

Abstract
We demonstrate selective labeling of cell surface proteins using fluorogen-activating proteins conjugated to standard immunoglobulins (IgGs). Conjugation was achieved with a polypeptide reagent comprised of an N-terminal photo-activatable Fc-binding domain and a C-terminal FAP domain. The resulting FAP-antibody conjugates were effective agents for protein detection and cell ablation in cultured mammalian cells, and for visualizing cell-cell contacts using a tethered fluorogen assay. Because our approach allows FAP-antibody conjugates to be generated for most currently available IgGs, it should have broad utility for experimental and therapeutic applications.
Graphical Abstract
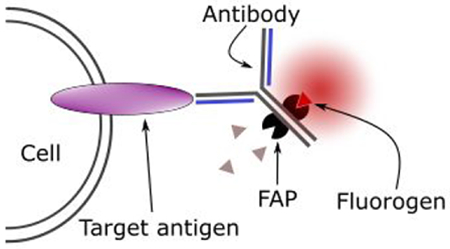
INTRODUCTION
Fluorogen-activating proteins (FAPs) bind organic dyes (fluorogens) that are not fluorescent in solution but become strongly fluorescent when bound to the FAP1. Since their initial description in 2008, FAPs have been used to probe a wide variety of biological processes including receptor trafficking, membrane translocation, and changes in intracellular pH2–7. When used in conjunction with membrane-impermeant fluorogens, FAPs have proved to be particularly valuable for labeling membrane proteins at the cell surface and monitoring their subsequent trafficking to and from the cell interior3,4,8–10.
A given FAP can activate the fluorescence of multiple chemically-related fluorogens, each with distinct biochemical and spectral properties4,10–15. The dL5**/MG pair used here, for instance, emits light in the far red spectrum where tissue autofluorescence and phototoxicity are low1,5. dL5** is also able to bind the green-emitting fluorogen MHN-ester14, which enables two-color assays and pulse-chase experiments. The modularity of the FAP/fluorogen system thereby allows a single peptide scaffold to serve multiple purposes for cell imaging or manipulation, including compartment and sub-population selectivity through the use of membrane permeant vs. impermeant dyes17.
Also used here is the iodinated fluorogen MG-2I, a Targeted and Activatable PhotoSensitizer (TAPS) molecule, that generates cell-destructive singlet oxygen molecules when bound to the FAP and irradiated with far-red light. It can thus be used to selectively ablate FAP-carrying cells16.
Published studies using FAP reporters have generally employed transgenic cell lines that express recombinant FAP-tagged proteins. These allow high-resolution assays in transfected cultured cells, but do not provide an easy path to performing equivalent assays in living organisms, where transgenic production is challenging and risky18. A more translatable alternative is to couple the FAPs to antibodies with binding specificities for proteins of interest, thereby enabling FAP labeling of endogenous native proteins.
In one approach, we produced a chimeric fusion protein with a FAP domain and an affibody domain, and we showed that it delivered the FAP to endogenously expressed antigen in cultured cells17,19. This approach is of limited general utility, however, because it requires the generation of a new chimeric reagent for each new target.
In a second approach, we generated reagents comprised of FAPs linked to known Fc-binding domains derived from Protein-A or Protein-G, and we showed that mouse and rabbit IgGs formed complexes with these reagents that specifically delivered the FAPs to antigens in fixed and live mammalian cells11. A limitation of this approach, however, is that the non-covalent FAP-antibody complexes are subject to spontaneous dissociation during storage or use.
As described here, we have overcome this limitation by generating a FAP reagent that is readily photo-crosslinked to IgGs in a site-specific manner20, yielding stable conjugates with the antigen-recognition properties of the antibody and the detection/ablation properties of the FAP. We demonstrate the use of these conjugates to (1) label cell surface antigens in cultured human cells, (2) detect zones of close proximity between one cell and another, and (3) photo-ablate antigen-expressing cells. Since thousands of validated primary antibodies are readily available for FAP-conjugation21, such conjugates should be widely applicable for experimental and therapeutic purposes.
RESULTS
Generation of HTB1-FAP reagent and conjugation to antibodies.
A fusion protein with an N-terminal HTB1(A24BPA) domain20 and a C-terminal FAP (dL5**, MBIC6) domain was expressed and purified as described in Experimental Procedures. HTB1 is a thermally stable variant of the B1 domain of Protein G with a photoreactive amino acid, benzoylphenylalanine, in its Fc-binding region. FAP dL5** binds fluorogens that are derivatives of the triaryl methane dye malachite green22. The HTB1-FAP reagent was photo-crosslinked to two monoclonal antibodies: cetuximab (Erbitux), whose target is the human receptor tyrosine kinase EGFR/HER1, and trastuzumab (Herceptin), whose target is the human receptor tyrosine kinase HER2. As shown for cetuximab in Figure S1, the procedure led to near complete covalent crosslinking of the HTB1-FAP polypeptide to the antibody.
Visualization of EGFR and HER2 in human cell lines.
HaCaT and A431 cells, which are known to express EGFR23,24, were incubated with FAP-conjugated cetuximab and membrane impermeant MG-fluorogen. Strong fluorescent signal was observed at the cell surface (Figure 1). Some internalized signal was also observed, due to the internalization of some receptor molecules by endocytosis during the incubation25. HEK-293 cells, which express little or no EGFR23,26, were not labeled, nor were HaCaT or A431 cells that were treated as in Figure 1 but without conjugated antibody.
Figure 1.
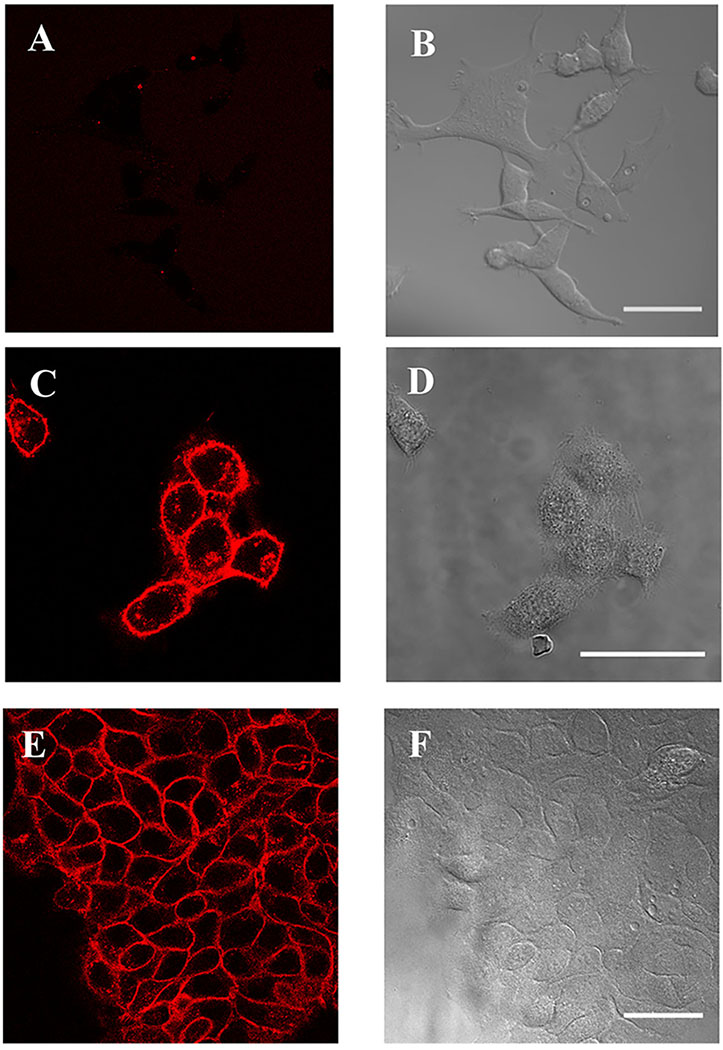
dL5**-cetuximab labels EGFR on HaCaT and A431 cells. (A-B) HEK-293 cells are not labeled by the reagent. (C-D) HaCaT and (E-F) A431 cells are labeled with dL5**-cetuximab and MG fluorogen. MG fluorescence is shown on the left, DIC transmitted light images on the right. Scale bars = 50μm.
Fixed cells were also probed with the labeled antibody (Figure 2). As expected, signal was confined to the surface in unpermeabilized cells, and was seen on the surface and more diffusely inside the cell in fixed and permeabilized cells.
Figure 2.
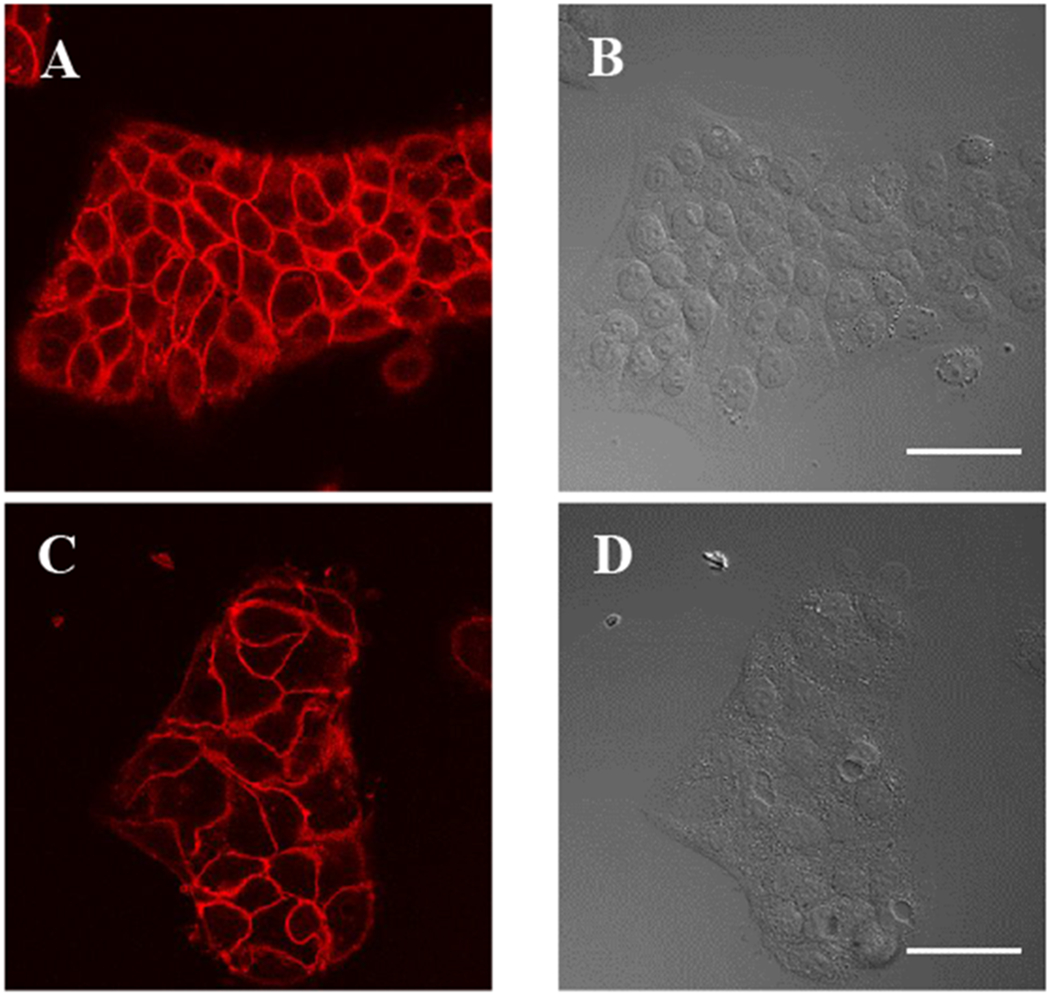
dL5**-cetuximab labels EGFR on fixed HaCaT cells. (A-B) Permeabilized and (C-D) unpermeabilized HaCaT cells labeled with dL5**-cetuximab and MG fluorogen. MG fluorescence is shown on the left, DIC transmitted light images on the right. Scale bars = 50μm.
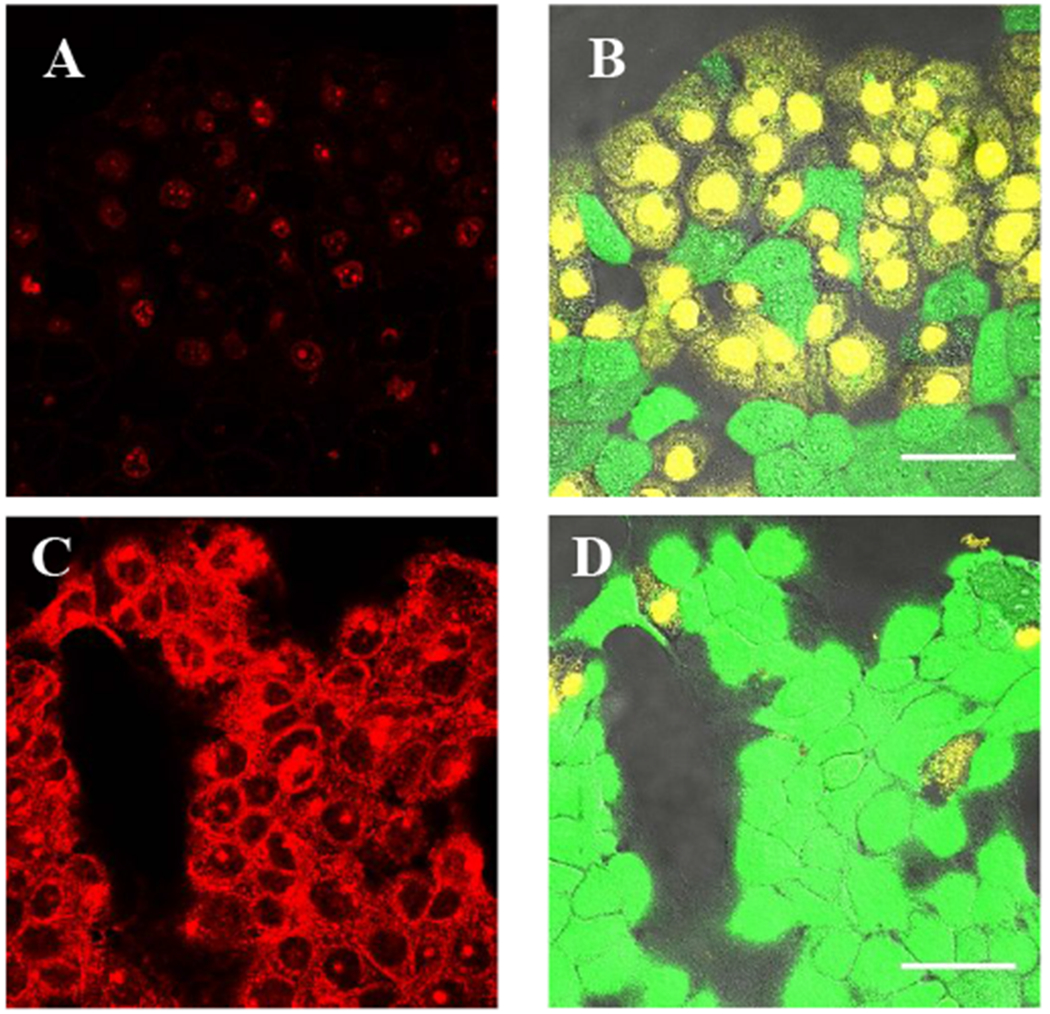
The procedures described above to produce FAP-conjugated cetuximab were also applied to trastuzumab (Herceptin), an antibody whose target is the human tyrosine kinase HER2/ERBB2, and the conjugate was used to probe SK-BR-3 cells, which natively express high levels of HER227. As shown in Figure 3, FAP-conjugated trastuzumab labeled SK-BR-3 cells with similar effect as the cetuximab reagent on EGFR-expressing cells. HEK-293 cells, which express minimal HER2, were not labeled by dL5**-trastuzumab.
Figure 3.
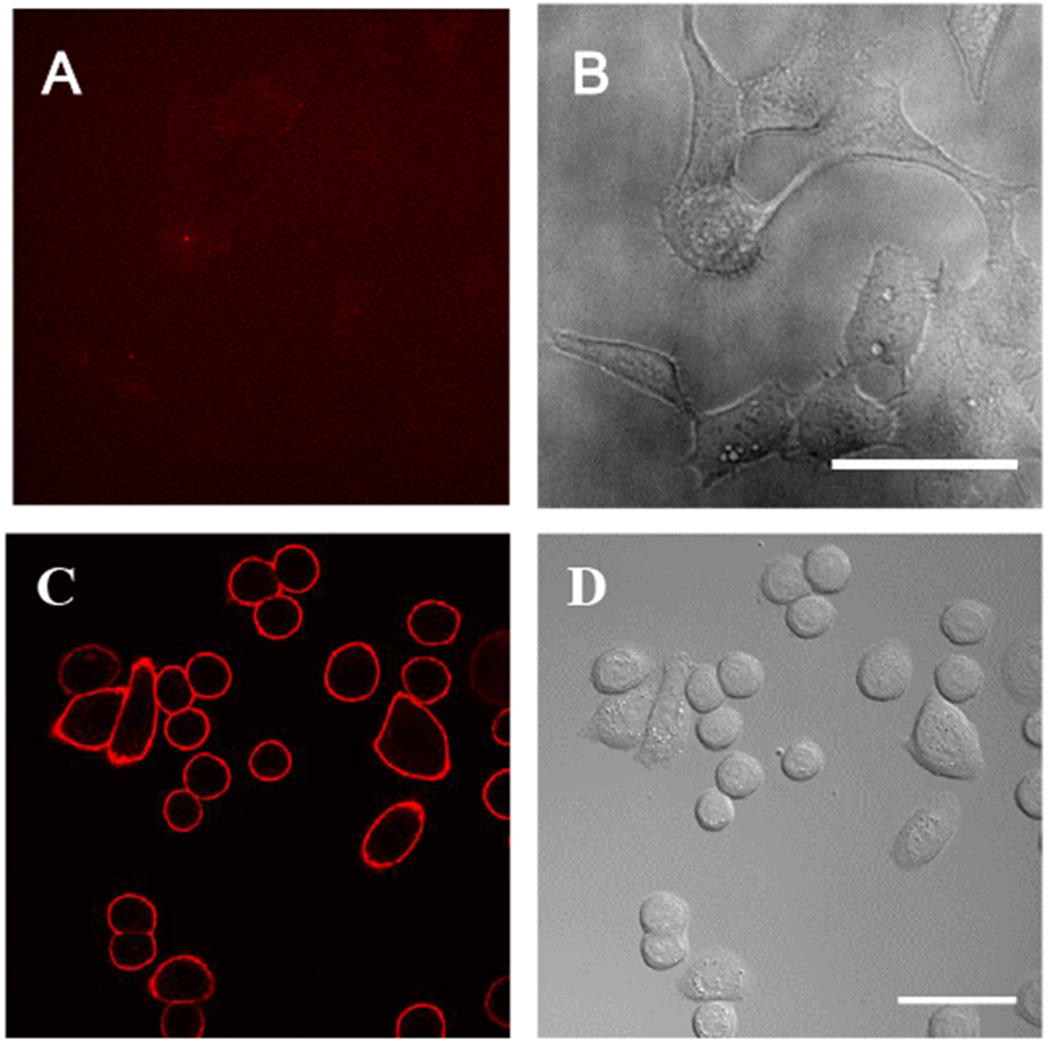
dL5**-trastuzumab labels HER2 on SK-BR-3 cells. (A-B) HEK-293 cells are not labeled by the reagent. (C-D) SK-BR-3 cells are labeled with dL5**-cetuximab and MG fluorogen. MG fluorescence is shown on the left, DIC transmitted light images on the right. Scale bars = 50μm.
Visualization of cell-cell contacts using FAP-tagged antibodies.
Previously28,29, we developed a technology called trans-TEFLA (tethered fluorogen assay), in which contact points between two FAP-expressing cell lines exhibit bright fluorescence when labeled with a bivalent fluorogen (MG-PEG3500-TO1). One cell line, expressing dL5**, produces red fluorescence when it binds the MG moiety of the bivalent reagent, while the other, expressing scFv1, yields green fluorescence upon binding the TO1 moiety. The “red” FAP binds its fluorogen with a very low Kd (~100 pM), whereas the “green” FAP binds its fluorogen with a much higher Kd (~350 nM). When bivalent fluorogen is provided at an intermediate concentration (10 nM), the red FAP is saturated with fluorogen and thus gives red fluorescence over the entire cell surface, but the green FAP only gives fluorescence when it is very close to the red FAP, where the local concentration of the TO1 moiety is very high. This leads to distinct red plus green (yellow) fluorescence at intercellular contact sites when the cells are co-cultured. There, both cell lines were required to be transgenic in order to express the FAP constructs. However, ease of use and biological applicability would both increase for the trans-TEFLA technology if it did not require the use of transgenic cells.
Here, we sought to advance on this goal by using dL5**-cetuximab to replace the transgenic FAP construct on one member of the trans-TEFLA pair (Figure 4A). Specifically, we used dL5**-cetuximab to label wild-type HaCaTs, which were co-cultured with transgenic scFv1-expressing HEK-293 cells. In these cultures, bright yellow fluorescence was apparent at intercellular contact points (Figure 4B, C), in a manner identical to that previously seen between two transgenic cell lines28. When the cells were incubated with separate, unlinked MG and TO1 fluorogens (Figure 4D, E), no fluorescence enhancement at contact sites was observed.
Figure 4.
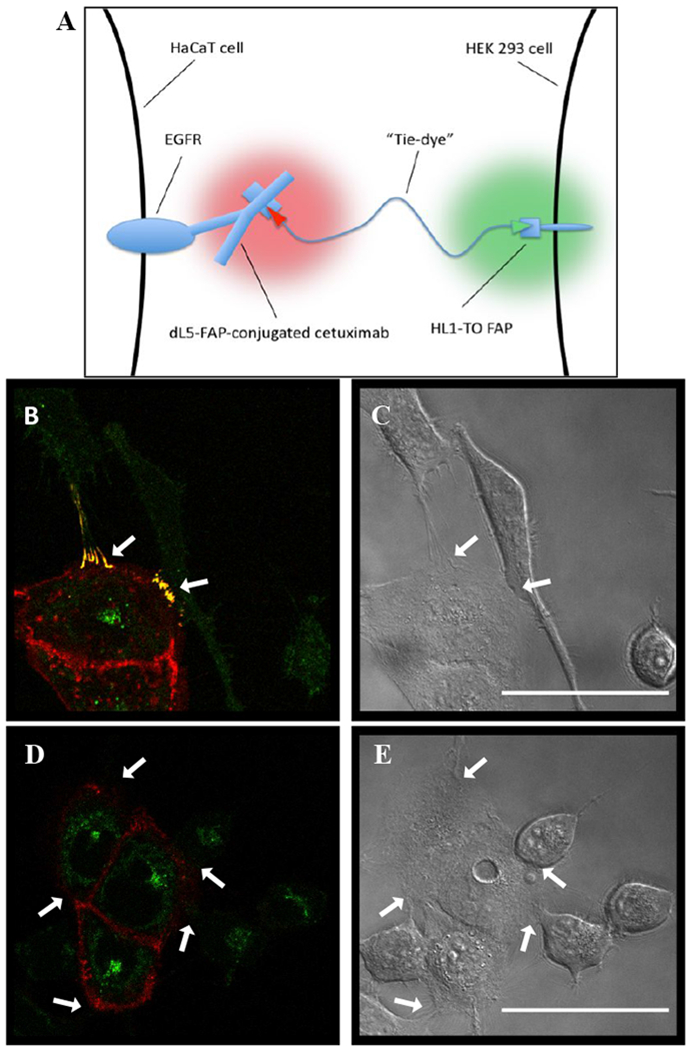
dL5**-cetuximab facilitates trans-TEFLA with only one partner expressing transgenic FAP. (A) Schematic illustrating the trans-TEFLA principle applied to the dL5**-antibody system. (B) and (C) Co-cultured wild-type HaCaT and scFv1-expressing HEK-293 cells. HaCaTs are uniformly surface-labeled by dL5**-cetuximab bound to MG-PEG3500-TO1 dual fluorogen, with faint green fluorescence on the scFv1-HEKs. Bright fluorescence enhancement in both channels is observed at sites of intercellular contact (white arrows). (D) and (E) Equivalent cells incubated with single dyes (MG and TO1 fluorogens) exhibit similar labeling but without enhancement at contact sites. Green signal inside HaCaT cells represents background autofluorescence. Scale bars = 50μm.
FAP-directed photo-ablation.
We also asked whether the FAP-conjugated antibodies could be used to photo-ablate cells using the iodinated photosensitizing fluorogen MG-2I16. Antigen-expressing cells grown on optical surfaces were treated with conjugated antibody, followed by addition of either the photosensitizing fluorogen or control fluorogen (MG Ester). Cultures were then illuminated with high-intensity 640nm light for 7.5 minutes - conditions that had previously been shown to produce severe photodamage in HEK-293 cells that expressed the dL5** FAP16. To aid in detecting and quantifying cell injury, we applied 1 μM calcein AM (stains cytoplasm of intact cells) and 5 μM ethidium homodimer-1 (EthD-1, a DNA stain that reaches the nucleus of membrane-damaged cells) to the cultures.
By 30 minutes post-illumination, the cells with the photosensitizing fluorogen showed clear evidence of injury, including surface blebbing and contraction, EthD-1 staining of the nucleus, and diminished calcein signal (Figure 5A–B). Cells that received the control fluorogen were largely unaffected (Figure 5C–D).
Figure 5.
dL5**-cetuxiumab and MG-2I mediate cell death in EGFR-expressing cells. (A-B) Red channel and merged green and yellow channels (calcein AM and EthD-1, respectively) are shown for HaCaT cells labeled with dL5**-cetuximab and MG-2I. (C-D) Red signal shown for non-photosensitizing control fluorogen MG Ester, along with the calcein AM and EthD-1 channels. Scale bars = 50μm.
To provide a quantitative counterpart to the microscopy data shown in Figure 5, we conducted a fluorescence-based cell viability assay (alamarBlue) on irradiated and non-irradiated cells (HEK-293, HaCaT, and A431) to confirm that irradiation of MG-2I bound to dL5**-cetuximab on endogenous EDGR leads to cell death in high-expressing cells. Results are shown in Table 1. In this assay, living cells reduce resazurin to resorufin, which is highly fluorescent. Therefore, cell damage and death are indicated by a reduced level of overall fluorescent signal. Substantial cell damage and/or death were evident in irradiated A431s, which express high levels of EGFR. This was not observed in EGFR-negative HEK-293 cells. HaCaT cells, which have an intermediate EGFR expression profile, showed a downward trend in signal after irradiation, though less dramatic than for A431 cells.
Table 1.
Fluorescence intensity data from plate reader analysis of alamarBlue-treated HEK-293, HaCaT, and A431 cells after treatment with dL5**-cetuximab +MG-2I and photoirradiation. Numbers shown are averages and standard deviations, in arbitrary units, from 5 replicates for each condition. Background fluorescence (media + alamarBlue only) has been subtracted out. Irr. = cells irradiated at 640nm. Non-irr. = cells not irradiated. Ester/2I only = only fluorogen, no FAP/antibody. Cetux+Ester/2I = antibody and indicated fluorogen.
| HEK-293 cells | ||||
|---|---|---|---|---|
| Irr. | Non-irr. | |||
| Avg. | StdDev. | Avg. | StdDev. | |
| Neg. ctrl. | 20420 | 3273 | 20997 | 8501 |
| Ester only | 15206 | 4080 | 16790 | 3229 |
| 2I only | 18512 | 2912 | 20633 | 9015 |
| Cetux+Ester | 16882 | 3158 | 20088 | 3802 |
| Cetux+2I | 16946 | 2957 | 19855 | 4996 |
| HaCaT cells | ||||
| Irr. | Non-irr. | |||
| Avg. | StdDev. | Avg. | StdDev. | |
| Neg. ctrl. | 24870 | 1813 | 30172 | 5507 |
| Ester only | 27844 | 1673 | 37122 | 12212 |
| 2I only | 24552 | 2331 | 30150 | 7381 |
| Cetux+Ester | 25932 | 959 | 30473 | 2935 |
| Cetux+2I | 17724 | 1174 | 25108 | 6241 |
| A431 cells | ||||
| Irr. | Non-irr. | |||
| Avg. | StdDev. | Avg. | StdDev. | |
| Neg. ctrl. | 20365 | 1771 | 34047 | 16013 |
| Ester only | 20910 | 1557 | 21852 | 4806 |
| 2I only | 18931 | 1371 | 25351 | 5603 |
| Cetux+Ester | 18380 | 1574 | 23188 | 2619 |
| Cetux+2I | 10427 | 1096 | 32145 | 8698 |
A flow cytometry-based viability assay (calcein AM/ethidium homodimer) was also performed on these cell types as an orthogonal test of the same variable. The results of this assay are shown in Figures S2 and S3. There, a large increase in cell damage in death, as indicated by increased ethidium fluorescence, was observed in irradiated EGFR-expressing cells (HaCaT and A431) treated with antibody and MG-2I, but not in EGFR-negative cells (HEK-293). For A431 cells, (Figure S2C), the great majority (>98%) of the dL5**-cetuximab+MG-2I-treated cells were so damaged by irradiation that they did not survive the analysis intact (Table S1). This is likely due to the higher expression of EGFR in A431s compared to HaCaT cells21, leading to more singlet oxygen production and consequent cell damage.
A test of dL5**-trastuzumab on SK-BR-3 cells provided similar photoablation results. These cells were also labeled with dL5**-cetuximab and either MG-2I (Figure 6A–B) or non-photosensitizing MG Ester (Figure 6C–D). After irradiation, MG-2I-labeled SK-BR-3 cells were observed to have a disrupted and granular appearance, and vital staining revealed many cells that were dead or dying.
Figure 6.
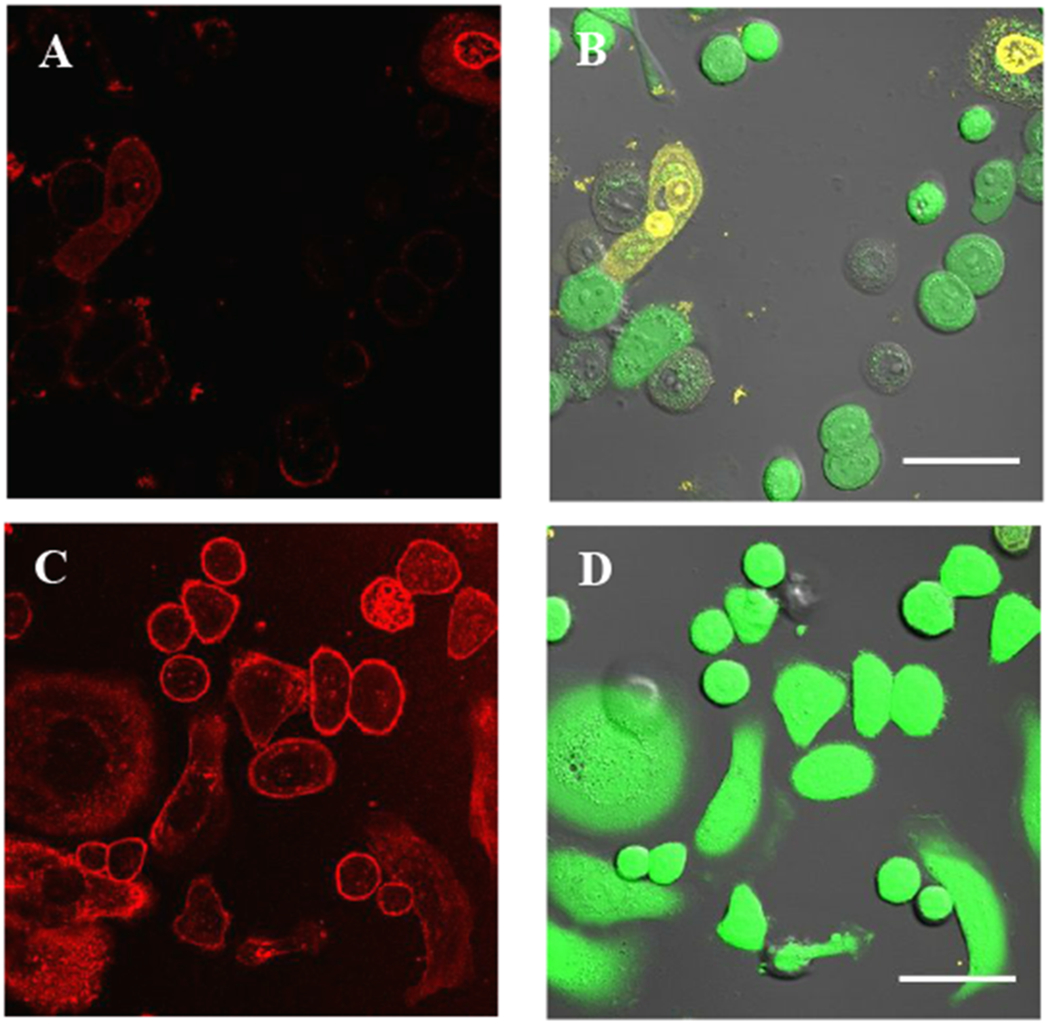
dL5**-trastuzumab and MG-2I mediate cell death in HER2-expressing SK-BR-3 cells. (A-B) Red channel (MG-2I signal) and merged green and yellow channels (calcein AM and EthD-1, respectively) are shown for cells labeled with dL5**-trastuzumab and MG-2I. (C-D) Red signal shown for non-photosensitizing control fluorogen MG-Ester, along with the calcein AM and EthD-1 channels. Scale bars = 50μm.
DISCUSSION
Efficient photo-crosslinking between the HTB1-FAP reagent and an IgG requires (1) interaction between the Fc portion of the antibody and the HTB1 domain of the reagent (which is derived from the Fc binding domain of protein G20), and (2) the presence of a crosslink-accepting methionine near the CH2-CH3 junction in the Fc domain of the antibody20. Such a residue is present in all human IgGs, all rabbit IgGs and three of the four subclasses of mouse IgG (IgG2a, 2b, 3). Thus, each antibody conjugate has a well-defined structure, with two FAP moieties at locations well removed from the antigen binding site. This is in distinction to antibody-fluorophore conjugates made by most other methods, where both the number and the locations of the fluorophores vary from one antibody molecule to another.
Additionally, each FAP represents a multifunctional module that, depending on the fluorogen used, provides a range of spectral and photochemical properties that can be exploited without the need to generate different antibody conjugates for different uses. The current study illustrates this versatility, with the dL5** FAP enabling the detection of cell-surface antigens, the detection of cell-cell proximity, and the photoablation of antigen expressing cells.
The opportunity to FAP-tag almost any IgG, coupled with the modularity of the FAPs themselves, gives the FAP-antibody technology broad potential application for use in multicellular organisms. Labeling and ablation of FAP-tagged cardiac cells has already been demonstrated in larval and adult transgenic zebrafish 16, and it would be reasonable to attempt to use FAP-tagged antibodies for labeling and /or ablation of other targets in this organism. Performing such experiments in mice could be problematic due to limited penetration of near infrared photons, but penetration should not be an issue for cells in cutaneous or subcutaneous locations, including subcutaneous tumors. Likewise, there is potential for therapeutic use of FAP-antibody conjugates to photoablate human tumor cells at the time of tumor resection, which is typically when photodynamic therapy is applied in cancer treatment.
Another opportunity, which extends results reported here, is to use pairs of antibody-conjugated FAPs to visualize locations where two antigens are in close proximity, as in neural or immunological synapses. FAPs expressed from recombinant genes have already been shown to enable such “trans-TEFLA” experiments in cultured cells28,and, it would be of interest to attempt trans-TEFLA labeling using FAP-tagged antibodies as both partners to investigate intercellular contacts in vitro and in vivo.
EXPERIMENTAL PROCEDURES
Mammalian cell culture and fixation.
HEK-293, HaCaT, A431, and SK-BR-3 cells were cultured under standard conditions in a tissue culture incubator at 37°C and 5% CO2. Growth media was DMEM (with 4.5g/L glucose, L-glutamine, and sodium pyruvate) supplemented with 10% fetal bovine serum and 10U/mL penicillin and streptomycin. For imaging, cells were lifted with 0.05% trypsin+EDTA and plated on glass-bottom MatTek dishes (MatTek, Ashland MA, #P35GC-0-14) to allow confocal microscopy.
For experiments on fixed cells, HaCaTs were incubated in 2% paraformaldehyde for 15 minutes at room temperature, followed by blocking with 3% BSA for one hour (if permeabilized, this was preceded by 15-minute incubation in 0.5% Triton X-100). Cells were then labeled with 50nM antibody for an hour and subsequently labeled with MG fluorogen.
Photoablation.
For photoablation experiments, cells were immersed in PBS and placed in a custom-built LED light-box emitting at 669nm16 and illuminated for 7.5 minutes at approximately 50mW/cm2. Subsequently, cells were incubated for 1-2 hours to allow time for photoablation to have an impact on cellular processes. All samples were then labeled with 1μM calcein AM (ThermoFisher, Waltham, MA) to label live cells and 5μM ethidium homodimer-1 (EthD-1; ThermoFisher) to label dead cells. Thirty minutes later, cells were prepared for microscopy and imaged as described below.
Fluorescence microscopy and image analysis.
Fluorescence microscopy was performed on a Carl Zeiss LSM 880 or Andor Revolution XD Spinning Disk confocal microscope, both equipped with heated, humidified culture chambers at 5% CO2. For all experiments, cells were incubated with 50nM antibody for at least 15 minutes prior to washing and labeling with fluorogens to allow adequate binding to surface antigen. Following dye labeling, cells were washed and imaged using an argon 488nm and solid-state 633nm laser were bandpass emission filters adjusted to catch maximum signal from TO1-2p and MG fluorogens, respectively. Microscopy on the LSM 880 used an oil immersion 40X plan-neofluar objective and an oil immersion 63X plan apochromat objective. Microscopy on the Revolution XD used a dry 40X objective. Microscopy settings were adjusted to minimize signal saturation and zero value intensities and were then held constant throughout each experiment. The resulting images were analyzed using ImageJ30.
Plate reader cell viability assay and data analysis.
For plate reader experiments, HEK-293, HaCaT, and A431 cells were plated at 10,000 cells/well in 96-well plates. Approximately 24 hours post-plating, cells were labeled with cetuximab (50nM) and either MG Ester or MG-2I (500nM), with wash steps in between to remove unbound reagent. One plate was then irradiated with red light as described above, while the other remained non-irradiated. Cells were incubated at 37°C for one hour and then labeled with alamarBlue cell viability stain (ThermoFisher) for 4 hours. Analysis of fluorescence was performed on a Tecan Infinite M1000 plate reader. Excitation was provided in the 540-570nm range and emission collected from 580-610nm as per manufacturer instructions. Gain for each cell type was optimized against the negative controls and held constant throughout the experiment. Resulting data was collated and analyzed in Microsoft Excel 2016 and GraphPad Prism 6.01 (GraphPad Software, La Jolla, CA).
Flow cytometry and data analysis.
For flow cytometry experiments, HEK-293, HaCaT, and A431 cells were prepared exactly as described above for the plate reader assays. Thirty minutes post-irradiation, all cells were labeled with 5μM EthD-1 to distinguish dead cells. After another thirty minutes, cells were lifted into Accutase (CELLnTEC, Bern, Switzerland) and analyzed using an Accuri C6 flow cytometer (BD Biosciences, San Jose, CA). The FL2 channel (488nm laser for excitation with a 585/40 bandpass filter to collect emission) was used to determine signal from the ethidium homodimer-1. The FL4 channel (640nm excitation, 670 longpass emission) was used to determine signal from the MG fluorogens. Data was collected in ForeCyt software (IntelliCyt, Albuquerque, NM) and analyzed using FlowJo ver. 10.2 (FlowJo LLC, Ashland, OR). Briefly, cells were gated and singlets discriminated to remove cell clumps from analysis. Median FL2 signals were calculated for each well, and averages and standard deviations calculated. Graphs were produced in GraphPad Prism 6.01.
Details of dL5 FAP.
Plasmid pG-2DL5 encoding the dL5** FAP and a C-terminal HTB1 peptide derived from Streptococcal Protein G was designed with codon usage appropriate for expression in E. coli. A UAG codon at HTB1 position A24 was included to direct incorporation of a p-benzoyl-L-phenylalanine (pBpa) residue , as described in Hui et al.20
Plasmids pEVOL-pBpF (Addgene) which carries the tRNA/aminoacyl transferase pair and the pG-2DL5 were co-transformed into Origami B(DE3) Competent Cells (EMD Millipore).
Bacterial starter cultures were grown in 2 mL LB + 100μg/mL ampicillin + 25 μg/mL chloramphenicol at 37°C in a shaker overnight. Starter cultures were added at a 1:1000 dilution to Autoinduction Media LB Broth Base Including Trace Elements (Formedium), 100 μg/mL ampicillin and 25 μg/mL chloramphenicol. For BP A incorporation, L-benzoylphenylalanine (Bachem, King of Prussia, PA) was added into the culture to a final concentration of 500 pM and arabinose was added to a final concentration of 0.1% to begin the inductions of the pEVOL plasmid. pG-2DL5 was expressed at 25°C in an open-air shaker for 72h.
Following expression, cultures were pelleted by centrifugation (5,500g for 15 min at 4°C) and the HTB1-FAP peptide was purified as previously described in Hui etal.20. SDS-PAGE electrophoresis was performed on Bolt 4-12% Bis-Tris Plus Gels (Thermo Fisher Scientific) with a Mini Gel Tank (Thermo Fisher Scientific), stained with SimplyBlue SafeStain (Invitrogen), and imaged using a Gel Logic 100 system (Kodak).
Photocrosslinking.
To create antibody conjugates, HTB1-FAP peptide was incubated with the IgG and exposed to long wavelength UV light (365nm) for 2 hours20. Crosslinked products were purified from access proteins by using 100 kDa molecular weight cut-off (MWCO) filter (Amicon Ultra, Milipore, Temecula, CA) and then they were analyzed directly using SDS-PAGE electrophoresis.
Analysis of proteins.
Protein products were analyzed by SDS-PAGE electrophoresis. SDS-PAGE was performed on Bolt 4-12% Bis-Tris Plus Gels (Thermo Fisher Scientific) with a Mini Gel Tank. Proteins were boiled 5 minutes with 1:1 volume of loading buffer (Biorad, Hercules, CA) containing 1:20 dilution of β-mercaptoethanol (Biorad). Samples were loaded and run for 30 mins at constant 200 volt. The gel was stained using SimplyBlue Coomassie stain (Invitrogen). Image of the gel was taken using a Kodak Gel Logic 100 system (Rochester, NY).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Haibing Teng for training and assistance with confocal microscopy, and Yehuda Creeger for training and assistance with flow cytometry. This work was supported in part by NIH/NffiIB R21-EB018863 (AT) and NIH/NCI R21-CA187657 (AT), NIH/NIBIB R01EB017236 (MPB, DK), and NIH/NIEHS R33ES025606 (MPB, DK).
ABBREVIATIONS
- EGFR
epidermal growth factor receptor
- EthD-1
ethidium homodimer-1
- FAP
fluorogen-activating protein
- IgG
immunoglobulin
- MG
malachite green
- PBS
phosphate-buffered saline
- PEG
polyethylene glycol
- TAPS
Targeted and Activatable PhotoSensitizer
- TEFLA
tethered fluorogen assay
- TO
thiazole orange
Footnotes
SUPPORTING INFORMATION
Sequence and purification data on HTB1-dL5**; raw data and analysis from flow cytometry-based cell viability assay.
REFERENCES
- (1).Szent-Gyorgyi C, Schmidt BF, Creeger Y, Fisher GW, Zakel KL, Adler S, Fitzpatrick JAJ, Woolford CA, Yan Q, Vasilev KV, et al. (2008) Fluorogen-activating single-chain antibodies for imaging cell surface proteins. Nat. Biotechnol 26, 235–240. [DOI] [PubMed] [Google Scholar]
- (2).Saurabh S, Perez AM, Comerci CJ, Shapiro L, and Moerner WE (2016) Superresolution imaging of live bacteria cells using a genetically directed, highly photostable fluoromodule. J. Am. Chem. Soc 138, 10398–10401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Holleran JP, Zeng J, Frizzell RA, and Watkins SC (2013) Regulated recycling of mutant CFTR is partially restored by pharmacological treatment. J. Cell Sci 126, 2692–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Fisher GW, Fuhrman MH, Adler SA, Szent-Gyorgyi C, Waggoner AS, and Jarvik JW (2014) Self-checking cell-based assays for GPCR desensitization and resensitization. J. Biomol. Screen 19, 1220–1226. [DOI] [PubMed] [Google Scholar]
- (5).Telmer CA, Verma R, Teng H, Andreko S, Law L, and Bruchez MP (2015) Rapid, Specific, No-wash, Far-red Fluorogen Activation in Subcellular Compartments by Targeted Fluorogen Activating Proteins. ACS Chem. Biol 10, 1239–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Schwartz SL, Yan Q, Telmer CA, Lidke KA, Bruchez MP, and Lidke DS (2015) Fluorogen-activating proteins provide tunable labeling densities for tracking FceRI independent of IgE. ACS Chem. Biol 10, 539–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Lorenz-Guertin JM, Wilcox MR, Zhang M, Larsen MB, Pilli J, Schmidt BF, Bruchez MP, Johnson JW, Waggoner AS, Watkins SC, et al. (2017) A versatile optical tool for studying synaptic GABA A receptor trafficking. J. Cell Sci 130, 3933–3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Fisher GW, Adler SA, Fuhrman MH, Waggoner AS, Bruchez MP, and Jarvik JW (2010) Detection and quantification of β2AR internalization in living cells using FAP-based biosensor technology. J. Biomol. Screen 15, 703–709. [DOI] [PubMed] [Google Scholar]
- (9).Wu Y, Tapia PH, Fisher GW, Simons PC, Strouse JJ, Foutz T, Waggoner AS, Jarvik J, and Sklar LA (2012) Discovery of regulators of receptor internalization with high-throughput flow cytometry. Mol. Pharmacol 82, 645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Perkins LA, Yan Q, Schmidt BF, Kolodieznyi D, Saurabh S, Larsen MB, Watkins SC, Kremer L, and Bruchez MP (2018) Genetically Targeted Ratiometric and Activated pH Indicator Complexes (TRApHIC) for Receptor Trafficking. Biochemistry 57, 861–871. [DOI] [PubMed] [Google Scholar]
- (11).Gallo E, Vasilev KV, and Jarvik J (2014) Fluorogen-activating-proteins as universal affinity biosensors for immunodetection. Biotechnol. Bioeng, 111, 475–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Gallo E, and Jarvik JW (2017) Breaking the color barrier – a multi-selective antibody reporter offers innovative strategies of fluorescence detection. J. Cell Sci 130, 2644–2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Pham HH, Szent-Gyorgyi C, Brotherton WL, Schmidt BF, Zanotti KJ, Waggoner AS, and Armitage BA (2015) Biochromophoric Dyes for Wavelength Shifting of Dye-Protein Fluoromodules. Org Biomol Chem 13, 3699–3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Pratt CP, He J, Wang Y, Barth AL, and Bruchez MP (2015) Fluorogenic green-inside red-outside (GIRO) labeling approach reveals adenylyl cyclase-dependent control of BKα surface expression. Bioconjug. Chem 26, 1963–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Bruchez MP (2015) Dark dyes-bright complexes: fluorogenic protein labeling. Curr. Opin. Chem. Biol 27, 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).He J, Wang Y, Missinato MA, Onuoha E, Perkins LA, Watkins SC, St. Croix CM, Tsang M, and Bruchez MP (2016) A near-infrared genetically targetable and activatable photosensitizer. Nat. Methods 13, 263–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Wang Y, Telmer CA, Schmidt BF, Franke JD, Ort S, Arndt-jovin DJ, and Bruchez MP (2014) Fluorogen Activating Protein - Affibody Probes : Modular , No-wash Measurement of Epidermal Growth Factor Receptors. Bioconjug. Chem 26, 137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Escors D, and Breckpot K (2011) Lentiviral vectors in gene therapy: Their current status and future potential. Arch. Immunol. Ther. Exp. (Warsz) 58, 107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Wang Y, Ballou B, Schmidt BF, Andreko S, St. Croix CM, Watkins SC, and Bruchez MP (2017) Affibody-targeted fluorogen activating protein for in vivo tumor imaging. Chem. Commun 53, 2001–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Hui JZ, Tamsen S, Song Y, and Tsourkas A (2015) LASIC : Light Activated Site-Specific Conjugation of Native IgGs. Bioconjug. Chem 26, 1456–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Thul PJ, Åkesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, Alm T, Asplund A, Björk L, Breckels LM, et al. (2017) A subcellular map of the human proteome. Science 356, eaa13321. [DOI] [PubMed] [Google Scholar]
- (22).Szent-Gyorgyi C, Stanfield RL, Andreko S, Dempsey A, Ahmed M, Capek S, Waggoner A, Wilson IA, and Bruchez MP (2013) Malachite green mediates homodimerization of antibody VL domains to form a fluorescent ternary complex with singular symmetric interfaces. J. Mol. Biol 425, 4595–4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Zhang F, Wang S, Yin L, Yang Y, Guan Y, Wang W, Xu H, and Tao N (2015) Quantification of Epidermal Growth Factor Receptor Expression Level and Binding Kinetics on Cell Surfaces by Surface Plasmon Resonance Imaging. Anal Chem 87, 9960–9965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ockenga W, Kühne S, Bocksberger S, Banning A, and Tikkanen R (2014) Epidermal growth factor receptor transactivation is required for mitogen-activated protein kinase activation by muscarinic acetylcholine receptors in HaCaT keratinocytes. Int. J. Mol. Sci 15, 21433–21454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Yewale C, Baradia D, Vhora I, Patil S, and Misra A (2013) Epidermal growth factor receptor targeting in cancer: A review of trends and strategies. Biomaterials 34, 8690–8707. [DOI] [PubMed] [Google Scholar]
- (26).Chen M, Chen LM, Lin CY, and Chai KX (2008) The epidermal growth factor receptor (EGFR) is proteolytically modified by the Matriptase-Prostasin serine protease cascade in cultured epithelial cells. Biochim. Biophys. Acta - Mol. Cell Res 1783, 896–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Holliday DL, and Speirs V (2011) Choosing the right cell line for breast cancer research. Breast Cancer Res 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ackerman DS, Vasilev KV, Schmidt BF, Cohen LB, and Jarvik JW (2017) Tethered Fluorogen Assay to Visualize Membrane Apposition in Living Cells. Bioconjug. Chem 28, 1356–1362. [DOI] [PubMed] [Google Scholar]
- (29).Vasilev KV, Gallo E, Shank N, and Jarvik JW (2016) Novel biosensor of membrane protein proximity based on fluorogen activated proteins. Comb. Chem. High Throughput Screen. 19, 392–399. [DOI] [PubMed] [Google Scholar]
- (30).Collins TJ (2007) ImageJ for microscopy. Biotechniques 43, 25–30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.