

Abstract
Since the discovery of sickle cell disease (SCD) in 1910, enormous strides have been made in the elucidation of the pathogenesis of its protean complications, which has inspired recent advances in targeted molecular therapies. In SCD, a single amino acid substitution in the β-globin chain leads to polymerization of mutant hemoglobin S, impairing erythrocyte rheology and survival. Clinically, erythrocyte abnormalities in SCD manifest in hemolytic anemia and cycles of microvascular vaso-occlusion leading to end-organ ischemia-reperfusion injury and infarction. Vaso-occlusive events and intravascular hemolysis promote inflammation and redox instability that lead to progressive small- and large-vessel vasculopathy. Based on current evidence, the pathobiology of SCD is considered to be a vicious cycle of four major processes, all the subject of active study and novel therapeutic targeting: (a) hemoglobin S polymerization, (b) impaired biorheology and increased adhesion-mediated vaso-occlusion, (c) hemolysis-mediated endothelial dysfunction, and (d) concerted activation of sterile inflammation (Toll-like receptor 4– and inflammasome-dependent innate immune pathways). These molecular, cellular, and biophysical processes synergize to promote acute and chronic pain and end-organ injury and failure in SCD. This review provides an exhaustive overview of the current understanding of the molecular pathophysiology of SCD, how this pathophysiology contributes to complications of the central nervous and cardiopulmonary systems, and how this knowledge is being harnessed to develop current and potential therapies.
Keywords: sickle cell anemia, hemolysis, inflammation, reperfusion injury, oxidative stress, infarction
INTRODUCTION
Sickle cell disease (SCD) is an autosomal-recessive genetic disorder that affects approximately 100,000 people in the United States and millions worldwide (1–3). According to the systematic analysis of the Global Burden of Disease Study (4), 3.2 million people live with SCD, 43 million people have sickle cell trait (i.e., are carriers of the mutation), and 176,000 people die of SCD-related complications per year. SCD is an umbrella term for all mutations in the β-globin gene that precipitate the same clinical syndrome (1). Sickle cell anemia (discussed together with other sickling disorders for the reader’s convenience in this review) is the most common form and accounts for 70% of cases of SCD in patients of African ethnicity (2, 3). Sickle cell anemia is caused by homozygosity of the beta-S (βS) allele (located on chromosome 11p15.5), which differs from the wild-type β-allele by a single nucleotide polymorphism dbSNP Rs334(T;T) in which GTG is substituted for GAG in the sixth codon of the β-globin gene (1, 3, 5, 6). This leads to replacement of a hydrophilic glutamic acid residue (Glu) with a hydrophobic valine residue (Val) at the sixth position in the β-globin chain, resulting in a mutated hemoglobin tetramer HbS (α2βs2) in the erythrocytes of individuals with sickle cell anemia (7, 8). Homozygous inheritance of the βS mutation (HbSS) or coinheritance of βS with other mutations such as βC (HbSC), βD (HbSD), βO (HbSO/Arab), βE (HbSE), or a β-thalassemia allele (HbS/β-thal0 or HbS/β-thal+) leads to other forms of SCD via multiple interlinked molecular and cellular mechanisms, which are described in the following sections. As shown in Figure 1, over the past 7 decades, scientists have characterized three major pathobiological processes (HbS polymerization, vaso-occlusion, and hemolysis-mediated endothelial dysfunction) that drive clinical disease; recently, a fourth pathway, sterile inflammation, has emerged.
Figure 1.
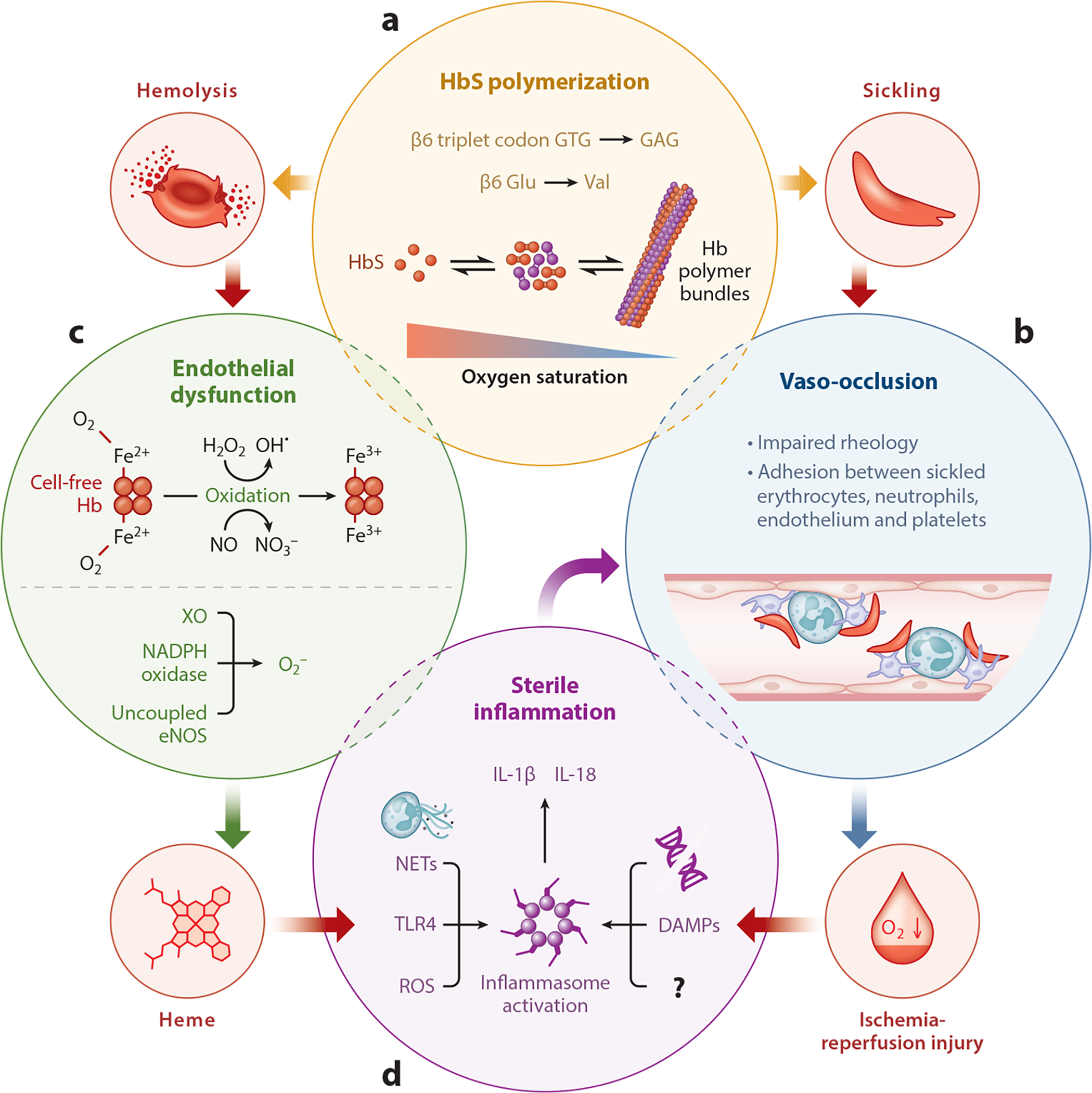
Molecular pathophysiology of sickle cell disease. (a) A single-nucleotide polymorphism in the β-globin gene leads to substitution of valine for glutamic acid at the sixth position in the β-globin chain. Following deoxygenation, the mutated hemoglobin (HbS) molecules polymerize to form bundles. The polymer bundles result in erythrocyte sickling (clockwise), which in turn results in (b) impaired rheology of the blood and aggregation of sickle erythrocytes with neutrophils, platelets, and endothelial cells to promote stasis of blood flow, referred to as vaso-occlusion. Vaso-occlusion promotes ischemia-reperfusion (I-R) injury (clockwise). (a) Hemoglobin (Hb) polymer bundles also promote hemolysis or lysis of erythrocytes (counterclockwise), which (c) releases cell-free Hb into the blood circulation. Oxygenated Hb (Fe2+) promotes endothelial dysfunction by depleting endothelial nitric oxide (NO) reserves to form nitrate (NO3−) and methemoglobin (Fe3+). Alternatively, Hb can also react with H2O2 through the Fenton reaction to form hydroxyl free radical (OH•) and methemoglobin (Fe3+). Also, NADPH oxidase, xanthine oxidase (XO), and uncoupled endothelial NO synthase (eNOS) generate oxygen free radicals to promote endothelial dysfunction. Methemoglobin (Fe3+) degrades to release cell-free heme (counterclockwise), which is a major erythrocyte damage-associated molecular pattern (DAMP). (d) Reactive oxygen species (ROS) generation, Toll-like receptor 4 (TLR4) activation, neutrophil extracellular trap (NET) generation, release of tissue or cell-derived DAMPs, DNA, and other unknown factors (?) triggered by cell-free heme or I-R injury can contribute to sterile inflammation by activating the inflammasome pathway in vascular and inflammatory cells to release IL-1β. Finally, sterile inflammation further promotes vaso-occlusion through a feedback loop by promoting adhesiveness of neutrophils, platelets, and endothelial cells.
HEMOGLOBIN S POLYMERIZATION
Intraerythrocytic HbS deoxygenation in tissues with high oxygen demand promotes the exposure of hydrophobic motifs on individual deoxygenated (T-state) HbS tetramers (1, 7). As a result, βS-globin chains on different deoxygenated HbS tetramers bind to each other to hide the hydrophobic motifs, thus initiating the nucleation of an HbS polymer. These HbS polymers grow rapidly to form long fibers that increase cellular rigidity and distort the erythrocyte membrane, leading to erythrocyte sickling, cellular energetic failure and stress, dehydration, impaired rheology and premature hemolysis (1, 7, 9) (Figure 1a). The rate of polymerization is proportional to the intraerythrocytic concentration of HbS (to the 34th power) and inversely proportional to the concentration of fetal Hb (HbF), which both replaces HbS and interferes with HbS polymerization (7, 10, 11). Co-inheritance of certain genetic factors or mutations such as hereditary persistence of HbF or α-thalassemia or βC-allele alongside βS may modulate disease severity (1, 12). As shown in Figure 2 and discussed in the section titled Current and Future Therapies Targeting Sickle Cell Disease Pathobiology, the improved understanding of the biophysical and biomolecular mechanism of HbS polymerization has inspired the development of several therapeutic strategies for SCD that interfere at different stages of intraerythrocyte HbS polymerization and altered biorheology.
Figure 2.
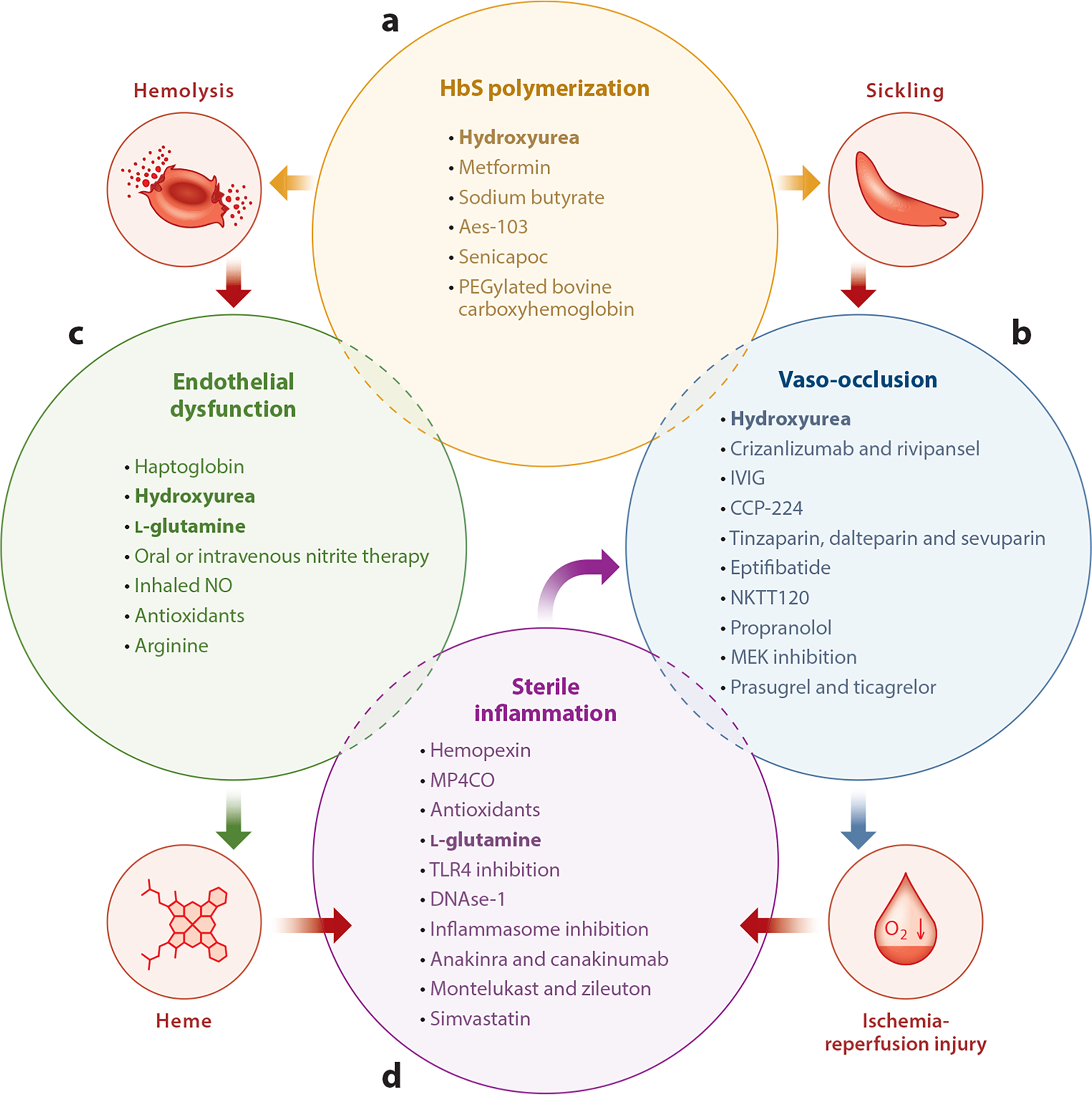
Current and future therapies targeting molecular pathobiology of sickle cell disease. (a) Drugs capable of modulating hemoglobin (Hb) polymerization, erythrocyte dehydration, and Hb oxygen affinity. (b) Drugs capable of preventing vaso-occlusion by inhibiting adhesive interactions between leukocytes, platelets, or endothelial cells and erythrocytes. (c) Drugs capable of preventing endothelial dysfunction by scavenging Hb and reactive oxygen species (ROS) or promoting nitric oxide (NO) synthesis. (d) Drugs capable of preventing sterile inflammation by scavenging heme and ROS, digesting neutrophil extracellular traps (NETs), inhibiting Toll-like receptor 4 (TLR4) or inflammasome activation, and inhibiting IL-1β-dependent innate immune signaling. Drugs approved by the US Food and Drug Administration (hydroxyurea and l-glutamine) are shown in bold font.
VASO-OCCLUSION
Vaso-occlusion, or blood vessel occlusion, leading to ischemia is the predominant pathophysiology responsible for acute systemic painful vaso-occlusive crisis (VOC) and the requirement for emergency medical care by SCD patients (13). Intravital imaging studies done in transgenic humanized SCD mice and in vitro flow chamber studies done with SCD human blood over the past decade have contributed to the current understanding of vaso-occlusion as the interplay among impaired blood rheology, increased adhesiveness of erythrocytes with inflammatory cells and vascular endothelium, and hemostatic activation (14). The blood rheology is dictated by the hematocrit, plasma viscosity, and erythrocyte deformability (9). The increased plasma viscosity, which occurs as a result of chronic hemolysis and reduced sickle erythrocyte deformability due to Hb polymerization and dehydration, contributes to impaired flow of blood through capillaries and postcapillary venules of tissues with high oxygen demand (9). Poorly deformable sickle erythrocytes may become mechanically sequestered in the microcirculation to promote transient vaso-occlusion (1, 7). Importantly, sickling-dependent damage of erythrocyte membranes also promotes exposure of adhesion molecules and binding motifs not normally expressed on erythrocytes, such as phosphatidyl serine (PS), basal cell adhesion molecule-1/Lutheran (B-CAM-1/Lu), integrin-associated protein (IAP), and intercellular-adhesion-molecule-4 (ICAM-4) (7, 9, 15). As a result of chronic anemia, the bone marrow undergoes stress reticulocytosis and releases immature erythrocytes or reticulocytes (1), which are decorated with adhesion molecules such as α4β1 integrin (VLA-4) and CD36 (15). Recent studies performed in SCD mice have also established a major role for adhesive interactions of erythrocytes and reticulocytes with inflammatory and endothelial cells in promoting vaso-occlusion in SCD (13, 14, 16, 17).
Endothelial dysfunction and sterile inflammation (discussed below), which are hallmarks of SCD, may contribute to upregulation of selectins (P- and E-), vascular-cell-adhesion-molecule-1 (VCAM-1), ICAM-1, and major leukocyte chemoattractants such as KC (in mice) or interleukin-8 (IL-8) (in humans) on endothelial cells (14, 17, 18). The inflammatory milieu in SCD may also promote activation of neutrophils, monocytes, and platelets, leading to their increased adhesion to each other and to activated endothelium (14, 17, 18). Indeed, SCD patients are known to have elevated levels of neutrophils, monocytes and platelets at baseline, and elevated levels of circulating neutrophil-platelet and monocyte-platelet aggregates in SCD human blood correlate with disease severity (19–28). Also, thrombocytopenia is a major predictor of progression of VOC in SCD patients to the potentially lethal lung injury known as acute chest syndrome (ACS) (29), suggesting a role for platelet sequestration at sites of vaso-occlusion (29–31). These clinical findings supported a role for inflammatory cells in vaso-occlusion and served as the impetus for several in vivo studies in transgenic SCD mice that led to the development of the current multicellular paradigm of vaso-occlusion (14, 17).
Epidemiological evidence (1, 32) indicates that VOC is frequently initiated by an inflammatory or environmental stimulus, including infection, hypoxia, dehydration, acidosis, or other unidentified factors. Inspired by this clinical evidence, in vivo studies have been primarily conducted by challenging SCD mice with an inflammatory stimulus such as TNFα (33), heme (34), Hb (34), hypoxia (35, 36), epinephrine (37), or lipopolysaccharide (LPS) (16, 34) to trigger vaso-occlusion. Importantly, these in vivo studies suggest that the cellular and molecular mechanisms of vaso-occlusion are also dictated by the type of organ or vascular bed. Using intravital imaging, Frenette and coworkers found that vaso-occlusion in the cremaster muscle microcirculation of TNFα-challenged SCD mice occurred primarily in postcapillary venules (13, 14, 38). Cremaster vaso-occlusion was initiated by P-/E-selectin-dependent neutrophil rolling followed by CD11a-CD18 (LFA-1) and CD11b-CD18 (Mac-1) β2-integrin-mediated firm arrest, E-selectin-dependent clustering of Mac-1 on arrested neutrophils, and capture of sickle erythrocytes by adhered neutrophils through binding of Mac-1 clusters to an unknown ligand on erythrocytes (13, 14, 38). Inhibition or deletion of endothelial E-selectin (33, 39), neutrophil Mac-1 (33, 40), CXCR2 receptor for endothelial-expressed chemokine KC (CXCL-1) (41), or reduction in circulating neutrophil counts using hydroxyurea (42) attenuated vaso-occlusion in the cremaster microcirculation of SCD mice. These studies suggested a role for erythrocyte–neutrophil–endothelium adhesion in promoting vaso-occlusion in the systemic microcirculation. However, recent evidence also supports a role for platelet–neutrophil–endothelium adhesion in promoting systemic vaso-occlusion. Platelet nucleation on arrested neutrophils leading to platelet–neutrophil aggregation was also shown to promote vaso-occlusion in the cremaster microcirculation of TNFα-challenged SCD mice, which was mediated by P-selectin and Mac-1 on activated platelets and neutrophils, respectively (43). P-selectin upregulation and Mac-1 activation on platelets and neutrophils, respectively, was shown to be dependent on phosphorylation of serine/threonine kinase AKT2, as well as nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2 (NOX2)-mediated reactive oxygen species (ROS) production (44).
The dependency of the cellular and molecular mechanisms of vaso-occlusion on the type of vascular bed is further supported by in vivo studies of skin, intestinal, and bone marrow circulation in mice. Unlike cremaster microcirculation, intravital imaging studies performed in the mucosal–intestinal microcirculation of SCD mice revealed P-selectin-dependent direct adhesion of sickle erythrocytes to activated endothelium in the postcapillary venules (45). In some studies (36, 37, 46), sickle erythrocyte adhesion to inflamed endothelium was studied in the bone marrow or skin microcirculation of non-SCD strains of mice bearing adoptively transferred, fluorescently labeled human or mice sickle erythrocytes. In one adoptive transfer study, sickle erythrocytes were observed to undergo P-selectin-dependent adhesion to endothelium in the bone marrow venules of eNOS-deficient mice exposed to hypoxia (36). In a different adoptive transfer study, cAMP-dependent protein kinase A and MEK-dependent ERK1/2 activation, which lead to binding of ICAM-4 on sickle erythrocytes to αvβ3 integrin on activated endothelium, were shown to mediate sickle erythrocyte sequestration in the skin microcirculation of epinephrine- or TNFα-challenged nude mice (37, 47–50). Interestingly, αvβ3 integrin also appeared to contribute to erythrocyte adhesion to endothelium in the skin microcirculation of nude mice (37, 47–50); however, inhibition of αvβ3 integrin was ineffective in preventing sickle erythrocyte adhesion in the bone marrow microcirculation of eNOS-deficient mice (36). Although studies done with adoptively transferred, fluorescently labeled sickle erythrocytes in non-SCD mice suggest a primary role for erythrocyte–endothelium adhesion over neutrophil–platelet or neutrophil–erythrocyte adhesion, they did not address the likelihood of erythrocytes also binding to neutrophils or platelets bound to endothelium. Regardless of these limitations, these studies suggest that the cellular and molecular paradigm of vaso-occlusion is not identical in all vascular beds.
The role for neutrophils in vaso-occlusive pathophysiology is further supported by the recent finding that translocation of Toll-like receptor 4 (TLR4) and TLR2 ligands or LPS from the gut into blood circulation contributes to increases in circulating numbers of proinflammatory neutrophils and to Mac-1-dependent neutrophil–erythrocyte aggregation in the cremaster microcirculation of TNFα-treated SCD mice (51). In support of this finding (16), our group has shown that intravenous challenge with nanogram levels of LPS promoted vaso-occlusion in the lung arterioles of SCD but not control mice. We found that vaso-occlusion in the lung involved entrapment of P-selectin-dependent platelet–neutrophil aggregates in bottle-necks located at the junction of pulmonary arterioles and capillaries (16). These aggregates may form in situ or arrive in the lung as microemboli and impair pulmonary blood flow, and were observed to consist of erythrocytes trapped within these platelet–neutrophil aggregates (16). Remarkably, therapeutic blockade of P-selectin with an inhibitory Ab prevented platelet–neutrophil aggregate–mediated lung vaso-occlusion and restored pulmonary blood flow (16). Another study identified a role for endothelial E-selectin in promoting lung injury in SCD mice following pneumococcal pneumonia and sepsis (52). In addition to the molecular interactions discussed above, in vitro flow chamber studies performed with SCD human blood or isolated cells also suggest a role for VLA-4 on reticulocytes binding to VCAM-1 on endothelial cells, as well as for GPIbα on platelets binding to Mac-1 on neutrophils, in promoting vaso-occlusion; however, in vivo evidence supporting a role for these interactions is scarce (15, 16, 53). To date, we have been unable to directly evaluate a vaso-occlusive event at the cellular and microvascular levels in humans.
Based on the above discussion, there seem to be diverse cellular and molecular mechanisms contributing to vaso-occlusion of hemoglobin S polymer–containing erythrocytes. The relative roles of leukocytes and platelets versus direct endothelial interactions with sickle erythrocytes, as well as the roles of different adhesion molecules, likely vary across vascular beds and with different inflammatory stimuli. The varied roles of platelets, neutrophils, or sickle erythrocytes in initiating and propagating vaso-occlusion in the lung versus muscle or bone marrow could be a consequence of the hypoxic environment of these vascular beds compared to the oxygen-rich environment in the lung. This diversity in cellular and molecular pathophysiology also suggests the need for intravital imaging studies to identify the cellular and molecular mechanisms of vaso-occlusion in other hypoxic organs, such as the liver, kidney, brain, or heart, that are affected in SCD, both in mouse models and, ultimately, in humans, using novel translational imaging methodologies (32).
The current understanding of vaso-occlusive pathophysiology has inspired several therapeutic approaches (Figure 2b) to prevent vaso-occlusive morbidity in SCD; these approaches are discussed in the section titled Current and Future Therapies Targeting Sickle Cell Disease Pathobiology. In addition to impaired rheology and cellular adhesion, activation of both extrinsic and intrinsic pathways of coagulation has also been shown to contribute to vaso-occlusion in SCD, and activated leukocytes, platelets, and endothelial cells have been implicated in progression of SCD-related coagulopathy (54). The discussion on coagulopathy is beyond the scope of this review, and the reader is advised to refer to recent reviews on this topic for more details (54–56). Also, a recent study has identified a protective role for a hemeoxygenase-1 (HO-1)-containing subset of circulating monocytes in vaso-occlusion, suggesting that elevated numbers of HO-1-rich patrolling monocytes in blood may provide protection from VOC (57); however, additional studies are needed to understand the molecular mechanism behind this protection and how it can be harnessed to attenuate the vaso-occlusive morbidity of SCD.
ENDOTHELIAL DYSFUNCTION
As patients with SCD live longer in high-income countries, the chronic impact of sustained hemolytic anemia and episodic vaso-occlusive events results in the progressive development of end-organ complications (58–70). As described above, HbS-containing erythrocytes with intra-cellular Hb polymer are less deformable and become entrapped within the microcirculation, resulting in episodic and sustained vaso-occlusion (1, 71, 72). Additionally, the polymer-containing erythrocytes are subject to intravascular and extravascular hemolysis, causing chronic anemia with Hb levels ranging from 6–11 g/dl (65, 73, 74). As discussed below, the process of intravascular hemolysis directly damages blood vessels (17, 75), and the resulting anemia exerts additional stress on the cardiovascular system (Figure 3) by chronically increasing cardiac output, ventricular chamber dilation, and ventricular wall stress (76, 77). The intrinsic rate of hemolytic anemia is relatively stable within an individual patient with SCD under steady-state (non-crisis) condition and is largely determined by the hemoglobin genotype (HbS, C, etc.) and HbF levels (74, 78). Patients with higher rates of hemolysis have lower steady-state Hb levels and are more likely to develop vascular injury and organ dysfunction as they age, manifesting as pulmonary hypertension, diastolic left heart disease, and renal dysfunction (proteinuria, albuminuria, and chronic kidney dysfunction) (62, 64, 65, 70, 79). Over time, patients develop vascular stiffness, which, combined with the high stroke volume in the setting of anemia, increases systolic systemic blood pressure and pulse pressure. Elevated systolic systemic blood pressures have been identified as an independent risk factor for the development of pulmonary hypertension, hypoxemia, diastolic heart dysfunction, chronic kidney injury, silent cerebral infarcts (SCI), and infarctive stroke (63, 67, 80–82). In addition to the effects of chronic anemia, intravascular hemolysis directly causes vascular injury and endothelial dysfunction (Figure 1c) and is linked to elevated pulse pressure (67). Because oxy-Hb reacts with nitric oxide (NO) in an extremely fast and essentially irreversible reaction to form inert nitrate, the release of intraerythrocytic Hb into plasma during intravascular hemolysis promotes NO scavenging reactions and impairs NO-dependent basal vasodilation. Equation 1 shows the dioxygenation reaction of NO with oxy-Hb to form nitrate and methemoglobin:
| 1. |
Cell-free Hb also promotes ROS formation, critically altering the vascular redox balance of steady-state NO production to ROS production (decreasing NO–ROS balance). NO is required for vasodilation and regulates platelet function, inflammation, cellular smooth muscle proliferation, and oxidative stress (83), and NO scavenging by cell-free plasma hemoglobin impairs endothelial function and promotes proliferative vasculopathy of the pulmonary and systemic vasculature (74, 84–87). The dysregulated redox balance may also oxidize critical enzymes in the vasculature, such as soluble guanylate cyclase, the target for NO (88). This may promote further endothelial dysfunction as the target for NO signaling is blocked. In addition to its primary effects on endothelial function and chronic vascular injury, Hb is also oxidized and degrades to release free heme and heme iron. Hb and heme activate innate immune pathways, through TLR4 and inflammasome signaling, discussed in the next section (34, 89, 90). These hemolysis products are considered erythrocyte damage-associated molecular patterns (eDAMPs) that promote and propagate sterile inflammation and oxidative stress, further impairing the redox balance (18). Release of Hb and erythrocyte ADP during hemolysis stimulates platelet activation and activates coagulation pathways, further contributing to vascular thrombosis and pulmonary hypertension (91–94). Interestingly, both hemolysis—through the release of eDAMPs—and SCD-related vaso-occlusive events—through tissue injury and release of cellular DAMPs—have the potential to activate sterile inflammation pathways (discussed in the next section), an active area of current investigation (95, 96). Based on the current understanding of hemolysis-mediated endothelial dysfunction in SCD, several therapeutic approaches have been proposed or approved by the US Food and Drug Administration (FDA) (Figure 2c) and are discussed below.
Figure 3.
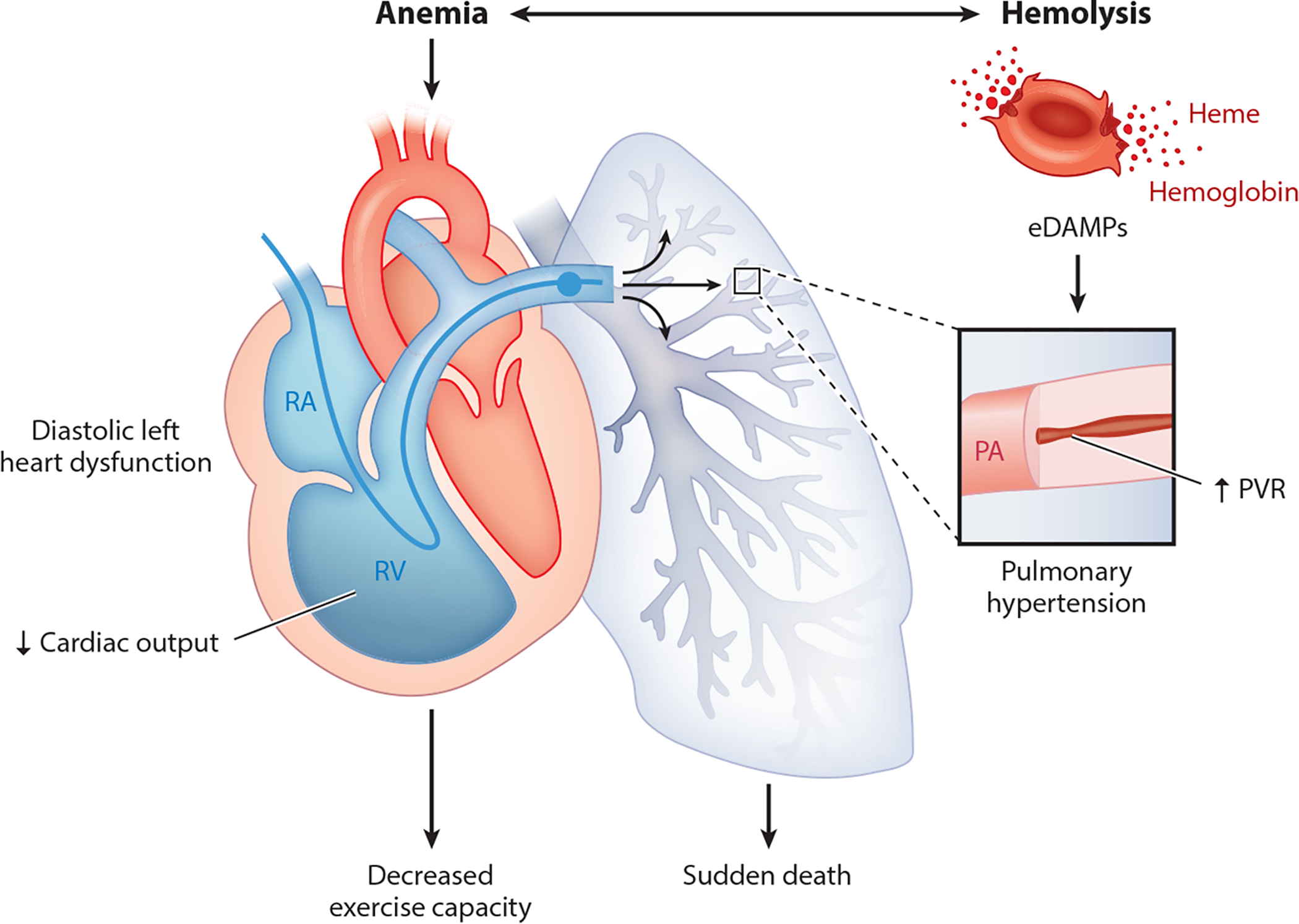
Endothelial dysfunction in sickle cell disease. Anemia and intravascular hemolysis lead to pulmonary vascular disease and diastolic heart dysfunction, both of which contribute to morbidity (reduced exercise capacity) and death (75). Figure adapted with permission from Reference 75. Abbreviations: eDAMP, erythrocyte damage-associated molecular pattern; PA, pulmonary artery; PVR, pulmonary vascular resistance; RA, right atrium; RV, right ventricle.
STERILE INFLAMMATION
Vaso-occlusion contributes to ischemia-reperfusion injury, which, along with release of eDAMPs, promotes the progression of sterile inflammation (Figure 1d) in SCD (14, 17, 18). Heme (ferrous protoporphyrin IX) and its oxidized form, hemin (ferric protoporphyrin IX), released following oxidation of Hb (discussed in the previous section), are potent TLR4 agonists that contribute to a proinflammatory and procoagulant state in SCD, characterized by activated leukocytes, platelets, endothelial cells, tissue factor, cytokine storm, NO depletion, and generation of ROS (18, 34, 54, 89, 97–99). Intravenous administration of cell-free heme has been shown to promote acute lung injury and pulmonary vascular congestion in SCD mice, which were prevented by therapeutic inhibition or genetic deletion of endothelial TLR4 (89). In another study (34), heme was shown to promote endothelial activation, leading to increased neutrophil adhesion and vaso-occlusion in skin venules, NOX-mediated ROS generation, and death in SCD mice, all of which were also dependent on endothelial TLR4. Heme-laden erythrocyte-derived microparticles have been shown to promote endothelial activation, ROS generation, and vaso-occlusion in the kidney of SCD mice by adhering to and delivering heme to endothelial cells (100). Heme has also been shown to activate TLR4 in macrophages to promote release of TNFα, KC, and leukotriene B4 (LTB4) (99, 101, 102). Thus, heme seems to promote sterile inflammation in SCD by stimulating TLR4-dependent innate immune signaling in endothelial and mononuclear cells.
Interestingly, heme appears to act through G-protein-coupled-receptor (GPCR)-dependent signaling to promote neutrophil migration, oxidative burst, neutrophil extracellular trap (NET) generation, IL-8 production, and increased neutrophil survival (103–106); however, the GPCR receptor for heme on neutrophils remains unknown (99). Activated neutrophils are known to release NETs, mesh-like structures composed of decondensed chromatin decorated with neutrophil proteases and citrullinated histones (107). NETs are released from neutrophils under diverse inflammatory conditions and promote the activation of innate immune responses, leading to tissue injury (107). Most recently (97), heme was shown to promote an oxidative burst leading to release of NETs by neutrophils in the lung microcirculation of TNFα-challenged SCD mice; NET release was inhibited following administration of the plasma heme scavenger hemopexin. Indeed, circulating markers of NETs, such as nucleosomes and elastase-α1-antitrypsin, are significantly elevated in the plasma of SCD patients at steady state, and the levels are further increased following VOC (108).
In a recent study (16), we showed that TLR4 inhibition led to reduction of P-selectin-PSGL-1-dependent platelet–neutrophil aggregation in SCD human blood flowing through microfluidic flow channels in vitro. Although it is unclear how heme promotes platelet activation in SCD, one study showed that heme enhances ADP- and epinephrine-dependent platelet aggregation (109). SCD patients are also known to be at higher risk for contracting bacterial infections compared to healthy control humans (110, 111). However, the molecular pathophysiology that contributes to this susceptibility to infections remains incompletely understood. Recently, heme was shown to promote cytoskeletal disruption leading to impaired bacterial clearance; phagocytosis; and migration by monocytes, macrophages and neutrophils. These alterations were dependent on guanine nucleotide exchange factor DOCK8-mediated activation of the GTP-binding Rho family protein Cdc42, suggesting a role for heme in promoting susceptibility to bacterial infections in SCD (112). Taken together, these studies suggest that cell-free heme contributes to TLR4 activation in mononuclear leukocytes and endothelial cells, generation of ROS by vascular cells, and NET generation by neutrophils in SCD (Figure 1d).
Besides release of cell-free heme, vaso-occlusion also contributes to progression of sterile inflammation in SCD (25, 35, 71, 72, 113–115). Repeated episodes of vaso-occlusion and reperfusion contribute to ischemia-reperfusion injury by promoting transient hypoxia, ROS generation, microvascular dysfunction, activation of innate and adaptive immune responses, and cell death (25, 35, 71, 72, 113–115). ROS-dependent damage of cellular proteins, lipids, DNA, and ribonucleic acids contributes to activation of cell death programs such as apoptosis, necrosis, autophagy, and NETosis (release of NETs by neutrophils). This in turn contributes to release of various tissue- and cell-derived DAMPs (115–117). These DAMPs promote the innate immune response by priming TLR signaling in endothelial cells and leukocytes, leading to activation of NF-κB, mitogen-activated-protein-kinase (MAPK), and type-I interferon pathways; this results in induction of proinflammatory cytokines and chemokines (95). For example, the DAMP HMGB1 is significantly elevated in the plasma of SCD patients and mice, and its levels further increase following VOC or hypoxia–reoxygenation in SCD patients and mice, respectively (118). The elevated levels of HMGB1 were also shown to promote TLR4 activity in the plasma of both SCD patients and mice (118).
Studies conducted over the past decade have identified inflammasome pathways as key regulators of sterile inflammation (95, 119, 120). Nucleotide-binding domain and leucine-rich repeat receptors (NLRs) or absent in melanoma 2 (AIM2)-like receptors (ALRs) are major components of the inflammasome complex. Inflammasomes are multimeric cytoplasmic pattern recognition receptor complexes that are activated by cell- and tissue-derived DAMPs, ROS, TLR4 activation, double-stranded DNA, NET fragments, and several unknown cell- or tissue-derived danger signals. Following activation, inflammasomes process and release activated IL-1β and IL-18 (119–121). Once released, IL-1β binds to IL-1 receptor (IL-1R) on leukocytes and vascular cells, promoting a cascade of downstream events that lead to activation of neutrophils and platelets and upregulation of E-selectin, P-selectin, VCAM-1, ICAM-1, and chemokines such as IL-8 in endothelial cells, all of which promote vaso-occlusion (14, 17) (Figure 1). Readers are advised to refer to more detailed reviews on the role of inflammasomes in sterile inflammation (119, 120). The NOD-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome is the most widely studied inflammasome complex; it consists of NLRP3, apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), and caspase-1 (119). Recently (96), cell-free heme was shown to promote NLRP3 inflammasome activation in LPS-primed macrophages, leading to IL-1β release. Deletion of NLRP3, ASC, caspase-1, or IL-1R attenuated hemolysis-induced lethality in mice, suggesting a role for NLRP3 inflammasome activation and systemic release of IL-1β in promoting hemolysis-dependent sterile inflammation (96). NLRP3 inflammasome activation in macrophages was dependent on heme-induced NOX2 activation, mitochondrial ROS production, and K+ efflux (96). In a recent study, NLRP3 and IL-1β were significantly elevated in peripheral blood mononuclear cells (PBMCs) of SCD patients compared to control human subjects, and incubation of control human PBMCs with sickle erythrocytes led to significant increases in expression of NLRP3, caspase-1, IL-1β, and IL-18 (122). Serum levels of IL-1β, IL-6 and IL-8 have been shown to be significantly elevated in SCD patients compared to healthy control subjects (123). Although NLRP3 and other inflammasome complexes are expressed in monocytes, macrophages, neutrophils, platelets, and endothelial cells (96, 124–127); heme is a potent inflammasome activator (96); and IL-1β is significantly elevated in the serum of SCD patients (122, 123), the contribution of inflammasome activation and IL-1β release by these different cell types in promoting sterile inflammation in SCD remains poorly understood. Improved understanding of these pathways in SCD can be harnessed to design improved therapies, as described in Figure 2d and the next section.
CURRENT AND FUTURE THERAPIES TARGETING SICKLE CELL DISEASE PATHOBIOLOGY
As shown in Figure 2, the current understanding of the cellular, molecular, and biophysical pathobiology of SCD has inspired several current and potential future therapeutic approaches to prevent disease morbidity. Readers are advised to refer to detailed reviews on these potential therapies (13, 47, 50, 53, 128). These therapies attenuate disease severity by interfering with different facets of SCD pathobiology, described above and in Figure 1. As shown in Figure 2a, some of the approved or potential therapies prevent HbS polymerization and rescue erythrocyte deformability by inducing HbF production (hydroxyurea, metformin, and sodium butyrate), allosterically modifying HbS oxygen affinity (5-hydoxymethyl-2-furfural or Aes-103), preventing erythrocyte dehydration (senicapoc), or serving as carbon monoxide (CO) donors (PEGylated bovine carboxyhemoglobin) (128). In addition to antipolymerization or antisickling therapies, several antiadhesion therapies are approved or being tested that seek to inhibit the multicellular adhesion cascade of vaso-occlusion (Figure 2b). These targeted therapies are variously directed at P-selectin (crizanlizumab), E-selectin (rivipansel), Mac-1 (intravenous immunoglobulin), platelet glycoprotein Ibα (CCP-224), or mitogen-activated-protein-kinase inhibitors (MEK inhibitors) to prevent erythrocyte adhesion. Other proposed or FDA-approved therapies may prevent endothelial dysfunction by scavenging cell-free Hb (haptoglobin), promoting NO production (hydroxyurea, oral or IV nitrite, inhaled NO, and oral arginine), or reducing oxidative stress (l-glutamine and antioxidants). The emerging role of sterile inflammation in SCD-associated morbidity suggests that anti-inflammatory approaches, such as therapies that induce heme degradation enzyme hemeoxygenase-1 (MP4CO), scavenge ROS (antioxidants and l-glutamine), inhibit TLR4 signaling, degrade NETs (DNase-1), inhibit leukotrienes, or inhibit inflammasome- or IL-1β-dependent signaling, could be beneficial in SCD (128, 129). Interestingly, IL-1RA-blocking Ab (anakinra) and IL-1β-blocking Ab (canakinumab) are already FDA approved as anti-inflammatory biologics for the treatment of rheumatoid arthritis (130) and NLRP3-inflammasome-mediated cryopyrin-associated periodic syndrome (CAPS) (131), respectively. The existing evidence justifies the need for clinical trials to test the safety and efficacy of repurposing these drugs for SCD and also highlights the need for more studies to refine our understanding of the role of inflammasome pathways in SCD.
THE PERFUSION PARADOX OF SICKLE CELL DISEASE: PATHOLOGY IN THE CENTRAL NERVOUS AND CARDIOPULMONARY SYSTEMS
The complex pathways described above converge to cause large- and small-vessel vasculopathy in SCD. From the standpoint of organ perfusion, both hypoperfusion of the microcirculation (due to microvascular occlusion or altered vasoregulation) and hyperperfusion of the systemic macrocirculation and major organ systems (due to anemia and fixed stenosis) coexist, a phenomenon that has been referred to as the perfusion paradox of SCD (132). While perfusion abnormalities are widespread (Table 1), their effects on the central nervous system (CNS) and cardiopulmonary system are profound and responsible for hallmark, devastating complications in children and adults with SCD. The following sections describe the most important pathology affecting the CNS and the cardiopulmonary system.
Table 1.
Main complications of SCD by organ system
| System | Complication | Pathology and imaging findings | Proposed mechanisms |
|---|---|---|---|
| Central nervous system | Stroke |
|
|
| Cognitive impairment |
|
|
|
|
Cardiopulmonary system |
Pulmonary hypertension |
|
|
| Acute chest syndrome |
|
|
|
| Restrictive lung disease |
|
|
|
|
Genitourinary system |
Priapism |
|
|
| Chronic kidney disease |
|
|
|
| Papillary necrosis of the kidney |
|
|
|
| Hepatic system | Hepatic sequestration |
|
|
| Hepatic crisis and intrahepatic cholestasis |
|
|
Abbreviations: CBF, cerebral blood flow; CT, computed tomography; CTEPH, chronic thromboembolic pulmonary hypertension; DLCO, diffusive capacity of the lungs for carbon monoxide; ICA, internal carotid artery; MCA, middle carotid artery; NO, nitric oxide; PDE-5, phosphodiesterase-5; RPG, retrograde pyelography; SCD, sickle cell disease; TLC, total lung capacity; TLR4, Toll-like receptor 4.
Large-Vessel Vasculopathy in the Central Nervous System
The CNS is severely affected in SCD, with both children and adults suffering from cerebrovascular complications. One of the most striking manifestations of SCD is the high incidence of stroke in young children (aged 2–9) with sickle cell anemia; stroke affected up to 10% of children prior to the implementation of screening programs (81, 133). Numerous autopsy case reports and series dating back several decades have outlined the major pathological lesions of pediatric stroke in SCD. These studies have shown that the large and medium-sized branches of the internal carotid artery are affected. Lesions include intimal hyperplasia leading to obliteration of the lumen, degeneration of the internal elastic lamina, and intraluminal thrombosis (134, 135). It is unclear if vasospasm occurs in the acute setting, but such a process could be analogous to the cerebral vasospasm seen in the setting of subarachnoid hemorrhage. Stenosis of the large branches of the internal carotid artery predisposes children to devastating strokes.
The main mechanism underlying the high susceptibility to stroke of children with SCD involves the phenomenon known as decreased cerebrovascular reserve. Highly metabolically active areas of the developing brain, such as cortical gray areas, are very dependent on the cerebral metabolic rate of oxygen utilization. This, in turn, is a product of cerebral arterial oxygen content, cerebral blood flow, and cerebral oxygen extraction. Cerebral blood flow is increased at baseline in children with SCD (136) to compensate for anemia and hemodynamically significant stenosis. Furthermore, both magnetic resonance imaging (MRI) and near infrared spectroscopy studies have shown that the cerebral autoregulatory capacity, i.e., the capacity to adjust vessel volume in response to carbon monoxide challenge or systemic blood pressure changes, is impaired in SCD (137, 138). Finally, the oxygen extraction fraction is also increased at baseline (139, 140). As a result of maximized compensatory mechanisms at steady state, when events characterized by acute or chronic anemia, such as aplastic crises from parvovirus B19, occur, children with SCD may have no residual reserve to meet the acutely increased oxygen demand. In this setting, cortical brain areas and cortical and white matter watershed (border zone) areas (134, 141, 142), which are particularly vulnerable to ischemia, may undergo acute infarction. In cortical areas, the ischemic insult may be evident in MRI as gray matter atrophy, cortical thinning (142, 143), and atrophy of specific subcortical regions (144). There is recent evidence that cortical atrophy progresses in children with SCD at a rate similar to that of adults without SCD but with small-vessel disease (0.6–1% per year)—a worrisome finding that suggests accelerated brain aging in SCD (145).
Over the past three decades, epidemiological studies have shed light on the natural history and risk factors of pediatric stroke in SCD, and advances in neuroimaging methods have allowed better characterization of the neurovascular phenotype in SCD. Children with sickle cell anemia have a 100-fold risk of developing stroke as compared to children without SCD (146), and 70% of children with SCD will experience a stroke recurrence (81). The fundamental role of anemia in ischemic stroke in children with SCD has been confirmed by clinical practice, where red blood cell transfusion rapidly improves stroke symptoms and cerebral tissue oxygen saturation in children (147), and by a landmark clinical trial showing that chronic prophylactic transfusions prevent stroke in 90% of children with high cerebral blood velocities as measured by transcranial Doppler (148). In addition to anemia, stroke is also associated with a number of other risk factors. Observational studies have shown that reticulocytosis (149), low HbF levels, leukocytosis (150, 151), and complications such as ACS (81) are associated with an increased risk of stroke. In addition, therapy with hydroxyurea, which induces HbF and reduces white blood cell count, reticulocyte count, and hemolysis, is effective in the primary prevention of stroke (152). The beneficial effects of transfusion, therefore, may be both an increase in cerebral arterial oxygen carrying capacity and a reduction of HbS and its downstream deleterious effects on blood rheology.
While transcranial Doppler screening and prophylactic transfusions both represent major breakthroughs in the care of patients with SCD, very little progress has been made in the elucidation of the mechanisms leading to the primary vascular lesions, particularly large-vessel stenosis. This knowledge gap is partly due to the lack of adequate animal models of stroke in SCD. Young sickle mice do not commonly develop large-vessel vasculopathy and stroke (153, 154), and SCD mouse models that employ additional ischemic stimuli, such as carotid ligation or acute hypoxia, do not fully recapitulate human pathology. SCD mouse models do, however, exhibit certain specific aspects of the cerebral pathology seen with sickle cell anemia, including decreased brain oxygen tension, increased cerebral blood flow (155), decreased blood flow regulation, and microinfarcts (156, 157). In addition, elevations of hypoxia inducible factor-1α (HIF-1α) expression have confirmed the presence of tissue-level hypoxia (157). One unifying hypothesis that takes into consideration both anemia and other sickle-specific factors was proposed by Hillery & Panepinto (158) and posits that increased shear stress from chronic anemia–induced high carotid blood velocity and systemic endotheliopathy leads to endothelial injury, particularly at sites of bifurcations. Endothelial dysfunction from hemolysis, NO deficiency, increased adhesiveness, and oxidative stress compound the injury and lead to hyperplasia of the vessel wall. Platelet recruitment and hemostatic activation then further contribute to obliteration of the lumen (158).
Among the SCD-specific factors, the role of hemolysis has garnered particular attention based on several observations. The finding of a similar pattern of lesions in the pulmonary arteries (described below) and large internal carotid branches in SCD (159) is intriguing, particularly because there is also an overlap in the risk factors that lead to pulmonary and cerebrovascular pathology, namely elevated systolic blood pressure and anemia. Thus, a unique mechanism explaining both lesions may be hypothesized. Since hemolysis has been strongly implicated in the pathogenesis of pulmonary hypertension in SCD, it is intuitive that it would also be responsible for large-vessel stenosis in the CNS. This hypothesis is supported by the results of a logistic regression analysis of the risk factors of elevated transcranial Doppler velocity in children, showing that elevated lactate dehydrogenase is independently associated with increased risk of transcranial Doppler ≥2 m/sec (OR per IU/L = 1.001, 95% CI 1.000–1.002; P = 0.047) (160). Studies in adults with SCD lend further support to the role of hemolysis by showing a link between the hyperhemolysis phenotype of SCD and stroke risk (17). It was therefore unexpected that normalization of transcranial Doppler velocity with transfusion in children was not associated with a reduction in markers of hemolysis in the Optimizing Primary Stroke Prevention in Sickle Cell Anemia (STOP 2) trial (161). This discordance may be explained by the observation that the approximately 22% of children who did not achieve normalization of transcranial Doppler velocity in spite of optimal transfusion in the trial were also protected from stroke (162). In these children, the beneficial effect of transfusion may have been mitigation of hemolysis-induced endothelial dysfunction rather than reduction of hyperperfusion.
Small-Vessel Vasculopathy in the Central Nervous System
Another significant limitation in the understanding of the cerebral pathology in SCD relates to the etiology of cerebral small-vessel vasculopathy. This is an important area of focus because MRI imaging has revealed that small-vessel lesions are the most common cerebrovascular lesions in SCD, with a prevalence of 13% at 14 months of age (163) that progressively rises throughout childhood (164) such that, by adulthood, it is approximately 50% (165, 166). In the pediatric SCD literature, these lesions have been commonly referred to as SCIs and have been defined as hyperintense lesions by T2-weighted MRI, >3 mm (167) or, alternatively, >5 mm in size (165) and detected in two orthogonal planes. In the non-SCD literature and in adult patients with SCD, SCIs have been commonly referred to as white matter hyperintensities or lacunar infarcts; all share partially overlapping MRI findings. White matter hyperintensities may be a more appropriate term since the association of SCI burden with decreased cognitive function (168–171) implies that SCIs are not clinically silent. As further evidence of the clinical importance of SCIs, the Silent Infarct Transfusion (SIT) trial has shown that, without transfusion, SCI burden at baseline predicts further accrual of ischemic lesions and stroke (172), confirming the Cooperative Study of Sickle Cell Disease (CSSCD) report of a higher risk of stroke in children with SCI (173). Anemia clearly also plays a role in the development of SCI, as demonstrated by studies in children without SCD, where an acute, severe drop in hemoglobin has been associated with the development of new white matter hyperintensities. In children with SCD, the SIT trial has shown that transfusion may reduce incident SCI, although SCIs were part of a composite endpoint that also included stroke, thus hampering the analysis of the effect of transfusion on SCI alone (172). Regardless of the effect of anemia, the cerebral arterial and venous microcirculation may be directly involved by the processes of vaso-occlusion, ischemia-reperfusion injury, and endothelial dysfunction. Old autopsy studies did show diffuse thickening and sclerosis of intracerebral arterioles (174), and more recent MRI evidence shows potential involvement of deep medullary veins (175, 176). Alternatively, decreased autoregulation may allow the detrimental effects of elevated cerebral blood flow, pulse pressure (67), and velocity to be transmitted to the microcirculation and cause damage, as occurs in older individuals with atherosclerosis.
One particularly subtle cerebral manifestation of SCD is cognitive impairment. While the link between cognitive impairment and overt and silent infarction is well established, there is evidence that children without MRI evidence of small-vessel disease also develop cognitive impairment (177). In adults, the link between conventional measures of small-vessel disease, such as lacunar infarcts, and cognitive function is even less clear. The largest study to date of cognitive function in adults with SCD has shown that 33% of a cohort of patients with homozygous SCD and without severe complications performed 1 standard deviation worse than the population mean in cognitive tests and had a much higher prevalence of lacunar infarcts (13% versus 2%). However, there was no association between lacunae and cognitive function (178). More sophisticated MRI protocols that include both global and regional assessment of cortical and white matter areas and better resolution offered by higher magnet fields [e.g., 7 Tesla (175, 179)] may be needed to identify the neuroimaging signature of cognitive impairment in adults with SCD and children with seemingly normal MRI scans.
Vasculopathy as a Risk Factor for Intracerebral Hemorrhage
Intracerebral hemorrhage preferentially affects young adults with SCD and is particularly common in those suffering from aneurysms and Moyamoya syndrome (180). Moyamoya syndrome refers to a pattern of vascularization that develops after occlusion of the large intracranial segments of the internal carotids and compensatory engorgement of perforator vessels to internal brain structures; the pattern appears angiographically as a puff of smoke. Both lesions occur with high prevalence in SCD; the prevalence of aneurysms may be as high as 15% in women with SCD aged 30–39 (181), and the prevalence of Moyamoya syndrome may be as high as 43% in patients who have experienced a pediatric stroke (180). Aneurysms are typically saccular and <5 mm in size and may coexist with vasculopathy and a Moyamoya pattern of cerebral vascularization (182). Autopsy review of patients with Moyamoya syndrome revealed that all three layers of the large intracerebral arteries were affected, with intimal hyperplasia, medial atrophy with fibrosis, and adventitial fibrosis (135).
Development of Pulmonary Hypertension and Left Ventricular Diastolic Heart Dysfunction in Adults with Sickle Cell Disease
There is a surprising similarity between the arteriopathy of the CNS and pulmonary arterial vasculature in SCD, and both are epidemiologically and mechanistically related to the severity of the hemolytic anemia (183). Pulmonary arterial hypertension is caused by progressive smooth muscle and intimal proliferation and in situ thrombosis, ultimately obliterating the pulmonary arterioles and increasing pulmonary vascular resistance (PVR) (184). Over time, the right heart begins to fail as afterload increases, leading to progressive dyspnea, reduced exercise capacity, and increased risk of acute cor pulmonale and sudden cardiac death. Pulmonary arterial hypertension is defined by a mean pulmonary artery pressure of ≥25 mm Hg, with a left ventricular end-diastolic pressure of ≤15 mm Hg and a PVR value of >3 Wood units, indicating an increase in the precapillary pulmonary pressures [World Health Organization (WHO) group 1 classification] (185). However, several recent studies suggest that a mean pulmonary artery pressure between 20 and 24 mm Hg is associated with impaired exercise capacity and higher risk of death, suggesting that even borderline increases in pulmonary pressure are relevant (186, 187). Pulmonary venous hypertension is caused by increases in pressures downstream of the pulmonary arterioles and capillaries, typically related to increases in left heart filling pressures caused by diastolic or systolic heart failure (WHO group 2 classification). Both hemodynamic forms of pulmonary hypertension are independent predictors of death in the adult SCD patient population (66).
Adult patients with SCD are screened for pulmonary hypertension using noninvasive Doppler echocardiography, which can be used to estimate pulmonary artery systolic pressure, or with a blood test to measure the plasma level of N-terminal pro–brain natriuretic peptide (NT-proBNP) (75, 184). Patients for whom these screening tests or clinical signs and symptoms of right heart failure indicate a high risk of pulmonary hypertension should undergo definitive testing with invasive right heart catheterization.
Doppler-echocardiographic measurement of the tricuspid regurgitant jet velocity is used to estimate the pulmonary artery systolic pressure. The velocity (V) of blood flowing backward from the right ventricle to the right atrium can be quantified by Doppler and is related to right ventricular systolic pressure (P = 4V2). The measured tricuspid regurgitant jet velocity has been evaluated in numerous studies and even mild increases of >2.5–2.7 m/s are associated with increased risk of death (58, 77, 110, 188–190). A meta-analysis of 45 screening studies from 15 countries of more than 6,000 patients indicates that the prevalence of elevated tricuspid regurgitant jet velocity ≥2.5 m/s is 30% (range 26–35%) in adults. Patients with elevated tricuspid regurgitant jet velocity walked an estimated 30.4 (6.9–53.9) meters less than those without elevated tricuspid regurgitant jet velocity, with an associated hazard ratio for death of 4.9 (2.4–9.7) (191). Another large population screening study confirmed that a tricuspid regurgitant jet velocity ≥2.5 m/s was associated with a hazard ratio of 6.81 in multivariate analysis and rose linearly above this value (50% of death at tricuspid regurgitant jet velocity of 3.2 m/s) (192).
NT-proBNP, a prepro hormone released from cardiac myocytes of the left and right ventricles in response to pressure overload and wall stress, also identifies patients at higher risk of having pulmonary hypertension, with lower exercise capacity and increased mortality risk. This has been shown in archived samples from the historic Multi-centers Trial of Hydroxyurea (193) and the CSSCD cohorts (193), as well as the more recent National Institutes of Health-Pulmonary Hypertension (NIH-PH) and Treatment of Pulmonary Hypertension and Sickle Cell Disease with Sildenafil Treatment cohorts (62). A value of ≥160 pg/ml identifies SCD patients at higher risk of having pulmonary hypertension and death.
Three large prospective screening studies performed in adult patients with SCD evaluated hemodynamic parameters by right heart catheterization in subjects at risk for pulmonary hypertension and their relationship to prospective mortality rates (66, 194–196). The largest study with the longest follow-up is the NIH-PH cohort study, which screened 531 patients and followed them for a median of 4.4 years (66, 194). Of these patients, 84 received right heart catheterizations due to suspected pulmonary hypertension, and 55 (10.4%) were diagnosed with pulmonary hypertension based on a mean pulmonary artery pressure >25 mm Hg. Among the group with pulmonary hypertension, 56.4% had precapillary pulmonary arterial hypertension, with a pulmonary artery occlusion pressure ≤15 mm Hg, and the remainder had pulmonary venous hypertension, with elevated pulmonary artery occlusion pressures >15 mm Hg. The diagnosis of both forms of pulmonary hypertension was associated with a high risk of death, and multivariate analysis of hemodynamic variables identified systolic pulmonary artery pressure, pulmonary pulse pressure, transpulmonary gradient, and PVR as predictors of mortality (66).
While the development of pulmonary hypertension is related to pulmonary vascular disease in the setting of intravascular hemolysis and thrombotic events, it can also occur secondary to diastolic left heart disease (77, 82). Measures of diastolic dysfunction obtained by cardiac echocardiography are associated with excess mortality, even after adjustment for tricuspid regurgitant jet velocity, with a risk ratio of 3.5 (82). Remarkably, the presence of both diastolic dysfunction and an elevated tricuspid regurgitant jet velocity is associated with a risk ratio for death of 12.0 (82). Recent studies in SCD mice further highlight the importance of diastolic dysfunction in SCD, and myocardial fibrosis is reported to occur both in these mice and in patients (the latter measured by extracellular volume using cardiac MRI imaging) (197–199). The reason that patients with SCD develop diastolic left heart disease is the subject of current research, with various studies suggesting that it stems from direct toxic effects of heme or hemoglobin on the myocardium (200), dilation caused by anemia that reduces the ability of the heart to relax during diastole (77, 82), or myocardial fibrosis secondary to lifelong episodic microinfarctions from vaso-occlusive events (197, 199).
While not a cardiac complication, the development of chronic kidney disease is a risk factor for the development of pulmonary hypertension and appears to occur as a consequence of chronic hemolysis and the injurious effects of filtered cell-free hemoglobin. The kidneys are among the most commonly affected organs in patients with SCD (70, 201), and the presence of chronic kidney disease is an independent predictor of pulmonary vascular disease and early mortality in adults with SCD (68, 202). Proposed mechanisms for SCD nephropathy include hemoglobinuria, ischemia-reperfusion injury, hyperfiltration, and hypertension (203).
CONVERGENCE OF MULTIPLE PATHOGENIC PATHWAYS: ACUTE CHEST SYNDROME
ACS is an acute lung injury syndrome that affects children and adults with any of the major SCD subtypes. While its clinical presentation is dramatic and the diagnosis straightforward (204), its pathogenesis is complex and the exact cause frequently unknown. Epidemiologically, ACS is a complication of VOC, based on its typical development 2–3 days after the onset of vaso-occlusive pain (111); it is more common in children and, if untreated, has a high mortality [9% in adults (111)]. The link to VOC is also mechanistic, since inhibition of HbS polymerization with hydroxyurea reduces the incidence of ACS (205). The fundamental lesion of ACS is a hyperinflammatory event leading to classic acute lung injury (Figure 4), defined by alveolar-capillary leak and neutrophilic inflammation, most commonly in two or more dependent lobes, which may be radiographically and clinically indistinguishable from multilobar pneumonia (206). Unlike pneumonia, ACS is the result of intrinsic SCD pathology. Even in those cases where a microbial trigger is identified, infarction is the product of the interplay between the infectious agent and SCD-related host vulnerability. As in acute lung injury observed in other conditions (e.g., sepsis, blood transfusion, trauma) (207), an initial pathogenic stimulus leads to a hyperinflammatory response in the lungs, with release of cytokines, engagement and massive recruitment of neutrophils, and sterile inflammation that leads to breakdown of the endothelial–epithelial barrier (gap formation), alveolar capillary leak, and disruption of the oxygen exchange (Figure 4). In its most extreme manifestation (207), acute lung injury presents with or evolves into acute respiratory distress syndrome.
Figure 4.
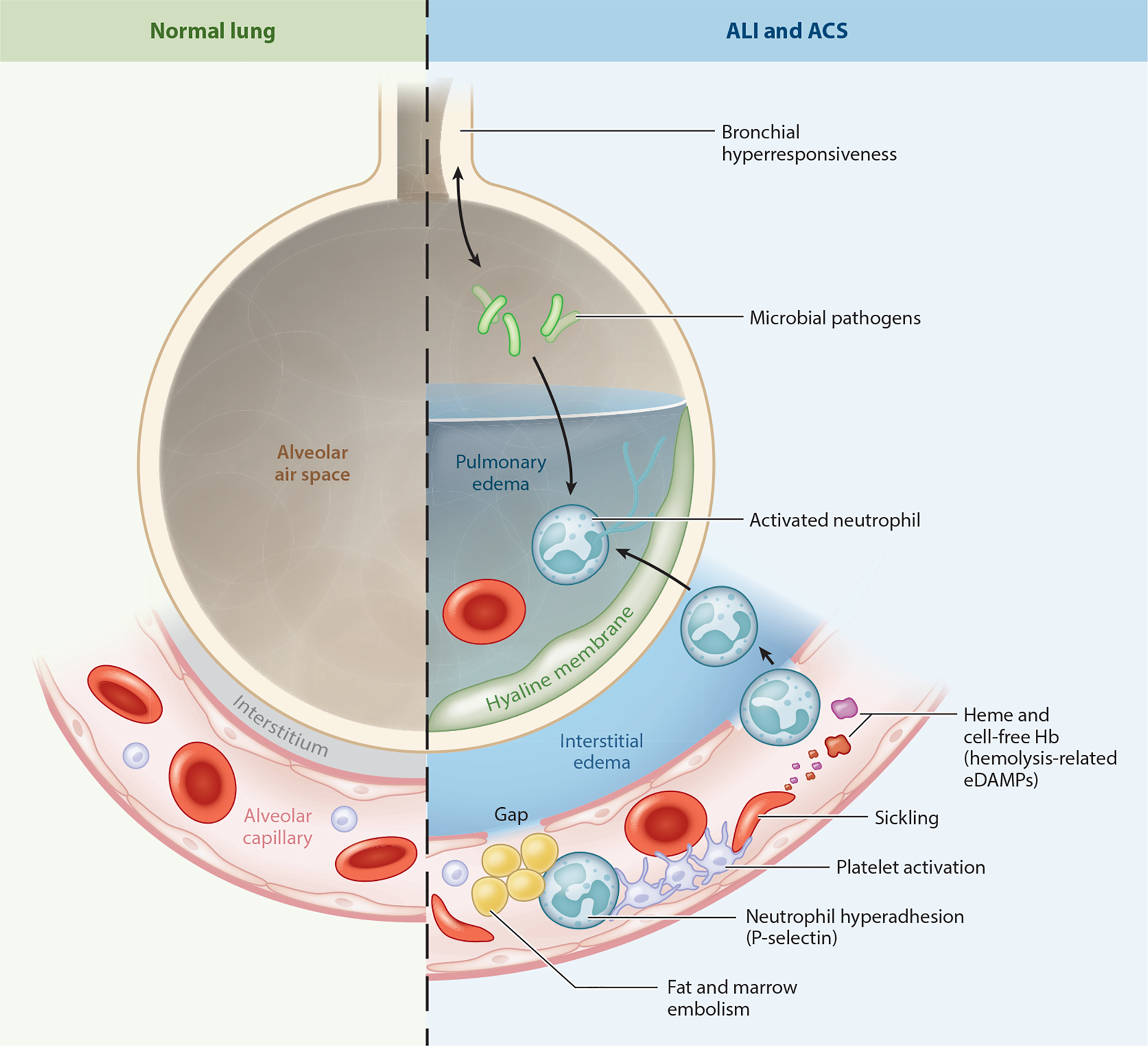
Mechanisms leading to the development of acute lung injury (ALI) and acute chest syndrome (ACS). Microbial pathogens interact with alveolar epithelial and inflammatory cells to promote release of proinflammatory cytokines. Heme and cell-free hemoglobin released from lysed sickle erythrocytes function as erythrocyte damage-associated molecular patterns (eDAMPs) to trigger Toll-like receptor 4 and inflammasome signaling in vascular and inflammatory cells. P-selectin-dependent platelet–neutrophil aggregates promote vaso-occlusion and microthrombosis in lung arterioles, leading to loss of pulmonary blood flow. Fat and marrow emboli released from necrotic bones obstruct the microcirculation and stimulate further inflammation by activating phospholipase A and other enzymes. Lung vaso-occlusion promotes ischemia-reperfusion injury, failure of the blood–air barrier, infarction, alveolar flooding, neutrophil recruitment, degranulation, release of neutrophil extracellular traps (NETosis), and oxidative burst, leading to epithelial injury, formation of hyaline membranes, and respiratory failure, all of which are hallmarks of ALI and ACS.
Five major pathologic mechanisms leading to ACS have been identified; as a whole, these mechanisms represent a compendium of SCD pathology and are instructive of the heterogeneity of the SCD phenotype.
Infection is the most common ACS trigger in children; common isolates include atypical microorganisms and Streptococcus pneumoniae (208). While some microorganisms are particularly virulent because of the functional asplenia that develops in infancy in sickle cell anemia, the lung, in general, is more vulnerable to infection in children with SCD. Bronchial hyper-reactivity, asthma, and chronic airway inflammation (209) are highly prevalent in children with SCD and may result in a proinflammatory milieu that amplifies cellular and humoral immunity to airway pathogens, thus leading to a paroxysmal, detrimental response (210, 211).
Acute bouts of bone ischemia during VOC lead, in severe cases, to necrosis of the bone marrow. Edema and increased intraosteal pressure in turn lead to embolization of fat and bone marrow to the microcirculation. As in the case of other sources of embolism (e.g., thrombi, amniotic fluid), fat and marrow emboli lodge in the pulmonary microcirculation, resulting in infarction (212). Alternatively, fat emboli may activate alveolar phospholipase A2 and other proinflammatory and pro-oxidant lung enzymes, leading to injury (213). Fat embolism is suspected when abundant lipid-laden macrophages are detected in the bronchoalveolar lavage, a sensitive but nonspecific test (214). In fulminant cases, collectively referred to as fat emboli syndrome, miliary dissemination of emboli to multiple organ systems may ensue. Fat emboli syndrome is often fatal, even with aggressive transfusion and supportive care (215).
The role of hemolysis in ACS is underscored by clinical observations linking severe hemolysis to increased risk (216, 217) and by animal models showing that heme is directly responsible for acute lung injury in SCD mouse models (89). This observation is compounded by the finding that high levels of hemopexin, the enzyme that scavenges heme, can rescue acute lung injury (89), while HO-1 deficiency potentiates it (57).
Many lines of evidence point to thrombosis as a significant ACS trigger. Recent autopsy findings have revealed platelet thrombi and increased endothelial von Willebrand factor deposition in the lung microvasculature of patients who died from ACS (218). This finding mirrors observations from our group of in vivo platelet–neutrophil aggregates and microthrombi-mediated occlusion of pulmonary arterioles in SCD mice (16). Interestingly, patients with evidence of platelet thrombi in lung arterioles post mortem had a higher platelet count at the onset of ACS (218). Elevated initial platelet count and dramatic drops during VOC have been found to portend a poor prognosis in ACS (29, 208, 219). To lend further support to the role of thrombosis in ACS, in situ pulmonary thrombosis has been found to complicate approximately 17% of ACS cases (219). Taken together and in the context of the well-described hemostatic activation at baseline and its increase during VOC in SCD (91), these findings suggest that the prothrombotic environment of VOC and ACS is conducive to pulmonary thrombosis.
Hypoventilation, as a result of pain with inspiration deriving from rib infarction (220) or oversedation from opiate analgesia, may lead to atelectasis, which is a known risk factor for pneumonia and lung injury (221).
The five mechanisms described above may coexist or independently cause ACS; any initial lung insult is bound to result in lung ventilation–perfusion mismatch, hypoxemia, and a potentiation of the initial pathogenic triggers, a phenomenon that has been described as “the vicious cycle of ACS” (110). It is also likely that ACS triggers or insults that activate neutrophils and platelets, in the setting of intensifying hemolytic anemia with release of eDAMPs, will propagate sterile inflammatory pathways through TLR4 and inflammasome signaling (discussed in Figure 1).
There have been attempts to link ACS to pulmonary fibrosis, another known manifestation of chronic lung disease in SCD, as it is intuitive that repeated episodes of parenchymal injury would result in deposition of fibrotic tissue. While ACS may accelerate restrictive lung disease (222), there is evidence that other pathologic insults are necessary for the development of lung fibrosis. Most recently, studies have found that elevated baseline levels of circulating fibrocytes, a type of mesenchymal, bone marrow–derived cell, are responsible for lung fibrogenesis in SCD mouse models (223). In humans, circulating fibrocytes are present in high numbers; are activated in SCD, particularly in VOC; and are associated with restrictive pulmonary function test patterns (224). These observations suggest that the contribution of chronic pathology may compound the role of recurrent ACS in the development of restrictive lung disease in SCD.
CONCLUSION
The interplay among genetics, HbS polymerization–dependent hemolysis and sickling, vaso-occlusion-dependent ischemia-reperfusion injury, endothelial dysfunction–dependent vasculopathy, and sterile inflammation contributes to the pathophysiology of SCD, which promotes acute and chronic complications of the CNS, heart, lung, kidney, liver, and other organs. Basic science and clinical studies over past decade have led to the understanding of the cellular, molecular, and biophysical mechanisms that promote these pathophysiological events and inspired the development of several prophylactic therapies that are either FDA approved or currently in clinical trials. However, recent findings showing a potential role for innate immune pathways in promoting sterile inflammation in SCD suggest that our current understanding of the SCD pathophysiology is still incomplete, and future studies should be aimed at harnessing the innate immune pathways to design new therapies for SCD.
DISCLOSURE STATEMENT
M.T.G. is a co-inventor of pending patent applications and planned patents directed to the use of recombinant neuroglobin and heme-based molecules as antidotes for CO poisoning, which have recently been licensed by Globin Solutions, Inc. He is a shareholder, advisor, and director at Globin Solutions, Inc. Additionally, and unrelated to CO poisoning, he is a co-inventor on patents directed to the use of nitrite salts in cardiovascular diseases, which have been licensed by United Therapeutics and Hope Pharmaceuticals, and is a co-investigator in a research collaboration with Bayer Pharmaceuticals to evaluate riociguat as a treatment for patients with SCD. M.T.G. has served as a consultant for Epizyme, Inc., Actelion Clinical Research, Inc., Acceleron Pharma, Inc., Catalyst Biosciences, Inc., Modus Therapeutics, and United Therapeutics Corporation. M.T.G. is also on Bayer HealthCare, LLC’s Heart and Vascular Disease Research Advisory Board. The other authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
ACKNOWLEDGMENTS
This work was supported by a National Heart, Lung and Blood Institute (NHLBI) grant to P.S. (1R01HL128297); an American Heart Association grant to P.S. (18TPA34170588); NHLBI grants to M.T.G. (2R01HL098032, 1R01HL125886, 5P01HL103455, and T32 HL110849); Institute for Transfusion Medicine and the Hemophilia Center of Western Pennsylvania funding to M.T.G.; and NHLBI grants to E.M.N. (1K23HL112848-01A1 and 5R01HL127107).
LITERATURE CITED
- 1.Rees DC, Williams TN, Gladwin MT. 2010. Sickle-cell disease. Lancet 376:2018–31 [DOI] [PubMed] [Google Scholar]
- 2.Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, et al. 2013. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet 381:142–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, et al. 2010. Global distribution of the sickle cell gene and geographical confirmation of the malaria hypothesis. Nat. Commun 1:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mortal GBD. Causes Death Collab. 2015. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 385:117–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ingram VM. 1956. A specific chemical difference between the globins of normal human and sickle-cell anaemia haemoglobin. Nature 178:792–94 [DOI] [PubMed] [Google Scholar]
- 6.Ingram VM. 1957. Gene mutations in human haemoglobin: the chemical difference between normal and sickle cell haemoglobin. Nature 180:326–28 [DOI] [PubMed] [Google Scholar]
- 7.Bunn HF. 1997. Pathogenesis and treatment of sickle cell disease. N. Engl. J. Med 337:762–69 [DOI] [PubMed] [Google Scholar]
- 8.Pauling L, Itano HA, Singer SJ, Wells IC. 1949. Sickle cell anemia, a molecular disease. Science 110:543–48 [DOI] [PubMed] [Google Scholar]
- 9.Barabino GA, Platt MO, Kaul DK. 2010. Sickle cell biomechanics. Annu. Rev. Biomed. Eng 12:345–67 [DOI] [PubMed] [Google Scholar]
- 10.Noguchi CT, Rodgers GP, Serjeant G, Schechter AN. 1988. Levels of fetal hemoglobin necessary for treatment of sickle cell disease. N. Engl. J. Med 318:96–99 [DOI] [PubMed] [Google Scholar]
- 11.Brittenham GM, Schechter AN, Noguchi CT. 1985. Hemoglobin S polymerization: primary determinant of the hemolytic and clinical severity of the sickling syndromes. Blood 65:183–89 [PubMed] [Google Scholar]
- 12.Ware RE, de Montalembert M, Tshilolo L, Abboud MR. 2017. Sickle cell disease. Lancet 390:311–23 [DOI] [PubMed] [Google Scholar]
- 13.Manwani D, Frenette PS. 2013. Vaso-occlusion in sickle cell disease: pathophysiology and novel targeted therapies. Blood 122:3892–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang D, Xu C, Manwani D, Frenette PS. 2016. Neutrophils, platelets, and inflammatory pathways at the nexus of sickle cell disease pathophysiology. Blood 127:801–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaul DK, Finnegan E, Barabino GA. 2009. Sickle red cell-endothelium interactions. Microcirculation 16:97–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bennewitz MF, Jimenez MA, Vats R, Tutuncuoglu E, Jonassaint J, et al. 2017. Lung vaso-occlusion in sickle cell disease mediated by arteriolar neutrophil-platelet microemboli. JCI Insight 2:e89761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kato GJ, Steinberg MH, Gladwin MT. 2017. Intravascular hemolysis and the pathophysiology of sickle cell disease. J. Clin. Invest 127:750–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gladwin MT, Ofori-Acquah SF. 2014. Erythroid DAMPs drive inflammation in SCD. Blood 123:3689–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kenny MW, George AJ, Stuart J. 1980. Platelet hyperactivity in sickle-cell disease: a consequence of hyposplenism. J. Clin. Pathol 33:622–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mohan JS, Lip GY, Bareford D, Blann AD. 2006. Platelet P-selectin and platelet mass, volume and component in sickle cell disease: relationship to genotype. Thromb. Res 117:623–29 [DOI] [PubMed] [Google Scholar]
- 21.Westwick J, Watson-Williams EJ, Krishnamurthi S, Marks G, Ellis V, et al. 1983. Platelet activation during steady state sickle cell disease. J. Med 14:17–36 [PubMed] [Google Scholar]
- 22.Curtis SA, Danda N, Etzion Z, Cohen HW, Billett HH. 2015. Elevated steady state WBC and platelet counts are associated with frequent emergency room use in adults with sickle cell anemia. PLOS ONE 10:e0133116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frelinger AL 3rd, Jakubowski JA, Brooks JK, Carmichael SL, Berny-Lang MA, et al. 2014. Platelet activation and inhibition in sickle cell disease (pains) study. Platelets 25:27–35 [DOI] [PubMed] [Google Scholar]
- 24.Dominical VM, Samsel L, Nichols JS, Costa FF, McCoy JP Jr., et al. 2014. Prominent role of platelets in the formation of circulating neutrophil-red cell heterocellular aggregates in sickle cell anemia. Haematologica 99:e214–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Polanowska-Grabowska R, Wallace K, Field JJ, Chen L, Marshall MA, et al. 2010. P-selectin-mediated platelet-neutrophil aggregate formation activates neutrophils in mouse and human sickle cell disease. Arterioscler. Thromb. Vasc. Biol 30:2392–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wun T, Cordoba M, Rangaswami A, Cheung AW, Paglieroni T. 2002. Activated monocytes and platelet-monocyte aggregates in patients with sickle cell disease. Clin. Lab. Haematol 24:81–88 [DOI] [PubMed] [Google Scholar]
- 27.Miller ST, Sleeper LA, Pegelow CH, Enos LE, Wang WC, et al. 2000. Prediction of adverse outcomes in children with sickle cell disease. N. Engl. J. Med 342:83–89 [DOI] [PubMed] [Google Scholar]
- 28.Wongtong N, Jones S, Deng Y, Cai J, Ataga KI. 2015. Monocytosis is associated with hemolysis in sickle cell disease. Hematology 20:593–97 [DOI] [PubMed] [Google Scholar]
- 29.Chaturvedi S, Ghafuri DL, Glassberg J, Kassim AA, Rodeghier M, DeBaun MR. 2016. Rapidly progressive acute chest syndrome in individuals with sickle cell anemia: a distinct acute chest syndrome phenotype. Am. J. Hematol 91:1185–90 [DOI] [PubMed] [Google Scholar]
- 30.Gardner K, Thein SL. 2015. Super-elevated LDH and thrombocytopenia are markers of a severe subtype of vaso-occlusive crisis in sickle cell disease. Am. J. Hematol 90:E206–7 [DOI] [PubMed] [Google Scholar]
- 31.Alhandalous CH, Han J, Hsu L, Gowhari M, Hassan J, et al. 2015. Platelets decline during vaso-occlusive crisis as a predictor of acute chest syndrome in sickle cell disease. Am. J. Hematol 90:E228–29 [DOI] [PubMed] [Google Scholar]
- 32.Novelli EM, Gladwin MT. 2016. Crises in sickle cell disease. Chest 149:1082–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hidalgo A, Chang J, Jang JE, Peired AJ, Chiang EY, Frenette PS. 2009. Heterotypic interactions enabled by polarized neutrophil microdomains mediate thromboinflammatory injury. Nat. Med 15:384–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Belcher JD, Chen C, Nguyen J, Milbauer L, Abdulla F, et al. 2014. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 123:377–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wallace KL, Linden J. 2010. Adenosine A2A receptors induced on iNKT and NK cells reduce pulmonary inflammation and injury in mice with sickle cell disease. Blood 116:5010–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gutsaeva DR, Montero-Huerta P, Parkerson JB, Yerigenahally SD, Ikuta T, Head CA. 2014. Molecular mechanisms underlying synergistic adhesion of sickle red blood cells by hypoxia and low nitric oxide bioavailability. Blood 123:1917–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zennadi R, Moeller BJ, Whalen EJ, Batchvarova M, Xu K, et al. 2007. Epinephrine-induced activation of LW-mediated sickle cell adhesion and vaso-occlusion in vivo. Blood 110:2708–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turhan A, Weiss LA, Mohandas N, Coller BS, Frenette PS. 2002. Primary role for adherent leukocytes in sickle cell vascular occlusion: a new paradigm. PNAS 99:3047–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang J, Patton JT, Sarkar A, Ernst B, Magnani JL, Frenette PS. 2010. GMI-1070, a novel pan-selectin antagonist, reverses acute vascular occlusions in sickle cell mice. Blood 116:1779–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chang J, Shi PA, Chiang EY, Frenette PS. 2008. Intravenous immunoglobulins reverse acute vaso-occlusive crises in sickle cell mice through rapid inhibition of neutrophil adhesion. Blood 111:915–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jang JE, Hod EA, Spitalnik SL, Frenette PS. 2011. CXCL1 and its receptor, CXCR2, mediate murine sickle cell vaso-occlusion during hemolytic transfusion reactions. J. Clin. Invest 121:1397–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Almeida CB, Scheiermann C, Jang JE, Prophete C, Costa FF, et al. 2012. Hydroxyurea and a cGMP-amplifying agent have immediate benefits on acute vaso-occlusive events in sickle cell disease mice. Blood 120:2879–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li J, Kim K, Hahm E, Molokie R, Hay N, et al. 2014. Neutrophil AKT2 regulates heterotypic cell-cell interactions during vascular inflammation. J. Clin. Invest 124:1483–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim K, Li J, Tseng A, Andrews RK, Cho J. 2015. NOX2 is critical for heterotypic neutrophil-platelet interactions during vascular inflammation. Blood 126:1952–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Embury SH, Matsui NM, Ramanujam S, Mayadas TN, Noguchi CT, et al. 2004. The contribution of endothelial cell P-selectin to the microvascular flow of mouse sickle erythrocytes in vivo. Blood 104:3378–85 [DOI] [PubMed] [Google Scholar]
- 46.Gutsaeva DR, Parkerson JB, Yerigenahally SD, Kurz JC, Schaub RG, et al. 2011. Inhibition of cell adhesion by anti-P-selectin aptamer: a new potential therapeutic agent for sickle cell disease. Blood 117:727–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zennadi R 2014. MEK inhibitors, novel anti-adhesive molecules, reduce sickle red blood cell adhesion in vitro and in vivo, and vasoocclusion in vivo. PLOS ONE 9:e110306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zennadi R, Hines PC, De Castro LM, Cartron JP, Parise LV, Telen MJ. 2004. Epinephrine acts through erythroid signaling pathways to activate sickle cell adhesion to endothelium via LW-alphavbeta3 interactions. Blood 104:3774–81 [DOI] [PubMed] [Google Scholar]
- 49.Zennadi R, Whalen EJ, Soderblom EJ, Alexander SC, Thompson JW, et al. 2012. Erythrocyte plasma membrane-bound ERK1/2 activation promotes ICAM-4-mediated sickle red cell adhesion to endothelium. Blood 119:1217–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao Y, Schwartz EA, Palmer GM, Zennadi R. 2016. MEK1/2 inhibitors reverse acute vascular occlusion in mouse models of sickle cell disease. FASEB J. 30:1171–86 [DOI] [PubMed] [Google Scholar]
- 51.Zhang D, Chen G, Manwani D, Mortha A, Xu C, et al. 2015. Neutrophil ageing is regulated by the microbiome. Nature 525:528–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lebensburger JD, Howard T, Hu Y, Pestina TI, Gao G, et al. 2012. Hydroxyurea therapy of a murine model of sickle cell anemia inhibits the progression of pneumococcal disease by down-modulating E-selectin. Blood 119:1915–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jimenez MA, Novelli E, Shaw GD, Sundd P. 2017. Glycoprotein Ibα inhibitor (CCP-224) prevents neutrophil-platelet aggregation in sickle cell disease. Blood Adv. 1:1712–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sparkenbaugh E, Pawlinski R. 2013. Interplay between coagulation and vascular inflammation in sickle cell disease. Br. J. Haematol 162:3–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pakbaz Z, Wun T. 2014. Role of the hemostatic system on sickle cell disease pathophysiology and potential therapeutics. Hematol. Oncol. Clin. North Am 28:355–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Noubouossie D, Key NS, Ataga KI. 2016. Coagulation abnormalities of sickle cell disease: relationship with clinical outcomes and the effect of disease modifying therapies. Blood Rev. 30:245–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu Y, Jing F, Yi W, Mendelson A, Shi P, et al. 2018. HO-1hi patrolling monocytes protect against vaso-occlusion in sickle cell disease. Blood 131:1600–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ataga KI, Moore CG, Jones S, Olajide O, Strayhorn D, et al. 2006. Pulmonary hypertension in patients with sickle cell disease: a longitudinal study. Br. J. Haematol 134:109–15 [DOI] [PubMed] [Google Scholar]
- 59.Bartolucci P, Brugnara C, Teixeira-Pinto A, Pissard S, Moradkhani K, et al. 2012. Erythrocyte density in sickle cell syndromes is associated with specific clinical manifestations and hemolysis. Blood 120:3136–41 [DOI] [PubMed] [Google Scholar]
- 60.Day TG, Drasar ER, Fulford T, Sharpe CC, Thein SL. 2012. Association between hemolysis and albuminuria in adults with sickle cell anemia. Haematologica 97:201–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fitzhugh CD, Lauder N, Jonassaint JC, Telen MJ, Zhao X, et al. 2010. Cardiopulmonary complications leading to premature deaths in adult patients with sickle cell disease. Am. J. Hematol 85:36–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gladwin MT, Barst RJ, Gibbs JS, Hildesheim M, Sachdev V, et al. 2014. Risk factors for death in 632 patients with sickle cell disease in the United States and United Kingdom. PLOS ONE 9:e99489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gladwin MT, Sachdev V. 2012. Cardiovascular abnormalities in sickle cell disease. J. Am. Coll. Cardiol 59:1123–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gladwin MT, Sachdev V, Jison ML, Shizukuda Y, Plehn JF, et al. 2004. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N. Engl. J. Med 350:886–95 [DOI] [PubMed] [Google Scholar]
- 65.Kato GJ, McGowan V, Machado RF, Little JA, Taylor J, et al. 2006. Lactate dehydrogenase as a biomarker of hemolysis-associated nitric oxide resistance, priapism, leg ulceration, pulmonary hypertension, and death in patients with sickle cell disease. Blood 107:2279–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mehari A, Alam S, Tian X, Cuttica MJ, Barnett CF, et al. 2013. Hemodynamic predictors of mortality in adults with sickle cell disease. Am. J. Respir. Crit. Care Med 187:840–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Novelli EM, Hildesheim M, Rosano C, Vanderpool R, Simon M, et al. 2014. Elevated pulse pressure is associated with hemolysis, proteinuria and chronic kidney disease in sickle cell disease. PLOS ONE 9:e114309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, et al. 1994. Mortality in sickle cell disease: life expectancy and risk factors for early death. N. Engl. J. Med 330:1639–44 [DOI] [PubMed] [Google Scholar]
- 69.Powars DR, Elliott-Mills DD, Chan L. 1991. Chronic renal failure in sickle cell disease: risk factors, clinical course, and mortality. Ann. Intern. Med 115:614–20 [DOI] [PubMed] [Google Scholar]
- 70.Saraf SL, Zhang X, Kanias T, Lash JP, Molokie RE, et al. 2014. Haemoglobinuria is associated with chronic kidney disease and its progression in patients with sickle cell anaemia. Br. J. Haematol 164:729–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Belcher JD, Bryant CJ, Nguyen J, Bowlin PR, Kielbik MC, et al. 2003. Transgenic sickle mice have vascular inflammation. Blood 101:3953–59 [DOI] [PubMed] [Google Scholar]
- 72.Osarogiagbon UR, Choong S, Belcher JD, Vercellotti GM, Paller MS, Hebbel RP. 2000. Reperfusion injury pathophysiology in sickle transgenic mice. Blood 96:314–20 [PubMed] [Google Scholar]
- 73.Connes P, Lamarre Y, Waltz X, Ballas SK, Lemonne N, et al. 2014. Haemolysis and abnormal haemorheology in sickle cell anaemia. Br. J. Haematol 165:564–72 [DOI] [PubMed] [Google Scholar]
- 74.Nouraie M, Lee JS, Zhang Y, Kanias T, Zhao X, et al. 2013. The relationship between the severity of hemolysis, clinical manifestations and risk of death in 415 patients with sickle cell anemia in the US and Europe. Haematologica 98:464–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gladwin MT. 2016. Cardiovascular complications and risk of death in sickle-cell disease. Lancet 387:2565–74 [DOI] [PubMed] [Google Scholar]
- 76.Poludasu S, Ramkissoon K, Salciccioli L, Kamran H, Lazar JM. 2013. Left ventricular systolic function in sickle cell anemia: a meta-analysis. J. Card. Fail 19:333–41 [DOI] [PubMed] [Google Scholar]
- 77.Sachdev V, Kato GJ, Gibbs JS, Barst RJ, Machado RF, et al. 2011. Echocardiographic markers of elevated pulmonary pressure and left ventricular diastolic dysfunction are associated with exercise intolerance in adults and adolescents with homozygous sickle cell anemia in the United States and United Kingdom. Circulation 124:1452–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Milton JN, Rooks H, Drasar E, McCabe EL, Baldwin CT, et al. 2013. Genetic determinants of haemolysis in sickle cell anaemia. Br. J. Haematol 161:270–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kato GJ, Gladwin MT, Steinberg MH. 2007. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 21:37–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.DeBaun MR, Armstrong FD, McKinstry RC, Ware RE, Vichinsky E, Kirkham FJ. 2012. Silent cerebral infarcts: a review on a prevalent and progressive cause of neurologic injury in sickle cell anemia. Blood 119:4587–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ohene-Frempong K, Weiner SJ, Sleeper LA, Miller ST, Embury S, et al. 1998. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood 91:288–94 [PubMed] [Google Scholar]
- 82.Sachdev V, Machado RF, Shizukuda Y, Rao YN, Sidenko S, et al. 2007. Diastolic dysfunction is an independent risk factor for death in patients with sickle cell disease. J. Am. Coll. Cardiol 49:472–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lundberg JO, Gladwin MT, Weitzberg E. 2015. Strategies to increase nitric oxide signalling in cardiovascular disease. Nat. Rev. Drug Discov 14:623–41 [DOI] [PubMed] [Google Scholar]
- 84.Reiter CD, Wang X, Tanus-Santos JE, Hogg N, Cannon RO III, et al. 2002. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat. Med 8:1383–89 [DOI] [PubMed] [Google Scholar]
- 85.Rother RP, Bell L, Hillmen P, Gladwin MT. 2005. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. JAMA 293:1653–62 [DOI] [PubMed] [Google Scholar]
- 86.Morris CR, Kato GJ, Poljakovic M, Wang X, Blackwelder WC, et al. 2005. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA 294:81–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hsu LL, Champion HC, Campbell-Lee SA, Bivalacqua TJ, Manci EA, et al. 2007. Hemolysis in sickle cell mice causes pulmonary hypertension due to global impairment in nitric oxide bioavailability. Blood 109:3088–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gladwin MT. 2006. Deconstructing endothelial dysfunction: soluble guanylyl cyclase oxidation and the NO resistance syndrome. J. Clin. Invest 116:2330–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ghosh S, Adisa OA, Chappa P, Tan F, Jackson KA, et al. 2013. Extracellular hemin crisis triggers acute chest syndrome in sickle mice. J. Clin. Invest 123:4809–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Almeida CB, Souza LE, Leonardo FC, Costa FT, Werneck CC, et al. 2015. Acute hemolytic vascular inflammatory processes are prevented by nitric oxide replacement or a single dose of hydroxyurea. Blood 126:711–20 [DOI] [PubMed] [Google Scholar]
- 91.Villagra J, Shiva S, Hunter LA, Machado RF, Gladwin MT, Kato GJ. 2007. Platelet activation in patients with sickle disease, hemolysis-associated pulmonary hypertension, and nitric oxide scavenging by cell-free hemoglobin. Blood 110:2166–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cardenes N, Corey C, Geary L, Jain S, Zharikov S, et al. 2014. Platelet bioenergetic screen in sickle cell patients reveals mitochondrial complex V inhibition, which contributes to platelet activation. Blood 123:2864–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Helms CC, Marvel M, Zhao W, Stahle M, Vest R, et al. 2013. Mechanisms of hemolysis-associated platelet activation. J. Thromb. Haemost 11:2148–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ataga KI. 2009. Hypercoagulability and thrombotic complications in hemolytic anemias. Haematologica 94:1481–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen GY, Nunez G. 2010. Sterile inflammation: sensing and reacting to damage. Nat. Rev. Immunol 10:826–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dutra FF, Alves LS, Rodrigues D, Fernandez PL, de Oliveira RB, et al. 2014. Hemolysis-induced lethality involves inflammasome activation by heme. PNAS 111:E4110–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chen G, Zhang D, Fuchs TA, Wagner DD, Frenette PS. 2014. Heme-induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood 123:3818–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Setty BN, Betal SG, Zhang J, Stuart MJ. 2008. Heme induces endothelial tissue factor expression: potential role in hemostatic activation in patients with hemolytic anemia. J. Thromb. Haemost 6:2202–9 [DOI] [PubMed] [Google Scholar]
- 99.Dutra FF, Bozza MT. 2014. Heme on innate immunity and inflammation. Front. Pharmacol 5:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Camus SM, De Moraes JA, Bonnin P, Abbyad P, Le Jeune S, et al. 2015. Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease. Blood 125:3805–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Monteiro AP, Pinheiro CS, Luna-Gomes T, Alves LR, Maya-Monteiro CM, et al. 2011. Leukotriene B4 mediates neutrophil migration induced by heme. J. Immunol 186:6562–67 [DOI] [PubMed] [Google Scholar]
- 102.Figueiredo RT, Fernandez PL, Mourao-Sa DS, Porto BN, Dutra FF, et al. 2007. Characterization of heme as activator of Toll-like receptor 4. J. Biol. Chem 282:20221–29 [DOI] [PubMed] [Google Scholar]
- 103.Arruda MA, Rossi AG, de Freitas MS, Barja-Fidalgo C, Graca-Souza AV. 2004. Heme inhibits human neutrophil apoptosis: involvement of phosphoinositide 3-kinase, MAPK, and NF-κB. J. Immunol 173:2023–30 [DOI] [PubMed] [Google Scholar]
- 104.Kono M, Saigo K, Takagi Y, Takahashi T, Kawauchi S, et al. 2014. Heme-related molecules induce rapid production of neutrophil extracellular traps. Transfusion 54:2811–19 [DOI] [PubMed] [Google Scholar]
- 105.Graca-Souza AV, Arruda MA, de Freitas MS, Barja-Fidalgo C, Oliveira PL. 2002. Neutrophil activation by heme: implications for inflammatory processes. Blood 99:4160–65 [DOI] [PubMed] [Google Scholar]
- 106.Porto BN, Alves LS, Fernandez PL, Dutra TP, Figueiredo RT, et al. 2007. Heme induces neutrophil migration and reactive oxygen species generation through signaling pathways characteristic of chemotactic receptors. J. Biol. Chem 282:24430–36 [DOI] [PubMed] [Google Scholar]
- 107.Jorch SK, Kubes P. 2017. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med 23:279–87 [DOI] [PubMed] [Google Scholar]
- 108.Schimmel M, Nur E, Biemond BJ, van Mierlo GJ, Solati S, et al. 2013. Nucleosomes and neutrophil activation in sickle cell disease painful crisis. Haematologica 98:1797–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Malik Z, Creter D, Cohen A, Djaldetti M. 1983. Haemin affects platelet aggregation and lymphocyte mitogenicity in whole blood incubations. Cytobios 38:33–38 [PubMed] [Google Scholar]
- 110.Gladwin MT, Vichinsky E. 2008. Pulmonary complications of sickle cell disease. N. Engl. J. Med 359:2254–65 [DOI] [PubMed] [Google Scholar]
- 111.Vichinsky EP, Neumayr LD, Earles AN, Williams R, Lennette ET, et al. 2000. Causes and outcomes of the acute chest syndrome in sickle cell disease. National Acute Chest Syndrome Study Group. N. Engl. J. Med 342:1855–65 [DOI] [PubMed] [Google Scholar]
- 112.Martins R, Maier J, Gorki AD, Huber KV, Sharif O, et al. 2016. Heme drives hemolysis-induced susceptibility to infection via disruption of phagocyte functions. Nat. Immunol 17:1361–72 [DOI] [PubMed] [Google Scholar]
- 113.Kaul DK, Hebbel RP. 2000. Hypoxia/reoxygenation causes inflammatory response in transgenic sickle mice but not in normal mice. J. Clin. Invest 106:411–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Belcher JD, Mahaseth H, Welch TE, Vilback AE, Sonbol KM, et al. 2005. Critical role of endothelial cell activation in hypoxia-induced vasoocclusion in transgenic sickle mice. Am. J. Physiol. Heart Circ. Physiol 288:H2715–25 [DOI] [PubMed] [Google Scholar]
- 115.Eltzschig HK, Eckle T. 2011. Ischemia and reperfusion—from mechanism to translation. Nat. Med 17:1391–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. 2009. Cell death. N. Engl. J. Med 361:1570–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, et al. 2011. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 32:157–64 [DOI] [PubMed] [Google Scholar]
- 118.Xu H, Wandersee NJ, Guo Y, Jones DW, Holzhauer SL, et al. 2014. Sickle cell disease increases high mobility group box 1: a novel mechanism of inflammation. Blood 124:3978–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Elliott EI, Sutterwala FS. 2015. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol. Rev 265:35–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Man SM, Kanneganti TD. 2015. Regulation of inflammasome activation. Immunol. Rev 265:6–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. 2015. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 349:316–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pitanga TN, Oliveira RR, Zanette DL, Guarda CC, Santiago RP, et al. 2016. Sickle red cells as danger signals on proinflammatory gene expression, leukotriene B4 and interleukin-1 beta production in peripheral blood mononuclear cell. Cytokine 83:75–84 [DOI] [PubMed] [Google Scholar]
- 123.Alagbe AE, Justo AS Jr., Ruas LP, Tonasse WV, Santana RM, et al. 2017. Interleukin-27 and interleukin-37 are elevated in sickle cell anemia patients and inhibit in vitro secretion of interleukin-8 in neutrophils and monocytes. Cytokine 107:85–92 [DOI] [PubMed] [Google Scholar]
- 124.Bakele M, Joos M, Burdi S, Allgaier N, Poschel S, et al. 2014. Localization and functionality of the inflammasome in neutrophils. J. Biol. Chem 289:5320–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hottz ED, Lopes JF, Freitas C, Valls-de-Souza R, Oliveira MF, et al. 2013. Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation. Blood 122:3405–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hottz ED, Monteiro AP, Bozza FA, Bozza PT. 2015. Inflammasome in platelets: allying coagulation and inflammation in infectious and sterile diseases? Mediat. Inflamm 2015:435783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Xia M, Boini KM, Abais JM, Xu M, Zhang Y, Li PL. 2014. Endothelial NLRP3 inflammasome activation and enhanced neointima formation in mice by adipokine visfatin. Am. J. Pathol 184:1617–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Telen MJ. 2016. Beyond hydroxyurea: new and old drugs in the pipeline for sickle cell disease. Blood 127:810–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Belcher JD, Young M, Chen C, Nguyen J, Burhop K, et al. 2013. MP4CO, a pegylated hemoglobin saturated with carbon monoxide, is a modulator of HO-1, inflammation, and vaso-occlusion in transgenic sickle mice. Blood 122:2757–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fleischmann RM, Tesser J, Schiff MH, Schechtman J, Burmester GR, et al. 2006. Safety of extended treatment with anakinra in patients with rheumatoid arthritis. Ann. Rheum. Dis 65:1006–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, Leslie KS, Hachulla E, et al. 2009. Use of canakinumab in the cryopyrin-associated periodic syndrome. N. Engl. J. Med 360:2416–25 [DOI] [PubMed] [Google Scholar]
- 132.Nath KA, Katusic ZS, Gladwin MT. 2004. The perfusion paradox and vascular instability in sickle cell disease. Microcirculation 11:179–93 [DOI] [PubMed] [Google Scholar]
- 133.Powars D, Wilson B, Imbus C, Pegelow C, Allen J. 1978. The natural history of stroke in sickle cell disease. Am. J. Med 65:461–71 [DOI] [PubMed] [Google Scholar]
- 134.Rothman SM, Fulling KH, Nelson JS. 1986. Sickle cell anemia and central nervous system infarction: a neuropathological study. Ann. Neurol 20:684–90 [DOI] [PubMed] [Google Scholar]
- 135.Merkel KH, Ginsberg PL, Parker JC Jr., Post MJ. 1978. Cerebrovascular disease in sickle cell anemia: a clinical, pathological and radiological correlation. Stroke 9:45–52 [DOI] [PubMed] [Google Scholar]
- 136.Prohovnik I, Pavlakis SG, Piomelli S, Bello J, Mohr JP, et al. 1989. Cerebral hyperemia, stroke, and transfusion in sickle cell disease. Neurology 39:344–48 [DOI] [PubMed] [Google Scholar]
- 137.Kim YS, Nur E, van Beers EJ, Truijen J, Davis SC, et al. 2009. Dynamic cerebral autoregulation in homozygous sickle cell disease. Stroke 40:808–14 [DOI] [PubMed] [Google Scholar]
- 138.Nur E, Kim YS, Truijen J, van Beers EJ, Davis SC, et al. 2009. Cerebrovascular reserve capacity is impaired in patients with sickle cell disease. Blood 114:3473–78 [DOI] [PubMed] [Google Scholar]
- 139.Fields ME, Guilliams KP, Ragan DK, Binkley MM, Eldeniz C, et al. 2018. Regional oxygen extraction predicts border zone vulnerability to stroke in sickle cell disease. Neurology 90:e1134–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jordan LC, Gindville MC, Scott AO, Juttukonda MR, Strother MK, et al. 2016. Non-invasive imaging of oxygen extraction fraction in adults with sickle cell anaemia. Brain 139:738–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Adams RJ, Nichols FT, McKie V, McKie K, Milner P, Gammal TE. 1988. Cerebral infarction in sickle cell anemia: mechanism based on CT and MRI. Neurology 38:1012–17 [DOI] [PubMed] [Google Scholar]
- 142.Guilliams KP, Fields ME, Ragan DK, Chen Y, Eldeniz C, et al. 2017. Large-vessel vasculopathy in children with sickle cell disease: a magnetic resonance imaging study of infarct topography and focal atrophy. Pediatr. Neurol 69:49–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kirk GR, Haynes MR, Palasis S, Brown C, Burns TG, et al. 2009. Regionally specific cortical thinning in children with sickle cell disease. Cereb. Cortex 19:1549–56 [DOI] [PubMed] [Google Scholar]
- 144.Kawadler JM, Clayden JD, Kirkham FJ, Cox TC, Saunders DE, Clark CA. 2013. Subcortical and cerebellar volumetric deficits in paediatric sickle cell anaemia. Br. J. Haematol 163:373–76 [DOI] [PubMed] [Google Scholar]
- 145.Kawadler JM, Clark CA, McKinstry RC, Kirkham FJ. 2017. Brain atrophy in paediatric sickle cell anaemia: findings from the silent infarct transfusion (SIT) trial. Br. J. Haematol 177:151–53 [DOI] [PubMed] [Google Scholar]
- 146.Earley CJ, Kittner SJ, Feeser BR, Gardner J, Epstein A, et al. 1998. Stroke in children and sickle-cell disease: Baltimore-Washington Cooperative Young Stroke Study. Neurology 51:169–76 [DOI] [PubMed] [Google Scholar]
- 147.Dhabangi A, Ainomugisha B, Cserti-Gazdewich C, Ddungu H, Kyeyune D, et al. 2016. Cerebral oximetry in Ugandan children with severe anemia: clinical categories and response to transfusion. JAMA Pediatr. 170:995–1002 [DOI] [PubMed] [Google Scholar]
- 148.Adams RJ, McKie VC, Hsu L, Files B, Vichinsky E, et al. 1998. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N. Engl. J. Med 339:5–11 [DOI] [PubMed] [Google Scholar]
- 149.Meier ER, Wright EC, Miller JL. 2014. Reticulocytosis and anemia are associated with an increased risk of death and stroke in the newborn cohort of the Cooperative Study of Sickle Cell Disease. Am. J. Hematol 89:904–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Balkaran B, Char G, Morris JS, Thomas PW, Serjeant BE, Serjeant GR. 1992. Stroke in a cohort of patients with homozygous sickle cell disease. J. Pediatr 120:360–66 [DOI] [PubMed] [Google Scholar]
- 151.Belisario AR, Sales RR, Toledo NE, Muniz MB, Velloso-Rodrigues C, et al. 2016. Reticulocyte count is the most important predictor of acute cerebral ischemia and high-risk transcranial Doppler in a newborn cohort of 395 children with sickle cell anemia. Ann. Hematol 95:1869–80 [DOI] [PubMed] [Google Scholar]
- 152.Ware RE, Davis BR, Schultz WH, Brown RC, Aygun B, et al. 2016. Hydroxycarbamide versus chronic transfusion for maintenance of transcranial Doppler flow velocities in children with sickle cell anaemia—TCD With Transfusions Changing to Hydroxyurea (TWiTCH): a multicentre, open-label, phase 3, non-inferiority trial. Lancet 387:661–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Manci EA, Hillery CA, Bodian CA, Zhang ZG, Lutty GA, Coller BS. 2006. Pathology of Berkeley sickle cell mice: similarities and differences with human sickle cell disease. Blood 107:1651–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Paszty C, Brion CM, Manci E, Witkowska HE, Stevens ME, et al. 1997. Transgenic knockout mice with exclusively human sickle hemoglobin and sickle cell disease. Science 278:876–78 [DOI] [PubMed] [Google Scholar]
- 155.Cui MH, Suzuka SM, Branch NA, Ambadipudi K, Thangaswamy S, et al. 2017. Brain neurochemical and hemodynamic findings in the NY1DD mouse model of mild sickle cell disease. NMR Biomed 30:e3692. [DOI] [PubMed] [Google Scholar]
- 156.Hyacinth HI, Sugihara CL, Spencer TL, Archer DR, Shih AY. 2017. Higher prevalence of spontaneous cerebral vasculopathy and cerebral infarcts in a mouse model of sickle cell disease. J. Cereb. Blood Flow Metab 10.1177/0271678X17732275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Cahill LS, Gazdzinski LM, Tsui AK, Zhou YQ, Portnoy S, et al. 2017. Functional and anatomical evidence of cerebral tissue hypoxia in young sickle cell anemia mice. J. Cereb. Blood Flow Metab 37:994–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hillery CA, Panepinto JA. 2004. Pathophysiology of stroke in sickle cell disease. Microcirculation 11:195–208 [DOI] [PubMed] [Google Scholar]
- 159.Kato GJ, Hsieh M, Machado R, Taylor JVI, Little J, et al. 2006. Cerebrovascular disease associated with sickle cell pulmonary hypertension. Am. J. Hematol 81:503–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Bernaudin F, Verlhac S, Chevret S, Torres M, Coic L, et al. 2008. G6PD deficiency, absence of alpha-thalassemia, and hemolytic rate at baseline are significant independent risk factors for abnormally high cerebral velocities in patients with sickle cell anemia. Blood 112:4314–17 [DOI] [PubMed] [Google Scholar]
- 161.Kwiatkowski JL, Yim E, Miller S, Adams RJ, STOP 2 Study Investig. 2011. Effect of transfusion therapy on transcranial Doppler ultrasonography velocities in children with sickle cell disease. Pediatr. Blood Cancer 56:777–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Adams RJ, Brambilla D, Optim. Prim. Stroke Prev. Sickle Cell Anemia STOP 2 Trial Investig. 2005. Discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease. N. Engl. J. Med 353:2769–78 [DOI] [PubMed] [Google Scholar]
- 163.Wang WC, Pavlakis SG, Helton KJ, McKinstry RC, Casella JF, et al. 2008. MRI abnormalities of the brain in one-year-old children with sickle cell anemia. Pediatr. Blood Cancer 51:643–46 [DOI] [PubMed] [Google Scholar]
- 164.Bernaudin F, Verlhac S, Arnaud C, Kamdem A, Chevret S, et al. 2011. Impact of early transcranial Doppler screening and intensive therapy on cerebral vasculopathy outcome in a newborn sickle cell anemia cohort. Blood 117:1130–40; quiz 1436 [DOI] [PubMed] [Google Scholar]
- 165.Calvet D, Tuilier T, Mele N, Turc G, Habibi A, et al. 2017. Low fetal hemoglobin percentage is associated with silent brain lesions in adults with homozygous sickle cell disease. Blood Adv. 1:2503–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kassim AA, Pruthi S, Day M, Rodeghier M, Gindville MC, et al. 2016. Silent cerebral infarcts and cerebral aneurysms are prevalent in adults with sickle cell anemia. Blood 127:2038–40 [DOI] [PubMed] [Google Scholar]
- 167.Casella JF, King AA, Barton B, White DA, Noetzel MJ, et al. 2010. Design of the silent cerebral infarct transfusion (SIT) trial. Pediatr. Hematol. Oncol 27:69–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.DeBaun MR, Schatz J, Siegel MJ, Koby M, Craft S, et al. 1998. Cognitive screening examinations for silent cerebral infarcts in sickle cell disease. Neurology 50:1678–82 [DOI] [PubMed] [Google Scholar]
- 169.King AA, Rodeghier MJ, Panepinto JA, Strouse JJ, Casella JF, et al. 2014. Silent cerebral infarction, income, and grade retention among students with sickle cell anemia. Am. J. Hematol 89:E188–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Schatz J, Brown RT, Pascual JM, Hsu L, DeBaun MR. 2001. Poor school and cognitive functioning with silent cerebral infarcts and sickle cell disease. Neurology 56:1109–11 [DOI] [PubMed] [Google Scholar]
- 171.van der Land V, Hijmans CT, de Ruiter M, Mutsaerts HJ, Cnossen MH, et al. 2015. Volume of white matter hyperintensities is an independent predictor of intelligence quotient and processing speed in children with sickle cell disease. Br. J. Haematol 168:553–56 [DOI] [PubMed] [Google Scholar]
- 172.DeBaun MR, Gordon M, McKinstry RC, Noetzel MJ, White DA, et al. 2014. Controlled trial of transfusions for silent cerebral infarcts in sickle cell anemia. N. Engl. J. Med 371:699–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Miller ST, Macklin EA, Pegelow CH, Kinney TR, Sleeper LA, et al. 2001. Silent infarction as a risk factor for overt stroke in children with sickle cell anemia: a report from the Cooperative Study of Sickle Cell Disease. J. Pediatr 139:385–90 [DOI] [PubMed] [Google Scholar]
- 174.Koshy M, Thomas C, Goodwin J. 1990. Vascular lesions in the central nervous system in sickle cell disease (neuropathology). J. Assoc. Acad. Minor. Phys 1:71–78 [PubMed] [Google Scholar]
- 175.Novelli EM, Sarles CE, Aizenstein HJ, Ibrahim TS, Butters MA, et al. 2015. Brain venular pattern by 7T MRI correlates with memory and haemoglobin in sickle cell anaemia. Psychiatry Res. 233:18–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Winchell AM, Taylor BA, Song R, Loeffler RB, Grundlehner P, et al. 2014. Evaluation of SWI in children with sickle cell disease. AJNR Am. J. Neuroradiol 35:1016–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Steen RG, Fineberg-Buchner C, Hankins G, Weiss L, Prifitera A, Mulhern RK. 2005. Cognitive deficits in children with sickle cell disease. J. Child Neurol 20:102–7 [DOI] [PubMed] [Google Scholar]
- 178.Vichinsky EP, Neumayr LD, Gold JI, Weiner MW, Rule RR, et al. 2010. Neuropsychological dysfunction and neuroimaging abnormalities in neurologically intact adults with sickle cell anemia. JAMA 303:1823–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.van der Land V, Zwanenburg JJ, Fijnvandraat K, Biemond BJ, Hendrikse J, et al. 2015. Cerebral lesions on 7 tesla MRI in patients with sickle cell anemia. Cerebrovasc. Dis 39:181–89 [DOI] [PubMed] [Google Scholar]
- 180.Dobson SR, Holden KR, Nietert PJ, Cure JK, Laver JH, et al. 2002. Moyamoya syndrome in childhood sickle cell disease: a predictive factor for recurrent cerebrovascular events. Blood 99:3144–50 [DOI] [PubMed] [Google Scholar]
- 181.Birkeland P, Gardner K, Kesse-Adu R, Davies J, Lauritsen J, et al. 2016. Intracranial aneurysms in sickle-cell disease are associated with the hemoglobin SS genotype but not with Moyamoya syndrome. Stroke 47:1710–13 [DOI] [PubMed] [Google Scholar]
- 182.Nabavizadeh SA, Vossough A, Ichord RN, Kwiatkowski J, Pukenas BA, et al. 2016. Intracranial aneurysms in sickle cell anemia: clinical and imaging findings. J. Neurointerv. Surg 8:434–40 [DOI] [PubMed] [Google Scholar]
- 183.Kato GJ, Hebbel RP, Steinberg MH, Gladwin MT. 2009. Vasculopathy in sickle cell disease: biology, pathophysiology, genetics, translational medicine, and new research directions. Am. J. Hematol 84:618–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lai YC, Potoka KC, Champion HC, Mora AL, Gladwin MT. 2014. Pulmonary arterial hypertension: the clinical syndrome. Circ. Res 115:115–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Hoeper MM, Bogaard HJ, Condliffe R, Frantz R, Khanna D, et al. 2013. Definitions and diagnosis of pulmonary hypertension. J. Am. Coll. Cardiol 62:D42–50 [DOI] [PubMed] [Google Scholar]
- 186.Maron BA, Brittain EL, Choudhary G, Gladwin MT. 2018. Redefining pulmonary hypertension. Lancet Respir. Med 6:168–70 [DOI] [PubMed] [Google Scholar]
- 187.Maron BA, Wertheim BM, Gladwin MT. 2018. Under pressure to clarify pulmonary hypertension clinical risk. Am. J. Respir. Crit. Care Med 197:423–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.De Castro LM, Jonassaint JC, Graham FL, Ashley-Koch A, Telen MJ. 2008. Pulmonary hypertension associated with sickle cell disease: clinical and laboratory endpoints and disease outcomes. Am. J. Hematol 83:19–25 [DOI] [PubMed] [Google Scholar]
- 189.Lorch D, Spevack D, Little J. 2011. An elevated estimated pulmonary arterial systolic pressure, whenever measured, is associated with excess mortality in adults with sickle cell disease. Acta Haematol. 125:225–29 [DOI] [PubMed] [Google Scholar]
- 190.Klings ES, Machado RF, Barst RJ, Morris CR, Mubarak KK, et al. 2014. An official American Thoracic Society clinical practice guideline: diagnosis, risk stratification, and management of pulmonary hypertension of sickle cell disease. Am. J. Respir. Crit. Care Med 189:727–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Caughey MC, Poole C, Ataga KI, Hinderliter AL. 2015. Estimated pulmonary artery systolic pressure and sickle cell disease: a meta-analysis and systematic review. Br. J. Haematol 170:416–24 [DOI] [PubMed] [Google Scholar]
- 192.Damy T, Bodez D, Habibi A, Guellich A, Rappeneau S, et al. 2016. Haematological determinants of cardiac involvement in adults with sickle cell disease. Eur. Heart J 37:1158–67 [DOI] [PubMed] [Google Scholar]
- 193.Machado RF, Anthi A, Steinberg MH, Bonds D, Sachdev V, et al. 2006. N-terminal pro-brain natriuretic peptide levels and risk of death in sickle cell disease. JAMA 296:310–18 [DOI] [PubMed] [Google Scholar]
- 194.Mehari A, Gladwin MT, Tian X, Machado RF, Kato GJ. 2012. Mortality in adults with sickle cell disease and pulmonary hypertension. JAMA 307:1254–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Parent F, Bachir D, Inamo J, Lionnet F, Driss F, et al. 2011. A hemodynamic study of pulmonary hypertension in sickle cell disease. N. Engl. J. Med 365:44–53 [DOI] [PubMed] [Google Scholar]
- 196.Fonseca GH, Souza R, Salemi VM, Jardim CV, Gualandro SF. 2012. Pulmonary hypertension diagnosed by right heart catheterisation in sickle cell disease. Eur. Respir. J 39:112–18 [DOI] [PubMed] [Google Scholar]
- 197.Bakeer N, James J, Roy S, Wansapura J, Shanmukhappa SK, et al. 2016. Sickle cell anemia mice develop a unique cardiomyopathy with restrictive physiology. PNAS 113:E5182–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Niss O, Quinn CT, Lane A, Daily J, Khoury PR, et al. 2016. Cardiomyopathy with restrictive physiology in sickle cell disease. JACC Cardiovasc. Imaging 9:243–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Niss O, Fleck R, Makue F, Alsaied T, Desai P, et al. 2017. Association between diffuse myocardial fibrosis and diastolic dysfunction in sickle cell anemia. Blood 130:205–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Ingoglia G, Sag CM, Rex N, De Franceschi L, Vinchi F, et al. 2017. Hemopexin counteracts systolic dysfunction induced by heme-driven oxidative stress. Free Radic. Biol. Med 108:452–64 [DOI] [PubMed] [Google Scholar]
- 201.Guasch A, Navarrete J, Nass K, Zayas CF. 2006. Glomerular involvement in adults with sickle cell hemoglobinopathies: prevalence and clinical correlates of progressive renal failure. J. Am. Soc. Nephrol 17:2228–35 [DOI] [PubMed] [Google Scholar]
- 202.Powars DR, Chan LS, Hiti A, Ramicone E, Johnson C. 2005. Outcome of sickle cell anemia: a 4-decade observational study of 1056 patients. Medicine 84:363–76 [DOI] [PubMed] [Google Scholar]
- 203.da Silva GB Jr., Liborio AB, Daher Ede F. 2011. New insights on pathophysiology, clinical manifestations, diagnosis, and treatment of sickle cell nephropathy. Ann. Hematol 90:1371–79 [DOI] [PubMed] [Google Scholar]
- 204.Charache S, Scott JC, Charache P. 1979. “Acute chest syndrome” in adults with sickle cell anemia: microbiology, treatment, and prevention. Arch. Intern. Med 139:67–69 [PubMed] [Google Scholar]
- 205.Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, et al. 1995. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N. Engl. J. Med 332:1317–22 [DOI] [PubMed] [Google Scholar]
- 206.Mekontso Dessap A, Deux JF, Habibi A, Abidi N, Godeau B, et al. 2014. Lung imaging during acute chest syndrome in sickle cell disease: computed tomography patterns and diagnostic accuracy of bedside chest radiograph. Thorax 69:144–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Matthay MA, Ware LB, Zimmerman GA. 2012. The acute respiratory distress syndrome. J. Clin. Invest 122:2731–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Vichinsky EP, Styles LA, Colangelo LH, Wright EC, Castro O, Nickerson B. 1997. Acute chest syndrome in sickle cell disease: clinical presentation and course. Cooperative Study of Sickle Cell Disease. Blood 89:1787–92 [PubMed] [Google Scholar]
- 209.Leong MA, Dampier C, Varlotta L, Allen JL. 1997. Airway hyperreactivity in children with sickle cell disease. J. Pediatr 131:278–83 [DOI] [PubMed] [Google Scholar]
- 210.DeBaun MR, Strunk RC. 2016. The intersection between asthma and acute chest syndrome in children with sickle-cell anaemia. Lancet 387:2545–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Willen SM, Rodeghier M, Strunk RC, Bacharier LB, Rosen CL, et al. 2018. Aeroallergen sensitization predicts acute chest syndrome in children with sickle cell anaemia. Br. J. Haematol 180:571–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Vichinsky E, Williams R, Das M, Earles AN, Lewis N, et al. 1994. Pulmonary fat embolism: a distinct cause of severe acute chest syndrome in sickle cell anemia. Blood 83:3107–12 [PubMed] [Google Scholar]
- 213.Styles LA, Aarsman AJ, Vichinsky EP, Kuypers FA. 2000. Secretory phospholipase A(2) predicts impending acute chest syndrome in sickle cell disease. Blood 96:3276–78 [PubMed] [Google Scholar]
- 214.Godeau B, Schaeffer A, Bachir D, Fleury-Feith J, Galacteros F, et al. 1996. Bronchoalveolar lavage in adult sickle cell patients with acute chest syndrome: value for diagnostic assessment of fat embolism. Am. J. Respir. Crit. Care Med 153:1691–96 [DOI] [PubMed] [Google Scholar]
- 215.Castro O 1996. Systemic fat embolism and pulmonary hypertension in sickle cell disease. Hematol. Oncol. Clin. North Am 10:1289–303 [DOI] [PubMed] [Google Scholar]
- 216.Adisa OA, Hu Y, Ghosh S, Aryee D, Osunkwo I, Ofori-Acquah SF. 2013. Association between plasma free haem and incidence of vaso-occlusive episodes and acute chest syndrome in children with sickle cell disease. Br. J. Haematol 162:702–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Stankovic Stojanovic K, Steichen O, Lefevre G, Bachmeyer C, Avellino V, et al. 2012. High lactate dehydrogenase levels at admission for painful vaso-occlusive crisis is associated with severe outcome in adult SCD patients. Clin. Biochem 45:1578–82 [DOI] [PubMed] [Google Scholar]
- 218.Anea CB, Lyon M, Lee IA, Gonzales JN, Adeyemi A, et al. 2016. Pulmonary platelet thrombi and vascular pathology in acute chest syndrome in patients with sickle cell disease. Am. J. Hematol 91:173–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Mekontso Dessap A, Deux JF, Abidi N, Lavenu-Bombled C, Melica G, et al. 2011. Pulmonary artery thrombosis during acute chest syndrome in sickle cell disease. Am. J. Respir. Crit. Care Med 184:1022–29 [DOI] [PubMed] [Google Scholar]
- 220.Rucknagel DL. 2001. The role of rib infarcts in the acute chest syndrome of sickle cell diseases. Pediatr. Pathol. Mol. Med 20:137–54 [PubMed] [Google Scholar]
- 221.Bellet PS, Kalinyak KA, Shukla R, Gelfand MJ, Rucknagel DL. 1995. Incentive spirometry to prevent acute pulmonary complications in sickle cell diseases. N. Engl. J. Med 333:699–703 [DOI] [PubMed] [Google Scholar]
- 222.Knight-Madden JM, Forrester TS, Lewis NA, Greenough A. 2010. The impact of recurrent acute chest syndrome on the lung function of young adults with sickle cell disease. Lung 188:499–504 [DOI] [PubMed] [Google Scholar]
- 223.Field JJ, Burdick MD, DeBaun MR, Strieter BA, Liu L, et al. 2012. The role of fibrocytes in sickle cell lung disease. PLOS ONE 7:e33702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Mehrad B, Burdick MD, Wandersee NJ, Shahir KS, Zhang L, et al. 2017. Circulating fibrocytes as biomarkers of impaired lung function in adults with sickle cell disease. Blood Adv. 1:2217–24 [DOI] [PMC free article] [PubMed] [Google Scholar]