

Abstract
Rationale
Associations between urban (outdoor) airborne particulate matter (PM) exposure and TB and potential biological mechanisms are poorly explored.
Objectives
To examine whether in vivo exposure to urban outdoor PM in Mexico City and in vitro exposure to urban outdoor PM2.5 (< 2.5μm median aerodynamic diameter) alters human host immune cell responses to Mycobacterium tuberculosis.
Methods
Cellular toxicity (flow cytometry, proliferation assay (MTS assay)), M. tuberculosis and PM2.5 phagocytosis (microscopy), cytokine-producing cells (Enzyme-linked immune absorbent spot (ELISPOT)), and signalling pathway markers (western blot) were examined in bronchoalveolar cells (BAC) and peripheral blood mononuclear cells (PBMC) from healthy, non-smoking, residents of Mexico City (n=35; 13 female, 22 male). In vivo-acquired PM burden in alveolar macrophages (AM) was measured by digital image analysis.
Measurements and main results
In vitro exposure of AM to PM2.5 did not affect M. tuberculosis phagocytosis. High in vivo-acquired AM PM burden reduced constitutive, M. tuberculosis and PM-induced interleukin-1β production in freshly isolated BAC but not in autologous PBMC while it reduced constitutive production of tumour necrosis factor-alpha in both BAC and PBMC. Further, PM burden was positively correlated with constitutive, PM, M. tuberculosis and purified protein derivative (PPD)-induced interferon gamma (IFN-γ) in BAC, and negatively correlated with PPD-induced IFN-γ in PBMC.
Conclusions
Inhalation exposure to urban air pollution PM impairs important components of the protective human lung and systemic immune response against M. tuberculosis. PM load in AM is correlated with altered M. tuberculosis-induced cytokine production in the lung and systemic compartments. Chronic PM exposure with high constitutive expression of proinflammatory cytokines results in relative cellular unresponsiveness.
INTRODUCTION
Fifty-five per cent of the world’s population currently reside and 68% are projected to reside in urban areas in 2050.1 With these rapid global urbanisation trends, exposure to urban airborne particulate matter (PM) represents a major global public health concern. The WHO reports that 4.3 million people die prematurely annually from illness attributable to household air pollution and an additional 3.3 million people due to outdoor air pollution exposures.2 Air pollution exposure is correlated with a multitude of pulmonary and systemic ailments including respiratory infections, asthma, COPD, cancers, cardiovascular and metabolic diseases.3,4 Following inhalation exposure, PM can be found systemically and detected in urines of exposed children.5
In low/middle-income countries, large populations in TB endemic regions live in urban centres, such as Mexico City where air pollution exposures are frequent. With an estimated 10.4 million new cases worldwide and 1.8 million deaths (including 0.4 million with HIV), TB is the leading cause of mortality among infectious diseases globally.6
Mycobacterium tuberculosis infection and TB development are associated with inhalation exposure to silica,7 tobacco smoke8–11 and household air pollution.12,13 Whether exposure to urban outdoor air PM, similarly, increases the risk of M. tuberculosis infection and TB development is subject of ongoing research.14 A recent study showed increased mortality from TB associated with living near major roadways in the USA.15 Mechanistic studies examining PM effects on antimycobacterial immunity, particularly in human immune cells, are limited16,17 and rare in human primary lung immune cells to date.
We had previously shown that diesel exhaust particles and urban PM suppress host immune responses to M. tuberculosis in human peripheral blood mononuclear cells (PBMC)17 and respiratory epithelial cell A549,16 respectively.
The current study examined whether inhalation exposure to PM in Mexico City and in vitro exposure to urban outdoor PM2.5 (<2.5 μm median aerodynamic diameter) alters immune responses to M. tuberculosis in bronchoalveolar cells (BAC), alveolar macrophages (AM) and autologous PBMC of inhabitants of two neighbouring municipalities, Iztapalapa and Iztacalco, in Mexico City. PM2.5 is the most relevant component of air pollution PM, a good proxy for an air pollutant that predominately derives from anthropogenic sources and reaches terminal bronchioles and alveoli where it induces and/or modifies local immune responses.18
We demonstrate that the in vivo-acquired PM burden in AM is negatively correlated with M. tuberculosis-induced interleukin (IL)-1β expression, positively correlated with M. tuberculosis and purified protein derivative (PPD)-induced interferon gamma (IFN-γ) expression in BAC and negatively correlated with PPD-induced IFN-γ expression in PBMC.
The findings from this study suggest that PM burden of AM resulting from in vivo inhalation exposure to urban air pollution impairs the magnitude of inflammatory cytokine production in BAC and PBMC in response to M. tuberculosis. PM-induced modulations of the immune cell functions may weaken protective antimycobacterial human host immune responses in urbanised areas with high levels of outdoor air pollution supporting epidemiological study findings of increased risk of TB development and altered therapy outcomes in air-polluted urban environments.
MATERIALS AND METHODS
Study participants
Venipunctures and bronchoalveolar lavages were conducted between October 2013 and September 2017. Thirty-five healthy, HIV-1 seronegative, non-smoking (urine cotinine-negative) study participants (13 women, 37%; 22 men, 63%; median age 29 years [min 21, max 51]) were recruited at the Instituto Nacional de Enfermedades Respiratorias. None of the study participants described exposures to smoke from biomass fuel in their living or work environment. All of the study participants’ home cooking was done with butane and natural gas. Further details are provided in the online supplementary material.
Primary human immune cells
PBMC and BAC were obtained as described previously.19 By cytospin analysis (n=32), BAC consisted of 91.5% AM (mean; max 99.3%, min 76.3%), 8.5% alveolar lymphocytes (AL, mean; max 23.7%, min 0.7%) and scarce neutrophils. By additional flow cytometry (n=8), BAC consisted of 85.64% AM (mean; max 89%, min 81.9%) and 10.8% AL (mean; max 16.4%, min 5.7%).
Collection of urban outdoor PM in Mexico City and preparation for in vitro studies
PM2.5 was collected on the rooftop of the National Institute of Ecology and Climate Change (Centro Nacional de Investigación y Capacitación Ambiental) at the Universidad Autónoma Metropolitana/Iztapalapa. Immediately prior to in vitro exposure experiments, PM2.5 suspensions were prepared. Details are provided in the online supplementary material.
Preparation of M. tuberculosis for in vitro infections
Preparation of M. tuberculosis H37Ra (ATCC 25177, Manassas, VA) for in vitro infection experiments was done as described previously.19–21 Details are provided in the online supplementary material.
Phagocytosis of M. tuberculosis by human AM
Proportions of cells containing one or more intracellular M. tuberculosis were determined by bright field microscopy (1000×, oil immersion) of cytospin preparations in a total of 300 randomly selected AMs from each participant at each experimental condition. Details are provided in the online supplementary material.
Cell viability, necrosis and apoptosis
Cell viability was assessed by LIVE/DEAD Fixable Far Red Dead Cell Stain (Life Technologies, Frederick, MD, USA) and GFP-Certified Apoptosis/Necrosis detection kit (Enzo Life Sciences, Farmingdale, NY, USA) according to the manufacturers’ instructions. Details are provided in the online supplementary material.
Assessment of in vivo PM burden in AM
Uptake of PM by AM was assessed microscopically on thin-layer cytospin slides (Wescor, Logan, UT) following Accustain Wright staining (Sigma Aldrich, St Louis, MO). Proportions of AM containing PM (%AM with PM) were examined in 300 randomly selected AMs from 120 000 BACs from each participant. Colour photographs of 50 randomly selected PM-containing AMs from each participant, obtained by digital bright field microscopy (1000×, Olympus BX51, Olympus DP71 camera), were evaluated with ImageJ software (V.1.49, https://imagej.nih.gov/ij/, for details see the online supplement material) to determine the area of AM occupied by PM relative to the total AM area (PM load per AM).
Cytokine ELISPOT assays
Frequencies of IL-1β, tumour necrosis factor alpha (TNF-α), IL-6 and IFN-γ-producing cells were assessed by enzyme-linked immune absorbent spot (ELISPOT) assays as described previously.17,21,22 Details are provided in the online supplementary material.
Western blotting
Activation of signalling pathways was assessed by western blotting. Details are provided in the online supplementary material.
Statistical analysis
For comparing conditions while controlling for repeated measures on study participants, we used analysis of variance (ANOVA) Friedman tests followed by Dunn’s multiple comparisons. Friedman tests, non-parametric repeated measures ANOVA, were used because viability and phagocytosis data were not normally distributed. We performed a Mann-Whitney test for cytokine production and comparison between individuals with higher and lower PM load or %AM with PM. Correlations between %AM with PM and cytokine production were analysed using Spearman’s rank correlation test. A CI of 95% (α = 0.05) was used for all conditions. All statistical analyses in this study were performed with GraphPad Prism software (for Mac OS X V.6.0).
RESULTS
Cell viability upon PM2.5 exposure
A significant loss of cell viability was observed in AM at exposures to 25 μg/mL PM2.5 for 24 (p=0.025) and 48 hours (p=0.014) (figure 1A), while no PM-induced decrease of cell viability was noted in peripheral blood monocytes (PBMN) (figure 1B), alveolar lymphocytes (AL) (figure 1C) or peripheral blood lymphocytes (PBL; figure 1D) using LIVE/DEAD reagents and flow cytometry.
Figure 1.
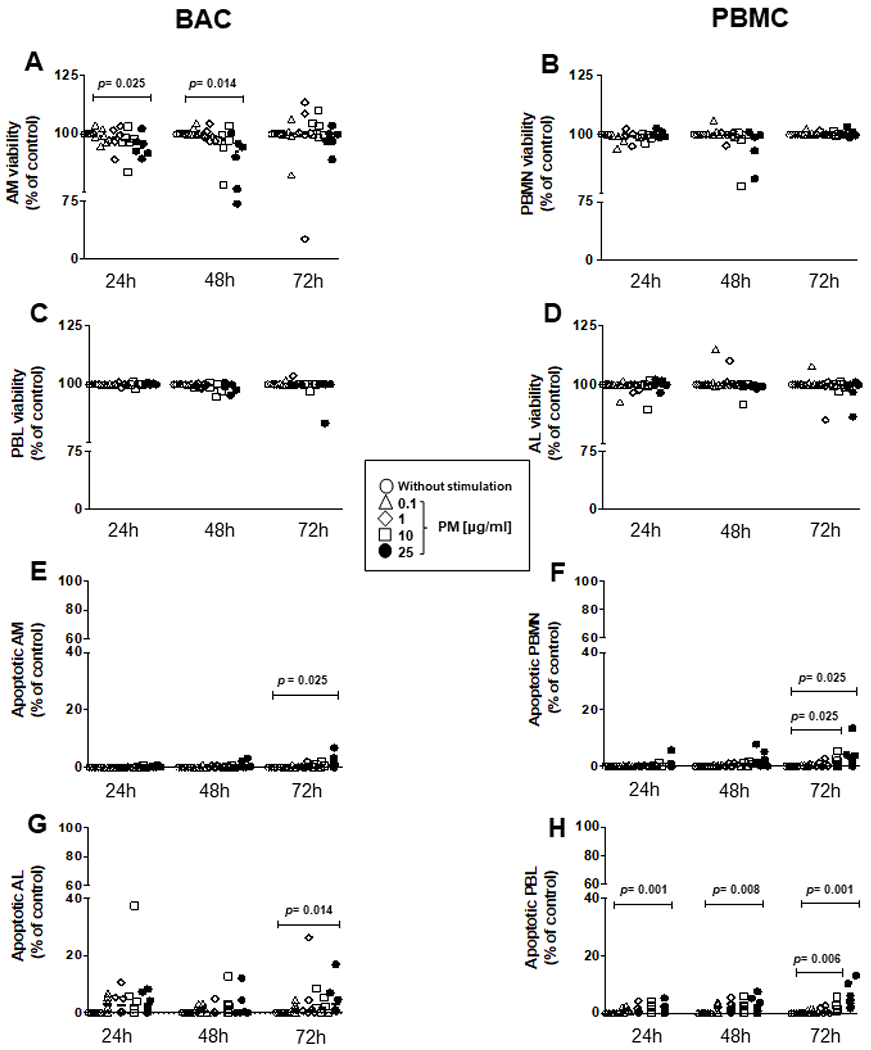
Cytotoxic effects of PM2.5 are observed only at high PM2.5 concentrations. BAC and PBMC (each n=6) were exposed to 0, 0.1, 1, 10 and 25 μg/mL of PM2.5 and incubated for 24–72 hours to evaluate particulate matter (PM) exposure effects on cell viability (A–D) and induction of apoptosis (E–H). (A) Proportions of live alveolar macrophages (AM), (B) peripheral blood monocytes (PBMN), (C) alveolar lymphocytes (AL), and (D) peripheral blood lymphocytes (PBL) were examined by a LIVE/DEAD Fixable Far Red Dead Cell Stain and analysed by flow cytometry with fluorescence detection at 660 nm (A–D). Apoptosis was identified with a far red-emitting DNA-intercalating dye by flow cytometry using a 488 nm laser with fluorescence detection at 570 nm (Apoptosis Detection Reagent) in a flourescence-activated cell sorting (FACS) fusion cytometer. (E) AM, (F) PBMN, (G) AL and (H) PBL. Friedman and Dunn’s post hoc test for multiple comparison was performed. The p values above horizontal lines represent the indicated significant differences comparison. BAC, bronchoalveolar cell; PBMC, peripheral blood mononuclear cell.
Apoptosis, assessed by GFP-Apoptosis/Necrosis detection kit and flow cytometry, was induced on exposure to 25 μg/mL of PM2.5 at 72 hours only in AM (figure 1E) (p=0.025), PBMN (figure 1F) (p=0.025), AL (figure 1G) (p=0.014) and PBL (figure 1H) (p=0.01). In addition, induction of apoptosis was observed in PBMN exposed to PM2.5 at 10 μg/mL at 72 hours only (figure 1F) (p=0.006), in PBL exposed to 25 μg/mL PM2.5 for 24 (p=0.001) and 48 hours (p=0.008) and in PBL exposed 10 μg/mL PM2.5 at 72 hours (data not shown). In conclusion, our results indicate that PM effects on viability and apoptosis are increased at higher PM concentrations and longer experimental exposure times.
No significant decrease in cell viability was observed in BAC figure 1G and PBMC figure 1H exposed to 0.1, 1 and 10 μg/mL of PM2.5 for 24 and 48 hours (data not shown). As only minor alterations in the cell viability were observed with these experimental conditions, all subsequent experiments assessing cellular immune responses to M. tuberculosis were done using PM2.5 exposures at 0.1 and 10 μg/mL for time periods between 0 and 48 hours only.
Flow cytometry gating strategy
Cell viability was assessed by gating on the side scatter (SSC-H) versus SSC-A to select single cells (online supplementary figure 1A), followed by analysis of cell subpopulations by size and granularity (online supplementary figure 1B) and development of single histogram plots identifying live cells using LIVE/DEAD Fixable Far Red Dead Cell Stain (online supplementary figure 1C,D)
M. tuberculosis phagocytosis by in vitro PM2.5-exposed AM
In an earlier study by coauthor AOV,23 ambient PM-induced inflammatory cytokine responses in THP-1 cells varied on exposures to PM collected in different weather seasons at multiple sites in Mexico City due to differences in the chemical composition of the PM.23 We therefore examined whether PM2.5 from cold-dry, warm-dry and rainy seasons in Mexico City differentially affects M. tuberculosis phagocytosis (online supplementary figure 2). No significant effects of pre-exposure to PM2.5 from any of the three weather seasons were observed on M. tuberculosis phagocytosis. As expected, the greater the multiplicity of infection (MOI) the higher were the proportions of AM containing M. tuberculosis (online supplementary figure 2).
Since there was no significant effect of seasonal PM on M. tuberculosis phagocytosis in AM, all subsequent experiments were performed with annual bulk PM2.5, that is, PM2.5 from the three weather seasons proportionally combined (see Materials and methods section).
Effect of in vitro PM2.5 exposure on M. tuberculosis-induced inflammatory cytokine production
To examine whether PM exposure modulates key proinflammatory cytokine responses required for protective host immunity to M. tuberculosis,24,25 BAC and PBMC were exposed to PM2.5 in vitro and infected with M. tuberculosis, and assessed for IL-1β, TNF-α, IL-6 and IFN-γ release by ELISPOT assay (online supplementary figure 3A–F).
Representative images of IL-1β, IL-6, TNF-α and IFN-γ-spot-forming cells in BAC are presented in online supplement figure 3A. We observed significantly higher frequencies of constitutively TNF-α and IL-6-producing cells in BAC than in the autologous PBMC (online supplement figure 3B).
No relevant significant effects of PM2.5 exposures on IL-1β, TNF-α, IL-6 and IFN-γ production were noted in BAC or PBMC (online supplement figure 3C–F). Infection with M. tuberculosis at MOI1 and MOI10 did not induce a significant effect on BAC or PBMC. Levels of cytokine production were not significantly higher in BAC compared with PBMC.
Frequencies of IFN-γ-producing PBMC increased significantly compared with unexposed control PBMC on M. tuberculosis infection at MOI1 and MOI5 (online supplement figure 3F).
Consistent with high constitutive frequencies of TNF-α and IL-6-producing BAC, constitutive phosphorylation of IκB-α (indicating activation of nuclear factor [NF]-κB, a transcription factor controlling IL-1β, IL-6 and TNF-α synthesis) and ERK1/2, but not STAT1, was observed in BAC. No such constitutive phosphorylation of IκB-α or ERK1/2 was observed in autologous PBMC (online supplement figure 4). These findings suggest that several molecular pathways are constitutively activated in BAC, but not in autologous PBMC, and led us to examine whether in vivo-acquired PM load of AM modulates M. tuberculosis-induced proinflammatory responses.
AM PM burden
BACs from the participants (n=30) residing in Iztapalapa or Iztacalco municipalities were examined on cytospins for AM PM inclusions (figure 2A,B). %AM with PM (figure 2A) and the area of AM occupied by PM relative to the total AM area (PM load per AM, figure 2B) were used to assess AM PM burden. %AM with PM (figure 2A) and PM load per AM (figure 2B) for all participants ranged from 26% to 62.3% (median 42.6%) and from 0.42% to 6.1% (median 1.6%), respectively. Of note, there was a wide range of PM loads per AM observed among the 50 AMs examined from each study participant (lowest 0.014%, participant CC072; highest 35%, participant CC049, figure 2B). Interestingly, there was no statistically significant correlation between %AM with PM (figure 2A) and PM load per AM (figure 2B). Levels of ambient PM have been reported to be high26,27 in the study area. During the study period, the yearly average for the monthly 24 hours’ average PM2.5 concentrations was 18.8 μg/m3 (max 31.6, min 8.2). BACs that were assessed by ELISPOT (figures 3 and 4) are indicated with grey bars.
Figure 2.
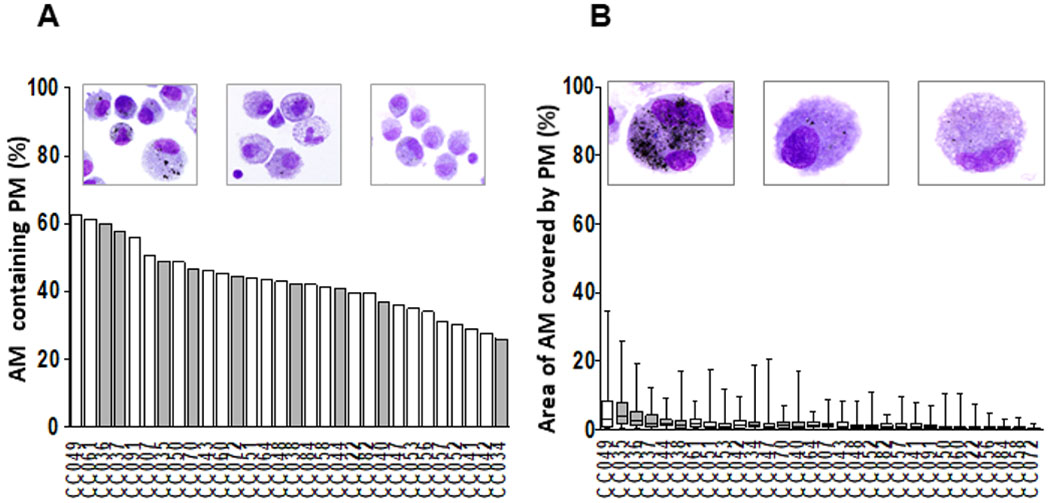
Assessment of PM burden in AM. Wright-stained cytospin preparations of bronchoalveolar cells (BAC) (120 000 BACs per cytospin) from 30 participants (CC #) were examined to assess the proportions of AM containing PM (%AM with PM) (A) and the mean and minimum and maximum areas of the total AM area occupied by PM (PM load per AM) (B) in each study participant. Digital colour images of 50 alveolar macrophages per study participant were obtained using 1000× magnified digital bright field microscopy (Olympus BX51 microscope, Olympus, Waltham, MA). PM load per AM (areas of AM occupied by PM) was determined using ImageJ V.1.49. The areas occupied by PM within each of the AMs are expressed as per cent of the total (100%) AM area. The grey bars represent the study participants whose BACs were assessed by enzyme-linked immune absorbent spot (ELISPOT) (see figures 3–5). AM, alveolar macrophage; PM, particulate matter.
Figure 3.
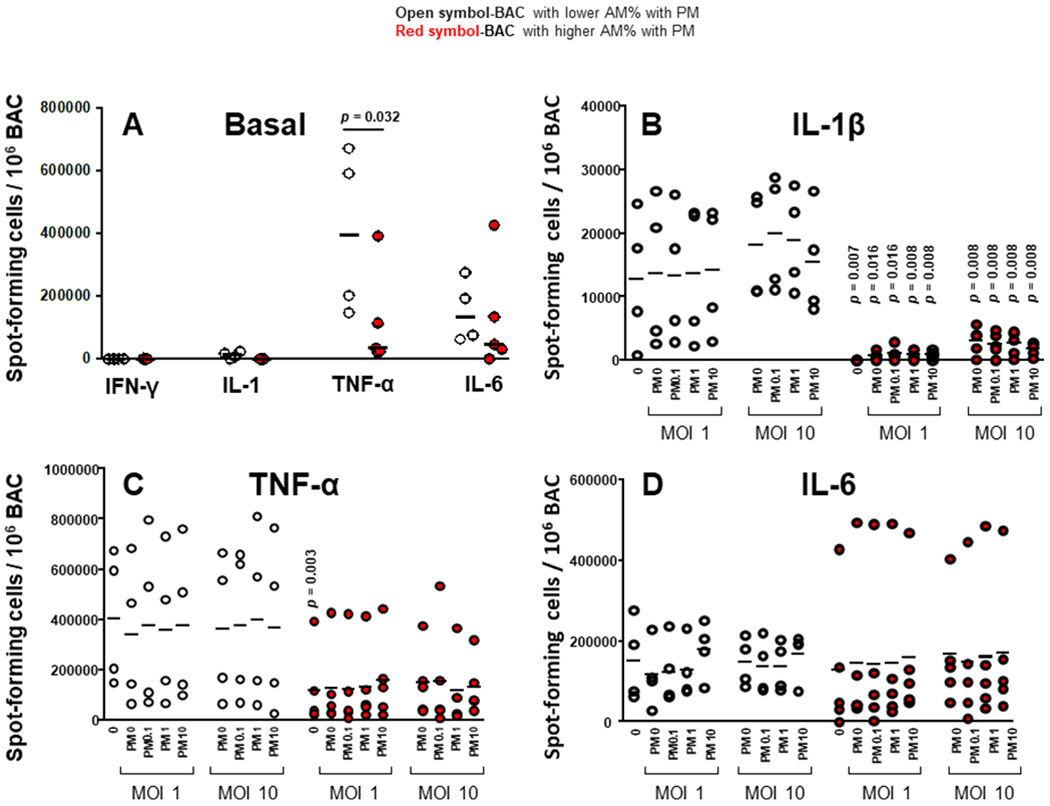
Proinflammatory cytokine production in BAC and peripheral blood mononuclear cells (PBMC) grouped by in vivo-acquired PM burden of AM. Frequencies of constitutive BAC-spot-forming cells (A) and frequencies of IL-1β (B), TNF-α (C) and IL-6 (D) spot-forming cells in PM exposed and/ or Mycobacterium tuberculosis infected, BAC (from n=9 participants) evaluated by enzyme-linked immune absorbent spot (ELISPOT) assay. Closed red symbols represent participants with ≥44.3% AM with PM and open symbols represent participants with <42.6% AM with PM (range: high proportion 44.3%–60%, low proportion 26%–42.6%). Mann-Whitney test was performed using the GraphPad Prism software for Mac OS X V.6.0. P values above symbols represent significant differences in frequencies of cytokine-producing cells between groups with higher and lower %AM with PM for the corresponding experimental condition. For example, in panel B, frequencies of IL-1β-producing BAC following M. tuberculosis infection (MOI1) and PM2.5 exposure from study participants with higher %AM with PM (red symbols) are compared with BAC following M. tuberculosis infection (MOI1) and PM2.5 exposure from study participants with lower %AM with PM (open symbols). AM, alveolar macrophage; BAC, bronchoalveolar cell; IFN, interferon; IL, interleukin; PM, particulate matter; TNF, tumour necrosis factor.
Figure 4.
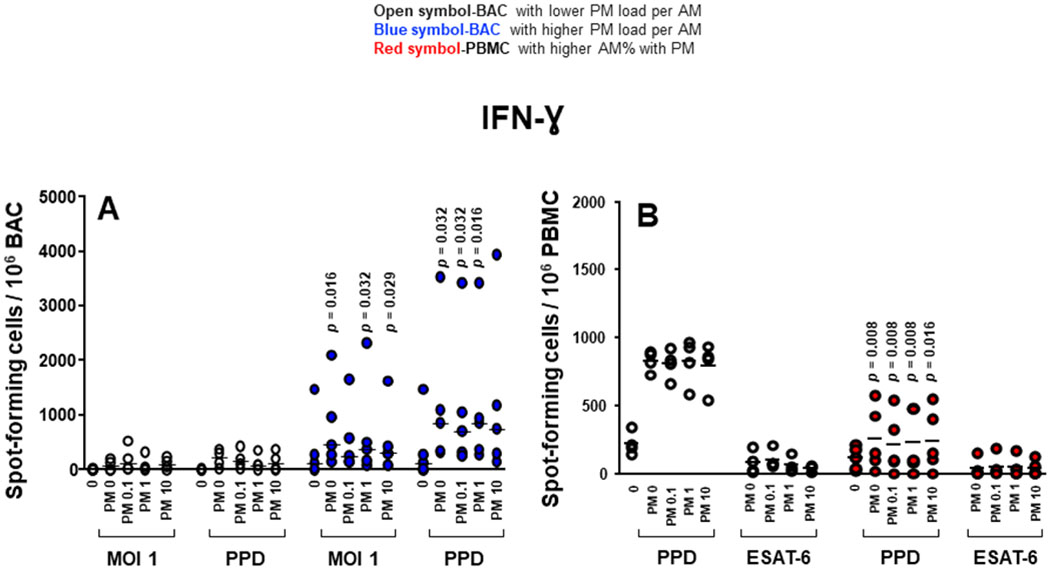
Proinflammatory cytokine production in BAC and PBMC grouped by in vivo-acquired PM burden of AM. (A) Frequencies of IFN-γ-spot-forming cells in PM exposed and/or Mycobacterium tuberculosis infected, PPD and 6 kDa early secretory antigenic target (ESAT-6)-stimulated BAC (from nine participants). (B) Frequencies of IFN-γ-spot-forming cells in PM exposed and/or M. tuberculosis infected, PPD and ESAT-6-stimulated PBMC evaluated by enzyme-linked immune absorbent spot (ELISPOT) assay. Closed blue symbols (A) represent high (≥2.24%) in vivo inhalation-acquired PM load (PM load per AM) and open symbols represent participants with low PM load per AM (<2.24%). Closed red symbols (B) represent participants with ≥44.3% AM with PM and open symbols represent participants with <42.6% AM with PM (range: high proportion 44.3%–60%, low proportion 26%–42.6%). Mann-Whitney test was performed using the GraphPad Prism software for Mac OS X V.6.0. P values above symbols represent significant differences in frequencies of cytokine-producing cells between groups with higher and lower PM load per AM (blue symbols) (A) or higher and lower %AM% with PM (red symbols) (B) for the corresponding experimental condition. For example, in panel A, frequencies of IFN-γ-producing BAC following M. tuberculosis infection (MOI1) and PM2.5 exposure from individuals with higher in vivo inhalation-acquired PM load per AM (blue symbols) were compared with BAC from individuals with lower in vivo inhalation-acquired PM load per AM (open symbols). AM, alveolar macrophages; BAC, bronchoalveolar cells; IFN, interferon; PBMC, peripheral blood mononuclear cells; PM, particulate matter; PPD, purified protein derivative.
Effects of AM PM burden on M. tuberculosis-induced inflammatory cytokine production
To assess whether the in vivo inhalation-acquired PM burden in AM differentially affects the responsiveness of BAC and PBMC, we re-examined frequencies of PM2.5 exposure and/or M. tuberculosis-infection-induced cytokine-producing cells (figure 3). Participants were grouped according to their median %AM with PM and PM load per AM values (figure 3). Frequencies of cytokine-producing cells for each cytokine were then compared in participants with lower and higher %AM with PM and PM load per AM. The cut-off values distinguishing lower and higher %AM with PM and lower and higher PM load per AM were 42.6% (%AM with PM lower range: 26%–42.6%, higher range 44.3%–60%) and 2.24% (PM load per AM lower range: 0.42%–1.90%, higher range 2.24%–5.49%), respectively. The cut-off values were defined on the basis of the median of all %AM with PM data (42.6%) (figures 3, 4 and 5) and the median of all PM load per AM (2.24%, figures 4 and 5).
Figure 5.
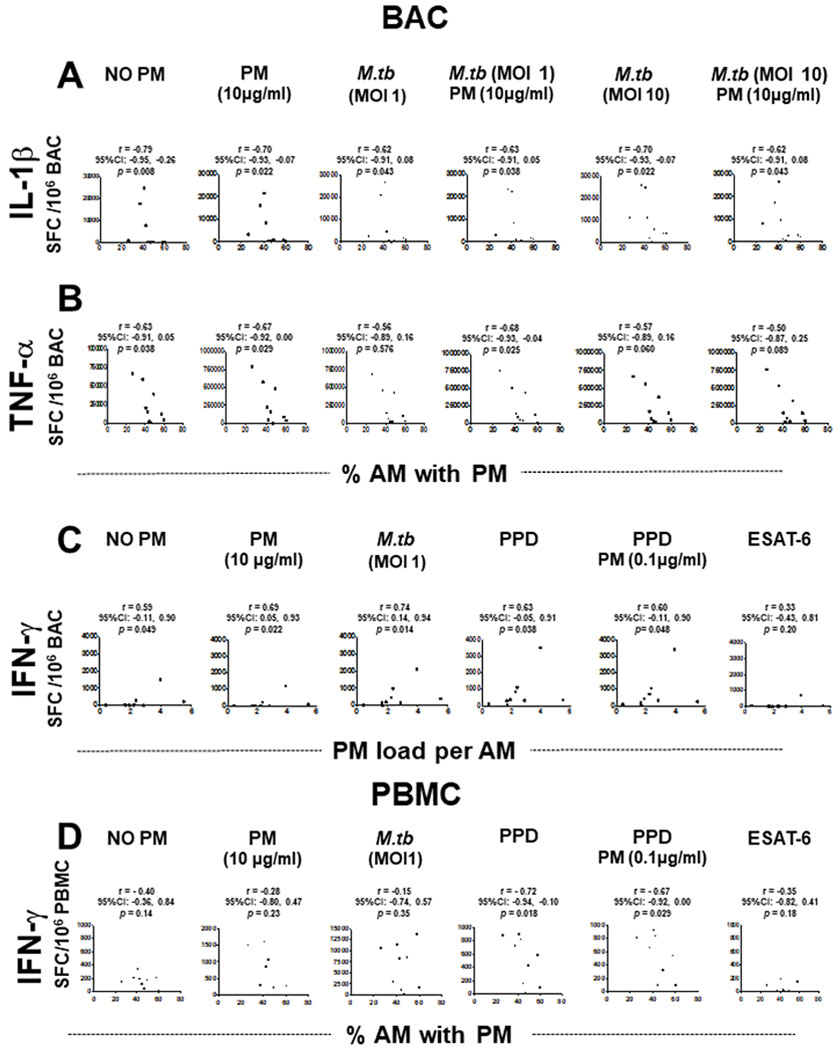
Correlations between in vivo-acquired PM burden in AM (%AM with PM or PM load per AM) and frequencies of cytokine-producing BAC and PBMC. Frequencies of IL-1β (A), TNF-α (B) and IFN-γ (C) spot-forming cells (SFC) in BAC (n=9) and frequencies of IFN-γ-SFC in PBMC (n=9) (D) are shown as a function of PM burden in AM following exposure to PM2.5 and/or M.tuberculosis infection, or PPD and ESAT-6 stimulation. Data points represent participants and slopes’ positive or negative correlations. Spearman’s r correlation coefficient test was performed using the GraphPad Prism software for Mac OS X V.6.0. A CI for all statistics test of 95% (α=0.05) was used. The p values, r values and CIs are indicated in all the experimental conditions. AM, alveolar macrophages; BAC, bronchoalveolar cells; IFN, interferon; IL, interleukin; M.tuberculosis, Mycobacterium tuberculosis; PBMC, peripheral blood mononuclear cells; PM, particulate matter; PPD, purified protein derivative; TNF, tumour necrosis factor; ESAT-6, 6kDa secretory antigenic target.
Frequencies of BAC producing IFN-γ, IL-1β and IL-6 constitutively were comparable in BAC from study participants with higher and lower %AM with PM. On the other hand, frequencies of BAC producing TNF-α constitutively were significantly greater in BAC with higher %AM with PM than with lower %AM with PM (figure 3A). Frequencies of IL-1β-producing BAC following M. tuberculosis infection and in vitro PM2.5 exposure were significantly lower in BAC with higher %AM with PM than with lower %AM with PM (figure 3B). This observation suggests that inhalation-acquired PM in AM suppresses IL-1β production in BAC on M. tuberculosis infection in the absence or presence of PM2.5 exposure. Thus, in vivo-acquired AM PM burden modulates immune responses in the lung microenvironment.
Frequencies of TNF-α (figure 3C) and IL-6 (figure 3D) producing BAC following M. tuberculosis infection and in vitro PM2.5 exposure were not significantly lower in BAC from participants with higher %AM with PM. This is probably due to the variability between individuals.
Frequencies of BAC producing IFN-γ, IL-1β, TNF-α and IL-6 constitutively were comparable in BAC with higher and lower PM loads per AM (data not shown).
Frequencies of IFN-γ-producing BAC in response to M. tuberculosis (MOI1+PM2.5 0, 1 and 10 μg/mL) and PPD (±PM2.5 0.1, 1 and 10 μg/mL) were significantly higher in BAC with higher PM load per AM than that with lower PM load per AM (figure 4A). Interestingly, frequencies of IFN-γ-producing PBMC induced by PPD stimulation+PM2.5 0, 0.1, 1 and 10 μg/mL were significantly lower in the group with higher %AM with PM (figure 4B). These findings suggest that PM burden of AM modulates IFN-γ production, which in turn potentially alters the cytokine milieu in autologous PBMC. Frequencies of IL-1β, TNF-α and IL-6-producing PBMC were not significantly different between high and low %AM with PM samples whether constitutive or following M. tuberculosis infection or in vitro PM2.5 exposure (data not shown).
In addition, we analysed whether correlations existed between in vivo inhalation-acquired PM burden in AM and cytokine frequencies. Frequencies of constitutively IL-1β and TNF-α-producing BAC (unexposed, controls) were negatively correlated with %AM with PM (IL-1β, r=−0.79, 95% CI −0.95 to 0.26, p=0.008; TNF-α, r=−0.63, 95% CI −0.91 to 0.05, p=0.038) (figure 5A,B). Similarly, frequencies of IL-1β-producing BAC were negatively correlated with %AM with PM when exposed to PM2.5 (10 μg/mL, r=−0.70, 95% CI −0.93 to 0.07, p=0.022) or infected with M. tuberculosis MOI1 in the absence (r=−0.62, 95% CI −0.91 to 0.08 p=0.043) or presence of PM2.5 (10 μg/mL) (r=−0.63, 95% CI −0.91 to 0.05, p=0.038) or with M. tuberculosis MOI10 in the absence (r=−0.70, 95% CI −0.93 to 0.07, p=0.022) or presence of PM (10 μg/mL) (r=−0.62, 95% CI −0.91 to 0.08, p=0.043) (figure 5A). Frequencies of TNF-α-producing BAC were negatively correlated with %AM with PM when cells were exposed to PM, (10 μg/mL) (r=−0.67, 95% CI −0.92 to 0.00, p=0.029) or infected with M. tuberculosis MOI1 (r=−0.68, 95% CI −0.93 to 0.04, p=0.025) in the presence of PM (10 μg/mL) (figure 5B).
Unlike IL-1β and TNF-α, which are negatively correlated with the %AM with PM, the frequencies of constitutively IFN-γ-producing BAC in absence of PM (r=0.59, 95% CI −0.11 to 0.90, p=0.049) exposed to PM2.5 (10 μg/mL) (r=0.69, 95% CI 0.05 to 0.93, p=0.022), infected with M. tuberculosis (MOI1) (r=0.74, 95% CI 0.14 to 0.94, p=0.014), stimulated with PPD (r=0.63, 95% CI −0.05 to 0.91, p=0.038) and stimulated with PPD and exposed to PM2.5 (0.1 μg/mL) (r=0.60, 95% CI −0.11 to 0.90, p=0.048) were positively correlated with the PM load per AM (figure 5C). Interestingly, frequencies of IFN-γ-producing PBMC on PPD stimulation (r=−0.72, 95% CI −0.94 to 0.10, p=0.018) and PPD exposed to PM2.5 (0.1 μg/mL) (r=−0.67, 95% CI −0.92 to 0.00, p=0.029) were negatively correlated with %AM with PM (figure 5D). These findings indicate that increased PM load in AM (presumably from increased exposure to PM from real-world inhalation air pollution exposure) impairs peripheral IFN-γ production in response to PPD. However, neither the %AM with PM nor the PM load per AM correlated with the frequencies of IL-1β and TNF-α-producing PBMC.
In summary, these findings (figures 3–5) indicate that inhaled PM burden in AM alters protective human host immune responses to M. tuberculosis infection by a PM-load-dependent modulation of constitutive, M. tuberculosis or PPD-induced IL-1β, TNF-α and IFN-γ production in BAC and PBMC.
DISCUSSION
This is the first study to explore the effects of urban air pollution exposure and PM load in AM on immune responses to M. tuberculosis by human primary lung and blood immune cells. In earlier studies, carbon load in human AM has been used as a biomarker for personal exposure to combustion-derived PM28–30 and PM10,31 and to assess innate immune responses to M. tuberculosis in household air pollution-exposed individuals.32
Here we examined how inhaled urban PM from a polluted municipality in Mexico City, where TB is still endemic, affects host immune responses to M. tuberculosis in human autologous BAC and PBMC. Constitutive production of proinflammatory IL-1β, TNF-α and IL-6 was observed in BAC with no further increase in frequencies of BAC producing these cytokines on in vitro PM2.5 exposure and/or M. tuberculosis infection (MOI1). Consistent with the constitutive expression of proinflammatory cytokines, we observed constitutive phosphorylation of IκB-α indicating activation of the transcription factor NF-κB in BAC (online supplement figure 4). This high constitutive expression of cytokines in the first 24 hours may explain the observed lack of cellular response to M. tuberculosis on infection with M. tuberculosis at MOI1, which, however, was not seen with M. tuberculosis at MOI10. This relative cellular unresponsiveness may resemble a state of cellular tolerance that has also been observed during long-term endotoxin exposures33,34 and was recently described as ‘anergy’ in AM exposed to household air pollution PM.32 Interestingly, minimal NF-κB activation identified in freshly isolated, quiescent AM from healthy, non-smoking individuals in the USA35 implies that the higher NF-κB activation observed in BAC from our participants is due to the greater air pollution exposure in Mexico City. Of note, the negative correlation between PM burden and the frequency of IL-1β-producing BAC (figure 5A) observed shortly after BAC removal from the human lung environment disappeared on 48 hours’ BAC cultures, consistent with lack of phosphorylation of IκB-α in BAC after 48 hours in cell culture (data not shown).
To determine the individual exposure levels of participants, we examined the in vivo-acquired PM burden of their BAC by microscopically examining the percentage of AM with PM and the PM load in AM (figure 2A,B). In BAC, the constitutive IL-1β and TNF-α expression, and the PM and M. tuberculosis-induced IL-1β expression were strongly suppressed in study participants with higher PM burden (figure 3A,B). Interestingly, the AM PM burden modified the IFN-γ responses of BAC to M. tuberculosis and PPD, and was correlated to those of the autologous PBMC. It is reasonable to speculate that the systemic effect of inhalation exposure to PM noted in this study may be related to the same processes that precede the development of adverse cardiovascular effects36,37 or allow the detection of PM in urine of air pollution-exposed children.5 The mechanisms that may underlie the apparent interaction between the lung and blood compartments during inhalational PM exposure could not be explored in this study but may be related to transepithelial translocation of PM38 or indirect effects from exosomes39 or cytokines.
This study has limitations that are intrinsic to the nature of in vitro experimental work (ie, the modelling of processes that in reality are far more complex) and a limited number of study participants able to provide both lung and blood cell samples. Given the widespread urban air pollution40 and the resulting lack of unexposed populations in the current study location, this study relied on comparing study participants with stratified exposure levels as defined by higher and lower AM PM burden. A separate analysis of study participant exposures considering their exposure behaviours and movements, for example, between home and workplace, is underway. It was beyond the scope of this analysis to assess which behavioural or environmental factors underlay the observed differences in PM load in AM. Inclusion of human controls from low PM2.5 locations was not feasible and considered to raise concerns about differences in the genetic background of the study participants, their diets (that may affect body oxidative stress) and also exposures to air pollution PM2.5 with different chemical compositions. Due to the limited number of available cells (BAC and PBMC) from the study participants, experiments with PM10 in parallel were not feasible. Since most TB occurs in low-income countries, frequently impacted by sources of larger PM (ie, PM10), future evaluation of this PM fraction would be useful.
Given the lack of data on PM concentrations in the human circulation following real-world urban air pollution exposures, we have chosen PM2.5 concentrations (0.1–10 μg/mL) that reflect the range of real-world inhalational PM exposure levels in Mexico City16 and are frequently chosen in comparable in vitro studies. Further justifying the chosen PM concentration ranges, we have noted on in vitro exposure of monocyte-derived macrophages to PM2.5 at 1, 5 and 10 μg/mL that 10%–30% (median range) of these macrophages (n=5 experiments, data not shown) present comparable amounts of PM inclusions as found in the AM of many of the air pollution-exposed study participants (figure 5) in the current study.
With increasing urbanisation and industrialisation globally and in TB endemic areas, our findings and the growing body of evidence that air pollution increases the risk of TB, indicate that the success of global TB control efforts will depend on environmental improvements in TB endemic areas worldwide. Holistic approaches to improving TB control globally will have to be interdisciplinary and integrate, for example, policymakers and urban planners to focus on poverty alleviation, improved housing, generation and provision of clean energy technology affordable public transportation and the regulation of air pollution from industry, home and vehicular traffic sources.
In summary, given the critical roles of IL-1β and TNF-α in human antimycobacterial host immunity,41,42 the current novel findings suggest that increased air pollution levels in urba environments of TB endemic areas may lead to increased M. tuberculosis susceptibility resulting from PM-induced alterations of protective host immune responses. Indeed, impairments of other innate immune mechanisms, such as decreased production of antimicrobial peptides observed earlier by our group in M. tuberculosis-infected respiratory epithelial cells on PM exposure,16 may in parallel contribute to greater susceptibility to M. tuberculosis infection, or other respiratory infections for that matter. Furthermore, as in vivo-acquired PM load in AM correlated negatively with peripheral blood IFN-γ expression in response to PPD, one may speculate that air pollution exposure could reduce the sensitivity of M. tuberculosis immunodiagnostic IFN-γ release assays that quantify IFN-γ release from blood T cells on M. tuberculosis antigen exposures (figure 5D).
Taken together, we propose that the PM effects on local lung and systemic cytokine responses to M. tuberculosis, observed in the current study, may represent functional correlates of air pollution effects that in earlier epidemiological studies were found to be correlated with increased incidence rates of TB and M. tuberculosis infection14 as well as adverse TB treatment outcomes.15
Supplementary Material
Key messages.
What is the key question?
With urbanisation growing globally and in TB endemic countries, does urban airborne particulate matter (PM) exposure adversely affect protective immune responses against Mycobacterium tuberculosis?
What is the bottom line?
Urban air pollution PM load in human alveolar macrophages suppresses protective immune responses to M. tuberculosis in both lung immune cells as well as autologous blood cells.
Why read on?
This study is the first to show that urban air pollution PM alters functional immune cell responses to M. tuberculosis, findings that are in accordance with epidemiological data of increased risk for TB development and altered TB treatment outcomes in air-polluted environments.
Acknowledgements
We are indebted to and thank all the study participants for their participation in this study, Ms Janet Nuñez and Ms Laura Fonseca for their diligence with recruitment of study participants and clinical data entry, and Ms Michelle Ruidiaz Santiago at Rutgers University School of Public Health for her diligent, reliable and forthcoming management support of this project. We thank Drs Rogelio Perez Padilla and Debra Laskin for their facilitation of this work. We appreciate the support from Drs Mauricio Castañón Arreola and Eduardo Carrillo Tapia and access to microscope and photographic services at the Universidad Autónoma de la Ciudad de México (UACM). We also thank the staff at INER and UAM for their help with this project.
Funding NIEHS RO1 Grant R01ES020382 (S. Schwander) and NIH-NIEHS P30 ES005022.
Footnotes
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval This research protocol was approved by the Scientific and Bioethics Committee of the instituto Nacional de Enfermedades Respiratorias ismael Cosio Villegas in Mexico City, Mexico (INER, protocol B22-12) and Rutgers University Institutional Review Board (protocol 2012001383) in New Brunswick, NJ.
Provenance and peer review Not commissioned; externally peer reviewed.
REFERENCES
- 1.Nations U. World urbanization prospects: the 2018 revision, 2018.
- 2.Organization WH. Who fact sheet No. 292, household air pollution and health 2016. Available: http://www.who.int/mediacentre/factsheets/fs292/en/
- 3.Bourdrel T, Bind M-A, Béjot Y, et al. Cardiovascular effects of air pollution. Arch Cardiovasc Dis 2017;110:634–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bowe B, Xie Y, Li T, et al. Global and national burden of diabetes mellitus attributable to PM2.5 air pollution. Lancet Planet Health 2016;2018:e301–12. [DOI] [PubMed] [Google Scholar]
- 5.Saenen ND, Bové H, Steuwe C, et al. Children’s urinary environmental carbon load. A novel marker reflecting residential ambient air pollution exposure? Am J Respir Crit Care Med 2017;196:873–81. [DOI] [PubMed] [Google Scholar]
- 6.Organization WH. Global tuberculosis report 2016, 2016.
- 7.Rees D, Murray J, Silica MJ. Silica, silicosis and tuberculosis. Int J Tuberc Lung Dis 2007;11:474–84. [PubMed] [Google Scholar]
- 8.van Zyl Smit RN, Pai M, Yew WW, et al. Global lung health: the colliding epidemics of tuberculosis, tobacco smoking, HIV and COPD. Eur Respir J 2010;35:27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Altet-Gómez MN, Alcaide J, Godoy P, et al. Clinical and epidemiological aspects of smoking and tuberculosis: a study of 13,038 cases. Int J Tuberc Lung Dis 2005;9:430–6. [PubMed] [Google Scholar]
- 10.Bates MN, Khalakdina A, Pai M, et al. Risk of tuberculosis from exposure to tobacco smoke: a systematic review and meta-analysis. Arch Intern Med 2007;167:335–42. [DOI] [PubMed] [Google Scholar]
- 11.Davies PDO, Yew WW, Ganguly D, et al. Smoking and tuberculosis: the epidemiological association and immunopathogenesis. Trans R Soc Trop Med Hyg 2006;100:291–8. [DOI] [PubMed] [Google Scholar]
- 12.Kolappan C, Subramani R. Association between biomass fuel and pulmonary tuberculosis: a nested case-control study. Thorax 2009;64:705–8. [DOI] [PubMed] [Google Scholar]
- 13.Pérez-Padilla R, Pérez-Guzmán C, Báez-Saldaña R, et al. Cooking with biomass stoves and tuberculosis: a case control study. Int J Tuberc Lung Dis 2001;5:441–7. [PubMed] [Google Scholar]
- 14.Lai T-C, Chiang C-Y, Wu C-F, et al. Ambient air pollution and risk of tuberculosis: a cohort study. Occup Environ Med 2016;73:56–61. [DOI] [PubMed] [Google Scholar]
- 15.Blount RJ, Pascopella L, Catanzaro DG, et al. Traffic-related air pollution and all-cause mortality during tuberculosis treatment in California. Environ Health Perspect 2017;125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rivas-Santiago CE, Sarkar S, Cantarella P, et al. Air pollution particulate matter alters antimycobacterial respiratory epithelium innate immunity. Infect Immun 2015;83:2507–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sarkar S, Song Y, Sarkar S, et al. Suppression of the NF-κB pathway by diesel exhaust particles impairs human antimycobacterial immunity. J Immunol 2012;188:2778–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma Q-Y, Huang D-Y, Zhang H-J, et al. Exposure to particulate matter 2.5 (PM2.5) induced macrophage-dependent inflammation, characterized by increased Th1/Th17 cytokine secretion and cytotoxicity. Int Immunopharmacol 2017;50:139–45. [DOI] [PubMed] [Google Scholar]
- 19.Carranza C, Juárez E, Torres M, et al. Mycobacterium tuberculosis growth control by lung macrophages and CD8 cells from patient contacts. Am J Respir Crit Care Med 2006;173:238–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sarkar S, Zhang L, Subramaniam P, et al. Variability in bioreactivity linked to changes in size and zeta potential of diesel exhaust particles in human immune cells. PLoS One 2014;9:e97304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sarkar S, Leo BF, Carranza C, et al. Modulation of human macrophage responses to Mycobacterium tuberculosis by silver nanoparticles of different size and surface modification. PLoS One 2015;10:e0143077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakamura T, Schwander SK, Donnelly R, et al. Cytokines across the night in chronic fatigue syndrome with and without fibromyalgia. Clin Vaccine Immunol 2010;17:582–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Manzano-León N, Serrano-Lomelin J, Sánchez BN, et al. TNFα and IL-6 responses to particulate matter in vitro: variation according to pM size, season, and polycyclic aromatic hydrocarbon and soil content. Environ Health Perspect 2016;124:406–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mayer-Barber KD, Barber DL, Shenderov K, et al. Cutting Edge: Caspase-1 Independent IL-1 Production Is Critical for Host Resistance to Mycobacterium tuberculosis and Does Not Require TLR Signaling In Vivo. J Immunol 2010;184:3326–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flynn JL, Goldstein MM, Chan J, et al. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity 1995;2:561–72. [DOI] [PubMed] [Google Scholar]
- 26.Barraza-Villarreal A, Escamilla-Nuñez MC, Schilmann A, et al. Lung function, airway inflammation, and polycyclic aromatic hydrocarbons exposure in Mexican schoolchildren: a pilot study. J Occup Environ Med 2014;56:415–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.SdMAdlCd M Calidad del AIRE en La Ciudad de México, informe 2015. México: Dirección General de Gestión de la Calidad del Aire. Dirección de Monitoreo Atmosférico, 2016. [Google Scholar]
- 28.Bai Y, Brugha RE, Jacobs L, et al. Carbon loading in airway macrophages as a biomarker for individual exposure to particulate matter air pollution - A critical review. Environ Int 2015;74:32–41. [DOI] [PubMed] [Google Scholar]
- 29.Kulkarni N, Pierse N, Rushton L, et al. Carbon in airway macrophages and lung function in children. N Engl J Med 2006;355:21–30. [DOI] [PubMed] [Google Scholar]
- 30.Kulkarni NS, Prudon B, Panditi SL, et al. Carbon loading of alveolar macrophages in adults and children exposed to biomass smoke particles. Sci Total Environ 2005;345:23–30. [DOI] [PubMed] [Google Scholar]
- 31.Brauer M, Avila-Casado C, Fortoul TI, et al. Air pollution and retained particles in the lung. Environ Health Perspect 2001;109:1039–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rylance J, Fullerton DG, Scriven J, et al. Household air pollution causes dose-dependent inflammation and altered phagocytosis in human macrophages. Am J Respir Cell Mol Biol 2015;52:584–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Medvedev AE, Piao W, Shoenfelt J, et al. Role of TLR4 tyrosine phosphorylation in signal transduction and endotoxin tolerance. J Biol Chem 2007;282:16042–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Medvedev AE, Sabroe I, Hasday JD, et al. Tolerance to microbial TLR ligands: molecular mechanisms and relevance to disease. J Endotoxin Res 2006;12:133–50. [DOI] [PubMed] [Google Scholar]
- 35.Fawer CF, Raychaudhuri B, Buhrow LT, et al. Constitutive NF-kappaB levels in human alveolar macrophages from normal volunteers. Cytokine 1998;10:868–71. [DOI] [PubMed] [Google Scholar]
- 36.Mills NL, Donaldson K, Hadoke PW, et al. Adverse cardiovascular effects of air pollution. Nat Clin Pract Cardiovasc Med 2009;6:36–44. [DOI] [PubMed] [Google Scholar]
- 37.Rich DQ, Kipen HM, Huang W, et al. Association between changes in air pollution levels during the Beijing Olympics and biomarkers of inflammation and thrombosis in healthy young adults. JAMA 2012;307:2068–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oberdörster G, Oberdörster E, Oberdörster J. Nanotoxicology: an emerging discipline evolving from studies of ultrafine particles. Environ Health Perspect 2005;113:823–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fujita Y, Kadota T, Araya J, et al. Extracellular vesicles: new players in lung immunity. Am J Respir Cell Mol Biol 2017. 10.3390/ijms17111801. [DOI] [PubMed] [Google Scholar]
- 40.Rivera-González LO, Zhang Z, Sánchez BN, et al. An assessment of air pollutant exposure methods in Mexico City, Mexico. J Air Waste Manag Assoc 2015;65:581–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mayer-Barber KD, Barber DL, Shenderov K, et al. Caspase-1 independent IL-1beta production is critical for host resistance to Mycobacterium tuberculosis and does not require TLR signaling in vivo. J Immunol 2010;184:3326–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keane J, Gershon S, Wise RP, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med 2001;345:1098–104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.