

Abstract
Antisense oligonucleotides (ASO) are short synthetic DNA molecules designed to inhibit translation of a targeted gene to protein via interaction with messenger RNA. More recently, small interfering (si)RNA have been developed as potent tools to specifically inhibit gene expression. ASO directed against signaling molecules, cytokine receptors, and transcription factors involved in allergic immune and inflammatory responses, have been applied in experimental models of asthma and demonstrate potential as therapeutics. Several ASO-based drugs directed against oncogenes have been developed for therapy of lung cancer, and some have recently reached clinical trials. ASO and siRNA to respiratory syncytial virus infection have demonstrated good potential to treat this condition, particularly in combination with an antiviral drug. Although ASO-based therapeutics are promising for lung diseases, issues of specificity, identification of correct molecular targets, delivery and carrier systems, as well as potential adverse effects must be carefully evaluated before clinical application.
Keywords: Asthma, Respiratory Syncytial Virus, Severe Acute Respiratory Syndrome, Allergic Asthma, Cationic Liposome
The original idea to use antisense oligonucleotides (ASO) to specifically inhibit gene expression was proposed over 25 years ago.[1] Advantages of ASO as a therapeutic tool were immediately obvious. In contrast to traditional drugs designed to inhibit disease-related proteins already synthesized, ASO prevent translation of a protein by interaction with its messenger (m)RNA. However, it took almost 20 years to develop this concept into the first (and currently only) ASO-based drug in clinical use. Fomivirsen (Vitravene®)1, a cytomegalovirus (CMV)-directed ASO, is used topically to treat CMV retinitis, a severe complication of AIDS.[2] Presently, more than 30 ASO-based drugs are in different phases of clinical trials, and hundreds are in development and in preclinical studies.[3] Despite the attraction of the antisense concept, there remain several important issues relating to clinical application of ASO. This review will discuss these problems, summarize published data on ASO strategies in respiratory diseases, and emphasize recent developments and future prospects.
1. Principles of Antisense Oligonucleotides (ASO), Mechanisms of Action, and Related Issues
ASO are short synthetic DNA molecules, designed to interact by Watson-Crick base-pairing with mRNA encoding a target protein. When single-stranded DNA complementary to a target mRNA is introduced into a cell, it binds the mRNA and prevents translation of the protein. While this approach appears straightforward, initial attempts to introduce ASO into cells were unsuccessful because: (i) oligonucleotides are large molecules that are highly negatively charged and do not penetrate cell membranes well; and (ii) oligonucleotides are easily degraded by endo]and exonucleases before they can bind corresponding mRNA.
Thus, critical issues in the development of ASO-based therapy of respiratory diseases include:
target selection and ASO specificity;
ASO stability;
delivery of ASO to target organ/cells.
To overcome the problem of oligonucleotide degradation and to ensure efficient cellular delivery, several chemical modifications of ASO have been developed. The most commonly used and best studied is the phosphorothioate backbone modification (figure 1).
Fig. 1.
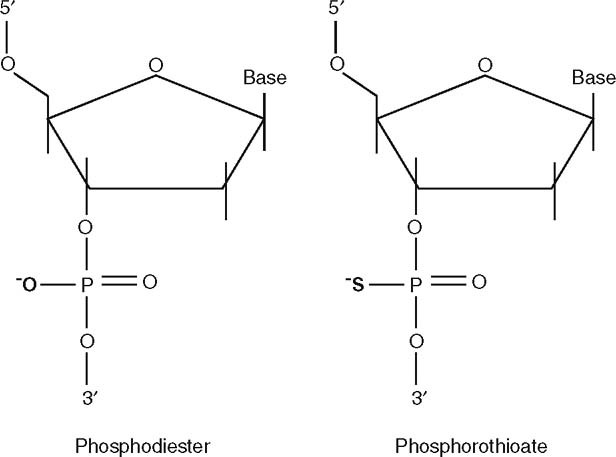
Phosphorothioate backbone modification of oligonucleotides that inhibits nuclease degradation of antisense.
1.1 ASO Mechanisms of Action
Despite intensive studies, mechanisms of ASO action on mRNA are still incompletely understood. Current concepts suggest that the major mechanism of action of phosphorothioate ASO is activation of endonuclease RNAse H when ASO binds to mRNA.[4] This results in mRNA degradation and prevents translation of a specific protein. ASO binding to mRNA can also prevent assembly of the ribosomal complex (e.g. via steric blocking) or inhibit RNA splicing[5] (figure 2).
Fig. 2.
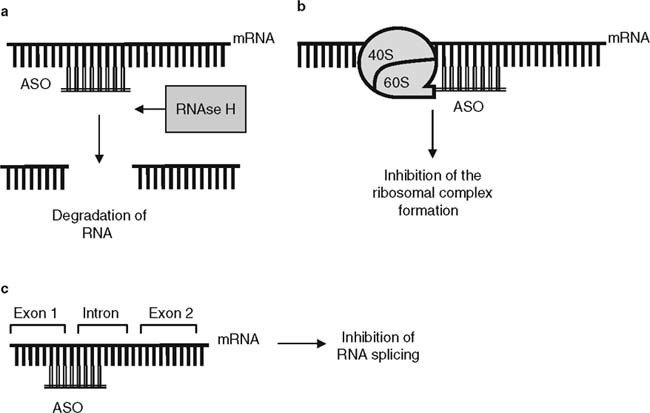
Major mechanisms of action of antisense oligonucleotides (ASO). (a) Activation of endonuclease RNAse H, leading to messenger (m)RNA degradation. (b) Inhibition of the ribosomal complex formation via steric blocking. (c) Inhibition of mRNA splicing.
1.2 Different ASO Structures and Modifications: Stability Issues
The phosphorothioate backbone modification represents a replacement of a non-bridging oxygen by a sulfur atom at each phosphorus[6] (figure 1). This modification, commonly referred to as the ‘first-generation’, greatly increases resistance to nucleases. However, it can render undesired biological activity to some ASO, unrelated to their antisense properties (see section 1.7).
Several other antisense formulations, such as methyl-oligonucleotides, morpholino, peptide nucleic acids, locked nucleic acids, ribozymes, and more recently, small interfering (si)RNA, have been developed.[7–12] Some have improved stability against nucleases and increased binding affinity to mRNA, however, they can have drawbacks such as low cell penetrance and lack of RNAse H recruitment. Despite new generations of ASO, these disadvantages can be significant[3] and thus phosphorothioate-modified ASO are still commonly used.
1.3 Small Interfering RNA
Since its discovery in 1998,[13] the natural phenomenon of RNA interference (RNAi) has been intensively studied. There is much enthusiasm about its potential to be a new, powerful therapeutic tool to specifically inhibit gene expression.[14] RNAi is a part of the innate antiviral defense in lower eukaryotes. It is induced by double-stranded (ds)RNA that is processed to 21–23 nucleotide siRNA (figure 3). RNAi results in degradation of homologous endogenous mRNA complementary to the antisense strand of siRNA. Although transfection of mammalian cells with dsRNA induces a strong interferon (IFN)-like response eventually leading to apoptosis, treatment with siRNA initiates RNAi without causing cell death.[15,16] siRNA has promise for therapy of genetic diseases, since siRNA can target single nucleotide polymorphisms, and thus specifically target selected oncogenes.[14]
Fig. 3.
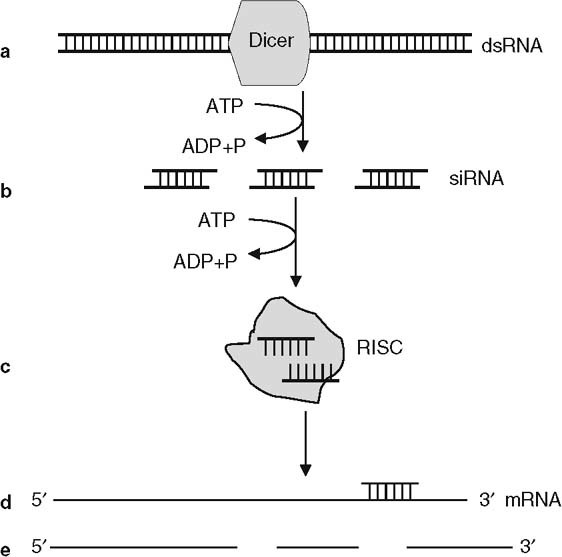
Hypothetical model of RNA interference. (a) When introduced into a cell, double-stranded (ds)RNA is cleaved into small interfering (si)RNAs by a Dicer nuclease in an adenosine triphosphate (ATP)-dependent process. (b) Duplex siRNAs are recruited by several intracellular proteins, forming the RNA-induced silencing complex (RISC). (c) Unwinding of duplex siRNA occurs in an ATP-dependent manner. (d) The antisense strand of the siRNA binds to the messenger (m)RNA. (e) Activation of nuclease activity leads to degradation of the target mRNA. ADP = adenosine diphosphate; P = phosphate.
Recent studies demonstrated that siRNA could selectively silence a disease-associated allele bearing a single mismatch.[17,18] However, clinical application of siRNA is still problematic because we do not fully understand mechanisms of RNAi action in higher eukaryotes. For example, exogenously applied siRNA may interfere with endogenous RNAi pathways and induce potentially dangerous off-target effects.[19,20] In addition, it is more difficult to ensure efficient delivery and cellular uptake of siRNA compared with ASO, because double-stranded siRNA do not bind plasma proteins and rapidly degrade in tissue environments.[19] Although published reports of siRNA use in in vivo in models of lung disease are limited to respiratory syncytial virus (RSV) and parainfluenza virus (PIV) infections,[21,22] siRNA-based approaches to inhibit oncogene expression, pro-inflammatory molecules or pro-fibrotic targets in lung disease are in development.
1.4 Target Selection and Specificity of ASO
Correct target selection is critical in development of ASO-based therapy of respiratory diseases. The targeted molecule must be important in disease pathogenesis and, as ASO can be extremely potent, it is essential to ensure both lung and disease specificity/selectivity of the target to avoid potential adverse effects.
Once a clinically relevant target protein has been selected, specificity of the ASO is a critical issue; it must inhibit expression of the target gene, but not other genes with similar sequences, i.e. the targeted mRNA sequence should not have homology to other genes. In design of ASO, the genome should be carefully checked for possible hybridization of the ASO to sequences in non-targeted genes. Sequences common to several molecules of the same family or domains expressed in many genes must be avoided. Self-complementary regions, four or more contiguous guanine residues, or regions rich in guanines may form complexes with ASO or secondary structures that prevent efficient Watson-Crick hybridization to targeted mRNA and should be avoided.[23] The presence of immunostimulatory cytosine-guanine phosphate-linked dinucleotide (CpG) motifs within ASO is normally undesirable as they can stimulate Toll-like receptor (TLR) 9 on several cell types.[24] However, in some instances they may be included because of additional beneficial effects on the immune system. In vitro controls for ASO specificity, such as nonsense or mismatched oligonucleotide sequences, are important as they assess specificity of hybridization to the selected targeted sequence.
1.5 Delivery Issues
To ensure delivery of ASO to target cells, cationic liposomes are often used in complexes with ASO that can be internalized by pinocytosis/endocytosis.[25–27] Liposome delivery systems have been extensively used for intravenous and local application of ASO to the airways. Upon systemic application for cancer therapy, ASO-liposome complexes preferentially enter tumor tissues because of increased permeability of blood vessels in tumors.[28]
However, the role of cationic lipids in ASO delivery is not limited by their carrier function. Complexes of DNA oligonucleotides with cationic lipids can greatly enhance immunostimulatory properties of DNA.[29,30] These properties can provide an additional therapeutic effect, for example, in cancer. However, systemic release of high levels of pro-inflammatory cytokines tumor necrosis factor (TNF), interleukin (IL)-12 and IFNγ, as well as activation of natural killer (NK) cells following application of DNA-cationic lipid complexes can induce adverse effects.[29] A novel cationic cardiolipin analog-based liposome appeared to be less toxic and more effective for transfection of DNA and siRNA both in vitro and in vivo, compared with a commercially available DOTAP (1,2-dioleoyl-3-trimethylammonium-propane)-based liposome.[31]
In recent years, new carriers such as polyethylenimine (PEI) have been developed with enhanced efficiency of ASO delivery to target cells in vitro and in vivo.[32] Despite enhanced delivery to airway epithelial cells, PEI has toxicity and can be detrimental to lung function.[33] A new strategy using chitosan-DNA nanospheres for intranasal delivery of DNA recently showed advantages over lipid cationic carriers.[34–36] These nanospheres can protect DNA from nuclease degradation, and multiple compounds can be incorporated into nanospheres to achieve additional effects.[37] However, this delivery system has never been assessed for antisense treatment and requires further study
Given carrier-related adverse effects, an attractive method for ASO delivery to the lung involves the use of a natural surfactant with cationic properties.[38] Several studies on local ASO application to the airways have relied on surfactant rather than using artificial carrier systems.[39–42] In a recent study, a single instillation of siRNA mixed with surfactant and elastase decreased expression of a targeted protein in mouse lungs by 50–70% during 7 days of examination.[43]
1.6 ASO Pharmacokinetics
The pharmacokinetics and toxicology of phosphorothioate modified ASO have been intensively studied.[44,45] Effective doses of ASO for in vivo application depend both on the efficiency of the delivery system and mode of administration (systemic versus local). Following systemic application, phosphorothioate oligonucleotides bind to plasma proteins, ensuring their prolonged effect.[44] Various ASO doses for systemic (intravenous or subcutaneous) application in humans were carefully evaluated in several anti-cancer therapy trials, and no significant toxicity was observed at clinically relevant doses.[3]
The pharmacokinetic properties of aerosolized ASO were studied in several animal models; following ASO delivery to the lung, limited systemic distribution was detected.[46] Local delivery of ASO to the airways allows administration of lower doses compared with intravenous therapy of lung diseases. In a study of phosphorothioate ASO to the type 1 adenosine receptor (ADORA1), a single effective inhaled dose was 50 µg/kg and duration of the effect in the lung was 6.8 days.[46] In our studies, 250 µg/rat/day of ASO to spleen tyrosine kinase (SYK) was nebulized into an enclosed chamber on each of three consecutive days prior to allergen challenge (the precise dose each rat received was not determined). This exposure suppressed antigen-induced airway inflammation during the following 48 hours.[47] These examples demonstrate that the effective ASO dose and duration of its effect depend on the target characteristics, in addition to the importance of an efficient delivery system. Quantity and half-life of both target mRNA and encoded protein are critical determinants of ASO pharmacokinetics and pharmacodynamics in in vivo applications.
1.7 Toxicity Issues: Sources of Adverse Effects of Antisense Therapy
Adverse effects of ASO therapy (table I) can result from hybridization of ASO to nonspecific sequences in mRNA, rather than the targeted sequence. Assessing expression of the targeted gene at both mRNA and protein levels following ASO treatment is important to confirm the specificity of the ASO effect.
Table I.
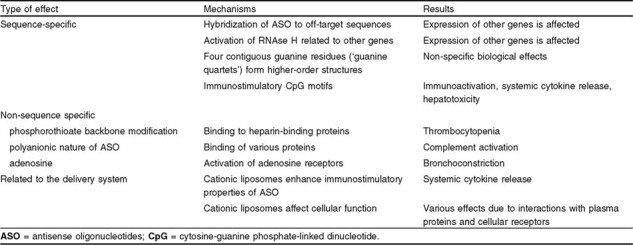
Adverse effects of antisense therapy
Upon DNA-RNA duplex formation, the endonuclease RNAse H is recruited to degrade the RNA in the duplex. As a result of this recruitment, there may be nonspecific activation of RNAse H, resulting in ‘irrelevant cleavage’ and effects on expression of other genes.[48]
A potential source of non-sequence-specific effects of ASO is the backbone modification of oligonucleotides. Phosphorothioate-modified oligonucleotides bind to a family of heparin-binding proteins including some growth factors and their receptors, extracellular matrix proteins and adhesion molecules.[49,50] This mechanism at least partially explains some adverse effects of systemic ASO application such as thrombocytopenia and hypotension.[51,52] This protein binding is due to the polyanionic nature of oligonucleotides, which is also responsible for their ability to activate complement.[44] As outlined in section 1.4, immunostimulatory CpG motifs in ASO sequences can also be an important source of adverse effects related to systemic cytokine release, such as fatigue, fever, and flu-like syndrome.[53] siRNA can also exert non-target-related biological effects, in particular, induction of pro-inflammatory cytokines. Such effects are related to the ability of dsRNA to bind TLRs present on immune cells and induce cellular activation,[54] and must be carefully assessed for each sequence used.
Local delivery of ASO offers advantages over systemic (intravenous) application because it allows lower doses to be used and thus minimizes systemic toxicity. An important consideration in using ASO in the airways is that adenosine can be released as an oligonucleotide degradation product.[46] It can activate adenosine receptors that induce bronchoconstriction. Adenosine receptors are up-regulated in certain clinical conditions, particularly asthma,[55,56] and they themselves have been targeted in studies of ASO treatment of experimental asthma (see section 2.1.2).
Another source of potential adverse effects is related to ASO delivery systems. As discussed in section 1.5, enhanced immunostimulatory properties of ASO applied in cationic liposome complexes may release pro-inflammatory cytokines,[29,30] or cationic liposomes may affect cellular functions directly, e.g. by inhibiting TNF-induced endothelial vascular cell adhesion molecule-1 expression.[57] Cytotoxicity of cationic liposomes is dose-dependent and requires careful evaluation when liposomes are used.[58]
2. Antisense Treatment of Lung Disease
2.1 Experimental Asthma
As asthma is a complex heterogeneous disease, a major challenge is to identify appropriate molecular targets for ASO, and to identify delivery systems that target the lung, and minimize systemic distribution and related adverse effects. There are several examples of such approaches in experimental models.
2.1.1 Tyrosine Kinase Targets
The tyrosine kinase SYK mediates early signaling events important in the pathophysiology of allergic asthma and initiated by cross-linking high affinity receptors for IgE (FcεRI) on mast cells and basophils.[59–61] A 60 bp ASO directed against the SYK gene was constructed as a stem-loop structure, containing three phosphorothioate modifications,[62] and delivered by aerosolization in vivo using SYK ASO-liposome complexes combining cationic liposomes (1,2-dioleoyl-3-trimethylammonium-propane, DOTAP) with a neutral carrier lipid (dioleoylphosphatidyl-ethanol-amine, DOPE). Treatment of rats with nebulized SYK ASO-liposome complexes inhibited SYK mRNA and protein expression in alveolar macrophages.[63]
We studied the effects of aerosolized SYK ASO-liposome complexes in two models: (i) an infectious model of airway inflammation induced by the helminth Nippostrongylus brasiliensis; and (ii) IgE-mediated allergic inflammation induced by ovalbumin in sensitized Brown Norway rats, a model of allergic asthma. SYK ASO significantly inhibited inflammatory cell infiltration in the airways, lung eosinophilia and the rise in TNF in broncho-alveolar lavage induced by antigenic challenge. SYK ASO also suppressed antigen-induced tracheal contraction.[47,63] We have also aerosolized siRNA to SYK in rat ovalbumin-induced asthma and obtained promising down-regulation of inflammation (unpublished observations). Thus, aerosolized SYK ASO inhibited many central components of allergic asthma and inflammation.
Although SYK is a promising molecular target for ASO therapy of asthma and other inflammatory conditions such as acute lung injury, more studies are needed to assess potential risks related to SYK inhibition. For example, recent studies implicated SYK as a tumor suppressor gene in breast and gastric cancer.[64–66] Additionally, we established that SYK is abundantly expressed in lung epithelial cells and is involved in their production of pro-inflammatory molecules.[67] Thus, while SYK ASO may have advantages as a short-term local therapy of severe lung conditions, e.g. acute respiratory distress syndrome, long-term application of SYK ASO raises potential safety issues that must be further assessed.
Another signaling molecule that is a potential target for ASO therapy of asthma is LYN, a Src-family kinase.[68] LYN phosphorylation occurs as an immediate result of conformational changes in cytoplasmic domains of FcεRI after allergen-mediated cross-linking. LYN then interacts with SYK and induces its activation.[69,70] In eosinophils, LYN is associated with IL-5 receptor α subunit (IL5RA) and is important for IL-5-induced differentiation from bone marrow stem cells.[71] In vitro studies demonstrated that ASO directed against LYN blocked eosinophil differentiation from stem cells.[71] Although LYN ASO has never been applied in experimental models of asthma, the importance of LYN for eosinophil differentiation in vivo was confirmed in LYN knockout mice.[71]
2.1.2 Other Targets in Inflammatory Cell Activation
Recent studies of allergic asthma attempted to inhibit intracellular pathways involved in inflammatory cell activation. Inhaled ASO to p38α mitogen-activated protein kinase (MAPK14) down-regulated ovalbumin-induced pulmonary eosinophilia, mucus hypersecretion, and airway hyper-responsiveness (AHR) in a murine model of asthma.[72] ASO to the p65 subunit of the transcription factor NF-κB (RELA) that regulates expression of pro-inflammatory genes[73] applied intravenously significantly inhibited allergic responses in a mouse model.[74] Despite this proof of principle study, systemic application of ASO to NF-κB does not seem to be feasible for treatment of asthma given crucial involvement of this transcription factor in regulation of immune responses.
Given the important role of T helper type 2 (Th2) cytokines and their receptors in allergic asthma, they have been targeted by ASO therapy. For example, ASO to IL-5, applied intravenously in a murine model of asthma, inhibited eosinophilia and AHR.[75] Allakhverdi and colleagues[42] used intratracheal injection of ASO to the common β chain of IL-3, IL-5, and granulocyte-macrophage colony-stimulating factor (GM-CSF) receptors and demonstrated significant reduction of eosinophilia and AHR in a rat model of asthma; this is moving forward to clinical testing. Intranasal application of ASO to stem cell factor (KIT ligand [KITLG]), essential for the development of mast cells,[76] decreased lung inflammation in a murine model of asthma.[40] ASO to the transcription factor GATA-3, essential for development of Th2 responses,[77] also reduced lung inflammation and AHR.[41] In a recent study, the signal transducer and activator of transcription (STAT)-1 was targeted using intranasal application of decoy oligonucleotides in a mouse model of ovalbumin-induced asthma. This study demonstrated inhibition of antigen-induced airway inflammation and hyperreactivity.[78]
To directly target bronchial smooth muscle contraction in asthma, ASO to ADORA1 was developed and administered in aerosol form to rabbits. There was a significant reduction in both broncho-constriction and airway inflammation.[39] This ASO-based therapy is currently in a phase II clinical trial.[79]
2.1.3 Alternative Approaches
All these ASO were applied as phosphorothioate-modified oligonucleotides in liposome delivery systems or as naked DNA. However, alternative approaches have recently been developed, including an adenoviral-mediated expression of ASO to Gob-5 (CLCA1) mRNA, a Ca2+-dependent chloride channel,[80] in the bronchial epithelium in ovalbumin-sensitized mice. This approach decreased AHR and mucus production.[81] Using recombinant adenovirus for ASO delivery to target cells offers some advantages over other methods, such as selectivity to airway epithelium and prolonged expression of transfected genes (>1 week following instillation).[82,83] However, adenovirus-mediated gene delivery induces immune responses to adenovirus that preclude repeated applications[84] and there are several safety concerns such as the potential for oncogenic transformation.
Importantly, in animal models of asthma, ASO to various target molecules were applied prior to antigenic challenge. Whether or not ASO-mediated targeting of molecules involved in asthma pathogenesis will also be efficient during ongoing allergic inflammation requires further studies.
Recently, a ribozyme targeting the human IL-4 receptor α chain (IL4R, also known as IL4RA) was developed.[85] This approach offers some advantages over ‘traditional’ antisense because mechanisms of action of ribozymes rely on activation of RNAse P, which is ubiquitously present in cells. The construct can recycle after inducing the complementary mRNA cleavage, and therefore appears to act more efficiently than DNA-based antisense.[86] Another recent study used siRNA to silence gene expression of STAT6, a transcriptional regulator of Th2 cytokines. In vitro-applied siRNA down-regulated STAT6 protein expression, as well as IL-4-dependent eotaxin production in human bronchial epithelial cells.[87]
Studies of the genetics of asthma have identified several potential asthma susceptibility genes, such as inflammatory mediators,[88] a disintegrin and metalloprotease domain 33 (AD-AM33)[89] and G protein-coupled receptor for asthma susceptibility (GPRA),[90] which may be new targets for antisense therapy. Although biochemical mechanisms linking many of these candidate genes to asthma pathogenesis are poorly understood,[90] ASO strategies may help to elucidate such pathways.
2.2 Lung Cancer
In lung cancer, many oncogenes have been identified and studied as targets for antisense therapy. Several ASO-based drugs have reached phase II–III trials, and it is anticipated that some will soon be approved for clinical use (see Stahel and Zangemeister-Wittke[91] for review). In particular, the anti-apoptotic protein BCL-2 is a promising target for ASO therapy in non-small cell lung cancer.[92,93] ASO directed against the BCL2 gene induces apoptosis of cancer cells and potentiates effects of chemotherapy.[91,94,95]
Other molecular targets for ASO therapy of lung cancer include signal transduction molecules: protein kinase C-α (PRKCA),[96] the regulatory subunit R1-α of protein kinase A (PRKAR1A),[97] RAF kinase (RAF1),[98] and the protein encoded by the HRAS oncogene.[99] Numerous pre-clinical studies are focused on targets in regulation of apoptosis, cell cycle progression, angiogenesis and metastasis, such as the apoptosis suppressor survivin (BIRC5);[100,101] the cytochrome c oxidase assembly protein COX17;[102] several growth factor receptors and receptor tyrosine kinases, as well as transcription factors.[103–107] ASO-based drugs in clinical trials in lung cancer are short oligonucleotide sequences (18–26-mer) with phosphorothioate backbone modifications. Recently, a locked nucleic acid-modified oligonucleotide with bispecific activity against BCL-2 and BCL-xL, another anti-apoptotic BCL protein, was developed and showed enhanced anti-tumor activity in cancer cells.[108]
Since human cancer cell lines preserve their RNAi machinery, use of siRNA to silence oncogenes involved in cancer pathogenesis has been suggested.[109] Indeed, siRNA against S-phase kinase-associated protein 2 (SKP2), a molecule involved in cell cycle regulation that is over-expressed in various cancers including small-cell lung carcinoma, was applied using lentiviral or adenoviral vectors. This strategy significantly inhibited tumor growth in vitro and in vivo.[110] Although there are several in vitro studies using siRNA to various target molecules potentially important in tumorigenesis,[111–114] siRNA-based strategies require further investigation with regard to efficiency, effectiveness and potential adverse effects.
When infused intravenously, ASO preparations showed moderate dose-dependent systemic toxicity such as thrombocytopenia, flu-like syndrome, hypotension and asthenia.[51] In some clinical trials, ASO-based therapeutics were combined with chemotherapeutic agents,[96] a strategy that also needs extensive study. Unfortunately, there are no published reports on local application of ASO in lung cancer.
2.3 Infectious Diseases
An antisense strategy intended to specifically cleave genomic RNA of RSV has recently been developed.[115] Application of ASO with 2′–5′ linked tetra-adenylates was shown to recruit the cellular nuclease RNAseL and successfully inhibit RSV replication.[116] Importantly, a combination of subtherapeutic doses of ASO with the antiviral drug ribavirin demonstrated a potent inhibitory effect.[116] A recent study described use of siRNA to RSV in vivo. Mice treated intranasally with siRNA nanoparticles to RSV protein NS1 before or after infection with RSV showed substantially decreased virus titers in the lung and decreased inflammation and airway hyper-reactivity compared with control animals.[21] Inhibition of both RSV and PIV following intranasal instillation of siRNA in the mouse was also demonstrated.[22] In addition, recent in vitro studies showed the ability of siRNA to inhibit replication of the newly discovered coronavirus SARS-CoV, the causative agent of severe acute respiratory syndrome (SARS).[117] This is the beginning of a potentially important research area, with opportunities to develop innovative ASO therapies for infectious diseases of the lung (table II).
Table II.
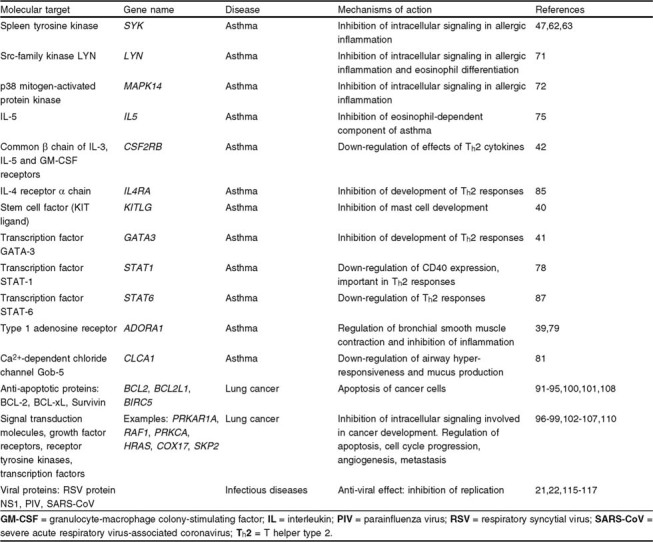
Clinically relevant targets of antisense-based therapy in respiratory diseases
3. Conclusions
ASO are promising therapeutic tools for various respiratory diseases ranging from infection to asthma, lung cancer, fibrosis and acute respiratory distress syndrome. A major advantage of ASO over conventional drugs is their capacity to specifically block synthesis of a disease-associated protein, thus preventing participation in pathogenesis.
ASO can be highly potent and specific and therefore it is essential that correct molecular targets be identified for therapy. Applying ASO in complex heterogeneous diseases such as asthma presents a major challenge, since this condition involves several pathways, numerous genes, and poorly understood gene-environment interactions. Some approaches to specifically target critical molecules in asthma have been successful, and development of ASO-based drugs for clinical application can be anticipated in the near future. The genetics of asthma is a rapidly developing field and important discoveries of susceptibility genes will be important. Such genes, when targeted by antisense therapy, may provide an important contribution to the treatment of this common disease.
ASO therapy also has the potential to become a powerful tool against lung cancer. Successes are anticipated based on intensive molecular studies and discovery of causal oncogenes in lung cancer. Based on published observations, targeted delivery of ASO to the lung is feasible and has significant advantages over systemic application because it can minimize therapeutic doses and thus reduce systemic adverse effects.
There are several critical challenges in the development of ASO-based therapeutics. In addition to selection of the correct target protein, specificity of the ASO effect is essential and inhibition of other genes must be avoided. Both sequence-specific (e.g. CpG-mediated) and sequence-nonspecific (phosphorothioate-mediated) adverse effects should be carefully assessed. New formulations of ASO, such as siRNA, are promising for therapeutic application, but require more studies on mechanisms of action and safety. New methodology for delivery of ASO to selected target cells and optimization of carrier systems are likely to provide important advances for therapeutic uses of ASO.
Acknowledgment
This work was supported by grants from the Canadian Institutes for Health Research, National Institutes of Health HL-27068 and HL-69498, and postdoctoral fellowships from the Canadian Society of Allergy and Clinical Immunology/Merck Frosst and the Alberta Heritage Foundation for Medical Research.
The authors have no conflicts of interest that are directly relevant to the content of this review.
Footnotes
The use of trade names is for product identification purposes only and does not imply endorsement.
References
- 1.Zamecnik P.C., Stephenson M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc Natl Acad Sci U S A. 1978;75(1):280–4. doi: 10.1073/pnas.75.1.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crooke S.T. Vitravene: another piece in the mosaic. Antisense Nucleic Acid Drug Dev. 1998;8(4):vii–viii. doi: 10.1089/oli.1.1998.8.vii. [DOI] [PubMed] [Google Scholar]
- 3.Pirollo K.F., Rait A., Sleer L.S., et al. Antisense therapeutics: from theory to clinical practice. Pharmacol Ther. 2003;99(1):55–77. doi: 10.1016/S0163-7258(03)00053-6. [DOI] [PubMed] [Google Scholar]
- 4.Dash P., Lotan I., Knapp M., et al. Selective elimination of mRNAs in vivo: complementary oligodeoxynucleotides promote RNA degradation by an RNase H-like activity. Proc Natl Acad Sci U S A. 1987;84(22):7896–900. doi: 10.1073/pnas.84.22.7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mercatante D.R., Kole R. Control of alternative splicing by antisense oligonucleotides as a potential chemotherapy: effects on gene expression. Biochim Biophys Acta. 2002;1587(2–3):126–32. doi: 10.1016/s0925-4439(02)00075-3. [DOI] [PubMed] [Google Scholar]
- 6.Matzura H., Eckstein F. A polyribonucleotide containing alternation P = O and P = S linkages. Eur J Biochem. 1968;3(4):448–52. doi: 10.1111/j.1432-1033.1967.tb19551.x. [DOI] [PubMed] [Google Scholar]
- 7.Nielsen P.E., Egholm M., Berg R.H., et al. Peptide nucleic acids (PNAs): potential antisense and anti-gene agents. Anticancer Drug Des. 1993;8(1):53–63. [PubMed] [Google Scholar]
- 8.Wahlestedt C., Salmi P., Good L., et al. Potent and nontoxic antisense oligonucleotides containing locked nucleic acids. Proc Natl Acad Sci U S A. 2000;97(10):5633–8. doi: 10.1073/pnas.97.10.5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doherty E.A., Doudna J.A. Ribozyme structures and mechanisms. Annu Rev Biochem. 2000;69:597–615. doi: 10.1146/annurev.biochem.69.1.597. [DOI] [PubMed] [Google Scholar]
- 10.Braasch D.A., Corey D.R. Novel antisense and peptide nucleic acid strategies for controlling gene expression. Biochemistry. 2002;41(14):4503–10. doi: 10.1021/bi0122112. [DOI] [PubMed] [Google Scholar]
- 11.Heasman J. Morpholino oligos: making sense of antisense? Dev Biol. 2002;243(2):209–14. doi: 10.1006/dbio.2001.0565. [DOI] [PubMed] [Google Scholar]
- 12.Stevenson M. Therapeutic potential of RNA interference. N Engl J Med. 2004;351(17):1772–7. doi: 10.1056/NEJMra045004. [DOI] [PubMed] [Google Scholar]
- 13.Fire A., Xu S., Montgomery M.K., et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391(6669):806–11. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 14.Ichim T.E., Li M., Qian H., et al. RNA interference: a potent tool for gene-specific therapeutics. Am J Transplant. 2004;4(8):1227–36. doi: 10.1111/j.1600-6143.2004.00530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caplen N.J., Mousses S. Short interfering RNA (siRNA)-mediated RNA interference (RNAi) in human cells. Ann N Y Acad Sci. 2003;1002:56–62. doi: 10.1196/annals.1281.007. [DOI] [PubMed] [Google Scholar]
- 16.Morris K.V., Chan S.W., Jacobsen S.E., et al. Small interfering RNA-induced transcriptional gene silencing in human cells. Science. 2004;305(5688):1289–92. doi: 10.1126/science.1101372. [DOI] [PubMed] [Google Scholar]
- 17.Miller V.M., Xia H., Marrs G.L., et al. Allele-specific silencing of dominant disease genes. Proc Natl Acad Sci U S A. 2003;100(12):7195–200. doi: 10.1073/pnas.1231012100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding H., Schwarz D.S., Keene A., et al. Selective silencing by RNAi of a dominant allele that causes amyotrophic lateral sclerosis. Aging Cell. 2003;2(4):209–17. doi: 10.1046/j.1474-9728.2003.00054.x. [DOI] [PubMed] [Google Scholar]
- 19.Hannon G.J., Rossi J.J. Unlocking the potential of the human genome with RNA interference. Nature. 2004;431(7006):371–8. doi: 10.1038/nature02870. [DOI] [PubMed] [Google Scholar]
- 20.Scacheri P.C., Rozenblatt-Rosen O., Caplen N.J., et al. Short interfering RNAs can induce unexpected and divergent changes in the levels of untargeted proteins in mammalian cells. Proc Natl Acad Sci U S A. 2004;101(7):1892–7. doi: 10.1073/pnas.0308698100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang W., Yang H., Kong X., et al. Inhibition of respiratory syncytial virus infection with intranasal siRNA nanoparticles targeting the viral NS1 gene. Nat Med. 2005;11(1):56–62. doi: 10.1038/nm1174. [DOI] [PubMed] [Google Scholar]
- 22.Bitko V., Musiyenko A., Shulyayeva O., et al. Inhibition of respiratory viruses by nasally administered siRNA. Nat Med. 2005;11(1):50–5. doi: 10.1038/nm1164. [DOI] [PubMed] [Google Scholar]
- 23.Dagle J.M., Weeks D.L. Oligonucleotide-based strategies to reduce gene expression. Differentiation. 2001;69(2-3):75–82. doi: 10.1046/j.1432-0436.2001.690201.x. [DOI] [PubMed] [Google Scholar]
- 24.Krieg A.M. CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol. 2002;20:709–60. doi: 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- 25.Bennett C.F., Chiang M.Y., Chan H., et al. Cationic lipids enhance cellular uptake and activity of phosphorothioate antisense oligonucleotides. Mol Pharmacol. 1992;41(6):1023–33. [PubMed] [Google Scholar]
- 26.Capaccioli S., Di Pasquale G., Mini E., et al. Cationic lipids improve antisense oligonucleotide uptake and prevent degradation in cultured cells and in human serum. Biochem Biophys Res Commun. 1993;197(2):818–25. doi: 10.1006/bbrc.1993.2552. [DOI] [PubMed] [Google Scholar]
- 27.Zelphati O., Szoka F.C., Jr Intracellular distribution and mechanism of delivery of oligonucleotides mediated by cationic lipids. Pharm Res. 1996;13(9):1367–72. doi: 10.1023/A:1016026101195. [DOI] [PubMed] [Google Scholar]
- 28.Gabizon A., Catane R., Uziely B., et al. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res. 1994;54(4):987–92. [PubMed] [Google Scholar]
- 29.Freimark B.D., Blezinger H.P., Florack V.J., et al. Cationic lipids enhance cytokine and cell influx levels in the lung following administration of plasmid: cationic lipid complexes. J Immunol. 1998;160(9):4580–6. [PubMed] [Google Scholar]
- 30.Dow S.W., Fradkin L.G., Liggitt D.H., et al. Lipid-DNA complexes induce potent activation of innate immune responses and antitumor activity when administered intravenously. J Immunol. 1999;163(3):1552–61. [PubMed] [Google Scholar]
- 31.Chien P.Y., Wang J., Carbonaro D., et al. Novel cationic cardiolipin analogue-based liposome for efficient DNA and small interfering RNA delivery in vitro and in vivo. Cancer Gene Ther. 2005;12(3):321–8. doi: 10.1038/sj.cgt.7700793. [DOI] [PubMed] [Google Scholar]
- 32.Wiseman J.W., Goddard C.A., McLelland D., et al. A comparison of linear and branched polyethylenimine (PEI) with DCChol/DOPE liposomes for gene delivery to epithelial cells in vitro and in vivo. Gene Ther. 2003;10(19):1654–62. doi: 10.1038/sj.gt.3302050. [DOI] [PubMed] [Google Scholar]
- 33.Chollet P., Favrot M.C., Hurbin A., et al. Side-effects of a systemic injection of linear polyethylenimine-DNA complexes. J Gene Med. 2002;4(1):84–91. doi: 10.1002/jgm.237. [DOI] [PubMed] [Google Scholar]
- 34.Kumar M., Behera A.K., Lockey R.F., et al. Intranasal gene transfer by chitosan-DNA nanospheres protects BALB/c mice against acute respiratory syncytial virus infection. Hum Gene Ther. 2002;13(12):1415–25. doi: 10.1089/10430340260185058. [DOI] [PubMed] [Google Scholar]
- 35.Ravi K. M., Bakowsky U., Lehr C.M. Preparation and characterization of cationic PLGA nanospheres as DNA carriers. Biomaterials. 2004;25(10):1771–7. doi: 10.1016/j.biomaterials.2003.08.069. [DOI] [PubMed] [Google Scholar]
- 36.Mohapatra S.S. Mucosal gene expression vaccine: a novel vaccine strategy for respiratory syncytial virus. Pediatr Infect Dis J. 2003;22(2Suppl.):S100–3. doi: 10.1097/01.inf.0000053894.31944.26. [DOI] [PubMed] [Google Scholar]
- 37.Leong K.W., Mao H.Q., Truong L., et al. DNA-polycation nanospheres as non-viral gene delivery vehicles. J Control Release. 1998;53(1–3):183–93. doi: 10.1016/S0168-3659(97)00252-6. [DOI] [PubMed] [Google Scholar]
- 38.Haitsma J.J., Lachmann U., Lachmann B. Exogenous surfactant as a drug delivery agent. Adv Drug Deliv Rev. 2001;47(2–3):197–207. doi: 10.1016/S0169-409X(01)00106-5. [DOI] [PubMed] [Google Scholar]
- 39.Nyce J.W., Metzger W.J. DNA antisense therapy for asthma in an animal model. Nature. 1997;385(6618):721–5. doi: 10.1038/385721a0. [DOI] [PubMed] [Google Scholar]
- 40.Finotto S., Buerke M., Lingnau K., et al. Local administration of antisense phosphorothioate oligonucleotides to the c-kit ligand, stem cell factor, suppresses airway inflammation and IL-4 production in a murine model of asthma. J Allergy Clin Immunol. 2001;107(2):279–86. doi: 10.1067/mai.2001.113049. [DOI] [PubMed] [Google Scholar]
- 41.Finotto S., De Sanctis G.T., Lehr H.A., et al. Treatment of allergic airway inflammation and hyperresponsiveness by antisense-induced local blockade of GATA-3 expression. J Exp Med. 2001;193(11):1247–60. doi: 10.1084/jem.193.11.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Allakhverdi Z., Allam M., Renzi P.M. Inhibition of antigen-induced eosinophilia and airway hyperresponsiveness by antisense oligonucleotides directed against the common beta chain of IL-3, IL-5, GM-CSF receptors in a rat model of allergic asthma. Am J Respir Crit Care Med. 2002;165(7):1015–21. doi: 10.1164/ajrccm.165.7.2109095. [DOI] [PubMed] [Google Scholar]
- 43.Massaro D., Massaro G.D., Clerch L.B. Noninvasive delivery of small inhibitory RNA and other reagents to pulmonary alveoli in mice. Am J Physiol Lung Cell Mol Physiol. 2004;287(5):L1066–70. doi: 10.1152/ajplung.00067.2004. [DOI] [PubMed] [Google Scholar]
- 44.Levin A.A. A review of the issues in the pharmacokinetics and toxicology of phosphorothioate antisense oligonucleotides. Biochim Biophys Acta. 1999;1489(1):69–84. doi: 10.1016/S0167-4781(99)00140-2. [DOI] [PubMed] [Google Scholar]
- 45.Crooke S.T. Progress in antisense technology. Annu Rev Med. 2004;55:61–95. doi: 10.1146/annurev.med.55.091902.104408. [DOI] [PubMed] [Google Scholar]
- 46.Tanaka M., Nyce J.W. Respirable antisense oligonucleotides: a new drug class for respiratory disease. Respir Res. 2001;2(1):5–9. doi: 10.1186/rr32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stenton G.R., Ulanova M., Dery R.E., et al. Inhibition of allergic inflammation in the airways using aerosolized antisense to Syk kinase. J Immunol. 2002;169(2):1028–36. doi: 10.4049/jimmunol.169.2.1028. [DOI] [PubMed] [Google Scholar]
- 48.Stein C.A. Is irrelevant cleavage the price of antisense efficacy? Pharmacol Ther. 2000;85(3):231–6. doi: 10.1016/S0163-7258(99)00053-4. [DOI] [PubMed] [Google Scholar]
- 49.Guvakova M.A., Yakubov L.A., Vlodavsky I., et al. Phosphorothioate oligodeoxynucleotides bind to basic fibroblast growth factor, inhibit its binding to cell surface receptors, and remove it from low affinity binding sites on extracellular matrix. J Biol Chem. 1995;270(6):2620–7. doi: 10.1074/jbc.270.6.2620. [DOI] [PubMed] [Google Scholar]
- 50.Fennewald S.M., Rando R.F. Inhibition of high affinity basic fibroblast growth factor binding by oligonucleotides. J Biol Chem. 1995;270(37):21718–21. doi: 10.1074/jbc.270.37.21718. [DOI] [PubMed] [Google Scholar]
- 51.Cotter F.E. Antisense oligonucleotides for haematological malignancies. Haematologica. 1999;84(SupplEHA-4):19–22. [PubMed] [Google Scholar]
- 52.Sereni D., Tubiana R., Lascoux C., et al. Pharmacokinetics and tolerability of intravenous trecovirsen (GEM 91), an antisense phosphorothioate oligonucleotide, in HIV-positive subjects. J Clin Pharmacol. 1999;39(1):47–54. doi: 10.1177/00912709922007552. [DOI] [PubMed] [Google Scholar]
- 53.Klinman D.M. Immunotherapeutic uses of CpG oligodeoxynucleotides. Nat Rev Immunol. 2004;4(4):249–58. doi: 10.1038/nri1329. [DOI] [PubMed] [Google Scholar]
- 54.Agrawal S., Kandimalla E.R. Role of Toll-like receptors in antisense and siRNA. Nat Biotechnol. 2004;22(12):1533–7. doi: 10.1038/nbt1042. [DOI] [PubMed] [Google Scholar]
- 55.Bjorck T., Gustafsson L.E., Dahlen S.E. Isolated bronchi from asthmatics are hyperresponsive to adenosine, which apparently acts indirectly by liberation of leukotrienes and histamine. Am Rev Respir Dis. 1992;145(5):1087–91. doi: 10.1164/ajrccm/145.5.1087. [DOI] [PubMed] [Google Scholar]
- 56.Ali S., Mustafa S.J., Metzger W.J. Adenosine-induced bronchoconstriction and contraction of airway smooth muscle from allergic rabbits with late-phase airway obstruction: evidence for an inducible adenosine Al receptor. J Pharmacol Exp Ther. 1994;268(3):1328–34. [PubMed] [Google Scholar]
- 57.Maus U., Rosseau S., Mandrakas N., et al. Cationic lipids employed for antisense oligodeoxynucleotide transport may inhibit vascular cell adhesion molecule-1 expression in human endothelial cells: a word of caution. Antisense Nucleic Acid Drug Dev. 1999;1:71–80. doi: 10.1089/oli.1.1999.9.71. [DOI] [PubMed] [Google Scholar]
- 58.Lysik M.A., Wu-Pong S. Innovations in oligonucleotide drug delivery. J Pharm Sci. 2003;92(8):1559–73. doi: 10.1002/jps.10399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Darby C., Geahlen R.L., Schreiber A.D. Stimulation of macrophage Fc gamma RIIIA activates the receptor-associated protein tyrosine kinase Syk and induces phosphorylation of multiple proteins including p95Vav and p62/GAP-associated protein. J Immunol. 1994;152(11):5429–37. [PubMed] [Google Scholar]
- 60.Costello P.S., Turner M., Walters A.E., et al. Critical role for the tyrosine kinase Syk in signalling through the high affinity IgE receptor of mast cells. Oncogene. 1996;13(12):2595–605. [PubMed] [Google Scholar]
- 61.Kiefer F., Brumell J., Al-Alawi N., et al. The Syk protein tyrosine kinase is essential for Fc gamma receptor signaling in macrophages and neutrophils. Mol Cell Biol. 1998;18(7):4209–20. doi: 10.1128/mcb.18.7.4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Matsuda M., Park J.G., Wang D.C., et al. Abrogation of the Fc gamma receptor IIA-mediated phagocytic signal by stem-loop Syk antisense oligonucleotides. Mol Biol Cell. 1996;7(7):1095–106. doi: 10.1091/mbc.7.7.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stenton G.R., Kim M.K., Nohara O., et al. Aerosolized Syk antisense suppresses Syk expression, mediator release from macrophages, and pulmonary inflammation. J Immunol. 2000;164(7):3790–7. doi: 10.4049/jimmunol.164.7.3790. [DOI] [PubMed] [Google Scholar]
- 64.Coopman P.J., Do M.T., Barth M., et al. The Syk tyrosine kinase suppresses malignant growth of human breast cancer cells. Nature. 2000;406(6797):742–7. doi: 10.1038/35021086. [DOI] [PubMed] [Google Scholar]
- 65.Moroni M., Soldatenkov V., Zhang L., et al. Progressive loss of Syk and abnormal proliferation in breast cancer cells. Cancer Res. 2004;64(20):7346–54. doi: 10.1158/0008-5472.CAN-03-3520. [DOI] [PubMed] [Google Scholar]
- 66.Wang S., Ding Y.B., Chen G.Y., et al. Hypermethylation of Syk gene in promoter region associated with oncogenesis and metastasis of gastric carcinoma. World J Gastroenterol. 2004;10(12):1815–8. doi: 10.3748/wjg.v10.i12.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ulanova M., Puttagunta L., Marcet-Palacios M., et al. Syk tyrosine kinase participates in β1 integrin signaling and inflammatory responses in airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2005;288(3):L297–507. doi: 10.1152/ajplung.00246.2004. [DOI] [PubMed] [Google Scholar]
- 68.Lowell C.A. Src-family kinases: rheostats of immune cell signaling. Mol Immunol. 2004;41(6–7):631–43. doi: 10.1016/j.molimm.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 69.Jouvin M.H., Adamczewski M., Numerof R., et al. Differential control of the tyrosine kinases Lyn and Syk by the two signaling chains of the high affinity immunoglobulin E receptor. J Biol Chem. 1994;269(8):5918–25. [PubMed] [Google Scholar]
- 70.Lin S., Cicala C., Scharenberg A.M., et al. The FcεRI β subunit functions as an amplifier of FcεRI γ-mediated cell activation signals. Cell. 1996;85(7):985–95. doi: 10.1016/S0092-8674(00)81300-8. [DOI] [PubMed] [Google Scholar]
- 71.Stafford S., Lowell C., Sur S., et al. Lyn tyrosine kinase is important for IL-5-stimulated eosinophil differentiation. J Immunol. 2002;168(4):1978–83. doi: 10.4049/jimmunol.168.4.1978. [DOI] [PubMed] [Google Scholar]
- 72.Duan W., Chan J.H., McKay K., et al. Inhaled p38α mitogen-activated protein kinase antisense oligonucleotide attenuates asthma in mice. Am J Respir Crit Care Med. 2004;171(6):571–8. doi: 10.1164/rccm.200408-1006OC. [DOI] [PubMed] [Google Scholar]
- 73.Wulczyn F.G., Krappmann D., Scheidereit C. The NF-kappa B/Rel and I kappa B gene families: mediators of immune response and inflammation. J Mol Med. 1996;74(12):749–69. doi: 10.1007/s001090050078. [DOI] [PubMed] [Google Scholar]
- 74.Choi I.W., Kim D.K., Ko H.M., et al. Administration of antisense phosphorothioate oligonucleotide to the p65 subunit of NF-kappaB inhibits established asthmatic reaction in mice. Int Immunopharmacol. 2004;4(14):1817–28. doi: 10.1016/j.intimp.2004.07.030. [DOI] [PubMed] [Google Scholar]
- 75.Karras J.G., McGraw K., McKay R.A., et al. Inhibition of antigen-induced eosinophilia and late phase airway hyperresponsiveness by an IL-5 antisense oligonucleotide in mouse models of asthma. J Immunol. 2000;164(10):5409–15. doi: 10.4049/jimmunol.164.10.5409. [DOI] [PubMed] [Google Scholar]
- 76.Zsebo K.M., Williams D.A., Geissler E.N., et al. Stem cell factor is encoded at the Sl locus of the mouse and is the ligand for the c-kit tyrosine kinase receptor. Cell. 1990;63(1):213–24. doi: 10.1016/0092-8674(90)90302-U. [DOI] [PubMed] [Google Scholar]
- 77.Zheng W., Flavell R.A. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89(4):587–96. doi: 10.1016/S0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- 78.Quarcoo D., Weixler S., Groneberg D., et al. Inhibition of signal transducer and activator of transcription 1 attenuates allergen-induced airway inflammation and hyperreactivity. J Allergy Clin Immunol. 2004;114(2):288–95. doi: 10.1016/j.jaci.2004.03.055. [DOI] [PubMed] [Google Scholar]
- 79.Popescu F.D. New asthma drugs acting on gene expression. J Cell Mol Med. 2003;7(4):475–86. doi: 10.1111/j.1582-4934.2003.tb00251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Komiya T., Tanigawa Y., Hirohashi S. Cloning and identification of the gene gob-5, which is expressed in intestinal goblet cells in mice. Biochem Biophys Res Commun. 1999;255(2):347–51. doi: 10.1006/bbrc.1999.0168. [DOI] [PubMed] [Google Scholar]
- 81.Nakanishi A., Morita S., Iwashita H., et al. Role of gob-5 in mucus overproduction and airway hyperresponsiveness in asthma. Proc Natl Acad Sci U S A. 2001;98(9):5175–80. doi: 10.1073/pnas.081510898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sime P.J., Xing Z., Graham F.L., et al. Adenovector-mediated gene transfer of active transforming growth factor-betal induces prolonged severe fibrosis in rat lung. J Clin Invest. 1997;100(4):768–76. doi: 10.1172/JCI119590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xing Z., Braciak T., Ohkawara Y., et al. Gene transfer for cytokine functional studies in the lung: the multifunctional role of GM-CSF in pulmonary inflammation. J Leukoc Biol. 1996;59(4):481–8. doi: 10.1002/jlb.59.4.481. [DOI] [PubMed] [Google Scholar]
- 84.Knowles M.R., Hohneker K.W., Zhou Z., et al. A controlled study of adenoviral-vector-mediated gene transfer in the nasal epithelium of patients with cystic fibrosis. N Engl J Med. 1995;333(13):823–31. doi: 10.1056/NEJM199509283331302. [DOI] [PubMed] [Google Scholar]
- 85.Dreyfus D.H., Matczuk A., Fuleihan R. An RNA external guide sequence ribozyme targeting human interleukin-4 receptor alpha mRNA. Int Immunopharmacol. 2004;4(8):1015–27. doi: 10.1016/j.intimp.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 86.Plehn-Dujowich D., Altman S. Effective inhibition of influenza virus production in cultured cells by external guide sequences and ribonuclease P. Proc Natl Acad Sci U S A. 1998;95(13):7327–32. doi: 10.1073/pnas.95.13.7327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rippmann J.F., Schnapp A., Weith A., et al. Gene silencing with STAT6 specific siRNAs blocks eotaxin release in IL-4/TNFalpha stimulated human epithelial cells. FEBS Lett. 2005;579(1):173–8. doi: 10.1016/j.febslet.2004.11.071. [DOI] [PubMed] [Google Scholar]
- 88.Allen M., Heinzmann A., Noguchi E., et al. Positional cloning of a novel gene influencing asthma from chromosome 2ql4. Nat Genet. 2003;35(3):258–63. doi: 10.1038/ng1256. [DOI] [PubMed] [Google Scholar]
- 89.Van Eerdewegh P., Little R.D., Dupuis J., et al. Association of the ADAM33 gene with asthma and bronchial hyperresponsiveness. Nature. 2002;418(6896):426–30. doi: 10.1038/nature00878. [DOI] [PubMed] [Google Scholar]
- 90.Laitinen T., Polvi A., Rydman P., et al. Characterization of a common susceptibility locus for asthma-related traits. Science. 2004;304(5668):300–4. doi: 10.1126/science.1090010. [DOI] [PubMed] [Google Scholar]
- 91.Stahel R.A., Zangemeister-Wittke U. Antisense oligonucleotides for cancer therapy: an overview. Lung Cancer. 2003;41(Suppl.1):S81–8. doi: 10.1016/S0169-5002(03)00147-8. [DOI] [PubMed] [Google Scholar]
- 92.Ziegler A., Luedke G.H., Fabbro D., et al. Induction of apoptosis in small-cell lung cancer cells by an antisense oligodeoxynucleotide targeting the Bcl-2 coding sequence. J Natl Cancer Inst. 1997;89(14):1027–36. doi: 10.1093/jnci/89.14.1027. [DOI] [PubMed] [Google Scholar]
- 93.Zangemeister-Wittke U., Leech S.H., Olie R.A., et al. A novel bispecific antisense oligonucleotide inhibiting both bcl-2 and bcl-xL expression efficiently induces apoptosis in tumor cells. Clin Cancer Res. 2000;6(6):2547–55. [PubMed] [Google Scholar]
- 94.Kim R., Emi M., Tanabe K., et al. Therapeutic potential of antisense Bcl-2 as a chemosensitizer for cancer therapy. Cancer. 2004;101(11):2491–502. doi: 10.1002/cncr.20696. [DOI] [PubMed] [Google Scholar]
- 95.Herbst R.S., Frankel S.R. Oblimersen sodium (Genasense bcl-2 antisense oligonucleotide): a rational therapeutic to enhance apoptosis in therapy of lung cancer. Clin Cancer Res. 2004;10:4245s–8s. doi: 10.1158/1078-0432.CCR-040018. [DOI] [PubMed] [Google Scholar]
- 96.Villalona-Calero M.A., Ritch P., Figueroa J.A., et al. A phase I/II study of LY900003, an antisense inhibitor of protein kinase C-alpha, in combination with cisplatin and gemcitabine in patients with advanced non-small cell lung cancer. Clin Cancer Res. 2004;10:6086–93. doi: 10.1158/1078-0432.CCR-04-0779. [DOI] [PubMed] [Google Scholar]
- 97.Wang H., Cai Q., Zeng X., et al. Antitumor activity and pharmacokinetics of a mixed-backbone antisense oligonucleotide targeted to the RI alpha subunit of protein kinase A after oral administration. Proc Natl Acad Sci U S A. 1999;96(24):13989–94. doi: 10.1073/pnas.96.24.13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Coudert B., Anthoney A., Fiedler W., et al. Phase II trial with ISIS 5132 in patients with small-cell (SCLC) and non-small cell (NSCLC) lung cancer: a European Organization for Research and Treatment of Cancer (EORTC) Early Clinical Studies Group report. Eur J Cancer. 2001;37(17):2194–8. doi: 10.1016/S0959-8049(01)00286-6. [DOI] [PubMed] [Google Scholar]
- 99.Jansen B., Zangemeister-Wittke U. Antisense therapy for cancer: the time of truth. Lancet Oncol. 2002;3(11):672–83. doi: 10.1016/S1470-2045(02)00903-8. [DOI] [PubMed] [Google Scholar]
- 100.Ambrosini G., Adida C., Altieri D.C. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3(8):917–21. doi: 10.1038/nm0897-917. [DOI] [PubMed] [Google Scholar]
- 101.Olie R.A., Simoes-Wust A.P., Baumann B., et al. A novel antisense oligonucleotide targeting survivin expression induces apoptosis and sensitizes lung cancer cells to chemotherapy. Cancer Res. 2000;60(11):2805–9. [PubMed] [Google Scholar]
- 102.Suzuki C., Daigo Y., Kikuchi T., et al. Identification of COX17 as a therapeutic target for non-small cell lung cancer. Cancer Res. 2003;63(21):7038–41. [PubMed] [Google Scholar]
- 103.Sak A., Wurm R., Elo B., et al. Increased radiation-induced apoptosis and altered cell cycle progression of human lung cancer cell lines by antisense oligodeox-ynucleotides targeting p53 and p21 (WAF1/CIP1) Cancer Gene Ther. 2003;10(12):926–34. doi: 10.1038/sj.cgt.7700649. [DOI] [PubMed] [Google Scholar]
- 104.Raben D., Helfrich B. Angiogenesis inhibitors: a rational strategy for radiosensitization in the treatment of non-small-cell lung cancer? Clin Lung Cancer. 2004;6(1):48–57. doi: 10.3816/CLC.2004.n.021. [DOI] [PubMed] [Google Scholar]
- 105.Pold M., Krysan K., Pold A., et al. Cyclooxygenase-2 modulates the insulin-like growth factor axis in non-small-cell lung cancer. Cancer Res. 2004;64(18):6549–55. doi: 10.1158/0008-5472.CAN-04-1225. [DOI] [PubMed] [Google Scholar]
- 106.Kuhn H., Kopff C., Konrad J., et al. Influence of basic fibroblast growth factor on the proliferation of non-small cell lung cancer cell lines. Lung Cancer. 2004;44(2):167–74. doi: 10.1016/j.lungcan.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 107.Stabile L.P., Lyker J.S., Huang L., et al. Inhibition of human non-small cell lung tumors by a c-Met antisense/U6 expression plasmid strategy. Gene Ther. 2004;11(3):325–35. doi: 10.1038/sj.gt.3302169. [DOI] [PubMed] [Google Scholar]
- 108.Hopkins-Donaldson S., Cathomas R., Simoes-Wust A.P., et al. Induction of apoptosis and chemosensitization of mesothelioma cells by Bcl-2 and Bcl-xL antisense treatment. Int J Cancer. 2003;106(2):160–6. doi: 10.1002/ijc.11209. [DOI] [PubMed] [Google Scholar]
- 109.Yin J.Q., Gao J., Shao R., et al. siRNA agents inhibit oncogene expression and attenuate human tumor cell growth. J Exp Ther Oncol. 2003;3(4):194–204. doi: 10.1046/j.1359-4117.2003.01092.x. [DOI] [PubMed] [Google Scholar]
- 110.Sumimoto H., Yamagata S., Shimizu A., et al. Gene therapy for human small-cell lung carcinoma by inactivation of Skp-2 with virally mediated RNA interference. Gene Ther. 2005;12(1):95–100. doi: 10.1038/sj.gt.3302391. [DOI] [PubMed] [Google Scholar]
- 111.Spankuch-Schmitt B., Bereiter-Hahn J., Kaufmann M., et al. Effect of RNA silencing of polo-like kinase-1 (PLK1) on apoptosis and spindle formation in human cancer cells. J Natl Cancer Inst. 2002;94(24):1863–77. doi: 10.1093/jnci/94.24.1863. [DOI] [PubMed] [Google Scholar]
- 112.Brummelkamp T.R., Bernards R., Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296(5567):550–3. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 113.Martinez L.A., Naguibneva I., Lehrmann H., et al. Synthetic small inhibiting RNAs: efficient tools to inactivate oncogenic mutations and restore p53 pathways. Proc Natl Acad Sci U S A. 2002;99(23):14849–54. doi: 10.1073/pnas.222406899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nieth C., Priebsch A., Stege A., et al. Modulation of the classical multidrug resistance (MDR) phenotype by RNA interference (RNAi) FEBS Lett. 2003;545(2–3):144–50. doi: 10.1016/S0014-5793(03)00523-4. [DOI] [PubMed] [Google Scholar]
- 115.Maggon K., Barik S. New drugs and treatment for respiratory syncytial virus. Rev Med Virol. 2004;14(3):149–68. doi: 10.1002/rmv.423. [DOI] [PubMed] [Google Scholar]
- 116.Xu Z., Kuang M., Okicki J.R., et al. Potent inhibition of respiratory syncytial virus by combination treatment with 2–5A antisense and ribavirin. Antiviral Res. 2004;61(3):195–206. doi: 10.1016/j.antiviral.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 117.Wu C.J., Huang H.W., Liu C.Y., et al. Inhibition of SARS-CoV replication by siRNA. Antiviral Res. 2005;65(1):45–8. doi: 10.1016/j.antiviral.2004.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]