

Abstract

A class of imidazoisoindole (III) heme-binding indoleamine-2,3-dioxygenase (IDO1) inhibitors were optimized via structure-based drug design into a series of tryptophan-2,3-dioxygenase (TDO)-selective inhibitors. Kynurenine pathway modulation was demonstrated in vivo, which enabled evaluation of TDO as a potential cancer immunotherapy target. As means of mitigating the risk of drug–drug interactions arising from cytochrome P450 inhibition, a novel property-based drug design parameter, herein referred to as the CYP Index, was implemented for the design of inhibitors with appreciable selectivity for TDO over CYP3A4. We anticipate the CYP Index will be a valuable design parameter for optimizing CYP inhibition of any small molecule inhibitor containing a Lewis basic motif capable of binding heme.
Keywords: Tryptophan-2,3-dioxygenase; indoleamine-2,3-dioxygenase; CYP inhibition; property-based drug design
Targeting the cellular mechanisms whereby tumors evade detection and clearance by the host immune response comprises an area of intense research effort.1 The kynurenine pathway, which involves the rate-limiting oxidative metabolism of tryptophan (TRP) to kynurenine (KYN) via N-formylkynurenine (NFK) by the cytosolic enzymes indoleamine-2,3-dioxygenase (IDO1) and tryptophan-2,3-dioxygenase (TDO), has been posited as a viable site for small molecule intervention.2−4 Genetic knockouts of IDO1 and TDO have implicated complementary roles for these enzymes in metabolism.5,6 Specifically, IDO1–/– mice exhibit a marked decrease in KYN concentration with minimal effect on TRP concentrations. TDO–/– animals, on the other hand, are characterized by a profound increase systemic TRP.7,8 Since both tryptophan and kynurenine serve as independent modulators of immune function, selectively targeting either IDO1 or TDO is an attractive point of intervention.
Selective inhibition of IDO1 has been of particular interest to the pharmaceutical community, exemplified by numerous small molecule clinical9−12 and preclinical13,14 entities. In the context of human tumors, populations of cancer patients are characterized by highly IDO1 expressing tumors, and increased translation has been observed as a potential mechanism of resistance to immunotherapies in the clinic.3,4,15−17
Only within the past decade has the transcriptional regulation and potential role of TDO in immune evasion by cancer cells been reported.6,18 TDO is predominantly expressed in the liver and responsible for regulation of systemic TRP concentrations and hepatic KYN concentrations. As with IDO1, abnormal TDO expression profiles have been observed in several cancers and are inversely correlated with overall survival rates.19 One proposed mechanism for acquired immunosuppression involves upregulation of TDO within the tumor microenvironment leading to localized depletion of TRP. The consequent amino acid deprivation leads to accumulation of uncharged tRNAs, activating general control non-derepressible 2 (GCN2) kinase, thereby promoting tumor angiogenesis.20 Direct immune inactivation is promoted by GCN2 in T cells: blockade of CD4+ T cell differentiation to T helper cells (TH17) and cell cycle arrest induced in CD8+ T cells are two reported mechanisms.21,22
We pursued a medicinal chemistry program aimed at identifying a potent and selective inhibitor of TDO, which would be compatible for coadministration in the clinic with an IDO1-selective inhibitor. Due to the interconnected biological function of the two enzymes, the substrate TRP, and the metabolite KYN, it was anticipated that the ability to independently modulate IDO1 versus TDO enzymatic activity might be beneficial for both patient safety and efficacy. By utilizing the IDO1 inhibitor GDC-0919 as a starting point for chemistry, we envisioned the added benefit of informing any future efforts to identify an IDO1/TDO dual inhibitor. In contrast to its structurally diverse counterparts, GDC-0919 is unique in its relative potency against both IDO1 and TDO. For example, clinical and preclinical inhibitors INCB24360, BMS-986205, and PF-06840003 bind IDO1 with low nanomolar affinity but exhibit minimal inhibitory activity against TDO (IC50 > 10 μM). By comparison, GDC-0919 shows half-maximal inhibition of both enzymes at concentrations within one logarithm (IDO1 EC50 = 0.45 μM; TDO EC50 = 2.0 μM). Encouraged by these observations, a combined structure- and property-based design approach was initiated to identify a TDO selective small molecule inhibitor.
We recognized that engaging the dioxygenase catalytic heme imparts significant inhibitory activity to GDC-0919. Unlike IDO1, which exclusively contains ferrous iron as the catalytic engine, the metal center in TDO exists in equilibrium between ferrous and ferric states. Thus, we considered whether the imidazoisoindole (III) heme-binder would prove optimal for inhibition of TDO. Wishing to maintain the general features of the III motif, which provided intrinsic TDO inhibitory activity, we explored isosteric replacements of the Lewis basic imidazole, tuning the electronics of the iron-coordinating nitrogen atom, to determine if enhancements in binding could be identified. Designs were triaged by estimating the propensity of the core to coordinate to the heme iron using quantum mechanical calculations of the partial charge of the iron-binding nitrogen (see the Supporting Information). Atoms with more negative partial charges might coordinate more strongly with the iron leading to increased affinity as has been investigated previously with IDO1 inhibitors. A number of 5-ethoxy substituted tricyclic heme-binders, a putative minimal pharmacophore, were then synthesized to cover a range of predicted partial negative character (Table 1). The correlation was validated by comparison to biochemical inhibition. Of the analogues for which a partial charge was calculated, the imidazoindazole and III analogues 1 and 2 were predicted the most Lewis basic (QM charge = −0.526 and −0.527 coulomb, respectively). The near equal partial charges translated into equipotent TDO inhibitory activity. Transposition of the nonbinding nitrogen to afford imidazoindole 3, with its lone pair of electrons in immediate conjugation with the benzenoid ring, resulted in a subtle decrease in partial negative character and a corresponding loss of binding affinity. Similarly, more significant reductions of partial charge, as with isothiazoloindene 4 and 1,2,4-triazolobenzimidazole 5, were correlated with substantial losses of inhibitory activity (IC50 = 7.0 and 17 μM, respectively). Still further attenuation of the heterocycle Lewis basicity in the form of 1,2,3-triazoloindole 6 resulted in a loss of measurable inhibition. Thus, we pursued optimization of the ancillary portion of the ligand with the imidazoisoindole core for the combination of potency and relative ease of chemical synthesis.
Table 1. Correlation of QM Calculated Partial Negative Charge and Inhibitor Activity of Tricyclic Heme-Bindersa.

Calculated partial charge (coulomb) of the iron-binding nitrogen (red) by quantum mechanical calculations. IC50: Half-maximal inhibitory concentration for recombinant human enzyme in biochemical assay, reported in μM (see the Supporting Information).
Despite the functional homology of IDO1 and TDO, turning our attention to X-ray structures alluded to subtle differences that we anticipated were exploitable handles for selectivity (Figure 1). While a few amino acid residue differences present along the inner sphere of the active site adjacent the tricyclic core, we focused attention on the extraneous portion of the ligand, where affinity might be modulated independent of heme-binding potential. Alignment of the cocrystal structure of GDC-0919 bound to IDO1 with a structure of TDO about the ferrous center suggested that the van der Waals surface of TDO along the hydrophobic channel could accommodate a larger ligand than IDO1. Despite conserved inner sphere residues, we targeted the spacious lipophilic pocket available adjacent to TDO F140 (IDO1 F226) to improve TDO affinity. Introducing a methyl group at C(10) of 7, a simplified GDC-0919 analogue, resulted in concomitant gain in TDO potency, loss in IDO1 activity, and net ∼20× improvement in selectivity (cf. Table 2, entries 1 and 2). By truncating the ligand into a cyclohexanol ring (entry 3, 9), still further improvements in IDO1 selectivity were observed. Due to apparent subtle changes in the rotameric disposition of the heme propionate, we then considered optimizing the presumed hydrogen bonding interaction. The apparent mobility of that motif led us to question its significance for potency or selectivity. While eliminating the interaction by removing the alcohol was tolerated for both potency and selectivity (entry 4, 10), the increased lipophilicity was accompanied by reduced in vitro stability as measured by turnover in liver microsomes. Modulating the vector by introducing a cyclopentanol or cyclobutanol ring (entries 5 and 6, 11 and 12, respectively) improved both the IDO1 selectivity and in vitro stability relative to 9.
Figure 1.
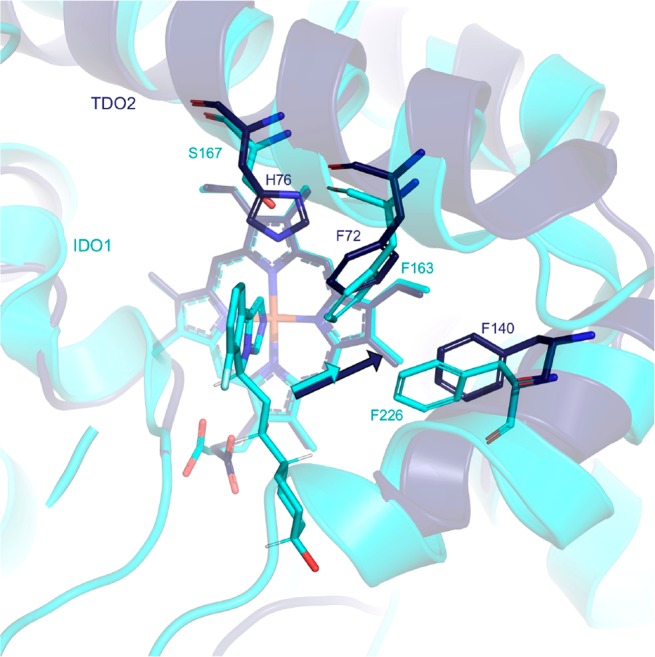
Overlay of the cocrystal structure of GDC-0919 in IDO1 (cyan) with TDO (navy).
Table 2. Structure-Guided Design of C(10)-Cyclic Inhibitors.
![]()

Half-maximal efficacious concentration in SW48 and A172 cellular assays, reported in μM (see the Supporting Information).
F.S. = Fold selectivity.
Clearance in human, rat, and mouse liver microsomes, reported in mL × min–1 × kg–1.
Upon comparison of TDO potency and selectivity for IDO1 across a range of ligands of varied ring size, there appeared a modest preference of TDO for four-membered alcohols (Figure 2). By contrast, five-to-seven membered alcohols exhibited trivial distinctions in cellular activity. All subseries of inhibitors, however, displayed a propensity for reversible inhibition of the cytochrome P450 subfamily member 3A4. In that regard, the four-membered cyclic alcohols also differentiated in their exquisitely potent CYP3A4 inhibition. We envisioned remedying reversible CYP inhibition by reducing lipophilicity via introduction of heteroatom functionality into the ancillary portion of the ligand. Due to the limited vectors available for modifying the four-membered ring inhibitors, our attention turned to the six-membered cyclic alcohols, offering a balance between lipophilicity and vectors for property-based optimization to improve selectivity and metabolic stability.
Figure 2.
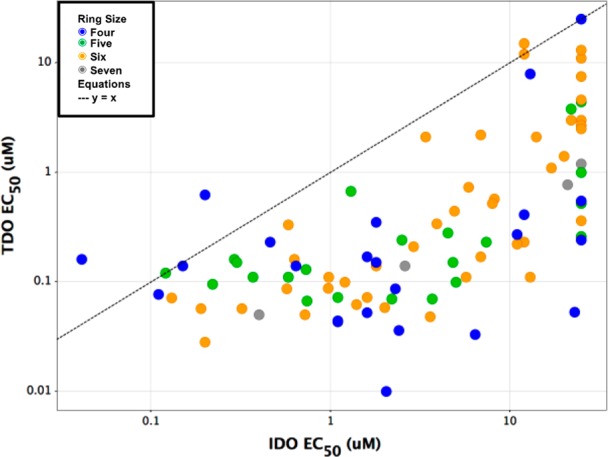
TDO vs IDO cellular potency of cyclic III inhibitors.
We decided to investigate the structure–activity relationships of tetrahydropyranols and piperidinols as readily accessible scaffolds for reducing the lipophilicity. While 3-tetrahydropyranol 13 was tolerated by TDO and improved IDO1 selectivity relative to 9, the compound–as with its cycloalkyl counterparts–was plagued by submicromolar CYP3A4 inhibition (Table 3, entry 1). The phenylalanine (F72, F140) and leucine (L46, L147) lined pocket of TDO presents a very lipophilic surface, and attempts to introduce polarity in that vector as with 4-tetrahydropyranol were less compatible (Table 3, entry 2, 14, TDO EC50 = 0.21 μM). Based on the cyclic ether structure–activity relationship (SAR), we targeted 3-piperidinols. Due to the proximity of the nitrogen atom to Arg144, it was not surprising to find the basic N-methyl-4-piperinol 15 was not accommodated. By reducing basicity, however, we were pleased to find that an N-methanesulfonylpiperidine analogue 16 provided comparable TDO potency and IDO1 selectivity to its carbocyclic counterpart. Consistent with the proposal of increasing polarity to mitigate CYP inhibition, 50% maximal inhibition of CYP3A4 was not observed at 10 μM of the compound. Nonetheless, we continued our efforts to identify an inhibitor with improved balance of IDO1 and CYP3A4 selectivity. Unfortunately, N-acetylpiperidine was not accommodated (entry 5, 17), potentially due to steric clash between the low energy conformer of the amide and the protein.
Table 3. C(10)-Heterocyclic Inhibitor SAR.
![]()

Half-maximal efficacious concentration in SW48 and A172 cellular assays, reported in μM (see the Supporting Information).
F.S. = Fold selectivity.
From a survey of the inhibitors profiled, we recognized that reversible CYP inhibition presented a significant challenge. Turning to the literature for guidance to mitigate CYP inhibition in the presence of a heme-binding motif, we identified two strategies: reduce lipophilicity (decrease cLogP) and introduce conformational rigidity (decrease rotatable bonds).23 A property-based analysis of III inhibitors, however, suggested that relying on traditional remedies would not suffice. Specifically, a comparison of TDO versus CYP3A4 inhibition as a function of lipophilicity indicated a lack of correlation between LogD and selectivity (Figure 3a). Although reduced rotatable bond count, as a surrogate for increased conformational rigidity, did track with improved chances of decreased CYP activity, the odds of achieving an IC50 > 3 μM were still very low for molecules with a single rotatable bond (Figure 3b). Further, we considered ring count, under the assumption that increased rings would correlate with increased conformational rigidity and, perhaps, improved CYP margins. Much to our dismay, ring count did not trend in a meaningful way with CYP inhibition (Figure 3c). Moreover, many promising leads were already well optimized with respect to conventional property-based refinements, such as 13 (LogD = 1.3, rotatable bonds = 1). We considered whether an intuitive, multiparameter approach might offer a solution to the challenge of promiscuous heme-binding. We hypothesized that simple ring count did not accurately predict CYP inhibition due to the propensity to also trend with increasing lipophilicity. Therefore, by introducing a lipophilicity correction, the relationship between rigidity and CYP binding might be realized. Accordingly, we defined the design parameter CYP Index as the quotient of ring count and cLogP (Figure 3; Figure 4).
Figure 3.

Property-based correlations with CYP3A4 inhibition. (a) CYP3A4 IC50/TDO EC50 fold selectivity vs lipophilicity; red = <3×, orange = 3–10×, yellow = 10–30×, green = >30×. CYP3A4 IC50 vs (b) rotatable bond count, (c) ring count, and (d) CYP Index; red = <0.3 μM, orange = 0.3–1.0 μM, yellow = 1.0–3.0 μM, green = >3.0 μM.
Figure 4.

Design strategy for increasing CYP index of III inhibitors.
Thus, increased rigidity without a proportionate increase in cLogP could improve the odds of introducing an offending interaction with the lipophilic CYP binding pocket. An analysis of reversible CYP3A4 inhibition as a function of the CYP Index provided a reasonable correlation, and guided future design space. Specifically, we noted that designs would need to meet the criteria of CYP Index > 2 for improved odds of mitigating submicromolar CYP3A4 binding.
For design compliance with the new property-based parameter, the team recognized that even for relatively nonlipophilic inhibitors (e.g., cLogP = 2.5), a minimum of five rings would be necessary to meet the CYP Index > 2 threshold. Not wishing to increase molecular complexity with additional contiguous stereocenters, we envisioned fusing an arene to the cycloalkanol inhibitor to both introduce an element of rigidity and a handle for modulating lipophilicity. From our seminal investigations, we observed that aryl alcohols (e.g., Figure 4, 18) were potent TDO inhibitors; though, from the outset they were less desirable due to increased lipophilicity compared to their aliphatic alcohol counterparts. Nonetheless, replacement of the solvent-exposed phenyl ring with a 2-oxazole analogue 19 demonstrated the ability to attenuate lipophilicity while maintaining TDO activity. Only by subsequent introduction of additional rigidity at comparable LogD (Figure 4, 20) did we observe appreciable CYP margins. Implementing a strategy of prioritizing molecules with increased CYP Index, the team devised to synthesize a series of fused tetrahydroarene analogues covering a range of lipophilicity in the hope of identifying an inhibitor with suitable margins over both CYP3A4 and IDO1 (Table 4). In addition to excellent TDO potency and an acceptable CYP3A4 margin, tetrahydroisoquinolin-8-ol analogue 20 exhibited desirable IDO1 selectivity and moderate liver microsome stability. As expected, increased lipophilicity and reduced CYP Index with tetrahydronaphthalene 21 was detrimental to CYP3A4 selectivity. Despite a comparable CYP Index, regioisomeric tetrahydroisoquinolin-5-ol 22 suffered a dramatic reduction in the CYP3A4 margin. We hypothesized that the slight decrease in steric encumbrance of the pyridyl nitrogen relative to 20 may be sufficient to increase its aptitude as a second heme-binding moiety. Substitutions of the tetrahydronaphthalene core with aim of reducing lipophilicity to improve IDO1 and CYP selectivity were considered.
Table 4. Tetrahydroarenol Inhibitor SAR.
![]()

Half-maximal efficacious concentration in SW48 and A172 cellular assays, reported in μM (see the Supporting Information).
F.S. = Fold selectivity.
Clearance in human, rat, and mouse liver microsomes, reported in mL × min–1 × kg–1.
Both 7- and 6-(methylsulfonyl)tetrahydronaphthalene-1-ol (23 and 24, respectively) were potent TDO inhibitors; however, the former regiosiomer exhibited a modest benefit with respect to both CYP3A4 margins and liver microsome stability. Although the dimethylphosphineoxide-substituted tetrahydronaphthalene 25 was promising due to excellent IDO1 selectivity and in vitro stability, poor CYP selectivity, due in part to reduced TDO affinity, impeded further advancement. The nitrile-substituted analogue 26 was surprisingly potent and selective over CYP3A4; however, the increased lipophilicity translated to rapid turnover in liver microsome incubations. Increasing polarity, as with the primary carboxamide-substituted compound 27, was well accommodated by TDO and restored CYP3A4 selectivity, as predicted by the increased CYP index, with a concomitant improvement in IDO1 selectivity. Due to increased hydrogen bond donor count and TPSA, however, the molecule suffered poor passive permeability in MDCK cell lines (Papp A:B = 1.7 × 10–6 cm × s–1). We shifted focus to introducing polarity via five-membered heteroaromatic rings in the solvent-exposed vector. Unfortunately, substantial decreases in lipophilicity with tetrahydrotriazolopyridin-8-ol 28 resulted in dramatic loss of TDO inhibitor activity. Nonetheless, we found that increased polarity beyond the tetrahydroisoquinolinol derivatives could be accommodated as with the tetrahydrobenzooxadiazol-4-ol 29. Moreover, the modest increase in CYP Index furnished improvements in CYP3A4 selectivity, and similarly, the reduced lipophilicity correlated with improved in vitro stability across species. Though the TDO potency was diminished with N-fused tetrahydropyrazolopyridin-4-ol 30, the increased CYP Index once again translated to improved CYP3A4 margins. Looking to restore some TDO potency to 24 by slight modulation of lipophilicity, N-methyl tetrahydroindazol-4-ol 31 was investigated. Despite generating a reasonable docking pose, the substitution pattern was not well tolerated by TDO, leading to further loss in potency.
Based on the combination of TDO potency, CYP3A4 and IDO1 selectivity and in vitro stability, we advanced compounds 20, 29, and 30 on to mouse in vivo pharmacokinetic studies to identify a suitable candidate for subsequent PK/PD studies. The three compounds exhibited complementary mouse liver microsome and hepatocyte predicted stability profiles, which we anticipated would be helpful for understanding higher species in vitro–in vivo correlations. Compound 20 was predicted to be labile by liver microsomes (CLhep MLM > 70% LBF) and moderately stable by hepatocyctes (CLhep MHep = 30–70% LBF), whereas the opposite was true for compound 29, exhibiting increased lability in hepatocytes relative to microsomes (Table 5). Only compound 30 binned as moderate stability in both liver microsomes and hepatocytes. All three inhibitors, however, categorically fell into high solubility (kinetic solubility > 100 μM) and high permeability (MDCK Papp A:B > 10 × 10–6 cm × s–1) space ideal for an orally bioavailable molecule. Upon discrete intravenous dosing in mice, both 20 and 30 exhibited moderate plasma clearance (CLp = 29 and 28 mL × min–1 × kg–1, respectively), whereas compound 29 was rapidly cleared (CLp = 63 mL × min–1 × kg–1). Correcting for differences in plasma protein binding, however, indicated the unbound clearances were all comparable. When dosed in oral cassette PK studies, furazan compound 29 was not detected (F below limit of quantification), while both pyridine 20 and pyrazole 30 showed good exposure, which was confirmed in subsequent discrete oral studies.
Table 5. Mouse In Vitro and In Vivo Pharmacokinetics of Compounds 20, 29, and 30.
| 20 | 29 | 30 | |
|---|---|---|---|
| CLhep MLM (mL × min–1 × kg–1) | 71 | 45 | 46 |
| CLhep MHep (mL × min–1 × kg–1) | 33 | 73 | 56 |
| MDCK Papp (A:B) (10–6 cm × s–1) | 11 | 23 | 13 |
| kinetic solubility (μM) | 139 | 165 | 149 |
| CLp (mL × min–1 × kg–1) | 29 | 63 | 28 |
| PPB fub | 7 | 27 | 9 |
| CLp,ub (mL × min–1 × kg–1) | 410 | 230 | 310 |
| Vss (L × kg–1) | 1.4 | 1.1 | 1.1 |
| T1/2 (h) | 0.62 | 0.21 | 0.78 |
| %F cassette (1 mg × kg–1) | 68 | <5 | 140 |
| AUC∞ (μM × h) | 0.34 | BLQa | 0.81 |
| %F discrete (5 mg × kg–1) | 78 | 70 |
BLQ = Below limit of quantification.
Due to the indistinguishable murine pharmacokinetic profiles of compounds 20 and 30, the former was advanced into a PK/PD study on the basis of superior TDO potency. As a consequence of poor mouse-to-human TDO homology, human inhibitors typically suffer one logarithm loss in potency against the murine isoform; thus, conducting pharmacodynamic studies with the more potent molecule was critical for proof-of-concept in vivo target engagement. Treatment with 20 resulted in a dose-dependent increase in blood–tryptophan concentrations (Figure 5). Above a 30 mg × kg–1 dose of 20, minimal increase in TRP Cmax was observed; however, a prolonged increase in TRP concentration resulted in a 2-fold increase in AUC0–6h upon dose escalation to 100 mg × kg–1 (Table 6). Moreover, in the maximum dose cohort, a nearly 4-fold increase in TRP AUC was observed relative to vehicle.
Figure 5.

Mouse pharmacodynamics of compound 20
Table 6. Dose–Response Summary for Mouse PK/PD Study of Compound 20.
|
20 |
TRP |
|||
|---|---|---|---|---|
| dosea | AUC∞ (μM × h) | Cmax (μM) | AUC0–6h (μM × h) | Cmax (μM) |
| 0 | 570 | 340 | ||
| 10 | 5.0 | 4.1 | 680 | 420 |
| 30 | 35 | 21.2 | 1,120 | 560 |
| 100 | 210 | 70 | 1,980 | 540 |
mg × kg–1 compound 20.
In conclusion, utilizing structure-based drug design, we designed a series of TDO-selective inhibitors from the scaffold of the IDO-selective inhibitor GDC-0919. A novel and intuitive design parameter, coined the CYP Index, was conceived and implemented to facilitate the design of heme-binding inhibitors with reduced CYP3A4 inhibition. Moreover, we believe the CYP Index may be utilized in any discovery program to optimize away from CYP inhibition due to presence of a heme-binding motif. A tool compound was identified suitable for demonstrating dose-dependent increases in blood tryptophan concentrations as a pharmacodynamic indicator of TDO inhibition in vivo. We believe that these tool molecules will be valuable in assessing TDO as a viable target for cancer immunotherapy as single agent or in combination with IDO inhibitors.
Acknowledgments
Crystallographic data were collected at the Stanford Synchrtotron Radiation Lightsource beamline 12-2. Use of the SSRL, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
Glossary
Abbreviations
- AUC
area under the curve
- CLhep
predictive hepatic clearance
- CLp
plasma clearance
- cLogP
calculated water–octanol partition coefficient
- fub
unbound fraction
- LogD
water–octanol partition coefficient at pH = 7.4
- LBF
liver blood flow
- MDCK
Madin-Darby canine kidney cell line
- MHep
mouse hepatocytes
- MLM
mouse liver microsomes
- Papp (A:B)
apparent permeability form the apical to basolateral chamber
- PPB
plasma protein binding
- QM
quantum mechanics
- T1/2
plasma half-life
- Vss
steady-state volume of distribution
- %F
percent fraction absorbed
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00004.
Assay conditions, experimental procedures, and crystallography data (PDF)
X-ray coordinates for 7 (CIF)
X-ray coordinates for 8 (CIF)
X-ray coordinates for 9 (CIF)
X-ray coordinates for 20 (CIF)
X-ray coordinates for ent-12 (CIF)
X-ray coordinates for ent-13 (CIF)
X-ray coordinates for ent-14 (CIF)
X-ray coordinates for ent-21 (CIF)
X-ray coordinates for ent-22 (CIF)
X-ray coordinates for ent-30 (CIF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Notes
Absolute stereochemical configuration of compounds 7, 8, 9, ent-12, ent-13, ent-14, 20, ent-21, ent-22, and ent-30 were determined by single crystal X-ray crystallographic analysis. Structure factors and coordinates are deposited with the CSD and PDB under accession code 6VBN. The configuration of all other molecules was tentatively assigned by analogy to the most TDO-potent isomer of the stereotetrad or stereo-octet.
Supplementary Material
References
- Mahoney K. M.; Rennert P. D.; Freeman G. J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discovery 2015, 14, 561–584. 10.1038/nrd4591. [DOI] [PubMed] [Google Scholar]
- Uyttenhove C.; Pilotte L.; Théate I.; Stroobant C.; Colau D.; Parmentier N.; Boon T.; Van den Eynde B. J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine-2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- Godin-Ethier J.; Hanafi L.-A.; Piccirillo C. A.; Lapointe R. Indoleamine-2,3-dioxygenase expression in human cancers: clinical and immunologic perspectives. Clin. Cancer Res. 2011, 17, 6985–6991. 10.1158/1078-0432.CCR-11-1331. [DOI] [PubMed] [Google Scholar]
- Platten M.; von Knebel Doeberitz N.; Oezen I.; Wick W.; Ochs K. Cancer immunotherapy by targetining IDO1/TDO and their downstream effectors. Front. Immunol. 2015, 5, 673. 10.3389/fimmu.2014.00673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koblish H. K.; Hansbury M. J.; Bowman K. J.; Yang G.; Neilan C. L.; Haley P. J.; Burn T. C.; Waeltz P.; Sparks R. B.; Yue E. W.; Combs A. P.; Scherle P. A.; Vaddi K.; Fridman J. S. Hydroxyamidine inhibitors of indoleamine-2,3-dioxygenase potently suppress systemic tryptophan catabolism and growth of IDO-expressing tumors. Mol. Cancer Ther. 2010, 9, 489–498. 10.1158/1535-7163.MCT-09-0628. [DOI] [PubMed] [Google Scholar]
- D’Amato N. C.; Rogers T. J.; Gordon M. A.; Greene L. I.; Cochrane D. R.; Spoelstra N. S.; Nemkov T. G.; D’Alessandro A.; Hansen K. C.; Richer J. K. A TDO2-AhR signaling axis facilitates anoikis resistance and metastasis in triple-negative breast cancer. Cancer Res. 2015, 75, 4651–4664. 10.1158/0008-5472.CAN-15-2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y.; Suda T.; Furuhashi K.; Suzuki M.; Fujie M.; Hahimoto D.; Nakamura Y.; Inui N.; Nakamura H.; Chida K. Increased serum kynurenine/tryptophan ratio correlates with disease progression in lung cancer. Lung Cancer 2010, 67, 361–365. 10.1016/j.lungcan.2009.05.001. [DOI] [PubMed] [Google Scholar]
- de Jong R. A.; Nijman H. W.; Boezen H. M.; Vomer M.; ten Hoor K. A.; Krijnen J.; van der Zee A. G. J.; Hollema H.; Kema I. P. Serum tryptophan and kynurenine concentrations as parameters for indoleamine 2,3-dioxygenase activity in patients with endometrial, ovarian, and vulvar cancer. Int. J. Gynecol. Cancer 2011, 21, 1320–1327. 10.1097/IGC.0b013e31822017fb. [DOI] [PubMed] [Google Scholar]
- Yue E. W.; Sparks R.; Polam P.; Modi D.; Douty B.; Wayland B.; Glass B.; Takvorian A.; Glenn J.; Zhu W.; Bower M.; Liu X.; Leffet L.; Wang Q.; Bowman K. J.; Hansbury M. J.; Wei M.; Li Y.; Wynn R.; Burn T. C.; Koblish H. K.; Fridman J. S.; Emm T.; Scherle P. A.; Metcalf B.; Combs A. P. INCB24360 (Epacadostat), a highly potent and selective indoleamine-2,3-dioxygenase 1 (IDO1) inhibitor for immuno-oncology. ACS Med. Chem. Lett. 2017, 8, 486–491. 10.1021/acsmedchemlett.6b00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosignani S.; Bingham P.; Bottemanne P.; Cannelle H.; Cauwenbergs S.; Cordonnier M.; Dalvie D.; Deroose F.; Feng J. L.; Gomes B.; Greasley S.; Kaiser S. E.; Kraus M.; Négrerie M.; Maegley K.; Miller N.; Murray B. W.; Schneider M.; Soloweij J.; Stewart A. E.; Tumang J.; Torti V. R.; Van Den Eynde B.; Wythes M. Discovery of a novel and selective indoleamine 2,3-dioxygenase (IDO-1) inhibitor 3-(5-fluoro-1H-indol-3-yl)pyrrolidine-2,5-dione (EOS200271/PF-06840003) and its characterization as a potential clinical candidate. J. Med. Chem. 2017, 60, 9617–9629. 10.1021/acs.jmedchem.7b00974. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Waldo J. P.; Jaipuri F. A.; Marcinowicz A.; Van Allen C.; Adams J.; Kesharwani T.; Zhang X.; Metz R.; Oh A. J.; Harris S. F.; Mautino M. R. Discovery of clinical candidate (1R,4r)-4-((R)-2-((S)-6-fluoro-5H-imidazo[5,1-a]isoindol-5-yl)-1-hydroxyethyl)cyclohexan-1-ol (Navoximod), a potent and selective inhibitor of indoleamine 2,3-dioxygenase 1. J. Med. Chem. 2019, 62, 6705–6733. 10.1021/acs.jmedchem.9b00662. [DOI] [PubMed] [Google Scholar]
- Prendergast G. C.; Malachowski W. P.; DuHadaway J. B.; Muller A. J. Discovery of IDO1 inhibitors: from bench to bedside. Cancer Res. 2017, 77, 6795–6811. 10.1158/0008-5472.CAN-17-2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S.; Li X.; Hu F.; Li Y.; Yang Y.; Yan J.; Kuang C.; Yang Q. Discovery of tryptanthrin derivatives as potent inhibitors of indoleamine 2,3-dioxygenase with therapeutic activity in lewis lung cancer (LLC) tumor-bearing mice. J. Med. Chem. 2013, 56, 8321–8331. 10.1021/jm401195n. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Liu K.; Pu Q.; Achab A.; Ardolino M. J.; Cheng M.; Deng Y.; Doty A. C.; Ferguson H.; Fradera X.; Knemeyer I.; Kurukulasuriya R.; Lam Y.-h.; Lesburg C. A.; Martinot T. A.; McGowan M. A.; Miller J. R.; Otte K.; Biju P. J.; Sciammetta N.; Solban N.; Yu W.; Zhou H.; Wang X.; Bennett D. J.; Han Y. Discovery of amino-cyclobutarene-derived indoleamine-2,3-dioxygenase 1 (IDO1) inhibitors for cancer immunotherapy. ACS Med. Chem. Lett. 2019, 10, 1530. 10.1021/acsmedchemlett.9b00344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn D. H.; Mellor A. L. Indoleamine 2,3-dioxygenase and metabolic control of immune responses. Trends Immunol. 2013, 34, 137–143. 10.1016/j.it.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallotta M. T.; Orabona C.; Volpi C.; Vacca C.; Belladonna M. L.; Bianchi R.; Servillo G.; Brunacci C.; Calvitti M.; Bicciato S.; Mazza E. M. C.; Boon L.; Grassi F.; Fioretti M. C.; Fallarino F.; Puccetti P.; Grohmann U. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat. Immunol. 2011, 12, 870–880. 10.1038/ni.2077. [DOI] [PubMed] [Google Scholar]
- Sharma M. D.; Hou D.-Y.; Baban B.; Koni P. K.; He Y.; Chandler P. R.; Blazar B. R.; Mellor A. L.; Munn D. H. Reprogrammed Foxp3+ regulatory T cells provide essential help to support cross-presentation and CDK8+ T cell priming in naïve mice. Immunity 2010, 33, 942–954. 10.1016/j.immuni.2010.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilotte L.; Larrieu P.; Stroobant V.; Colau D.; Dolušić E.; Frédérick R.; De Plaen E.; Uyttenhove C.; Wouters J.; Masereel B.; Van den Eynde B. J. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 2497–2502. 10.1073/pnas.1113873109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opitz C. A.; Litzenburger U. M.; Sahm F.; Ott M.; Tritschler I.; Trump S.; Schumacher T.; Jestaedt L.; Schrenk D.; Weller M.; Jugold M.; Guillemin G. J.; Miller C. L.; Lutz C.; Radlwimmer B.; Lehmann I.; von Deimling A.; Wick W.; Platten M. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. 10.1038/nature10491. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Ning Y.; Alam G. N.; Jankowski B. M.; Dong Z.; Nör J. E.; Polverini P. J. Amino acid deprivation promotes tumor angiogenesis through the GCN2/ATF4 pathway. Neoplasia 2013, 15, 989–997. 10.1593/neo.13262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn D. H.; Sharma M. D.; Baban B.; Harding H. P.; Zhang Y.; Ron D.; Mellor A. L. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 2005, 22, 633–642. 10.1016/j.immuni.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Eleftheriadis T.; Pissas G.; Antoniadi G.; Liakopoulos V.; Tsogka K.; Sounidaki M.; Stefanidis I. Differential effects of the two amino acid sensing systems, the GCN2 kinase and the mTOR complex 1, on primary human alloreactive CDK4+ T-cells. Int. J. Mol. Med. 2016, 37, 1412–1420. 10.3892/ijmm.2016.2547. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Sharma R.; Roychowdhury A. Modulation of cytochrome P450 inhibition (CYP) in drug discovery: a medicinal chemistry perspective. Curr. Med. Chem. 2012, 19, 3605–3621. 10.2174/092986712801323180. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.