX3 has two GLRX domains, with a conserved CGFS motif. The TXN-like domain is sufficient for GLRX3-PKCθ interaction, although it is unclear whether it is sufficient for the inhibitory effect GLRX3 has exhibited toward PKCθ (263). Similar to GLRX2, GLRX3 is a Fe/S cluster-binding protein (90). Holo GLRX3 is a homodimer, with two Fe/S clusters, with the active site cysteines (Cys159 and Cys216) at each GLRX domain contributing to the binding of respective Fe/S clusters. Like GLRX2, the GLRX3 holo complex is also unstable in aerobic conditions or in the presence of oxidants such as GSNO (90). It is currently unclear whether GLRX3 exhibits any deglutathionylating activity.
Glutaredoxin-5
Glutaredoxin-5 (GLRX5) is a single-domain monothiol glutaredoxin with 157 amino acids and a molecular mass of 16.6 kDa. The active site motif has a sequence of CGFS. GLRX5 possesses deglutathionylating activity, although the activity is multiple folds less than that of GLRX2 when glutathionylated BSA is used as the substrate (111). The GLRX5 holo complex is composed of four GLRX5 monomers, two Fe/S clusters, and four GSH molecules, with each Fe/S cluster binding coordinated by the active site cysteines (Cys67) from two GLRX5 monomers and two GSH molecules (111). GLRX5 translocates to mitochondria and plays an important role in Fe/S cluster biogenesis and cellular iron homeostasis, based on findings that ablation of GLRX5 led to a loss of cytosolic Fe and a concomitant mitochondrial Fe overload (33, 210, 273).
OTHER ENZYME SYSTEMS IMPLICATED IN S-GLUTATHIONYLATION AND DEGLUTATHIONYLATION REACTIONS
In addition to GLRX, a number of other enzymes have emerged as critical regulators of protein glutathionylation and deglutathionylation. These include members of the family of GSTs, which were originally described as enzymes that facilitate the conjugation of GSH to xenobiotics as part of phase II drug metabolism (25). More recently, GSTs have become recognized for their role in promoting PSSG, with GSTP being the prototypical S-glutathionylating enzyme (248, 281). In addition, glyoxalases (GLOs), which convert α-ketoaldehydes into D-hydroxyacids, also have been implicated in the catalysis of S-glutathionylation (59). Sulfiredoxin is an oxidoreductase that catalyzes the reduction of cysteine sulfinic acids of the active site cysteines of 2-cys PRDXs (21, 39). Sulfiredoxin has been demonstrated to catalyze deglutathionylation (32, 62, 185). Lastly, TXNs are oxidoreductases that catalyze protein disulfide reduction that share thioredoxin fold with glutaredoxins (149, 158). TXNs have also been shown to deglutathionylate GAPDH, PRDX3, the 20S proteasome, and eNOS by use of a dithiol reduction mechanism that is independent of GSH (16, 86, 226, 236). Due to space limitations, we will not further describe these other enzyme systems here.
DYSREGULATION OF THE S-GLUTATHIONYLATION/GLUTAREDOXIN REDOX AXIS: IMPLICATION FOR LUNG DISEASES
The lung, due to its biological function as the organ for gas exchange, is exposed to oxygen more than any other internal organ. It is estimated that we breathe in 10,000–20,000 liters of air daily (212). As a result, the lung experiences insults not just from oxidants derived from endogenous sources such as inflammatory cells or NADPH oxidases, mitochondria, etc., but also from exogenous sources, including cigarette smoke (discussed below), ozone, or other oxidant gases (13, 212). Oxidative stress has been linked to lung diseases for many years now, and the precise “redox mechanisms” that regulate lung function and/or pathophysiology are only slowly being teased apart owing to improved methodologies to detect oxidant-induced modifications. Similarly, the role of PSSG chemistry and glutaredoxins in the pathophysiology of lung diseases is only now emerging. In the next paragraphs we discuss relevant studies pertaining to PSSG/glutaredoxins in idiopathic pulmonary fibrosis, asthma, and chronic obstructive pulmonary disease.
IDIOPATHIC PULMONARY FIBROSIS
Idiopathic pulmonary fibrosis (IPF) is a lung disease with progressive scarring in the lung that is defined by the histopathological pattern of usual interstitial pneumonia with unknown etiology (159). The prevalence of IPF ranges from 6 to 10 per 100,000 people, with a median survival of 3–5 years after the initial diagnosis. Age is the greatest risk factor identified, with the disease being slightly predominant in males. At the cellular level, disruption of type 2 alveolar epithelial cells (AEC2s; cells that secrete surfactant to maintain alveolar structure) homeostasis has been shown to play important roles in IPF, as AEC2s in IPF lungs are found to be senescent, with genomic instability, telomere attrition, and mitochondrial dysfunction (122). A role of epithelial cells in the pathogenesis of IPF also has emerged, based on observations in patients with familial IPF-harboring mutations in the surfactant protein C (SFTPC) or ATP binding cassette member A3 (ABCA3) genes. SFTPC and ABCA3 are uniquely expressed in epithelial cells, and mutations in these genes induce endoplasmic reticulum (ER) stress and mitochondrial perturbations and culminate in pulmonary fibrosis (34, 177). Myofibroblasts, cells transdifferentiated from fibroblasts that secrete various growth factors, cytokines, and extracellular matrix during tissue repair, are also disproportionately activated, resulting in aberrant deposition of extracellular matrix proteins (122) (Fig. 5).
Fig. 5.

Summary of how an altered glutaredoxin-1/protein S-glutathionylation (GLRX/PSSG) status affects idiopathic pulmonary fibrosis (IPF), asthma, and chronic obstructive pulmonary disease (COPD). The main pathophysiological features that accompany these diseases are represented graphically. The text below summarizes what is currently known about GLRX status in these diseases. Potential PSSG targets are also indicated. Please note that many of these were derived from rodent models or cell-based studies with disease-relevant stimuli, but the majority will need to be validated in human specimens. As is described in the text, S-glutathionylated proteomes in human tissues also remain to be unraveled. Nonetheless, the proteins that were found to be S-glutathionylated are believed to have substantial implications for a number of processes and pathways that are listed and are known to control the pathogenesis of these chronic respiratory diseases. [Created with BioRender.]
Oxidative stress has been linked to the development of pulmonary fibrosis (44). However, the precise mechanisms whereby redox perturbations contribute to disease pathogenesis remain unclear, and, thus far, approaches that use the nonspecific low-molecular-weight thiol and glutathione precursor N-acetyl-l-cysteine (NAC), failed to show therapeutic benefit in patients with IPF (105). Epithelial cell death and the death receptor Fas (CD95) have been recognized as key drivers of pulmonary fibrosis. For instance, it has been demonstrated that administering FasL into mouse lungs causes epithelial cell apoptosis with an accompanying development of lung fibrosis (83, 130, 131). The amount of GSH has been documented to be significantly less in the epithelial lining fluids from IPF patients, indicating a change of redox environment and the potential role of protein S-glutathionylation (35). We have shown previously the importance of glutathionylation of Cys294 of Fas (Fas-SSG) for epithelial cell death (5) and that protein disulfide isomerase-3 (PDIA3) and GSTP both contributed to Fas-SSG in the ER. Fas-SSG promoted its trafficking to the surface, increased the assembly of the death-inducing signaling complex (DISC) and binding of FasL, leading to enhanced cell death (7) (Fig. 5). The interaction between GSTP and Fas was increased in lungs of IPF patients, with concomitant increased Fas-SSG, and similar findings were apparent in lungs from mice with bleomycin- or adenovirus-expressing active transforming growth factor-β1 (AdTGFβ)-induced fibrosis (163). The increase in Fas-SSG was attenuated in Gstp1−/− mice following treatment with TLK117, a selective GSTP inhibitor, in bleomycin- and AdTGFβ-induced mouse models of pulmonary fibrosis (163). The importance of PSSG in the pathology of pulmonary fibrosis is further substantiated by observations that, overall, PSSG was increased in the lungs from mice with bleomycin- or AdTGFβ-induced lung fibrosis, and in lungs of IPF patients (6, 163). Findings that Gstp1−/− mice or mice treated with TLK117 have attenuated collagen content and decreased overall PSSG also suggest the importance of PSSG in fibrogenesis (163). The increase in Fas-SSG was accompanied by caspase-3/8-dependent degradation of GLRX, sustaining the increases in Fas-SSG. Accordingly, epithelial cells lacking Glrx displayed more Fas-SSG and were more prone to FasL-induced cell death (5). Importantly, Glrx−/− mice were more susceptible to bleomycin/AdTGFβ-induced fibrosis, with exacerbated collagen content in the lungs and increased overall PSSG compared with their wild-type counterparts (6). Conversely, the overexpression of Glrx specifically in epithelial cells or direct airway administration of recombinant GLRX into the airways attenuated Fas-SSG, reduced caspase-3 activity, and decreased the collagen content in mice with existing bleomycin/AdTGFβ-induced fibrosis, accompanied by increased collagenase activity (6). Although the exact mechanisms whereby the collagen degradation activity was restored/enhanced remain unknown, our data showed an uptake of recombinant GLRX by epithelial cells and cells that resemble macrophages (6). Interestingly, Glrx mRNA expression increased in monocytes stimulated with IL-6 to induce monocyte-to-macrophage differentiation in association with enhanced phagocytosis (244). Increases in PSSG have been linked to macrophage dysfunction (10). Our work has demonstrated that absence of Glrx impaired lipopolysaccharide (LPS)-induced macrophage activation and phagocytosis (3). Taken together, these observations suggest that the impact of GLRX on macrophage function may be important in regulating tissue fibrogenesis.
The increases in overall PSSG in lungs of IPF patients were accompanied by a decrease in GLRX activity. Levels of GLRX protein and mRNA in IPF lungs were slightly decreased compared with lungs from healthy subjects. Moreover, GLRX protein in lungs from IPF patients was significantly S-glutathionylated (6). The same observation was made in the mouse model of bleomycin-induced lung fibrosis. S-glutathionylation of GLRX was reduced in mice administered recombinant GLRX, in association with an overall increase in GLRX activity in these lungs. Lastly, isolation of GLRX from IPF lungs followed by dithiothreitol reduction restored its activity (6). Collectively these findings suggest that endogenous GLRX is inactivated by its own substrate, glutathione, presumably due to oxidation of cysteines outside of the thioredoxin domain, leading to runaway glutathionylation (Fig. 5).
As mentioned above, IPF is a disease associated with aging, with age being the greatest risk factor for IPF development (159). Examination of 18- to 24-mo-old mice revealed decreased GLRX activity and enhanced susceptibility to bleomycin-induced lung fibrosis compared with young adult control groups (6). Three splice variants of human GLRX (A–C; see Fig. 4 for EST) exist, and analysis of the gene expression data of all splice variants of GLRX together, in lungs from nondiseased humans, as a function of age, revealed no difference between age groups (6). However, separate analysis of the individual splice variants of GLRX revealed an increase in expression of GLRX variant A with age, whereas GLRX variant B was decreased in lung tissues as a function of age, and expression GLRX splice variant C did not change with age (Fig. 4A). In contrast to these divergent patterns of expression of GLRX with age, GLRX2, GLRX3, and GLRX5 showed weak but statistically significant trends toward decreasing mRNA expression in lungs from older age groups (Fig. 4B). Mitochondrial dysfunction has been well linked to aging and pulmonary fibrosis, where decreased expression of PTEN-induced kinase-1 leads to dysfunction in electron transport chain, reduced mitophagy, and increased apoptosis (170). Although the glutathionylated proteome of IPF lungs and epithelial cells remains unknown, mitochondrial complex I and UCP2/3 have been demonstrated to be regulated by S-glutathionylation (154, 187) (Fig. 5). Thus, the reduced expression of GLRX2 in older age groups may increase S-glutathionylation of mitochondrial complex I and/or UCP2/3 and potentially as of yet-unknown mitochondrial proteins and contribute to mitochondrial dysfunction. Examination of GLRX expression in lung tissues from patients with IPF compared with controls revealed that GLRX2 and GLRX3 mRNA increased in patients with IPF compared with the nondisease control group, in contrast to the previously detected decreases in GLRX, whereas GLRX5 mRNA decreased slightly (Fig. 4C). All together, reduced overall deglutathionylation capacity caused by reduced expression of glutaredoxins with age might lower the “threshold” level of protein S-glutathionylation, rendering older age groups with increased susceptibility to the onset of pulmonary fibrosis. Increases in GLRX2 and GLRX3 mRNA in lungs from patients with IPF may reflect compensatory, adaptive responses that are nonetheless insufficient to protect against the development and/or progression of disease.
Fig. 4.

Glutaredoxins transcript levels in lung tissue from patients with lung diseases and different age groups. A: glutaredoxin-1 (GLRX) isoform-specific expression in the lung as a function of age. Splice variant A: ENST00000379979.4; Splice variant B: ENST00000237858.6; splice variant C: ENST00000512469.2. Expression of splice variants A and B demonstrates significant correlation with age, with no significant change occurring for splice variant C (n = 25, 20–29; n = 21, 30–39; n = 60, 40–49; n = 112, 50–59; n = 97, 60–69; n = 5, 70–79). Statistical significance was calculated using Welch’s t test comparing age bins 20–39, 40–59, and 60–79. *P < 0.05; ** P < 0.01. B: GLRX2, GLRX3, and GLRX5 transcripts evaluated as a function of age in nondiseased human lungs. Spearman correlation analysis revealed a significant negative correlation between age and expression of GLRX2, GLRX3, and GLRX5, with ρ values indicated in the figure. Data represent 342 normal lung specimens, 236 derived from male donors aged 21 to 70 yr (black dots), and 106 derived from female donors aged 21 to 70 yr (red dots). C: interstitial lung disease (ILD) and chronic obstructive pulmonary disease (COPD). GLRX transcripts were assessed using publicly available microarray gene expression data from the Lung Genomics Research Consortium (LGRC; http://www.lung-genomics.org) for ILD patients with a diagnosis of idiopathic pulmonary fibrosis (I; n = 255), COPD (CO; n = 219), and nondiseased control tissues (C; n = 137) (data are available in the Gene Expression Omnibus database accession no. GSE47460). [GLRX transcripts were previously reported (6) and are reformatted here for consistency]. Asthma: GLRX transcripts were assessed from publicly available RNA-seq data from RNA isolated from nasal brushing of asthmatic patients (As; n = 53) compared with healthy subjects (C; n = 97). [The full description of RNA isolation and processing, and processing of the raw RNA-seq data are published elsewhere (181)]. Results are shown as average ± SE. Statistical significance was calculated using a Mann-Whitney test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. A and B: RNA-seq data are from the GTExPortal v.6 (A) or v.7 (B), were analyzed (accession no. phs000424.v7.p2, 06/2018) after exclusion of patients with lung diseases (58). The Genotype-Tissue Expression (GTEx) Project is supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by the National Cancer Institute, National Human Genome Research Institute, National Heart, Lung, and Blood Institute, National Institute on Drug Abuse, National Institute of Mental Health, and National Institute of Neurological Disorders and Stroke.
ASTHMA
Asthma is a complex lung disorder characterized by chronic airway inflammation and remodeling (Fig. 5). The diagnosis of asthma requires symptoms including wheeze, shortness of breath, chest tightness, cough, together with reversible airflow obstruction and/or airway hyperresponsiveness (95). Risk factors include environmental allergens such as dust mites, viral infection, air pollution, tobacco smoke, and obesity. Type 2 inflammation is found in 50% of adult with asthma, where TH2 or innate lymphoid cells (ILCs) secrete cytokines such as IL-5 and IL-13 and is closely associated with eosinophilia (182). Separately, a subgroup of asthma patients is known to have high levels of IL17 (TH17-high asthma) in association with neutrophilic inflammation, steroid resistance and hard-to-control severe disease (91, 106). Structurally, the airways of people with severe asthma undergo substantial remodeling, marked by increased airway smooth muscle mass, thickening of the basement membrane, and subepithelial fibrosis (95). Stiff mucus plugs, described below, are also found in airways of severely asthmatic people, contributing to chronic airflow obstruction (55). Epithelial cells play important roles in shaping the immune response (85). Epithelial cells express pattern recognition receptors that, upon ligand binding, release proinflammatory mediators that recruit, activate, and/or promote differentiation and survival of immune cells, including dendritic cells, T cells, neutrophils, and eosinophils. Epithelial cell-derived IL-25, IL-33, and thymic stromal lymphopoietin (TSLP), the “alarmins”, induce type 2 responses that, among other processes, enhance release of IL-13, thus promoting goblet cell hyperplasia and mucus hypersecretion (85, 91).
A large number of studies have been conducted to examine changes in the GSH redox status in patients with asthma by examining total glutathione, GSH, and GSSG in lung lining fluid, bronchoalveolar lavage (BAL), induced sputum, and exhaled breath condensates or systemically via the analysis of glutathione in whole blood, plasma, or erythrocytes. These studies were conducted in children as well as adults with varying degrees of asthma, in some cases after challenge with allergen or ozone or after exacerbations and used different methods to collect the clinical sample and/or to detect glutathione. It is therefore not surprising that reported values vary tremendously, with variable changes reported, resulting in an incomplete picture of how the GSH/GSSG redox couple is affected in asthma and how this relates to the endotypes of asthma, age, and/or medication use (63). Intriguingly, corticosteroids, the mainstay therapy for managing inflammation in asthmatic patients, decrease levels of glutathione (87, 191). Therefore, prior medication use, or the aforementioned concerns about the requirements to chemically protect thiols in order to prevent oxidation of GSH and other oxidation reactions, might be a factor that contributed to these variable observations.
In settings where total levels of GSH were shown to be increased in asthmatic patients, this has been interpreted as being indicative of a response to increased oxidative stress in asthmatic patients (207). Indeed, a number of enzymes critical in the synthesis and metabolism of GSH are regulated by the oxidative stress-responsive transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2, encoded by the NFE2L2 gene), which in murine models of asthma has been strongly linked to protection against disease (137, 204, 242). One study of patients with severe asthma demonstrated that, despite increases in NFE2L2 mRNA and NRF2 protein, NRF2 activation and downstream targets of NRF2, including glutathione-dependent enzymes, were not different between asthmatic and control groups, in association with decreases in levels of GSH and cysteine (64). Dysregulation of the Nrf2 pathway in severe asthma may be linked to decreases in GSH levels in those patients and may explain the aforementioned fluctuations in GSH between the different studies (63), although this scenario requires further investigation. Additional studies in mouse models of allergic airway disease have corroborated a role for glutathione redox homeostasis. Notably, mice lacking Ggt, one of the enzymes that degrades GSH, as described earlier, have increases in GSH levels in the lung lining fluid and decreases in intracellular GSH. GGT-deficient mice showed attenuated allergic airway disease in response to IL-13 (148), and similar results were reported using a GGT inhibitor (254). Furthermore, depletion of glutathione with buthioneine sulfoximine (BSO) worsened allergen-induced airway reactivity and inflammation, while conversely, NAC dampened the responses to allergens in association with modulation of glutathione pools and markers of oxidative stress (174). Glutathione S-transferases have also been strongly implicated in the pathogenesis of asthma, and polymorphic variants of a number of GSTs have been reported in patients with asthma, with variable correlations to disease severity (49, 193). Similarly, glutathione peroxidases also have been shown to be affected in patients with asthma or rodent models of disease (15, 46, 47, 63).
A role for PSSG chemistry and glutaredoxins in settings of asthma is currently emerging. Analysis of sputum samples from asthmatic patients revealed an increased level of GLRX and a decrease in PSSG compared with controls. A concomitant increase in GLRX mRNA was detected in bronchial epithelial cells isolated from asthmatic patients compared with controls (128). In contrast to those observations, RNA isolated from nasal brushing of asthmatic patients showed a decrease in mRNA expression of all GLRXs (Fig. 4C) (181), perhaps reflecting the different sites of sampling. Based on these limited observations, it remains unclear to date how GLRX status relates to asthma endotypes. Studies using mouse models of allergic airway disease show that Glrx mRNA and protein levels in lung tissues were increased in response to exposure to ovalbumin (Ova) or house dust mites (HDM), whereas in response to Ova, Glrx2 mRNA remained unchanged. Despite increases in GLRX in these settings, PSSG was also increased, showing that the increases in GLRX were not sufficient to normalize the extent of PSSG within the time frame of investigation (93, 94, 209). A kinetic assessment of GLRX, GSH, and PSSG in airways and lung tissues from mice subjected to the Ova model showed that GLRX protein increased transiently in BAL fluid 6 h after the final Ova challenge and decreased in a time-dependent manner while remaining significantly higher than baseline level 8 days post-challenge. Coinciding with this were increases in PSSG within the airway epithelium and increases in GSH levels in the BAL, collectively suggesting that the GLRX and PSSG status changes rapidly in airways in response to challenge with antigens in a sensitized host. The increases in GLRX levels in the BAL were found to positively correlate with increases in cytokines, suggestive of a link between GLRX and airway inflammation. In contrast to the acute increases in BAL, increases in GLRX in lung tissues persisted (94, 155).
To directly address a causal link between GLRX and allergic airway disease, we have evaluated mice that globally lack Glrx in the Ova and HDM models of disease. Glrx−/− mice on a Balb/C background initially developed inflammation, mucus metaplasia, and increases in airway hyperresponsiveness in response to Ova, similar to WT littermates. However, Glrx−/− mice resolved the disease faster compared with the respective WT groups (94), suggesting that the GLRX/PSSG axis regulates the duration/resolution of allergic airway disease. As HDM is a more relevant antigen than Ova (a large proportion of asthmatic patients is allergic to HDM), using a HDM model of airway sensitization and challenge, our laboratories demonstrated a change in patterns of inflammatory profiles and cytokine secretion in Balb/C Glrx−/− mice in response to HDM, with a decrease of airway eosinophils and Th2 cytokines, HDM-specific IgE, an increase in airway macrophages and neutrophils, along with an increase in TH17 cytokine IL-17, compared with respective WT groups. Re-stimulation of single-cell suspensions, derived from the lung or spleen, with HDM led to an increase in IL-17 production and a decrease in IL-5 in Glrx−/− mice, consistent with a shift in T cell polarization (93, 94). In addition to these observations, Glrx−/− mice displayed altered profiles of airway hyperresponsiveness, with increases in central airway resistance, yet dampened peripheral elastance in response to HDM, compared with littermates. These findings indicate that GLRX and PSSG affect T cell polarization and patterns of airway hyperresponsiveness.
The NF-κB pathway plays a central role in immune response and asthma, with downstream target genes including cytokines, chemokines, apoptosis regulators, and inflammatory mediators (282). LPS is a key activator of the NF-κB pathway via activation of the Toll-like receptor 4 (TLR4) pathway. Importantly, TLR4 activation on airway epithelial cells is also a critical step in the initiation of allergic airway disease induced by HDM (84). Work from our laboratory and that of others has demonstrated the importance of epithelial NF-κB in the pathogenesis of allergic disease models of asthma (220, 252). In the canonical pathway, activation of inhibitory κB kinase (IKK), the holoenzyme consisting of IKKα, IKKβ, and NF-κB essential modulator (NEMO), leads to phosphorylation and polyubiquitination of IκB subunit, its degradation, and the release of p50/RelA for nuclear translocation, whereas in the noncanonical pathway IKKα and p52/RelB dictate the transcriptional output, and both of these pathways are engaged in response to LPS stimulation of lung epithelial cells (253). PSSG/glutaredoxin have been shown to regulate the NF-κB pathway at multiple levels. We have demonstrated that glutathionylation of IKKβ Cys179 inhibited its kinase activity, resulting in reduced RelA nuclear translocation and decreased levels of macrophage inflammatory protein 2 (MIP-2) and keratinocyte-derived chemokine (KC) in epithelial cells with LPS stimulation (208). Conversely, NF-κB activation led to transcriptional activation of GLRX, which is important in enhancing activation of NF-κB and LPS-induced proinflammatory responses in association with decreases in S-glutathionylation of IKKβ (4). We also demonstrated that GSTP constitutively interacted with the inhibitor of NF-κB, IκBα, in control cells and that after LPS stimulation this interaction was lost, whereas at later time points, when NF-κB activation decreased, GSTP showed an increased interaction with IKKβ in association with its S-glutathionylation (112) (Fig. 5). This scenario is suggestive of a multifaceted role of GSTP in the regulation of NF-κB, prevention of activation of NF-κB in the absence of stimulus through protein-protein interaction, and regulatory mechanism involving GSTP-mediated S-glutathionylation to shut down NF-κB activation. In a model of LPS-induced lung inflammation, Glrx−/− mice had attenuated secretion of IL-1β, IL-6, granulocyte-macrophage colony-stimulating factor (GM-CSF), and TNF-α but increased secretion of KC (3). Although the composition of the inflammatory profile in Glrx−/− mice remained the same, alveolar macrophages lacking Glrx appeared smaller than their wild-type counterparts. LPS stimulation of alveolar macrophages isolated from Glrx−/− mice had attenuated NF-κB activation, as demonstrated by decreased levels of RelB, IκBα, and IKKα, decreased RelA nuclear translocation, and decreased secretion of TNF-α and IL-6 (3).
Given the increases in IL-17 in lungs from HDM-exposed Glrx−/− mice, and the importance of IL-17 in the regulation of airway barrier function and remodeling, our laboratories assessed whether S-glutathionylation affected the responses of epithelial cells to IL-17 stimulation. Exposure of epithelial cells to IL-17 induced IKKα-mediated NF-κB activation, and glutathionylation of RelA and IKKα were observed. Epithelial cells lacking Glrx showed enhanced glutathionylation of RelA and IKKα, as well as decreased expression of the proinflammatory mediators KC and C-C ligand 20 (CCL20) mRNA, although, paradoxically, an increase in expression of IL-6. Absence of Glrx also led to a more robust increase in A20, a negative regulator of the NF-κB pathway, suggesting that S-glutathionylation contributed to cessation of some components of NF-κB signaling in IL-17-stimulated epithelial cells (176).
PSSG/glutaredoxins have also been implicated in the regulation of other adaptor proteins and downstream transcription factors that are relevant in allergen-induced immune responses. Notably, TNF receptor-associated factors (TRAFs) have been shown to be glutathionylated (40, 75). TRAF6 is an adaptor protein critical in transducing signals by members of the IL-1 receptor/TLR (IL-1R/TLR) family. TRAF6 was shown to be constitutively glutathionylated in various cell types and was deglutathionylated following stimulation with IL-1 (40). GLRX knockdown attenuated IL-1R1/TLR4-induced NF-kB activation, as TRAF6-SSG inhibited its polyubiquitination and failed to recruit and activate the transforming growth factor-β1 activation kinase-1 (TAK1) complex (40). In macrophages, herpes simplex virus infection led to ROS-dependent S-glutathionylation of TRAF3 (Cys455) and TRAF6 (Cys390), critical to the activation of NF-κB and secretion of IFN-α/β). Mutation of these cysteines, or overexpression of GLRX, decreased these herpes simplex virus-induced responses, suggesting an activating role of TRAF3/TRAF6 glutathionylation in the innate immune responses (75). Interestingly expression of a TRAF6 Cys390 mutant did not affect the ability of IL-1β to activate NF-kB (75), in contrast to the aforementioned inhibitory role of TRAF6 S-glutathionylation in IL-1β signaling (40), perhaps reflecting differences in cell types or sites of S-glutathionylation between these two studies. Furthermore, the adaptor protein MyD88 adaptor-like (MAL or TIRAP), important in TLR signaling, was also shown be glutathionylated, at Cys91, in response to LPS treatment of macrophages. MAL mutations (C91A and H92P), which are resistant to glutathionylation, resulted in dampened recruitment of and interaction with MyD88 and displayed decreased NF-κB activation (104), consistent with an activating role of Cys91 S-glutathionylation in proinflammatory signaling. All together, these findings point to an activating role of TRAF/MAL S-glutathionylation in TLR/IL-1-induced proinflammatory signaling (Fig. 5).
Dimethyl fumarate (DMF) is a compound with electrophilic properties that has garnered increasing attention after its FDA approval for the treatment of patients with multiple sclerosis (23). Relevant to the scope is this paper are findings demonstrating that DMF dampens HDM-induced allergic airway inflammation (107). DMF also has been anecdotally reported to reduce asthma symptoms and to improve quality of life of asthma patients (221). DMF decreased TNF-α-mediated NF-κB activation and cytokine secretion in primary human airway smooth muscle cells (221). Furthermore, in the same model, DMF reduced intracellular GSH and induced IκBα glutathionylation (IκBα-SSG), which in turn was associated with inhibition of IκBα degradation, NF-κB p65 nuclear entry and NF-κB/DNA binding, and attenuated cytokine responses induced by TNF-α (222).
As stated earlier, IL-33 is a key alarmin that is rapidly secreted from airway epithelial cells stimulated with a range of allergens or other insults. IL-33 contributes to type 2 inflammatory responses in asthma. It has been demonstrated that IL-33 secretion from airway epithelial cells occurs via a redox-based mechanism that requires the H2O2-producing enzyme dual oxidase-1 (DUOX) (103). A role of GLRX and PSSG in the response to IL-33 has become apparent recently in studies conducted in macrophages. Although in WT macrophages LPS led to increases in mRNA expression of IL33, this response was diminished in macrophages lacking GLRX, in association with enhanced S-glutathionylation of TRAF6, inhibiting downstream IKKβ and NF-κB activation, events required for the upregulation of IL33. Intriguingly, exposure of macrophages to recombinant IL-33 also induced IL33 mRNA expression, demonstrative of feed-forward signaling, which is also inhibited by GLRX knockdown. The importance of GLRX in IL-33 signaling was further corroborated using an intratracheal cockroach antigen model of allergic airway disease, wherein increases in GLRX protein were observed, analogous to previously described studies involving Ova or HDM. Intriguingly, GLRX induction in response to cockroach was attenuated in lungs from mice lacking IL33, demonstrating a requirement not only for GLRX in the induction of IL-33 but also a role for IL-33 signaling in the induction of GLRX in settings of allergen exposure (261), findings that collectively suggest a role for GLRX and PSSG in the regulation of type 2 responses in asthma through the regulation of the key alarmin IL-33.
Beside the aforementioned targets of S-glutathionylation important in the regulation of proinflammatory signals, asthma-relevant cytokines themselves, notably IL-1β, IL-6, and IL-17 are direct targets of S-glutathionylation (283). In particular, the activity of IL-1β was demonstrated to be inhibited following overoxidation of Cys188 to sulfinic and sulfonic acids. Importantly, S-glutathionylation of Cys188 protected IL-1β from oxidative inhibition and contributed to sustain the biological activity of IL-1β in vivo. Furthermore, GLRX was shown to be present and active in the extracellular space and to physiologically regulate IL-1β glutathionylation (283) (Fig. 5).
CHRONIC OBSTRUCTIVE PULMONARY DISEASE
Chronic obstructive pulmonary disease (COPD) is a progressive lung disease characterized by poorly reversible airway obstruction, in association with obstruction of the small airways (linked to mucus hypersecretion and fibrosis around the small airways), and emphysema, exemplified by a loss of alveolar integrity and airspace enlargement (14, 197) (Fig. 5). The global prevalence of COPD is estimated to be around 174 million, with ~3 million deaths attributed to it (197). COPD is currently the third leading cause of death worldwide (14). Inhalation of particulate matter and pollutants has been closely linked to the pathogenesis of COPD, with cigarette smoking the leading risk factor (14, 171, 197). However, additional factors are believed to contribute the risk of COPD development, including genetic factors. As described above in familial IPF, COPD tends to cluster in families, suggesting that there is significant heritability of COPD (14). The mechanisms that drive COPD remain incompletely understood, but the disease is associated with chronic inflammation of the peripheral airways and parenchyma, characterized by increases in diverse cell types including neutrophils, macrophages, and lymphocytes. Chronic exposure to inhaled pollutants leads to activation of pattern recognition receptors, triggering an innate immune response and activation of epithelial cells that are important in mucus hypersecretion. An adaptive immune response characterized by an increased presence of TH1, TH17, and CD8+ cytotoxic T cells, has also been described in patients with COPD. COPD-associated inflammation is frequently resistant to treatment with corticosteroids. Patients with COPD can have acute exacerbations triggered by viral or bacterial infections, and frequent exacerbations are linked to a poor prognosis. COPD airways have been shown to be colonized by Hemophilus influenzae and Streptococcus pneumoniae (14). Like IPF, emphysema is believed to be caused by accelerated aging of the lung parenchyma owing to the decreased function of sirtuins (201, 272), enhanced telomere shortening (172), increased cellular senescence (14, 251), stem cell exhaustion, and decreases in autophagy (198). Proteases derived from neutrophils, macrophages, and epithelial cells, including neutrophil elastase, matrix metalloproteinase-9 (MMP9), and MMP12 have been linked to mucus hypersecretion, extracellular matrix degradation, impaired bacterial clearance, enhanced bacterial exacerbations, and chronic inflammation (92, 171, 257). Neutrophil elastase is a major factor implicated in the pathogenesis of COPD. Administration of elastase into airways of rodents is sufficient to induce emphysema (237). α1-Antitrypsin (A1AT) is a major circulating inhibitor of neutrophil elastase (223), and importantly, humans with a genetic deficiency of A1AT are prone to the development of emphysema (50).
Oxidative stress has been considered a major driving mechanism in the development of COPD (14). This notion does not come as a surprise, as inhaled pollutants such as cigarette smoke contain a myriad of oxidants (56). Subsequent activation of epithelial cells and innate immune cells by inhaled pollutants triggers activation of NADPH oxidases, further contributing to a redox imbalance. Numerous studies have evaluated GSH in chronic smokers and COPD patients. Cigarette smoke directly reacts with GSH, leading to GSH depletion and adduct formation, notably with acrolein and crotonaldehyde (205). Although GSH levels initially decrease in response to cigarette smoke, GSH levels subsequently rebound leading to elevated levels in the lung lining fluid (76). Levels of GSH were shown to be increased in the epithelial lining fluid and BAL fluid from chronic smokers compared with nonsmokers (173), and this increase in GSH was correlated to an increased neutrophil count and increased myeloperoxidase activity (143). GSH levels were also increased in blood and sputum of smokers and COPD patients (20). These increases in GSH reflect adaptive responses, involving NRF2, to combat the disrupted redox environment (77), and, consistent with this notion, mice lacking NRF2 are more prone to the development of emphysema (203). As was stated earlier, COPD is a disease associated with aging. Of interest are findings that older individuals were shown to be deficient in the adaptive GSH response after exposure to cigarette smoke. More importantly, in a follow-up in vivo study by the same group, depletion of GSH in young mice to levels of GSH found in aged individuals led to an accelerated enlargement of airspace after exposure to cigarette smoke, along with increases in PSSG, inflammatory mediators, and elastase and MMP activities (77), strongly suggesting that defects in GSH synthesis or metabolism or increases in S-glutathionylation contribute to the development of emphysema. Indeed, it has been suggested that differences in lung glutathione metabolism that occur between rodent species may account for the variations in rodent susceptibility in elastase-induced emphysema development (24).
These and other observations have prompted a large number of clinical trials since the 1970s involving the small thiol compound and GSH precursor, NAC as potential therapeutic for COPD, reporting varying outcomes (22, 78, 250, 287, 289). However, meta-analysis of clinical studies involving NAC, and the related thiol compounds erdosteine and carbocysteine in COPD patients, illuminated a therapeutic effect of these compounds on acute exacerbations in COPD patients (38, 213, 224). It is of relevance to note that these compounds were evaluated with a putative mechanism of action as a mucolytic. Indeed, oxidation of cysteine thiol groups in mucin polymers contributes to mucus gel stiffening in the lung (277), and small thiol reductants, such as NAC, at high concentrations have the potential to reduce mucins, leading to their increased solubility. Tenacious mucus, found in plugged airways of people with asthma, has been linked to severe disease in association with formation of oxidants derived from eosinophil peroxidase, leading to persistent crosslinks (55), unlikely to be resolved with the doses of NAC, erdosteine, or carbocysteine that are clinically achievable. It remains unclear whether mucins are targets for S-glutathionylation and whether this impacts its biophysical/biological properties.
The importance of GLRX and PSSG in the pathogenesis of COPD is only now emerging. Slight decreases in GLRX protein were observed in lung lysates from COPD patients. GLRX was found to be predominantly expressed in macrophages and was decreased in patients with severe COPD compared with controls. Interestingly, the percentage of GLRX-positive macrophages directly correlated with lung function. GLRX protein was also detected in sputum supernatants, and levels were increased in COPD patients undergoing acute exacerbations, compared with nonsmoker controls (190). On the basis of these findings, it is tempting to speculate that GLRX expression in macrophages exerts a protective function and that its secretion may promote disease exacerbation. Assessment of transcript levels of GLRX mRNAs in lungs from patients with COPD showed that all GLRX mRNAs remained unchanged (Fig. 4C), pointing to a need for single-cell transcriptomics to evaluate GLRX in macrophages and other cell types from patients with COPD. In addition to the putative role of GLRX/GSSG in macrophages, GLRX/PSSG play a role in epithelial cell death in response to cigarette smoke. Exposure of lung epithelial cells to cigarette smoke extract led to a decrease in GLRX mRNA and protein levels and an increase in PSSG and cell death. In addition, cigarette smoke itself, or acrolein, an electrophile present in cigarette smoke, irreversibly oxidized GLRX, with a concomitant decrease in activity, showing that cigarette smoke affects GLRX at multiple levels (Fig. 5). Overexpression of GLRX afforded protection from cigarette-smoke-induced cell death while conversely, absence of GLRX enhanced the susceptibility of epithelial cells to cell death, in association with respective decreases and increases in S-glutathionylation (127). Paradoxically, cigarette smoke exposure in mice led to decreases in PSSG in lung tissues compared with the controls, in association with a decrease in free thiols and increases in carbonylation, suggestive of a decrease in reversible oxidation induced by cigarette smoke (126). A subsequent study by the same group reported increases in PSSG in the BAL fluid or lavaged cells (125), indicating discordant regulation of PSSG in different regions in the lung or airways after exposure to cigarette smoke. Increases in PSSG in BAL fluid induced by cigarette smoke were enhanced in Glrx−/− mice compared with WT animals, in association with altered numbers of inflammatory cells (increases in macrophages and decreases in neutrophils, lymphocytes, and dendritic cells) and decreases in proinflammatory cytokines (125). Macrophages and tracheal epithelial cells isolated from Glrx−/− mice exposed to cigarette smoke extract also had attenuated secretion of KC (125). In a separate study, GLRX protein expression in lung tissue was shown to be decreased in response to cigarette smoke exposure. Those authors also reported an increase in airway neutrophils and proinflammatory cytokines in Glrx−/− mice exposed to cigarette smoke (45), in contrast to the previous study (125), possibly due to the different time of evaluation post-cigarette smoke exposure. As was described above, IKKs are key regulatory kinases in the NF-κB pathway important for proinflammatory signaling, and S-glutathionylation of both IKKα and IKKβ have been reported (176, 208). Intriguingly, in Glrx−/− mice exposed to cigarette smoke, NF-κB activity, evaluated via phosphorylation and acetylation of RelA/p65, was enhanced compared with WT mice, in association with enhanced expression of IKKα and decreases in expression of IKKβ. Although S-glutathionylation of both IKKα and IKKβ were observed in these settings, phosphorylation of IKKα was enhanced (45). These findings point to discordant regulation of IKKs via the GLRX/PSSG axis, although additional studies will be required to elucidate the relative importance of S-glutathionylation of these kinases in regulating the proinflammatory responses of cigarette smoke (Fig. 5).
The mechanistic details whereby PSSG/GLRX regulate the pathophysiology of COPD remain unclear, and S-glutathionylation targets remain to the explored. As was mentioned above, A1AT deficiency leads to emphysema. Notably, A1AT has been shown to be prone to oxidation (249), and a number of oxidative modifications of A1AT have been shown, including S-glutathionylation (79), sulfenic acid formation (80) and nitrosylation of Cys232 (167), and oxidation of Met351/358 (36, 243) in association with alterations in or loss of protease inhibitor function. Consequently oxidation-resistant versions of A1AT have been created (225, 288) with the goal to provide improved therapeutic responses in patients with A1AT deficiency who receive weekly A1AT injections. A1AT can be taken up from the circulation and can protect against endothelial cell apoptosis, in association with binding to cysteine-dependent proteases, notably caspases (223). It remains unclear whether this process is affected via S-glutathionylation.
In addition to the aforementioned role of S-glutathionylation in promoting cigarette smoke-induced death of epithelial cells and affecting macrophage activation, other cell types are also affected via S-glutathionylation. Notably, PSSG and glutaredoxin regulate the biology of neutrophils, including their chemotactic migration, adhesion, and phagocytosis and bacterial clearance capacity. Glutathionylation/deglutathionylation of actin and L-plastin, a β-actin-bundling protein, regulate actin polymerization in neutrophils (54, 216). Specifically, actin and L-plastin S-glutathionylation inhibited actin polymerization and led to defects in pseudopod formation which in turn impaired migration toward chemoattractant and decreased phagocytic activity (54, 216). Glrx-deficient murine neutrophils showed impaired recruitment to sites of inflammation and reduced bactericidal capability (216). Neutrophil extracellular traps (NETs) are formed upon their degranulation and consist of granule proteins and DNA, forming net-like configurations that are critical in trapping and killing of bacteria. It has been demonstrated that NET formation was increased in patients with severe COPD in association with enhanced innate immune responses, more frequent exacerbations, and a loss of microbiota diversity (51, 264). Interestingly, due to defects in actin polymerization associated with increases in actin S-glutathionylation, neutrophils lacking Glrx also fail to degranulate or release DNA, leading to defects in NET formation. Actin Cys374 was identified as the target of actin glutathionylation, actin polymerization, and NET formation. Based on these findings, GLRX has been suggested to be “a master regulator of actin cytoskeleton rearrangement and DNA release” (233). Respiratory pathogens, including those that colonize the COPD airways, have been shown to evade NETs, and various mechanisms of NET evasion have emerged (234). Intriguingly expression of bifunctional peroxiredoxin-glutaredoxin, and catalase in nontypeable Hemophilus influenzae (NTHI) 86-028NP promote its resistance to oxidants, increase survival within neutrophil extracellular traps, and are determinants of bacterial persistence during chronic/recurrent NTHI infections (113). Collectively, these findings demonstrate not only the importance of PSSG/GLRX in the regulation of NET formation but also the importance of similar redox-based processes within microbes in regulating their persistence and resistance to antibiotics (Fig. 5).
Besides the regulation of the actin cytoskeleton, PSSG also regulates neutrophil migration through the glutathionylation of S100A9 (also known as MRP14) and α4-integrin (142, 275). S100A9 was shown to be glutathionylated in response to phorbol myristate acetate (PMA) or ionomycin stimulation. The S100A8/A9-SSG complex decreased neutrophil adhesion to fibronectin (142), with potential implications for neutrophil migration. Neutrophil α4-integrin plays an integral role in retention and release of neutrophils from bone marrow via binding to the ligand vascular cell adhesion molecule 1 (VCAM-1). Glutathionylation of α4-integrin increases the binding of neutrophil-associated α4-integrin to VCAM-1 on human endothelial cells. Furthermore, in an E. coli-induced acute peritonitis model, absence of GLRX dramatically elevated α4-glutathionylation and subsequently enhanced neutrophil egress from the bone marrow. Conversely, intravenous administration of recombinant GLRX inhibited α4-integrin glutathionylation and resulted in decreases of blood neutrophils (275).
SUMMARY AND FUTURE DIRECTIONS
Oxidative stress has been linked to myriad diseases, including chronic lung diseases. However, clinical trials using generic antioxidants, like NAC, as potential therapeutics have produced no or modest effects and have not resulted in a widely accepted FDA-approved treatment regimen for patients with chronic lung diseases. These failures occur in the face of a plethora of studies convincingly demonstrating the functional relevance of redox-based pathways in essentially every biological process. It is now clear that oxidants are not merely damaging molecules and in fact evoke orchestrated biological responses, in part due to targeted oxidation of protein cysteine residues, notably protein cysteine S-glutathionylation, reviewed herein. It is therefore conceivable that redox-based strategies have the potential to be powerful in the treatment of chronic lung diseases but have to be redesigned to be more specific, to prevent off-target effects, and to allow beneficial redox-based signals to occur. In this sense, protein S-glutathionylation and deglutathionylation are not dissimilar to phosphorylation and dephosphorylation reactions. Analogous to the specificity of phosphorylation and dephosphorylation reactions, general kinase or phosphatase inhibitors with off-target effects are replaced by designer drugs targeting specific phosphorylation sites in specific protein targets. Similar strategies are envisioned to combat adverse redox-based reactions. The protein S-glutathionylation/GLRX axis presents an ideal area of focus as this mechanism of redox-based signaling has emerged as a critical regulatory system that affects a number of processes relevant to chronic lung diseases (Fig. 5). A number of signaling molecules (TRAF3/6, STAT3, IKKα, IKKβ, IkBa), structural proteins (actin, L-plastin, S100A9, α4 integrin), mitochondrial proteins (UCP2 and -3, mitochondrial complex 1), the death receptor Fas, the protease inhibitor A1AT, and cytokines (IL-1β, IL-6, IL-17) are targets for S-glutathionylation and therefore have the potential to affect cell death, migration, mitochondrial function, release of alarmins, innate and adaptive immune responses, epithelial and immune cell activation, and protease activities. The aforementioned processes accompany IPF, severe asthma, and COPD. Therefore, additional studies to corroborate the exact PSSG targets in these diseases will be critical. This review focused on studies performed predominantly in mouse models and cell lines exposed to disease-relevant stimuli and stresses. The extent to which findings obtained from such studies bear relevance to humans with these disorders will necessitate translational studies to validate these PSSG targets. Indeed, S-glutathionylated proteomes remain to be discovered in specimens from patients, and the targets highlighted herein for the most part remain to be validated in human tissues. Recent advances in detection methods and mass spectrometry quantification techniques, as well as bioinformatic tools for predictions of glutathionylation sites, provide improved opportunities to identify glutathionylated proteins. In addition, the development of single-cell technologies such as single-cell RNA-seq and proteomics could aid in deciphering the roles that glutathione and glutaredoxins play in cell and tissue homeostasis. Most studies described herein have used mouse models that globally lack the Glrx gene. A recently developed conditional Glrx-deficient mouse model (unpublished observations) will enable the elucidation of the role of GLRX and indirectly, PSSG, within specific cell types, and these approaches will be informative toward an improved understanding of PSSG/GLRX in chronic lung diseases. Although GLRX2, GLRX3, and GLRX5 expressions are modified in lungs from patients with IPF and nasal brush obtained cells from asthmatics, and their expressions decrease in lung with aging, a setting where COPD and IPF predominate, GLRX2, 3 and 5 remain unstudied in these diseases to date. Therefore, investigation of these GLRXs, along with other redox enzymes described briefly, will be critical to gain an improved understanding about the processes that govern S-glutathionylation. Similarly, an improved understanding about the fate of GSH itself (for example its transport into the airways, degradation, formation of GSNO and GSSG), in settings of chronic diseases also will be critical, as GSH status governs PSSG chemistry and GLRX activity. Despite these current limitations, findings of diminished activity of GLRX in lungs from patients with IPF, coupled to the reversal of fibrosis in diverse mouse models following direct administration of GLRX into the lungs (6), offer an exciting platform for elucidation of mechanisms of action whereby GLRX exerts its protective function and offer an exciting opportunity for development of new therapeutics. Targeting redox-based chemistry in chronic lung disease may, in fact, be a treasure trove for drug development.
DISCLOSURES
Y. M. W. J-Heininger and V. Anathy hold patents: United States Patent No. 8,679,811: “Treatments Involving Glutaredoxins and Similar Agents” and United States Patent No. 8,877,447: “Detection of Glutathionylated Proteins”. Y. M. W. J-Heininger and V. Anathy are scientific founders of Celdara Medical LLC and have received consulting fees from Celdara Medical LLC. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
GRANTS
This work was supported by the National Institutes of Health Grant Nos. R35HL135828 (to Y. M. W. Janssen-Heininger); R01HL137268, R01HL085646, R01HL138708, and R21AG055325 (to A. van der Vliet); R01HL122383, R01HL141364, and RO1HL136917 (to V. Anathy); F31 HL144051 (to E. A. Elko); and HL076122 (to A. M. Manuel).
AUTHOR CONTRIBUTIONS
S.B.C., E.A.E., and D.J.S. prepared figures; S.B.C. drafted manuscript; S.B.C., E.A.E., R.A., A.M.M., C.v.d.W., J.E.D., J.v.d.V., D.J.S., V.A., C.G.I., Y.-W.L., A.v.d.V., and Y.M.W.J.-H. edited and revised manuscript; S.B.C., E.A.E., R.A., A.M.M., C.v.d.W., J.E.D., J.v.d.V., D.J.S., V.A., C.G.I., Y.-W.L., A.v.d.V., and Y.M.W.J.-H. approved final version of manuscript.
REFERENCES
- 1.Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schöneich C, Cohen RA. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med 10: 1200–1207, 2004. doi: 10.1038/nm1119. [DOI] [PubMed] [Google Scholar]
- 2.Aesif SW, Anathy V, Havermans M, Guala AS, Ckless K, Taatjes DJ, Janssen-Heininger YM. In situ analysis of protein S-glutathionylation in lung tissue using glutaredoxin-1-catalyzed cysteine derivatization. Am J Pathol 175: 36–45, 2009. doi: 10.2353/ajpath.2009.080736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aesif SW, Anathy V, Kuipers I, Guala AS, Reiss JN, Ho YS, Janssen-Heininger YM. Ablation of glutaredoxin-1 attenuates lipopolysaccharide-induced lung inflammation and alveolar macrophage activation. Am J Respir Cell Mol Biol 44: 491–499, 2011. doi: 10.1165/rcmb.2009-0136OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aesif SW, Kuipers I, van der Velden J, Tully JE, Guala AS, Anathy V, Sheely JI, Reynaert NL, Wouters EF, van der Vliet A, Janssen-Heininger YM. Activation of the glutaredoxin-1 gene by nuclear factor κB enhances signaling. Free Radic Biol Med 51: 1249–1257, 2011. doi: 10.1016/j.freeradbiomed.2011.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anathy V, Aesif SW, Guala AS, Havermans M, Reynaert NL, Ho YS, Budd RC, Janssen-Heininger YM. Redox amplification of apoptosis by caspase-dependent cleavage of glutaredoxin 1 and S-glutathionylation of Fas. J Cell Biol 184: 241–252, 2009. doi: 10.1083/jcb.200807019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anathy V, Lahue KG, Chapman DG, Chia SB, Casey DT, Aboushousha R, van der Velden JLJ, Elko E, Hoffman SM, McMillan DH, Jones JT, Nolin JD, Abdalla S, Schneider R, Seward DJ, Roberson EC, Liptak MD, Cousins ME, Butnor KJ, Taatjes DJ, Budd RC, Irvin CG, Ho YS, Hakem R, Brown KK, Matsui R, Bachschmid MM, Gomez JL, Kaminski N, van der Vliet A, Janssen-Heininger YMW. Reducing protein oxidation reverses lung fibrosis. Nat Med 24: 1128–1135, 2018. doi: 10.1038/s41591-018-0090-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anathy V, Roberson E, Cunniff B, Nolin JD, Hoffman S, Spiess P, Guala AS, Lahue KG, Goldman D, Flemer S, van der Vliet A, Heintz NH, Budd RC, Tew KD, Janssen-Heininger YM. Oxidative processing of latent Fas in the endoplasmic reticulum controls the strength of apoptosis. Mol Cell Biol 32: 3464–3478, 2012. doi: 10.1128/MCB.00125-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Appenzeller-Herzog C. Glutathione- and non-glutathione-based oxidant control in the endoplasmic reticulum. J Cell Sci 124: 847–855, 2011. doi: 10.1242/jcs.080895. [DOI] [PubMed] [Google Scholar]
- 9.Askelöf P, Axelsson K, Eriksson S, Mannervik B. Mechanism of action of enzymes catalyzing thiol-disulfide interchange. Thioltransferases rather than transhydrogenases. FEBS Lett 38: 263–267, 1974. doi: 10.1016/0014-5793(74)80068-2. [DOI] [PubMed] [Google Scholar]
- 10.Asmis R, Qiao M, Rossi RR, Cholewa J, Xu L, Asmis LM. Adriamycin promotes macrophage dysfunction in mice. Free Radic Biol Med 41: 165–174, 2006. doi: 10.1016/j.freeradbiomed.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 11.Axelsson K, Eriksson S, Mannervik B. Purification and characterization of cytoplasmic thioltransferase (glutathione:disulfide oxidoreductase) from rat liver. Biochemistry 17: 2978–2984, 1978. doi: 10.1021/bi00608a006. [DOI] [PubMed] [Google Scholar]
- 12.Axelsson K, Mannervik B. General specificity of cytoplasmic thioltransferase (thiol:disulfide oxidoreductase) from rat liver for thiol and disulfide substrates. Biochim Biophys Acta 613: 324–336, 1980. doi: 10.1016/0005-2744(80)90087-X. [DOI] [PubMed] [Google Scholar]
- 13.Bargagli E, Olivieri C, Bennett D, Prasse A, Muller-Quernheim J, Rottoli P. Oxidative stress in the pathogenesis of diffuse lung diseases: a review. Respir Med 103: 1245–1256, 2009. doi: 10.1016/j.rmed.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 14.Barnes PJ, Burney PG, Silverman EK, Celli BR, Vestbo J, Wedzicha JA, Wouters EF. Chronic obstructive pulmonary disease. Nat Rev Dis Primers 1: 15076, 2015. doi: 10.1038/nrdp.2015.76. [DOI] [PubMed] [Google Scholar]
- 15.Barnett SD, Buxton ILO. The role of S-nitrosoglutathione reductase (GSNOR) in human disease and therapy. Crit Rev Biochem Mol Biol 52: 340–354, 2017. doi: 10.1080/10409238.2017.1304353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bedhomme M, Adamo M, Marchand CH, Couturier J, Rouhier N, Lemaire SD, Zaffagnini M, Trost P. Glutathionylation of cytosolic glyceraldehyde-3-phosphate dehydrogenase from the model plant Arabidopsis thaliana is reversed by both glutaredoxins and thioredoxins in vitro. Biochem J 445: 337–347, 2012. doi: 10.1042/BJ20120505. [DOI] [PubMed] [Google Scholar]
- 17.Beer SM, Taylor ER, Brown SE, Dahm CC, Costa NJ, Runswick MJ, Murphy MP. Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins: implications for mitochondrial redox regulation and antioxidant DEFENSE. J Biol Chem 279: 47939–47951, 2004. doi: 10.1074/jbc.M408011200. [DOI] [PubMed] [Google Scholar]
- 18.Berndt C, Hudemann C, Hanschmann EM, Axelsson R, Holmgren A, Lillig CH. How does iron-sulfur cluster coordination regulate the activity of human glutaredoxin 2? Antioxid Redox Signal 9: 151–157, 2007. doi: 10.1089/ars.2007.9.151. [DOI] [PubMed] [Google Scholar]
- 19.Bibert S, Liu CC, Figtree GA, Garcia A, Hamilton EJ, Marassi FM, Sweadner KJ, Cornelius F, Geering K, Rasmussen HH. FXYD proteins reverse inhibition of the Na+-K+ pump mediated by glutathionylation of its beta1 subunit. J Biol Chem 286: 18562–18572, 2011. doi: 10.1074/jbc.M110.184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Biljak VR, Rumora L, Cepelak I, Pancirov D, Popović-Grle S, Sorić J, Grubisić TZ. Glutathione cycle in stable chronic obstructive pulmonary disease. Cell Biochem Funct 28: 448–453, 2010. doi: 10.1002/cbf.1675. [DOI] [PubMed] [Google Scholar]
- 21.Biteau B, Labarre J, Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature 425: 980–984, 2003. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- 22.Black PN, Morgan-Day A, McMillan TE, Poole PJ, Young RP. Randomised, controlled trial of N-acetylcysteine for treatment of acute exacerbations of chronic obstructive pulmonary disease [ISRCTN21676344]. BMC Pulm Med 4: 13, 2004. doi: 10.1186/1471-2466-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bomprezzi R. Dimethyl fumarate in the treatment of relapsing-remitting multiple sclerosis: an overview. Ther Adv Neurol Disorder 8: 20–30, 2015. doi: 10.1177/1756285614564152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Borzone GR, Liberona LF, Bustamante AP, Saez CG, Olmos PR, Vecchiola A, Villagrán A, Serrano C, Reyes TP. Differences in lung glutathione metabolism may account for rodent susceptibility in elastase-induced emphysema development. Am J Physiol Regul Integr Comp Physiol 296: R1113–R1123, 2009. doi: 10.1152/ajpregu.90361.2008. [DOI] [PubMed] [Google Scholar]
- 25.Boyland E, Chasseaud LF. Enzyme-catalysed conjugations of glutathione with unsaturated compounds. Biochem J 104: 95–102, 1967. doi: 10.1042/bj1040095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brennan JP, Miller JI, Fuller W, Wait R, Begum S, Dunn MJ, Eaton P. The utility of N,N-biotinyl glutathione disulfide in the study of protein S-glutathiolation. Mol Cell Proteomics 5: 215–225, 2006. doi: 10.1074/mcp.M500212-MCP200. [DOI] [PubMed] [Google Scholar]
- 27.Brigelius-Flohé R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta 1830: 3289–3303, 2013. doi: 10.1016/j.bbagen.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 28.Bushweller JH, Aaslund F, Wuethrich K, Holmgren A. Structural and functional characterization of the mutant Escherichia coli glutaredoxin (C14→S) and its mixed disulfide with glutathione. Biochemistry 31: 9288–9293, 1992. doi: 10.1021/bi00153a023. [DOI] [PubMed] [Google Scholar]
- 29.Bushweller JH, Billeter M, Holmgren A, Wüthrich K. The nuclear magnetic resonance solution structure of the mixed disulfide between Escherichia coli glutaredoxin(C14S) and glutathione. J Mol Biol 235: 1585–1597, 1994. doi: 10.1006/jmbi.1994.1108. [DOI] [PubMed] [Google Scholar]
- 30.Butturini E, Carcereri de Prati A, Chiavegato G, Rigo A, Cavalieri E, Darra E, Mariotto S. Mild oxidative stress induces S-glutathionylation of STAT3 and enhances chemosensitivity of tumoural cells to chemotherapeutic drugs. Free Radic Biol Med 65: 1322–1330, 2013. doi: 10.1016/j.freeradbiomed.2013.09.015. [DOI] [PubMed] [Google Scholar]
- 31.Butturini E, Darra E, Chiavegato G, Cellini B, Cozzolino F, Monti M, Pucci P, Dell’Orco D, Mariotto S. S-glutathionylation at Cys328 and Cys542 impairs STAT3 phosphorylation. ACS Chem Biol 9: 1885–1893, 2014. doi: 10.1021/cb500407d. [DOI] [PubMed] [Google Scholar]
- 32.Calderón A, Lázaro-Payo A, Iglesias-Baena I, Camejo D, Lázaro JJ, Sevilla F, Jiménez A. Glutathionylation of pea chloroplast 2-Cys Prx and mitochondrial Prx IIF affects their structure and peroxidase activity and sulfiredoxin deglutathionylates only the 2-Cys Prx. Front Plant Sci 8: 118, 2017. doi: 10.3389/fpls.2017.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Camaschella C, Campanella A, De Falco L, Boschetto L, Merlini R, Silvestri L, Levi S, Iolascon A. The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood 110: 1353–1358, 2007. doi: 10.1182/blood-2007-02-072520. [DOI] [PubMed] [Google Scholar]
- 34.Campo I, Zorzetto M, Mariani F, Kadija Z, Morbini P, Dore R, Kaltenborn E, Frixel S, Zarbock R, Liebisch G, Hegermann J, Wrede C, Griese M, Luisetti M. A large kindred of pulmonary fibrosis associated with a novel ABCA3 gene variant. Respir Res 15: 43, 2014. doi: 10.1186/1465-9921-15-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cantin AM, Hubbard RC, Crystal RG. Glutathione deficiency in the epithelial lining fluid of the lower respiratory tract in idiopathic pulmonary fibrosis. Am Rev Respir Dis 139: 370–372, 1989. doi: 10.1164/ajrccm/139.2.370. [DOI] [PubMed] [Google Scholar]
- 36.Carp H, Miller F, Hoidal JR, Janoff A. Potential mechanism of emphysema: alpha 1-proteinase inhibitor recovered from lungs of cigarette smokers contains oxidized methionine and has decreased elastase inhibitory capacity. Proc Natl Acad Sci USA 79: 2041–2045, 1982. doi: 10.1073/pnas.79.6.2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Casagrande S, Bonetto V, Fratelli M, Gianazza E, Eberini I, Massignan T, Salmona M, Chang G, Holmgren A, Ghezzi P. Glutathionylation of human thioredoxin: a possible crosstalk between the glutathione and thioredoxin systems. Proc Natl Acad Sci USA 99: 9745–9749, 2002. doi: 10.1073/pnas.152168599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cazzola M, Rogliani P, Calzetta L, Hanania NA, Matera MG. Impact of mucolytic agents on COPD exacerbations: a pair-wise and network meta-analysis. COPD 14: 552–563, 2017. doi: 10.1080/15412555.2017.1347918. [DOI] [PubMed] [Google Scholar]
- 39.Chang TS, Jeong W, Woo HA, Lee SM, Park S, Rhee SG. Characterization of mammalian sulfiredoxin and its reactivation of hyperoxidized peroxiredoxin through reduction of cysteine sulfinic acid in the active site to cysteine. J Biol Chem 279: 50994–51001, 2004. doi: 10.1074/jbc.M409482200. [DOI] [PubMed] [Google Scholar]
- 40.Chantzoura E, Prinarakis E, Panagopoulos D, Mosialos G, Spyrou G. Glutaredoxin-1 regulates TRAF6 activation and the IL-1 receptor/TLR4 signalling. Biochem Biophys Res Commun 403: 335–339, 2010. doi: 10.1016/j.bbrc.2010.11.029. [DOI] [PubMed] [Google Scholar]
- 41.Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MA, Chen YR, Druhan LJ, Zweier JL. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 468: 1115–1118, 2010. doi: 10.1038/nature09599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen W, Seefeldt T, Young A, Zhang X, Zhao Y, Ruffolo J, Kaushik RS, Guan X. Microtubule S-glutathionylation as a potential approach for antimitotic agents. BMC Cancer 12: 245, 2012. doi: 10.1186/1471-2407-12-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen YJ, Lu CT, Lee TY, Chen YJ. dbGSH: a database of S-glutathionylation. Bioinformatics 30: 2386–2388, 2014. doi: 10.1093/bioinformatics/btu301. [DOI] [PubMed] [Google Scholar]
- 44.Cheresh P, Kim SJ, Tulasiram S, Kamp DW. Oxidative stress and pulmonary fibrosis. Biochim Biophys Acta 1832: 1028–1040, 2013. doi: 10.1016/j.bbadis.2012.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chung S, Sundar IK, Yao H, Ho YS, Rahman I. Glutaredoxin 1 regulates cigarette smoke-mediated lung inflammation through differential modulation of IκB kinases in mice: impact on histone acetylation. Am J Physiol Lung Cell Mol Physiol 299: L192–L203, 2010. doi: 10.1152/ajplung.00426.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Comhair SA, Bhathena PR, Farver C, Thunnissen FB, Erzurum SC. Extracellular glutathione peroxidase induction in asthmatic lungs: evidence for redox regulation of expression in human airway epithelial cells. FASEB J 15: 70–78, 2001. doi: 10.1096/fj.00-0085com. [DOI] [PubMed] [Google Scholar]
- 47.Comhair SA, Erzurum SC. The regulation and role of extracellular glutathione peroxidase. Antioxid Redox Signal 7: 72–79, 2005. doi: 10.1089/ars.2005.7.72. [DOI] [PubMed] [Google Scholar]
- 48.Coppo L, Montano SJ, Padilla AC, Holmgren A. Determination of glutaredoxin enzyme activity and protein S-glutathionylation using fluorescent eosin-glutathione. Anal Biochem 499: 24–33, 2016. doi: 10.1016/j.ab.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 49.Dai X, Bowatte G, Lowe AJ, Matheson MC, Gurrin LC, Burgess JA, Dharmage SC, Lodge CJ. Do glutathione S-transferase genes modify the link between indoor air pollution and asthma, allergies, and lung function? A systematic review. Curr Allergy Asthma Rep 18: 20, 2018. doi: 10.1007/s11882-018-0771-0. [DOI] [PubMed] [Google Scholar]
- 50.DeMeo DL, Silverman EK. Alpha1-antitrypsin deficiency · 2: genetic aspects of alpha(1)-antitrypsin deficiency: phenotypes and genetic modifiers of emphysema risk. Thorax 59: 259–264, 2004. doi: 10.1136/thx.2003.006502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dicker AJ, Crichton ML, Pumphrey EG, Cassidy AJ, Suarez-Cuartin G, Sibila O, Furrie E, Fong CJ, Ibrahim W, Brady G, Einarsson GG, Elborn JS, Schembri S, Marshall SE, Palmer CNA, Chalmers JD. Neutrophil extracellular traps are associated with disease severity and microbiota diversity in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol 141: 117–127, 2018. doi: 10.1016/j.jaci.2017.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dong K, Wu M, Liu X, Huang Y, Zhang D, Wang Y, Yan LJ, Shi D. Glutaredoxins concomitant with optimal ROS activate AMPK through S-glutathionylation to improve glucose metabolism in type 2 diabetes. Free Radic Biol Med 101: 334–347, 2016. doi: 10.1016/j.freeradbiomed.2016.10.007. [DOI] [PubMed] [Google Scholar]
- 53.Du Y, Zhang H, Zhang X, Lu J, Holmgren A. Thioredoxin 1 is inactivated due to oxidation induced by peroxiredoxin under oxidative stress and reactivated by the glutaredoxin system. J Biol Chem 288: 32241–32247, 2013. doi: 10.1074/jbc.M113.495150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dubey M, Singh AK, Awasthi D, Nagarkoti S, Kumar S, Ali W, Chandra T, Kumar V, Barthwal MK, Jagavelu K, Sánchez-Gómez FJ, Lamas S, Dikshit M. L-Plastin S-glutathionylation promotes reduced binding to β-actin and affects neutrophil functions. Free Radic Biol Med 86: 1–15, 2015. doi: 10.1016/j.freeradbiomed.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 55.Dunican EM, Elicker BM, Gierada DS, Nagle SK, Schiebler ML, Newell JD, Raymond WW, Lachowicz-Scroggins ME, Di Maio S, Hoffman EA, Castro M, Fain SB, Jarjour NN, Israel E, Levy BD, Erzurum SC, Wenzel SE, Meyers DA, Bleecker ER, Phillips BR, Mauger DT, Gordon ED, Woodruff PG, Peters MC, Fahy JV; National Heart Lung and Blood Institute (NHLBI) Severe Asthma Research Program (SARP) . Mucus plugs in patients with asthma linked to eosinophilia and airflow obstruction. J Clin Invest 128: 997–1009, 2018. doi: 10.1172/JCI95693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eiserich JP, Vossen V, O’Neill CA, Halliwell B, Cross CE, van der Vliet A. Molecular mechanisms of damage by excess nitrogen oxides: nitration of tyrosine by gas-phase cigarette smoke. FEBS Lett 353: 53–56, 1994. doi: 10.1016/0014-5793(94)01011-0. [DOI] [PubMed] [Google Scholar]
- 57.Elko EA, Cunniff B, Seward DJ, Chia SB, Aboushousha R, van de Wetering C, van der Velden J, Manuel A, Shukla A, Heintz NH, Anathy V, van der Vliet A, Janssen-Heininger YMW. Peroxiredoxins and beyond; redox systems regulating lung physiology and disease. Antioxid Redox Signal 31: 1070–1091, 2019. doi: 10.1089/ars.2019.7752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Elko EA, Mahoney JM, Vacek P, van der Vliet A, Anathy V, van der Velden JLJL, Janssen-Heininger YMW, Seward DJ. Age-dependent dysregulation of redox genes may contribute to fibrotic pulmonary disease susceptibility. Free Radic Biol Med 141: 438–446, 2019. doi: 10.1016/j.freeradbiomed.2019.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ercolani L, Scirè A, Galeazzi R, Massaccesi L, Cianfruglia L, Amici A, Piva F, Urbanelli L, Emiliani C, Principato G, Armeni T. A possible S-glutathionylation of specific proteins by glyoxalase II: An in vitro and in silico study. Cell Biochem Funct 34: 620–627, 2016. doi: 10.1002/cbf.3236. [DOI] [PubMed] [Google Scholar]
- 60.Farré D, Roset R, Huerta M, Adsuara JE, Roselló L, Albà MM, Messeguer X. Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res 31: 3651–3653, 2003. doi: 10.1093/nar/gkg605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Feng S, Chen Y, Yang F, Zhang L, Gong Y, Adilijiang G, Gao Y, Deng H. Development of a clickable probe for profiling of protein glutathionylation in the central cellular metabolism of E. coli and Drosophila. Chem Biol 22: 1461–1469, 2015. doi: 10.1016/j.chembiol.2015.09.012. [DOI] [PubMed] [Google Scholar]
- 62.Findlay VJ, Townsend DM, Morris TE, Fraser JP, He L, Tew KD. A novel role for human sulfiredoxin in the reversal of glutathionylation. Cancer Res 66: 6800–6806, 2006. doi: 10.1158/0008-5472.CAN-06-0484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fitzpatrick AM, Jones DP, Brown LA. Glutathione redox control of asthma: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal 17: 375–408, 2012. doi: 10.1089/ars.2011.4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fitzpatrick AM, Stephenson ST, Hadley GR, Burwell L, Penugonda M, Simon DM, Hansen J, Jones DP, Brown LA. Thiol redox disturbances in children with severe asthma are associated with posttranslational modification of the transcription factor nuclear factor (erythroid-derived 2)-like 2. J Allergy Clin Immunol 127: 1604–1611, 2011. doi: 10.1016/j.jaci.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Förstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J 33: 829–837, 2012. doi: 10.1093/eurheartj/ehr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gallogly MM, Starke DW, Leonberg AK, Ospina SM, Mieyal JJ. Kinetic and mechanistic characterization and versatile catalytic properties of mammalian glutaredoxin 2: implications for intracellular roles. Biochemistry 47: 11144–11157, 2008. doi: 10.1021/bi800966v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gan ZR, Sardana MK, Jacobs JW, Polokoff MA. Yeast thioltransferase–the active site cysteines display differential reactivity. Arch Biochem Biophys 282: 110–115, 1990. doi: 10.1016/0003-9861(90)90093-E. [DOI] [PubMed] [Google Scholar]
- 68.Gan ZR, Wells WW. Identification and reactivity of the catalytic site of pig liver thioltransferase. J Biol Chem 262: 6704–6707, 1987. [PubMed] [Google Scholar]
- 69.García-Giménez JL, Òlaso G, Hake SB, Bönisch C, Wiedemann SM, Markovic J, Dasí F, Gimeno A, Pérez-Quilis C, Palacios O, Capdevila M, Viña J, Pallardó FV. Histone h3 glutathionylation in proliferating mammalian cells destabilizes nucleosomal structure. Antioxid Redox Signal 19: 1305–1320, 2013. doi: 10.1089/ars.2012.5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gergondey R, Garcia C, Marchand CH, Lemaire SD, Camadro JM, Auchère F. Modulation of the specific glutathionylation of mitochondrial proteins in the yeast Saccharomyces cerevisiae under basal and stress conditions. Biochem J 474: 1175–1193, 2017. doi: 10.1042/BCJ20160927. [DOI] [PubMed] [Google Scholar]
- 71.Giustarini D, Galvagni F, Tesei A, Farolfi A, Zanoni M, Pignatta S, Milzani A, Marone IM, Dalle-Donne I, Nassini R, Rossi R. Glutathione, glutathione disulfide, and S-glutathionylated proteins in cell cultures. Free Radic Biol Med 89: 972–981, 2015. doi: 10.1016/j.freeradbiomed.2015.10.410. [DOI] [PubMed] [Google Scholar]
- 72.Giustarini D, Milzani A, Aldini G, Carini M, Rossi R, Dalle-Donne I. S-nitrosation versus S-glutathionylation of protein sulfhydryl groups by S-nitrosoglutathione. Antioxid Redox Signal 7: 930–939, 2005. doi: 10.1089/ars.2005.7.930. [DOI] [PubMed] [Google Scholar]
- 73.Giustarini D, Tsikas D, Colombo G, Milzani A, Dalle-Donne I, Fanti P, Rossi R. Pitfalls in the analysis of the physiological antioxidant glutathione (GSH) and its disulfide (GSSG) in biological samples: an elephant in the room. J Chromatogr B Analyt Technol Biomed Life Sci 1019: 21–28, 2016. doi: 10.1016/j.jchromb.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gladyshev VN, Liu A, Novoselov SV, Krysan K, Sun QA, Kryukov VM, Kryukov GV, Lou MF. Identification and characterization of a new mammalian glutaredoxin (thioltransferase), Grx2. J Biol Chem 276: 30374–30380, 2001. doi: 10.1074/jbc.M100020200. [DOI] [PubMed] [Google Scholar]
- 75.Gonzalez-Dosal R, Horan KA, Rahbek SH, Ichijo H, Chen ZJ, Mieyal JJ, Hartmann R, Paludan SR. HSV infection induces production of ROS, which potentiate signaling from pattern recognition receptors: role for S-glutathionylation of TRAF3 and 6. PLoS Pathog 7: e1002250, 2011. doi: 10.1371/journal.ppat.1002250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gould NS, Min E, Gauthier S, Martin RJ, Day BJ. Lung glutathione adaptive responses to cigarette smoke exposure. Respir Res 12: 133, 2011. doi: 10.1186/1465-9921-12-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gould NS, Min E, Huang J, Chu HW, Good J, Martin RJ, Day BJ. Glutathione depletion accelerates cigarette smoke-induced inflammation and airspace enlargement. Toxicol Sci 147: 466–474, 2015. doi: 10.1093/toxsci/kfv143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Grassi C, Morandini GC. A controlled trial of intermittent oral acetylcysteine in the long-term treatment of chronic bronchitis. Eur J Clin Pharmacol 9: 393–396, 1976. doi: 10.1007/BF00606554. [DOI] [PubMed] [Google Scholar]
- 79.Grek CL, Townsend DM, Uys JD, Manevich Y, Coker WJ III, Pazoles CJ, Tew KD. S-glutathionylated serine proteinase inhibitors as plasma biomarkers in assessing response to redox-modulating drugs. Cancer Res 72: 2383–2393, 2012. doi: 10.1158/0008-5472.CAN-11-4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Griffiths SW, King J, Cooney CL. The reactivity and oxidation pathway of cysteine 232 in recombinant human alpha 1-antitrypsin. J Biol Chem 277: 25486–25492, 2002. doi: 10.1074/jbc.M203089200. [DOI] [PubMed] [Google Scholar]
- 81.Guo J, Gaffrey MJ, Su D, Liu T, Camp DG II, Smith RD, Qian WJ. Resin-assisted enrichment of thiols as a general strategy for proteomic profiling of cysteine-based reversible modifications. Nat Protoc 9: 64–75, 2014. doi: 10.1038/nprot.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gupta V, Carroll KS. Sulfenic acid chemistry, detection and cellular lifetime. Biochim Biophys Acta 1840: 847–875, 2014. doi: 10.1016/j.bbagen.2013.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hagimoto N, Kuwano K, Miyazaki H, Kunitake R, Fujita M, Kawasaki M, Kaneko Y, Hara N. Induction of apoptosis and pulmonary fibrosis in mice in response to ligation of Fas antigen. Am J Respir Cell Mol Biol 17: 272–278, 1997. doi: 10.1165/ajrcmb.17.3.2893. [DOI] [PubMed] [Google Scholar]
- 84.Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat Med 15: 410–416, 2009. doi: 10.1038/nm.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hammad H, Lambrecht BN. Barrier epithelial cells and the control of type 2 immunity. Immunity 43: 29–40, 2015. doi: 10.1016/j.immuni.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 86.Hanschmann EM, Lönn ME, Schütte LD, Funke M, Godoy JR, Eitner S, Hudemann C, Lillig CH. Both thioredoxin 2 and glutaredoxin 2 contribute to the reduction of the mitochondrial 2-Cys peroxiredoxin Prx3. J Biol Chem 285: 40699–40705, 2010. doi: 10.1074/jbc.M110.185827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hasbal C, Aksu BY, Himmetoglu S, Dincer Y, Koc EE, Hatipoglu S, Akcay T. DNA damage and glutathione level in children with asthma bronchiale: effect of antiasthmatic therapy. Pediatr Allergy Immunol 21: e674–e678, 2010. doi: 10.1111/j.1399-3038.2009.00959.x. [DOI] [PubMed] [Google Scholar]
- 88.Hashemy SI, Johansson C, Berndt C, Lillig CH, Holmgren A. Oxidation and S-nitrosylation of cysteines in human cytosolic and mitochondrial glutaredoxins: effects on structure and activity. J Biol Chem 282: 14428–14436, 2007. doi: 10.1074/jbc.M700927200. [DOI] [PubMed] [Google Scholar]
- 89.Hatakeyama M, Tanimoto Y, Mizoguchi T. Purification and some properties of bovine liver cytosol thioltransferase. J Biochem 95: 1811–1818, 1984. doi: 10.1093/oxfordjournals.jbchem.a134794. [DOI] [PubMed] [Google Scholar]
- 90.Haunhorst P, Berndt C, Eitner S, Godoy JR, Lillig CH. Characterization of the human monothiol glutaredoxin 3 (PICOT) as iron-sulfur protein. Biochem Biophys Res Commun 394: 372–376, 2010. doi: 10.1016/j.bbrc.2010.03.016. [DOI] [PubMed] [Google Scholar]
- 91.Hirose K, Iwata A, Tamachi T, Nakajima H. Allergic airway inflammation: key players beyond the Th2 cell pathway. Immunol Rev 278: 145–161, 2017. doi: 10.1111/imr.12540. [DOI] [PubMed] [Google Scholar]
- 92.Hoenderdos K, Condliffe A. The neutrophil in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 48: 531–539, 2013. doi: 10.1165/rcmb.2012-0492TR. [DOI] [PubMed] [Google Scholar]
- 93.Hoffman SM, Qian X, Nolin JD, Chapman DG, Chia SB, Lahue KG, Schneider R, Ather JL, Randall MJ, McMillan DH, Jones JT, Taatjes DJ, Aliyeva M, Daphtary N, Abdalla S, Lundblad LK, Ho YS, Anathy V, Irvin CG, Wouters EF, Reynaert NL, Dixon AE, van der Vliet A, Poynter ME, Janssen-Heininger YM. Ablation of glutaredoxin-1 modulates house dust mite-induced allergic airways disease in mice. Am J Respir Cell Mol Biol 55: 377–386, 2016. doi: 10.1165/rcmb.2015-0401OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hoffman SM, Tully JE, Lahue KG, Anathy V, Nolin JD, Guala AS, van der Velden JL, Ho YS, Aliyeva M, Daphtary N, Lundblad LK, Irvin CG, Janssen-Heininger YM. Genetic ablation of glutaredoxin-1 causes enhanced resolution of airways hyperresponsiveness and mucus metaplasia in mice with allergic airways disease. Am J Physiol Lung Cell Mol Physiol 303: L528–L538, 2012. doi: 10.1152/ajplung.00167.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Holgate ST, Wenzel S, Postma DS, Weiss ST, Renz H, Sly PD. Asthma. Nat Rev Dis Primers 1: 15025, 2015. doi: 10.1038/nrdp.2015.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Holmgren A. Glutaredoxin from Escherichia coli and calf thymus. Methods Enzymol 113: 525–540, 1985. doi: 10.1016/S0076-6879(85)13071-5. [DOI] [PubMed] [Google Scholar]
- 97.Holmgren A. Glutathione-dependent synthesis of deoxyribonucleotides. Characterization of the enzymatic mechanism of Escherichia coli glutaredoxin. J Biol Chem 254: 3672–3678, 1979. [PubMed] [Google Scholar]
- 98.Holmgren A. Glutathione-dependent synthesis of deoxyribonucleotides. Purification and characterization of glutaredoxin from Escherichia coli. J Biol Chem 254: 3664–3671, 1979. [PubMed] [Google Scholar]
- 99.Holmgren A. Hydrogen donor system for Escherichia coli ribonucleoside-diphosphate reductase dependent upon glutathione. Proc Natl Acad Sci USA 73: 2275–2279, 1976. doi: 10.1073/pnas.73.7.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Holtgrefe S, Gohlke J, Starmann J, Druce S, Klocke S, Altmann B, Wojtera J, Lindermayr C, Scheibe R. Regulation of plant cytosolic glyceraldehyde 3-phosphate dehydrogenase isoforms by thiol modifications. Physiol Plant 133: 211–228, 2008. doi: 10.1111/j.1399-3054.2008.01066.x. [DOI] [PubMed] [Google Scholar]
- 101.Höög JO, Jörnvall H, Holmgren A, Carlquist M, Persson M. The primary structure of Escherichia coli glutaredoxin. Distant homology with thioredoxins in a superfamily of small proteins with a redox-active cystine disulfide/cysteine dithiol. Eur J Biochem 136: 223–232, 1983. doi: 10.1111/j.1432-1033.1983.tb07730.x. [DOI] [PubMed] [Google Scholar]
- 102.Hopper S, Johnson RS, Vath JE, Biemann K. Glutaredoxin from rabbit bone marrow. Purification, characterization, and amino acid sequence determined by tandem mass spectrometry. J Biol Chem 264: 20438–20447, 1989. [PubMed] [Google Scholar]
- 103.Hristova M, Habibovic A, Veith C, Janssen-Heininger YM, Dixon AE, Geiszt M, van der Vliet A. Airway epithelial dual oxidase 1 mediates allergen-induced IL-33 secretion and activation of type 2 immune responses. J Allergy Clin Immunol 137: 1545–1556.e11, 2016. doi: 10.1016/j.jaci.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hughes MM, Lavrencic P, Coll RC, Ve T, Ryan DG, Williams NC, Menon D, Mansell A, Board PG, Mobli M, Kobe B, O’Neill LAJ. Solution structure of the TLR adaptor MAL/TIRAP reveals an intact BB loop and supports MAL Cys91 glutathionylation for signaling. Proc Natl Acad Sci USA 114: E6480–E6489, 2017. doi: 10.1073/pnas.1701868114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Martinez FJ, de Andrade JA, Anstrom KJ, King TE Jr, Raghu G; Idiopathic Pulmonary Fibrosis Clinical Research Network . Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med 370: 2093–2101, 2014. doi: 10.1056/NEJMoa1401739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Israel E, Reddel HK. Severe and difficult-to-treat asthma in adults. N Engl J Med 377: 965–976, 2017. doi: 10.1056/NEJMra1608969. [DOI] [PubMed] [Google Scholar]
- 107.Jaiswal AK, Sandey M, Suryawanshi A, Cattley RC, Mishra A. Dimethyl fumarate abrogates dust mite-induced allergic asthma by altering dendritic cell function. Immun Inflamm Dis 7: 201–213, 2019. doi: 10.1002/iid3.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jao SC, English Ospina SM, Berdis AJ, Starke DW, Post CB, Mieyal JJ. Computational and mutational analysis of human glutaredoxin (thioltransferase): probing the molecular basis of the low pKa of cysteine 22 and its role in catalysis. Biochemistry 45: 4785–4796, 2006. doi: 10.1021/bi0516327. [DOI] [PubMed] [Google Scholar]
- 109.Johansson C, Kavanagh KL, Gileadi O, Oppermann U. Reversible sequestration of active site cysteines in a 2Fe-2S-bridged dimer provides a mechanism for glutaredoxin 2 regulation in human mitochondria. J Biol Chem 282: 3077–3082, 2007. doi: 10.1074/jbc.M608179200. [DOI] [PubMed] [Google Scholar]
- 110.Johansson C, Lillig CH, Holmgren A. Human mitochondrial glutaredoxin reduces S-glutathionylated proteins with high affinity accepting electrons from either glutathione or thioredoxin reductase. J Biol Chem 279: 7537–7543, 2004. doi: 10.1074/jbc.M312719200. [DOI] [PubMed] [Google Scholar]
- 111.Johansson C, Roos AK, Montano SJ, Sengupta R, Filippakopoulos P, Guo K, von Delft F, Holmgren A, Oppermann U, Kavanagh KL. The crystal structure of human GLRX5: iron-sulfur cluster co-ordination, tetrameric assembly and monomer activity. Biochem J 433: 303–311, 2011. doi: 10.1042/BJ20101286. [DOI] [PubMed] [Google Scholar]
- 112.Jones JT, Qian X, van der Velden JL, Chia SB, McMillan DH, Flemer S, Hoffman SM, Lahue KG, Schneider RW, Nolin JD, Anathy V, van der Vliet A, Townsend DM, Tew KD, Janssen-Heininger YM. Glutathione S-transferase pi modulates NF-κB activation and pro-inflammatory responses in lung epithelial cells. Redox Biol 8: 375–382, 2016. doi: 10.1016/j.redox.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Juneau RA, Pang B, Armbruster CE, Murrah KA, Perez AC, Swords WE. Peroxiredoxin-glutaredoxin and catalase promote resistance of nontypeable Haemophilus influenzae 86-028NP to oxidants and survival within neutrophil extracellular traps. Infect Immun 83: 239–246, 2015. doi: 10.1128/IAI.02390-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jung CH, Thomas JA. S-glutathiolated hepatocyte proteins and insulin disulfides as substrates for reduction by glutaredoxin, thioredoxin, protein disulfide isomerase, and glutathione. Arch Biochem Biophys 335: 61–72, 1996. doi: 10.1006/abbi.1996.0482. [DOI] [PubMed] [Google Scholar]
- 115.Kambe T, Song T, Takata T, Hatano N, Miyamoto Y, Nozaki N, Naito Y, Tokumitsu H, Watanabe Y. Inactivation of Ca2+/calmodulin-dependent protein kinase I by S-glutathionylation of the active-site cysteine residue. FEBS Lett 584: 2478–2484, 2010. doi: 10.1016/j.febslet.2010.04.059. [DOI] [PubMed] [Google Scholar]
- 116.Kang PT, Zhang L, Chen CL, Chen J, Green KB, Chen YR. Protein thiyl radical mediates S-glutathionylation of complex I. Free Radic Biol Med 53: 962–973, 2012. doi: 10.1016/j.freeradbiomed.2012.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Katti SK, Robbins AH, Yang Y, Wells WW. Crystal structure of thioltransferase at 2.2 A resolution. Protein Sci 4: 1998–2005, 1995. doi: 10.1002/pro.5560041005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kaur A, Gautam R, Srivastava R, Chandel A, Kumar A, Karthikeyan S, Bachhawat AK. ChaC2, an enzyme for slow turnover of cytosolic glutathione. J Biol Chem 292: 638–651, 2017. doi: 10.1074/jbc.M116.727479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kekulandara DN, Samarasinghe KT, Munkanatta Godage DN, Ahn YH. Clickable glutathione using tetrazine-alkene bioorthogonal chemistry for detecting protein glutathionylation. Org Biomol Chem 14: 10886–10893, 2016. [Erratum in Org Biomol Chem 16: 2576, 2018.] doi: 10.1039/C6OB02050J. [DOI] [PubMed] [Google Scholar]
- 120.Kim HS, Ullevig SL, Nguyen HN, Vanegas D, Asmis R. Redox regulation of 14-3-3ζ controls monocyte migration. Arterioscler Thromb Vasc Biol 34: 1514–1521, 2014. doi: 10.1161/ATVBAHA.114.303746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kim HS, Ullevig SL, Zamora D, Lee CF, Asmis R. Redox regulation of MAPK phosphatase 1 controls monocyte migration and macrophage recruitment. Proc Natl Acad Sci USA 109: E2803–E2812, 2012. doi: 10.1073/pnas.1212596109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet 378: 1949–1961, 2011. doi: 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- 123.Kortemme T, Creighton TE. Ionisation of cysteine residues at the termini of model α-helical peptides. Relevance to unusual thiol pKa values in proteins of the thioredoxin family. J Mol Biol 253: 799–812, 1995. doi: 10.1006/jmbi.1995.0592. [DOI] [PubMed] [Google Scholar]
- 124.Kosower NS, Kosower EM, Wertheim B, Correa WS. Diamide, a new reagent for the intracellular oxidation of glutathione to the disulfide. Biochem Biophys Res Commun 37: 593–596, 1969. doi: 10.1016/0006-291X(69)90850-X. [DOI] [PubMed] [Google Scholar]
- 125.Kuipers I, Bracke KR, Brusselle GG, Aesif SW, Krijgsman R, Arts IC, Wouters EF, Reynaert NL. Altered cigarette smoke-induced lung inflammation due to ablation of Grx1. PLoS One 7: e38984, 2012. doi: 10.1371/journal.pone.0038984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kuipers I, Bracke KR, Brusselle GG, Wouters EF, Reynaert NL. Smoke decreases reversible oxidations S-glutathionylation and S-nitrosylation in mice. Free Radic Res 46: 164–173, 2012. doi: 10.3109/10715762.2011.647011. [DOI] [PubMed] [Google Scholar]
- 127.Kuipers I, Guala AS, Aesif SW, Konings G, Bouwman FG, Mariman EC, Wouters EF, Janssen-Heininger YM, Reynaert NL. Cigarette smoke targets glutaredoxin 1, increasing s-glutathionylation and epithelial cell death. Am J Respir Cell Mol Biol 45: 931–937, 2011. doi: 10.1165/rcmb.2010-0249OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kuipers I, Louis R, Manise M, Dentener MA, Irvin CG, Janssen-Heininger YM, Brightling CE, Wouters EF, Reynaert NL. Increased glutaredoxin-1 and decreased protein S-glutathionylation in sputum of asthmatics. Eur Respir J 41: 469–472, 2013. doi: 10.1183/09031936.00115212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kumar A, Tikoo S, Maity S, Sengupta S, Sengupta S, Kaur A, Bachhawat AK. Mammalian proapoptotic factor ChaC1 and its homologues function as γ-glutamyl cyclotransferases acting specifically on glutathione. EMBO Rep 13: 1095–1101, 2012. doi: 10.1038/embor.2012.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kuwano K, Hagimoto N, Kawasaki M, Yatomi T, Nakamura N, Nagata S, Suda T, Kunitake R, Maeyama T, Miyazaki H, Hara N. Essential roles of the Fas-Fas ligand pathway in the development of pulmonary fibrosis. J Clin Invest 104: 13–19, 1999. doi: 10.1172/JCI5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kuwano K, Miyazaki H, Hagimoto N, Kawasaki M, Fujita M, Kunitake R, Kaneko Y, Hara N. The involvement of Fas-Fas ligand pathway in fibrosing lung diseases. Am J Respir Cell Mol Biol 20: 53–60, 1999. doi: 10.1165/ajrcmb.20.1.2941. [DOI] [PubMed] [Google Scholar]
- 132.Lam BK, Austen KF. Leukotriene C4 synthase: a pivotal enzyme in cellular biosynthesis of the cysteinyl leukotrienes. Prostaglandins Other Lipid Mediat 68-69: 511–520, 2002. doi: 10.1016/S0090-6980(02)00052-7. [DOI] [PubMed] [Google Scholar]
- 133.Landino LM, Brown CM, Edson CA, Gilbert LJ, Grega-Larson N, Wirth AJ, Lane KC. Fluorescein-labeled glutathione to study protein S-glutathionylation. Anal Biochem 402: 102–104, 2010. doi: 10.1016/j.ab.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Larson K, Eriksson V, Mannervik B. Thioltransferase from human placenta. Methods Enzymol 113: 520–524, 1985. doi: 10.1016/S0076-6879(85)13070-3. [DOI] [PubMed] [Google Scholar]
- 135.Lee EK, Jeon WK, Chae MY, Hong HY, Lee YS, Kim JH, Kwon JY, Kim BC, Park SH. Decreased expression of glutaredoxin 1 is required for transforming growth factor-beta1-mediated epithelial-mesenchymal transition of EpRas mammary epithelial cells. Biochem Biophys Res Commun 391: 1021–1027, 2010. doi: 10.1016/j.bbrc.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 136.Li J, Huang FL, Huang KP. Glutathiolation of proteins by glutathione disulfide S-oxide derived from S-nitrosoglutathione. Modifications of rat brain neurogranin/RC3 and neuromodulin/GAP-43. J Biol Chem 276: 3098–3105, 2001. doi: 10.1074/jbc.M008260200. [DOI] [PubMed] [Google Scholar]
- 137.Li N, Nel AE. Role of the Nrf2-mediated signaling pathway as a negative regulator of inflammation: implications for the impact of particulate pollutants on asthma. Antioxid Redox Signal 8: 88–98, 2006. doi: 10.1089/ars.2006.8.88. [DOI] [PubMed] [Google Scholar]
- 138.Liao BC, Hsieh CW, Lin YC, Wung BS. The glutaredoxin/glutathione system modulates NF-kappaB activity by glutathionylation of p65 in cinnamaldehyde-treated endothelial cells. Toxicol Sci 116: 151–163, 2010. doi: 10.1093/toxsci/kfq098. [DOI] [PubMed] [Google Scholar]
- 139.Lillig CH, Berndt C, Vergnolle O, Lönn ME, Hudemann C, Bill E, Holmgren A. Characterization of human glutaredoxin 2 as iron-sulfur protein: a possible role as redox sensor. Proc Natl Acad Sci USA 102: 8168–8173, 2005. doi: 10.1073/pnas.0500735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lillig CH, Lönn ME, Enoksson M, Fernandes AP, Holmgren A. Short interfering RNA-mediated silencing of glutaredoxin 2 increases the sensitivity of HeLa cells toward doxorubicin and phenylarsine oxide. Proc Natl Acad Sci USA 101: 13227–13232, 2004. doi: 10.1073/pnas.0401896101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lillig CH, Prior A, Schwenn JD, Aslund F, Ritz D, Vlamis-Gardikas A, Holmgren A. New thioredoxins and glutaredoxins as electron donors of 3′-phosphoadenylylsulfate reductase. J Biol Chem 274: 7695–7698, 1999. doi: 10.1074/jbc.274.12.7695. [DOI] [PubMed] [Google Scholar]
- 142.Lim SY, Raftery MJ, Goyette J, Geczy CL. S-glutathionylation regulates inflammatory activities of S100A9. J Biol Chem 285: 14377–14388, 2010. doi: 10.1074/jbc.M109.075242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Linden M, Håkansson L, Ohlsson K, Sjödin K, Tegner H, Tunek A, Venge P. Glutathione in bronchoalveolar lavage fluid from smokers is related to humoral markers of inflammatory cell activity. Inflammation 13: 651–658, 1989. doi: 10.1007/BF00914309. [DOI] [PubMed] [Google Scholar]
- 144.Liu CC, Fry NA, Hamilton EJ, Chia KK, Garcia A, Karimi Galougahi K, Figtree GA, Clarke RJ, Bundgaard H, Rasmussen HH. Redox-dependent regulation of the Na+-K+ pump: new twists to an old target for treatment of heart failure. J Mol Cell Cardiol 61: 94–101, 2013. doi: 10.1016/j.yjmcc.2013.05.013. [DOI] [PubMed] [Google Scholar]
- 145.Liu CC, Karimi Galougahi K, Weisbrod RM, Hansen T, Ravaie R, Nunez A, Liu YB, Fry N, Garcia A, Hamilton EJ, Sweadner KJ, Cohen RA, Figtree GA. Oxidative inhibition of the vascular Na+-K+ pump via NADPH oxidase-dependent β1-subunit glutathionylation: implications for angiotensin II-induced vascular dysfunction. Free Radic Biol Med 65: 563–572, 2013. doi: 10.1016/j.freeradbiomed.2013.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Liu Y, Hyde AS, Simpson MA, Barycki JJ. Emerging regulatory paradigms in glutathione metabolism. Adv Cancer Res 122: 69–101, 2014. doi: 10.1016/B978-0-12-420117-0.00002-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lönn ME, Hudemann C, Berndt C, Cherkasov V, Capani F, Holmgren A, Lillig CH. Expression pattern of human glutaredoxin 2 isoforms: identification and characterization of two testis/cancer cell-specific isoforms. Antioxid Redox Signal 10: 547–557, 2008. doi: 10.1089/ars.2007.1821. [DOI] [PubMed] [Google Scholar]
- 148.Lowry MH, McAllister BP, Jean JC, Brown LA, Hughey RP, Cruikshank WW, Amar S, Lucey EC, Braun K, Johnson P, Wight TN, Joyce-Brady M. Lung lining fluid glutathione attenuates IL-13-induced asthma. Am J Respir Cell Mol Biol 38: 509–516, 2008. doi: 10.1165/rcmb.2007-0128OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lu J, Holmgren A. The thioredoxin antioxidant system. Free Radic Biol Med 66: 75–87, 2014. doi: 10.1016/j.freeradbiomed.2013.07.036. [DOI] [PubMed] [Google Scholar]
- 150.Lu J, Holmgren A. The thioredoxin superfamily in oxidative protein folding. Antioxid Redox Signal 21: 457–470, 2014. doi: 10.1089/ars.2014.5849. [DOI] [PubMed] [Google Scholar]
- 151.Lu SC. Glutathione synthesis. Biochim Biophys Acta 1830: 3143–3153, 2013. doi: 10.1016/j.bbagen.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lundberg M, Johansson C, Chandra J, Enoksson M, Jacobsson G, Ljung J, Johansson M, Holmgren A. Cloning and expression of a novel human glutaredoxin (Grx2) with mitochondrial and nuclear isoforms. J Biol Chem 276: 26269–26275, 2001. doi: 10.1074/jbc.M011605200. [DOI] [PubMed] [Google Scholar]
- 153.Luthman M, Eriksson S, Holmgren A, Thelander L. Glutathione-dependent hydrogen donor system for calf thymus ribonucleoside-diphosphate reductase. Proc Natl Acad Sci USA 76: 2158–2162, 1979. doi: 10.1073/pnas.76.5.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mailloux RJ, Seifert EL, Bouillaud F, Aguer C, Collins S, Harper ME. Glutathionylation acts as a control switch for uncoupling proteins UCP2 and UCP3. J Biol Chem 286: 21865–21875, 2011. doi: 10.1074/jbc.M111.240242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Maki K, Nagai K, Suzuki M, Inomata T, Yoshida T, Nishimura M. Temporal changes in glutaredoxin 1 and protein s-glutathionylation in allergic airway inflammation. PLoS One 10: e0122986, 2015. doi: 10.1371/journal.pone.0122986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Manevich Y, Feinstein SI, Fisher AB. Activation of the antioxidant enzyme 1-CYS peroxiredoxin requires glutathionylation mediated by heterodimerization with pi GST. Proc Natl Acad Sci USA 101: 3780–3785, 2004. doi: 10.1073/pnas.0400181101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Mannervik B, Axelsson K, Sundewall AC, Holmgren A. Relative contributions of thioltransferase-and thioredoxin-dependent systems in reduction of low-molecular-mass and protein disulphides. Biochem J 213: 519–523, 1983. doi: 10.1042/bj2130519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Martin JL. Thioredoxin—a fold for all reasons. Structure 3: 245–250, 1995. doi: 10.1016/S0969-2126(01)00154-X. [DOI] [PubMed] [Google Scholar]
- 159.Martinez FJ, Collard HR, Pardo A, Raghu G, Richeldi L, Selman M, Swigris JJ, Taniguchi H, Wells AU. Idiopathic pulmonary fibrosis. Nat Rev Dis Primers 3: 17074, 2017. doi: 10.1038/nrdp.2017.74. [DOI] [PubMed] [Google Scholar]
- 160.Martínez-Ruiz A, Cadenas S, Lamas S. Nitric oxide signaling: classical, less classical, and nonclassical mechanisms. Free Radic Biol Med 51: 17–29, 2011. doi: 10.1016/j.freeradbiomed.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 161.Maryam A, Mehmood T, Zhang H, Li Y, Khan M, Ma T. Alantolactone induces apoptosis, promotes STAT3 glutathionylation and enhances chemosensitivity of A549 lung adenocarcinoma cells to doxorubicin via oxidative stress. Sci Rep 7: 6242, 2017. doi: 10.1038/s41598-017-06535-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.McGarry DJ, Chen W, Chakravarty P, Lamont DL, Wolf CR, Henderson CJ. Proteome-wide identification and quantification of S-glutathionylation targets in mouse liver. Biochem J 469: 25–32, 2015. doi: 10.1042/BJ20141256. [DOI] [PubMed] [Google Scholar]
- 163.McMillan DH, van der Velden JL, Lahue KG, Qian X, Schneider RW, Iberg MS, Nolin JD, Abdalla S, Casey DT, Tew KD, Townsend DM, Henderson CJ, Wolf CR, Butnor KJ, Taatjes DJ, Budd RC, Irvin CG, van der Vliet A, Flemer S, Anathy V, Janssen-Heininger YM. Attenuation of lung fibrosis in mice with a clinically relevant inhibitor of glutathione-S-transferase π. JCI Insight 1: 1, 2016. doi: 10.1172/jci.insight.85717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Messeguer X, Escudero R, Farré D, Núñez O, Martínez J, Albà MM. PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 18: 333–334, 2002. doi: 10.1093/bioinformatics/18.2.333. [DOI] [PubMed] [Google Scholar]
- 165.Mieyal JJ, Starke DW, Gravina SA, Hocevar BA. Thioltransferase in human red blood cells: kinetics and equilibrium. Biochemistry 30: 8883–8891, 1991. doi: 10.1021/bi00100a023. [DOI] [PubMed] [Google Scholar]
- 166.Mitra G, Muralidharan M, Narayanan S, Pinto J, Srinivasan K, Mandal AK. Glutathionylation induced structural changes in oxy human hemoglobin analyzed by backbone amide hydrogen/deuterium exchange and MALDI-mass spectrometry. Bioconjug Chem 23: 2344–2353, 2012. doi: 10.1021/bc300291u. [DOI] [PubMed] [Google Scholar]
- 167.Miyamoto Y, Akaike T, Alam MS, Inoue K, Hamamoto T, Ikebe N, Yoshitake J, Okamoto T, Maeda H. Novel functions of human alpha(1)-protease inhibitor after S-nitrosylation: inhibition of cysteine protease and antibacterial activity. Biochem Biophys Res Commun 267: 918–923, 2000. doi: 10.1006/bbrc.1999.2046. [DOI] [PubMed] [Google Scholar]
- 168.Mohr S, Hallak H, de Boitte A, Lapetina EG, Brüne B. Nitric oxide-induced S-glutathionylation and inactivation of glyceraldehyde-3-phosphate dehydrogenase. J Biol Chem 274: 9427–9430, 1999. doi: 10.1074/jbc.274.14.9427. [DOI] [PubMed] [Google Scholar]
- 169.Montero D, Tachibana C, Rahr Winther J, Appenzeller-Herzog C. Intracellular glutathione pools are heterogeneously concentrated. Redox Biol 1: 508–513, 2013. doi: 10.1016/j.redox.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Mora AL, Bueno M, Rojas M. Mitochondria in the spotlight of aging and idiopathic pulmonary fibrosis. J Clin Invest 127: 405–414, 2017. doi: 10.1172/JCI87440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Morissette MC, Parent J, Milot J. Alveolar epithelial and endothelial cell apoptosis in emphysema: what we know and what we need to know. Int J Chron Obstruct Pulmon Dis 4: 19–31, 2009. [PMC free article] [PubMed] [Google Scholar]
- 172.Morlá M, Busquets X, Pons J, Sauleda J, MacNee W, Agustí AG. Telomere shortening in smokers with and without COPD. Eur Respir J 27: 525–528, 2006. doi: 10.1183/09031936.06.00087005. [DOI] [PubMed] [Google Scholar]
- 173.Morrison D, Rahman I, Lannan S, MacNee W. Epithelial permeability, inflammation, and oxidant stress in the air spaces of smokers. Am J Respir Crit Care Med 159: 473–479, 1999. doi: 10.1164/ajrccm.159.2.9804080. [DOI] [PubMed] [Google Scholar]
- 174.Nadeem A, Siddiqui N, Alharbi NO, Alharbi MM, Imam F, Sayed-Ahmed MM. Glutathione modulation during sensitization as well as challenge phase regulates airway reactivity and inflammation in mouse model of allergic asthma. Biochimie 103: 61–70, 2014. doi: 10.1016/j.biochi.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 175.Nagai S, Black S. A thiol-disulfide transhydrogenase from yeast. J Biol Chem 243: 1942–1947, 1968. [PubMed] [Google Scholar]
- 176.Nolin JD, Tully JE, Hoffman SM, Guala AS, van der Velden JL, Poynter ME, van der Vliet A, Anathy V, Janssen-Heininger YM. The glutaredoxin/S-glutathionylation axis regulates interleukin-17A-induced proinflammatory responses in lung epithelial cells in association with S-glutathionylation of nuclear factor κB family proteins. Free Radic Biol Med 73: 143–153, 2014. doi: 10.1016/j.freeradbiomed.2014.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Nureki SI, Tomer Y, Venosa A, Katzen J, Russo SJ, Jamil S, Barrett M, Nguyen V, Kopp M, Mulugeta S, Beers MF. Expression of mutant Sftpc in murine alveolar epithelia drives spontaneous lung fibrosis. J Clin Invest 128: 4008–4024, 2018. doi: 10.1172/JCI99287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Padilla CA, Bajalica S, Lagercrantz J, Holmgren A. The gene for human glutaredoxin (GLRX) is localized to human chromosome 5q14. Genomics 32: 455–457, 1996. doi: 10.1006/geno.1996.0141. [DOI] [PubMed] [Google Scholar]
- 179.Padilla CA, Spyrou G, Holmgren A. High-level expression of fully active human glutaredoxin (thioltransferase) in E. coli and characterization of Cys7 to Ser mutant protein. FEBS Lett 378: 69–73, 1996. doi: 10.1016/0014-5793(95)01413-6. [DOI] [PubMed] [Google Scholar]
- 180.Pal D, Sharma D, Kumar M, Sandur SK. Prediction of glutathionylation sites in proteins using minimal sequence information and their experimental validation. Free Radic Res 50: 1011–1021, 2016. doi: 10.1080/10715762.2016.1216551. [DOI] [PubMed] [Google Scholar]
- 181.Pandey G, Pandey OP, Rogers AJ, Ahsen ME, Hoffman GE, Raby BA, Weiss ST, Schadt EE, Bunyavanich S. A nasal brush-based classifier of asthma identified by machine learning analysis of nasal RNA sequence data. Sci Rep 8: 8826, 2018. doi: 10.1038/s41598-018-27189-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Papi A, Brightling C, Pedersen SE, Reddel HK. Asthma. Lancet 391: 783–800, 2018. doi: 10.1016/S0140-6736(17)33311-1. [DOI] [PubMed] [Google Scholar]
- 183.Papov VV, Gravina SA, Mieyal JJ, Biemann K. The primary structure and properties of thioltransferase (glutaredoxin) from human red blood cells. Protein Sci 3: 428–434, 1994. doi: 10.1002/pro.5560030307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Park JB, Levine M. The human glutaredoxin gene: determination of its organization, transcription start point, and promoter analysis. Gene 197: 189–193, 1997. doi: 10.1016/S0378-1119(97)00262-X. [DOI] [PubMed] [Google Scholar]
- 185.Park JW, Mieyal JJ, Rhee SG, Chock PB. Deglutathionylation of 2-Cys peroxiredoxin is specifically catalyzed by sulfiredoxin. J Biol Chem 284: 23364–23374, 2009. doi: 10.1074/jbc.M109.021394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Park JW, Piszczek G, Rhee SG, Chock PB. Glutathionylation of peroxiredoxin I induces decamer to dimers dissociation with concomitant loss of chaperone activity. Biochemistry 50: 3204–3210, 2011. doi: 10.1021/bi101373h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Passarelli C, Tozzi G, Pastore A, Bertini E, Piemonte F. GSSG-mediated Complex I defect in isolated cardiac mitochondria. Int J Mol Med 26: 95–99, 2010. doi: 10.3892/ijmm_00000439. [DOI] [PubMed] [Google Scholar]
- 188.Peltoniemi M, Kaarteenaho-Wiik R, Säily M, Sormunen R, Pääkkö P, Holmgren A, Soini Y, Kinnula VL. Expression of glutaredoxin is highly cell specific in human lung and is decreased by transforming growth factor-beta in vitro and in interstitial lung diseases in vivo. Hum Pathol 35: 1000–1007, 2004. doi: 10.1016/j.humpath.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 189.Peltoniemi MJ, Karala AR, Jurvansuu JK, Kinnula VL, Ruddock LW. Insights into deglutathionylation reactions. Different intermediates in the glutaredoxin and protein disulfide isomerase catalyzed reactions are defined by the gamma-linkage present in glutathione. J Biol Chem 281: 33107–33114, 2006. doi: 10.1074/jbc.M605602200. [DOI] [PubMed] [Google Scholar]
- 190.Peltoniemi MJ, Rytilä PH, Harju TH, Soini YM, Salmenkivi KM, Ruddock LW, Kinnula VL. Modulation of glutaredoxin in the lung and sputum of cigarette smokers and chronic obstructive pulmonary disease. Respir Res 7: 133, 2006. doi: 10.1186/1465-9921-7-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Pennings HJ, Borm PJ, Evelo CT, Wouters EF. Changes in levels of catalase and glutathione in erythrocytes of patients with stable asthma, treated with beclomethasone dipropionate. Eur Respir J 13: 1260–1266, 1999. doi: 10.1183/09031936.99.13612679. [DOI] [PubMed] [Google Scholar]
- 192.Peskin AV, Pace PE, Behring JB, Paton LN, Soethoudt M, Bachschmid MM, Winterbourn CC. Glutathionylation of the active site cysteines of peroxiredoxin 2 and recycling by glutaredoxin. J Biol Chem 291: 3053–3062, 2016. doi: 10.1074/jbc.M115.692798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Piacentini S, Polimanti R, Simonelli I, Donno S, Pasqualetti P, Manfellotto D, Fuciarelli M. Glutathione S-transferase polymorphisms, asthma susceptibility and confounding variables: a meta-analysis. Mol Biol Rep 40: 3299–3313, 2013. doi: 10.1007/s11033-012-2405-2. [DOI] [PubMed] [Google Scholar]
- 194.Prinz WA, Aslund F, Holmgren A, Beckwith J. The role of the thioredoxin and glutaredoxin pathways in reducing protein disulfide bonds in the Escherichia coli cytoplasm. J Biol Chem 272: 15661–15667, 1997. doi: 10.1074/jbc.272.25.15661. [DOI] [PubMed] [Google Scholar]
- 195.Qanungo S, Starke DW, Pai HV, Mieyal JJ, Nieminen AL. Glutathione supplementation potentiates hypoxic apoptosis by S-glutathionylation of p65-NFkappaB. J Biol Chem 282: 18427–18436, 2007. doi: 10.1074/jbc.M610934200. [DOI] [PubMed] [Google Scholar]
- 196.Qiao F, Xing K, Liu A, Ehlers N, Raghavachari N, Lou MF. Human lens thioltransferase: cloning, purification, and function. Invest Ophthalmol Vis Sci 42: 743–751, 2001. [PubMed] [Google Scholar]
- 197.Rabe KF, Watz H. Chronic obstructive pulmonary disease. Lancet 389: 1931–1940, 2017. doi: 10.1016/S0140-6736(17)31222-9. [DOI] [PubMed] [Google Scholar]
- 198.Racanelli AC, Kikkers SA, Choi AMK, Cloonan SM. Autophagy and inflammation in chronic respiratory disease. Autophagy 14: 221–232, 2018. doi: 10.1080/15548627.2017.1389823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Racker E. Glutathione-homocystine transhydrogenase. J Biol Chem 217: 867–874, 1955. [PubMed] [Google Scholar]
- 200.Rahman I, Kode A, Biswas SK. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat Protoc 1: 3159–3165, 2006. doi: 10.1038/nprot.2006.378. [DOI] [PubMed] [Google Scholar]
- 201.Rajendrasozhan S, Yang SR, Kinnula VL, Rahman I. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 177: 861–870, 2008. doi: 10.1164/rccm.200708-1269OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ralat LA, Manevich Y, Fisher AB, Colman RF. Direct evidence for the formation of a complex between 1-cysteine peroxiredoxin and glutathione S-transferase pi with activity changes in both enzymes. Biochemistry 45: 360–372, 2006. doi: 10.1021/bi0520737. [DOI] [PubMed] [Google Scholar]
- 203.Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, Kensler TW, Yamamoto M, Petrache I, Tuder RM, Biswal S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest 114: 1248–1259, 2004. doi: 10.1172/JCI200421146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Rangasamy T, Guo J, Mitzner WA, Roman J, Singh A, Fryer AD, Yamamoto M, Kensler TW, Tuder RM, Georas SN, Biswal S. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J Exp Med 202: 47–59, 2005. doi: 10.1084/jem.20050538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Reddy S, Finkelstein EI, Wong PS, Phung A, Cross CE, van der Vliet A. Identification of glutathione modifications by cigarette smoke. Free Radic Biol Med 33: 1490–1498, 2002. doi: 10.1016/S0891-5849(02)01079-1. [DOI] [PubMed] [Google Scholar]
- 206.Redler RL, Wilcox KC, Proctor EA, Fee L, Caplow M, Dokholyan NV. Glutathionylation at Cys-111 induces dissociation of wild type and FALS mutant SOD1 dimers. Biochemistry 50: 7057–7066, 2011. doi: 10.1021/bi200614y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Reynaert NL. Glutathione biochemistry in asthma. Biochim Biophys Acta 1810: 1045–1051, 2011. doi: 10.1016/j.bbagen.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 208.Reynaert NL, van der Vliet A, Guala AS, McGovern T, Hristova M, Pantano C, Heintz NH, Heim J, Ho YS, Matthews DE, Wouters EF, Janssen-Heininger YM. Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc Natl Acad Sci USA 103: 13086–13091, 2006. doi: 10.1073/pnas.0603290103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Reynaert NL, Wouters EF, Janssen-Heininger YM. Modulation of glutaredoxin-1 expression in a mouse model of allergic airway disease. Am J Respir Cell Mol Biol 36: 147–151, 2007. doi: 10.1165/rcmb.2006-0259RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Rodríguez-Manzaneque MT, Tamarit J, Bellí G, Ros J, Herrero E. Grx5 is a mitochondrial glutaredoxin required for the activity of iron/sulfur enzymes. Mol Biol Cell 13: 1109–1121, 2002. doi: 10.1091/mbc.01-10-0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Rodriguez-Rocha H, Garcia Garcia A, Zavala-Flores L, Li S, Madayiputhiya N, Franco R. Glutaredoxin 1 protects dopaminergic cells by increased protein glutathionylation in experimental Parkinson’s disease. Antioxid Redox Signal 17: 1676–1693, 2012. doi: 10.1089/ars.2011.4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Rogers LK, Cismowski MJ. Oxidative stress in the lung—the essential paradox. Curr Opin Toxicol 7: 37–43, 2018. doi: 10.1016/j.cotox.2017.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Rogliani P, Matera MG, Page C, Puxeddu E, Cazzola M, Calzetta L. Efficacy and safety profile of mucolytic/antioxidant agents in chronic obstructive pulmonary disease: a comparative analysis across erdosteine, carbocysteine, and N-acetylcysteine. Respir Res 20: 104, 2019. doi: 10.1186/s12931-019-1078-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Rouhier N. Plant glutaredoxins: pivotal players in redox biology and iron-sulphur centre assembly. New Phytol 186: 365–372, 2010. doi: 10.1111/j.1469-8137.2009.03146.x. [DOI] [PubMed] [Google Scholar]
- 215.Saaranen MJ, Salo KE, Latva-Ranta MK, Kinnula VL, Ruddock LW. The C-terminal active site cysteine of Escherichia coli glutaredoxin 1 determines the glutathione specificity of the second step of peptide deglutathionylation. Antioxid Redox Signal 11: 1819–1828, 2009. doi: 10.1089/ars.2008.2387. [DOI] [PubMed] [Google Scholar]
- 216.Sakai J, Li J, Subramanian KK, Mondal S, Bajrami B, Hattori H, Jia Y, Dickinson BC, Zhong J, Ye K, Chang CJ, Ho YS, Zhou J, Luo HR. Reactive oxygen species-induced actin glutathionylation controls actin dynamics in neutrophils. Immunity 37: 1037–1049, 2012. doi: 10.1016/j.immuni.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Samarasinghe KT, Munkanatta Godage DN, VanHecke GC, Ahn YH. Metabolic synthesis of clickable glutathione for chemoselective detection of glutathionylation. J Am Chem Soc 136: 11566–11569, 2014. doi: 10.1021/ja503946q. [DOI] [PubMed] [Google Scholar]
- 218.Samarasinghe KT, Munkanatta Godage DN, Zhou Y, Ndombera FT, Weerapana E, Ahn YH. A clickable glutathione approach for identification of protein glutathionylation in response to glucose metabolism. Mol Biosyst 12: 2471–2480, 2016. doi: 10.1039/C6MB00175K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med 30: 1191–1212, 2001. doi: 10.1016/S0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 220.Schuliga M. NF-kappaB signaling in chronic inflammatory airway disease. Biomolecules 5: 1266–1283, 2015. doi: 10.3390/biom5031266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Seidel P, Merfort I, Hughes JM, Oliver BG, Tamm M, Roth M. Dimethylfumarate inhibits NF-κB function at multiple levels to limit airway smooth muscle cell cytokine secretion. Am J Physiol Lung Cell Mol Physiol 297: L326–L339, 2009. doi: 10.1152/ajplung.90624.2008. [DOI] [PubMed] [Google Scholar]
- 222.Seidel P, Roth M, Ge Q, Merfort I, S’ng CT, Ammit AJ. IκBα glutathionylation and reduced histone H3 phosphorylation inhibit eotaxin and RANTES. Eur Respir J 38: 1444–1452, 2011. doi: 10.1183/09031936.00129610. [DOI] [PubMed] [Google Scholar]
- 223.Serban KA, Petrache I. Alpha-1 antitrypsin and lung cell apoptosis. Ann Am Thorac Soc 13, Suppl 2: S146–S149, 2016. doi: 10.1513/AnnalsATS.201505-312KV. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Shen Y, Cai W, Lei S, Zhang Z. Effect of high/low dose N-acetylcysteine on chronic obstructive pulmonary disease: a systematic review and meta-analysis. COPD 11: 351–358, 2014. doi: 10.3109/15412555.2013.858315. [DOI] [PubMed] [Google Scholar]
- 225.Silberstein DZ, Karuppanan K, Aung HH, Chen CH, Cross CE, McDonald KA. An oxidation-resistant, recombinant alpha-1 antitrypsin produced in Nicotiana benthamiana. Free Radic Biol Med 120: 303–310, 2018. doi: 10.1016/j.freeradbiomed.2018.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Silva GM, Netto LE, Discola KF, Piassa-Filho GM, Pimenta DC, Bárcena JA, Demasi M. Role of glutaredoxin 2 and cytosolic thioredoxins in cysteinyl-based redox modification of the 20S proteasome. FEBS J 275: 2942–2955, 2008. doi: 10.1111/j.1742-4658.2008.06441.x. [DOI] [PubMed] [Google Scholar]
- 227.Silva GM, Netto LE, Simões V, Santos LF, Gozzo FC, Demasi MA, Oliveira CL, Bicev RN, Klitzke CF, Sogayar MC, Demasi M. Redox control of 20S proteasome gating. Antioxid Redox Signal 16: 1183–1194, 2012. doi: 10.1089/ars.2011.4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Srinivasan U, Mieyal PA, Mieyal JJ. pH profiles indicative of rate-limiting nucleophilic displacement in thioltransferase catalysis. Biochemistry 36: 3199–3206, 1997. doi: 10.1021/bi962017t. [DOI] [PubMed] [Google Scholar]
- 229.Starke DW, Chen Y, Bapna CP, Lesnefsky EJ, Mieyal JJ. Sensitivity of protein sulfhydryl repair enzymes to oxidative stress. Free Radic Biol Med 23: 373–384, 1997. doi: 10.1016/S0891-5849(97)00009-9. [DOI] [PubMed] [Google Scholar]
- 230.Starke DW, Chock PB, Mieyal JJ. Glutathione-thiyl radical scavenging and transferase properties of human glutaredoxin (thioltransferase). Potential role in redox signal transduction. J Biol Chem 278: 14607–14613, 2003. doi: 10.1074/jbc.M210434200. [DOI] [PubMed] [Google Scholar]
- 231.Stincone A, Prigione A, Cramer T, Wamelink MM, Campbell K, Cheung E, Olin-Sandoval V, Grüning NM, Krüger A, Tauqeer Alam M, Keller MA, Breitenbach M, Brindle KM, Rabinowitz JD, Ralser M. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev Camb Philos Soc 90: 927–963, 2015. doi: 10.1111/brv.12140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Stipanuk MH, Dominy JE Jr, Lee JI, Coloso RM. Mammalian cysteine metabolism: new insights into regulation of cysteine metabolism. J Nutr 136, Suppl: 1652S–1659S, 2006. doi: 10.1093/jn/136.6.1652S. [DOI] [PubMed] [Google Scholar]
- 233.Stojkov D, Amini P, Oberson K, Sokollik C, Duppenthaler A, Simon HU, Yousefi S. ROS and glutathionylation balance cytoskeletal dynamics in neutrophil extracellular trap formation. J Cell Biol 216: 4073–4090, 2017. doi: 10.1083/jcb.201611168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Storisteanu DM, Pocock JM, Cowburn AS, Juss JK, Nadesalingam A, Nizet V, Chilvers ER. Evasion of neutrophil extracellular traps by respiratory pathogens. Am J Respir Cell Mol Biol 56: 423–431, 2017. doi: 10.1165/rcmb.2016-0193PS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Su D, Gaffrey MJ, Guo J, Hatchell KE, Chu RK, Clauss TR, Aldrich JT, Wu S, Purvine S, Camp DG, Smith RD, Thrall BD, Qian WJ. Proteomic identification and quantification of S-glutathionylation in mouse macrophages using resin-assisted enrichment and isobaric labeling. Free Radic Biol Med 67: 460–470, 2014. doi: 10.1016/j.freeradbiomed.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Subramani J, Kundumani-Sridharan V, Hilgers RH, Owens C, Das KC. Thioredoxin uses a GSH-independent route to deglutathionylate endothelial nitric-oxide synthase and protect against myocardial infarction. J Biol Chem 291: 23374–23389, 2016. doi: 10.1074/jbc.M116.745034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Suki B, Bartolák-Suki E, Rocco PRM. Elastase-induced lung emphysema models in mice. Methods Mol Biol 1639: 67–75, 2017. doi: 10.1007/978-1-4939-7163-3_7. [DOI] [PubMed] [Google Scholar]
- 238.Sullivan DM, Wehr NB, Fergusson MM, Levine RL, Finkel T. Identification of oxidant-sensitive proteins: TNF-alpha induces protein glutathiolation. Biochemistry 39: 11121–11128, 2000. doi: 10.1021/bi0007674. [DOI] [PubMed] [Google Scholar]
- 239.Sun C, Berardi MJ, Bushweller JH. The NMR solution structure of human glutaredoxin in the fully reduced form. J Mol Biol 280: 687–701, 1998. doi: 10.1006/jmbi.1998.1913. [DOI] [PubMed] [Google Scholar]
- 240.Sun C, Holmgren A, Bushweller JH. Complete 1H, 13C, and 15N NMR resonance assignments and secondary structure of human glutaredoxin in the fully reduced form. Protein Sci 6: 383–390, 1997. doi: 10.1002/pro.5560060214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Sun C, Shi ZZ, Zhou X, Chen L, Zhao XM. Prediction of S-glutathionylation sites based on protein sequences. PLoS One 8: e55512, 2013. doi: 10.1371/journal.pone.0055512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Sussan TE, Gajghate S, Chatterjee S, Mandke P, McCormick S, Sudini K, Kumar S, Breysse PN, Diette GB, Sidhaye VK, Biswal S. Nrf2 reduces allergic asthma in mice through enhanced airway epithelial cytoprotective function. Am J Physiol Lung Cell Mol Physiol 309: L27–L36, 2015. doi: 10.1152/ajplung.00398.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Taggart C, Cervantes-Laurean D, Kim G, McElvaney NG, Wehr N, Moss J, Levine RL. Oxidation of either methionine 351 or methionine 358 in alpha 1-antitrypsin causes loss of anti-neutrophil elastase activity. J Biol Chem 275: 27258–27265, 2000. [DOI] [PubMed] [Google Scholar]
- 244.Takashima Y, Hirota K, Nakamura H, Nakamura T, Akiyama K, Cheng FS, Maeda M, Yodoi J. Differential expression of glutaredoxin and thioredoxin during monocytic differentiation. Immunol Lett 68: 397–401, 1999. doi: 10.1016/S0165-2478(99)00087-5. [DOI] [PubMed] [Google Scholar]
- 245.Takata T, Tsuchiya Y, Watanabe Y. 90-kDa ribosomal S6 kinase 1 is inhibited by S-glutathionylation of its active-site cysteine residue during oxidative stress. FEBS Lett 587: 1681–1686, 2013. doi: 10.1016/j.febslet.2013.04.017. [DOI] [PubMed] [Google Scholar]
- 246.Tamarit J, Belli G, Cabiscol E, Herrero E, Ros J. Biochemical characterization of yeast mitochondrial Grx5 monothiol glutaredoxin. J Biol Chem 278: 25745–25751, 2003. doi: 10.1074/jbc.M303477200. [DOI] [PubMed] [Google Scholar]
- 247.Timme-Laragy AR, Goldstone JV, Imhoff BR, Stegeman JJ, Hahn ME, Hansen JM. Glutathione redox dynamics and expression of glutathione-related genes in the developing embryo. Free Radic Biol Med 65: 89–101, 2013. doi: 10.1016/j.freeradbiomed.2013.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Townsend DM, Manevich Y, He L, Hutchens S, Pazoles CJ, Tew KD. Novel role for glutathione S-transferase pi. Regulator of protein S-Glutathionylation following oxidative and nitrosative stress. J Biol Chem 284: 436–445, 2009. doi: 10.1074/jbc.M805586200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Travis J, Beatty K, Wong PS, Matheson NR. Oxidation of alpha 1-proteinase inhibitor as a major, contributing factor in the development of pulmonary emphysema. Bull Eur Physiopathol Respir 16, Suppl: 341–352, 1980. doi: 10.1016/b978-0-08-027379-2.50035-1. [DOI] [PubMed] [Google Scholar]
- 250.Tse HN, Raiteri L, Wong KY, Yee KS, Ng LY, Wai KY, Loo CK, Chan MH. High-dose N-acetylcysteine in stable COPD: the 1-year, double-blind, randomized, placebo-controlled HIACE study. Chest 144: 106–118, 2013. doi: 10.1378/chest.12-2357. [DOI] [PubMed] [Google Scholar]
- 251.Tsuji T, Aoshiba K, Nagai A. Alveolar cell senescence in patients with pulmonary emphysema. Am J Respir Crit Care Med 174: 886–893, 2006. doi: 10.1164/rccm.200509-1374OC. [DOI] [PubMed] [Google Scholar]
- 252.Tully JE, Hoffman SM, Lahue KG, Nolin JD, Anathy V, Lundblad LK, Daphtary N, Aliyeva M, Black KE, Dixon AE, Poynter ME, Irvin CG, Janssen-Heininger YM. Epithelial NF-κB orchestrates house dust mite-induced airway inflammation, hyperresponsiveness, and fibrotic remodeling. J Immunol 191: 5811–5821, 2013. doi: 10.4049/jimmunol.1301329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Tully JE, Nolin JD, Guala AS, Hoffman SM, Roberson EC, Lahue KG, van der Velden J, Anathy V, Blackwell TS, Janssen-Heininger YM. Cooperation between classical and alternative NF-κB pathways regulates proinflammatory responses in epithelial cells. Am J Respir Cell Mol Biol 47: 497–508, 2012. doi: 10.1165/rcmb.2012-0014OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Tuzova M, Jean JC, Hughey RP, Brown LA, Cruikshank WW, Hiratake J, Joyce-Brady M. Inhibiting lung lining fluid glutathione metabolism with GGsTop as a novel treatment for asthma. Front Pharmacol 5: 179, 2014. doi: 10.3389/fphar.2014.00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Ullevig S, Zhao Q, Lee CF, Seok Kim H, Zamora D, Asmis R. NADPH oxidase 4 mediates monocyte priming and accelerated chemotaxis induced by metabolic stress. Arterioscler Thromb Vasc Biol 32: 415–426, 2012. doi: 10.1161/ATVBAHA.111.238899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Vandeputte C, Guizon I, Genestie-Denis I, Vannier B, Lorenzon G. A microtiter plate assay for total glutathione and glutathione disulfide contents in cultured/isolated cells: performance study of a new miniaturized protocol. Cell Biol Toxicol 10: 415–421, 1994. doi: 10.1007/BF00755791. [DOI] [PubMed] [Google Scholar]
- 257.Vernooy JH, Küçükaycan M, Jacobs JA, Chavannes NH, Buurman WA, Dentener MA, Wouters EF. Local and systemic inflammation in patients with chronic obstructive pulmonary disease: soluble tumor necrosis factor receptors are increased in sputum. Am J Respir Crit Care Med 166: 1218–1224, 2002. doi: 10.1164/rccm.2202023. [DOI] [PubMed] [Google Scholar]
- 258.Wang J, Boja ES, Tan W, Tekle E, Fales HM, English S, Mieyal JJ, Chock PB. Reversible glutathionylation regulates actin polymerization in A431 cells. J Biol Chem 276: 47763–47766, 2001. doi: 10.1074/jbc.C100415200. [DOI] [PubMed] [Google Scholar]
- 259.Wang W, Wu Z, Dai Z, Yang Y, Wang J, Wu G. Glycine metabolism in animals and humans: implications for nutrition and health. Amino Acids 45: 463–477, 2013. doi: 10.1007/s00726-013-1493-1. [DOI] [PubMed] [Google Scholar]
- 260.Watanabe Y, Murdoch CE, Sano S, Ido Y, Bachschmid MM, Cohen RA, Matsui R. Glutathione adducts induced by ischemia and deletion of glutaredoxin-1 stabilize HIF-1α and improve limb revascularization. Proc Natl Acad Sci USA 113: 6011–6016, 2016. doi: 10.1073/pnas.1524198113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Weinberg EO, Ferran B, Tsukahara Y, Hatch MM, Han J, Murdoch CE, Matsui R. IL-33 induction and signaling are controlled by glutaredoxin-1 in mouse macrophages. PLoS One 14: e0210827, 2019. doi: 10.1371/journal.pone.0210827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Wells WW, Xu DP, Yang YF, Rocque PA. Mammalian thioltransferase (glutaredoxin) and protein disulfide isomerase have dehydroascorbate reductase activity. J Biol Chem 265: 15361–15364, 1990. [PubMed] [Google Scholar]
- 263.Witte S, Villalba M, Bi K, Liu Y, Isakov N, Altman A. Inhibition of the c-Jun N-terminal kinase/AP-1 and NF-kappaB pathways by PICOT, a novel protein kinase C-interacting protein with a thioredoxin homology domain. J Biol Chem 275: 1902–1909, 2000. doi: 10.1074/jbc.275.3.1902. [DOI] [PubMed] [Google Scholar]
- 264.Wright TK, Gibson PG, Simpson JL, McDonald VM, Wood LG, Baines KJ. Neutrophil extracellular traps are associated with inflammation in chronic airway disease. Respirology 21: 467–475, 2016. doi: 10.1111/resp.12730. [DOI] [PubMed] [Google Scholar]
- 265.Wu H, Lin L, Giblin F, Ho YS, Lou MF. Glutaredoxin 2 knockout increases sensitivity to oxidative stress in mouse lens epithelial cells. Free Radic Biol Med 51: 2108–2117, 2011. doi: 10.1016/j.freeradbiomed.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Xia TH, Bushweller JH, Sodano P, Billeter M, Björnberg O, Holmgren A, Wüthrich K. NMR structure of oxidized Escherichia coli glutaredoxin: comparison with reduced E. coli glutaredoxin and functionally related proteins. Protein Sci 1: 310–321, 1992. doi: 10.1002/pro.5560010302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Xie J, Potter A, Xie W, Lynch C, Seefeldt T. Evaluation of a dithiocarbamate derivative as a model of thiol oxidative stress in H9c2 rat cardiomyocytes. Free Radic Biol Med 70: 214–222, 2014. doi: 10.1016/j.freeradbiomed.2014.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Yang Y, Jao S, Nanduri S, Starke DW, Mieyal JJ, Qin J. Reactivity of the human thioltransferase (glutaredoxin) C7S, C25S, C78S, C82S mutant and NMR solution structure of its glutathionyl mixed disulfide intermediate reflect catalytic specificity. Biochemistry 37: 17145–17156, 1998. doi: 10.1021/bi9806504. [DOI] [PubMed] [Google Scholar]
- 269.Yang YC, Huang YT, Hsieh CW, Yang PM, Wung BS. Carbon monoxide induces heme oxygenase-1 to modulate STAT3 activation in endothelial cells via S-glutathionylation. PLoS One 9: e100677, 2014. doi: 10.1371/journal.pone.0100677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Yang YF, Wells WW. Catalytic mechanism of thioltransferase. J Biol Chem 266: 12766–12771, 1991. [PubMed] [Google Scholar]
- 271.Yang YF, Wells WW. Identification and characterization of the functional amino acids at the active center of pig liver thioltransferase by site-directed mutagenesis. J Biol Chem 266: 12759–12765, 1991. [PubMed] [Google Scholar]
- 272.Yao H, Chung S, Hwang JW, Rajendrasozhan S, Sundar IK, Dean DA, McBurney MW, Guarente L, Gu W, Rönty M, Kinnula VL, Rahman I. SIRT1 protects against emphysema via FOXO3-mediated reduction of premature senescence in mice. J Clin Invest 122: 2032–2045, 2012. doi: 10.1172/JCI60132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Ye H, Jeong SY, Ghosh MC, Kovtunovych G, Silvestri L, Ortillo D, Uchida N, Tisdale J, Camaschella C, Rouault TA. Glutaredoxin 5 deficiency causes sideroblastic anemia by specifically impairing heme biosynthesis and depleting cytosolic iron in human erythroblasts. J Clin Invest 120: 1749–1761, 2010. doi: 10.1172/JCI40372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Yelamanchi SD, Jayaram S, Thomas JK, Gundimeda S, Khan AA, Singhal A, Keshava Prasad TS, Pandey A, Somani BL, Gowda H. A pathway map of glutamate metabolism. J Cell Commun Signal 10: 69–75, 2016. doi: 10.1007/s12079-015-0315-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.You Y, Chen J, Zhu F, Xu Q, Han L, Gao X, Zhang X, Luo HR, Miao J, Sun X, Ren H, Du Y, Guo L, Wang X, Wang Y, Chen S, Huang N, Li J. Glutaredoxin 1 up-regulates deglutathionylation of α4 integrin and thereby restricts neutrophil mobilization from bone marrow. J Biol Chem 294: 2616–2627, 2019. doi: 10.1074/jbc.RA118.006096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Yu J, Zhang NN, Yin PD, Cui PX, Zhou CZ. Glutathionylation-triggered conformational changes of glutaredoxin Grx1 from the yeast Saccharomyces cerevisiae. Proteins 72: 1077–1083, 2008. doi: 10.1002/prot.22096. [DOI] [PubMed] [Google Scholar]
- 277.Yuan S, Hollinger M, Lachowicz-Scroggins ME, Kerr SC, Dunican EM, Daniel BM, Ghosh S, Erzurum SC, Willard B, Hazen SL, Huang X, Carrington SD, Oscarson S, Fahy JV. Oxidation increases mucin polymer cross-links to stiffen airway mucus gels. Sci Transl Med 7: 276ra27, 2015. doi: 10.1126/scitranslmed.3010525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Zaffagnini M, Michelet L, Massot V, Trost P, Lemaire SD. Biochemical characterization of glutaredoxins from Chlamydomonas reinhardtii reveals the unique properties of a chloroplastic CGFS-type glutaredoxin. J Biol Chem 283: 8868–8876, 2008. doi: 10.1074/jbc.M709567200. [DOI] [PubMed] [Google Scholar]
- 279.Zhang C, Rodriguez C, Circu ML, Aw TY, Feng J. S-Glutathionyl quantification in the attomole range using glutaredoxin-3-catalyzed cysteine derivatization and capillary gel electrophoresis with laser-induced fluorescence detection. Anal Bioanal Chem 401: 2165–2175, 2011. doi: 10.1007/s00216-011-5311-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Zhang H, Yang J, Wu S, Gong W, Chen C, Perrett S. Glutathionylation of the bacterial Hsp70 chaperone DnaK provides a link between oxidative stress and the heat shock response. J Biol Chem 291: 6967–6981, 2016. doi: 10.1074/jbc.M115.673608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Zhang J, Grek C, Ye ZW, Manevich Y, Tew KD, Townsend DM. Pleiotropic functions of glutathione S-transferase P. Adv Cancer Res 122: 143–175, 2014. doi: 10.1016/B978-0-12-420117-0.00004-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Zhang Q, Lenardo MJ, Baltimore D. 30 Years of NF-κB: a blossoming of relevance to human pathobiology. Cell 168: 37–57, 2017. doi: 10.1016/j.cell.2016.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Zhang X, Liu P, Zhang C, Chiewchengchol D, Zhao F, Yu H, Li J, Kambara H, Luo KY, Venkataraman A, Zhou Z, Zhou W, Zhu H, Zhao L, Sakai J, Chen Y, Ho YS, Bajrami B, Xu B, Silberstein LE, Cheng T, Xu Y, Ke Y, Luo HR. Positive regulation of interleukin-1β bioactivity by physiological ROS-mediated cysteine S-glutathionylation. Cell Reports 20: 224–235, 2017. doi: 10.1016/j.celrep.2017.05.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Zhao X, Ning Q, Ai M, Chai H, Yang G. Identification of S-glutathionylation sites in species-specific proteins by incorporating five sequence-derived features into the general pseudo-amino acid composition. J Theor Biol 398: 96–102, 2016. doi: 10.1016/j.jtbi.2016.03.030. [DOI] [PubMed] [Google Scholar]
- 285.Zhao X, Ning Q, Ai M, Chai H, Yin M. PGluS: prediction of protein S-glutathionylation sites with multiple features and analysis. Mol Biosyst 11: 923–929, 2015. doi: 10.1039/C4MB00680A. [DOI] [PubMed] [Google Scholar]
- 286.Zhao Y, Vanhoutte PM, Leung SW. Vascular nitric oxide: beyond eNOS. J Pharmacol Sci 129: 83–94, 2015. doi: 10.1016/j.jphs.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 287.Zheng JP, Wen FQ, Bai CX, Wan HY, Kang J, Chen P, Yao WZ, Ma LJ, Li X, Raiteri L, Sardina M, Gao Y, Wang BS, Zhong NS; PANTHEON study group . Twice daily N-acetylcysteine 600 mg for exacerbations of chronic obstructive pulmonary disease (PANTHEON): a randomised, double-blind placebo-controlled trial. Lancet Respir Med 2: 187–194, 2014. doi: 10.1016/S2213-2600(13)70286-8. [DOI] [PubMed] [Google Scholar]
- 288.Zhu W, Li L, Deng M, Wang B, Li M, Ding G, Yang Z, Medynski D, Lin X, Ouyang Y, Lin J, Li L, Lin X. Oxidation-resistant and thermostable forms of alpha-1 antitrypsin from Escherichia coli inclusion bodies. FEBS Open Bio 8: 1711–1721, 2018. doi: 10.1002/2211-5463.12515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Zuin R, Palamidese A, Negrin R, Catozzo L, Scarda A, Balbinot M. High-dose N-acetylcysteine in patients with exacerbations of chronic obstructive pulmonary disease. Clin Drug Investig 25: 401–408, 2005. doi: 10.2165/00044011-200525060-00005. [DOI] [PubMed] [Google Scholar]