

Abstract
Background and Aims
Hepatic ischemia‐reperfusion (I/R) injury remains a major challenge affecting the morbidity and mortality of liver transplantation. Effective strategies to improve liver function after hepatic I/R injury are limited. Six‐transmembrane epithelial antigen of the prostate 3 (Steap3), a key regulator of iron uptake, was reported to be involved in immunity and apoptotic processes in various cell types. However, the role of Steap3 in hepatic I/R‐induced liver damage remains largely unclear.
Approach and Results
In the present study, we found that Steap3 expression was significantly up‐regulated in liver tissue from mice subjected to hepatic I/R surgery and primary hepatocytes challenged with hypoxia/reoxygenation insult. Subsequently, global Steap3 knockout (Steap3‐KO) mice, hepatocyte‐specific Steap3 transgenic (Steap3‐HTG) mice, and their corresponding controls were subjected to partial hepatic warm I/R injury. Hepatic histology, the inflammatory response, and apoptosis were monitored to assess liver damage. The molecular mechanisms of Steap3 function were explored in vivo and in vitro. The results demonstrated that, compared with control mice, Steap3‐KO mice exhibited alleviated liver damage after hepatic I/R injury, as shown by smaller necrotic areas, lower serum transaminase levels, decreased apoptosis rates, and reduced inflammatory cell infiltration, whereas Steap3‐HTG mice had the opposite phenotype. Further molecular experiments showed that Steap3 deficiency could inhibit transforming growth factor‐β–activated kinase 1 (TAK1) activation and downstream c‐Jun N‐terminal kinase (JNK) and p38 signaling during hepatic I/R injury.
Conclusions
Steap3 is a mediator of hepatic I/R injury that functions by regulating inflammatory responses as well as apoptosis through TAK1‐dependent activation of the JNK/p38 pathways. Targeting hepatocytes, Steap3 may be a promising approach to protect the liver against I/R injury.
Abbreviations
- ALT
alanine aminotransferase
- ANOVA
analysis of variance
- ASK1
apoptosis signal–regulating kinase 1
- AST
aspartate aminotransferase
- Bad
BCL2‐associated agonist of cell death
- Bax
Bcl2‐associated x
- Bcl2
B‐cell lymphoma 2
- Ccl2
C‐C motif chemokine ligand 2
- CD11b
cluster of differentiation 11b
- cDNA
complementary DNA
- co‐IP
coimmunoprecipitation
- Cxcl10
chemokine (C‐X‐C motif) ligand 10
- DMSO
dimethyl sulfoxide
- ELISA
enzyme‐linked immunosorbent assay
- GAPDH
glyceraldehyde‐3‐phosphate dehydrogenase
- GST
glutathione S‐transferase
- HA
hemagglutinin
- H&E
hematoxylin and eosin
- H/R
hypoxia/reoxygenation
- HTG
hepatocyte‐specific transgenic mice
- Il
interleukin
- I/R
ischemia‐reperfusion
- JNK
c‐Jun N‐terminal kinase
- KO
knockout
- MAPK
mitogen‐activated protein kinase
- NF‐κB
nuclear factor kappa B
- NTG
nontransgenic
- Steap3
six‐transmembrane epithelial antigen of the prostate 3
- TAK1
transforming growth factor‐β–activated kinase 1
- Tnf‐α
tumor necrosis factor‐α
- TUNEL
terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick end labeling
- WT
wild type
Liver transplantation has become the only effective treatment for patients with end‐stage liver disease.1 Ischemia‐reperfusion (I/R) injury, a process whereby the blood supply is restored by reperfusion rather than by restoring liver function, aggravates hepatocyte dysfunction and structural damage and remains a major challenge affecting the outcome of liver transplantation.2 Approximately 10% of early graft failure cases are caused by I/R injury, and the incidences of acute and chronic rejection are higher.3 Moreover, with the rising demand for organs and steady improvement in immunosuppression strategies, an increasing number of marginal donors, who are more susceptible to I/R injury, will be used.4 Although great efforts have been made to explore a treatment strategy to alleviate acute liver I/R injury, no strategy has been proven to be absolutely effective in clinical practice.5, 6 Therefore, more insights into the mechanism of injury and therapeutic measures will need to be developed.
The progression of liver I/R injury is complex, involving cell damage directly caused by ischemia and more severe liver cell damage caused by the initial inflammation associated with reperfusion.7 The main changes in ischemia are metabolic acidosis and increased intracellular calcium levels as well as the corresponding damage. During reperfusion, the production of reactive oxygen species (ROS) and activation of Kupffer cells, lymphocytes, and neutrophils produce a series of damaging cellular responses that lead to apoptosis.8 Hepatocyte apoptosis induced by I/R injury subsequently leads to the release of proinflammatory mediators such as interleukin‐6 (Il6), Il1b, and tumor necrosis factor‐α (Tnf‐α). These molecules recruit myeloid cells, especially cluster of differentiation 11b (CD11b)–positive subsets, which in turn induce hepatocyte death and aggravate local tissue damage.9, 10 Therefore, it is of great significance to identify effective treatment methods and simultaneously target apoptosis and innate immune system activation to block the vicious cycle involving apoptosis and the inflammatory response in this process for improving clinical I/R injury. Mechanistically, several signaling pathways are activated in liver I/R injury, such as the nuclear factor kappa B (NF‐κB) and mitogen‐activated protein kinase (MAPK) signaling pathways that can mediate both apoptosis and inflammation during liver I/R injury.11, 12 Identification of pivotal regulators controlling these pathways is essential for the development of strategies for clinical intervention to reduce hepatic I/R injury.
The 6‐transmembrane epithelial antigen of prostate (Steap) family has been found to be involved in iron and copper uptake and reduction, endoplasmic reticulum and oxidative stress responses, inflammation, proliferation, and apoptosis. Among these proteins, Steap3 is the major ferric reductase in developing erythrocytes and is important in regulating iron homeostasis.13 Steap3 null mice display severe microcytic anemia due to reduced ferric reductase activity and abnormal erythroid maturation.14 Because iron is irreplaceable in the functions of ribonucleotide reductase and the mitochondrial respiratory chain, Steap3 may influence cellular fate toward proliferation or apoptosis by regulating the intracellular iron content.15 In addition, Steap3 can promote tumor cell apoptosis to inhibit tumor cell proliferation and metastasis through caspase‐3‐dependent pathways.16 Moreover, Steap3 deficiency is reported to impair the toll‐like receptor 4 (TLR4)–mediated inflammatory response.17 However, the function of Steap3 in hepatic I/R injury is largely unknown.
In our preliminary study, we observed that Steap3 expression was significantly up‐regulated in mouse liver subjected to hepatic I/R operation and primary hepatocytes challenged with hypoxia/reoxygenation (H/R) insult, suggesting a potential role for Steap3 in hepatic I/R injury. Considering the critical roles of inflammation and apoptosis in hepatic I/R injury, Steap3 may function as a key mediator in hepatic I/R injury by regulating these processes. To evaluate our hypothesis, we employed global Steap3 knockout (Steap3‐KO) and hepatocyte‐specific Steap3 transgenic (Steap3‐HTG) mice to evaluate the effects of Steap3 on liver I/R injury as well as the underlying mechanisms. Together, our in vivo and in vitro studies showed that Steap3 deficiency could reduce liver injury by alleviating apoptosis and inhibiting inflammation in hepatic I/R injury. Furthermore, our study identified the Steap3–transforming growth factor‐β‐activated kinase 1 (TAK1)–c‐Jun amino‐terminal kinase (JNK)/p38 pathway to be crucial during hepatic I/R injury.
Materials and Methods
Animals
Male mice (8‐10 weeks of age, 25 ± 2 g) were housed in a specific pathogen‐free and temperature‐controlled (23 ± 2°C) environment with a 12‐hour light/dark cycle. Food and water were available ad libitum. Humane care was given to the animals in adherence with the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institutes of Health (publication 86‐23, revised 1985). All animal procedures were approved by the Ethics Committee of The First Affiliated Hospital of Zhengzhou University. Steap3‐KO mice were purchased from the Texas A&M Institute for Genomic Medicine (IST13594C11; TIGM, Germany). Identification primer sequences of Steap3‐KO mice were F: 5′‐CTTGCAAAATGGCGTTACTTAAGC‐3′ and R: 5′‐CACGCTATAACACCGCCCT‐3′. Steap3‐HTG mice were generated using the following method: the full‐length consensus coding sequence of the mouse Steap3 (CCDS48343.1) gene was cloned downstream of the albumin (Alb) promoter, and an Alb‐Steap3 transgene vector was obtained. The vector was linearized and microinjected into pronuclear‐stage embryos. Two cell–stage embryos were transplanted into the oviducts of pseudo‐pregnant foster mothers. Genomic DNA was extracted from the toes or tail tissue of newborn mice for screening by PCR.
Mouse Liver I/R Injury Model
As described, we used an established partial (70%) liver warm ischemia model.18 In brief, mice were first anesthetized with pentobarbital sodium (50 mg/kg), and a midline laparotomy was performed. Then, the portal vein, hepatic artery, and bile duct above the branching to the right lateral lobe were clamped with microvascular clips to interrupt the blood supply to the left lateral/median lobes of the liver. After ischemia for 60 minutes and reperfusion for different times, the animals were sacrificed. Sham control mice underwent the same procedure without clamping the vasculature. At the end of the study, liver tissue and serum samples were collected for further analysis.
Liver Biochemical Measurement
Serum levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured by the ADVIA 2400 Chemistry System (Siemens, Tarrytown, NY), according to the manufacturer’s protocols, to evaluate mouse liver function. The inflammatory state was assessed by measuring serum cytokines using commercial enzyme‐linked immunosorbent assay (ELISA) kits (Murine Tnf‐α Standard ABTS ELISA Development Kit, 900‐T54, from PeproTech, Rocky Hill, NJ; Mouse/Rat Ccl2/JE/MCP‐1 Quantikine ELISA Kit, MJE00, from R&D Systems, Minneapolis, MN) according to the manufacturer’s protocol.
Histological and Immunohistochemical Staining
To assess necrosis in the liver, liver tissue samples were fixed in 10% formalin, embedded in paraffin, and sectioned (4‐5 μm per section). Then, the sections were stained with hematoxylin and eosin (H&E). Expression of Steap3 was investigated by immunohistochemistry.19
Immunofluorescence and Terminal Deoxynucleotidyl Transferase Deoxyuridine Triphosphate Nick End Labeling Staining
Paraffin‐embedded liver sections were also used for immunofluorescence and terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick end labeling (TUNEL) staining, as described.20 A primary antibody against mouse CD11b (ab75476; Abcam, Cambridge, UK) was used. The secondary antibody used was a donkey antirabbit immunoglobulin G (H+L) cross‐adsorbed secondary antibody (A‐10042; Thermo Fisher Scientific). Apoptosis in paraffin‐embedded liver sections was detected by the TUNEL method (Roche; 11684817910) according to the manufacturer’s protocol.
Quantitative Real‐Time PCR
Quantitative real‐time PCR was performed as described.21 Total mRNA was extracted from liver tissue and cultured cells using TRIZOL reagent (Invitrogen) according to the manufacturer's instructions and quantified using a Nanodrop 2000. Quantitative real‐time PCR was performed with SYBR Green PCR Master Mix (catalog no. 04887352001; Roche). The results were normalized against β‐actin expression. The primer sequences of the target genes for real‐time PCR are provided in Supporting Table S1.
Western Blot Analysis
Protein expression levels in mouse liver tissue samples and cells were detected by western blot analysis, as described.22 Protein expression was quantified by ImageJ software, and glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was used as a control. All antibodies used for western blot analysis in this study are shown in Supporting Table S2.
Isolation of Primary Hepatocytes and a Hepatocyte H/R Model
Primary hepatocytes were isolated from the liver as described, and cells with >80% viability were used for further experiments.23 Isolated cells were cultured in complete Dulbecco's modified Eagle's medium (DMEM) overnight. For H/R experiments, the medium was changed into sugar‐free, serum‐free DMEM. Cell hypoxia conditions (1% O2, 5% CO2, and 94% N2) were created in a modular incubator chamber (Biospherix, Lacona, NY). After 60 minutes of hypoxia, the cells were returned to normal air conditions (95% air, 5% CO2) and complete medium at the indicated time point to simulate liver I/R injury in vivo. The cells and related culture medium were collected for further experiments.
Plasmid Construction
The entire homo Steap3 complementary DNA (cDNA) was cloned into pcDNA5‐hemagglutinin (HA), pcDNA5‐Flag, and pHAGE‐3×flag plasmids to express HA‐tagged Steap3 and Flag‐tagged Steap3 recombinant proteins. pcDNA5‐HA‐TAK1, pcDNA5‐Flag‐TAK1, and Flag‐tagged TAK1 truncations were constructed using the same methods. Glutathione S‐transferase (GST)–tagged Steap3 and GST‐TAK1 were obtained by cloning Steap3 or TAK1 cDNA into the pcDNA5‐GST‐HA vector. Expression plasmids encoding truncated Steap3 (1‐258, 259‐488) or TAK1 (1‐300, 1‐390, 301‐579, and 391‐579) were amplified using PCR and cloned into pcDNA5‐flag and pcDNA5‐HA, respectively, using standard molecular biology techniques. The primers used in this study are listed in Supporting Table S3.
Coimmunoprecipitation and GST Pulldown Assays
Coimmunoprecipitation (co‐IP) assays were performed as described to identify the interactions of Steap3 with other factors. GST precipitation assays were performed to examine the direct interaction between Steap3 and TAK1.24
Statistical Analysis
All data analyzed in this study are expressed as the mean ± SD. A two‐tailed Student t test was used for comparisons between two groups, and one‐way analysis of variance (ANOVA) was used for comparisons between multiple groups. P < 0.05 was considered statistically significant. SPSS software (version 21.0) was used for all statistical analyses.
Please see the Supporting Information for more detailed information.
Results
Steap3 Expression is Significantly Up‐Regulated During Hepatic I/R Injury
To analyze whether Steap3 is associated with hepatic I/R injury, we firstly assessed Steap3 expression in wild‐type (WT) mice subjected to partial hepatic I/R injury. Western blotting revealed significantly increased protein expression of Steap3 after hepatic I/R injury (Fig. 1A). However, there was no significant change in the mRNA level (Supporting Fig. S1). Further immunohistochemistry of liver sections indicated an obvious localization of increased Steap3 in the hepatocytes (Fig. 1B). In accordance with the in vivo results, Steap3 expression was also higher in primary hepatocytes challenged by H/R administration than in untreated controls (Fig. 1C), suggesting that hepatocytes might be the target cells of Steap3. Taken together, the induced Steap3 expression in the hepatocytes suggested the possible involvement of Steap3 during hepatic I/R injury.
Figure 1.
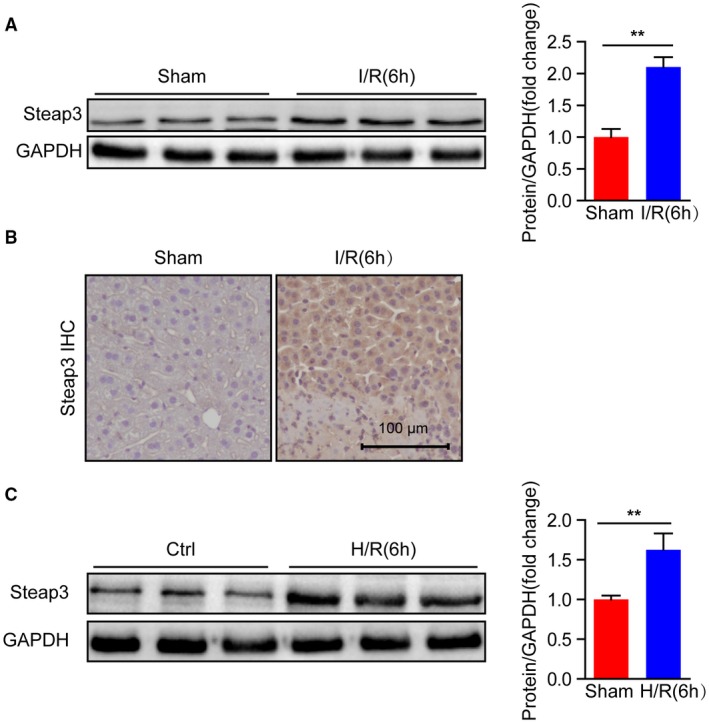
Steap3 expression is up‐regulated after hepatic I/R. (A) Steap3 protein expression in the liver of WT mice at 6 hours after hepatic I/R operation. GAPDH served as the loading control (n = 3/group). (B) Immunohistochemical staining of Steap3 in ischemic liver lobes from WT mice 6 hours after hepatic I/R injury (n = 4 per group). Scale bar, 100 μm. (C) Steap3 expression in cultured primary hepatocytes isolated from WT mice challenged by H/R. GAPDH served as the loading control. Representative of three independent experiments. All data are presented as the mean ± SD. The level of statistical significance is indicated as **P < 0.01. For statistical analysis, a two‐tailed Student t test was used.
Steap3 Deficiency Alleviates Liver Damage and Inflammatory Response During Hepatic I/R Injury
Based on the expression level of Steap3 being significantly up‐regulated in hepatic I/R injury, we employed Steap3‐KO mice to further investigate the role of Steap3 in liver I/R injury. Western blotting confirmed Steap3 deletion in the liver (Fig. 2A). Serum AST and ALT levels were significantly increased after hepatic I/R in WT mice and reached a peak at 6 hours after reperfusion, followed by a decrease. However, serum AST and ALT levels in the Steap3‐KO mice were lower than those in WT mice (Fig. 2B). In addition, H&E staining confirmed that the areas of necrosis in liver sections from the Steap3‐KO mice after hepatic I/R were smaller than those in the sections from the WT mice (Fig. 2C). These findings demonstrated that Steap3 deficiency protected against hepatic I/R injury.
Figure 2.
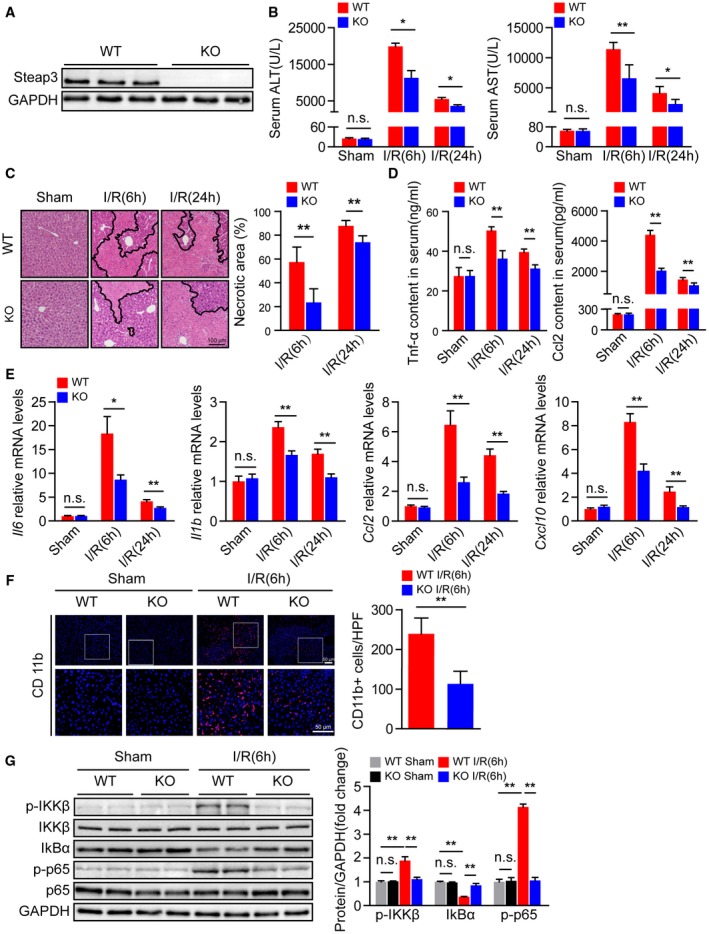
Steap3 deficiency alleviates liver damage and inflammatory responses during hepatic I/R injury. (A) Steap3 protein expression in the liver of WT and Steap3‐KO mice. GAPDH served as the loading control (n = 3/group). (B) Serum ALT/AST activities in WT and Steap3‐KO mice at 6 and 24 hours after hepatic I/R operation (n = 8/group). (C) Representative histological H&E‐stained images and statistics showing necrotic areas in liver tissue from WT and Steap3‐KO mice at 6 and 24 hours after hepatic I/R operation (n = 6/group). Scale bar, 100 μm. (D) Serum levels of inflammatory factors (Tnf‐α and Ccl2) in WT and Steap3‐KO mice at 6 and 24 hours after hepatic I/R operation (n = 6‐8/group). (E) mRNA levels of proinflammatory factors (Il6, Il1b, Ccl2, and Cxcl10) in the liver of WT and Steap3‐KO mice at 6 and 24 hours after hepatic I/R operation (n = 6/group). (F) Representative CD11b immunofluorescence staining in the liver lobes of WT and Steap3‐KO mice at 6 hours after hepatic I/R operation (n = 5/group). Scale bar, 50 μm. (G) Protein levels of NF‐κB signaling pathway molecules in the liver of WT and Steap3‐KO mice at 6 hours after hepatic I/R operation. GAPDH served as the loading control (n = 3/group). All data are presented as the mean ± SD. Levels of statistical significance are indicated as *P < 0.05, **P < 0.01. For statistical analysis, one‐way ANOVA with Bonferroni’s post hoc analysis or Tamhane’s T2 post hoc analysis and two‐tailed Student t test were used. Abbreviations: HPF, high‐power field; IκBα, inhibitory κBα; IKKβ, IκB kinase β; n.s., not statistically significant.
The inflammatory response is an essential factor during hepatic I/R injury.7 Therefore, we investigated the effect of Steap3 deficiency on the inflammatory response after hepatic I/R injury in mice. After hepatic I/R injury, serum levels of proinflammatory cytokines/chemokines such as Tnf‐α and C‐C motif chemokine ligand 2 (Ccl2) increased, reached a peak at 6 hours after reperfusion, and then decreased. Compared with those in the WT group, the expression levels of Tnf‐α and Ccl2 in the Steap3‐KO group were decreased (Fig. 2D). The expression trend of the mRNA levels of inflammatory cytokines (Il6 and Il1b) and chemokines (Ccl2 and chemokine [C‐X‐C motif] ligand 10 [Cxcl10]) were consistent with the trend in the serum (Fig. 2E). Moreover, immunofluorescence staining demonstrated that, compared to that in the WT mouse liver, the number of CD11b‐positive cells in the Steap3‐KO mouse liver was significantly decreased after liver I/R injury (Fig. 2F). NF‐κB plays a critical role in regulating the expression of many inflammatory genes in hepatic I/R injury, leading to the production of cytotoxic free radicals and inflammatory cytokines.25 We found decreased activation of NF‐κB signaling in the Steap3‐KO mice treated with I/R injury compared with the controls (Fig. 2G). In conclusion, these findings demonstrated that Steap3 deficiency suppressed inflammation in hepatic I/R injury.
Hepatocyte‐Specific Steap3 Overexpression Aggravates the Liver Damage and Inflammatory Response During Hepatic I/R Injury
Given that Steap3 deficiency in the liver protects against hepatic I/R injury, we hypothesized that Steap3 overexpression might promote I/R‐mediated liver insult. We constructed Steap3‐HTG mice, and western blotting confirmed that Steap3 was overexpressed in the Steap3‐HTG mouse liver (Fig. 3A). Compared with that in the nontransgenic (NTG) I/R group, the liver injury in the Steap3‐HTG group after hepatic I/R was aggravated, which manifested as higher serum ALT and AST activities (Fig. 3B). Histological analysis of liver tissue also demonstrated an increased necrotic area in Steap3‐HTG mice (Fig. 3C). In addition, Steap3 overexpression can promote the inflammatory response after hepatic I/R injury in mice, and this response is characterized by elevated serum Tnf‐α and Ccl2 levels (Fig. 3D) and liver tissue inflammatory cytokines (Il6 and Il1b) and chemokine (Ccl2 and Cxcl10) mRNA expression levels (Fig. 3E). Immunofluorescence staining also confirmed that, compared with that in the liver of mice in the NTG group, the number of activated CD11b‐positive cells in the liver of Steap3‐HTG mice increased after hepatic I/R injury (Fig. 3F). Moreover, we found enhanced activation of NF‐κB signaling in Steap3‐HTG mice treated with I/R challenge compared with the NTG group (Fig. 3G). These findings suggested that hepatocyte‐specific Steap3 overexpression aggravated inflammatory responses and liver damage during hepatic I/R injury.
Figure 3.
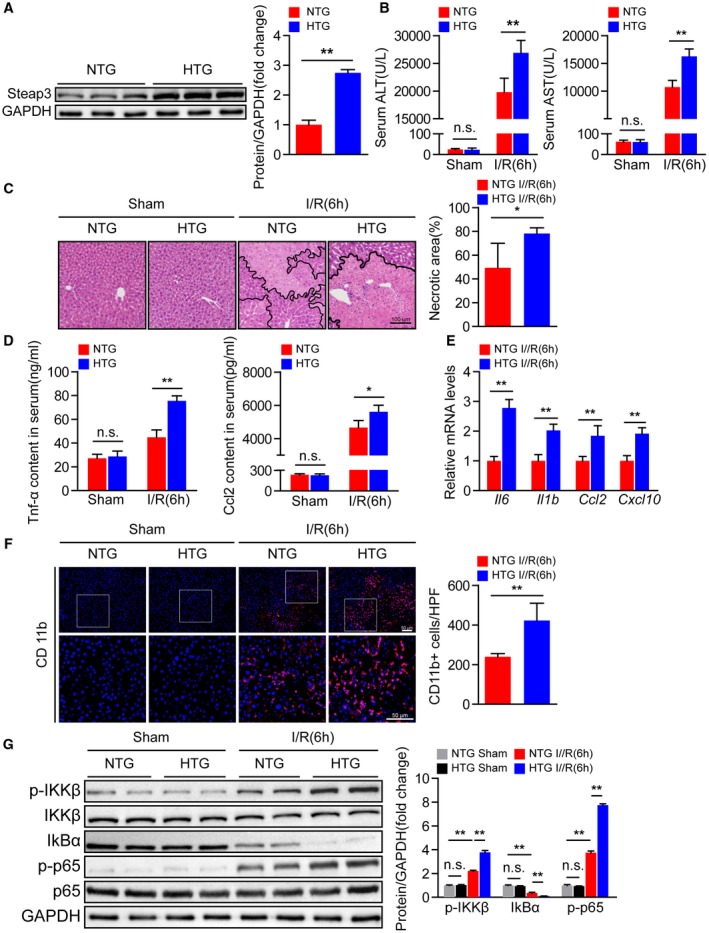
Hepatocyte‐specific Steap3 overexpression aggravates inflammatory responses and liver damage during hepatic I/R injury. (A) Steap3 protein expression in the liver of NTG and Steap3‐HTG mice. GAPDH served as the loading control (n = 3/group). (B) Serum ALT/AST activities in NTG and Steap3‐HTG mice at 6 hours after hepatic I/R operation (n = 8/group). (C) Representative histological H&E‐stained images and statistics showing necrotic areas in liver tissue from NTG and Steap3‐HTG mice at 6 hours after hepatic I/R operation (n = 6/group). Scale bar, 100 μm. (D) Serum levels of inflammatory factors (Tnf‐α and Ccl2) in NTG and Steap3‐HTG mice at 6 hours after hepatic I/R operation (n = 6‐8/group). (E) mRNA levels of proinflammatory factors (Il6, Il1b, Ccl2, and Cxcl10) in the liver of NTG and Steap3‐HTG mice at 6 hours after hepatic I/R operation (n = 6/group). (F) Representative CD11b immunofluorescence staining in the liver lobes of NTG and Steap3‐HTG mice at 6 hours after hepatic I/R operation (n = 5/group). Scale bar, 50 μm. (G) Protein levels of NF‐κB signaling pathway molecules in the liver of NTG and Steap3‐HTG mice at 6 hours after hepatic I/R operation. GAPDH served as the loading control (n = 3/group). All data are presented as the mean ± SD. Levels of statistical significance are indicated as *P < 0.05, **P < 0.01. For statistical analysis, one‐way ANOVA with Bonferroni’s post hoc analysis or Tamhane’s T2 post hoc analysis and two‐tailed Student t test were used. Abbreviations: HPF, high‐power field; IκBα, inhibitory κBα; IKKβ, IκB kinase β; n.s., not statistically significant.
Steap3 Promotes Apoptosis During Hepatic I/R Injury
During hepatic I/R injury, inflammatory mediators recruit neutrophils that induce apoptosis and aggravate local tissue damage.10 Therefore, we further explored whether Steap3 regulates this process. TUNEL staining demonstrated that the number of apoptotic cells was increased at 6 hours after hepatic I/R operation but was significantly reduced in Steap3‐KO mice compared with WT mice (Fig. 4A). Moreover, the levels of the proapoptotic molecules B‐cell lymphoma 2 (Bcl2)–associated x (Bax) and BCL2‐associated agonist of cell death (Bad) were decreased, while the level of the antiapoptotic molecule Bcl2 was up‐regulated in Steap3‐KO mice subjected to hepatic I/R injury compared with WT mice, as measured by quantitative real‐time PCR and western blot analysis (Fig. 4B,C). In contrast, Steap3‐HTG mice exhibited more apoptosis than NTG mice (Fig. 4D‐F). These observations revealed that Steap3 promoted apoptosis during hepatic I/R injury.
Figure 4.
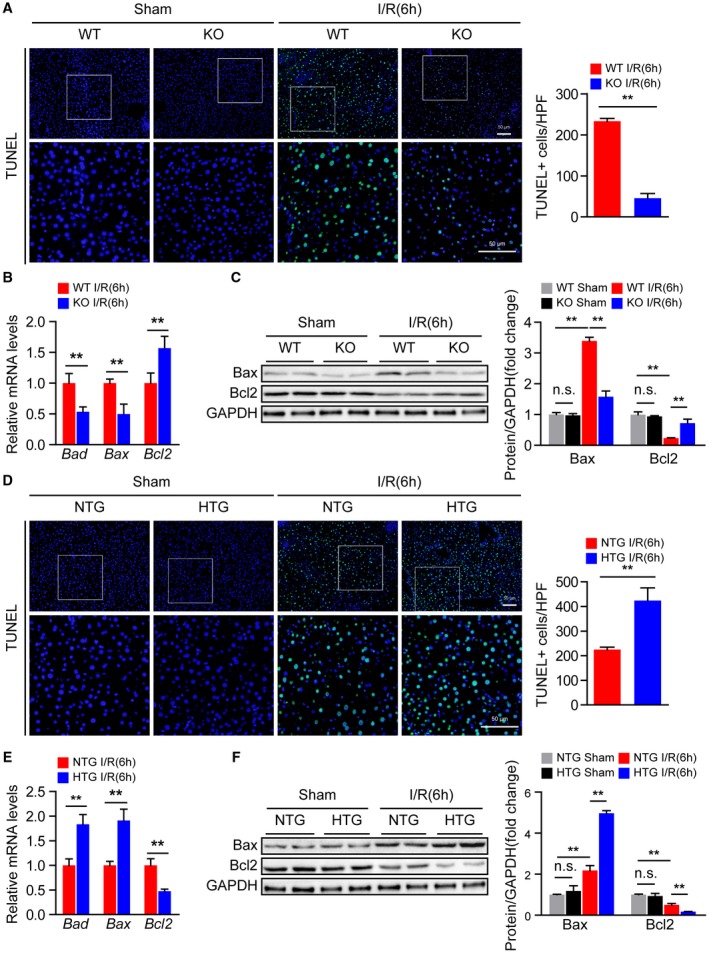
Steap3 promotes apoptosis during hepatic I/R injury. (A) TUNEL staining in liver sections from WT and Steap3‐KO mice at 6 hours after hepatic I/R operation (n = 4/group). Scale bar, 50 μm. (B) mRNA levels of apoptosis‐related genes (Bad, Bax, and Bcl2) in liver samples of WT and Steap3‐KO mice at 6 hours after hepatic I/R operation (n = 6/group). (C) Western blot analysis of the Bax and Bcl2 levels in the liver of WT and Steap3‐KO mice at 6 hours after hepatic I/R operation. GAPDH served as a loading control (n = 3/group). (D) TUNEL staining in liver sections of NTG and Steap3‐HTG mice at 6 hours after hepatic I/R operation (n = 4/group). Scale bar, 50 μm. (E) mRNA levels of apoptosis‐related genes (Bad, Bax, and Bcl2) in liver samples from NTG and Steap3‐HTG mice at 6 hours after hepatic I/R operation (n = 6/group). (F) Western blot analysis of the Bax and Bcl2 levels in the liver of NTG and Steap3‐HTG mice at 6 hours after hepatic I/R operation. GAPDH served as a loading control (n = 3/group). All data are shown as the mean ± SD. Levels of statistical significance are indicated as *P < 0.05, **P < 0.01. For statistical analysis, one‐way ANOVA with Bonferroni’s post hoc analysis or Tamhane’s T2 post hoc analysis and two‐tailed Student t test were used. Abbreviations: HPF, high‐power field; n.s., not statistically significant.
Steap3 Accelerates the Hepatocyte Inflammatory Response and Apoptosis During H/R Injury
To further determine the direct influences of Steap3 on hepatocytes, primary hepatocytes isolated from the livers of Steap3‐KO and Steap3‐HTG mice as well as their respective controls were subjected to H/R challenge. Similar to the results obtained from our in vivo experiments, the mRNA levels of inflammatory cytokines (Il6 and Il1b) and chemokines (Ccl2 and Cxcl10) in the hepatocytes from Steap3‐KO mice were significantly down‐regulated after H/R (Fig. 5A). Moreover, decreased activation of NF‐κB signaling was observed in the Steap3‐KO hepatocytes challenged by H/R treatment compared with the WT hepatocytes (Fig. 5B). In addition, Bcl2 expression was up‐regulated, whereas cleaved caspase‐3, Bad, and Bax levels were down‐regulated in the Steap3‐KO hepatocytes after H/R (Fig. 5C,D). Meanwhile, we found no difference in inflammatory and apoptotic molecules in hepatocytes from WT and Steap3‐KO mice without H/R challenge (Supporting Fig. S3), indicating that the protective effect of Steap3 on inflammation and apoptosis was H/R stimulation–dependent. In contrast, the Steap3‐HTG hepatocytes exhibited a promotional effect on inflammation and apoptosis after H/R injury (Fig. 5E‐H). These gain‐of‐function and loss‐of‐function data strongly demonstrated that Steap3 in hepatocytes was a key mediator of cell survival and promoter of cellular inflammation during hepatic I/R injury.
Figure 5.
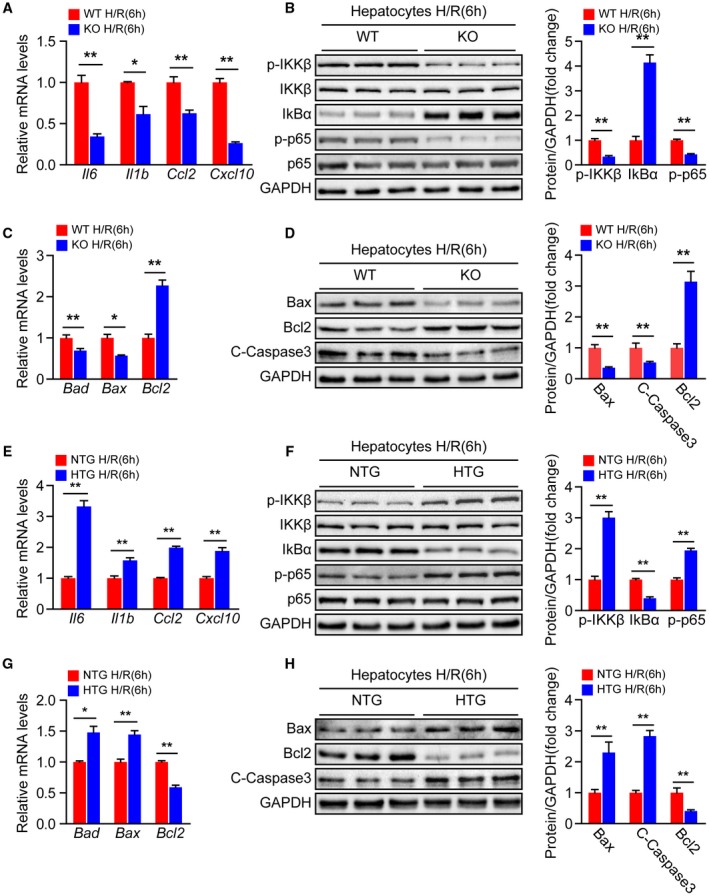
Steap3 accelerates hepatocyte inflammation and apoptosis during hepatic I/R injury. (A) mRNA levels of proinflammatory factors (Il6, Il1b, Ccl2, and Cxcl10) in cultured primary hepatocytes isolated from WT and Steap3‐KO mice that were subjected to H/R challenge (n = 3/group). (B) Protein levels of NF‐κB signaling pathway molecules in cultured primary hepatocytes isolated from WT and Steap3‐KO mice that were challenged by H/R insult. GAPDH served as the loading control. Representative of three independent experiments. (C) mRNA levels of apoptosis‐related genes (Bad, Bax, and Bcl2) in cultured primary hepatocytes isolated from WT and Steap3‐KO mice that were subjected to H/R challenge (n = 3/group). (D) Western blot analysis of the Bax, Bcl2, and cleaved caspase‐3 levels in cultured primary hepatocytes isolated from WT and Steap3‐KO mice that were challenged by H/R insult. GAPDH served as the loading control. Representative of three independent experiments. (E) mRNA levels of proinflammatory factors (Il6, Il1b, Ccl2, and Cxcl10) in cultured primary hepatocytes isolated from NTG and Steap3‐HTG mice that were subjected to H/R challenge (n = 3/group). (F) Protein levels of NF‐κB signaling pathway molecules in cultured primary hepatocytes isolated from NTG and Steap3‐HTG mice that were challenged by H/R insult. GAPDH served as the loading control. Representative of three independent experiments. (G) mRNA levels of apoptosis‐related genes (Bad, Bax, and Bcl2) in cultured primary hepatocytes isolated from NTG and Steap3‐HTG mice that were subjected to H/R challenge (n = 3/group). (H) Western blot analysis of the Bax, Bcl2, and cleaved caspase‐3 levels in cultured primary hepatocytes isolated from NTG and Steap3‐HTG mice that were challenged by H/R insult. GAPDH served as the loading control. Representative of three independent experiments. All data are shown as the mean ± SD. Levels of statistical significance are indicated as *P < 0.05, **P < 0.01. For statistical analysis, a two‐tailed Student t test was used. Abbreviations: IκBα, inhibitory κBα; IKKβ, IκB kinase β.
Steap3 Deficiency Inhibits TAK1 Activation and Downstream JNK and p38 Signaling During Hepatic I/R Injury
Having demonstrated that Steap3 promoted inflammatory response and apoptosis in hepatocytes during hepatic I/R injury, we next explored the underlying mechanism by which Steap3 regulates cellular functions. The MAPK signal transduction pathway plays a key role in regulating inflammatory and apoptotic processes during hepatic I/R injury.26 Therefore, we examined whether Steap3 modulates MAPK signaling during hepatic I/R injury. Interestingly, compared with endogenous Steap3 expression, Steap3 deficiency suppressed JNK and p38 phosphorylation, while Steap3 overexpression elevated JNK and p38 phosphorylation in mice suffering from hepatic I/R injury. However, extracellular signal–regulated protein kinase phosphorylation was not affected in either setting (Fig. 6A,B). In addition, the phosphorylation of TAK1 (p‐TAK1) and apoptosis signal–regulating kinase 1 (p‐ASK1), two upstream kinases that activate MAPK signaling in hepatic I/R injury, were measured to determine the upstream kinase acted upon by Steap3 to stimulate the JNK and p38 signaling pathways. The phosphorylation of TAK1 in liver tissue subjected to hepatic I/R injury was significantly suppressed by Steap3 deficiency but activated by Steap3 overexpression, while ASK1 phosphorylation was unaffected (Fig. 6C,D). Furthermore, similar results were obtained in primary hepatocytes isolated from Steap3‐KO and Steap3‐HTG mice in response to H/R (Fig. 6E‐H). Collectively, these results showed that Steap3 deficiency inhibited TAK1 activation and downstream JNK and p38 signaling in hepatocytes during hepatic I/R injury.
Figure 6.
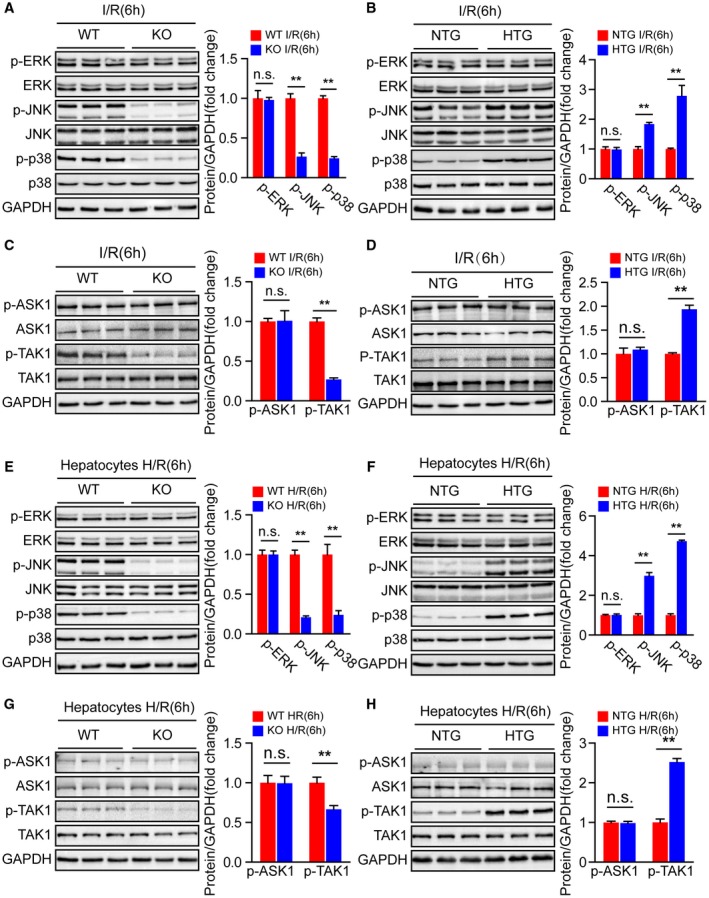
Steap3 deficiency inhibits TAK1/JNK/p38 signaling in liver injury. (A,B) Western blot analysis of the total and phosphorylated protein expression levels of classic MAPKs in the liver of Steap3‐KO (A) and Steap3‐HTG (B) mice after hepatic I/R operation (n = 3/group). (C,D) Western blot analysis of the total and phosphorylated protein expression levels of classic MAPK kinase kinases (MAP3Ks), including TAK1 and ASK1, in the liver of Steap3‐KO (C) and Steap3‐HTG (D) mice after hepatic I/R operation (n = 3/group). (E,F) Western blot analysis of the total and phosphorylated protein expression levels of classic MAPKs in cultured primary hepatocytes isolated from Steap3‐KO (E) and Steap3‐HTG (F) mice that were challenged by H/R insult. Representative of three independent experiments. (G,H) Western blot analysis of the total and phosphorylated protein expression levels of classic MAP3Ks, including TAK1 and ASK1, in cultured primary hepatocytes isolated from Steap3‐KO (G) and Steap3‐HTG (H) mice that were challenged by H/R insult. Representative of three independent experiments. For (A‐H), GAPDH served as the loading control. All data are shown as the mean ± SD. Levels of statistical significance are indicated as *P < 0.05, **P < 0.01. For statistical analysis, a two‐tailed Student t test was used. Abbreviations: ERK, extracellular signal–regulated kinase; n.s., not statistically significant.
Steap3 Directly Interacts With TAK1 and Promotes TAK1 Signaling Activation
To determine how Steap3 affects TAK1 signaling, we first performed an immunofluorescence colocalization experiment to detect the localization of Steap3 and TAK1. Interestingly, we found that Steap3 and TAK1 colocalized in the cytoplasm (Fig. 7A). Furthermore, co‐IP studies showed an interaction between exogenous Steap3 and TAK1 (Fig. 7B). In addition, a GST‐HA‐tagged TAK1 protein efficiently pulled down Flag‐tagged Steap3, and GST‐HA‐tagged Steap3 pulled down Flag‐tagged TAK1 (Fig. 7C). Additionally, co‐IP using serially truncated forms of TAK1 or Steap3 demonstrated that the N‐terminal region segment amino acids 1‐258 of Steap3 (Flag‐Steap3 [1‐258]) and the spanning 1‐300 amino acids of TAK1 (HA‐TAK1 [1‐300]) were the interacting regions of Steap3 and TAK1, respectively (Fig. 7D,E). To further confirm that the regulation of Steap3 on TAK1‐p38/JNK signaling was dependent on its interaction with TAK1 during H/R challenge, hepatocytes were transfected with plasmids expressing full‐length Steap3 or truncated Steap3 (Steap3 [1‐258], Steap3 [259‐488]). We noticed that the 1‐258 domain of Steap3 was the fragment responsible for its activation of TAK1‐p38/JNK signaling (Fig. 7F). These results demonstrated that Steap3 directly interacted with TAK1 and promoted TAK1 signaling activation.
Figure 7.
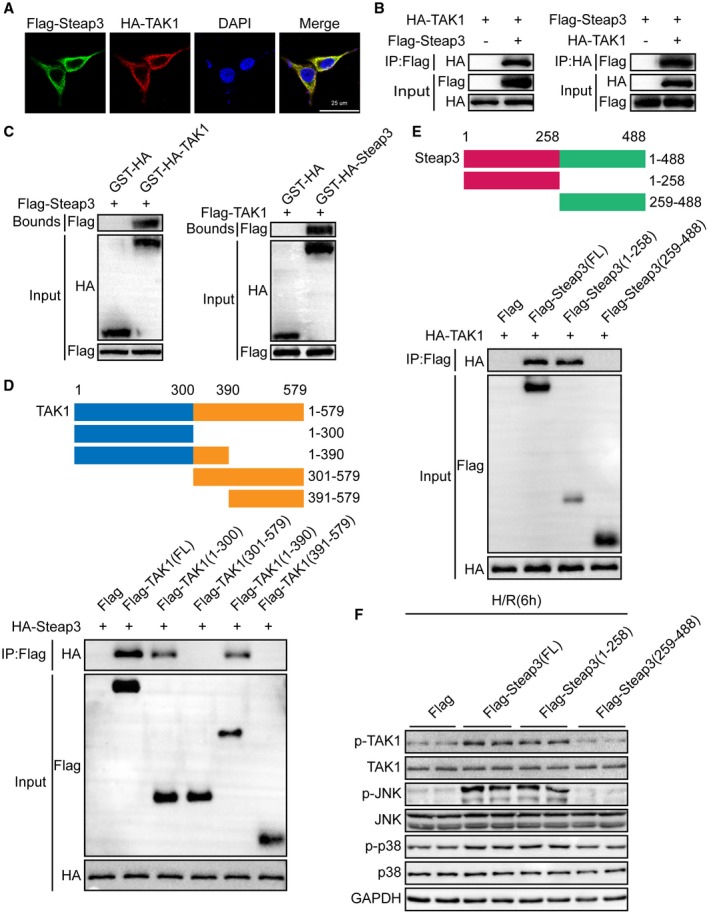
Steap3 directly interacts with TAK1 and promotes TAK1 signaling activation. (A) Representative confocal images showed 293T cells cotransfected with Flag‐tagged Steap3 and HA‐tagged TAK1. Proteins were visualized using anti‐Flag (green) and anti‐HA (red) antibodies, followed by fluorophore‐conjugated secondary antibodies after 24 hours of transfection. Nuclei were stained using 4′,6‐diamidino‐2‐phenylindole (blue). n = 3 independent experiments per group with 6 images per group. Scale bar, 25 μm. (B) Flag‐tagged Steap3 and HA‐tagged TAK1 plasmids were cotransfected into 293T cells. Anti‐Flag antibody (left panel) or anti‐HA antibody (right panel) were used for immunoprecipitation. Representative of three independent experiments. (C) Flag‐tagged Steap3 and GST‐HA‐tagged TAK1 plasmids or Flag‐tagged TAK1 and GST‐HA‐Steap3 TAK1 were cotransfected into 293T cells. Representative of three independent experiments. (D) Full‐length HA‐Steap3 and various truncated forms of Flag‐TAK1 were cotransfected into 293T cells. An anti‐Flag antibody was used for immunoprecipitation. Representative of three independent experiments. (E) Full‐length HA‐TAK1 and various truncated forms of Flag‐Steap3 were cotransfected into 293T cells. An anti‐Flag antibody was used for immunoprecipitation. Representative of three independent experiments. (F) Western blot analysis of total and phosphorylated TAK1, JNK, and p38 in hepatocytes transfected with various truncated forms of Flag‐Steap3 subjected to H/R challenge. GAPDH served as the loading control. Representative of three independent experiments. Abbreviations: DAPI, 4′,6‐diamidino‐2‐phenylindole; IP, immunoprecipitation.
TAK1 Mediated the Effect of Steap3 on Hepatocyte Inflammation and Apoptosis After H/R Treatment
To further evaluate whether TAK1 mediated the effect of Steap3 on liver I/R injury, we blocked TAK1 activity in L02 hepatocytes using a specific TAK1 inhibitor, NG25.27 Compared with dimethyl sulfoxide (DMSO), NG25 largely abolished the activation of TAK1 and downstream JNK/p38 in Steap3 overexpressed hepatocytes subjected to H/R challenge (Fig. 8A). Furthermore, inhibition of TAK1 activity also abrogated the inflammatory response and apoptosis induced by Steap3 overexpression in hepatocytes subjected to H/R challenge, as evidenced by decreased gene expression of proinflammatory factors (Tnf‐α, Il16, Il1b, and Ccl2), suppression of NF‐κB signaling, increased expression of Bcl2, and decreased expression of Bad, Bax, and cleaved caspase‐3 (Fig. 8B‐E). These results demonstrated that TAK1 mediated the effect of Steap3 on hepatocyte inflammation and apoptosis after H/R challenge.
Figure 8.
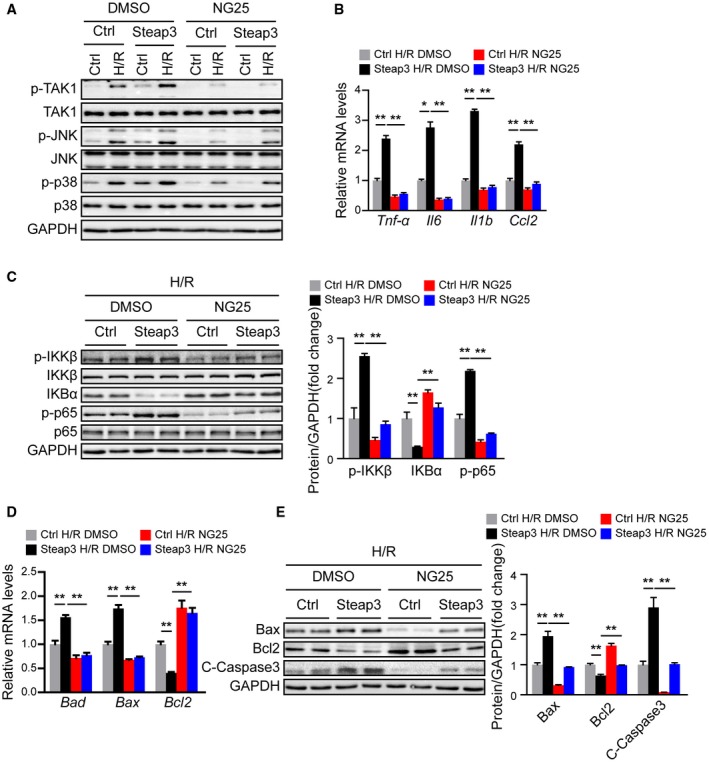
TAK1 mediated the effect of Steap3 on hepatocyte inflammation and apoptosis after H/R treatment. (A) Western blot analysis of total and phosphorylated TAK1, JNK, and p38 in Steap3 overexpressed hepatocytes treated with NG25 subjected to H/R challenge. GAPDH served as the loading control. Representative of three independent experiments. (B) mRNA levels of proinflammatory factors (Tnf‐α, Il6, Il1b, and Ccl2) in Steap3 overexpressed hepatocytes treated with NG25 subjected to H/R challenge. (C) Protein levels of NF‐κB signaling pathway molecules in Steap3 overexpressed hepatocytes treated with NG25 subjected to H/R challenge. GAPDH served as the loading control. Representative of three independent experiments. (D) mRNA levels of apoptosis‐related genes (Bad, Bax, and Bcl2) in Steap3 overexpressed hepatocytes treated with NG25 subjected to H/R challenge. (E) Western blot analysis of the Bax, Bcl2, and cleaved caspase‐3 levels in Steap3 overexpressed hepatocytes treated with NG25 subjected to H/R challenge. GAPDH served as the loading control. Representative of three independent experiments. All data are shown as the mean ± SD. Levels of statistical significance are indicated as *P < 0.05, **P < 0.01. For statistical analysis, one‐way ANOVAs with Bonferroni’s post hoc analysis or Tamhane’s T2 post hoc analysis were used. Abbreviations: IκBα, inhibitory κBα; IKKβ, IκB kinase β.
Discussion
Hepatic I/R injury includes a complex network and multiple molecular mechanisms and cellular interactions.2 Of these, iron‐mediated apoptosis and the inflammatory response play integral roles.28 As a member of the iron regulatory protein family, Steap3 functions as a ferric reductase that reduces ferric iron to ferrous iron in endosomes.29 More importantly, Steap3 is vital in apoptosis and cell‐cycle progression, especially in G2‐M progression.30 However, the biological and functional roles of Steap3 in I/R‐induced liver damage remain poorly understood. In the present study, we observed that the Steap3 protein level was up‐regulated after hepatic I/R. However, Steap3 mRNA levels were almost unchanged after hepatic I/R, suggesting that the changes of Steap3 after liver I/R injury mainly occur at the level of protein modification after translation, but the specific mechanism needs to be further explored. Meanwhile, experiments with Steap3‐KO and Steap3‐HTG mice revealed a harmful role for Steap3 in hepatic I/R injury. Further molecular research demonstrated that Steap3 physically binds to TAK1 and promotes the activation of TAK1–JNK/p38 signaling. Collectively, our results demonstrated that Steap3 is a key mediator of hepatic I/R injury and has important functions in the pathological process.
Inflammation has been considered one of the hallmarks of I/R injury, and inhibiting inflammation can effectively prevent or interfere with liver I/R injury.31 The inflammatory response during hepatic I/R injury is characterized by the induction of a cascade of proinflammatory mediators that culminates in the recruitment of leukocytes to the postischemic tissue, leading to parenchymal cell injury.32 During hepatic I/R injury, the activation of hepatocytes and Kupffer cells releases proinflammatory factors such as Tnf‐α, Il6, and Ccl2, which are transcriptionally regulated by NF‐κB activation, to attract neutrophil‐like CD11b+Ly6C+ monocytes, which egress from the bone marrow and migrate to the liver, ultimately resulting in liver damage.33 Additionally, melatonin pretreatment enhances the therapeutic effects of exogenous mitochondria on hepatic I/R injury in rats by suppressing the expression of inflammatory mediators (Tnf‐α/NF‐κB/Il1b/matrix metalloproteinase 9).6 Steap3 is reported to be important in regulating inflammatory responses, and deleting Steap3 causes impaired TLR4‐mediated inflammatory responses.17 Consistently, in the present study, we demonstrated that Steap3 deletion reduced the expression of Il6, Il1b, Ccl2, and Tnf‐α after hepatic I/R injury. Moreover, Steap3 deficiency could suppress NF‐κB activation and reduce CD11b‐positive inflammatory cell infiltration after hepatic I/R injury, whereas Steap3 overexpression displayed the opposite pattern. More importantly, through separating primary hepatocytes from Steap3‐KO and Steap3‐HTG mice, we found that Steap3 in hepatocytes regulated the inflammatory response after H/R through NF‐κB. Therefore, we speculate that Steap3 in hepatocytes activates the NF‐κB pathway, which promotes the secretion of a series of inflammatory factors; these inflammatory factors then recruit the CD11b+Ly6C+ monocytes to the liver during hepatic I/R injury.
During hepatic I/R injury, various stress factors can activate the apoptotic pathway mediated by mitochondria, which leads to an increase in the apoptosis rate and a decrease in parenchymal cells; thus, hepatic tissue and liver function are severely damaged.34 It is well known that Bax promotes intrinsic apoptosis by forming oligomers in the mitochondrial outer membrane, participating in the release of apoptogenic molecules. In contrast, Bcl2 inhibits mitochondrial apoptosis by blocking the release and oligomerization of Bax.35 In addition, antiapoptotic measures, including overexpressing Bcl2, knocking out Bax, and inhibiting caspase‐3, can protect the liver from I/R injury.36 In previous studies, Steap3 was originally identified as a p53‐inducible protein and an inhibitor of cell growth through the induction of apoptosis through a caspase‐3‐dependent pathway.37 Moreover, adenoviral delivery of the rat Steap3 homolog pHyde into human prostate cancer cells synergizes with cisplatin to cause growth suppression and apoptosis.16 Consistently, in this study, we found that Steap3 induced hepatocyte apoptosis, as indicated by the enhanced levels of cleaved caspase‐3, Bax, and Bad in the liver of Steap3‐HTG mice compared with that of NTG control mice, while the liver of Steap3‐KO mice exhibited the opposite effect. Taken together, our results illustrate that accumulated Steap3 promotes hepatocyte apoptosis after I/R injury.
The MAPK signaling pathway family members JNK and p38 play critical biological roles, including in inflammation, cell‐cycle regulation, and apoptosis, in the progression of hepatic I/R injury.38 During hepatic I/R, ROS directly act on cell membrane receptors to phosphorylate p38, which promotes the translocation of Bax, leading to the release of cytochrome c and the activation of caspase‐9 and caspase‐3.39 Phosphorylated p38 can also act on monocytes/macrophages or promote the transcription of NF‐κB, resulting in the release of more Tnf‐α, Il6, and Il1b.40 In addition, JNK is phosphorylated and activated by several types of stresses, including stimulation by cytokines such as Tnf‐α and IL‐1 and environmental stresses such as radiation and oxidative stress.41 The inhibition of JNK activation can ameliorate liver I/R injury through increased expression of antiapoptotic Bcl2 and decreased expression of proapoptotic Bax.42 However, there have been no reports on the function of Steap3 in the regulation of MAPK signaling. We observed significantly higher levels of phosphorylated JNK and p38 signaling in the liver and primary hepatocytes from Steap3‐HTG mice compared with those from NTG mice after I/R or H/R insult, respectively, whereas the liver and primary hepatocytes from Steap3‐KO mice showed the opposite trend. Together, these observations indicate that in hepatocytes Steap3 activates the phosphorylation of JNK and p38 during hepatic I/R‐induced liver injury.
TAK1, a MAPK kinase family serine threonine kinase, has been implicated in the regulation of a diverse range of cellular processes that include cellular death and inflammatory responses and acts as a key prime upstream molecular regulator of MAPK activation.21 A previous study showed that deletion of TAK1 results in autonomous apoptosis in hepatocytes and hematopoietic cells.43 Interestingly, other studies have shown that deletion of TAK1 can protect hepatocytes against I/R injury.44 In the present study, we demonstrated that TAK1 was a molecular target during the regulation of liver I/R injury by Steap3. We found that Steap3 physically interacted with TAK1 through specific binding domains and then activated TAK1 phosphorylation to aggravate hepatic I/R injury. Therefore, TAK1 may need to maintain a balanced level to support its molecular, cellular, and functional behaviors during hepatic I/R injury. Additionally, the different partners of TAK1 and their corresponding biological activities are responsible for the dual roles of TAK1. The results of the present study demonstrated that Steap3 interacted with the N‐terminal domain of TAK1. Further experiments demonstrated that TAK1 mediated the effect of Steap3 on hepatocyte inflammation and apoptosis after H/R treatment. Thus, targeting Steap3–TAK1 is a strategy for clinical intervention in hepatic I/R injury.
In conclusion, our study reveals that Steap3 is a regulator of hepatic I/R injury. The Steap3–TAK1 interaction activates the phosphorylation of MAPK signaling, subsequently leading to apoptosis and inflammation. In addition, these findings broaden our understanding of the direct regulatory role of Steap3 in hepatocytes. Therefore, the Steap3–TAK1–JNK/p38 regulatory axis could be an essential mechanism of hepatic I/R injury.
Author Contributions
W.‐Z.G, H.‐B.F., S.‐L.C., and S.‐J.Z. participated in research design; S.‐Y.C, J.L., J.‐H.S., H.‐W.T., Y.Z., P.‐H.W., J.‐K.Z., Z.‐H.W., X.‐Y.S., C.P., Y.H., and B.‐W.H. conducted experiments; all authors performed data analysis and interpretation; W.‐Z.G, H.‐B.F., and S.‐L.C. drafted the paper. S.‐J.Z supervised the study, and all authors read and approved the final manuscript.
Supporting information
Potential conflict of interest: Nothing to report.
Supported by grants from the National Natural Science Foundation of China (U1604282, 81571947, 81671958) and the Supporting Plan for Scientific and Technological Innovative Talents in Universities of Henan Province (19HASTIT003).
Contributor Information
Wen‐Zhi Guo, Email: guowz66@163.com, Email: zhangshuijun@zzu.edu.cn.
Shui‐Jun Zhang, Email: zhangshuijun@zzu.edu.cn.
References
Author names in bold designate shared co‐first authorship.
- 1. Nakamura K, Kageyama S, Yue S, Huang J, Fujii T, Ke B, et al. Heme oxygenase‐1 regulates sirtuin‐1‐autophagy pathway in liver transplantation: from mouse to human. Am J Transplant 2018;18:1110‐1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhai Y, Petrowsky H, Hong JC, Busuttil RW, Kupiec‐Weglinski JW. Ischaemia‐reperfusion injury in liver transplantation—from bench to bedside. Nat Rev Gastroenterol Hepatol 2013;10:79‐89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Casillas‐Ramírez A, Mosbah IB, Ramalho F, Roselló‐Catafau J, Peralta C. Past and future approaches to ischemia‐reperfusion lesion associated with liver transplantation. Life Sci 2006;79:1881‐1894. [DOI] [PubMed] [Google Scholar]
- 4. Motiño O, Francés DE, Casanova N, Fuertes‐Agudo M, Cucarella C, Flores JM, et al. Protective role of hepatocyte cyclooxygenase‐2 expression against liver ischemia‐reperfusion injury in mice. Hepatology 2019;70:650‐665. [DOI] [PubMed] [Google Scholar]
- 5. Ricca L, Lemoine A, Cauchy F, Hamelin J, Sebagh M, Esposti DD, et al. Ischemic postconditioning of the liver graft in adult liver transplantation. Transplantation 2015;99:1633‐1643. [DOI] [PubMed] [Google Scholar]
- 6. Chen HH, Chen YT, Yang CC, Chen KH, Sung PH, Chiang HJ, et al. Melatonin pretreatment enhances the therapeutic effects of exogenous mitochondria against hepatic ischemia‐reperfusion injury in rats through suppression of mitochondrial permeability transition. J Pineal Res 2016;61:52‐68. [DOI] [PubMed] [Google Scholar]
- 7. van Golen RF, Reiniers MJ, Olthof PB, van Gulik TM, Heger M. Sterile inflammation in hepatic ischemia/reperfusion injury: present concepts and potential therapeutics. J Gastroenterol Hepatol 2013;28:394‐400. [DOI] [PubMed] [Google Scholar]
- 8. Zhai Y, Busuttil RW, Kupiec‐Weglinski JW. Liver ischemia and reperfusion injury: new insights into mechanisms of innate‐adaptive immune‐mediated tissue inflammation. Am J Transplant 2011;11:1563‐1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Arrenberg P, Maricic I, Kumar V. Sulfatide‐mediated activation of type II natural killer T cells prevents hepatic ischemic reperfusion injury in mice. Gastroenterology 2011;140:646‐655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Elias‐Miró M, Jiménez‐Castro MB, Rodés J, Peralta C. Current knowledge on oxidative stress in hepatic ischemia/reperfusion. Free Radic Res 2013;47:555‐568. [DOI] [PubMed] [Google Scholar]
- 11. Tao X, Wan X, Xu Y, Xu L, Qi Y, Yin L, et al. Dioscin attenuates hepatic ischemia‐reperfusion injury in rats through inhibition of oxidative‐nitrative stress, inflammation and apoptosis. Transplantation 2014;98:604‐611. [DOI] [PubMed] [Google Scholar]
- 12. Zeng S, Dun H, Ippagunta N, Rosario R, Zhang QY, Lefkowitch J, et al. Receptor for advanced glycation end product (RAGE)–dependent modulation of early growth response‐1 in hepatic ischemia/reperfusion injury. J Hepatol 2009;50:929‐936. [DOI] [PubMed] [Google Scholar]
- 13. Kleven MD, Dlakić M, Lawrence CM. Characterization of a single b‐type heme, FAD, and metal binding sites in the transmembrane domain of six‐transmembrane epithelial antigen of the prostate (STEAP) family proteins. J Biol Chem 2015;290:22558‐22569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Blanc L, Papoin J, Debnath G, Vidal M, Amson R, Telerman A, et al. Abnormal erythroid maturation leads to microcytic anemia in the TSAP6/Steap3 null mouse model. Am J Hematol 2015;90:235‐241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Isobe T, Baba E, Arita S, Komoda M, Tamura S, Shirakawa T, et al. Human STEAP3 maintains tumor growth under hypoferric condition. Exp Cell Res 2011;317:2582‐2591. [DOI] [PubMed] [Google Scholar]
- 16. Grunewald TG, Bach H, Cossarizza A, Matsumoto I. The STEAP protein family: versatile oxidoreductases and targets for cancer immunotherapy with overlapping and distinct cellular functions. Biol Cell 2012;104:641‐657. [DOI] [PubMed] [Google Scholar]
- 17. Zhang F, Tao Y, Zhang Z, Guo X, An P, Shen Y, et al. Metalloreductase Steap3 coordinates the regulation of iron homeostasis and inflammatory responses. Haematologica 2012;97:1826‐1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sakai N, Van Sweringen HL, Schuster R, Blanchard J, Burns JM, Tevar AD, et al. Receptor activator of nuclear factor‐κB ligand (RANKL) protects against hepatic ischemia/reperfusion injury in mice. Hepatology 2012;55:888‐897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang C, Huang J, An W. Hepatic stimulator substance resists hepatic ischemia/reperfusion injury by regulating Drp1 translocation and activation. Hepatology 2017;66:1989‐2001. [DOI] [PubMed] [Google Scholar]
- 20. Sun P, Zhang P, Wang PX, Zhu LH, Du Y, Tian S, et al. Mindin deficiency protects the liver against ischemia/reperfusion injury. J Hepatol 2015;63:1198‐1211. [DOI] [PubMed] [Google Scholar]
- 21. Wang X, Mao W, Fang C, Tian S, Zhu X, Yang L, et al. Dusp14 protects against hepatic ischaemia‐reperfusion injury via Tak1 suppression. J Hepatol 2018;68:118‐129. [DOI] [PubMed] [Google Scholar]
- 22. Hou J, Xia Y, Jiang R, Chen D, Xu J, Deng L, et al. PTPRO plays a dual role in hepatic ischemia reperfusion injury through feedback activation of NF‐κB. J Hepatol 2014;60:306‐312. [DOI] [PubMed] [Google Scholar]
- 23. Zhang XJ, Cheng X, Yan ZZ, Fang J, Wang X, Wang W, et al. An ALOX12–12‐HETE–GPR31 signaling axis is a key mediator of hepatic ischemia‐reperfusion injury. Nat Med 2018;24:73‐83. [DOI] [PubMed] [Google Scholar]
- 24. Yang L, Wang W, Wang X, Zhao J, Xiao L, Gui W, et al. Creg in hepatocytes ameliorates liver ischemia/reperfusion injury in a TAK1‐dependent manner in mice. Hepatology 2019;69:294‐313. [DOI] [PubMed] [Google Scholar]
- 25. Glanemann M, Strenziok R, Kuntze R, Münchow S, Dikopoulos N, Lippek F, et al. Ischemic preconditioning and methylprednisolone both equally reduce hepatic ischemia/reperfusion injury. Surgery 2004;135:203‐214. [DOI] [PubMed] [Google Scholar]
- 26. Duarte S, Shen XD, Fondevila C, Busuttil RW, Coito AJ. Fibronectin‐α4β1 interactions in hepatic cold ischemia and reperfusion injury: regulation of MMP‐9 and MT1‐MMP via the p38 MAPK pathway. Am J Transplant 2012;12:2689‐2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang Z, Zhang H, Shi M, Yu Y, Wang H, Cao WM, et al. TAK1 inhibitor NG25 enhances doxorubicin‐mediated apoptosis in breast cancer cells. Sci Rep 2016;6:32737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Scindia Y, Dey P, Thirunagari A, Liping H, Rosin DL, Floris M, et al. Hepcidin mitigates renal ischemia‐reperfusion injury by modulating systemic iron homeostasis. J Am Soc Nephrol 2015;26:2800‐2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Han M, Xu R, Wang S, Yang N, Ni S, Zhang Q, et al. Six‐transmembrane epithelial antigen of prostate 3 predicts poor prognosis and promotes glioblastoma growth and invasion. Neoplasia 2018;20:543‐554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gomes IM, Maia CJ, Santos CR. STEAP proteins: from structure to applications in cancer therapy. Mol Cancer Res 2012;10:573‐587. [DOI] [PubMed] [Google Scholar]
- 31. Yoshida O, Kimura S, Jackson EK, Robson SC, Geller DA, Murase N, et al. CD39 expression by hepatic myeloid dendritic cells attenuates inflammation in liver transplant ischemia‐reperfusion injury in mice. Hepatology 2013;58:2163‐2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Husted TL, Lentsch AB. The role of cytokines in pharmacological modulation of hepatic ischemia/reperfusion injury. Curr Pharm Des 2006;12:2867‐2873. [DOI] [PubMed] [Google Scholar]
- 33. Song P, Zhang J, Zhang Y, Shu Z, Xu P, He L, et al. Hepatic recruitment of CD11b+Ly6C+ inflammatory monocytes promotes hepatic ischemia/reperfusion injury. Int J Mol Med 2018;41:935‐945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sun K, Liu ZS, Sun Q. Role of mitochondria in cell apoptosis during hepatic ischemia‐reperfusion injury and protective effect of ischemic postconditioning. World J Gastroenterol 2004;10:1934‐1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shen M, Lu J, Dai W, Wang F, Xu L, Chen K, et al. Ethyl pyruvate ameliorates hepatic ischemia‐reperfusion injury by inhibiting intrinsic pathway of apoptosis and autophagy. Mediators Inflamm 2013;2013:461536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Feng J, Zhang Q, Mo W, Wu L, Li S, Li J, et al. Salidroside pretreatment attenuates apoptosis and autophagy during hepatic ischemia‐reperfusion injury by inhibiting the mitogen‐activated protein kinase pathway in mice. Drug Des Devel Ther 2017;11:1989‐2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang X, Steiner MS, Rinaldy A, Lu Y. Apoptosis induction in prostate cancer cells by a novel gene product, pHyde, involves caspase‐3. Oncogene 2001;20:5982‐5990. [DOI] [PubMed] [Google Scholar]
- 38. Coulthard LR, White DE, Jones DL, McDermott MF, Burchill SA. p38(MAPK): stress responses from molecular mechanisms to therapeutics. Trends Mol Med 2009;15:369‐379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gibson EM, Henson ES, Haney N, Villanueva J, Gibson SB. Epidermal growth factor protects epithelial‐derived cells from tumor necrosis factor‐related apoptosis‐inducing ligand‐induced apoptosis by inhibiting cytochrome c release. Cancer Res 2002;62:488‐496. [PubMed] [Google Scholar]
- 40. Li J, Wang F, Xia Y, Dai W, Chen K, Li S, et al. Astaxanthin pretreatment attenuates hepatic ischemia reperfusion‐induced apoptosis and autophagy via the ROS/MAPK pathway in mice. Mar Drugs 2015;13:3368‐3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Uehara T, Bennett B, Sakata ST, Satoh Y, Bilter GK, Westwick JK, et al. JNK mediates hepatic ischemia reperfusion injury. J Hepatol 2005;42:850‐859. [DOI] [PubMed] [Google Scholar]
- 42. Ocuin LM, Zeng S, Cavnar MJ, Sorenson EC, Bamboat ZM, Greer JB, et al. Nilotinib protects the murine liver from ischemia/reperfusion injury. J Hepatol 2012;57:766‐773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tang M, Wei X, Guo Y, Breslin P, Zhang S, Zhang S, et al. TAK1 is required for the survival of hematopoietic cells and hepatocytes in mice. J Exp Med 2008;205:1611‐1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hu J, Zhu XH, Zhang XJ, Wang PX, Zhang R, Zhang P, et al. Targeting TRAF3 signaling protects against hepatic ischemia/reperfusions injury. J Hepatol 2016;64:146‐159. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials