

Abstract
Background
Inherited retinal dystrophies (IRDs) are characterized by extreme genetic and clinical heterogeneity. There are many genes that are known to cause IRD which makes the identification of the underlying genetic causes quite challenging. And in view of the emergence of therapeutic options, it is essential to combine molecular and clinical data to correctly diagnose IRD patients. In this study, we aimed to identify the disease‐causing variants (DCVs) in four consanguineous Jordanian families with IRDs and describe genotype–phenotype correlations.
Methods
Exome sequencing (ES) was employed on the proband patients of each family, followed by segregation analysis of candidate variants in affected and unaffected family members by Sanger sequencing. Simulation analysis was done on one novel CLRN1 variant to characterize its effect on mRNA processing. Clinical evaluation included history, slit‐lamp biomicroscopy, and indirect ophthalmoscopy.
Results
We identified two novel variants in CLRN1 [(c.433+1G>A) and (c.323T>C, p.Leu108Pro)], and two recurrent variants in ABCA4 [(c.1648G>A, p.Gly550Arg) and (c.5460+1G>A)]. Two families with the same DCV were found to have different phenotypes and another family was shown to have sector RP. Moreover, simulation analysis for the CLRN1 splice donor variant (c.433+1G>A) showed that the variant might affect mRNA processing resulting in the formation of an abnormal receptor. Also, a family that was previously diagnosed with nonsyndromic RP was found to have Usher syndrome based on their genetic assessment and audiometry.
Conclusion
Our findings extend the spectrum of CLRN1‐ and ABCA4‐associated IRDs and describe new phenotypes for these genes. We also highlighted the importance of combining molecular and clinical data to correctly diagnose IRDs and the utility of simulation analysis to predict the effect of splice donor variants on protein formation and function.
Keywords: ABCA4, CLRN1, Exome sequencing, Inherited retinal dystrophy
We identified novel and recurrent CLRN1 and ABCA4 disease‐causing variants in four Jordanian families with inherited retinal dystrophies. One of the ABCA4 variants was found in two unrelated families but surprisingly caused two different phenotypes.

1. INTRODUCTION
Inherited retinal dystrophies (IRDs) are a group of clinically and genetically heterogeneous inherited retinal disorders with an estimated worldwide prevalence of about 1:3,000 (Hartong, Berson, & Dryja, 2006; Wang et al., 2018). IRDs can be either syndromic or nonsyndromic. Of the nonsyndromic IRDs, retinitis pigmentosa (RP) is the most common (Chizzolini et al., 2011; Werdich, Place, & Pierce, 2014; Wright, Chakarova, Abd El‐Aziz, & Bhattacharya, 2010), whereas Usher syndrome is the most common syndromic IRD that is characterized by hearing loss in addition to RP features (Cosgrove & Zallocchi, 2014; Mathur & Yang, 2015).
To date, over 300 genes were shown to be associated with IRDs (https://sph.uth.edu/Retnet/), and all the genetic inheritance patterns are involved (Astuti et al., 2018; Kajiwara, Berson, & Dryja, 1994; Wang et al., 2018). Not only variants in the same gene but also the same genetic variants have been shown to cause variable phenotypes in inter‐ and intrafamilial cases (Fahim, Daiger, & Weleber, 1993).
This broad genetic and clinical complexity warrants highly efficient and high‐throughput genetic testing approach to uncover the underlying genetic causes and reach to the accurate diagnosis of IRDs.
CLRN1 which is located on chromosome 3q25.1 and codes for clarin‐1 protein that has functions in the retina and the cochlea (Adato et al., 2002; Geng et al., 2009). Therefore, different DCVs in this gene were shown to cause RP and Usher syndrome type III in previous studies (Fields et al., 2002; Khan et al., 2011). ABCA4 is expressed in retinal photoreceptors and was implicated in different IRDs like cone‐rod dystrophy and Stargardt's disease (Jiang et al., 2016; Molday, Zhong, & Quazi, 2009).
Exome sequencing (ES) is an affordable technology that covers the coding regions and splice‐site variants and is powerful tool for identification of disease‐causing variants (DCVs) for Mendelian diseases (Dixon‐Salazar et al., 2012; Goldfeder, Wall, Khoury, Ioannidis, & Ashley, 2017; Petersen, Fredrich, Hoeppner, Ellinghaus, & Franke, 2017). Although the coding region represents only 1%–2% of the human genome, it carries 85% of the disease‐related variants (Gilissen, Hoischen, Brunner, & Veltman, 2012).
Therefore, in this study, we aimed to identify the DCVs in four Jordanian pedigrees with autosomal recessive IRDs using ES. Moreover, full ophthalmic examination was performed for genotype–phenotype correlations.
2. MATERIALS AND METHODS
2.1. Study subjects and clinical examination
This study was performed in accordance with the tenets of the Declaration of Helsinki and approved by the Institutional Review Board of the Cell Therapy Center, Amman, Jordan. Written informed consent was obtained from all participants prior to their inclusion in the study.
Thirteen patients from four unrelated Jordanian families with inherited visual problems were recruited into the study (Figure 1). Only family members labeled with a number in the pedigrees were included.
Figure 1.
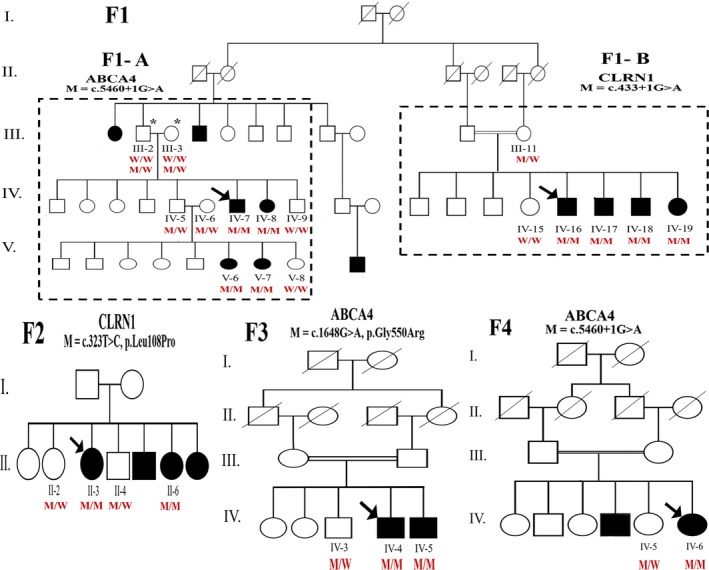
Pedigrees of the four Jordanian families participating in the study. Squares, circles, and dashes represent males, females, and deceased individuals, respectively. Arrows indicate probands. Solid symbols refer to affected individuals and open symbols to normal or carrier individuals. The two dashed squares in family F1 pedigree divide it into F2‐A and F2‐B. Double horizontal lines indicate consanguinity. M/M, homozygous mutated; M/W, heterozygous mutated; W/W, homozygous wild type. Zygosity for individuals III‐2 and III‐3 in family F1‐A is written in two lines, the above one for CLRN1 (c.433+1G>A), and the lower one for ABCA4 (c.5460+1G>A)
A detailed history was taken from all patients including their age at onset, first symptom, progression, and other co‐morbidities. All affected individuals underwent standard ophthalmic examination, including Snellen chart uncorrected visual acuity, best‐corrected visual acuity (BCVA), slit‐lamp biomicroscopy (Haag‐Streit BM 900, Switzerland), fundus photography (Optos 200Tx, Scotland), Optical Coherence Tomography (OCT), Keratometry (Oculus Pentacam Typ70700, Germany), and full‐field Electroretinography (ffERG) (Roland consult color Ganzfeld Q450 C, Germany). ERG recordings were performed according to the 2015 update of the International Society for Clinical Electrophysiology of Vision Standards (ISCEV) (McCulloch et al., 2015). Where necessary, pure‐tone audiometry was done to evaluate hearing status for suspected Usher syndrome cases.
2.2. Exome sequencing and bioinformatics analysis
Genomic DNA was isolated from peripheral blood samples of the patients and participating family members using QIAprep Spin Miniprep Kit. ES was performed on the proband patients of each family (Figure 1) by Partners HealthCare Personalized Medicine (PPM), Harvard, USA. Briefly, extracted DNA was sheared to 150–200 bp fragments. Exome enrichment was performed using Agilent SureSelect Human All Exon V5 kit. DNA was sequenced using 100 bp paired‐end reads on Hiseq 2500 platform (Illumina Inc.). Raw data were processed by Illumina base‐calling software 1.7 using default parameters. The sequencing reads were aligned to the NCBI reference sequence (GRCh37), using the Burrows‐Wheeler Aligner (BWA), and variant calls were made using the Genomic Analysis Tool Kit (GATK). The detected variants were annotated and filtered against four databases (i.e., NCBI CCDS [http://www.ncbi.nlm.nih.gov/CCDS/CcdsBrowse.cgi], RefSeq [http://www.ncbi.nlm.nih.-gov/RefSeq/], Ensembl [http://www.ensembl.org], and Encode [http://genome.ucsc.edu/ENCODE]).
2.3. Variants filtration and prioritization
To detect the candidate DCVs, multistep filtration approach was applied using Illumina basespace variant interpreter tool (https://variantinterpreter.informatics.illumina.com/) (Table S1). Variants were filtered based on (a) quality (total read depth ≥10), (b) genomic position (within exons and flanking intronic regions), (c) genes of interest (list of 186 genes) (Azab et al. 2019), (d) allele frequencies; we only included variants with MAF <0.01 in EXAC, gnomAD, 1000 Genomes project, or ESP, and (e) mode of inheritance; our four pedigrees follow an autosomal recessive mode of inheritance, therefore only homozygous or compound heterozygous variants were included (Table S1). Ensembl was also used as a reference dataset of population‐based allele frequencies.
To assess the pathogenicity of the identified variants, we used a combination of computational tools. The possible damaging impacts of missense variants on protein structure and/or function were predicted using SIFT (http://sift.jcvi.org), PolyPhen‐2 (http://genetics.bwh.harvard.edu/pph2), Provean (http://provean.jcvi.org/index.php), CONDEL (http://bg.upf.edu/fannsdb/) and MutationTaster2 (Schwarz, Cooper, Schuelke, & Seelow, 2014). Evolutionary conservations across species for missense variants were also assessed using Alamut Visual Prediction Software. For the impact of splice site variants, the following prediction tools were used: Human Splicer Finder (http://www.umd.be/HSF/), NNSPLICE (http://www.fruitfly.org/seq_tools/splice.html), SSF, and MaxEnt.
2.4. Variants validation and segregation analysis
Sanger sequencing was performed to verify the candidate DCVs identified by ES and for segregation analysis in all family members available for testing. Primers flanking the candidate loci were designed using Primer3 software and synthesized by IDT (S) (Table S2). The extracted DNA was PCR‐amplified by Platinum™ PCR SuperMix (Invitrogen), and purified by GeneJET PCR purification kit (Invitrogen). Sequencing was performed with the BigDye Terminator V3.1 Cycle Sequencing kit (Applied Biosystems) on an ABI 3500 genetic analyzer. The sequences data were analyzed by SeqA software (Applied Biosystems) and Chromas Pro software (Technolysium LTD). The variants were then classified as benign, likely benign, variant of unknown significance, likely pathogenic, and pathogenic according to the American College of Medical Genetics and Genomics (ACMG) standards and guidelines for the interpretation of sequence variants (Richards et al., 2015). Variants were submitted to ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/).
2.5. Simulation of CLRN1 (c.433+1G>A) splice donor variant
Simulation analysis including docking and molecular dynamic simulation was used to assess the effect of CLRN1 (c.433+1G>A) variant on mRNA splicing. The detailed explanation of the experiment methods is available in the supporting information.
3. RESULTS
3.1. Genetic findings
Aiming to identify the DCVs, ES was performed on five probands with autosomal recessive IRDs. Around six Gb of data was obtained, on average 80% of the target region was at least 10X covered, and about 70k variants were identified in each proband. To identify the DCV in each family, we did a multistep filtration approach as described in the methods section. In total, four pathogenic variants in CLRN1 and ABCA4 were identified in the four families (Table 1), in which two were novel CLRN1 variants and two were recurrent ABCA4 variants.
Table 1.
Candidate variants identified in four inherited retinal dystrophy pedigrees by exome sequencing
| Family Number | Gene |
Variant Coordinate (hg19) |
Transcript (Ensembl) |
dbSNP ID | Variation |
Total MAF (Gnomad) |
ClinVar | Classification of variantsa | Associated phenotype | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HGVS cDNA | HGVS aa | Type | Zygosity | |||||||||
| 1‐A & 4 | ABCA4 | chr1:94480098 | ENST00000370225.3 | rs61753030 | c.5460+1G>A | NA | Splice donor | Hom | 0.00003185 | Pathogenic | Pathogenic | RP in F1‐A and CRD in F4 |
| 1‐B | CLRN1 | chr3:150659368 | ENST00000327047.5 | rs201205811 | c.433+1G>A | NA | Splice donor | Hom | 0.000007970 | NA | Pathogenic | Usher syndrome type III |
| 2 | CLRN1 | chr3:150659479 | ENST00000327047.5 | NA | c.323T>C | p.Leu108Pro | Missense | Hom | NA | NA | Uncertain Significance | Usher syndrome type III |
| 3 | ABCA4 | chr1:94528780 | ENST00000370225.3 | rs61748558 | c.1648G>A | p.Gly550Arg | Missense | Hom | 0.000003977 | Likely Pathogenic | LikelyPathogenic | CRD |
Novel variants are highlighted in bold. Abbreviations: CRD, cone‐rod dystrophy; Hom, homozygous; MAF, minor allele frequency; NA, not available; RP, retinitis pigmentosa.
Classification is based on the ACMG guidelines for interpretation of sequence variants (See the text).
Family F1 was a large extended family that has two probands, one from each side of this pedigree (F1‐A and F1‐B) (Figure 1). ES revealed ABCA4 splice site donor variant (c.5460+1G>A) in F1‐A. Interestingly, this variant (c.5460+1G>A) was also found in family F4. On the other hand, for F1‐B novel splice site donor variant in CLRN1 (c.433+1G>A) was detected. The second novel CLRN1 variant was a missense variant (c.323T>C, p.Leu108Pro) identified in family F2. In F3, a missense variant in ABCA4 (c.1648G>A, p.Gly550Arg) was identified (Table 1; Figure S1).
3.1.1. ABCA4 variants
The splice site donor variant (c.5460+1G>A) in families F1‐A and F4 is predicted by the different in silico tools to affect splicing causing exon skipping resulting in out‐of‐frame transcripts (Table 2). This variant has been reported previously in the literature to be pathogenic (Rivera et al., 2000; Ścieżyńska et al., 2016; Xiong et al., 2015). The missense ABCA4 (c.1648G>A) variant introduces a substitution from glycine to arginine at amino acid residue 550 (p.Gly550Arg). It is located in a conserved genomic region and in silico analyses tools predicted it as pathogenic (Table 2). Moreover, this variant is already reported in ClinVar as likely pathogenic.
Table 2.
In silico prediction of the identified candidate pathogenic variants
| Gene | Variant (HGVS cDNA) | Variant type | Missense prediction | Splice site prediction | Overall prediction of pathogenicity | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SIFT | Polyphen | CONDEL | PROVEAN | MutationTaster2 | SSF | MaxEnt | NNSPLICE | HSF | ||||
| CLRN1 | c.433+1G>A | Splice donor | − | − | − | − | − | + | + | + | + | Pathogenic |
| ABCA4 | c.5460+1G>A | Splice donor | − | − | − | − | − | + | + | + | + | Pathogenic |
| ABCA4 | c.1648G>A | Missense | D | PD | D | DE | Disease causing | − | − | − | − | Pathogenic |
| CLRN1 | c.323T>C | Missense | D | PD | D | NA | Disease causing | − | − | − | − | Pathogenic |
Abbreviations: D, damaging; DE, deleterious; NA, not available; PD, probably damaging.
3.1.2. CLRN1 variants
All splicing‐related in silico platforms we utilized unanimously predicted the novel CLRN1 splice site variant (c.433+1G>A) to break the splice donor site (Table 2). To further assess the effect of this novel variant, Sanger sequencing revealed that the four identified candidate variants completely segregated with disease phenotypes following an autosomal recessive inheritance pattern (i.e., DCVs were homozygous in affected individuals, heterozygous in their parents, and normal/heterozygous in unaffected relatives) (Figure 1). According to the ACMG guidelines, we classified the two novel CLRN1 ((c.433+1G>A) and (c.323T>C)) as pathogenic and uncertain significance, respectively. We also confirmed the previous ClinVar classification of ABCA4 (c.5460+1G>A) and ABCA4 (c.1648G>A, p.Gly550Arg) as pathogenic and likely pathogenic, respectively.
3.2. Simulation analysis
To characterize the effect of the splice‐donor variant (c.433+1G>A) in CLRN1 on splicing and the function of the transmembrane protein clarin‐1, simulation analysis was performed. Docking simulation was used to assess the interaction between U1 snRNP and RNA splice site in both the wild‐type and mutated forms. Figure 2a shows the best oriented pose of the wild‐type RNA and U1 snRNP. Molecular dynamics simulation showed that mutating G21 to A in the best‐docked pose between wild‐type RNA and U1 snRNP would result in loss of noncanonical interactions between certain atoms in wild‐type RNA and U1 snRNP. Figure 2b shows the interacting atoms between G21 (from the wild‐type RNA strand) with C8 (from U1 snRNP) as annotated spheres. These noncanonical interactions include hydrogen bonds, and interactions among C–H and O/N groups. These interacting atoms have not been found when mutating G21 to A, indicating the importance of G21 for such binding interactions (Figure 2c). Further details of the simulation analysis results are illustrated in the supporting information.
Figure 2.
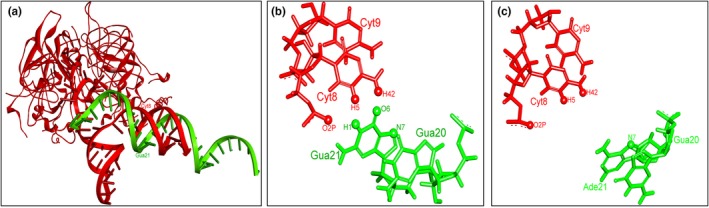
Simulation analysis of the splice donor variant results CLRN1 (c.433+1G>A). (a) Best oriented docked pose of the wild‐type RNA with U1 snRNP. The wild‐type RNA and the U1 snRNP are shown in green and red colors, respectively. The wild‐type polymorphism (Guanine) is shown as G21, it is binding with Cytosine 8 of U1 snRNP, (b) The main noncanonical interacting atoms between Guanine (G21) from wild‐type RNA with Cytosine (C8) from U1 snRNP. (c) Noncanonical interactions became less frequent when G21 is mutated to A. Noncanonical interacting atoms are shown as annotated spheres
3.3. Clinical results
Thirteen patients from four consanguineous Jordanian families with autosomal recessive visual problems were recruited to this study for further evaluation and molecular diagnosis. The detailed clinical information of all patients is summarized in Table 3.
Table 3.
Clinical data of all participated inherited retinal dystrophy patients in the study
| Family | Affected members | Age | First symptom | BCVA | ffERG | Fundus examination | Slit‐lamp biomicroscopy | OCT | Keratoconus |
Hearing status (audiometry) |
Inherited retinopathy phenotype | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| At exam | Onset | OD | OS | Scotopic response | Photopic response | OD | OS | OD | OS | |||||||
| 1A | IV‐7 | 49 | 10 | Low VA | CF at 1.25 m | 0.05 | Severely reduced | Severely reduced | Typical RP | Typical RP | No cataract | Central atrophic maculopathy with no CME | Central atrophic maculopathy with no CME | Topographic keratoconus in both eyes | Normal | RP |
| IV‐8 | 37 | 10 | Low VA | HM | HM | Severely reduced | Severely reduced | Typical RP | Typical RP |
No cataract Horizontal nystagmus |
Central atrophic maculopathy with no CME | Central atrophic maculopathy with no CME | No keratoconus | Normal | RP | |
| V‐6 | 11 | 9 | Low VA | 0.1 | 0.1 | Slightly reduced | Normal | Atypical RP | Atypical RP | No cataract | Central atrophic maculopathy with no CME | Central atrophic maculopathy with no CME | No keratoconus | Normal | RP | |
| V‐7 | 12 | 6 | Low VA | 0.1 | 0.1 | Slightly reduced | Normal | Atypical RP | Atypical RP | No cataract | Central atrophic maculopathy with no CME | Central atrophic maculopathy with no CME | No keratoconus | Normal | RP | |
| 1B | IV‐16 | 48 | 5 | Nyctalopia | HM | CF closely | Severely reduced | Severely reduced | Typical RP | Typical RP | PSCC in both eyes | Blurred OCT due to cataract | Blurred OCT due to cataract | No keratoconus | Bilateral moderate‐to‐severe hearing loss | USH III |
| IV‐17a | 54 | 7 | Nyctalopia | No LP | 0.4 | Severely reduced | Severely reduced | Typical RP | Typical RP | Had cataract surgery in the left eye | Central atrophic maculopathy with no CME | Central atrophic maculopathy with no CME |
OD: cannot comment due to trauma OS: no keratoconus |
Bilateral moderate‐to‐severe hearing loss | USH III | |
| IV‐18 | 44 | 6 | Nyctalopia | CF at 1 m | CF at 1.5 m | Severely reduced | Severely reduced | Typical RP | Typical RP | Posterior polar cataract with nuclear sclerosis | Mild central atrophic maculopathy with no CME | Mild central atrophic maculopathy with no CME | No keratoconus | Bilateral mild‐to‐moderate hearing loss | USH III | |
| IV‐19 | 48 | 7 | Nyctalopia | CF at 2 m | CF at 2 m | Severely reduced | Severely reduced | Typical RP | Typical RP | PSCC in both eyes | Mild central atrophic maculopathy with no CME | Poor quality due to cataract | Keratometry not available | Not available | USH III | |
| 2 | II‐3 | 41 | 36 | Nyctalopia | 0.3 | CF at 2 m | Severely reduced | Severely reduced | Typical RP | Typical RP | PSCC in both eyes | Mild central atrophic maculopathy with no CME |
Mild central atrophic maculopathy CME with epiretinal membrane |
No keratoconus | Bilateral mild hearing loss | USH III |
| 3 | IV‐4 | NA | 11 | Low VA | CF at 1 m | 0.1 |
OD: severely reduced OS: moderately reduced |
Severely reduced | Atypical RP | Atypical RP | Congenital cataract in the left eye | Central atrophic maculopathy with no CME | Central atrophic maculopathy with no CME | No keratoconus | Normal | CRD |
| 4 | IV‐4 | 19 | 8 | Low VA | CF at 2 m | CF at 2 m | Slightly reduced | Moderately reduced | Atypical RP | Atypical RP | Had cataract surgery | Central atrophic maculopathy with no CME | Central atrophic maculopathy with no CME | No keratoconus | Normal | CRD |
| IV‐6 | 24 | 8 | Low VA | CF at 2 m | CF at 2 m | Moderately reduced | Severely reduced | Atypical RP | Atypical RP | No cataract | Central atrophic maculopathy with no CME | Central atrophic maculopathy with no CME | No keratoconus | Normal | CRD | |
Typical RP denotes the presence of the classical triad (attenuated arterioles, disc pallor, and bone spicules) in fundus photography. Atypical RP means that a part of the triad is missing.
Abbreviations: BCVA, best‐corrected visual acuity; CF, counting fingers; CME, cystoid macular edema; CRD, cone‐rod dystrophy; ffERG, full‐field electroretinography; HM, hand motion; LP, light perception; NA, not available; OCT, optical coherence tomography; OD, right rye; OS, left eye; PSCC, posterior subcapsular cataract; RP, retinitis pigmentosa; USH III, Usher syndrome III.
This patient had cataract surgery in the left eye, and his right eye was lost due to trauma.
3.4. Clinical consequences of the identified CLRN1 variants
Two families (F1B and F2) were found to have their DCVs in CLRN1 gene (Table 1) (Figure 1). The patients' ages in F1B and F2 ranged between 35 and 54 years old at the time of examination. Almost all of them were legally blind at time of examination (best‐corrected visual acuity <3/60 (0.05) in the better‐seeing eye, according to WHO) (World Health Organization, 2016). Patients in F1‐B started to have night blindness during early childhood, followed by progressive constriction of peripheral vision and central vision later. Fundus examination showed typical RP features (Figure 3; Figure S2). Clinically, F1‐B patients had no associated symptoms with RP and were thus considered to have nonsyndromic RP. However, ES results showed CLRN1 as disease causing and this gene has been classically associated with Usher syndrome. This warranted further audiometric evaluation to rule in/out hearing problems.
Figure 3.
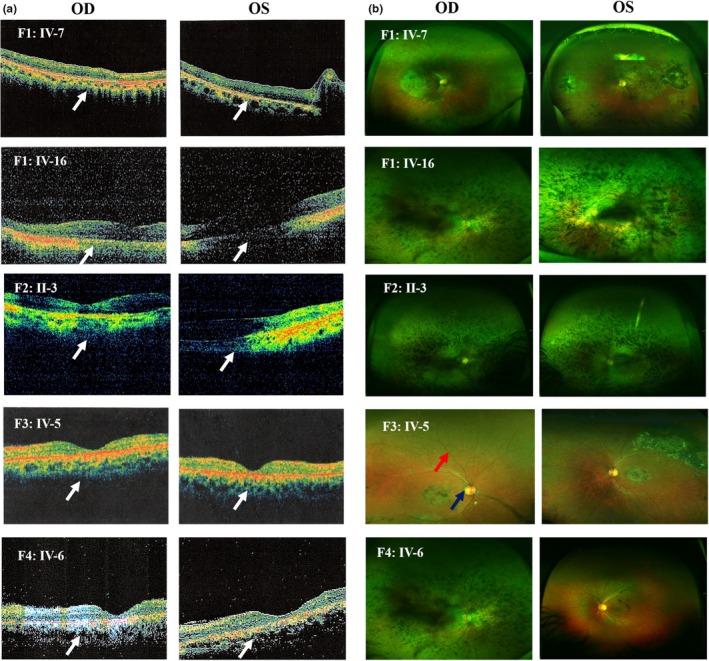
Optical coherence tomography (OCT) (a) and (b) fundus photography of probands of all families (OD and OS). White arrows show central atrophic maculopathy with no cystoid macular edema, blue arrow indicates waxy pallor of optic disc, and red arrow indicates attenuation of retinal arterioles. Fundus images also show bone spicules
Audiometry showed different degrees of bilateral sensorineural deafness in all patients, confirming the diagnosis of Usher syndrome (Figure S3). Patients' hearing was normal at birth, their speech is normal, and they do not have balance problems, all suggesting type III Usher syndrome (Mathur & Yang, 2015). Being at late stages, ffERG in all patients showed severe reduction in both scotopic and photopic responses (Figure 4; Figure S5). OCT revealed variable degrees of central macular atrophy (Figure 3; Figure S4). Besides, they have cataract in both eyes (Table 3).
Figure 4.
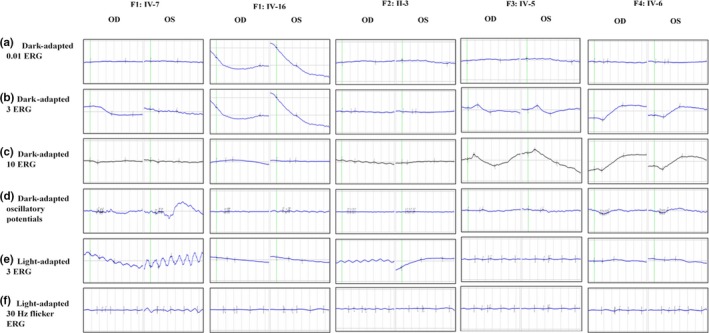
Electroretinograms (ERGs) of both eyes in all probands including the standard six recordings denoted beside each row
On the other hand, patient F2: II‐3 had a late‐onset RP (at the age of 36), unlike patients in F1‐B family. Although her ffERG showed severe reduction in both photopic and scotopic responses in both eyes, her visual acuity is 0.3 in the better‐seeing eye (Figure 4; Figure S5). Audiometry was also done for this patient revealing mild bilateral sensorineural deafness. Her affected sister (F2: II‐3) declined to participate in the clinical assessments.
3.5. Clinical consequences of the identified ABCA4 variants
The molecular assessment for families; F1‐A, F3, and F4 revealed pathogenic variants in ABCA4 (Figure 1). Patients in F1‐A and F4 share the same DCV (ABCA4, c.5460+1G>A) (Table 1). F1‐A has four affected members; two older individuals (IV‐7 and IV‐8) whose both night vision and visual acuity are affected, and two younger individuals (V‐6 and V‐7) who are still at an early stage of the disease (Figure 1; Table 3). The presenting symptom for all of them was decreased visual acuity in the first decade of life. ffERG showed severe reduction in both photopic and scotopic responses for the patients (F1‐A: IV‐7 and IV‐8) (Figure 4; Figure S5). Whereas, those of the younger siblings (F1‐A: V‐6 and V‐7) showed slight impairment of scotopic responses only. Hence, the disease in this family (F1‐A) started in the rods followed by the cones (rod‐cone dystrophy (RCD).
Family F4 patients (IV‐4 and IV‐6) who harbor the same DCV as family F1: A also reported decreased visual acuity as the first symptom. However, their ffERGs showed more severe impairment of photopic responses than scotopic responses (Figure 4). Additionally, fundus photography showed atypical RP features; RP sine pigmentoin addition to the presence of bone spicules that were restricted to the central part of the retina (Figure 3; Figure S2). OCT of all patients showed central macular atrophy (Figure 3; Figure S4). Therefore, this family was diagnosed with cone‐rod dystrophy (CRD).
In the third family (F3) with ABCA4 DCV (Figure 1), IV‐5 reported decreased visual acuity as the first symptom, which was followed by gradual constriction of peripheral vision. Funduscopic examination showed bone spicules only in the central macula and in the inferior part of the retina, suggesting sector RP features (Figure 3). OCT revealed central atrophic maculopathy in both retinae which was more severe in the left eye (Figure 3). FfERG showed severe reduction in both scotopic and photopic responses, except for scotopic responses in the left eye which were moderately reduced (Figure 4; Table 3). Both OCT and ERG results were consistent with visual acuity being better in the left eye than the right (0.1 and CF at 2 m, respectively). This family was diagnosed with CRD.
3.6. Associated ocular diseases
Among patients with available clinical data, seven of 12 patients either have cataract or underwent cataract surgery in the past. Three patients (25%) had posterior subcapsular cataract in both eyes, one has posterior polar cataract with nuclear sclerosis, and one with congenital cataract (Table 1). Keratometry showed topographic keratoconus only in patient IV‐7 in family F1‐A (Figure S6).
4. DISCUSSION
In this study, we combined clinical and molecular data to characterize the phenotype of patients with various forms of IRDs. We identified two novel DCVs in CLRN1; one splice donor variant (c.433+1G>A) and another missense variant (c.323T>C, p.Leu108Pro). Moreover, we added thorough clinical descriptions to two recurrent ABCA4 variants ((c.5460+1G>A), (c.1648G>A, p.Gly550Arg)).
CLRN1 codes for a four‐transmembrane domain protein called clarin‐1 that localizes to the plasma membrane of auditory hair cells and spiral ganglion cells in the retina (Adato et al., 2002). Clarin‐1 has multiple functions in the retina and auditory hair cells including organization of actin cytoskeleton, and sensory perception of visual and auditory stimuli (Aller et al., 2004; Tian et al., 2009). CLRN1 variants have been classically associated with Usher syndrome (Aller et al., 2004; Khan et al., 2011; Pennings, Kremer, Deutman, Kimberling, & Cremers, 2002; Roux, 2005). A study published in 2011 described two missense CLRN1 variants causing nonsyndromic IRD (Khan et al., 2011). This study suggested that missense CLRN1 variants located in the boundaries of transmembrane domains, and resulting in no change in the polarity of the amino acid are more likely to cause nonsyndromic IRD (Khan et al., 2011). In other studies, Usher syndrome was mostly caused by CLRN1 variants located in the extra‐ or intracellular domain of clarin‐1 (Garcia‐Garcia et al., 2012; Isosomppi et al., 2009). The CLRN1 (c.323T>C, p.Leu108Pro) missense variant identified in family F2 is located in the middle of the second transmembrane domain of clarin‐1 and caused Usher syndrome. This is similar to two missense variants (c.449T>C, p.Leu150Pro) and (c.313T>C, p.S105P) identified before and were causative of Usher syndrome (Fields et al., 2002; Sadeghi, Cohn, Kimberling, Tranebjaerg, & Moller, 2005). Therefore, our study and those of others suggest that the location of the substituted amino acid within clarin‐1 domains as well as the change in its polarity appear to be reliable predictors of the resultant phenotype. This variant was associated with late‐onset Usher syndrome (late onset of both RP and hearing loss). On the other hand, family F1‐B patients who harbor the splice donor CLRN1 DCV had a much earlier onset and more severe disease as evident by their lower BCVA. Noteworthy, cataract also contributes to the lower VA of some patients of family F1‐B compared to F2.
The story of patients in these two families highlights the importance of integrating ES into clinical diagnosis of IRDs. Prior to molecular assessment, all our patients with CLRN1 variants had no hearing or balance complaints and were thus diagnosed with nonsyndromic RP. Only after molecular testing showed CLRN1 as disease causing, audiometry was done and revealed sensorineural hearing loss in all patients. Consequently, all patients in families F1‐B and F2 were diagnosed with Usher syndrome III. This illustrates the utility of audiometry to diagnose asymptomatic hearing impairment as well as the importance of genetic testing to accurately diagnose IRD caused by CLRN1 variants and differentiate between syndromic and nonsyndromic phenotypes.
Simulation analysis including docking and molecular dynamics simulation was performed to the novel CLRN1 splice donor variant (c.433+1G>A) to study the effects of a splice donor variant in CLRN1 on mRNA processing. Docking simulations showed that mutating guanine to adenine at the splice site affects binding with U1 snRNP tremendously. Molecular dynamic simulations clarified that even when the mutated RNA binds U1 snRNP, there would be lessening of the main noncanonical contacting interactions between RNA and U1 snRNP. Taken together, these results indicate that this variant is predicted to affect splicing and thus produce an abnormal disease‐causing protein.
The other families (F1‐A, F3, and F4) have DCVs in ABCA4. ABCA4 is exclusively expressed in the retina, especially in its photoreceptors and functions to translocate diverse intermediates of visual cycle out of them (Allikmets et al., 1997). Reduced activity of ABCA4 protein leads to accumulation of its substrates in the photoreceptors and RPE which consequently leads to apoptosis (Cideciyan et al., 2009; Sun, Molday, & Nathans, 1999). Death of RPE cells occurs within and near the macula where cones and most of the rods are located, respectively (Martinez‐Mir et al., 1998; Tsybovsky, Molday, & Palczewski, 2010). This might explain why ABCA4 variants may cause both RCDs and CRDs. ABCA4 variants have been linked to Stargardt's disease, CRD, and RP (Lois, Holder, Bunce, Fitzke, & Bird, 2001). Of interest, we identified the same rare variant (ABCA4 (c.5460 G>A)), whose MAF is 0.00003185, as disease causing in two unrelated families (F1‐A and F4). More surprisingly, they had different clinical phenotypes. F1‐A patients have RCD, while F4 patients have CRD based on their ERGs (Table 3). This illustrates the extensive heterogeneity of IRDs as they do not only exhibit genetic heterogeneity and variable expressivity but also "variant pleiotropy". F3 has ABCA4 (c.1648G>A, p.Gly550Arg) as the DCV. This variant has been previously reported to cause CRD, Stargardt's disease, and generalized choriocapillaris dystrophy (Shroyer, Lewis, Yatsenko, Wensel, & Lupski, 2001; Thiadens et al., 2012). Here, we report this variant with CRD and sector RP; a form of atypical RP. To the best of our knowledge, this variant has never been reported to cause sector RP. Heterozygous ABCA4 variants were also associated with age‐related macular degeneration (ARMD) (Kjellström, 2015; Schulz et al., 2017). Hence, both affected and carrier individuals of families F1‐A, F3, and F4 might be at increased risk of ARMD in the future. In conclusion, our findings emphasize the importance of combining ophthalmic and genetic tests to correctly diagnose rare diseases like IRDs and provide proper counseling. It also characterizes detailed explanation of the effect of a splice donor variant in CLRN1 clinically and the level of mRNA processing. Furthermore, we emphasized that ABCA4 variants can cause a spectrum of retinal diseases rather than a single entity, even when the same DCV exists in two unrelated families. This might be an indication of the presence of other environmental or epigenetic factors that govern genotype–phenotype correlations.
CONFLICT OF INTEREST
All authors declare that they do not have any competing interests.
Supporting information
ACKNOWLEDGMENTS
Supported by a grant from the Deanship of Scientific Research at the University of Jordan (Grant No. 2013/39) and the Abdul Hameed Shoman Foundation (Grant No. 2017/05), Amman, Jordan.
Abu‐Ameerh M, Mohammad H, Dardas Z, et al. Extending the spectrum of CLRN1‐ and ABCA4‐associated inherited retinal dystrophies caused by novel and recurrent variants using exome sequencing. Mol Genet Genomic Med. 2020;8:e1123 10.1002/mgg3.1123
Contributor Information
Abdalla Awidi, Email: abdalla.awidi@gmail.com.
Belal Azab, Email: azabbm@mymail.vcu.edu.
REFERENCES
- Adato, A. , Vreugde, S. , Joensuu, T. , Avidan, N. , Hamalainen, R. , Belenkiy, O. , … Lancet, D. (2002). USH3A transcripts encode clarin‐1, a four‐transmembrane‐domain protein with a possible role in sensory synapses. European Journal of Human Genetics, 10(6), 339–350. 10.1038/sj.ejhg.5200831 [DOI] [PubMed] [Google Scholar]
- Aller, E. , Jaijo, T. , Oltra, S. , Alió, J. , Galán, F. , Nájera, C. , … Millán, J. M. (2004). Mutation screening of USH3 gene (clarin‐1) in Spanish patients with Usher syndrome: Low prevalence and phenotypic variability. Clinical Genetics, 66(6), 525–529. 10.1111/j.1399-0004.2004.00352.x [DOI] [PubMed] [Google Scholar]
- Allikmets, R. , Singh, N. , Sun, H. , Shroyer, N. F. , Hutchinson, A. , Chidambaram, A. , … Lupski, J. R. (1997). A photoreceptor cell‐specific ATP‐binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nature Genetics, 15(3), 236–246. 10.1038/ng0397-236 [DOI] [PubMed] [Google Scholar]
- Astuti, G. , van den Born, L. , Khan, M. , Hamel, C. , Bocquet, B. , Manes, G. , … Roosing, S. (2018). Identification of inherited retinal disease‐associated genetic variants in 11 candidate genes. Genes (Basel), 9(1), 10.3390/genes9010021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azab, B. , Barham, R. , Ali, D. , Dardas, Z. , Rashdan, L. , Bijawi, M. , … Awidi, A. (2019). Novel CERKL variant in consanguineous Jordanian pedigrees with inherited retinal dystrophies. Canadian Journal of Ophthalmology, 54(1), 51–59. 10.1016/j.jcjo.2018.02.018 [DOI] [PubMed] [Google Scholar]
- Chizzolini, M. , Galan, A. , Milan, E. , Sebastiani, A. , Costagliola, C. , & Parmeggiani, F. (2011). Good epidemiologic practice in retinitis pigmentosa: From phenotyping to biobanking. Current Genomics, 12(4), 260–266. 10.2174/138920211795860071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan, A. V. , Swider, M. , Aleman, T. S. , Tsybovsky, Y. , Schwartz, S. B. , Windsor, E. A. M. , … Palczewski, K. (2009). ABCA4 disease progression and a proposed strategy for gene therapy. Human Molecular Genetics, 18(5), 931–941. 10.1093/hmg/ddn421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosgrove, D. , & Zallocchi, M. (2014). Usher protein functions in hair cells and photoreceptors. The International Journal of Biochemistry & Cell Biology, 46, 80–89. 10.1016/j.biocel.2013.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon‐Salazar, T. J. , Silhavy, J. L. , Udpa, N. , Schroth, J. , Bielas, S. , Schaffer, A. E. , … Gleeson, J. G. (2012). Exome sequencing can improve diagnosis and alter patient management. Science Translational Medicine, 4(138), 138ra178 10.1126/scitranslmed.3003544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahim, A. T. , Daiger, S. P. , & Weleber, R. G. (1993). Nonsyndromic retinitis pigmentosa overview In Pagon R. A., Adam M. P., Ardinger H. H., Wallace S. E., Amemiya A., Bean L. J. H., Bird T. D., Ledbetter N., Mefford H. C., Smith R. J. H., & Stephens K. (Eds.), GeneReviews(R) (pp. 7–9). Seattle, Washington: University of Washington. [Google Scholar]
- Fields, R. R. , Zhou, G. , Huang, D. , Davis, J. R. , Möller, C. , Jacobson, S. G. , … Sumegi, J. (2002). Usher syndrome type III: Revised genomic structure of the USH3 gene and identification of novel mutations. American Journal of Human Genetics, 71(3), 607–617. 10.1086/342098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Garcia, G. , Aparisi, M. J. , Rodrigo, R. , Sequedo, M. D. , Espinos, C. , Rosell, J. , … Millan, J. M. (2012). Two novel disease‐causing mutations in the CLRN1 gene in patients with Usher syndrome type 3. Molecular Vision, 18, 3070–3078. [PMC free article] [PubMed] [Google Scholar]
- Geng, R. , Geller, S. F. , Hayashi, T. , Ray, C. A. , Reh, T. A. , Bermingham‐McDonogh, O. , … Flannery, J. G. (2009). Usher syndrome IIIA gene clarin‐1 is essential for hair cell function and associated neural activation. Human Molecular Genetics, 18(15), 2748–2760. 10.1093/hmg/ddp210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilissen, C. , Hoischen, A. , Brunner, H. G. , & Veltman, J. A. (2012). Disease gene identification strategies for exome sequencing. European Journal of Human Genetics: EJHG, 20(5), 490–497. 10.1038/ejhg.2011.258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfeder, R. L. , Wall, D. P. , Khoury, M. J. , Ioannidis, J. P. A. , & Ashley, E. A. (2017). Human genome sequencing at the population scale: A primer on high‐throughput DNA sequencing and analysis. American Journal of Epidemiology, 186(8), 1000–1009. 10.1093/aje/kww224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartong, D. T. , Berson, E. L. , & Dryja, T. P. (2006). Retinitis pigmentosa. Lancet, 368(9549), 1795–1809. 10.1016/s0140-6736(06)69740-7 [DOI] [PubMed] [Google Scholar]
- Isosomppi, J. , Västinsalo, H. , Geller, S. F. , Heon, E. , Flannery, J. G. , & Sankila, E.‐M. (2009). Disease‐causing mutations in the CLRN1 gene alter normal CLRN1 protein trafficking to the plasma membrane. Molecular Vision, 15, 1806–1818. [PMC free article] [PubMed] [Google Scholar]
- Jiang, F. , Pan, Z. , Xu, K. , Tian, L. , Xie, Y. , Zhang, X. , … Li, Y. (2016). Screening of ABCA4 gene in a Chinese cohort with Stargardt disease or cone‐rod dystrophy with a report on 85 novel mutations. Investigative Ophthalmology & Visual Science, 57(1), 145–152. 10.1167/iovs.15-18190 [DOI] [PubMed] [Google Scholar]
- Kajiwara, K. , Berson, E. L. , & Dryja, T. P. (1994). Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science, 264(5165), 1604–1608. 10.1126/science.8202715 [DOI] [PubMed] [Google Scholar]
- Khan, M. I. , Kersten, F. F. J. , Azam, M. , Collin, R. W. J. , Hussain, A. , Shah, S.‐A. , … den Hollander, A. I. (2011). CLRN1 mutations cause nonsyndromic retinitis pigmentosa. Ophthalmology, 118(7), 1444–1448. 10.1016/j.ophtha.2010.10.047 [DOI] [PubMed] [Google Scholar]
- Kjellström, U. (2015). Reduced macular function in ABCA4 carriers. Molecular Vision, 21, 767–782. [PMC free article] [PubMed] [Google Scholar]
- Lois, N. , Holder, G. E. , Bunce, C. , Fitzke, F. W. , & Bird, A. C. (2001). Phenotypic subtypes of stargardt macular dystrophy–fundus flavimaculatus. Archives of Ophthalmology, 119(3), 359–369. 10.1001/archopht.119.3.359 [DOI] [PubMed] [Google Scholar]
- Martínez‐Mir, A. , Paloma, E. , Allikmets, R. , Ayuso, C. , Río, T. D. , Dean, M. , … Balcells, S. (1998). Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nature Genetics, 18(1), 11–12. 10.1038/ng0198-11 [DOI] [PubMed] [Google Scholar]
- Mathur, P. , & Yang, J. (2015). Usher syndrome: Hearing loss, retinal degeneration and associated abnormalities. Biochimica et Biophysica Acta, 1852(3), 406–420. 10.1016/j.bbadis.2014.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch, D. L. , Marmor, M. F. , Brigell, M. G. , Hamilton, R. , Holder, G. E. , Tzekov, R. , & Bach, M. (2015). ISCEV Standard for full‐field clinical electroretinography (2015 update). Documenta Ophthalmologica, 130(1), 1–12. 10.1007/s10633-014-9473-7 [DOI] [PubMed] [Google Scholar]
- Molday, R. S. , Zhong, M. , & Quazi, F. (2009). The role of the photoreceptor ABC transporter ABCA4 in lipid transport and Stargardt macular degeneration. Biochimica et Biophysica Acta, 1791(7), 573–583. 10.1016/j.bbalip.2009.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennings, R. J. , Kremer, H. , Deutman, A. F. , Kimberling, W. J. , & Cremers, C. W. (2002). From gene to disease; genetic causes of hearing loss and visual impairment sometimes accompanied by vestibular problems (Usher syndrome). Nederlands Tijdschrift voor Geneeskunde, 146(49), 2354–2358. [PubMed] [Google Scholar]
- Petersen, B.‐S. , Fredrich, B. , Hoeppner, M. P. , Ellinghaus, D. , & Franke, A. (2017). Opportunities and challenges of whole‐genome and –exome sequencing. BMC Genetics, 18(1), 14–14. 10.1186/s12863-017-0479-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera, A. , White, K. , Stöhr, H. , Steiner, K. , Hemmrich, N. , Grimm, T. , … Weber Bernhard, H. F. (2000). A Comprehensive Survey of Sequence Variation in the ABCA4 (ABCR) Gene in Stargardt Disease and Age-Related Macular Degeneration. The American Journal of Human Genetics, 67(4), 800–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux, A. F. (2005). Molecular updates on Usher syndrome. Journal Francais D'ophtalmologie, 28(1), 93–97. 10.1016/s0181-5512(05)81030-7 [DOI] [PubMed] [Google Scholar]
- Sadeghi, M. , Cohn, E. S. , Kimberling, W. J. , Tranebjaerg, L. , & Moller, C. (2005). Audiological and vestibular features in affected subjects with USH3: A genotype/phenotype correlation. International Journal of Audiology, 44(5), 307–316. 10.1080/14992020500060610 [DOI] [PubMed] [Google Scholar]
- Schulz, H. L. , Grassmann, F. , Kellner, U. , Spital, G. , Rüther, K. , Jägle, H. , … Stöhr, H. (2017). Mutation spectrum of the ABCA4 gene in 335 Stargardt disease patients from a multicenter German ohort—Impact of selected deep intronic variants and common SNPs. Investigative Ophthalmology & Visual Science, 58(1), 394–403. 10.1167/iovs.16-19936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz, J. M. , Cooper, D. N. , Schuelke, M. , & Seelow, D. (2014). MutationTaster2: Mutation prediction for the deep‐sequencing age. Nature Methods, 11, 361–362. 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- Ścieżyńska, A. , Oziębło, D. , Ambroziak, A. M. , Korwin,M. , Szulborski, K. , Krawczyński M., … Ołdak, M . (2016). Next-generation sequencing of ABCA4: High frequency of complex alleles and novel mutations in patients with retinal dystrophies from Central Europe. Experimental Eye Research, 145, 93–99. [DOI] [PubMed] [Google Scholar]
- Shroyer, N. F. , Lewis, R. A. , Yatsenko, A. N. , Wensel, T. G. , & Lupski, J. R. (2001). Cosegregation and functional analysis of mutant ABCR (ABCA4) alleles in families that manifest both Stargardt disease and age‐related macular degeneration. Human Molecular Genetics, 10(23), 2671–2678. 10.1093/hmg/10.23.2671 [DOI] [PubMed] [Google Scholar]
- Sun, H. , Molday, R. S. , & Nathans, J. (1999). Retinal stimulates ATP hydrolysis by purified and reconstituted ABCR, the photoreceptor‐specific ATP‐binding cassette transporter responsible for Stargardt disease. Journal of Biological Chemistry, 274(12), 8269–8281. 10.1074/jbc.274.12.8269 [DOI] [PubMed] [Google Scholar]
- Thiadens, A. A. H. J. , Phan, T. M. L. , Zekveld‐Vroon, R. C. , Leroy, B. P. , van den Born, L. I. , Hoyng, C. B. , … Lotery, A. J. (2012). Clinical course, genetic etiology, and visual outcome in cone and cone‐rod dystrophy. Ophthalmology, 119(4), 819–826. 10.1016/j.ophtha.2011.10.011 [DOI] [PubMed] [Google Scholar]
- Tian, G. , Zhou, Y. , Hajkova, D. , Miyagi, M. , Dinculescu, A. , Hauswirth, W. W. , … Imanishi, Y. (2009). Clarin‐1, encoded by the Usher syndrome III causative gene, forms a membranous microdomain: Possible role of clarin‐1 in organizing the actin cytoskeleton. Journal of Biological Chemistry, 284(28), 18980–18993. 10.1074/jbc.M109.003160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsybovsky, Y. , Molday, R. S. , & Palczewski, K. (2010). The ATP‐binding cassette transporter ABCA4: Structural and functional properties and role in retinal disease. Advances in Experimental Medicine and Biology, 703, 105–125. 10.1007/978-1-4419-5635-4_8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Zhang, J. , Chen, N. , Wang, L. , Zhang, F. , Ma, Z. , … Yang, L. (2018). Application of whole exome and targeted panel sequencing in the clinical molecular diagnosis of 319 Chinese families with inherited retinal dystrophy and comparison study. Genes, 9(7), 360 10.3390/genes9070360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werdich, X. Q. , Place, E. M. , & Pierce, E. A. (2014). Systemic diseases associated with retinal dystrophies. Seminars in Ophthalmology, 29(5–6), 319–328. 10.3109/08820538.2014.959202 [DOI] [PubMed] [Google Scholar]
- World Health Organization (2016). International statistical classification of diseases and related health problems (5th edition, Vol 1, pp. 402-404). Geneva, Switzerland: World Health Organization, ; [Google Scholar]
- Wright, A. F. , Chakarova, C. F. , Abd El‐Aziz, M. M. , & Bhattacharya, S. S. (2010). Photoreceptor degeneration: Genetic and mechanistic dissection of a complex trait. Nature Reviews Genetics, 11(4), 273–284. 10.1038/nrg2717 [DOI] [PubMed] [Google Scholar]
- Xiong H. Y., Alipanahi, B. , Lee, L. J. , Bretschneider, H. , Merico, D. , Yuen, R. K. C. , … Frey, B. J . (2015). The human splicing code reveals new insights into the genetic determinants of disease. Science, 347(6218), 1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials