

Abstract
Fragment-based methods for drug discovery are increasingly popular because they provide drug leads with greater ligand efficiency than conventional high-throughput screening. However, established methods for fragment detection do not address the central question in fragment-based ligand discovery: how can a primary ligand be optimally extended by a secondary fragment? Dynamic screening methods solve this issue by using a protein target as a template for ligand assembly, thus yielding high-affinity binders from low-affinity fragments. This review summarizes recent work on dynamic screening methodology, which resulted in the development of several high-affinity binders for various targets. Strengths and limitations of the published approaches are discussed and possible contributions of dynamic screening methodology to the drug discovery process are highlighted.
Introduction
Since an ever-increasing number of proteins have been discovered or postulated as potential new drug targets [1], the demand of the pharmaceutical industry for novel drug candidates and of the chemical biology field for chemical tools has been strongly stimulated. However, the actual output of research and development (R&D) has been described as insufficient in proportion to the investment made. For example, in 2006 the overall spending on biopharmaceutical R&D reached a record of $55.2 billion in North America. In the same year the US Food and Drug Administration (FDA) approved only 22 new molecular entities (NMEs) and biologics. In contrast, in 1996 some 53 NMEs were approved for clinical use, although R&D expenditure in 1996 was less than half the amount spent in 2006.1 This discrepancy has been termed a ‘compound crisis’ and it has been postulated that it results from inefficient methods in drug discovery summarized as an ‘innovation deficit’ 2, 3, 4, 5. Thus, it is apparent that the pharmaceutical industry must improve its R&D productivity by using new technologies for the development of NMEs.
The concept of fragment-based drug discovery: small is beautiful
Fragment-based drug discovery concepts have been developed since the mid-1990s as an alternative to conventional combinatorial chemistry and high-throughput screening (HTS) approaches (Figure 1a). As opposed to combinatorial chemistry and HTS, which use large chemical libraries (>106) requiring considerable effort, expensive library storage, quality control and data handling, fragment-based approaches require only small libraries (often <1000 members) as a starting point.
Figure 1.
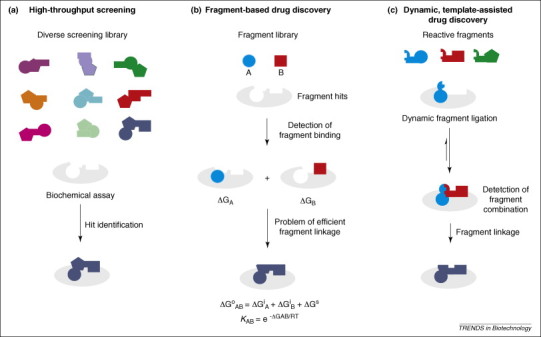
Concepts in lead discovery. (a) High-throughput screening (HTS). A diverse library of chemical compounds is collected and tested against the drug target. (b) Fragment-based lead discovery. The binding of small molecular fragments to the protein is detected. Low-affinity fragments can be linked to provide high-affinity ligands. However, detection and linkage of these fragments are difficult tasks. The physicochemical basis of fragment-based drug discovery is indicated below. The free binding energy ΔG°AB of a protein ligand results from the additive contributions of its molecular components (A and B) [6]. The binding constant KAB is an exponential function of the binding energy. (c) Dynamic strategies in fragment-based drug discovery. Reactive fragments are incubated with the protein and form specific combinations of fragments on the protein template, which facilitates fragment detection and linkage to a new ligand.
Conceptually, fragment-based drug discovery is based on the consideration that the free binding energy of a protein ligand results from the contributions of its molecular components. Therefore, small contributions from molecular fragments can add up to yield a high-affinity protein ligand (Figure 1b) [6]. First, a small molecule fragment that binds to the protein pocket of interest is identified. The starting fragment is then chemically modified to generate a binder of higher affinity, which is subsequently further optimized to a lead structure (Figure 1b). The concept has become very popular for two main reasons. First, there are much fewer fragments than drug-sized molecules. Rough estimates indicate that approximately 107 fragments with up to 12 heavy atoms do exist (excluding 3- and 4-ring-containing structures) [7], whereas there are 1063 possible small drug-like molecules with up to 30 heavy atoms [8]. For comparison, only approximately 108 molecules have been synthesized to date [9]. Thus, initial screening of fragment libraries is expected to sample the chemical space much more efficiently than traditional approaches ever could.
The second reason is that fragment-derived lead structures have significantly higher ligand efficiency (free binding energy per non-hydrogen atom of the ligand) than molecules discovered by screening of large compound libraries. An investigation of 150 known natural and synthetic ligands revealed that the free binding energy increased in proportion to ligand size up to a maximum of 15 atoms 10, 11. The maximum average free-energy contribution per heavy atom was –1.5 kcal/mol. For molecules larger than this, no further increase in ligand efficiency was observed [12]. These observations confirm how crucial limiting the molecular size is for the efficiency of protein ligands, thereby supporting the preference of fragment hits (<12 heavy atoms per molecule) over typical HTS hits. These results also indicate easier optimization and hit-to-lead development of fragment hits relative to that of HTS hits.
Dynamic template-assisted strategies in fragment-based drug discovery
The major challenges of fragment-based drug discovery are the identification of low-affinity fragments and efficient and biologically active linkage of the fragments identified [13]. Weakly binding ligands are difficult to detect. In general, four main biophysical techniques have been used for this task: NMR spectroscopy 14, 15, X-ray crystallography 16, 17, surface plasmon resonance [18] and isothermal titration calorimetry 19, 20. Major bottlenecks of these biophysical techniques are their high protein consumption, the need for expensive detection equipment and their limited throughput. Moreover, the methods do not solve the other challenging question in fragment-based discovery, i.e. how can two low-affinity binders be linked optimally to yield a high-affinity binder [21]? Biophysical methods do not provide information regarding the optimal bioactive combination of fragments.
To address this problem, alternative so-called dynamic and template-assisted strategies have been proposed for fragment-based drug discovery and are discussed here. All these methods use a target protein as a template for selection and/or assembly of optimal fragment combinations. All dynamic template-assisted approaches covered in this review have in common that a chemical reaction, which can be reversible or irreversible and enzymatic or non-enzymatic, is exploited for detection of the best fragment combination.
The first step in this direction was to shift chemical equilibria by introducing proteins, as realized in dynamic combinatorial chemistry (DCC) (Figure 2 ) 22, 23, 24, which has been conceptually extended in tethering 25, 26, 27, 28, dynamic combinatorial resolution (DCR) 29, 30, 31, and pseudo-dynamic combinatorial chemistry (pDCC) 32, 33, 34 approaches. A second subcategory of fragment detection comprises template-assisted strategies such as target-guided synthesis (TGS), in which the chemical reaction used for detection is catalyzed by the protein 31, 35, 36, 37, 38, 39, 40. Substrate activity screening (SAS) 41, 42, 43, which is discussed below, can be used for the transfer of fragment information from substrates to non-substrate protein ligands. Finally, dynamic ligation screening (DLS) 44, 45 uses classical bioassays, such as fluorogenic substrate competition or fluorescence polarization [43], for fragment detection and can thus be used for HTS fragment-based drug discovery.
Figure 2.
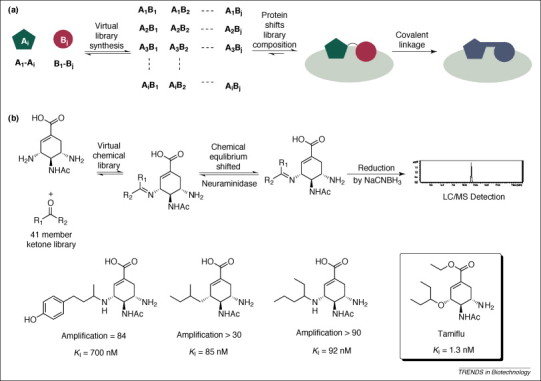
Dynamic combinatorial chemistry. (a) Schematic representation of the selection process in DCC. Receptor-bound library members are amplified in a virtual chemical library and identified following covalent fixation. (b) Condensation of diamine 1 with 41 different ketones yielded a library of 41 potential neuramidase inhibitors. Reduction followed by HPLC analysis led to the identification of several inhibitors (bottom panel). However, species showing the greatest amplification were not the strongest binders (data not shown) 46, 55.
There are significant differences in dynamic template-assisted strategies, which largely determine their practical use in drug discovery. One major criterion is the amount of protein required for fragment detection. Some of the strategies, such as DCC 23, 24, pDCC [32], and TGS [40], require near-stoichiometric or stoichiometric protein quantities, whereas more recently developed strategies, such as DCR [29], SAS [41] and DLS [44], use the protein template in only catalytic amounts. A second relevant criterion is the detection technology required. In DCC, tethering, DCR, pDCC, and TGS, fragment identification relies on NMR spectroscopy [29], chromatography 22, 25, 32, 40, 46, X-ray crystallography [47] or mass spectrometry 25, 26, 27, 28. Recently, fragment detection consistent with established high-throughput methodologies that use standardized microtiter plates and detection by fluorescence, absorption, or fluorescence polarization in DLS have been developed [43]. Finally, the methods described here differ significantly in their scope. Whereas some can be used for detection of a wide range of protein binders, others are more specific for one protein or for a class of proteins. Thus, the aim of our review is to provide an orientation guide for potential users of dynamic template-assisted fragment-based methodology.
Dynamic combinatorial chemistry (DCC)
If a mixture of compounds formed in a reversible chemical equilibrium interacts with a protein, a shift in equilibrium towards the best binding components can be expected in accordance with the law of mass action. The first application of this principle for the discovery of protein binders was reported by Huc and Lehn in 1997 and termed DCC [22].
A DCC application was demonstrated by Hochgürtel et al. in 2002 46, 48. They created an imine library by condensing a diamine (Figure 2b) that was derived from Tamiflu — a known inhibitor of neuraminidase, a key influenza virus enzyme — with 41 different ketones. For detection of enzyme activity, the imines formed were reduced to the corresponding amines by addition of NaCNBH3 and the resulting set of amines was analyzed by HPLC. High-affinity binders of neuraminidase were identified by comparing HPLC chromatograms for reactions containing the template with controls. However, in this particular application of the DCC concept the relative amplification of fragment combinations did not correspond to the binding affinities of the compounds identified (Figure 2b) 46, 48. For example, the compound exhibiting the greatest amplification was not a potent inhibitor, whereas amplification of the strongest inhibitor obtained from this screen was three-fold less. There are a number of possible explanations for this observation. In particular, the final reduction reaction can distort the results in two ways. Amines, which are derived from chemically more stable imines, are expected to be preferentially formed, irrespective of the protein template. Second, reduction of a strongly binding imine can yield an amine product with poorer binding properties.
Although in principle DCC should be applicable to different types of proteins or enzymes, the method is limited by the near-stoichiometric amounts of protein required. Second, the reaction time is long, ranging from 1 day to 1 week. Third, larger libraries are difficult to screen because HPLC cannot easily be used to separate considerably larger libraries [48]. In addition, the detection of preferentially formed library members via reduction does not correlate with the affinity of the originally binder. Thus, differences in amplification and the affinity of the reduction products are likely to occur.
Tethering
Tethering constitutes a specific form of DCC and was been introduced by the Sunesis company (www.sunesis.com). Their approach exploits a reversible disulfide exchange reaction on the surface of a protein 25, 49. For disulfide ligation, native cysteine residues can be used; alternatively, these can be engineered in proximity to the site of interest 26, 28 or added by a cross-linking reaction [27]. Thiol fragments that bind tightly in the screened pocket will form a stable disulfide with the cysteine residue of the protein or the thiol-containing cross-linker. The key advantage of tethering is the ability to focus on a particular region of the template. For example, in the case of caspase-3, the cross-linker, a derivative of aspartate, was bound covalently to the enzyme and the cross-linked capase-3 was incubated with 7000 disulfides. The thiol group of the cross-linker interacted with the disulfides by exchange reaction. Disulfides that fit to capase-3 are preferentially formed. The most stabile disulfide–enzyme complex could be detected by high-resolution mass spectrometry, from which a potent caspase-3 inhibitor could be obtained (Figure 3 ).
Figure 3.
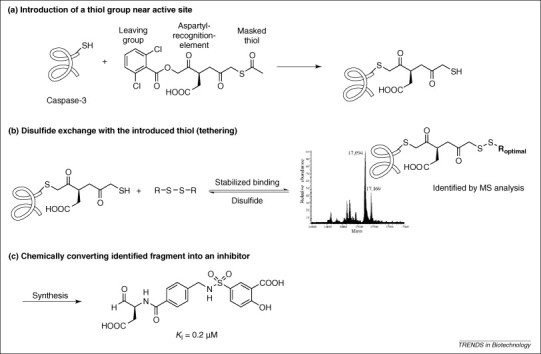
Example of tethering. (a) Introduction of a masked thiol group near the active site by cross-linking a known irreversible inhibitor to the target, caspase-3. (b) After deprotection of the thiol a disulfide library is added. The disulfide with the highest affinity is stabilized by the template. The most stabile disulfide can be identified by mass spectrometric analysis of the entire protein–cross-linker–fragment complex. (c) Conversion of the identified fragment into a potent inhibitor of caspase-3 [27].
Disadvantages of the tethering approach are the requirement for near-stoichiometric amounts of template, the need for a specialized disulfide library, which is not commercially available, and fragment detection by high-resolution mass spectrometry. In addition, because tethering products are template-linked disulfides, the fragments need to be converted into a chemically stable ligand, which needs to retain the affinity of the disulfide, before further information can be obtained. Nevertheless, the tethering approach enables site-directed screening via cross-linking and, in contrast to DCR, pDCC and SAS, is broadly applicable to every type of protein and enzyme.
Dynamic combinatorial resolution (DCR) and pseudo-dynamic combinatorial chemistry (pDCC)
Both DCR and pDCC rely on the concept of DCC. A dynamic combinatorial library (An–Bm) is generated and incubated with an enzyme that catalyzes an irreversible reaction of the library members (An–Bm) to Cnm with a certain degree of selectivity (Figure 4a). The selection process can be performed using enzymes that either catalyze bond formation (in DCR) (Figure 4b) or cleavage (in pDCC) (Figure 4c).
Figure 4.
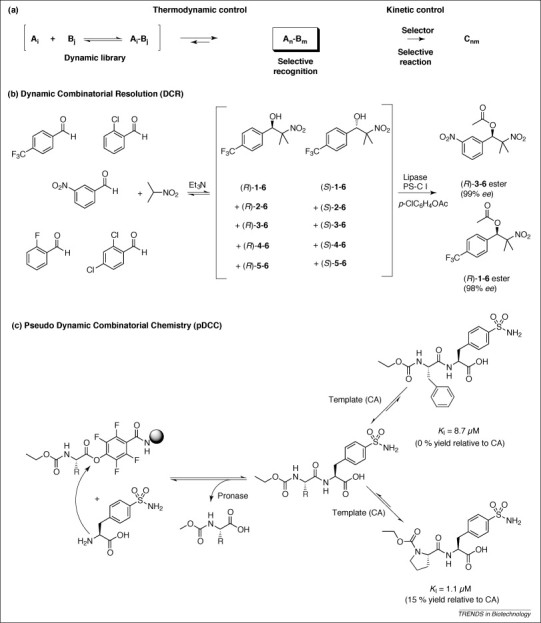
Dynamic combinatorial resolution (DCR) and pseudo-dynamic combinatorial chemistry (pDCC). (a) Both methods rely on a dynamically formed library of products that are further converted in a selective reaction with template-derived specificity. (b) In an example of DCR, a dynamic nitroaldol library was formed by Henry reaction for five aldehydes and one nitroalkane component. Selective recognition of one of the secondary alcohols formed by the selector lipase PS-C I led to enantioselective formation of two ester products [29]. (c) In pDCC, a library of dipeptides was formed from resin-bound active esters (left). All dipeptides could be cleaved by the non-selective peptidase pronase. Only dipeptides binding to the carbonic anhydrase protein template were partially protected against hydrolysis and were amplified in an iterative process [32].
Inspired by dynamic resolution, a method for separation of a racemic mixture [41], Ramström's group introduced the concept of DCR 29, 30, 31. The proof of principle for this approach was provided by a nitroaldol reaction (the so-called Henry reaction), in which β-nitro alcohols were reversibly formed by addition of nitroalkanes to carbonyl compounds. On addition of the selector, in this case the lipase PS-C I from Pseudomonas cepacia, library members that could fit into the active site of the lipase were acylated (Figure 4b) and subsequently identified by 1H-NMR [29].
pDCC was developed by the Kazlauskas group 32, 34. They replaced the equilibrium used for selection in DCC by combining an irreversible reaction used for library formation with enzymatic cleavage of library members. Here, the selection protein template protects better-binding fragment combinations from enzymatic cleavage, whereas unbound compounds are rapidly removed from the mixture. The concept was successfully demonstrated for a carbonic anhydrase template using a synthetic dipeptide library (Figure 4c). After incubation with the protein template, the non-specific protease pronase from Streptomyces griseus was added to hydrolyze all unbound dipeptides. As expected, the cleavage reaction was strongly inhibited for the best binder compared to weaker binders, allowing detection by NMR.
In both approaches, amplification correlated directly with the binding affinity of the compounds identified. However, as above, one major disadvantage is the difficulty in detecting the best fragment combinations by NMR, which limits the possible library size. Another limitation is the restricted applicability, which is confined to chemical libraries for which a cleavage reaction can be established. For example, in the case of DCR, the choice of selector templates is confined to relatively few enzymes such as lipases, preventing broader applicability of this particular approach for drug discovery. However, only catalytic amounts of the selector protein are required, which constitutes a clear advantage over DCC. Similar to DCR, pDCC is also confined to specific chemicals. Dipeptides, as demonstrated here, are ideal for establishing the concept; however, they are not very likely drug leads. Thus, in summary these approaches, although conceptually interesting, are less likely to be widely used in drug lead discovery.
Target-guided synthesis (TGS)
A logical and straightforward extension of template-assisted shifts in chemical equilibria is the direct use of protein targets as either templates or catalysts in irreversible chemical reactions, a concept termed TGS 31, 50. In TGS, the protein template binds two reagents that are in close proximity, thus accelerating their chemical reaction, usually independently of the actual protein function.
Several examples of TGS have been demonstrated 36, 37, 38. The most widely used reaction for TGS is the so-called in situ click chemistry 39, 40, 51. Click reactions of azides with alkines regioselectively yield 1,4-disubstituted triazoles by copper(I) catalysis [52]. In the presence of a suitable protein template, however, a well-bound azide and alkine can react in the absence of the copper salt. For example, in the case of HIV protease, an alkine (IC50 >100 μM) and an azide (IC50 4.2 μM) were incubated in the presence of the enzyme for 24 h. HPLC analysis revealed that the triazole was formed, which is an inhibitor of the wild-type HIV1 protease (IC50 6 nM) [40]. Because of the growing importance of triazoles in drug discovery [52], in situ click chemistry could become a powerful tool in lead discovery.
In principle, TGS is suitable for the development of highly active molecules. For broader applicability, however, some issues have to be addressed. For template-assisted reaction and detection of the desired products by HPLC, NMR, or mass spectrometry, they need to be present in relatively high concentrations (≥100 μM). Second, the need for a near-stoichiometric amounts of the template protein limits the feasibility of TGS. We anticipate that the set of reactions for which TGS is particularly suited will not be fully explored until further research has been carried out.
Substrate activity screening (SAS)
SAS was introduced by the Ellman group and uses substrate libraries that are screened in standard enzyme assays to determine the best binding fragments [41]. Initially, this method was demonstrated for proteases. In this particular application, a fluorophore was coupled to a variety of low-molecular-weight fragments, resulting in a so-called fluorogenic substrate library of ∼100 members that was screened using several proteases (such as cathepsin S shown in Figure 5a) to identify the best protease substrates via cleavage of the amide bond, which released the fluorescent dye. Here, high substrate turnover indicated high affinity of the respective substrate fragments. In the case of cathepsin S, a non-peptidic substrate could be identified. The best substrate identified in this reaction was then converted into an inhibitor by introducing an aldehyde at the C-terminus, providing a non-peptidic nanomolar inhibitor of cathepsin S [42].
Figure 5.

Overview of the first applications of substrate activity screening (SAS). The reaction schemes for (a) cathepsin S and (b) Mycobacterium tuberculosis protein tyrosine phosphatase B (MptpB) are shown 42, 43.
SAS was also used to identify protein tyrosine phosphatase inhibitors (Figure 5b). A substrate library of O-aryl phosphates was prepared and screened with Mycobacterium tuberculosis protein tyrosine phosphatase B (MptpB), a secreted virulence factor and potential target against tuberculosis (TB). A biphenyl scaffold could be identified as a potential lead fragment. The phosphate group in the molecule was then replaced by a phosphate mimetic and, following optimization, a potent and selective MptpB inhibitor was obtained (Figure 5b) [53].
Several molecules acting on therapeutically relevant enzymes have already been identified by SAS 42, 53. The strength of this method is its ability to efficiently identify substrate structures that bind to the active site. The use of protein in only catalytic amounts contributes to its efficiency. However, SAS requires the catalytic function of enzymatic drug targets, such as proteases or protein tyrosine phosphatases, and therefore cannot be applied more generally to other types of drug targets. The approach is further restricted by the limited availability of substrate libraries, which are not commercially available and have to be synthesized on a case-by-case basis. Moreover, the classical rationale of fragment-based drug discovery, i.e. to connect several low-affinity binders for specific pockets, is not applied in SAS. Another drawback is that the substrates discovered have to be converted into useful inhibitors. For example, in the case of cathepsin S, the substrate fluorophore was replaced with an aliphatic aldehyde, which is less suitable in a drug. Similarly, for phosphatase inhibitors, replacement of the phosphate by a mimetic such as isothiazolidine or isoxazole carboxylic acid [53] resulted in adverse effects, such as lower selectivity and cell permeability.
Dynamic ligation screening (DLS)
Considering the limitations of different dynamic fragment-based methods for drug discovery, DLS was developed as an approach that combines dynamic target-assisted formation of inhibitory species with detection via a biochemical assay [44]. For example, an enzyme reaction can be used for amplified detection of the fragment, thus drastically reducing the amount of protein required and allowing HTS (Figure 6 ). In addition, the use of chemically reactive protein ligands that bind to a defined pocket on the protein surface makes it possible to test for inhibitors fragments acting at the pocket of interest.
Figure 6.
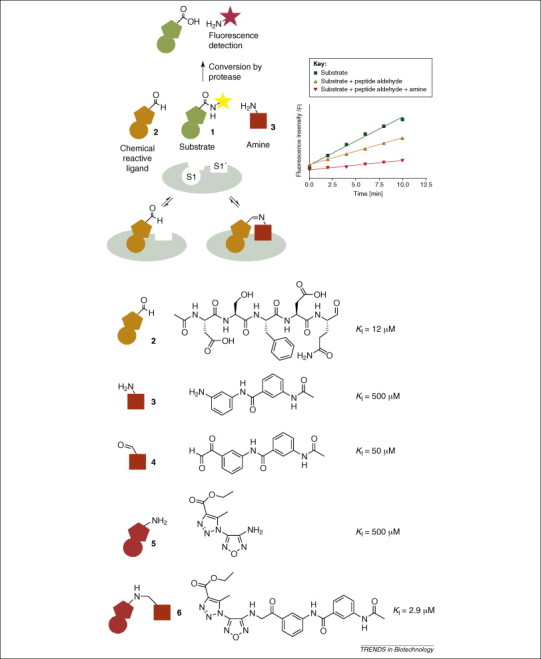
Concept of dynamic ligation screening (DLS) illustrated by the development of a non-peptidic SARS-CoV Mpro inhibitor. The substrate 1 (shown in green) competes with a peptide aldehyde inhibitor 2 (orange) targeting the S1 pocket of the SARS-CoV main protease (gray). An active amine fragment 3 (red) binds to the S1′ pocket, leading to increased inhibition via dynamic ligation in the active protein site [44]. Based on the active amine fragment 3 (red), an analogous electrophilic aldehyde fragment 4 (red) that also binds to S1′ was synthesized and used for DLS of the S1 pocket, yielding active fragment 5 as a hit. Finally, both hit fragments were chemically linked to yield the non-peptide inhibitor 6 with a KI value of 2.9 μM [44].
The DLS approach was first demonstrated for the main protease of SARS coronavirus (SARS-CoV Mpro) as the protein target. For site-directed identification of inhibitory fragments, an activity assay for SARS-CoV Mpro was first developed using a peptidic substrate (Figure 6). Enzymatic cleavage of the peptidic substrate released a fluorophore, which served as an indicator for protease activity. The chemically reactive protein ligand, a peptide aldehyde 45, 54, was then incubated with an excess of a nucleophilic fragment in the presence of the enzyme. Following addition of the fluorogenic substrate, rate differences in substrate turnover were quantified to identify active inhibitory fragments. For SARS-CoV Mpro, one fragment was identified that showed considerably stronger inhibition in presence of the peptide aldehyde than the peptide aldehyde inhibitor alone. The fragment alone showed no significant activity against the SARS enzyme. To verify the result, the fragment identified was converted by chemical synthesis into an electrophile, which was shown to be an active inhibitor of SARS-CoV Mpro and was used as a new chemically reactive protein ligand. Thus, the DLS approach was repeated with the newly identified electrophile to identify a new nucleophile fragment. Covalent linkage of the new electrophile to the fragment identified yielded an active, non-peptidic SARS-CoV Mpro inhibitor with a K I value of 2.9 μM (Figure 6).
In conclusion, DLS provides site-directed detection of low-affinity fragments. The sensitivity of the method is therefore higher than for conventional and other dynamic fragment-based approaches. The method can be applied in a high-throughput format. No additional equipment besides a standard microtiter plate reader is needed. Most importantly, DLS can be operated iteratively in an evolutionary process and has been shown to succeed in transforming a moderately active peptidic inhibitor into an entirely non-peptidic inhibitor [44]. Although successful DLS has only been demonstrated for the development of protease inhibitors to date 43, 44, DLS assays could easily be extended to other proteases and to other enzyme classes, as well as to any type of protein–protein interaction, so that standard biochemical assays, including binding assays, can easily be adapted to the particular strategy required [43].
Outlook
Methodological innovations in the drug discovery process are required to alleviate the dwindling success of current drug development efforts in the pharmaceutical industry. Recent advances in dynamic fragment-based screening methods have the potential to contribute significantly to this process because they combine the power of high-throughput screening with the advantages of fragment-derived hits. With the development of dynamic ligation screening approaches, fragment-based drug discovery has become more competitive with traditional methods because only catalytic amounts of the target protein are required and standard HTS methods can be adapted. Considering recent breakthroughs in the field, dynamic template-assisted strategies are now facing the next challenge: it must be demonstrated that they can significantly support the development of an approved drug. It is our firm belief that successful application of these approaches will yield positive results in the near future.
Acknowledgements
We thank Dr. Samuel Beligny, Markus Matthaei and Jörn Saupe for helpful feedback and comments on this manuscript. J.R. thanks the Fonds der Chemischen Industrie for continuous support.
Footnotes
PricewaterhouseCoopers. Pharma 2020: The vision, which path will you take? (2007) http://www.pwc.com/extweb/pwcpublications.nsf/docid/91BF330647FFA402852572F2005ECC22
References
- 1.Schreiber S.L. Small molecules: the missing link in the central dogma. Nat. Chem. Biol. 2005;1:64–66. doi: 10.1038/nchembio0705-64. [DOI] [PubMed] [Google Scholar]
- 2.Drews J. Strategic trends in the drug industry. Drug Discov. Today. 2003;8:411–420. doi: 10.1016/s1359-6446(03)02690-4. [DOI] [PubMed] [Google Scholar]
- 3.Drews J. Innovation deficit revisited: reflections on the productivity of pharmaceutical R&D. Drug Discov. Today. 1998;3:491–494. [Google Scholar]
- 4.Drews J., Ryser S. The role of innovation in drug development. Nat. Biotechnol. 1997;15:1318–1319. doi: 10.1038/nbt1297-1318. [DOI] [PubMed] [Google Scholar]
- 5.Brown D. Future pathways for combinatorial chemistry. Mol. Divers. 1996;2:217–222. doi: 10.1007/BF01715637. [DOI] [PubMed] [Google Scholar]
- 6.Jencks W.P. On the attribution and additivity of binding energies. Proc. Natl. Acad. Sci. U. S. A. 1981;78:4046–4050. doi: 10.1073/pnas.78.7.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fink T. Virtual exploration of the small-molecule chemical universe below 160 daltons. Angew. Chem. Int. Ed. 2005;44:1504–1508. doi: 10.1002/anie.200462457. [DOI] [PubMed] [Google Scholar]
- 8.Bohacek R.S. The art and practice of structure-based drug design: a molecular modeling perspective. Med. Res. Rev. 1996;16:3–50. doi: 10.1002/(SICI)1098-1128(199601)16:1<3::AID-MED1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 9.Hann M.M., Opera T.I. Pursuing the lead likeness concept in pharmaceutical research. Curr. Opin. Chem. Biol. 2004;8:255–263. doi: 10.1016/j.cbpa.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 10.Andrews P.R. Functional group contributions to drug–receptor interactions. J. Med. Chem. 1984;27:1648–1657. doi: 10.1021/jm00378a021. [DOI] [PubMed] [Google Scholar]
- 11.Hopkins A.L. Ligand efficiency: a useful metric for lead selection. Drug Discov. Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 12.Kuntz I.D. The maximal affinity of ligands. Proc. Natl. Acad. Sci. U. S. A. 1999;96:9997–10002. doi: 10.1073/pnas.96.18.9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hajduk P.J., Greer J. A decade of fragment-based drug design: strategic advances and lessons learned. Nat. Rev. Drug Discov. 2007;6:211–219. doi: 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]
- 14.Shuker S.B. Discovery high-affinity ligands for proteins: SAR by NMR. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 15.Nienaber V.L. Discovering novel ligands for macromolecules using X-ray crystallographic screening. Nat. Biotechnol. 2000;18:1105–1108. doi: 10.1038/80319. [DOI] [PubMed] [Google Scholar]
- 16.Blundell T.L. High-throughput crystallography for lead discovery in drug design. Nat. Rev. Drug Discov. 2002;1:45–54. doi: 10.1038/nrd706. [DOI] [PubMed] [Google Scholar]
- 17.Blundell T.L., Patel S. High-throughput X-ray crystallography for drug discovery. Curr. Opin. Pharmacol. 2004;4:490–496. doi: 10.1016/j.coph.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 18.Neumann T. SPR-based fragment screening: advantages and applications. Curr. Top. Med. Chem. 2007;7:1630–1642. doi: 10.2174/156802607782341073. [DOI] [PubMed] [Google Scholar]
- 19.Freire E. Isothermal titration calorimetry: controlling binding forces in lead optimization. Drug Discov. Today Technol. 2004;1:295–299. doi: 10.1016/j.ddtec.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 20.Ciulli A. Inhibition of Mycobacterium tuberculosis pantothenate synthetase by analogues of the reaction intermediate. ChemBioChem. 2008;9:2606–2611. doi: 10.1002/cbic.200800437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whitty A. Cooperativity and biological complexity. Nat. Chem. Biol. 2008;4:435–439. doi: 10.1038/nchembio0808-435. [DOI] [PubMed] [Google Scholar]
- 22.Huc I., Lehn J-M. Virtual combinatorial libraries: dynamic generation of molecular and supramolecular diversity by self-assembly. Proc. Natl. Acad. Sci. U. S. A. 1997;94:2106–2110. doi: 10.1073/pnas.94.6.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lehn J-M. Dynamic combinatorial chemistry and virtual libraries. Chem. Eur. J. 1999;5:2455–2463. [Google Scholar]
- 24.Corbett P.T. Dynamic combinatorial chemistry. Chem. Rev. 2006;106:3652–3711. doi: 10.1021/cr020452p. [DOI] [PubMed] [Google Scholar]
- 25.Erlanson D.A. Tethering: fragment-based drug discovery. Annu. Rev. Biophys. Biomol. Struct. 2004;33:199–223. doi: 10.1146/annurev.biophys.33.110502.140409. [DOI] [PubMed] [Google Scholar]
- 26.Erlanson D.A. Site-directed ligand discovery. Proc. Natl. Acad. Sci. U. S. A. 2000;97:9367–9372. doi: 10.1073/pnas.97.17.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Erlanson D.A. In situ assembly of enzyme inhibitors using extended tethering. Nat. Biotechnol. 2003;21:308–314. doi: 10.1038/nbt786. [DOI] [PubMed] [Google Scholar]
- 28.Cancilla M.T. Discovery of an Aurora kinase inhibitor through site-specific dynamic combinatorial chemistry. Bioorg. Med. Chem. Lett. 2008;18:3978–3981. doi: 10.1016/j.bmcl.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 29.Vongvilai P. Dynamic combinatorial resolution: direct asymmetric lipase-mediated screening of a dynamic nitroaldol library. Angew. Chem. Int. Ed. 2007;46:948–950. doi: 10.1002/anie.200603740. [DOI] [PubMed] [Google Scholar]
- 30.Angelin M. Tandem driven dynamic combinatorial resolution via Henry-iminolactone rearrangement. Chem. Commun. 2008:768–770. doi: 10.1039/b716521h. [DOI] [PubMed] [Google Scholar]
- 31.Maly D.J. Combinatorial target-guided ligand assembly: identification of potent subtype-selective c-Src inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2000;97:2419–2424. doi: 10.1073/pnas.97.6.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Corbett A.D. Pseudodynamic combinatorial libraries: a receptor-assisted approach for drug discovery. Angew. Chem. Int. Ed. 2004;43:2432–2436. doi: 10.1002/anie.200453769. [DOI] [PubMed] [Google Scholar]
- 33.Cheeseman J.D. Amplification of screening sensitivity through selective destruction: theory and screening of a library of carbonic anhydrase inhibitors. J. Am. Chem. Soc. 2002;124:5693–5701. doi: 10.1021/ja017099+. [DOI] [PubMed] [Google Scholar]
- 34.Corbett A.D., Gleason J.L. Preparation of active esters on solid support for aqueous-phase peptide coupling. Tetrahedron Lett. 2002;43:1369–1372. [Google Scholar]
- 35.Angelin M. Crystallization-induced secondary selection from a tandem driven dynamic combinatorial resolution process. J. Org. Chem. 2008;73:3593–3595. doi: 10.1021/jo8002453. [DOI] [PubMed] [Google Scholar]
- 36.Nicolaou K.C. Target-accelerated combinatorial synthesis and discovery of highly potent antibiotics effective against vancomycin-resistant bacteria. Angew. Chem. Int. Ed. 2000;39:3823–3828. doi: 10.1002/1521-3773(20001103)39:21<3823::AID-ANIE3823>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 37.Hu X. Bcl-XL-templated assembly of its own protein–protein interaction modulator from fragments decorated with thio acids and sulfonyl azides. J. Am. Chem. Soc. 2008;130:13820–13821. doi: 10.1021/ja802683u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nguyen R., Huc I. Using an enzyme's active site to template inhibitors. Angew. Chem. Int. Ed. 2001;40:1774–1776. [PubMed] [Google Scholar]
- 39.Bourne Y. Freeze-frame inhibitor captures acetylcholinesterase in a unique conformation. Proc. Natl. Acad. Sci. U. S. A. 2003;101:1449–1454. doi: 10.1073/pnas.0308206100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Whiting M. Inhibitors of HIV-1 protease by using in situ click chemistry. Angew. Chem. Int. Ed. 2006;45:1435–1439. doi: 10.1002/anie.200502161. [DOI] [PubMed] [Google Scholar]
- 41.Patterson A.W. Substrate activity screening (SAS): a general procedure for the preparation and screening of a fragment-based non-peptidic protease substrate library for inhibitor discovery. Nat. Protoc. 2007;2:424–433. doi: 10.1038/nprot.2007.28. [DOI] [PubMed] [Google Scholar]
- 42.Wood W.J. Substrate activity screening: a fragment-based method for the rapid identification of non peptidic proteasae inhibitors. J. Am. Chem. Soc. 2005;127:15521–15527. doi: 10.1021/ja0547230. [DOI] [PubMed] [Google Scholar]
- 43.Schmidt, M.F. et al. (2009) Selective identification of cooperatively binding fragments in a high-throughput ligation assay enables development of a picomolar caspase-3 inhibitor. Angew. Chem. Int. Ed. 48, July 23 [Epub ahead of print] [DOI] [PubMed]
- 44.Schmidt M.F. Sensitized detection of inhibitory fragments and iterative development of non-peptidic protease inhibitors by dynamic ligation screening. Angew. Chem. Int. Ed. 2008;47:3275–3278. doi: 10.1002/anie.200704594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Al-Gharabli S.I. An efficient method for the synthesis of peptide aldehyde libraries employed in the discovery of reversible SARS coronavirus main protease (SARS-CoV Mpro) inhibitors. ChemBioChem. 2006;7:1048–1055. doi: 10.1002/cbic.200500533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hochgürtel M. Ketones as building blocks for dynamic combinatorial libraries: highly active neuramidase inhibitors generated via selection pressure of the biological target. J. Med. Chem. 2003;46:356–358. doi: 10.1021/jm025589m. [DOI] [PubMed] [Google Scholar]
- 47.Congreve M.S. Detection of ligands from a dynamic combinatorial library by X-ray crystallography. Angew. Chem. Int. Ed. 2003;42:4479–4482. doi: 10.1002/anie.200351951. [DOI] [PubMed] [Google Scholar]
- 48.Ludlow R.F., Otto S. Two-vial. LC-MS identification of ephedrine receptors from a solution-phase dynamic combinatorial library of over 9000 compounds. J. Am. Chem. Soc. 2008;130:12218–12219. doi: 10.1021/ja803317k. [DOI] [PubMed] [Google Scholar]
- 49.Ramström O., Lehn J.-M. In situ generation and screening of a dynamic combinatorial carbohydrate library against Concanavalin A. ChemBioChem. 2000;1:41–48. doi: 10.1002/1439-7633(20000703)1:1<41::AID-CBIC41>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 50.Inglese J., Benkovic S.J. Multisubstrate adduct inhibitors of glycinamide ribonucleotide transformylase: synthetic and enzyme-assembled. Tetrahedron. 1991;47:2351–2364. [Google Scholar]
- 51.Lewis W.G. Click chemistry in situ: acetylcholinesterase as a reaction vessel for the selective assembly of a femtomolar inhibitor from an array of building blocks. Angew. Chem. Int. Ed. 2002;41:1053–1057. doi: 10.1002/1521-3773(20020315)41:6<1053::aid-anie1053>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 52.Kolb H.C. Click chemistry: diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 53.Soellner M.B. Fragment-based substrate activity screening method for the identification of potent inhibitors of the Mycobacterium tuberculosis phosphatase PtpB. J. Am. Chem. Soc. 2007;126:9613–9615. doi: 10.1021/ja0727520. [DOI] [PubMed] [Google Scholar]
- 54.El-Dashan A. C-Acylation of polymeric phosphoranylidene acetates for C-terminal variation of peptide carboxylic acids. Org Lett. 2007;15:949–952. doi: 10.1021/ol062754+. [DOI] [PubMed] [Google Scholar]
- 55.Hochgürtel M. Target-induced formation of neuramidase inhibitors from in vitro virtual combinatorial libraries. Proc. Natl. Acad. Sci. U. S. A. 2002;99:3382–3387. doi: 10.1073/pnas.052703799. [DOI] [PMC free article] [PubMed] [Google Scholar]