

Abstract
There are numerous lines of clinical evidence that inhibition of the renin–angiotensin system (RAS) can prevent and delay the development of diabetes. Also, the role of RAS in the pathogenesis of diabetes, including insulin resistance and β‐cell dysfunction, has been extensively investigated. Nevertheless, this role had not yet been fully shown. A variety of possible protective mechanisms for RAS blockers in the regulation of glucose homeostasis have been suggested. However, the direct effect on pancreatic islet fibrosis has only recently been spotlighted. Various degrees of islet fibrosis are often observed in the islets of patients with type 2 diabetes mellitus, which can be associated with a decrease in β‐cell mass and function in these patients. Pancreatic stellate cells are thought to be deeply involved in this islet fibrosis. In this process, the activation of RAS in islets is shown to transform quiescent pancreatic stellate cells into the activated form, stimulates their proliferation and consequently leads to islet fibrotic destruction. In this article, we introduce existing clinical and experimental evidence for diabetes prevention through inhibition of RAS, and review the responsible local RAS signaling pathways in pancreatic stellate cells. Finally, we propose possible targets for the prevention of islet fibrosis.
Keywords: Islet fibrosis, Pancreatic stellate cells, Renin–angiotensin system
In this review, we introduce existing clinical and experimental evidence for diabetes prevention through inhibition of the renin–angiotensin system, and review the responsible local renin–angiotensin system signaling pathways in pancreatic stellate cells involved in islet fibrosis. Finally, we propose possible targets for the prevention of islet fibrosis.
Introduction
Type 2 diabetes mellitus is a heterogeneous metabolic disease entity that shares its phenotype of hyperglycemia, and fundamental pathophysiology of insulin resistance and impaired β‐cell function. Over the past decades, insulin resistance was thought to be the main contributor to type 2 diabetes mellitus, and decreased insulin secretion was considered as a part of the late manifestation in the course of the disease1. However, there has been abundant evidence that individuals with type 2 diabetes mellitus already have both a significant decrease in β‐cell function2, 3 and a loss of 30–40% β‐cell mass, even at the time of diagnosis4, 5. It is now well recognized that decompensation of β‐cells to increase insulin secretion compared with insulin resistance leads to overt hyperglycemia, indicating that β‐cell dysfunction is critical to the development of type 2 diabetes mellitus6, 7. Furthermore, as β‐cell dysfunction accelerates over time, it should be focused on as a significant determinant of the progression rate of type 2 diabetes mellitus8.
Recent evidence suggests that there are ethnic differences in the pathophysiological characteristics of type 2 diabetes mellitus. Asians typically have lower levels of obesity, but a more significantly reduced incremental insulin release in response to insulin resistance compared with Caucasians9, 10. Combining these backgrounds, we should emphasize the preservation of β‐cell mass and function to preventing the development and progression of type 2 diabetes mellitus, especially in Asians.
The causes of the loss of β‐cell mass and function are numerous. Glucolipotoxicity, chronic inflammation, endoplasmic reticulum stress, oxidative stress, amyloid deposition and epigenetic modifications have all been linked to the initiation and progression of β‐cell impairment and loss11, 12, 13. Also, there is evidence that islet disorganization through progressive fibrosis might also be an important mechanism of β‐cell dysfunction and loss14, 15. Fibrosis is the well‐known pathogenic process that results in progressive loss of the structure and function of the affected organs16. Progressive fibrosis and the subsequent loss of functional tissue (replaced by extracellular matrix [ECM]‐rich connective tissue containing amylin) is also well demonstrated in the pathogenesis of chronic pancreatitis and pancreatic ductal adenocarcinoma17. In this process of pancreatic fibrogenesis, pancreatic stellate cells (PSCs) have been identified as a major source of ECM proteins18.
Besides exocrine pancreatic fibrosis, pancreatic islet fibrosis has also been identified in both animal models and individuals with diabetes5, 19. Interestingly, pancreatic fibrosis in diabetes is mostly restricted to pancreatic islets compared with the fibrosis of exocrine pancreatic tissue in chronic pancreatitis, suggesting that islet fibrosis in diabetes is linked to activating pathways that are different from those of the exocrine pancreatic disease20, 21. In our basic research, we found that activation of PSCs played a crucial role in the process of islet fibrosis through the angiotensin II (Ang II) signaling pathway20, 22. However, the exact mechanism of fibrogenesis in pancreatic islets and the involvement of PSCs in this process have been much less studied and highlighted, despite their importance.
In the present review, we introduce the existing clinical and experimental evidence of diabetes prevention through inhibition of the renin–angiotensin system (RAS), and discuss the responsible RAS signaling pathways in PSCs. Finally, we introduce possible targets for the prevention of islet fibrosis.
Clinical studies showing a preventive effect of RAS inhibition on diabetes
Diabetes represents a global epidemic whose incidence is steadily increasing, and therefore, there are ongoing efforts worldwide aimed at preventing or delaying the onset of type 2 diabetes mellitus. The two main strategies are lifestyle modifications and pharmacological interventions. Most of the pharmacological evidence comes from the use of antidiabetic drugs, such as metformin, thiazolidinediones, α‐glucosidase inhibitors and glucagon‐like peptide‐1 (GLP‐1) receptor agonist23, or antiobesity drugs, such as orlistat24. Another potential pharmacological strategy is the use of RAS blockers, which are antihypertensive drugs. Although other antihypertensive drugs, such as diuretics and β‐adrenergic blockers, have adverse effects on insulin resistance and glucose tolerance25, RAS blockers have shown favorable results by preventing the onset of diabetes in numerous clinical trials on individuals with hypertension or other cardiovascular risk factors (Table 1).
Table 1.
Summary of clinical studies showing the effect of RAS inhibition on new‐onset type 2 diabetes mellitus
| Trial | Characteristics of participants | Comparison groups | No. participants in analysis† | Duration of follow up (years) | Relative risk (95% confidence interval) | Reference | |
|---|---|---|---|---|---|---|---|
| Treatment group | Control group | ||||||
| CAPPP | Aged 25–66 years with diastolic hypertension | Captopril | Diuretics, β‐blockers | 5,183 vs 5,230 | Mean 6.1 | 0.86 (0.74–0.99) | 26 |
| HOPE | Aged ≥55 years with coronary artery disease, stroke, peripheral vascular disease | Ramipril | Placebo | 2,837 vs 2,883 | Median 4.5 | 0.66 (0.51–0.85) | 27 |
| ALLHAT | Aged ≥55 years with hypertension and at least one other coronary heart disease risk factor | Lisinopril | Chlorthalidone | 2,567 vs 4,543 | Median 4.9 | 0.70 (0.63–0.77) | 28, 29 |
| Amlodipine | 2,567 vs 2,692 | 0.83 (0.74–0.93) | |||||
| LIFE | Aged 55–80 years with hypertension and left ventricular hypertrophy | Losartan | Atenolol | 4,019 vs 3,979 | Mean 4.8 | 0.75 (0.63–0.88) | 30 |
| SCOPE | Aged 70–89 years with hypertension | Candesartan | Placebo | 2,167 vs 2,175 | Mean 3.7 | 0.75 (NA), P = 0.09 | 31 |
| CHARM | Aged >18 years with heart failure NYHA grade II–IV | Candesartan | Placebo | 2,715 vs 2,721 | Median 3.1 | 0.78 (0.64–0.96) | 88 |
| PEACE | Aged ≥50 years with stable coronary artery disease and left ventricular ejection fraction >40% | Trandolapril | Placebo | 3,432 vs 3,472 | Median 4.8 | 0.83 (0.72–0.96) | 89 |
| VALUE | Aged ≥50 years with hypertension and high risk of cardiovascular events | Valsartan | Amlodipine | 5,032 vs 4,963 | Mean 4.2 | 0.77 (0.69–0.87) | 32 |
| DREAM‡ | Aged ≥30 years without cardiovascular disease but with impaired fasting glucose or impaired glucose tolerance | Ramipril | Placebo | 2,623 vs 2,646 | Median 3.0 | 0.91 (0.81–1.03) | 33 |
| ONTARGET | Aged ≥55 years with coronary, peripheral artery or cerebrovascular disease | Ramipril +Telmisartan | Ramipril | 5,280 vs 5,427 | Median 4.7 | 0.91 (0.78–1.06) | 90 |
| TRANSCEND | Aged ≥55 years with coronary, peripheral artery or cerebrovascular disease | Telmisartan | Placebo | 1,895 vs 1,913 | Median 4.7 | 0.85 (0.71–1.02) | 90 |
| NAVIGATOR‡ | Aged ≥50 years with impaired glucose tolerance and established cardiovascular disease or cardiovascular risk factors | Valsartan | Placebo | 4,631 vs 4,675 | Median 5.0 | 0.86 (0.80–0.92) | 34 |
| CASE‐J Ex | 20–Aged 85 years, Japanese with hypertension and at least one risk factor for cardiovascular events | Candesartan | Amlodipine | 636 vs 620 | Mean 4.5 | 0.71 (0.51–1.00) | 91 |
| ANBP2 | Aged 65–84 years with hypertension, but having no recent cardiovascular morbidity (within 6 months) | Enalapril | Hydrochlorothiazide | 2,815 vs 2,827 | Median 6.9 | 0.70 (0.56–0.86) | 92, 93 |
Number of participants included in the analysis of secondary outcomes was estimated as the number of total participants – the number of participants with type 2 diabetes mellitus at baseline, if there was no information in the original article.
Only Diabetes Reduction Approaches With Ramipril and Rosiglitazone Medications (DREAM) and Nateglinide and Valsartan in Impaired Glucose Tolerance Outcomes Research (NAVIGATOR) were the double‐blind, placebo‐controlled, randomized trials whose primary outcome was the development of type 2 diabetes mellitus. CAPPP, Captopril Prevention Project; HOPE, Heart Outcomes Prevention Evaluation; LIFE, Losartan Intervention For End Point Reduction in Hypertension; NA, not available; NYHA, New York Heart Association; SCOPE, Study on Cognition and Prognosis in the Elderly; VALUE, Valsartan Antihypertensive Long‐Term Use Evaluation.
The Captopril Prevention Project was a randomized and open‐label study that compared captopril, an angiotensin‐converting enzyme inhibitor (ACEi), with conventional antihypertensive agents (diuretics and β‐blockers)26. The incidence of diabetes was lower in the captopril group than in the conventional treatment group (relative risk 0.86, 95% confidence interval [CI] 0.74–0.99; P = 0.039). Similar results have been observed in other trials. The Heart Outcomes Prevention Evaluation study was a randomized, placebo‐controlled trial27 in which ramipril, an ACEi, showed a risk reduction for diabetes (relative risk 0.66, 95% CI 0.51–0.85; P < 0.001). The Antihypertensive and Lipid‐Lowering Treatment to Prevent Heart Attack Trial28, 29 and the Losartan Intervention For End Point Reduction in Hypertension trial30 were randomized, double‐blind studies that compared the ACEi, lisinopril, with either the diuretic, chlorthalidone, or calcium channel blocker, amlodipine, and the angiotensin receptor blockers (ARB), losartan, with the β‐blocker, atenolol, respectively. The risk reduction for diabetes in both trials was ~30% (lisinopril vs chlorthalidone, P < 0.001), 17% (lisinopril vs amlodipine, P < 0.01) and 25% (losartan vs atenolol, P < 0.001), respectively. The Study on Cognition and Prognosis in the Elderly (SCOPE)31, which compared the ARB candesartan with a placebo, showed a tendency toward risk reduction for diabetes in elderly hypertensive patients (25%, P = 0.09). More recently, the Valsartan Antihypertensive Long‐Term Use Evaluation trial32 was a randomized, double‐blinded study that compared the ARB, valsartan, with amlodipine. The Valsartan Antihypertensive Long‐Term Use Evaluation trial also produced positive results for the onset of diabetes (odds ratio 0.77, 95% CI 0.69–0.87; P < 0.0001). Taken together, these clinical studies suggest that inhibition of RAS with either an ACEi or an ARB might protect high‐risk individuals (with hypertension or other cardiovascular risk factors) from the development of type 2 diabetes mellitus. Among these studies, the Heart Outcomes Prevention Evaluation and Valsartan Antihypertensive Long‐Term Use Evaluation trials provided the strongest evidence, because these trials were carried out using a placebo or calcium channel blocker, which is considered metabolically neutral.
Because all the positive data obtained in these clinical studies came from comparisons with other antihypertensive drugs or resulted from the use of a secondary end‐point or post‐hoc analysis, more direct clinical proof of the preventive effect of RAS blockade on the onset of diabetes was required. This situation resulted in the initiation of two large, double‐blind, placebo‐controlled, randomized trials whose primary outcome was the development of type 2 diabetes mellitus: the Diabetes Reduction Approaches With Ramipril and Rosiglitazone Medications (DREAM) trial using the ACEi, ramipril, as well as the Nateglinide and Valsartan in Impaired Glucose Tolerance Outcomes Research (NAVIGATOR) trial using the ARB, valsartan33, 34. Frustratingly, the DREAM trial failed to show any difference in the incidence of new‐onset diabetes (hazard ratio for the ramipril group 0.91, 95% CI 0.81–1.03; P = 0.15)33. In contrast, the NAVIGATOR trial showed a protective effect of ARB as compared with the placebo group (hazard ratio for the valsartan group 0.86, 95% CI 0.80–0.92; P < 0.001)34.
Except for the administration of an ACEi or an ARB, these two large trials differed in several respects. First, unlike the other trials, DREAM included only patients who had impaired fasting glucose (IFG) or impaired glucose tolerance (IGT), but no known cardiovascular disease (CVD). In contrast, the NAVIGATOR trial, like the previous trials, included patients with IFG and also with established CVD or cardiovascular risk factors. Nearly 99% of the participants in the NAVIGATOR trial had at least one cardiovascular risk factor, including a history of CVD (24.3%) and hypertension (79.6%). Thus, the participants in the NAVIGATOR trial were expected to have a higher RAS activity, a risk factor for the development of diabetes, as compared with the patients in the DREAM trial who were not at the same risk. Second, the study period differed between the two trials. The median follow‐up period was 3.0 and 5.0 years in DREAM and NAVIGATOR, respectively. Perhaps due to the relatively short period of the study, the DREAM trial could only show the potential for efficacy. In this regard, although the DREAM trial failed to yield positive results in terms of the effect on the incidence of new diabetes, 42.5% of patients in the ramipril group converted from IFG or IGT to normoglycemia, and this improvement was higher than the 38.2% conversion seen in the placebo group (P = 0.001).
In the meta‐analyses of clinical studies, both ACEis and ARBs showed a consistent significant risk reduction in new‐onset diabetes in the various subgroups of combining CVDs, except for the individuals with IFG or IGT, due to the negative results of the DREAM trial35, 36. Interestingly, the reduced risk of new‐onset diabetes was irrespective of achieved blood pressure levels in both ACEis and ARBs.
Hypothetical mechanisms explaining the preventive effects of RAS inhibition on diabetes
In addition to clinical evidence, there is abundant in vitro and in vivo evidence that has clearly shown the adverse effects of RAS activation on insulin secretion and sensitivity, and also the possibility of reversal of these effects through RAS blockade. In a study with db/db mice, the upregulated pancreatic islet Ang II receptor type 1 (AT1R) was accompanied with deleterious effects on insulin secretion and (pro)insulin biosynthesis37. In rodents, exposure of isolated islets to Ang II induced a dose‐dependent inhibition of glucose‐stimulated insulin secretion and (pro)insulin biosynthesis. This inhibitory action was completely prevented by pretreatment with losartan38. In humans, a 3‐month treatment with candesartan was shown to increase first‐phase insulin secretion during an oral glucose tolerance test39. Additionally, in a study on a hyperinsulinemic‐euglycemic and hyperglycemic clamp, 26 weeks of treatment with valsartan increased both glucose‐stimulated insulin secretion and insulin sensitivity in normotensive individuals with IGT40.
Although abundant evidence has been accumulated, the precise molecular mechanism by which RAS inhibition affects the pathogenesis of diabetes has not yet been elucidated. A variety of potential mechanisms have, therefore, been proposed to explain the preventive and delaying effects of RAS inhibition on diabetes. These include physiological changes that might improve insulin sensitivity and secretion, and also direct effects on changes in islet morphology.
First, RAS inhibition‐mediated vasodilation might facilitate insulin secretion and insulin action by improving muscular and pancreatic blood flow. Both ACEis and ARBs have been reported to enhance blood flow in peripheral tissues, such as skeletal muscle, and this change might then improve insulin sensitivity and facilitate glucose disposal41, 42. Second, RAS inhibition could directly affect insulin signaling and improve insulin sensitivity in skeletal muscle. Alterations in post‐receptor insulin signaling in type 2 diabetes mellitus have been shown, including anomalies in phosphatidylinositol‐3 kinase–protein kinase B signaling. There is evidence that Ang II aggravates these abnormalities. Therefore, RAS inhibition might have a direct effect on insulin signaling and regulation of glucose transporters43. Third, RAS inhibition could also improve cellular insulin signaling and insulin sensitivity by reducing the levels of free fatty acids, and by inhibiting Ang II‐mediated oxidative stress. Given that Ang II activates nicotinamide adenine dinucleotide phosphate oxidase, a major source of reactive oxygen species (ROS), increased RAS activity in β‐cells may aggravate oxidative stress‐induced β‐cell dysfunction and apoptosis. ARBs have been reported to attenuate fatty acid‐induced oxidative stress and nicotinamide adenine dinucleotide phosphate oxidase activity in pancreatic β‐cells44, 45, 46. In addition, a subset of AT1R blockers have been shown to induce peroxisome proliferator‐activated receptor‐gamma (PPAR‐γ) activity by interaction with the PPAR‐γ ligand‐binding domain, thus they can improve insulin sensitivity47. For example, telmisartan, one of the selective AT1R blockers, shares structural similarity with pioglitazone, and is known to act as a partial PPAR‐γ agonist48, 49. However, it is unclear whether this effect is a class effect of ARBs. Finally, it has been shown that Ang II promotes lipid deposition in adipose tissue by inhibiting lipolysis and promoting lipogenesis, and also increases secretion of adipose tissue‐derived proinflammatory cytokines. Thus, RAS inhibition might selectively alleviate insulin resistance in adipose tissue50.
As discussed, many studies explaining the participation of RAS in the modulation of glucose homeostasis have focused on insulin sensitivity, but recently, the possibility of RAS having a direct influence on pancreatic islet fibrosis has gained attention. There has been enough evidence of the presence of Ang II receptors on the surface of pancreatic islet β‐cells in rats and humans51. Thus, we initially hypothesized that RAS blockers might have a direct influence on pancreatic islets, eventually conserving the β‐cell mass and function. Although the restoring of insulin sensitivity is essential, this mechanism alone could not fully explain the findings of increased insulin secretion after the administration of RAS blockers.
Effect of RAS inhibition on islet fibrosis and preservation of β‐cell mass and function
The RAS is well‐known for its classic function in the systemic regulation of blood pressure, fluid retention and electrolyte balance through action on vascular smooth muscle cells and aldosterone secretion. In the RAS signaling cascade, Ang II is the main effector peptide that acts mainly through AT1R. Also, Ang II acts as a growth factor that promotes cell growth and tissue inflammation, by which it plays a pivotal role in various disease‐associated target organ fibrosis and dysfunction52, 53. For example, it has been well demonstrated that inhibition of RAS with ACEis or ARBs can delay the progression of diabetic nephropathy and prevent renal fibrosis54. Also, inhibition of RAS has been consistently shown to reduce endothelial dysfunction and atherosclerosis by suppression of inflammation, fibrosis and oxidative stress55. Like other organ damage processes, we observed some similar destructive fibrotic changes in the pancreatic islets of both animal models of type 2 diabetes mellitus and patients with type 2 diabetes mellitus5, 21 and thought the local RAS might be the main contributor to this phenomenon. There is abundant evidence that overactivation of local RAS also exists in the pancreas like other organs and tissues, including the brain, heart, kidney, adrenal glands, adipose tissue and skeletal muscle51, 56.
Otsuka Long Evans Tokushima fatty (OLETF) rats are an established rat model of type 2 diabetes mellitus in which extensive connective tissue proliferation in the pancreas is associated with pancreatic islet cell atrophy57. We initially found that long‐term ramipril treatment (24 weeks) prevented islet destruction caused by fibrosis in diabetic OLETF rats. These effects were accompanied with a decreased expression of transforming growth factor‐beta (TGF‐β) and its downstream signaling molecules, such as connective tissue growth factor (CTGF), fibronectin and alpha‐smooth muscle actin (α‐SMA)20. However, although long‐term ramipril treatment was shown to alleviate pancreatic islet fibrosis, the involvement of the islet RAS in islet fibrosis was not evident in OLETF rats. Upregulation of intra‐islet AT1R and ACE was then shown by immunohistochemistry and real‐time polymerase chain reaction analysis of the whole pancreas from another type 2 diabetes mellitus rat model, Zucker diabetic fatty rats58. The increased intra‐islet expression of components of RAS correlated with increased intra‐islet fibrosis, apoptosis and oxidative stress. In addition, the RAS blockade significantly reduced islet fibrogenesis and improved islet architecture. These effects were associated with the attenuation of TGF‐β and profibrotic pathways. Finally, improvements in structural parameters were associated with a significant improvement of first‐phase insulin secretion.
In conclusion, RAS blockers appear to have a direct effect on pancreatic islet cells, such as the prevention of islet fibrosis and the maintenance of cellular architecture, which ultimately conserves the β‐cell mass and function, thereby exerting a beneficial action on glucose tolerance. Thus, we highlight the reduction in islet fibrosis as an important and promising mechanism behind the long‐term protective effects of RAS blockers on the development and progression of diabetes.
Role of PSCs in islet fibrosis and relevant signaling pathways
Role of PSCs in pancreatic fibrosis
PSCs were identified in the early 1980s through the characteristic of sharing similarities with hepatic stellate cells59. PSCs are estimated to constitute 4% of total pancreatic cells, but are essential for maintaining the normal pancreatic architecture by regulating ECM turnover. Quiescent PSCs are characterized by the presence of intracellular fat droplets, desmin and glial fibrillary acidic protein, but the absence of α‐SMA17. When activated, PSCs are transformed into the myofibroblast‐like phenotype characterized by the disappearance of intracellular fat droplets and the expression of α‐SMA60. The expression of glial fibrillary acidic protein is specific to PSCs in the pancreas, and the presence of lipid droplets in the cytoplasm defines the quiescent phenotype of PSCs. In contrast, the expression of α‐SMA represents the transdifferentiation of the quiescent PSCs to an activated phenotype, and thus, is used as a marker of PSC activation61. Activated PSCs abundantly produce collagen and other ECM proteins, such as fibronectin (Figure 1)18, and are shown to maintain their activated phenotype through an autocrine loop involving different cytokines. Accordingly, PSCs have been widely researched as the major effector cells in the exocrine pancreatic fibrogenesis, but their role in islet fibrosis has been unclear.
Figure 1.
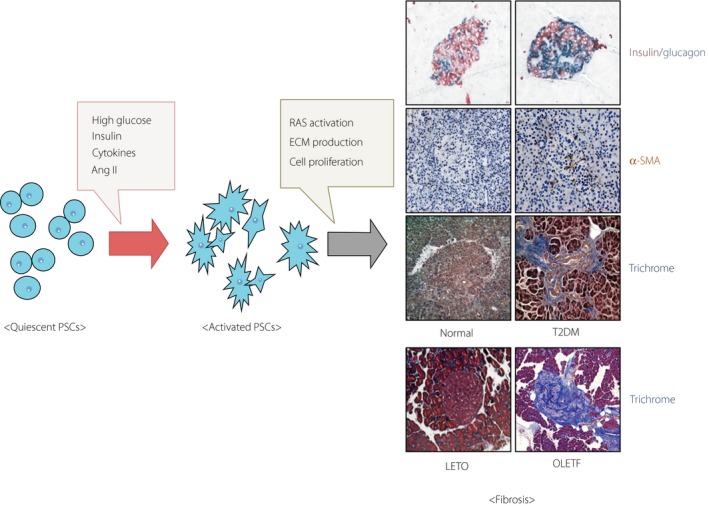
Pancreatic stellate cell activation and islet fibrosis in type 2 diabetes mellitus (T2DM). High‐glucose levels, insulin and angiotensin II (Ang II), and a release of pro‐inflammatory cytokines induce pancreatic stellate cell (PSC) activation. Activated PSCs cause extracellular matrix production and cell proliferation. These phenomena appear to drive fibrosis within the pancreatic islets in type 2 diabetes mellitus. Islet fibrosis can be detected by immunostaining for alpha‐smooth muscle actin (α‐SMA) and trichrome. LETO, Long‐Evans Tokushima Otsuka; OLETF, Otsuka Long Evans Tokushima fatty; RAS, renin–angiotensin system.
Direct effect of RAS on PSC proliferation and related signaling pathway
Initial studies showed that PSCs were localized to the interlobular and interacinar regions of the pancreas, but not in the islets61, 62. In our study on diabetic OLETF rats, α‐SMA expression was observed in the peri‐islet area of advanced fibrotic islets, not only in the vascular smooth muscle cells and exocrine tissue20. This result was consistent with similar findings of another study of OLETF rats63. We also observed that an ACEi had suppressive effects on ECM protein expression, accompanied by the downregulation of α‐SMA20. Overall, PSCs seemed to play a major role in fibrotic islet destruction in animal models of type 2 diabetes mellitus. Nevertheless, it was not clear whether ACE inhibitors directly affected PSCs or had secondary effects through systemic alterations.
At the next step, we isolated PSCs from male Sprague–Dawley rats and cultured PSCs under hyperglycemic conditions22. As a result, PSC proliferation significantly increased at high‐glucose concentrations in a dose‐dependent manner, up to fourfold, as compared with that under low glucose conditions. The real‐time polymerase chain reaction analysis of local‐RAS components in PSCs showed that AT1R, angiotensinogen and ACE messenger ribonucleic acids were significantly upregulated in response to increased glucose concentrations, but there was no change in Ang II type 2 receptor level. We also found that TGF‐β expression rose after the increase in Ang II levels. Then, the expression of CTGF and type IV collagen proteins increased markedly, and this elevation was effectively attenuated by candesartan and ramipril.
In conclusion, we can explain that Ang II induced by high levels of glucose might stimulate TGF‐β synthesis, which subsequently leads to ECM protein synthesis (Figure 2). Furthermore, these results suggest that RAS has a direct impact on PSC proliferation. Thus, ACE inhibitors or ARBs might be effective in preventing or attenuating islet fibrosis in hyperglycemic individuals by directly suppressing PSCs.
Figure 2.
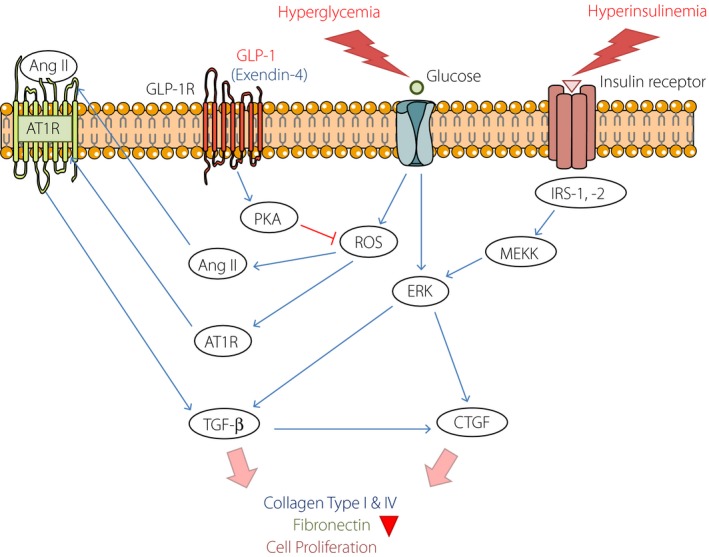
Mechanism of pancreatic stellate cell activation. Hyperglycemia and hyperinsulinemia: A high‐glucose‐ and insulin‐activated pancreatic stellate cell is induced to proliferate through two independent pathways (extracellular signal‐regulated kinase [ERK] and angiotensin II [Ang II] pathways). Glucose and insulin independently enhance ERK activation and increase connective tissue growth factor (CTGF) expression. High‐glucose concentration stimulates Ang II production and Ang II receptor type 1(AT1R) expression and upregulates transforming growth factor‐beta (TGF‐β) through the binding of Ang II to AT1R. These pathways ultimately lead to the production of TGF‐β1 and expression of CTGF, an important downstream mediator of TGF‐β1 activity. Finally, these activation events increase collagen and fibronectin formation, and induce cell proliferation. As an antifibrotic effect, the GLP‐1 receptor agonist exendin‐4 reduces Ang II and TGF‐β1 production through inhibition of protein kinase A (PKA)‐related reactive oxygen species (ROS) formation. GLP‐1, glucagon‐like peptide‐1; GLP‐1R, GLP‐1 receptor; IRS, insulin receptor substrate; MEKK, mitogen‐activated protein kinase kinase.
Additive effects of hyperglycemia and hyperinsulinemia on PSCs: An explanation of the islet‐specific fibrosis
Interestingly, a hyperglycemic environment can influence the entire pancreas, but PSCs activation and fibrosis are limited mainly to the islets in the OLETF rat model21, 63. Therefore, it remains to be determined why this fibrosis is restricted to islets in individuals with diabetes and animal models of diabetes, even though the whole pancreatic tissue is exposed to hyperglycemia. One possible explanation is that PSCs in the islets might be exposed not only to hyperglycemia, but also to local hyperinsulinemia. Because insulin, a well‐known growth factor to various cells in the body64, is continuously secreted into the capillaries of the islets at a relatively high concentration, PSCs in islets might be predisposed to activation and proliferation under the influence of hyperinsulinemia. In addition, there is evidence that hepatic stellate cells, functionally similar to PSCs, are highly sensitive to insulin and insulin‐like growth factor‐1, resulting in mitogenesis and collagen synthesis65.
To confirm this, we stimulated the isolated PSCs with both glucose and insulin, and each66. Both stimuli promoted PSCs proliferation and extracellular signal‐regulated kinase (ERK) 1/2 phosphorylation independently, and the additive effect was also identified. Blocking of ERK signaling by a mitogen‐activated protein kinase kinase inhibitor, U0126, inhibited both glucose‐ and insulin‐induced ERK 1/2 phosphorylation and PSC proliferation. In addition, we showed that the glucose‐ and insulin‐induced ERK 1/2 phosphorylation stimulated CTGF gene expression (Figure 2). Thus, we can conclude that hyperglycemia and hyperinsulinemia are two crucial mitogenic factors that activate and proliferate the PSCs, providing a possible explanation for the islet‐specific fibrosis in type 2 diabetes mellitus compared with chronic pancreatitis.
Other possible strategies for prevention of islet fibrosis
Antidiabetic drugs
Besides using RAS blockers, there have been various other attempts at prevention of islet fibrosis (Table 2). In recent years, GLP‐1 analog exendin‐4 was shown to improve β‐cell function by upregulating key genes involved in insulin secretion67. The exendin‐4 treatment prevented the development of diabetes in a partial pancreatectomy rat model of type 2 diabetes mellitus, and resulted in a 40% expansion of β‐cell mass through the combined effect of differentiation and neogenesis of precursor cells, as well as β‐cell proliferation68, 69. Furthermore, exendin‐4 has been shown to have anti‐inflammatory effects, and acts as an antifibrotic agent in mesangial cells70, 71. Exendin‐4 has been shown to be capable of inhibiting the proliferation of human mesangial cells and downregulating the high‐glucose‐induced expression of TGF‐β1 and CTGF72. Recently, our team reported that exendin‐4 significantly reduced Ang II and TGF‐β1 production through inhibition of ROS formation, but not ERK phosphorylation73. These inhibitory effects of exendin‐4 were related mainly to the activation of the cyclic adenosine monophosphate–protein kinase A signaling pathway (Figure 2). Consequently, these data suggest that GLP‐1 analogs might be useful not only as of the antidiabetic agent, but also as the antifibrotic therapeutic in type 2 diabetes mellitus.
Table 2.
Possible strategies for prevention of islet fibrosis
| Class | Agent | Effects | Reference |
|---|---|---|---|
| Antidiabetic agents | GLP‐1 agonist (exendin‐4) | Inhibition of ROS production | 73 |
| PPAR‐γ agonist (troglitazone) | Reduction of PSC proliferation, Downregulation of TGF‐β | 94, 95 | |
| SGLT2 inhibitor (luseogliflozin) | Downregulation of TGF‐β, fibronectin, collagen I and collagen III | 77 | |
| Antioxidants | Taurine | Downregulation of collagen I and TGF‐β | 96, 97 |
| Tempol | Downregulation of collagen I and TGF‐β | 81 | |
| Thioredoxin‐1 | Attenuation of PSC activation and fibrosis, Downregulation of TGF‐β | 98 | |
| Ascorbic acid† | |||
| Polyphenols | Resveratrol | Inhibition of ROS production | 80 |
| Rhein | Downregulation of collagen I, α‐SMA and fibronectin | 99 | |
| Emodin | Inhibition of PSC activation | 99 | |
| Curcumin | Inhibition of cell proliferation | 99, 100 | |
| Epigallocatechin‐3‐gallate | Downregulation of TGF‐β | 101 | |
| Vitamins | Tocotrienols | Inhibition of PSC activation | 102 |
| Retinoic acid | Inhibition of PSC activation, Downregulation of α‐SMA and collagen I | 103, 104, 105 | |
| Palm oil | Downregulation of TGF‐β, α‐SMA and fibronectin | 106 | |
| α‐Tocopherol | Attenuation of fibrosis | 107 | |
| RAS blockers (ACEis & ARBs) | Ramipril | Downregulation of TGF‐β | 20, 22 |
| Candesartan | Downregulation of TGF‐β | 22, 108, 109 | |
| Lisinopril | Downregulation of TGF‐β | 108, 110 | |
| Losartan | Downregulation of TGF‐β | 111 | |
| MEK inhibitors | Trametinib | Downregulation of TGF‐β | 83 |
| Dactolisib | Downregulation of α‐SMA and collagen I | 83 | |
| Antifibrotic agents | Pirfenidone | Downregulation of PSC proliferation and collagen I | 112 |
Not yet confirmed. α‐SMA, alpha‐smooth muscle actin; ACEi, angiotensin‐converting enzyme inhibitor; ARB, angiotensin receptor blocker; GLP‐1, glucagon‐like peptide‐1; MEK, mitogen‐activated protein kinase kinase; PPAR‐γ, peroxisome proliferator‐activated receptor‐gamma; PSC, pancreatic stellate cell; RAS, renin–angiotensin system; ROS, reactive oxygen species; SGLT2, sodium–glucose cotransporter 2; TGF‐β, transforming growth factor‐beta.
Sodium–glucose cotransporter 2 (SGLT2) inhibitors, the new oral hypoglycemic agents, function in an insulin‐independent manner, and lower blood glucose levels by enhancing urinary glucose excretion. SGLT2 is found to be mainly present in the S1 segment of the proximal renal tubules, and accounts for ~90% of total renal glucose reabsorption. It has been shown that SGLT2 expression in kidneys increases under diabetic conditions74. Recently, the SGLT2 inhibitor, dapagliflozin, was found to suppress the expression of components of the renal RAS system and interstitial fibrosis in OLETF rats75. Although these effects were confined to the local RAS system and fibrosis in kidneys, it has also been reported that lowering of glucose levels by dapagliflozin attenuates the decline of pancreatic function and the disruption of normal islet morphology in female Zucker diabetic fatty rats76.
Nevertheless, it remains unclear how the SGLT2 inhibitor shows protective effects on pancreatic β ‐cells. Recently, Okauchi et al.77 reported the effects of another SGLT2 inhibitor, luseogliflozin, on β‐cell function and mass in the obese db/db mouse model of type 2 diabetes mellitus. Luseogliflozin was shown to increase insulin biosynthesis and secretion accompanied by an increased expression of important β‐cell‐related factors, and was also shown to increase β‐cell mass through augmentation of β‐cell proliferation and reduction in β‐cell apoptosis. Furthermore, the expression levels of fibrosis‐related genes, such as TGF‐β, fibronectin, collagen I and collagen III, were significantly lower in luseogliflozin‐treated animals77. These findings suggest that SGLT2 inhibitors might also be promising agents that protect β‐cells from islet fibrosis.
Finally, a possible relationship between PPAR‐γ agonists and AT1R has been suggested by the finding that activation of PPAR‐γ decreases AT1R promoter activity and expression78. In addition, the PPAR‐γ agonist, troglitazone, has been shown to inhibit the profibrogenic activity of PSCs79.
Anti‐oxidants, polyphenols and vitamins
One of the other possible therapeutic agents is resveratrol, a natural polyphenolic compound. In a study with db/db mice, chronic treatment with resveratrol improved glucose tolerance, attenuated high‐glucose‐induced oxidative stress, and decreased ROS and islet fibrosis80. Taurine and tempol, anti‐oxidants, might also be possible agents that show an antifibrotic effect. They decreased high‐glucose‐induced PSC activation and islet fibrosis in OLETF rats81, 82.
Mitogen‐activated protein kinase kinase inhibitors and antifibrotic agents
Mitogen‐activated protein kinase kinase inhibitors, which have shown the suppression of both glucose‐ and insulin‐induced ERK 1/2 phosphorylation and PSC proliferation in our previous study, might also be good candidates, although further studies are required to identify their effects66, 83. In addition, a recent study on the antifibrotic agent, pirfenidone, showed decreased expression of α‐SMA and a smaller extent of islet fibrosis in the pancreas of OLETF rats84. However, there was no accompanying improvement in glucose tolerance and insulin secretion84. Although that study showed a limited effect, agents that directly target PSCs might also be a targeted strategy either in combination with other drugs or alone.
Targeting of the Mas receptor axis
In addition to the classic RAS pathway, the roles of the Ang II type 2 receptor and the ACE2‐Ang (1‐7)‐Mas receptor axis have been investigated. In a study with high‐fat diet C57BL/6 mice, the ACEi, enalapril, enhanced islet remodeling, normalized both α‐ and β‐cell mass, and finally sustained β‐cell function85. These effects were associated with increased expression of ACE2 and of the MAS receptor, which has the effects opposite to Ang II actions mediated by AT1R86, 87. Thus, enhancing the alternative MAS receptor pathway might be a valid strategy to counter islet fibrosis.
Conclusion
After substantial clinical evidence, many new and improved insights into the effects of RAS on diabetes have accumulated. Furthermore, new evidence has emerged regarding the involvement of PSCs in endocrine pancreatic function and islet fibrosis. Like antineoplastic agents, aimed at multiple targets to achieve maximal efficacy, the strategies for the prevention of islet fibrosis might offer a benefit by targeting multiple parts in the pathway to disrupt the ominous cross‐talk among the related organs. One possible reason why the DREAM trial did not show definitive prevention of diabetes might be that systemic RAS activation was reduced, but the local RAS blockade was incomplete. More potent interventions might, therefore, be required to achieve more definitive results.
In addition, it is important to select appropriate targets according to the risks of patients to attain maximal beneficial effects. Based on the evidence from numerous mega clinical trials, RAS blockers should be considered as the first option to preserve β‐cell mass and function in patients with a history of CVD or with cardiovascular risk factors. If individuals have higher levels of systemic inflammation markers, such as elevated serum C‐reactive protein levels, an anti‐oxidant can be the right choice, although further clinical evidence is required. For patients with prediabetes or with overt type 2 diabetes mellitus, who are thought to have hyperinsulinemia, GLP‐1 agonist, SGLT2 inhibitor and PPAR‐γ agonist can be good options for slowing the progression of diabetes. In the future, it will be essential to find biomarkers that reflect the status of individual risk factors through studies using genetics and big data. These research advances could provide a more tailored choice of medical treatments based on individual characteristics and thus enabling more specific effects.
Disclosure
The authors declare no conflict of interest.
J Diabetes Investig 2020; 11: 268–280
References
- 1. Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 1988; 37: 1595‐1607. [DOI] [PubMed] [Google Scholar]
- 2. Group UKPDS . U.K. prospective diabetes study 16. Overview of 6 years' therapy of type II diabetes: a progressive disease. U.K. Prospective Diabetes Study Group. Diabetes 1995; 44: 1249–1258. [PubMed] [Google Scholar]
- 3. Levy J, Atkinson AB, Bell PM, et al Beta‐cell deterioration determines the onset and rate of progression of secondary dietary failure in type 2 diabetes mellitus: the 10‐year follow‐up of the Belfast Diet Study. Diabet Med 1998; 15: 290–296. [DOI] [PubMed] [Google Scholar]
- 4. Butler AE, Janson J, Bonner‐Weir S, et al Beta‐cell deficit and increased beta‐cell apoptosis in humans with type 2 diabetes. Diabetes 2003; 52: 102–110. [DOI] [PubMed] [Google Scholar]
- 5. Yoon KH, Ko SH, Cho JH, et al Selective beta‐cell loss and alpha‐cell expansion in patients with type 2 diabetes mellitus in Korea. J Clin Endocrinol Metab 2003; 88: 2300–2308. [DOI] [PubMed] [Google Scholar]
- 6. Weyer C, Bogardus C, Mott DM, et al The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J Clin Investig 1999; 104: 787–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Defronzo RA. Banting Lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 2009; 58: 773–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Prentki M, Nolan CJ. Islet beta cell failure in type 2 diabetes. J Clin Investig 2006; 116: 1802–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yoon KH, Lee JH, Kim JW, et al Epidemic obesity and type 2 diabetes in Asia. Lancet 2006; 368: 1681–1688. [DOI] [PubMed] [Google Scholar]
- 10. Chan JC, Malik V, Jia W, et al Diabetes in Asia: epidemiology, risk factors, and pathophysiology. JAMA 2009; 301: 2129–2140. [DOI] [PubMed] [Google Scholar]
- 11. Poitout V, Robertson RP. Glucolipotoxicity: fuel excess and beta‐cell dysfunction. Endocr Rev 2008; 29: 351–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jurgens CA, Toukatly MN, Fligner CL, et al Beta‐cell loss and beta‐cell apoptosis in human type 2 diabetes are related to islet amyloid deposition. Am J Pathol 2011; 178: 2632–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Keating ST, El‐Osta A. Epigenetic changes in diabetes. Clin Genet 2013; 84: 1–10. [DOI] [PubMed] [Google Scholar]
- 14. Weir GC, Bonner‐Weir S. Five stages of evolving beta‐cell dysfunction during progression to diabetes. Diabetes 2004; 53(Suppl 3): S16–S21. [DOI] [PubMed] [Google Scholar]
- 15. Kim JW, Ko SH, Cho JH, et al Loss of beta‐cells with fibrotic islet destruction in type 2 diabetes mellitus. Front Biosci 2008; 13: 6022–6033. [DOI] [PubMed] [Google Scholar]
- 16. Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Investig 2007; 117: 524–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Omary MB, Lugea A, Lowe AW, et al The pancreatic stellate cell: a star on the rise in pancreatic diseases. J Clin Investig 2007; 117: 50–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Apte MV, Haber PS, Darby SJ, et al Pancreatic stellate cells are activated by proinflammatory cytokines: implications for pancreatic fibrogenesis. Gut 1999; 44: 534–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Homo‐Delarche F, Calderari S, Irminger JC, et al Islet inflammation and fibrosis in a spontaneous model of type 2 diabetes, the GK rat. Diabetes 2006; 55: 1625–1633. [DOI] [PubMed] [Google Scholar]
- 20. Ko SH, Kwon HS, Kim SR, et al Ramipril treatment suppresses islet fibrosis in Otsuka Long‐Evans Tokushima fatty rats. Biochem Biophys Res Commun 2004; 316: 114–122. [DOI] [PubMed] [Google Scholar]
- 21. Cho JH, Kim JW, Shin JA, et al Beta‐cell mass in people with type 2 diabetes. J Diabetes Investig 2011; 2: 6–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ko SH, Hong OK, Kim JW, et al High glucose increases extracellular matrix production in pancreatic stellate cells by activating the renin‐angiotensin system. J Cell Biochem 2006; 98: 343–355. [DOI] [PubMed] [Google Scholar]
- 23. le Roux CW, Astrup A, Fujioka K, et al 3 years of liraglutide versus placebo for type 2 diabetes risk reduction and weight management in individuals with prediabetes: a randomised, double‐blind trial. Lancet 2017; 389: 1399–1409. [DOI] [PubMed] [Google Scholar]
- 24. Torgerson JS, Hauptman J, Boldrin MN, et al XENical in the prevention of diabetes in obese subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care 2004; 27: 155–161. [DOI] [PubMed] [Google Scholar]
- 25. Jacob S, Rett K, Henriksen EJ. Antihypertensive therapy and insulin sensitivity: do we have to redefine the role of beta‐blocking agents? Am J Hypertens 1998; 11: 1258–1265. [DOI] [PubMed] [Google Scholar]
- 26. Hansson L, Lindholm LH, Niskanen L, et al Effect of angiotensin‐converting‐enzyme inhibition compared with conventional therapy on cardiovascular morbidity and mortality in hypertension: the Captopril Prevention Project (CAPPP) randomised trial. Lancet 1999; 353: 611–616. [DOI] [PubMed] [Google Scholar]
- 27. Yusuf S, Sleight P, Pogue J, et al Effects of an angiotensin‐converting‐enzyme inhibitor, ramipril, on cardiovascular events in high‐risk patients. N Engl J Med 2000; 342: 145–153. [DOI] [PubMed] [Google Scholar]
- 28. ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group. The Antihypertensive and Lipid‐Lowering Treatment to Prevent Heart Attack Trial . Major outcomes in high‐risk hypertensive patients randomized to angiotensin‐converting enzyme inhibitor or calcium channel blocker vs diuretic: the Antihypertensive and Lipid‐Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA 2002; 288: 2981–2997. [DOI] [PubMed] [Google Scholar]
- 29. Barzilay JI, Davis BR, Cutler JA, et al Fasting glucose levels and incident diabetes mellitus in older nondiabetic adults randomized to receive 3 different classes of antihypertensive treatment: a report from the Antihypertensive and Lipid‐Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). Arch Intern Med 2006; 166: 2191–2201. [DOI] [PubMed] [Google Scholar]
- 30. Lindholm LH, Ibsen H, Borch‐Johnsen K, et al Risk of new‐onset diabetes in the Losartan intervention for endpoint reduction in hypertension study. J Hypertens 2002; 20: 1879–1886. [DOI] [PubMed] [Google Scholar]
- 31. Lithell H, Hansson L, Skoog I, et al The Study on Cognition and Prognosis in the Elderly (SCOPE): principal results of a randomized double‐blind intervention trial. J Hypertens 2003; 21: 875–886. [DOI] [PubMed] [Google Scholar]
- 32. Kjeldsen SE, Julius S, Mancia G, et al Effects of valsartan compared to amlodipine on preventing type 2 diabetes in high‐risk hypertensive patients: the VALUE trial. J Hypertens 2006; 24: 1405–1412. [DOI] [PubMed] [Google Scholar]
- 33. Bosch J, Yusuf S, Gerstein HC, et al Effect of ramipril on the incidence of diabetes. N Engl J Med 2006; 355: 1551–1562. [DOI] [PubMed] [Google Scholar]
- 34. NAVIGATOR Study Group , McMurray JJ, Holman RR, et al Effect of valsartan on the incidence of diabetes and cardiovascular events. N Engl J Med 2010; 362: 1477–1490. [DOI] [PubMed] [Google Scholar]
- 35. Geng DF, Jin DM, Wu W, et al Angiotensin receptor blockers for prevention of new‐onset type 2 diabetes: a meta‐analysis of 59,862 patients. Int J Cardiol 2012; 155: 236–242. [DOI] [PubMed] [Google Scholar]
- 36. Geng DF, Jin DM, Wu W, et al Angiotensin converting enzyme inhibitors for prevention of new‐onset type 2 diabetes mellitus: a meta‐analysis of 72,128 patients. Int J Cardiol 2013; 167: 2605–2610. [DOI] [PubMed] [Google Scholar]
- 37. Chu KY, Lau T, Carlsson PO, et al Angiotensin II type 1 receptor blockade improves beta‐cell function and glucose tolerance in a mouse model of type 2 diabetes. Diabetes 2006; 55: 367–374. [DOI] [PubMed] [Google Scholar]
- 38. Lau T, Carlsson PO, Leung PS. Evidence for a local angiotensin‐generating system and dose‐dependent inhibition of glucose‐stimulated insulin release by angiotensin II in isolated pancreatic islets. Diabetologia 2004; 47: 240–248. [DOI] [PubMed] [Google Scholar]
- 39. Suzuki K, Nakagawa O, Aizawa Y. Improved early‐phase insulin response after candesartan treatment in hypertensive patients with impaired glucose tolerance. Clin Exp Hypertens 2008; 30: 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. van der Zijl NJ, Moors CC, Goossens GH, et al Valsartan improves {beta}‐cell function and insulin sensitivity in subjects with impaired glucose metabolism: a randomized controlled trial. Diabetes Care 2011; 34: 845–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Paolisso G, Tagliamonte MR, Gambardella A, et al Losartan mediated improvement in insulin action is mainly due to an increase in non‐oxidative glucose metabolism and blood flow in insulin‐resistant hypertensive patients. J Hum Hypertens 1997; 11: 307–312. [DOI] [PubMed] [Google Scholar]
- 42. Olsen MH, Fossum E, Hoieggen A, et al Long‐term treatment with losartan versus atenolol improves insulin sensitivity in hypertension: ICARUS, a LIFE substudy. J Hypertens 2005; 23: 891–898. [DOI] [PubMed] [Google Scholar]
- 43. Carvalho CR, Thirone AC, Gontijo JA, et al Statement of Retraction. Effect of captopril, losartan, and bradykinin on early steps of insulin action. Diabetes 1997;46: 1950–1957. Diabetes 2016; 65: 1128. [DOI] [PubMed] [Google Scholar]
- 44. Lupi R, Del Guerra S, Bugliani M, et al The direct effects of the angiotensin‐converting enzyme inhibitors, zofenoprilat and enalaprilat, on isolated human pancreatic islets. Eur J Endocrinol 2006; 154: 355–361. [DOI] [PubMed] [Google Scholar]
- 45. Saitoh Y, Hongwei W, Ueno H, et al Telmisartan attenuates fatty‐acid‐induced oxidative stress and NAD(P)H oxidase activity in pancreatic beta‐cells. Diabetes Metab 2009; 35: 392–397. [DOI] [PubMed] [Google Scholar]
- 46. Saitoh Y, Hongwei W, Ueno H, et al Candesartan attenuates fatty acid‐induced oxidative stress and NAD(P)H oxidase activity in pancreatic beta‐cells. Diabetes Res Clin Pract 2010; 90: 54–59. [DOI] [PubMed] [Google Scholar]
- 47. Schupp M, Janke J, Clasen R, et al Angiotensin type 1 receptor blockers induce peroxisome proliferator‐activated receptor‐gamma activity. Circulation 2004; 109: 2054–2057. [DOI] [PubMed] [Google Scholar]
- 48. Benson SC, Pershadsingh HA, Ho CI, et al Identification of telmisartan as a unique angiotensin II receptor antagonist with selective PPARgamma‐modulating activity. Hypertension 2004; 43: 993–1002. [DOI] [PubMed] [Google Scholar]
- 49. Schupp M, Clemenz M, Gineste R, et al Molecular characterization of new selective peroxisome proliferator‐activated receptor gamma modulators with angiotensin receptor blocking activity. Diabetes 2005; 54: 3442–3452. [DOI] [PubMed] [Google Scholar]
- 50. Yvan‐Charvet L, Quignard‐Boulange A. Role of adipose tissue renin‐angiotensin system in metabolic and inflammatory diseases associated with obesity. Kidney Int 2011; 79: 162–168. [DOI] [PubMed] [Google Scholar]
- 51. Tahmasebi M, Puddefoot JR, Inwang ER, et al The tissue renin‐angiotensin system in human pancreas. J Endocrinol 1999; 161: 317–322. [DOI] [PubMed] [Google Scholar]
- 52. Suzuki Y, Ruiz‐Ortega M, Lorenzo O, et al Inflammation and angiotensin II. Int J Biochem Cell Biol 2003; 35: 881–900. [DOI] [PubMed] [Google Scholar]
- 53. Danser AH. Local renin‐angiotensin systems: the unanswered questions. Int J Biochem Cell Biol 2003; 35: 759–768. [DOI] [PubMed] [Google Scholar]
- 54. Boffa JJ, Lu Y, Placier S, et al Regression of renal vascular and glomerular fibrosis: role of angiotensin II receptor antagonism and matrix metalloproteinases. J Am Soc Nephrol 2003; 14: 1132–1144. [DOI] [PubMed] [Google Scholar]
- 55. Durante A, Peretto G, Laricchia A, et al Role of the renin‐angiotensin‐aldosterone system in the pathogenesis of atherosclerosis. Curr Pharm Des 2012; 18: 981–1004. [DOI] [PubMed] [Google Scholar]
- 56. Engeli S, Gorzelniak K, Kreutz R, et al Co‐expression of renin‐angiotensin system genes in human adipose tissue. J Hypertens 1999; 17: 555–560. [DOI] [PubMed] [Google Scholar]
- 57. Kawano K, Hirashima T, Mori S, et al OLETF (Otsuka Long‐Evans Tokushima Fatty) rat: a new NIDDM rat strain. Diabetes Res Clin Pract 1994; 24(Suppl): S317–320. [DOI] [PubMed] [Google Scholar]
- 58. Tikellis C, Wookey PJ, Candido R, et al Improved islet morphology after blockade of the renin‐ angiotensin system in the ZDF rat. Diabetes 2004; 53: 989–997. [DOI] [PubMed] [Google Scholar]
- 59. Watari N, Hotta Y, Mabuchi Y. Morphological studies on a vitamin A‐storing cell and its complex with macrophage observed in mouse pancreatic tissues following excess vitamin A administration. Okajimas Folia Anat Jpn 1982; 58: 837–858. [DOI] [PubMed] [Google Scholar]
- 60. Bynigeri RR, Jakkampudi A, Jangala R, et al Pancreatic stellate cell: Pandora's box for pancreatic disease biology. World J Gastroenterol 2017; 23: 382–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bachem MG, Schneider E, Gross H, et al Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology 1998; 115: 421–432. [DOI] [PubMed] [Google Scholar]
- 62. Apte MV, Haber PS, Applegate TL, et al Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut 1998; 43: 128–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yoshikawa H, Kihara Y, Taguchi M, et al Role of TGF‐beta1 in the development of pancreatic fibrosis in Otsuka Long‐Evans Tokushima Fatty rats. Am J Physiol Gastrointest Liver Physiol 2002; 282: G549–G558. [DOI] [PubMed] [Google Scholar]
- 64. De Meyts P, Wallach B, Christoffersen CT, et al The insulin‐like growth factor‐I receptor. Structure, ligand‐binding mechanism and signal transduction. Horm Res 1994; 42: 152–169. [DOI] [PubMed] [Google Scholar]
- 65. Svegliati‐Baroni G, Ridolfi F, Di Sario A, et al Insulin and insulin‐like growth factor‐1 stimulate proliferation and type I collagen accumulation by human hepatic stellate cells: differential effects on signal transduction pathways. Hepatology 1999; 29: 1743–1751. [DOI] [PubMed] [Google Scholar]
- 66. Hong OK, Lee SH, Rhee M, et al Hyperglycemia and hyperinsulinemia have additive effects on activation and proliferation of pancreatic stellate cells: possible explanation of islet‐specific fibrosis in type 2 diabetes mellitus. J Cell Biochem 2007; 101: 665–675. [DOI] [PubMed] [Google Scholar]
- 67. Nielsen LL, Young AA, Parkes DG. Pharmacology of exenatide (synthetic exendin‐4): a potential therapeutic for improved glycemic control of type 2 diabetes. Regul Pept 2004; 117: 77–88. [DOI] [PubMed] [Google Scholar]
- 68. Xu G, Stoffers DA, Habener JF, et al Exendin‐4 stimulates both beta‐cell replication and neogenesis, resulting in increased beta‐cell mass and improved glucose tolerance in diabetic rats. Diabetes 1999; 48: 2270–2276. [DOI] [PubMed] [Google Scholar]
- 69. Chung Kim le T, Hosaka T, Yoshida M, et al Exendin‐4, a GLP‐1 receptor agonist, directly induces adiponectin expression through protein kinase A pathway and prevents inflammatory adipokine expression. Biochem Biophys Res Commun 2009; 390: 613–618. [DOI] [PubMed] [Google Scholar]
- 70. Hirata K, Kume S, Araki S, et al Exendin‐4 has an anti‐hypertensive effect in salt‐sensitive mice model. Biochem Biophys Res Commun 2009; 380: 44–49. [DOI] [PubMed] [Google Scholar]
- 71. Li W, Cui M, Wei Y, et al Inhibition of the expression of TGF‐beta1 and CTGF in human mesangial cells by exendin‐4, a glucagon‐like peptide‐1 receptor agonist. Cell Physiol Biochem 2012; 30: 749–757. [DOI] [PubMed] [Google Scholar]
- 72. Jhala US, Canettieri G, Screaton RA, et al cAMP promotes pancreatic beta‐cell survival via CREB‐mediated induction of IRS2. Genes Dev 2003; 17: 1575–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kim JW, Park SY, You YH, et al Suppression of ROS production by exendin‐4 in PSC attenuates the high glucose‐induced islet fibrosis. PLoS One 2016; 11: e0163187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rahmoune H, Thompson PW, Ward JM, et al Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non‐insulin‐dependent diabetes. Diabetes 2005; 54: 3427–3434. [DOI] [PubMed] [Google Scholar]
- 75. Shin SJ, Chung S, Kim SJ, et al Effect of sodium‐glucose co‐transporter 2 inhibitor, dapagliflozin, on renal renin‐angiotensin system in an animal model of type 2 diabetes. PLoS One 2016; 11: e0165703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Macdonald FR, Peel JE, Jones HB, et al The novel sodium glucose transporter 2 inhibitor dapagliflozin sustains pancreatic function and preserves islet morphology in obese, diabetic rats. Diabetes Obes Metab 2010; 12: 1004–1012. [DOI] [PubMed] [Google Scholar]
- 77. Okauchi S, Shimoda M, Obata A, et al Protective effects of SGLT2 inhibitor luseogliflozin on pancreatic beta‐cells in obese type 2 diabetic db/db mice. Biochem Biophys Res Commun 2016; 470: 772–782. [DOI] [PubMed] [Google Scholar]
- 78. Tham DM, Martin‐McNulty B, Wang YX, et al Angiotensin II is associated with activation of NF‐kappaB‐mediated genes and downregulation of PPARs. Physiol Genomics 2002; 11: 21–30. [DOI] [PubMed] [Google Scholar]
- 79. Shimizu K, Shiratori K, Kobayashi M, et al Troglitazone inhibits the progression of chronic pancreatitis and the profibrogenic activity of pancreatic stellate cells via a PPARgamma‐independent mechanism. Pancreas 2004; 29: 67–74. [DOI] [PubMed] [Google Scholar]
- 80. Lee YE, Kim JW, Lee EM, et al Chronic resveratrol treatment protects pancreatic islets against oxidative stress in db/db mice. PLoS One 2012; 7: e50412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lee E, Ryu GR, Ko SH, et al Antioxidant treatment may protect pancreatic beta cells through the attenuation of islet fibrosis in an animal model of type 2 diabetes. Biochem Biophys Res Commun 2011; 414: 397–402. [DOI] [PubMed] [Google Scholar]
- 82. Ryu GR, Lee E, Chun HJ, et al Oxidative stress plays a role in high glucose‐induced activation of pancreatic stellate cells. Biochem Biophys Res Commun 2013; 439: 258–263. [DOI] [PubMed] [Google Scholar]
- 83. Witteck L, Jaster R. Trametinib and dactolisib but not regorafenib exert antiproliferative effects on rat pancreatic stellate cells. Hepatobiliary Pancreat Dis Int 2015; 14: 642–650. [DOI] [PubMed] [Google Scholar]
- 84. Lee E, Ryu GR, Ko SH, et al A role of pancreatic stellate cells in islet fibrosis and beta‐cell dysfunction in type 2 diabetes mellitus. Biochem Biophys Res Commun 2017; 485: 328–334. [DOI] [PubMed] [Google Scholar]
- 85. Frantz ED, Crespo‐Mascarenhas C, Barreto‐Vianna AR, et al Renin‐angiotensin system blockers protect pancreatic islets against diet‐induced obesity and insulin resistance in mice. PLoS One 2013; 8: e67192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Santos RA, Simoes e Silva AC, Maric C, et al Angiotensin‐(1–7) is an endogenous ligand for the G protein‐coupled receptor Mas. Proc Natl Acad Sci USA 2003; 100: 8258–8263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Giani JF, Mayer MA, Munoz MC, et al Chronic infusion of angiotensin‐(1–7) improves insulin resistance and hypertension induced by a high‐fructose diet in rats. Am J Physiol Endocrinol Metab 2009; 296: E262–271. [DOI] [PubMed] [Google Scholar]
- 88. Pfeffer MA, Swedberg K, Granger CB, et al Effects of candesartan on mortality and morbidity in patients with chronic heart failure: the CHARM‐Overall programme. Lancet 2003; 362: 759–766. [DOI] [PubMed] [Google Scholar]
- 89. Braunwald E, Domanski MJ, Fowler SE, et al Angiotensin‐converting‐enzyme inhibition in stable coronary artery disease. N Engl J Med 2004; 351: 2058–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Yusuf S, Teo KK, Pogue J, et al Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med 2008; 358: 1547–1559. [DOI] [PubMed] [Google Scholar]
- 91. Ogihara T, Ueshima K, Nakao K, et al Long‐term effects of candesartan and amlodipine on cardiovascular morbidity and mortality in Japanese high‐risk hypertensive patients: the Candesartan Antihypertensive Survival Evaluation in Japan Extension Study (CASE‐J Ex). Hypertens Res 2011; 34: 1295–1301. [DOI] [PubMed] [Google Scholar]
- 92. Wing LM, Reid CM, Ryan P, et al A comparison of outcomes with angiotensin‐converting–enzyme inhibitors and diuretics for hypertension in the elderly. N Engl J Med 2003; 348: 583–592. [DOI] [PubMed] [Google Scholar]
- 93. Chowdhury EK, Owen A, Ademi Z, et al Short‐ and long‐term survival in treated elderly hypertensive patients with or without diabetes: findings from the Second Australian National Blood Pressure study. Am J Hypertens 2014; 27: 199–206. [DOI] [PubMed] [Google Scholar]
- 94. van Westerloo DJ, Florquin S, de Boer AM, et al Therapeutic effects of troglitazone in experimental chronic pancreatitis in mice. Am J Pathol 2005; 166: 721–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Lee BJ, Lee HS, Kim CD, et al The effects of combined treatment with an HMG‐CoA reductase inhibitor and PPARgamma agonist on the activation of rat pancreatic stellate cells. Gut Liv 2012; 6: 262–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Shirahige A, Mizushima T, Matsushita K, et al Oral administration of taurine improves experimental pancreatic fibrosis. J Gastroenterol Hepatol 2008; 23: 321–327. [DOI] [PubMed] [Google Scholar]
- 97. Matsushita K, Mizushima T, Shirahige A, et al Effect of taurine on acinar cell apoptosis and pancreatic fibrosis in dibutyltin dichloride‐induced chronic pancreatitis. Acta Med Okayama 2012; 66: 329–334. [DOI] [PubMed] [Google Scholar]
- 98. Ohashi S, Nishio A, Nakamura H, et al Overexpression of redox‐active protein thioredoxin‐1 prevents development of chronic pancreatitis in mice. Antioxid Redox Signal 2006; 8: 1835–1845. [DOI] [PubMed] [Google Scholar]
- 99. Lin Z, Zheng LC, Zhang HJ, et al Anti‐fibrotic effects of phenolic compounds on pancreatic stellate cells. BMC Complement Altern Med 2015; 15: 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Sun XD, Liu XE, Huang DS. Curcumin reverses the epithelial‐mesenchymal transition of pancreatic cancer cells by inhibiting the Hedgehog signaling pathway. Oncol Rep 2013; 29: 2401–2407. [DOI] [PubMed] [Google Scholar]
- 101. Asaumi H, Watanabe S, Taguchi M, et al Green tea polyphenol (‐)‐epigallocatechin‐3‐gallate inhibits ethanol‐induced activation of pancreatic stellate cells. Eur J Clin Investig 2006; 36: 113–122. [DOI] [PubMed] [Google Scholar]
- 102. Rickmann M, Vaquero EC, Malagelada JR, et al Tocotrienols induce apoptosis and autophagy in rat pancreatic stellate cells through the mitochondrial death pathway. Gastroenterology 2007; 132: 2518–2532. [DOI] [PubMed] [Google Scholar]
- 103. McCarroll JA, Phillips PA, Santucci N, et al Vitamin A inhibits pancreatic stellate cell activation: implications for treatment of pancreatic fibrosis. Gut 2006; 55: 79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Guan J, Zhang H, Wen Z, et al Retinoic acid inhibits pancreatic cancer cell migration and EMT through the downregulation of IL‐6 in cancer associated fibroblast cells. Cancer Lett 2014; 345: 132–139. [DOI] [PubMed] [Google Scholar]
- 105. Xiao W, Jiang W, Shen J, et al Retinoic acid ameliorates pancreatic fibrosis and inhibits the activation of pancreatic stellate cells in mice with experimental chronic pancreatitis via suppressing the Wnt/beta‐catenin signaling pathway. PLoS One 2015; 10: e0141462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Gonzalez AM, Garcia T, Samper E, et al Assessment of the protective effects of oral tocotrienols in arginine chronic‐like pancreatitis. Am J Physiol Gastrointest Liver Physiol 2011; 301: G846–G855. [DOI] [PubMed] [Google Scholar]
- 107. Li XC, Lu XL, Chen HH. alpha‐Tocopherol treatment ameliorates chronic pancreatitis in an experimental rat model induced by trinitrobenzene sulfonic acid. Pancreatology 2011; 11: 5–11. [DOI] [PubMed] [Google Scholar]
- 108. Yamada T, Kuno A, Ogawa K, et al Combination therapy with an angiotensin‐converting enzyme inhibitor and an angiotensin II receptor blocker synergistically suppresses chronic pancreatitis in rats. J Pharmacol Exp Ther 2005; 313: 36–45. [DOI] [PubMed] [Google Scholar]
- 109. Yamada T, Kuno A, Masuda K, et al Candesartan, an angiotensin II receptor antagonist, suppresses pancreatic inflammation and fibrosis in rats. J Pharmacol Exp Ther 2003; 307: 17–23. [DOI] [PubMed] [Google Scholar]
- 110. Kuno A, Yamada T, Masuda K, et al Angiotensin‐converting enzyme inhibitor attenuates pancreatic inflammation and fibrosis in male Wistar Bonn/Kobori rats. Gastroenterology 2003; 124: 1010–1019. [DOI] [PubMed] [Google Scholar]
- 111. Chauhan VP, Martin JD, Liu H, et al Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun 2013; 4: 2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Kozono S, Ohuchida K, Eguchi D, et al Pirfenidone inhibits pancreatic cancer desmoplasia by regulating stellate cells. Cancer Res 2013; 73: 2345–2356. [DOI] [PubMed] [Google Scholar]