

Abstract
Purpose:
Adrenocortical carcinoma (ACC) is a rare, aggressive malignancy with few therapies; however, patients with locoregional disease have variable outcomes. The Cancer Genome Atlas project on ACC (ACC-TCGA) identified that cancers of patients with homogeneously rapidly recurrent or fatal disease bear a unique CpG island hypermethylation phenotype, “CIMP-high.” We sought to identify a biomarker that faithfully captures this subgroup.
Experimental Design:
We analyzed ACC-TCGA data to characterize differentially regulated biological processes, and identify a biomarker that is methylated and silenced exclusively in CIMP-high ACC. In an independent cohort of 114 adrenocortical tumors (80 treatment-naive primary ACC, 22 adrenocortical adenomas, and 12 non-naive/non-primary ACC), we evaluated biomarker methylation by a restriction digest/qPCR-based approach, validated by targeted bisulfite sequencing. We evaluated expression of this biomarker and additional prognostic markers by qPCR.
Results:
We show that CIMP-high ACC is characterized by upregulation of cell cycle and DNA damage response programs, and identify that hypermethylation and silencing of G0S2 distinguishes this subgroup. We confirmed G0S2 hypermethylation and silencing is exclusive to 40% of ACC, and independently predicts shorter disease-free and overall survival (median 14 and 17 months, respectively). Finally, G0S2 methylation combined with validated molecular markers (BUB1B-PINK1) stratifies ACC into three groups, with uniformly favorable, intermediate, and uniformly dismal outcomes.
Conclusions:
G0S2 hypermethylation is a hallmark of rapidly recurrent or fatal ACC, amenable to targeted assessment using routine molecular diagnostics. Assessing G0S2 methylation is straightforward, feasible for clinical decision-making, and will enable the direction of efficacious adjuvant therapies for patients with aggressive ACC.
Keywords: adrenocortical carcinoma, DNA methylation, CpG island methylator phenotype, biomarker, epigenetics
Introduction
Adrenocortical carcinoma (ACC) is a rare cancer of the adrenal cortex affecting 0.5–2 individuals/million/year globally (1,2). Though rare, ACC is frequently aggressive with 35% 5-year survival (3). Therapies for metastatic ACC are primarily palliative, limited to administration of adrenolytic drug mitotane and/or cytotoxic chemotherapy (3). Patients with locoregional ACC routinely receive surgery and adjuvant mitotane, but 50–70% recur and develop metastases even after complete (R0) resection (4,5). Retrospective studies suggest adjuvant mitotane prolongs recurrence free survival (6,7), but its efficacy is limited by its poor pharmacokinetic properties and dose-limiting toxicities. Obtaining therapeutic serum levels of mitotane may take several months to achieve if at all (8). Furthermore, there is a substantial proportion of ACC patients who experience rapid recurrence (<12 months) (4,5,9), whose aggressive disease course may preclude response to mitotane. These patients may instead benefit from more rapidly acting therapies; however, prospectively identifying this subgroup remains challenging.
Histological grade based on cellular proliferation is the strongest predictor of recurrence following R0 resection in ACC; high grade disease is associated with higher risk of recurrence (10–12). Despite its clinical utility, significant caveats complicate interpretation of grade on an individual basis. Evaluation of grade is prone to high inter-rater variability (13), and outcomes of patients with low and high grade disease remain heterogeneous, with rapidly recurrent patients in both strata (10,14). While some studies indicate clinical factors may be predictive of recurrence (9,15), molecular profiling studies suggest biomarkers may better resolve this heterogeneity by identifying patients with homogeneously dismal outcomes.
We and others have shown that transcriptomes of aggressive ACC are characterized by pronounced cell cycle activation (16), and a score based on mRNA levels of mitotic regulator BUB1B (BUB1 Mitotic Checkpoint Serine/Threonine Kinase B) and mitochondrial kinase PINK1 (PTEN Induced Putative Kinase 1) discriminates uniformly favorable from poor clinical outcomes (17,18). Recent studies have implicated aberrant epigenetic patterning in ACC pathogenesis, highlighting that aggressive carcinomas bear widespread promoter CpG island hypermethylation (19,20). Notably, the most comprehensive molecular study on ACC to date, The Cancer Genome Atlas project on ACC (ACC-TCGA), similarly identified that rapidly recurrent ACC is distinguished by a CpG island hypermethylation phenotype, “CIMP-high” (21).
While these studies have illuminated molecular programs core to aggressive ACC biology, clinical translation of “big data”-derived biomarkers remains challenging. Thresholds for continuous data, e.g. mRNA-based biomarkers, vary across patient cohorts (17,18), compromising biomarker utility for prospective clinical management of a rare malignancy. Furthermore, while targeted assessment of DNA methylation appears promising for prognosticating ACC (20,22), measurement strategies frequently rely on several genomic loci, complicated data normalization procedures, and reference benign lesions (22). Finally, it remains unclear if validated biomarkers identify uniform ACC molecular subtypes amenable to clinical assessment of subtype-specific therapeutic approaches. It is therefore not surprising that mRNA- and DNA methylation-based biomarkers have yet to be successfully translated clinically to prognosticate ACC, and highlights a strong need for identifying novel biomarkers with simplified, binary readouts and therapeutic import.
Here, we present a new analysis of ACC-TCGA data in which we demonstrate that CIMP-high ACC is a unique, rapidly recurrent ACC molecular subtype, bearing upregulation of cell cycle- and DNA damage-associated cellular programs. We identify that uniform hypermethylation and silencing of the gene G0S2 (G0/G1 Switch 2) is largely exclusive to CIMP-high ACC. We show in an independent cohort that targeted assessment of G0S2 methylation using an overnight assay independently identifies a subgroup of ACC patients with rapidly progressive or fatal disease course. Our data demonstrates that G0S2 methylation status is essentially binary, and thereby has high potential to enable clinicians to prospectively identify ACC patients unlikely to exhibit durable response to standard adjuvant therapy. We also propose that rapidly acting adjuvant cytotoxic agents may benefit patients with this ACC subtype. Finally, our study demonstrates the utility of comprehensive databases like TCGA, and illustrates a pipeline to identify and test clinically relevant biomarkers for ACC that may be extended to other cancers.
Materials and Methods
Data mining from ACC-TCGA.
We downloaded the ACC-TCGA RNA-seq count table and raw data (IDAT files) from the Infinium HumanMethylation450 BeadChip (“450k”) platform from the GDC legacy archive (https://portal.gdc.cancer.gov/legacy-archive). We used R (23)/Bioconductor packages limma (24) and minfi (25) to obtain log2-normalized counts per million (CPM) values for gene expression and beta and M values for methylation arrays. We used limma to nominate differentially expressed genes (Benjamini-Hochberg FDR-corrected p-value<0.05) between CIMP-high and non CIMP-high ACC. We used goana (24,26) to identify gene ontology terms enriched among differentially expressed genes in CIMP-high vs. non-CIMP-high ACC. REVIGO (27) is an online tool that enables non-redundant visualization of large sets of GO terms based on semantic similarity. We used REVIGO with SimRel semantic similarity algorithm to plot the 200 most significant biological processes up (ranked by increasing P.Up, P.Up<0.05) or down (ranked by increasing P.Down, P.Down<0.05) in Figure 1C. We used DMRcate (28) to interrogate differentially methylated regions (Stouffer-corrected p-value<0.05) across groups. We used logistic regression on the RNA-seq data to identify transcripts predictive of CIMP-high status. We used pheatmap (29) to perform unsupervised complete hierarchical clustering, and caret (30) to perform k-fold cross validation.
Figure 1.

ACC-TCGA demonstrates that rapidly recurrent, CIMP-high carcinomas are a unique molecular subtype associated with upregulation of pharmacologically targetable cell cycle- and DNA damage-associated processes. A. ACC-TCGA reveals that disease-free survival (DFS) of ACC patients with locoregional disease following R0/RX resection can be stratified by CpG island methylator phenotype (“CIMP”) status. Patients with CIMP-high carcinoma status have dismal outcomes, with median DFS of 13.6 months compared to failure to achieve median DFS in CIMP-low + CIMP-intermediate group. B. ACC-TCGA reveals that CIMP-high ACC is a unique molecular subtype, associated with “Steroid-high + prolif” transcriptional program (mRNA group), “Noisy” chromosomal landscape with frequent focal copy number gains and losses (SCNA group), and higher incidence of somatic alterations leading to constitutive cell cycle activation. Notably, CIMP-high ACC is not associated with an increased incidence of somatic alterations leading to activation of the Wnt pathway. C. Gene ontology (GO) analysis of 3,558 differentially expressed genes (1688 up, 1870 down; Benjamini-Hochberg FDR-corrected p-value < 0.05) in CIMP-high vs. CIMP-low + CIMP-intermediate (“non-CIMP-high”) ACC reveals that CIMP-high carcinomas bear transcriptional activation of several pharmacologically targetable cell cycle- and DNA damage-associated biological processes (left panel). Interestingly, several immunological processes are concordantly downregulated in CIMP-high ACC (right panel). 200 most significant biological processes up (left panel) or down (right panel) were plotted using REVIGO. Circle size indicates set size and color indicates p-value according to legend right; axes units are arbitrary, but smaller distance between circles reflects higher degree of semantic similarity (measured by simRel score) between sets.
Patients.
Our study includes 114 adrenocortical tumors evaluated from 1989–2017. 42 treatment-naive primary ACC, 1 primary ACC from a patient who received neoadjuvant etoposide/doxorubicin/cisplatin+mitotane, 3 non-primary ACC, and 14 cortisol-secreting adrenocortical adenomas (ACA) are from Faculdade de Medicina da Universidade de São Paulo (FMUSP), São Paulo, Brazil; 38 primary ACC, 8 non-primary ACC, 4 aldosterone-secreting ACA, and 4 cortisol-secreting ACA are from the University of Michigan (UM), Ann Arbor, MI, USA. Diagnosis of ACA/ACC was established by expert pathological assessment (M.C.N.Z., T.J.G.) of surgical specimen using Weiss criteria (12). Diagnosis of ACA or ACC was assigned to samples with Weiss score <3 or ≥3, respectively. Informed consent was obtained from all participants, and studies were conducted in accordance with the Declaration of Helsinki with study protocols approved by FMUSP and UM Institutional Review Boards. Clinical, hormonal, and demographic data were collected retrospectively.
Tissue processing, nucleic acid extraction, and quantification of mRNA expression.
FMUSP:
Immediately following surgical resection, samples were collected by an institutional pathologist and snap frozen in liquid nitrogen. Frozen tumor tissue was cryotome sectioned (6 μm) under RNase-free conditions to acquire ≥30 mg tissue per sample. ≥3 random non-contiguous sections from each tumor were prepared for rapid hematoxylin and eosin (H&E) staining to evaluate sample quality and tumor purity. Samples with >50% acellular material in >2/3 sections were excluded from downstream processing. Slides from samples included in downstream processing were assessed by T.J.G. to confirm typical ACC histology. Genomic DNA (gDNA) and total RNA were simultaneously extracted with AllPrep DNA/RNA Mini Kit (Qiagen, USA; 80204) and optional on-column RNase A (Qiagen, USA; 19101) and DNase I (RNase-free DNase Set, Qiagen, USA; 79254) digests, respectively.
UM:
Samples collected immediately following surgical resection were snap frozen in liquid nitrogen, embedded in OCT freezing media (Miles Scientific, USA), cryotome sectioned (5 μm), and evaluated by routine H&E by surgical pathologists. When possible, corresponding H&E sections from paraffin blocks were also evaluated. Areas of pure tumor (≥70% tumor cells) were selected for nucleic acid extraction. gDNA and RNA were extracted using one of the following methods: TRIzol (Invitrogen/Thermo Fisher Scientific, USA; 15596026) with acid-phenol:chloroform cleanup, RNeasy Mini Kit (Qiagen, USA; 74104) or DNeasy Blood and Tissue Kit (Qiagen, USA; 69504), or AllPrep DNA/RNA/Protein Mini Kit (Qiagen, USA; 80004).
For all samples, RNA integrity was evaluated by agarose gel electrophoresis; purity (260/280, 260/230 ratios) and quantity of gDNA and RNA were measured by spectrophotometry (NanoDrop 2000 Spectrophotometer, Thermo Fisher Scientific, USA; Cat. No. ND-2000). cDNA was synthesized (High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor, Applied Biosciences/Thermo Fisher Scientific, USA; 4374966) from high integrity and high quality RNA (visual 28S:18S rRNA ratio 2:1 and 260/280 ratio≥2.00). Quantitative real-time polymerase chain reaction (qPCR) was performed in the QuantStudio 3 Real-Time PCR System (Applied Biosciences/Thermo Fisher Scientific, USA; A28136), using TaqMan Fast Advanced Master Mix (Applied Biosciences/Thermo Fisher Scientific, USA; 4444557) and FAM-MGB-labeled TaqMan Gene Expression Assays (Applied Biosciences/Thermo Fisher Scientific, USA) to evaluate expression of G0S2 (Hs00274783_s1), BUB1B (Hs01084828_m1), PINK1 (Hs00260868_m1), and housekeeping gene GUSB (Hs00939627_m1). TaqMan Gene Expression Assays were performed in triplicate. Gene expression levels were calculated using the ΔCt method where ΔCt(X)=Ct(X)–Ct(GUSB), and BUB1B-PINK1 score calculated as ΔCt(BUB1B)–ΔCt(PINK1).
Measurement of G0S2 methylation.
Targeted bisulfite sequencing:
Assessment of G0S2 methylation by targeted bisulfite sequencing in physiological tissues, ACA, and ACC was performed by Zymo Research Corporation, Irvine, CA, USA. Zymo Research Co. designed/validated primers to amplify the G0S2 locus, chr1:209,848,443-chr1:209,848,900 (hg19), using a proprietary pipeline. Submitted gDNA with 260/280≥1.7, intact genomic band (≥5 kb) by gel electrophoresis, and sufficient quantity (≥100 ng) was subject to bisulfite conversion, targeted amplification, next generation sequencing library indexing, and sequencing on Illumina MiSeq. Sequence data was demultiplexed and assessed for bisulfite conversion rate, read coverage, mapping efficiency, and CpG coverage. Bisulfite conversion rate was ≥99% for all samples. Average CpG coverage ranged from 5,000–50,000X. Methylation at each CpG was calculated from the ratio of methylated to total CpG count.
Methylation-sensitive restriction digest/qPCR:
Available gDNA from ACC and ACA was subject to methylation-sensitive restriction digestion using EpiTect II DNA Methylation Enzyme Kit (Qiagen, USA; Cat. No. 335452). This kit contains two enzymes: Methylation Sensitive Enzyme A (cannot cleave gDNA in the presence of CpG methylation in the proprietary restriction site) and Methylation Dependent Enzyme B (can cleave gDNA only in the presence of CpG methylation in the proprietary restriction site). Per manufacturer protocol, gDNA from each tumor was subject to four digests: “mock” digest (Mo, containing no restriction enzymes), methylation-sensitive digest (Ms, containing only Enzyme A), methylation-dependent digest (Md, containing only Enzyme B), and double digest (Msd, containing both enzymes). To measure intact gDNA following overnight restriction digestion, gDNA was amplified by qPCR using the EpiTect Methyl II PCR Primer Assay for Human G0S2 (Qiagen, USA; Cat. No. EPHS101235–1A) and RT2 SYBR Green ROX qPCR Mastermix (Qiagen, USA; 330521). Percent G0S2 methylation was calculated arithmetically from Mo, Ms, Md, and Msd Ct values according to manufacturer instructions, using a Microsoft Excel spreadsheet provided by Qiagen.
Statistical Analysis.
We used Chi-square test to evaluate associations between categorical variables, Mann-Whitney test or Pearson correlation to compare continuous data from 2 groups, and Kruskal-Wallis with Dunn’s multiple comparisons test to compare continuous data from >2 groups. We used pheatmap (29) to perform unsupervised complete hierarchical clustering. We used caret (30) to perform k-fold cross validation. We used receiver operating characteristic (ROC) curve analysis to estimate a cutoff of G0S2 expression that predicts methylation. We used Kaplan-Meier analysis with pairwise log-rank test to compare overall survival (OS) and disease-free survival (DFS), and Cox proportional hazards regression models to estimate hazard ratios for clinical/molecular variables. p-value<0.05 was significant for all analyses. Statistical analyses were performed in GraphPad Prism, MedCalc, and R (23).
Results
ACC-TCGA reveals CIMP-high defines a rapidly recurrent molecular subtype.
In ACC-TCGA, comprehensive DNA methylome profiling of 79 treatment-naive primary ACC using the 450k platform clustered ACC into three DNA-methylation-based subtypes: “CIMP-low,” “CIMP-intermediate,” and “CIMP-high” (21). While patients with CIMP-low and CIMP-intermediate carcinomas exhibited indistinguishable disease course (log-rank p=0.22 for disease-free survival of CIMP-low vs. CIMP-intermediate, Supplementary Figure S1A), patients with CIMP-high carcinomas characteristically exhibited rapidly recurrent or deadly disease course with median disease-free survival (DFS) following R0/RX resection of 13.6 months (Figure 1A) and median overall survival (OS) of 36 months (Supplementary Figure S1B). Given the striking clinical phenotype associated with the CIMP-high signature, we sought to determine if other molecular classes and somatic alterations identified by ACC-TCGA were associated with this epigenetic program. We performed association tests between CIMP status and ACC-TCGA-defined transcriptome class (mRNA group), somatic copy number alteration profile (SCNA group), or somatic alterations. We observed that CIMP-high carcinomas were distinguished by a transcriptional signature featuring increased expression of steroidogenic and proliferative machinery (“Steroid-high + prolif.” transcriptional program), and a chromosomally “noisy” genomic landscape with numerous arm-level breaks and focal copy number gains and losses (Figure 1B). CIMP-high ACC also frequently bore somatic alterations leading to activation of the cell cycle; however, CIMP-high status was not associated with an increased incidence of alterations leading to activation of Wnt signaling, present in ~40% of ACC (21) (Figure 1B).
We next analyzed RNA-seq data (n=78) from ACC-TCGA to identify differentially expressed genes in CIMP-high compared to non-CIMP-high (CIMP-low + CIMP-intermediate) carcinomas (Supplementary Table S1). We performed gene ontology analysis on differentially expressed genes and identified that CIMP-high ACC exhibited transcriptional upregulation of numerous cell cycle- and DNA damage-associated biological processes, consistent with the enrichment of cell cycle-activating somatic alterations and chromosomal “noisiness” in this subgroup (Figure 1C, left panel). Intriguingly, CIMP-high carcinomas exhibited transcriptional downregulation of a wide array of immunological processes (Figure 1C, right panel), suggesting that CIMP-high ACC are relatively immune poor. The convergence of this unique transcriptional program, somatic alterations targeting the cell cycle, and “noisy” chromosomal landscape in CIMP-high carcinomas demonstrates that CIMP-high status defines a distinct molecular subtype of ACC characterized by rapidly recurrent or fatal disease course. Therefore, prospectively identifying CIMP-high carcinomas using targeted molecular markers may have strong clinical utility.
Analysis of ACC-TCGA nominates G0S2.
We sought to identify a single biomarker with strong discriminatory power between CIMP-high and non-CIMP-high ACC, straightforward to measure and interpret without reference samples or extensive data manipulation. We were therefore interested in genomic loci that are methylated and silenced exclusively in CIMP-high ACC. We analyzed DNA methylation data from ACC-TCGA to identify regions hypermethylated in CIMP-high compared to non-CIMP-high carcinomas (Supplementary Table S2). Among the top ten most hypermethylated regions in our analysis was a 2kb region on chromosome 1 (chr1:209847618–209849445, hg19; Supplementary Figure S2), encompassing 13 contiguous 450k probes and spanning the G0S2 gene locus (non-CIMP-high v. CIMP-high: max beta fold-change −0.709, mean beta fold-change −0.508, Stouffer-corrected p-value 4.32×10−134). Our analysis of differentially expressed genes in CIMP-high compared to non-CIMP-high ACC also revealed G0S2 was among the top five down-regulated genes, nearly silenced in CIMP-high carcinomas (CIMP-high v. non-CIMP-high: log2 fold change −5.21, Benjamini-Hochberg FDR-corrected p-value 2.31×10−10), and highly predictive of CIMP-high status (logistic regression coefficient −0.925, p-value 2.10×10−5); Supplementary Table S1. These results suggested G0S2 is silenced by hypermethylation in a subgroup of ACC as reported in a smaller ACC cohort (20), and that low G0S2 expression and hypermethylation predict CIMP-high status. This observation was particularly intriguing as analysis of GTEx RNA-seq data (31) revealed G0S2 is highly expressed in the physiological adrenal gland (Supplementary Figure S3).
We then plotted all 450k probes spanning the G0S2 locus in each tumor sample from ACC-TCGA, ranked by decreasing G0S2 expression. Strikingly, tumors exhibited an “all or none,” binary pattern of methylation, with uniform hypermethylation (probe beta value>0.5) across the gene locus nearly restricted to CIMP-high carcinomas, and associated with reduced G0S2 expression (Figure 2A). Indeed, average methylation level of probes residing in the G0S2 CpG island is significantly higher in CIMP-high compared to non-CIMP-high ACC (p<0.0001, Kruskall-Wallis with Dunn’s multiple comparisons test; Figure 2B), expression of G0S2 is significantly lower in CIMP-high compared to non-CIMP-high ACC (p<0.0001, Kruskall-Wallis with Dunn’s multiple comparisons test; Figure 2C), and both metrics are strongly inversely correlated (p<2.2×10−16, r=−0.82, R2=0.68, Pearson correlation; Figure 2D). The inverse correlation between G0S2 methylation and expression in ACC-TCGA suggested that measurement of G0S2 methylation (or expression in the absence of genomic DNA) can enable identification of CIMP-high ACC.
Figure 2.

Hypermethylation of the G0S2 locus and decreased G0S2 expression are hallmarks of CIMP-high ACC. A. Methylation level (beta values reflecting % methylation) of all CpG dinucleotides spanning the G0S2 locus including 2 distal CpGs (Illumina Infinium HumanMethylation450 BeadChip, “450k”), and scaled G0S2 expression data (RNA-seq) from ACC-TCGA (n=78) are plotted. Coordinates along chromosome 1 are hg19. Each row represents a sample, and samples are ranked in decreasing order of G0S2 expression (displayed as “Scaled Expression”; RNA-seq CPM scaled to fall in the range of 0–1), with assignment to CIMP status indicated right. Note that hypermethylation of the entire G0S2 locus is largely exclusive to CIMP-high ACC, and that hypermethylation is associated with reduced or absent expression of G0S2 transcript. Indicated by the pink bar at the bottom of the figure are probes lying within the G0S2-associated CpG island. B. Dot plot displaying average beta value of probes indicated in pink from A in ACC-TCGA samples by CIMP group demonstrates that methylation of the G0S2 CpG island distinguishes CIMP-high ACC, and is significantly higher in CIMP-high ACC (clustered at >0.5) compared to CIMP-low or CIMP-intermediate ACC (clustered close to 0). C. Expression of G0S2 in ACC-TCGA samples by CIMP group demonstrates that reduced G0S2 expression is a striking feature of CIMP-high ACC. D. Scatterplot displaying the relationship between logit-transformed average beta value from B and G0S2 expression from C demonstrates that G0S2 methylation and expression are inversely correlated, with CIMP-high ACC bearing the highest levels of G0S2 methylation and lowest levels of G0S2 expression. In B-C, mean and 95% CI of the mean are represented by bar and whiskers, respectively.
Finally, we sought to evaluate the ability of G0S2 methylation alone to classify ACC-TCGA samples by CIMP status. We performed unsupervised hierarchical clustering analysis using the logit-transformed beta values of 450k probes lying within the G0S2 CpG island (Supplementary Figure S4A). This analysis identified two distinct clusters of samples: one cluster with samples bearing either no or low levels of G0S2 methylation (“G0S2 Unmethylated”) corresponding to 2/3 of ACC-TCGA, and one with samples bearing high levels of uniform or heterogeneous G0S2 methylation (“G0S2 Methylated”) corresponding to 1/3 of ACC-TCGA. The G0S2 Methylated cluster was strongly enriched for CIMP-high ACC (p<0.0001, Fisher’s exact test), capturing 18/19 CIMP-high samples. To evaluate the performance of a logistic regression model utilizing G0S2 methylation to discriminate CIMP-high from non-CIMP-high ACC, we performed an internal k-fold cross validation (k=5, 20 repeats) on the average of the logit-transformed beta values of probes residing in the G0S2 CpG island. Our fitted logistic regression model is described in Supplementary Table S3, and the ROC curve (ROC AUC=0.928, 95% CI: 0.8235 to 1) is depicted in Supplementary Fig. S4B. At average G0S2 methylation >0.5200819 (measured by 450k array), we can predict assignment to CIMP-high using G0S2 methylation alone at 94.87% accuracy, with 94.74% sensitivity, 94.92% specificity, 85.71% positive predictive value and 98.25% negative predictive value. This analysis demonstrates that G0S2 hypermethylation has high discriminatory power to distinguish CIMP-high from non-CIMP-high ACC, and shows that unsupervised clustering of G0S2 CpG island methylation enables reliable identification of CIMP-high samples. Taken together, our analysis of ACC-TCGA suggests that assessment of G0S2 methylation and/or expression can reliably identify CIMP-high ACC without comprehensive DNA methylome data.
G0S2 hypermethylation and silencing is exclusive to ACC.
We sought to evaluate G0S2 methylation in an independent ACC cohort, and determine if physiological tissues and ACA exhibit G0S2 methylation. We collected gDNA and mRNA from a retrospective cohort of 80 treatment-naive primary ACC, 22 ACA, and 12 non-naive/non-primary ACC, summarized in Supplementary Table S4. We also collected gDNA from extra-adrenal tissues, microdissected adult adrenal cortex, and total adult adrenal cortex. We performed targeted bisulfite sequencing of G0S2 and determined that uniform hypermethylation throughout the locus is pathological, exclusive to a subset of primary ACC and non-primary/recurrent ACC (Figure 3A, Supplementary Table S5). These findings are supported by unsupervised hierarchical clustering analysis on logit-transformed targeted bisulfite sequencing data (Supplementary Figure S5A), in which we recapitulate G0S2 Unmethylated and G0S2 Methylated clusters we identified in ACC-TCGA. We also demonstrate that physiological tissue and benign adrenocortical tumors cluster with G0S2 Unmethylated ACC, while only ACC with high levels of uniform or heterogeneous G0S2 methylation reside in the G0S2 Methylated cluster. The association of physiological adrenal cortex samples with G0S2 Unmethylated ACC is consistent with the high expression of G0S2 in the physiological adrenal gland (Supplementary Figure S3).
Figure 3.
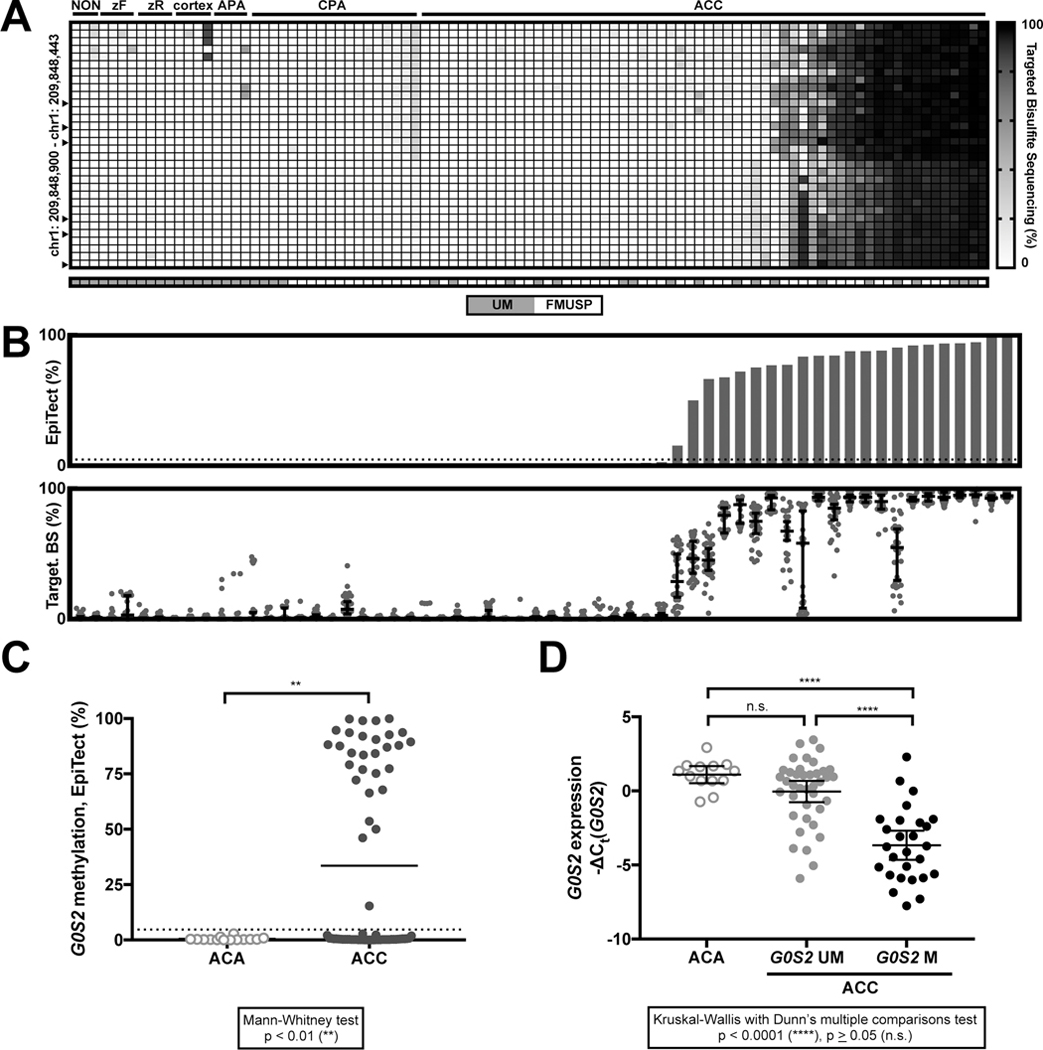
Hypermethylation of the G0S2 locus is binary, exclusive to a subset of ACC, amenable to targeted assessment, and associated with decreased G0S2 expression. A. Heatmap depicting results of bisulfite sequencing of the G0S2 locus in physiological tissues and adrenal tumors. Each row is a CpG position in the G0S2 locus, and any sequenced CpGs in positions corresponding to probes on the 450k array are indicated by arrowheads. Each column is a sample; “NON” refers to human extra-adrenal tissues (from left: kidney, lung, and corpus luteum); “zF” refers to the cortisol-secreting zona fasciculata layer of the adrenal cortex, microdissected from adult adrenal cortex; “zR” refers to the androgen-secreting zona reticularis layer of the adrenal cortex, microdissected from adult adrenal cortex; “cortex” refers to an entire adult adrenal cortex; “APA” refers to an aldosterone-producing adrenocortical adenoma (ACA) and “CPA” refers to a cortisol-producing ACA. Only treatment-naive primary ACC samples are shown here. All tumor samples in this panel are from FMUSP+UM ACA and Primary ACC Cohorts. The G0S2 locus is unmethylated in extra-adrenal tissues, the physiological adrenal cortex and ACA evaluated here. Hypermethylation of the entire G0S2 locus is exclusive to a subset of ACC. B. Targeted assessment of G0S2 methylation by EpiTect (Qiagen, USA; upper panel) in treatment-naive primary ACC (n=60) recapitulates results of bisulfite sequencing (lower panel; each dot represents the methylation level of a single CpG, and median and 95% CI are represented by bar and whiskers, respectively). We performed an internal k-fold cross-validation (k=5, 20 repeats) on all samples with paired EpiTect and targeted bisulfite sequencing data (n=74; 14 ACA, 60 ACC) to identify the appropriate EpiTect cutoff to classify a sample as bearing G0S2 hypermethylation (Supplementary Fig. S5 and Supplementary Table S6). This analysis established a threshold of >4.696% methylation by EpiTect as a cutoff for pathological hypermethylation of the G0S2 locus, which is depicted here by the dotted line. C. Dot plot displaying distribution of G0S2 methylation as measured by EpiTect assay in FMUSP+UM Primary ACC and ACA Cohorts demonstrates that G0S2 methylation is clustered at 0% in ACA (n=14) and is bimodally clustered at 0% and >50% in ACC (n=70) with few intermediate values, consistent with ACC-TCGA. Mean of each group is indicated by the bar, and EpiTect cutoff is indicated by the dotted line. D. Evaluation of G0S2 methylation in ACA and primary ACC without methylation of the G0S2 locus (“G0S2 UM”) or with hypermethylation of the G0S2 locus (“G0S2 M”) demonstrates that G0S2 Methylated tumors have lower expression of G0S2 compared to other adrenocortical tumors, consistent with ACC-TCGA. Note that plot depicts −ΔCt(G0S2), so a smaller value indicates lower expression. Mean and 95% CI of the mean are represented by bar and whiskers, respectively.
The uniform pattern of G0S2 methylation in ACC-TCGA and our cohort indicated that locus methylation may be accurately measured by methylation-sensitive restriction digestion/qPCR-based methods instead of bisulfite-based approaches. We evaluated G0S2 methylation using one such approach, EpiTect (Qiagen, USA). EpiTect and targeted bisulfite sequencing were highly concordant (Figure 3B, Supplementary Figure S5C), demonstrating that ACA have no measurable G0S2 methylation, while ACC have a bimodal distribution (Figure 3C; 40% of ACC in FMUSP+UM Primary ACC Cohort have G0S2 hypermethylation). We then sought to evaluate the concordance between EpiTect and binary G0S2 methylation status defined by unsupervised hierarchical clustering analysis (Supplementary Figure S5A). For all samples with paired EpiTect and targeted bisulfite sequencing data (n=74; 60 ACC, 14 ACA), we performed an internal k-fold cross validation (k=5, 20 repeats) to evaluate a logistic regression model utilizing EpiTect measurements to discriminate these two classes. Our fitted logistic regression model is described in Supplementary Table S6 and ROC curve (ROC AUC=1) depicted in Supplementary Figure S5B, and enables us to obtain a perfect classification with an EpiTect cutoff of 4.696%. These analyses demonstrate that EpiTect enables accurate assessment of binary G0S2 methylation status defined by gold-standard targeted bisulfite sequencing, reinforcing its potential clinical utility.
As in ACC-TCGA, tumors with G0S2 hypermethylation have minimal transcript expression compared to ACA or ACC without G0S2 methylation (Figure 3D). Interestingly, non-primary/non-naive ACC also exhibited the G0S2 methylation/expression inverse relationship (Supplementary Table S7). Finally, we used ROC curve analysis to identify a threshold of G0S2 expression that reliably predicts G0S2 hypermethylation (ROC AUC=0.8557, p<0.0001; Supplementary Figure S6). At ΔCt(G0S2)>3.944, we could predict G0S2 hypermethylation with 92.31% specificity (95% CI: 79.13–98.38%) and 48.15% sensitivity (95% CI: 28.67–68.05%); we used this cutoff to infer G0S2 methylation status of 10 primary ACC for which gDNA was unavailable.
Together with ACC-TCGA, these data illustrate that uniform G0S2 hypermethylation and silencing is exclusive to a subset of ACC, and that G0S2 methylation can be accurately measured using restriction digest/qPCR-based methods or inferred from G0S2 expression when gDNA is unavailable.
G0S2 hypermethylation independently predicts rapid recurrence and death.
High histological grade is an established predictor of dismal outcomes in ACC (10–12). In the FMUSP+UM Primary ACC Cohort, patients with high grade tumors accordingly exhibited rapidly recurrent disease following R0/RX resection (median DFS of 7.8 months). However, 3/10 of patients with high grade tumors remain disease free after >48 months follow-up and 11/32 patients with low grade disease exhibited recurrence, demonstrating that proliferation-based grade alone stratifies patients into heterogeneous groups (Figure 4A). In striking contrast, stratification by G0S2 methylation (measured by EpiTect or inferred from G0S2 expression when gDNA unavailable) demonstrates that patients with tumors bearing G0S2 hypermethylation homogeneously exhibited rapidly recurrent or fatal disease course (median DFS following R0/RX resection of 14 months and median OS of 17 months; Figure 4B–C). Remarkably, G0S2 hypermethylation was identified at comparable frequency in low and high grade tumors (p=0.076, Fisher’s exact test), with G0S2 hypermethylation in 13/44 low grade tumors (Figure 4D), suggesting that G0S2 hypermethylation identifies aggressive disease in tumors inadequately stratified by tumor grade. Finally, carcinomas with G0S2 hypermethylation were identified at comparable frequency in patients with localized ACC (ENSAT I-II), localized ACC with locoregional invasion or lymph node involvement (ENSAT III), and ACC with distal metastases (ENSAT IV) at diagnosis (p=0.31, Chi-square test; Figure 4E). Notably, among 17 ENSAT I-III patients with R0/RX resection and G0S2 hypermethylation, only 1 patient remains disease free at >24 months.
Figure 4.

Hypermethylation of the G0S2 locus predicts rapid recurrence and death in an independent ACC cohort. A. Stratification of carcinomas from FMUSP+UM Primary ACC Cohort by grade (mitotic counts, where <20 mitotic counts/50 high-powered fields [HPF] is “low grade” and >20/50 HPF is “high grade”) identifies two subgroups of carcinomas with failure to achieve median DFS (low grade) and median DFS of 7.8 mo (high grade) following R0/RX resection. B. Stratification of primary ACC by measured or inferred G0S2 methylation status demonstrates that patients with G0S2 Methylated carcinomas have rapid recurrence and median DFS of 14 months following R0/RX resection. In contrast to patients with G0S2 Unmethylated carcinomas that fail to achieve median DFS, only 1 patient in the G0S2 Methylated group remains disease-free at >24 months, consistent with CIMP-high/G0S2 Methylated carcinomas from ACC-TCGA. C. Stratification of primary ACC by measured or inferred G0S2 methylation status demonstrates that patients with G0S2 Methylated carcinomas have dismal OS outcomes, with median OS of 17 months compared to failure to achieve median OS in the G0S2 Unmethylated group. D. G0S2 Methylated primary carcinomas were identified at statistically comparable frequency in patients with high grade disease (13/25) and in patients with low grade disease (13/44). E. G0S2 Methylated primary carcinomas were identified in patients with ENSAT II-IV disease at diagnosis without predilection for late stage disease.
We performed Cox proportional hazards regression analysis to evaluate the significance of G0S2 hypermethylation at predicting recurrence and death compared to other clinical metrics in the FMUSP+UM Primary ACC Cohort (Table 1). High grade and G0S2 hypermethylation were the only variables that significantly predicted recurrence as univariates (high grade v. low grade hazard ratio [HR]=3.15, G0S2 Methylated v. Unmethylated HR=6.91). In contrast, cortisol secretion, ENSAT IV, tumor size, tumor weight, high grade, and G0S2 hypermethylation all significantly predicted death as univariates (cortisol-secreting v. non-cortisol-secreting HR=2.86, ENSAT IV v. II & I HR=5.26, tumor size [cm] HR=1.16, tumor weight [g] HR=1.0007, high grade v. low grade HR=3.42, G0S2 Methylated v. Unmethylated HR=2.65). G0S2 hypermethylation remained significant in all multivariate models (Table 1). These observations demonstrate that G0S2 hypermethylation independently predicts rapidly recurrent disease course prior to detection of macroscopic disease spread, and routinely fatal disease course in the setting of disseminated disease.
Table 1.
Hypermethylation of the G0S2 locus independently predicts poor clinical outcomes.
| Recurrence | Death | ||||||
|---|---|---|---|---|---|---|---|
| Variable (Univariable model) | HR | 95% CI | p-value | Variable (Univariable model) | HR | 95% CI | p-value |
| Male v. Female (N = 47) | 0.560 | 0.216 to 1.45 | 0.233 | Male v. Female (N = 80) | 0.852 | 0.450 to 1.61 | 0.622 |
| Cortisol-secreting v. Non-cortisol-secreting (N = 47) | 2.58 | 0.996 to 6.66 | 0.0509 | Cortisol-secreting v. Non-cortisol-secreting (N = 80) | 2.86 | 1.36 to 5.99 | 5.45 × 10−3 |
| Stage at diagnosis (N = 47) | Stage at diagnosis (N = 80) | ||||||
| ENSAT III v. II & I | 1.49 | 0.602 to 3.70 | 0.388 | ENSAT III v. II & I | 2.09 | 0.937 to 4.66 | 0.0718 |
| ENSAT IV v. II & I | 5.26 | 2.52 to 11.0 | 1.03 × 10−5 | ||||
| Tumor size (cm) (N = 45) | 1.09 | 0.992 to 1.20 | 0.0739 | Tumor size (cm) (N = 75) | 1.16 | 1.08 to 1.24 | 7.71 × 10−5 |
| Tumor weight (g) (N = 36) | 1.0003 | 0.9995 to 1.0011 | 0.444 | Tumor weight (g) (N = 55) | 1.0007 | 1.0003 to 1.0011 | 4.59 × 10−4 |
| High grade v. Low grade (N = 42) | 3.15 | 1.21 to 8.16 | 0.0183 | High grade v. Low grade (N = 69) | 3.42 | 1.74 to 6.74 | 3.80 × 10−4 |
| G0S2 M v. UM (N = 47) | 6.91 | 2.74 to 17.5 | 4.31 × 10−5 | G0S2 M v. UM (N = 80) | 2.65 | 1.45 to 4.86 | 1.60 × 10−3 |
| Variable (Multivariable model) | HR | 95% CI | p-value | Variable (Multivariable model) | HR | 95% CI | p-value |
| (N = 47) | (N = 80) | ||||||
| Cortisol-secreting v. Non-cortisol-secreting | 2.51 | 0.964 to 6.53 | 0.0594 | Cortisol-secreting v. Non-cortisol-secreting | 2.43 | 1.14 to 5.16 | 0.0208 |
| G0S2 M v. UM | 6.88 | 2.71 to 17.5 | 5.02 × 10−5 | G0S2 M v. UM | 2.27 | 1.23 to 4.20 | 9.00 × 10−3 |
| (N = 47) | (N = 80) | ||||||
| ENSAT III v. II & I | 1.11 | 0.440 to 2.78 | 0.830 | ENSAT III v. II & I | 1.80 | 0.804 to 4.04 | 0.153 |
| ENSAT IV v. II & I | 5.44 | 2.57 to 11.5 | 9.30 × 10−6 | ||||
| G0S2 M v. UM | 6.81 | 2.67 to 17.4 | 5.98 × 10−5 | G0S2 M v. UM | 2.77 | 1.48 to 5.19 | 1.39 × 10−3 |
| (N = 45) | (N = 75) | ||||||
| Tumor size (cm) | 1.09 | 0.982 to 1.20 | 0.108 | Tumor size (cm) | 1.17 | 1.09 to 1.26 | 3.72 × 10−5 |
| G0S2 M v. UM | 6.95 | 2.71 to 17.8 | 5.33 × 10−5 | G0S2 M v. UM | 3.53 | 1.85 to 6.75 | 1.36 × 10−4 |
| (N = 36) | (N = 55) | ||||||
| Tumor weight (g) | 1.0005 | 0.9997 to 1.0012 | 0.208 | Tumor weight (g) | 1.0009 | 1.0005 to 1.0013 | 2.78 × 10−5 |
| G0S2 M v. UM | 7.10 | 2.55 to 19.8 | 1.78 × 10−4 | G0S2 M v. UM | 4.51 | 2.01 to 10.1 | 2.48 × 10−4 |
| (N = 42) | (N = 69) | ||||||
| High grade v. Low grade | 3.38 | 1.27 to 8.98 | 0.0147 | High grade v. Low grade | 3.10 | 1.56 to 6.17 | 1.25 × 10−3 |
| G0S2 M v. UM | 7.90 | 2.86 to 21.8 | 6.69 × 10−5 | G0S2 M v. UM | 3.05 | 1.54 to 6.05 | 1.43 × 10−3 |
NOTE. Hazard ratios (HR) and 95% confidence intervals (95% CI) were determined by Cox proportional hazards regression using available clinical and molecular data from all tumors in the FMUSP+UM Primary ACC Cohort. In each category, N indicates number of samples included in each univariable or multivariable model. The p-value is calculated from the Wald statistic using a chi-squared distribution. Grade is calculated on the basis of mitotic counts, where ≤ 20 mitoses/50 high-powered fields (HPF) is “Low grade” and > 20/50 HPF is “High grade.” “G0S2 M. v. UM” = G0S2 Methylated v. Unmethylated.
G0S2 hypermethylation facilitates ACC stratification in combination with BUB1B-PINK1.
Though G0S2 hypermethylation independently predicts uniformly dismal disease course, patients without G0S2 methylation exhibited heterogeneous outcomes (Figure 4B-C). We sought to determine if alternative molecular predictors could resolve this heterogeneity by separating patients with certain favorable prognosis from those with intermediate recurrence risk. We and others have shown that a score derived from expression of BUB1B and PINK1 (BUB1B-PINK1) can stratify ACC into “good prognosis” and “bad prognosis” groups (17,18). The disease course of “good prognosis” ACC has been likened to that of patients with ACA, as patients were primarily cured by surgery. Interestingly, “good prognosis” ACC had BUB1B-PINK1 indistinguishable from ACA (17).
We evaluated BUB1B-PINK1 in FMUSP+UM Primary ACC and ACA Cohorts. We then performed an internal k-fold cross validation (k=5, 20 repeats) on BUB1B-PINK1 score to evaluate the performance of a logistic regression model predicting any history of metastasis in G0S2 Unmethylated ACC (Supplementary Figure S7 depicts fitted logistic regression model ROC curve with ROC AUC 0.840, 95% CI: 0.7177 to 0.9619; model is described in Supplementary Table S8). At BUB1B-PINK1<5.200, we predicted metastasis in patients with G0S2 Unmethylated carcinomas with 100% sensitivity and 31.58% specificity. We assigned carcinomas from the FMUSP+UM Primary ACC Cohort to three groups: ACC I (G0S2 Unmethylated, BUB1B-PINK1>5.200), ACC II (G0S2 Unmethylated, BUB1B-PINK1<5.200), and ACC III (G0S2 Methylated).
ACA and ACC I tumors had no difference in BUB1B-PINK1 (p>0.05, Kruskal-Wallis with Dunn’s multiple comparisons test), while ACC II and ACC III had different BUB1B-PINK1 from ACA (p<0.0001) and ACC I (II v. I: p<0.005, III v. I: p<0.0001). ACC II and ACC III had indistinguishable BUB1B-PINK1 (p>0.05), suggesting BUB1B-PINK1 cannot further stratify G0S2 Methylated carcinomas (Figure 5A). Using this combination of BUB1B-PINK1 and G0S2 methylation status, we stratified the FMUSP+UM Primary ACC Cohort into three groups with variable risk of recurrence (Figure 5B) and death (Figure 5C). In patients with G0S2 Unmethylated carcinomas, we could now distinguish those who remain disease free and alive (ACC I) from those with history of recurrence and death (ACC II). All clinical and molecular data are summarized in Supplementary Table S9.
Figure 5.

Hypermethylation of the G0S2 locus facilitates stratification of ACC into good, intermediate, and poor prognostic groups when combined with BUB1B-PINK1 score. A. Application of an internal k-fold cross validation (k=5, 20 repeats) to BUB1B-PINK1 score in G0S2 Unmethylated primary samples from FMUSP+UM cohort enabled identification of a BUB1B-PINK1 score threshold (BUB1B-PINK1<5.200; Supplementary Fig. S7, Supplementary Table S8) with 100% sensitivity to identify any history of recurrence or metastatic disease. G0S2 Methylated carcinomas were assigned to ACC III. G0S2 Unmethylated carcinomas with BUB1B-PINK1 score above threshold were classified as ACC I, and below threshold were classified as ACC II. Importantly, ACC I carcinomas have BUB1B-PINK1 score indistinguishable from ACA. ACC II and ACC III (G0S2 Methylated) carcinomas have indistinguishable BUB1B-PINK1 scores. Mean and 95% CI of the mean are represented by bar and whiskers, respectively. B. Combined assessment of BUB1B-PINK1 score and G0S2 methylation facilitates stratification of ACC into three groups by DFS. Patients with ACC I carcinomas have no known history of recurrence, patients with ACC II carcinomas have heterogeneous outcomes (fail to achieve median DFS following R0/RX resection), and patients with ACC III (G0S2 Methylated) carcinomas have rapidly recurrent disease (median DFS of 14 months following R0/RX resection). C. Combined assessment of BUB1B-PINK1 score and G0S2 methylation also facilitates stratification of ACC into three groups by OS. Patients with ACC I carcinomas have no known history of mortality at the time of this study, patients with ACC II carcinomas have median OS of 36.3 months, and patients with ACC III carcinomas have median OS of 17 months. D. Proposed stratification and treatment workflow incorporating G0S2 methylation and other molecular markers. Patients with G0S2 Methylated carcinomas have homogenously dismal outcomes, and are unlikely to exhibit durable response to adjuvant mitotane therapy. We therefore propose the evaluation of adjuvant cytotoxic chemotherapy in this subgroup. Alternative predictors such as BUB1B-PINK1 facilitate stratification of patients with G0S2 Unmethylated carcinomas, and enable identification of a subgroup with uniformly favorable prognosis. We propose observation for this subgroup of patients, restricting adjuvant mitotane to patients with intermediate prognosis. Proposed treatment decisions for patients with ENSAT I-III ACC will need to be evaluated in prospective clinical trials prior to incorporation into clinical practice.
These results demonstrate the combined utility of G0S2 methylation and BUB1B-PINK1 score in stratifying patients into three groups, two of which have uniformly favorable or dismal outcomes. These data illustrate a strategy for implementing molecular biomarkers in series to precisely define risk categories in ACC, with high potential to impact clinical management.
Discussion
ACC is a rare cancer with variable outcomes inadequately stratified by clinical and histological metrics. ACC-TCGA identified 3 molecular subtypes of ACC and posited that clinical heterogeneity arises from unique transcriptional and epigenetic programs driving each class (21). We noted that the genomes of rapidly recurrent carcinomas are characterized by aberrant methylation directed to promoter CpG islands, “CIMP-high.” In this study, we also identified that CIMP-high carcinomas comprise a distinct molecular subtype of ACC, bearing upregulation of cell cycle- and DNA damage-associated cellular programs. However, prospective assessment of this complex signature is infeasible for routine molecular diagnostics.
Here, we identified that hypermethylation and silencing of G0S2 is a hallmark of ACC-TCGA CIMP-high carcinomas. In an independent cohort, we determined that G0S2 hypermethylation is restricted to 40% of ACC, absent from ACA and physiological tissues. We then demonstrated that measurement of G0S2 methylation using a straightforward, overnight assay independently identifies a homogeneous subgroup of ACC patients with rapidly recurrent and fatal disease course. G0S2 methylation is essentially binary (carcinomas are either G0S2 Methylated or G0S2 Unmethylated), subverting a requirement for complicated analytical strategies and reference samples. G0S2 hypermethylation almost invariably predicts rapidly recurrent and fatal disease in patients with localized, locoregional and disseminated ACC, including one third of patients with low grade disease. Interestingly, we observed only one patient with tumor G0S2 hypermethylation who remains disease free >24 months following R0/RX resection. Given that adjuvant mitotane therapy is the standard of care at FMUSP and UM, our data suggests that G0S2 hypermethylation predicts short-lived remission regardless, reinforcing the need to develop improved adjuvant therapies for high risk patients.
Expert opinion proposes that adjuvant cytotoxic chemotherapy should be considered as alternative to mitotane in high risk patients (32,33). However, a precise definition of “high risk” is lacking, relying on histological grade and subjective clinical assessment. Our study suggests that prospective assessment of G0S2 methylation would objectively identify uniformly high risk patients. Additionally, we illustrated that G0S2 methylation can be combined in series with validated biomarkers (BUB1B-PINK1) to stratify ACC into three groups, with uniformly favorable (recurrence free), intermediate, and uniformly dismal (inevitable recurrence) clinical outcomes. Such a strategy could dramatically improve clinical management and direct future trials on adjuvant therapies (Figure 5D). The major ongoing clinical trial evaluating the efficacy of adjuvant mitotane in low-intermediate risk ACC (“ADIUVO,” NCT00777244) defines risk using grade; our study suggests this criterion is inadequate, as up to one third of these patients will have tumor G0S2 hypermethylation and likely recur on adjuvant mitotane. As new clinical trials are designed to evaluate adjuvant therapies in high risk patients, we propose assessment of G0S2 methylation to determine risk as in Figure 5D.
High risk CIMP-high/G0S2 Methylated ACC is associated with a unique transcriptional, copy number and mutational landscape in ACC-TCGA, suggesting a common biological program underlies this aggressive ACC subtype (21). We demonstrated that CIMP-high carcinomas are chromosomally noisy, frequently bear somatic alterations leading to activation of cell cycle, and exhibit a transcriptional program characterized by increased expression of steroidogenic enzymes, proliferation machinery, and genes coordinating DNA damage-associated processes. Cell cycle and DNA damage-associated genes upregulated in CIMP-high ACC include MELK, AURKB, CDK6, PLK1, and TOP2A which have been successfully targeted in preclinical and translational models of ACC (34–38), and may even predict clinical responsiveness to combination therapy with etoposide, doxorubicin, cisplatin and mitotane (39). While there is currently little data to support a clinical trial evaluating utility of demethylating agents alone in ACC (40,41), studies in other solid tumors demonstrate that epigenetic priming with demethylating agents may increase efficacy of cytotoxic therapies and targeted DNA repair inhibitors (42–44). Together, these observations suggest that therapies targeting the cell cycle, DNA repair, and epigenetics may be efficacious in patients with CIMP-high/G0S2 Methylated ACC and warrant future study.
The molecular mechanisms driving CpG island hypermethylation in IDH1/2-wild type cancers including CIMP-high ACC are still poorly understood (45). Our data and studies identifying G0S2 hypermethylation in other cancer types (46,47) suggest that methylation of this locus is driven by the same unknown molecular programs supporting hypermethylation in other regions of the CIMP-high cancer genome. However, the high expression of G0S2 in lipid-rich tissues including the adrenal gland (Supplementary Figure S3) suggests that G0S2 may have tissue-specific tumor suppressor roles. While G0S2 has been best characterized as a regulator of lipid metabolism (48), recent studies have demonstrated that methylation-dependent silencing of G0S2 in breast cancer augments oncogenic PI3K/mTOR signaling (49) and MYC transcriptional activity (50). These studies suggest that G0S2 may have important roles in adrenocortical biology, including a similar tumor suppressor function worthy of future investigation.
In conclusion, our study is the first to reduce the complex genome-wide CpG island hypermethylation signature from ACC-TCGA to a single, binary molecular marker, amenable to targeted assessment using routine molecular diagnostics. Assessing G0S2 methylation as we have here is inexpensive, straightforward, compatible with a timeline feasible for clinical decision-making, and will enable the direction of efficacious adjuvant therapies for patients with uniformly aggressive ACC. Future studies will be directed towards evaluating G0S2 methylation prospectively, in circulating tumor DNA, and in readily available clinical samples including formalin-fixed paraffin-embedded tissues.
Supplementary Material
Supplementary Table S1. Results of differentially expressed genes analysis on ACC-TCGA CIMP-high v. non-CIMP-high and results of gene ontology analysis on ACC-TCGA CIMP-high v. non-CIMP-high.
Supplementary Table S2. Results of DMRcate analysis on ACC-TCGA non-CIMP-high v. CIMP-high.
Supplementary Table S5. Results of G0S2 targeted bisulfite sequencing.
Table S3. G0S2 hypermethylation predicts CIMP-high.
Supp. Table S4. Clinical characteristics of FMUSP+UM ACC and ACA Cohorts.
Supp. Table S6. EpiTect accurately measures binary G0S2 methylation status.
Supp. Table S7. Hypermethylation and silencing of G0S2 is heterogeneous in recurrent, metastatic, and non-treatment naive carcinomas.
Supp. Table S8. BUB1B-PINK1 can predict any history of metastasis in patients with G0S2 Unmethylated ACC.
Supplementary Table S9. Complete clinical and molecular data of all samples used in this study.
Supp. Fig. S1. ACC-TCGA patients with CIMP-low or CIMP-intermediate tumor status exhibit indistinguishable disease kinetics, while patients with CIMP-high tumor status have deadly disease course.
Supp. Fig. S2. Schematic of the G0S2 locus.
Supp. Fig. S3. G0S2 is highly expressed in the human adrenal gland.
Supp Fig. S4. Unsupervised hierarchical clustering of G0S2 CpG island methylaiton enables identification of CIMP-high ACC.
Supp. Fig. S5. G0S2 hypermethylation is exclusive to malignant adrenocortical tumors and can be reliably measured by EpiTect.
Supp. Fig. S6. G0S2 expression/methylation ROC curve.
Supp. Fig. S7. BUB1B-PINK1 score can predict metastasis in G0S2 Unmethylated ACC.
Supplemental legend.
Translational Relevance.
Adrenocortical carcinoma (ACC) is a rare, frequently aggressive malignancy with few therapies. Standard of care for patients with locoregional disease is surgery with adjuvant mitotane, but response is variable and unpredictable. The Cancer Genome Atlas project on ACC (ACC-TCGA) revealed that aberrant promoter CpG island hypermethylation (“CIMP-high”) independently predicts rapidly recurrent or fatal disease course. In this study, we analyze ACC-TCGA data and identify that uniform hypermethylation and silencing of G0S2 is a hallmark of CIMP-high ACC. We demonstrate in an independent cohort that G0S2 hypermethylation is exclusive to ACC, amenable to binary targeted assessment, and independently predictive of recurrence and death. We also show that CIMP-high ACC exhibit upregulation of pharmacologically targetable cell cycle and DNA damage response programs. Taken together, we demonstrate that evaluation of tumor G0S2 methylation identifies a subgroup of patients with rapidly progressive disease course who may benefit from aggressive adjuvant and surveillance approaches.
Acknowledgements
This work is supported by the University of Michigan Rogel Cancer Center (grant to G. D. Hammer), and supported (in part) by the National Institutes of Health through the University of Michigan’s Cancer Center Support Grant (5 P30 CA46592). D. R. Mohan was/is supported by the University of Michigan Medical Scientist Training Program (5 T32 GM7863), the University of Michigan Doctoral Program in Cancer Biology, the University of Michigan Rogel Cancer Center, and The Drew O’Donoghue Fund.
The authors would like to express their deepest gratitude to all study participants for their generous contribution, without whom advances in ACC research and clinical care would be impossible. The authors would also like to thank Sueli Oba-Shinjo, PhD; Stella Goncalves, BS; Isabele F. Moretti, BS; and Thais F. Galatro, PhD; for their assistance in processing patient samples from Faculdade de Medicina da Universidade de São Paulo.
Footnotes
Conflicts of interest: G.D.H. is cofounder of, has equity interest in, and is a consultant for Millendo Therapeutics.
References
- 1.Wajchenberg BL, Albergaria Pereira MA, Medonca BB, Latronico AC, Campos Carneiro P, Alves VA, et al. Adrenocortical carcinoma: clinical and laboratory observations. Cancer 2000;88(4):711–36. [PubMed] [Google Scholar]
- 2.Wooten MD, King DK. Adrenal cortical carcinoma. Epidemiology and treatment with mitotane and a review of the literature. Cancer 1993;72(11):3145–55. [DOI] [PubMed] [Google Scholar]
- 3.Else T, Kim AC, Sabolch A, Raymond VM, Kandathil A, Caoili EM, et al. Adrenocortical carcinoma. Endocr Rev 2014;35(2):282–326 doi 10.1210/er.2013-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Else T, Williams AR, Sabolch A, Jolly S, Miller BS, Hammer GD. Adjuvant therapies and patient and tumor characteristics associated with survival of adult patients with adrenocortical carcinoma. J Clin Endocrinol Metab 2014;99(2):455–61 doi 10.1210/jc.2013-2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glenn JA, Else T, Hughes DT, Cohen MS, Jolly S, Giordano TJ, et al. Longitudinal patterns of recurrence in patients with adrenocortical carcinoma. Surgery 2018. doi 10.1016/j.surg.2018.04.068. [DOI] [PubMed] [Google Scholar]
- 6.Berruti A, Grisanti S, Pulzer A, Claps M, Daffara F, Loli P, et al. Long-Term Outcomes of Adjuvant Mitotane Therapy in Patients With Radically Resected Adrenocortical Carcinoma. J Clin Endocrinol Metab 2017;102(4):1358–65 doi 10.1210/jc.2016-2894. [DOI] [PubMed] [Google Scholar]
- 7.Terzolo M, Angeli A, Fassnacht M, Daffara F, Tauchmanova L, Conton PA, et al. Adjuvant mitotane treatment for adrenocortical carcinoma. N Engl J Med 2007;356(23):2372–80 doi 10.1056/NEJMoa063360. [DOI] [PubMed] [Google Scholar]
- 8.Terzolo M, Baudin AE, Ardito A, Kroiss M, Leboulleux S, Daffara F, et al. Mitotane levels predict the outcome of patients with adrenocortical carcinoma treated adjuvantly following radical resection. Eur J Endocrinol 2013;169(3):263–70 doi 10.1530/EJE-13-0242. [DOI] [PubMed] [Google Scholar]
- 9.Kim Y, Margonis GA, Prescott JD, Tran TB, Postlewait LM, Maithel SK, et al. Nomograms to Predict Recurrence-Free and Overall Survival After Curative Resection of Adrenocortical Carcinoma. JAMA Surg 2016;151(4):365–73 doi 10.1001/jamasurg.2015.4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beuschlein F, Weigel J, Saeger W, Kroiss M, Wild V, Daffara F, et al. Major prognostic role of Ki67 in localized adrenocortical carcinoma after complete resection. J Clin Endocrinol Metab 2015;100(3):841–9 doi 10.1210/jc.2014-3182. [DOI] [PubMed] [Google Scholar]
- 11.Giordano TJ. The argument for mitotic rate-based grading for the prognostication of adrenocortical carcinoma. Am J Surg Pathol 2011;35(4):471–3 doi 10.1097/PAS.0b013e31820bcf21. [DOI] [PubMed] [Google Scholar]
- 12.Weiss LM, Medeiros LJ, Vickery AL Jr. Pathologic features of prognostic significance in adrenocortical carcinoma. Am J Surg Pathol 1989;13(3):202–6. [DOI] [PubMed] [Google Scholar]
- 13.Papathomas TG, Pucci E, Giordano TJ, Lu H, Duregon E, Volante M, et al. An International Ki67 Reproducibility Study in Adrenal Cortical Carcinoma. Am J Surg Pathol 2016;40(4):569–76 doi 10.1097/PAS.0000000000000574. [DOI] [PubMed] [Google Scholar]
- 14.Miller BS, Gauger PG, Hammer GD, Giordano TJ, Doherty GM. Proposal for modification of the ENSAT staging system for adrenocortical carcinoma using tumor grade. Langenbecks Arch Surg 2010;395(7):955–61 doi 10.1007/s00423-010-0698-y. [DOI] [PubMed] [Google Scholar]
- 15.Scollo C, Russo M, Trovato MA, Sambataro D, Giuffrida D, Manusia M, et al. Prognostic Factors for Adrenocortical Carcinoma Outcomes. Front Endocrinol (Lausanne) 2016;7:99 doi 10.3389/fendo.2016.00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giordano TJ, Kuick R, Else T, Gauger PG, Vinco M, Bauersfeld J, et al. Molecular classification and prognostication of adrenocortical tumors by transcriptome profiling. Clin Cancer Res 2009;15(2):668–76 doi 10.1158/1078-0432.CCR-08-1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Reynies A, Assie G, Rickman DS, Tissier F, Groussin L, Rene-Corail F, et al. Gene expression profiling reveals a new classification of adrenocortical tumors and identifies molecular predictors of malignancy and survival. J Clin Oncol 2009;27(7):1108–15 doi 10.1200/JCO.2008.18.5678. [DOI] [PubMed] [Google Scholar]
- 18.Fragoso MC, Almeida MQ, Mazzuco TL, Mariani BM, Brito LP, Goncalves TC, et al. Combined expression of BUB1B, DLGAP5, and PINK1 as predictors of poor outcome in adrenocortical tumors: validation in a Brazilian cohort of adult and pediatric patients. Eur J Endocrinol 2012;166(1):61–7 doi 10.1530/EJE-11-0806. [DOI] [PubMed] [Google Scholar]
- 19.Assie G, Letouze E, Fassnacht M, Jouinot A, Luscap W, Barreau O, et al. Integrated genomic characterization of adrenocortical carcinoma. Nat Genet 2014;46(6):607–12 doi 10.1038/ng.2953. [DOI] [PubMed] [Google Scholar]
- 20.Barreau O, Assie G, Wilmot-Roussel H, Ragazzon B, Baudry C, Perlemoine K, et al. Identification of a CpG island methylator phenotype in adrenocortical carcinomas. J Clin Endocrinol Metab 2013;98(1):E174–84 doi 10.1210/jc.2012-2993. [DOI] [PubMed] [Google Scholar]
- 21.Zheng S, Cherniack AD, Dewal N, Moffitt RA, Danilova L, Murray BA, et al. Comprehensive Pan-Genomic Characterization of Adrenocortical Carcinoma. Cancer Cell 2016;29(5):723–36 doi 10.1016/j.ccell.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jouinot A, Assie G, Libe R, Fassnacht M, Papathomas T, Barreau O, et al. DNA Methylation Is an Independent Prognostic Marker of Survival in Adrenocortical Cancer. J Clin Endocrinol Metab 2017;102(3):923–32 doi 10.1210/jc.2016-3205. [DOI] [PubMed] [Google Scholar]
- 23.Team RC. 2016. R: A language and environment for statistical computing. R Foundation for Statistical Computing; https://www.r-project.org/. [Google Scholar]
- 24.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43(7):e47 doi 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014;30(10):1363–9 doi 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol 2010;11(2):R14 doi 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Supek F, Bošnjak M, Škunca N, Šmuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 2011;6(7):e21800 doi 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peters TJ, Buckley MJ, Statham AL, Pidsley R, Samaras K, V Lord R, et al. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin 2015;8:6 doi 10.1186/1756-8935-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kolde R pheatmap: Pretty Heatmaps 2018. [Google Scholar]
- 30.Kuhn M, Wing J, Weston S, Williams A, Keefer C, Engelhardt A, et al. caret: Classification and Regression Training. R 2018. [Google Scholar]
- 31.Carithers LJ, Ardlie K, Barcus M, Branton PA, Britton A, Buia SA, et al. A Novel Approach to High-Quality Postmortem Tissue Procurement: The GTEx Project. Biopreserv Biobank 2015;13(5):311–9 doi 10.1089/bio.2015.0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Varghese J, Habra MA. Update on adrenocortical carcinoma management and future directions. Curr Opin Endocrinol Diabetes Obes 2017;24(3):208–14 doi 10.1097/MED.0000000000000332. [DOI] [PubMed] [Google Scholar]
- 33.Fassnacht M, Dekkers OM, Else T, Baudin E, Berruti A, de Krijger R, et al. European Society of Endocrinology Clinical Practice Guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 2018;179(4):G1–G46 doi 10.1530/EJE-18-0608. [DOI] [PubMed] [Google Scholar]
- 34.Kiseljak-Vassiliades K, Zhang Y, Kar A, Razzaghi R, Xu M, Gowan K, et al. Elucidating the Role of the Maternal Embryonic Leucine Zipper Kinase in Adrenocortical Carcinoma. Endocrinology 2018;159(7):2532–44 doi 10.1210/en.2018-00310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Borges KS, Andrade AF, Silveira VS, Marco Antonio DS, Vasconcelos EJR, Antonini SRR, et al. The aurora kinase inhibitor AMG 900 increases apoptosis and induces chemosensitivity to anticancer drugs in the NCI-H295 adrenocortical carcinoma cell line. Anticancer Drugs 2017;28(6):634–44 doi 10.1097/CAD.0000000000000504. [DOI] [PubMed] [Google Scholar]
- 36.Fiorentini C, Fragni M, Tiberio GAM, Galli D, Roca E, Salvi V, et al. Palbociclib inhibits proliferation of human adrenocortical tumor cells. Endocrine 2018;59(1):213–7 doi 10.1007/s12020-017-1270-0. [DOI] [PubMed] [Google Scholar]
- 37.Hadjadj D, Kim SJ, Denecker T, Ben Driss L, Cadoret JC, Maric C, et al. A hypothesis-driven approach identifies CDK4 and CDK6 inhibitors as candidate drugs for treatments of adrenocortical carcinomas. Aging (Albany NY) 2017;9(12):2695–716 doi 10.18632/aging.101356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bussey KJ, Bapat A, Linnehan C, Wandoloski M, Dastrup E, Rogers E, et al. Targeting polo-like kinase 1, a regulator of p53, in the treatment of adrenocortical carcinoma. Clin Transl Med 2016;5(1):1 doi 10.1186/s40169-015-0080-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roca E, Berruti A, Sbiera S, Rapa I, Oneda E, Sperone P, et al. Topoisomerase 2α and thymidylate synthase expression in adrenocortical cancer. Endocr Relat Cancer 2017;24(7):319–27 doi 10.1530/ERC-17-0095. [DOI] [PubMed] [Google Scholar]
- 40.Liu J, Li XD, Vaheri A, Voutilainen R. DNA methylation affects cell proliferation, cortisol secretion and steroidogenic gene expression in human adrenocortical NCI-H295R cells. J Mol Endocrinol 2004;33(3):651–62 doi 10.1677/jme.1.01560. [DOI] [PubMed] [Google Scholar]
- 41.Suh I, Weng J, Fernandez-Ranvier G, Shen WT, Duh QY, Clark OH, et al. Antineoplastic effects of decitabine, an inhibitor of DNA promoter methylation, in adrenocortical carcinoma cells. Arch Surg 2010;145(3):226–32 doi 10.1001/archsurg.2009.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matei D, Fang F, Shen C, Schilder J, Arnold A, Zeng Y, et al. Epigenetic resensitization to platinum in ovarian cancer. Cancer Res 2012;72(9):2197–205 doi 10.1158/0008-5472.CAN-11-3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matei D, Ghamande S, Roman L, Alvarez Secord A, Nemunaitis J, Markham MJ, et al. A Phase I Clinical Trial of Guadecitabine and Carboplatin in Platinum-Resistant, Recurrent Ovarian Cancer: Clinical, Pharmacokinetic, and Pharmacodynamic Analyses. Clin Cancer Res 2018;24(10):2285–93 doi 10.1158/1078-0432.CCR-17-3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pulliam N, Fang F, Ozes AR, Tang J, Adewuyi A, Keer H, et al. An Effective Epigenetic-PARP Inhibitor Combination Therapy for Breast and Ovarian Cancers Independent of BRCA Mutations. Clin Cancer Res 2018;24(13):3163–75 doi 10.1158/1078-0432.CCR-18-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller BF, Sánchez-Vega F, Elnitski L. The Emergence of Pan-Cancer CIMP and Its Elusive Interpretation. Biomolecules 2016;6(4) doi 10.3390/biom6040045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tokumaru Y, Yamashita K, Osada M, Nomoto S, Sun DI, Xiao Y, et al. Inverse correlation between cyclin A1 hypermethylation and p53 mutation in head and neck cancer identified by reversal of epigenetic silencing. Cancer Res 2004;64(17):5982–7 doi 10.1158/0008-5472.CAN-04-0993. [DOI] [PubMed] [Google Scholar]
- 47.Chang X, Monitto CL, Demokan S, Kim MS, Chang SS, Zhong X, et al. Identification of hypermethylated genes associated with cisplatin resistance in human cancers. Cancer Res 2010;70(7):2870–9 doi 10.1158/0008-5472.CAN-09-3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang X, Lu X, Lombès M, Rha GB, Chi YI, Guerin TM, et al. The G(0)/G(1) switch gene 2 regulates adipose lipolysis through association with adipose triglyceride lipase. Cell Metab 2010;11(3):194–205 doi 10.1016/j.cmet.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yim CY, Bikorimana E, Khan E, Warzecha JM, Shin L, Rodriguez J, et al. G0S2 represses PI3K/mTOR signaling and increases sensitivity to PI3K/mTOR pathway inhibitors in breast cancer. Cell Cycle 2017;16(21):2146–55 doi 10.1080/15384101.2017.1371884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yim CY, Sekula DJ, Hever-Jardine MP, Liu X, Warzecha JM, Tam J, et al. G0S2 Suppresses Oncogenic Transformation by Repressing a MYC-Regulated Transcriptional Program. Cancer Res 2016;76(5):1204–13 doi 10.1158/0008-5472.CAN-15-2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1. Results of differentially expressed genes analysis on ACC-TCGA CIMP-high v. non-CIMP-high and results of gene ontology analysis on ACC-TCGA CIMP-high v. non-CIMP-high.
Supplementary Table S2. Results of DMRcate analysis on ACC-TCGA non-CIMP-high v. CIMP-high.
Supplementary Table S5. Results of G0S2 targeted bisulfite sequencing.
Table S3. G0S2 hypermethylation predicts CIMP-high.
Supp. Table S4. Clinical characteristics of FMUSP+UM ACC and ACA Cohorts.
Supp. Table S6. EpiTect accurately measures binary G0S2 methylation status.
Supp. Table S7. Hypermethylation and silencing of G0S2 is heterogeneous in recurrent, metastatic, and non-treatment naive carcinomas.
Supp. Table S8. BUB1B-PINK1 can predict any history of metastasis in patients with G0S2 Unmethylated ACC.
Supplementary Table S9. Complete clinical and molecular data of all samples used in this study.
Supp. Fig. S1. ACC-TCGA patients with CIMP-low or CIMP-intermediate tumor status exhibit indistinguishable disease kinetics, while patients with CIMP-high tumor status have deadly disease course.
Supp. Fig. S2. Schematic of the G0S2 locus.
Supp. Fig. S3. G0S2 is highly expressed in the human adrenal gland.
Supp Fig. S4. Unsupervised hierarchical clustering of G0S2 CpG island methylaiton enables identification of CIMP-high ACC.
Supp. Fig. S5. G0S2 hypermethylation is exclusive to malignant adrenocortical tumors and can be reliably measured by EpiTect.
Supp. Fig. S6. G0S2 expression/methylation ROC curve.
Supp. Fig. S7. BUB1B-PINK1 score can predict metastasis in G0S2 Unmethylated ACC.
Supplemental legend.