

Abstract
Synthesis of the negative‐strand ((−)‐strand) counterpart is the first step of coronavirus (CoV) replication; however, the detailed mechanism of the early event and the factors involved remain to be determined. Here, using bovine coronavirus (BCoV)‐defective interfering (DI) RNA, we showed that (a) a poly(A) tail with a length of 15 nucleotides (nt) was sufficient to initiate efficient (−)‐strand RNA synthesis and (b) substitution of the poly(A) tail with poly(U), (C) or (G) only slightly decreased the efficiency of (−)‐strand synthesis. The findings indicate that in addition to the poly(A) tail, other factors acting in trans may also participate in (−)‐strand synthesis. The BCoV nucleocapsid (N) protein, an RNA‐binding protein, was therefore tested as a candidate. Based on dissociation constant (Kd) values, it was found that the binding affinity between N protein, but not poly(A)‐binding protein, and the 3′‐terminal 55 nt plus a poly(A), poly(U), poly(C) or poly(G) tail correlates with the efficiency of (−)‐strand synthesis. Such an association was also evidenced by the binding affinity between the N protein and 5′‐ and 3′‐terminal cis‐acting elements important for (−)‐strand synthesis. Further analysis demonstrated that N protein can act as a bridge to facilitate interaction between the 5′‐ and 3′‐ends of the CoV genome, leading to circularization of the genome. Together, the current study extends our understanding of the mechanism of CoV (−)‐strand RNA synthesis through involvement of N protein and genome circularization and thus may explain why the addition of N protein in trans is required for efficient CoV replication.
Keywords: (−)‐strand synthesis, cis‐acting element, coronavirus, genome circularization, nucleocapsid protein, replication
In the initial stage of coronavirus (CoV) (−)‐strand RNA synthesis, binding of the CoV N protein to the 5′‐ and 3′‐terminal structures of (+)‐strand CoV genome leads to circularization of the genome, serving as a platform to recruit cellular proteins and viral replicase proteins including RNA‐dependent RNA polymerase. This then initiates (−)‐strand RNA synthesis.
Abbreviations
- BCoV
bovine coronavirus
- DI RNA
defective interfering RNA
- HCoV‐OC43
human coronavirus OC43
- HVR
hypervariable region
- Kd
dissociation constant
- MHV
mouse hepatitis virus
- N
nucleocapsid
- PABP
poly(A)‐binding protein
- PK
pseudoknot
- sgmRNA
subgenomic mRNA
Introduction
Coronaviruses (CoVs), which belong to the subfamily Coronavirinae, family Coronaviridae, order Nidovirales, are single‐stranded, positive‐sense RNA viruses with a genome size of 26–32 kilobases (kb) 1, 2, 3. The subfamily contains four genera: Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus 1, 3. The 5′‐capped CoV genome consists of a 5′‐untranslated region (UTR), open reading frames (ORFs), a 3′‐UTR and a 3′‐poly(A) tail. The 5′ two‐thirds of the genome encode replicase‐related nonstructural proteins (nsps); the other one‐third of the genome mostly encodes structural proteins 2, 4. In infected cells, in addition to replication of genomic RNA, a 3′ coterminal nested set of subgenomic mRNAs (sgmRNAs) is synthesized; these sgmRNAs are also 5′ coterminal with the leader sequence of the genome 2, 4.
Using defective interfering RNAs (DI RNAs) and reverse‐genetics systems, the cis‐acting RNA elements required for CoV replication have been identified; these elements are largely located at the 5‐ and 3′‐terminal regions of the genome (2, 4, 5, 6, 7 and references therein). In addition, several 5′‐terminal structures have been suggested to function as cis‐acting RNA elements essential for replication (interpreted as (+)‐strand RNA synthesis). Among these structures, the leader sequence and stem‐loop I (SLI) in betacoronaviruses are also critical in (−)‐strand RNA synthesis 8, 9, 10. Regarding the cis‐acting RNA elements at the 3′‐terminal region of the genome, the 5′‐most bulged stem‐loop (BSL) in betacoronaviruses is conserved and required for replication 11, 12. Downstream of BSL is a pseudoknot (PK) structure present in both beta‐ and alphacoronaviruses that is also essential for replication 13. The region next to the 3′‐end of PK, referred to as the hypervariable region (HVR) is less conserved. In mouse hepatitis virus (MHV), the HVR contains a secondary structure that has been shown to be dispensable for replication but that has an important role in pathogenesis 14, 15. Furthermore, the 3′‐most 55 nucleotides (nt) and poly(A) tail are also important for both (−)‐ and (+)‐strand RNA synthesis in DI RNA systems of bovine coronavirus (BCoV) and MHV 16, 17, 18. Although it is known that a minimum poly(A) tail length of ~ 5–10 nt is required for (+)‐strand RNA synthesis of BCoV 18, the length required for efficient (−)‐strand RNA synthesis has yet to be determined. More recently, Züst et al. 19 proposed that a stem structure formed by base pairing between the 3′‐most nt and loop 1 of the PK stem has an important function in the initiation of (−)‐strand RNA synthesis. Using reverse‐genetic approaches, a subsequent study by Liu et al. 20 showed that disrupting the stem structure by mutation generated a lethal virus.
The ~ 50‐kDa CoV nucleocapsid (N) protein contains RNA‐binding domains and binds to various regions of the genome with different affinities 21, 22, 23, 24, 25. In addition to its riboprotein activity, N protein is also able to interact with the replication‐transcription complex, which contains coronaviral replicase and cellular proteins 25, 26, 27, 28, 29. Several lines of evidence suggest that the N protein is required for or able to enhance (+)‐strand RNA synthesis 30, 31, 32. However, the stepwise mechanism by which N protein participates in both (−)‐ and (+)‐strand RNA synthesis remains to be elucidated.
Interactions between the 5′‐ and 3′‐ends of the genome, which leads to genome circularization either via RNA‐RNA, RNA‐protein or protein‐protein interaction, have been suggested in (+)‐strand RNA viruses 33, 34, 35. In addition, Zuniga et al. showed that in CoVs, long‐distance RNA‐RNA interaction between the 5′‐ and 3′‐ends of the transmissible gastroenteritis coronavirus (TGEV) genome is required for sgmRNA synthesis 32. Genetic evidence from Li et al. also supports that the crosstalk between the 5′‐ and 3′‐ends of the MHV genome is linked to sgmRNA synthesis 9. Contact between the 5′‐ and 3′‐ends of the CoV genome mediated by RNA‐protein interaction has also been suggested 6, 36 and may play a role in replication 36. However, whether such 5′‐ and 3′‐end interaction is required for (−)‐strand RNA synthesis remains to be determined.
Synthesis of the (−)‐strand RNA is the first step in CoV replication, though the detailed mechanism is still not fully understood. In the current study, we examined the requirement of the sequence and length of the poly(A) tail for efficient (−)‐strand RNA synthesis and the association of N protein with (−)‐strand RNA synthesis. We also assessed whether the N protein can serve as a bridge for interaction between the 5′‐ and 3′‐ends of the CoV genome. Our results show the involvement of N protein and genome circularization in (−)‐strand RNA synthesis and thus extend the Züst model for (−)‐strand RNA synthesis 19.
Results
The sequence and length of poly(A) tail are involved in the efficient (−)‐strand RNA synthesis
In CoVs, (−)‐strand RNA synthesis is presumably initiated from the poly(A) tail 37, 38, an RNA element that is required for both translation and replication in CoVs 18, 38. Although the poly(A) tail at the end of 3′ UTR of the CoV genome has been proposed to be required for efficient (−)‐strand RNA synthesis 17, the length needed to initiate efficient synthesis remains to be determined. Additionally, it is also not known whether the efficiency of (−)‐strand synthesis increases as a linear function of poly(A) tail length. To address these questions, BM‐DI RNA 16, 39 derived from BCoV DI RNA and containing the MHV 3′ UTR as a marker (Fig. 1A) was employed. Because the poly(A) tail length on BM‐DI RNA in the early infection of virus passage 1 (VP1) is ~ 25 nt 38 and a poly(A) tail of 25 nt has been shown to be efficient for (−)‐strand RNA synthesis 16, the BM‐DI RNA, which has a replication efficiency similar to that of BCoV DI RNA 39, was then engineered to contain poly(A) tails ranging from 0 to 25 nt in length (Fig. 1B). Note that the DI RNA marked with the MHV 3′ UTR allows us to distinguish it from the BCoV helper virus genome for subsequent (−)‐strand detection by RT‐PCR. For (−)‐strand RNA detection, head‐to‐tail ligation of RNA followed by RT‐PCR 10, 16, 39 was employed to differentiate the coronaviral polymerase‐generated (−)‐strand DI RNA from the T7 RNA polymerase‐generated artifact (−)‐strand DI RNA; this is because a product cannot be yielded from copy‐back (−)‐strand transcripts after ligation with the primers used for RT‐PCR (Controls 3 and 4 in Fig. 1B, right panel). After 2 h of infection, DI RNA transcript was transfected into BCoV‐infected human rectum tumor (HRT)‐18 cells. RNA was then collected at 8 h post‐transfection (hpt) and subjected to RT‐qPCR to evaluate the effect of poly(A) tail length on (−)‐strand synthesis efficiency. It should be noted that there is no possibility of DI RNA‐BCoV genome recombinant synthesized at 8 hpt because no RT‐PCR product was observed (data not shown) using previously described methods 8, 16, 39. Surprisingly, (−)‐strand RNA synthesis was still detected using DI RNA with no poly(A) tail (BM0A, Fig. 1B, right panel), and the efficiency of (−)‐strand RNA synthesis for BM0A and BM5A was ~ 60% of that for BM25A. Sequencing results suggested that the poly(A) tail length for these DI RNA constructs was not altered at the time of sample collection. In addition, the level of (−)‐strand synthesis was almost the same for BM15A and BM25A, suggesting that DI RNA with a 15‐nt poly(A) tail is sufficient for efficient synthesis of its (−)‐strand counterpart. To further confirm the results of (−)‐strand RNA synthesis shown in Fig. 1B, RT‐qPCR was also performed using RNA samples without head‐to‐tail ligation 10. Although the detection of (−)‐strand RNA caused by T7 RNA polymerase‐generated copy‐back (−)‐transcripts may occur, the levels of detected (−)‐strand RNA by RT‐qPCR from uninfected cells transfected with transcripts can be used as the background levels. Thus, compared to the background levels, the increase in the detected (−)‐strand RNA synthesis can be due to the activity of viral RNA‐dependent RNA polymerase. Using the method to detect (−)‐strand RNA synthesis, it was determined that the efficiency of the (−)‐strand RNA synthesis for BM0A and BM5A was ~ 50% of that for BM15A and BM25A (data not shown), consistent with the results shown in Fig. 1B in which RNA samples were head‐to‐tail ligated prior to RT‐qPCR. Accordingly, the results suggest that the length of the poly(A) tail on CoV DI RNA can affect the efficiency of (−)‐strand RNA synthesis; however, the (−)‐strand RNA synthesis did not increase as a linear function of DI RNA poly(A) tail length ranging from 0 to 25 nt. In addition, since the results from the two methods are similar, the head‐to‐tail ligation prior to RT‐qPCR for detection of (−)‐strand synthesis was employed for the following experiments.
Figure 1.
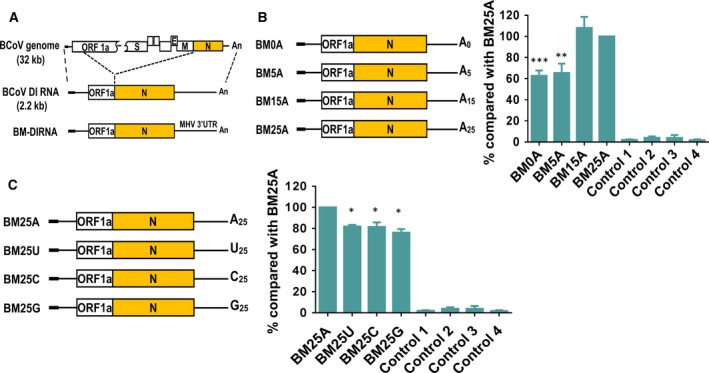
Effect of the length and sequence of the poly(A) tail on (−)‐strand RNA synthesis. (A) Schematic diagram depicting the structure of the BCoV genome, BCoV DI RNA and BM‐DI RNA, which is derived from BCoV DI RNA but carrying the MHV 3′ UTR. BCoV DI RNA contains only 288 nt of ORF1a gene. (B) Left panel: BM‐DI RNA constructs engineered to contain poly(A) tails of various lengths. Right panel: relative efficiency of (−)‐strand RNA synthesis from BM‐DI RNA constructs (left panel), as measured by RT‐qPCR. (C) Left panel: BM‐DI RNA constructs engineered to replace the 25‐nt poly(A) tail with a 25‐nt poly(U), poly(C) or poly(G) tail. Right panel: relative efficiency of (−)‐strand RNA synthesis from BM‐DI RNA constructs (left panel), as measured by RT‐qPCR. Controls in (B) and (C): Control 1 – total cellular RNA from mock‐infected cells. Control 2 – total cellular RNA from BCoV‐infected cells. Control 3 – total cellular RNA from BM25A‐transfected mock‐infected cells. Control 4 – a mixture of BCoV‐infected cellular RNA extracted at 8 hpt and 200 ng of BM25A transcript. The values in the right panel of (B) and (C) represent the mean ± SD of three individual experiments. The statistical significance was evaluated using a t‐test versus BM25A: *P < 0.05, **P < 0.01, ***P < 0.001.
As we found that despite decreased efficiency, (−)‐strand RNA can be produced from DI RNA with no poly(A) tail (Fig. 1B), we next sought to assess whether the sequence of the poly(A) tail is also a factor for (−)‐strand RNA synthesis. To this end, the 25‐nt poly(A) tail at the 3′‐terminus of DI RNA was replaced with a 25‐nt poly(U), poly(C) or poly(G) tail (Fig. 1C, left panel). As shown in Fig. 1C, right panel, (−)‐strand RNA was still synthesized from these DI RNA constructs and the efficiency decreased by ~ 20% compared to BM25A. Note that no recombination between the DI RNA construct and helper virus genome occurred and that the length and sequence (i.e., 25‐nt poly(U), (C) and (G)) remained the same in these mutated DI RNAs and had not reversed to BM25A at the time of RNA collection (i.e., 8 hpt) (data not shown). Therefore, based on the statistical significance between these DI RNA constructs (Fig. 1C, right panel), it was concluded that the sequence of the poly(A) tail, albeit not essential, is able to influence the efficiency of (−)‐strand RNA synthesis.
Involvement of the N protein in (−)‐strand RNA synthesis
It appeared that (−)‐strand DI RNA can be produced from DI RNA with no poly(A) tail (Fig. 1B) or with a non‐poly(A) sequence (i.e., poly(U), (C) or (G)) (Fig. 1C), although the efficiency was reduced. These findings led to the hypothesis that in addition to poly(A) tail length and sequence, other factors such as proteins interacting with the 3′‐terminal sequence of DI RNA may also play an important role in (−)‐strand RNA synthesis. We speculated that the CoV N protein is a candidate factor involved in this interaction because it can bind to CoV genome 21, 22, 23, 24 including poly(A) tail 25 and is required for CoV replication 30, 31, 32. Because similar efficiencies of (−)‐strand RNA synthesis were observed for DI RNA with a poly(U), poly(C) or poly(G) tail (Fig. 1C), we hypothesized that the interaction of N protein with the DI RNA 3′‐terminal sequence may correlate with (−)‐strand RNA synthesis if N protein binds to poly(U), poly(C) and poly(G) tails with similar affinities. To test the hypothesis, 32P‐labeled RNA containing the CoV 3′‐terminal 55 nt plus a 25‐nt poly(A), poly(U), poly(C) or poly(G) tail was generated (Fig. 2A, left panel) and the binding affinity between BCoV N protein (if not specified, the following N protein indicates BCoV N protein) and RNA was determined by the dissociation constant (Kd) derived from electrophoretic mobility shift assays (EMSAs). As shown in Fig. 2B–E, Kd values for the RNA containing CoV 3′‐terminal 55 nt plus a 25‐nt poly(A), poly(U), poly(C) or poly(G) tail were calculated to be 31.1 ± 1.2, 32.2 ± 2.1, 31.7 ± 3.7 and 30.2 ± 2.2 nm, respectively, suggesting similar binding affinities (Fig. 2F). To highlight the biological relevance of such binding between the N protein and these RNA elements, we tested further whether the N protein exhibits different binding affinities with other RNA species. To this end, 80‐nt RNA probes derived from the 3′‐terminus of the β‐actin mRNA and TOPO‐XL plasmid (Invitrogen, Carlsbad, CA, USA) were synthesized. As shown in Fig. 2G, H, Kd for the two RNA species was ~ 3–5‐fold that of the RNA containing the CoV 3′‐terminal 55 nt plus a 25‐nt poly(A), poly(U), poly(C) or poly(G) tail. Since BCoV DI RNA used for (−)‐strand RNA synthesis assay contained MHV 3′ UTR (Fig. 1), the binding affinity between MHV N protein and MHV 3′‐terminal 55 nt plus a 25‐nt poly(A) was also determined. As shown in Fig. 2I, Kd for MHV N protein was 35.6 ± 9.2 nm and similar to that (31.1 ± 1.2 nm) for BCoV N protein, suggesting BCoV and MHV N proteins have similar binding affinity with MHV 3′ terminus sequence. Together, the result supports our hypothesis that the N protein may be involved in (−)‐strand RNA synthesis by interacting with DI RNA 3′‐terminal sequence. In addition, as it is well characterized that poly(A)‐binding protein (PABP) binds to poly(A) tails with high affinity, the same RNA probes were also examined for their ability to interact with PABP (Fig. 3A) to evaluate whether PABP is also involved in the (−)‐strand RNA synthesis. As shown in Fig. 3B‐E, we calculated Kd values of 14.9 ± 2.0 nm, 30.1 ± 2.2 nm and 93.4 ± 4.3 for RNA containing the CoV 3′‐terminal 55 nt plus the 25‐nt poly(A), poly(U) and poly(C), respectively. However, we were unable to determine the Kd for RNA containing the CoV 3′‐terminal 55 nt plus the 25‐nt poly(G), suggesting that the value may be greater than 150 nm. The results suggest that the binding of PABP to the CoV 3′‐terminal sequence is less likely correlated with (−)‐strand RNA synthesis. Because (a) N protein but not PABP binds with similar affinity to the CoV 3′‐terminal 55 nt plus a 25‐nt poly(U), poly(C) or poly(G) tail, (b) the efficiency of (−)‐strand RNA synthesis is similar between DI RNA with a poly(U), poly(C) or poly(G) tail and (c) N protein binds with various affinities to other RNA probes containing different sequences (Fig. 2G,H), the results (Figs 1, 2, 3) together suggest that the N protein is involved in (−)‐strand RNA synthesis.
Figure 2.
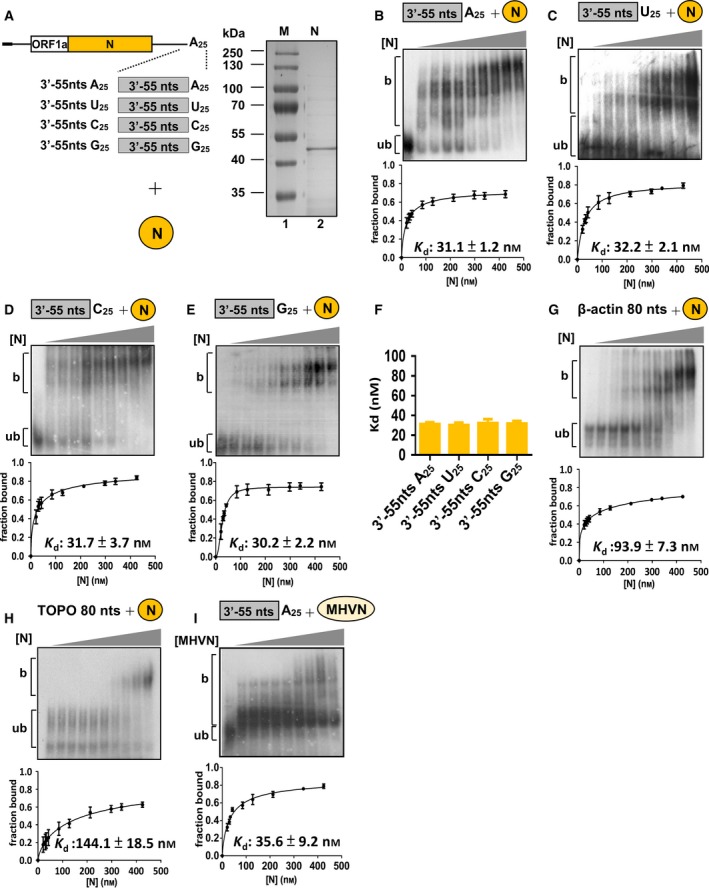
Determination of binding affinity between N protein and RNA elements by Kd. (A) Schematic diagrams depicting the structure of RNA probes (left panel) and E. coli‐expressed BCoV N protein (right panel) for determination of N protein‐binding affinities. (B–E) Upper panel: EMSA showing binding experiments using a fixed concentration of 0.2 nm 32P‐labeled RNA probe with increasing amount of N protein. Lower panel: a plot of a fraction of bound RNA against protein concentration based on the EMSA results shown in the upper panel, which was used for Kd determination using the Hill equation. (F) Kd values for the RNA probes shown in (A) with N protein. (G and H) Determination of Kd for RNA probes derived from the 3′‐terminus of the β‐actin mRNA and TOPO‐XL plasmid, respectively, using the same methods described for (B–E). (I) Determination of the bonding affinity between MHV N protein and RNA probe containing the CoV 3′‐terminal 55 nt plus a 25‐nt poly(A) tail by EMSA and Kd. The values in (B–I) represent the mean ± SD (n = 3) of three independent experiments. b, protein‐bound RNA; ub, unbound RNA.
Figure 3.
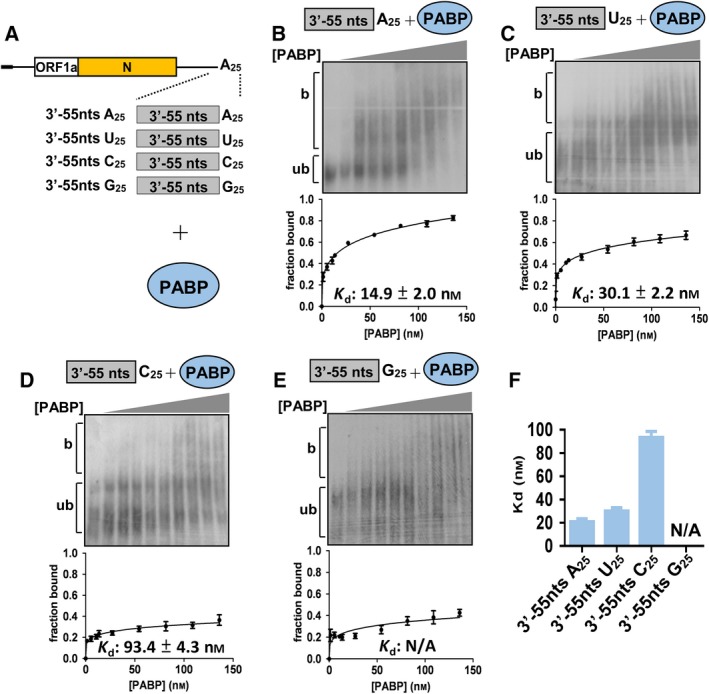
Determination of binding affinity between PABP and RNA elements by Kd. (A) Schematic diagrams depicting the structure of RNA probes used for determination of PABP binding affinities. (B–E) Upper panel: EMSA showing binding experiments using a fixed concentration of 0.2 nm 32P‐labeled RNA probe with increasing amount of PABP. Lower panel: a plot of a fraction of bound RNA against protein concentration based on the EMSA results shown in the upper panel, which was used for Kd determination using the Hill equation. (F) Kd values for the RNA probes shown in (A) with PABP. The values in (B–F) represent the mean ± SD (n = 3) of three independent experiments. b, protein‐bound RNA; ub, unbound RNA; N/A, not available.
Binding affinity between the N protein and the 3′‐terminal structure correlates with the efficiency of (−)‐strand RNA synthesis
It has been shown that the CoV poly(A) tail length (Fig. 1B) and the 3′‐terminal 55 nt 16 are factors affecting (−)‐strand RNA synthesis. To further examine whether the binding of N protein to the 3′‐terminal sequence is associated with (−)‐strand RNA synthesis, RNA probes (Fig. 4A) containing the 3′‐terminal 55 nt and various poly(A) tail lengths or only the 25‐nt poly(A) tail were constructed. In comparison with the binding affinity between the N protein and the 3′‐terminal 55 nt plus a 25‐nt poly(A) tail (Kd: ~ 33 nm) (Fig. 4E), the binding affinity between N protein and the 3′‐terminal 55 nt plus a 15‐nt poly(A) tail (Kd: ~ 40 nm) was slightly decreased (Fig. 4D). In addition, the binding affinity between N protein and only the 3′‐terminal 55 nt, 3′‐terminal 55 nt plus a 5‐nt poly(A) tail, or only the 25‐nt poly(A) tail was also decreased (Kd: ~ 87, ~67 and 48 nm, respectively) (Figs. 4B,C,F). These results are in agreement with our hypothesis that the efficiency of (−)‐strand RNA synthesis (Fig. 1B and 16) correlates with the binding affinity between the N protein and poly(A) tail length (Fig. 4B‐E,G) and 3′‐terminal 55 nt (Fig. 4F,G).
Figure 4.
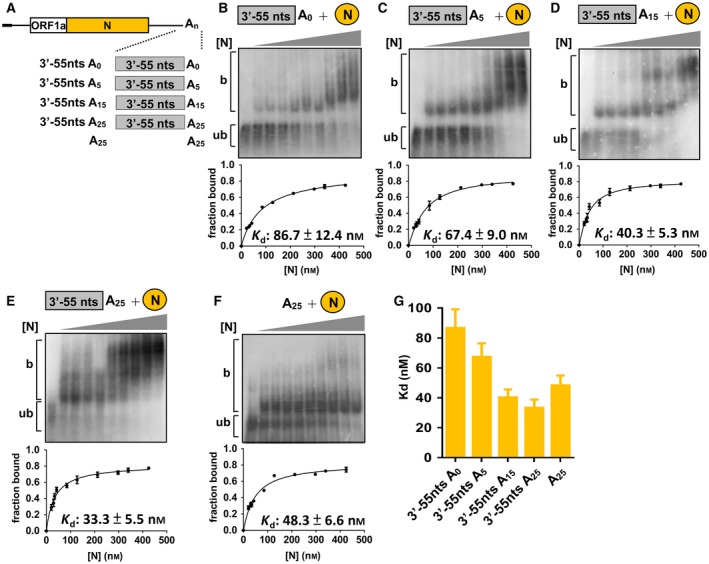
Correlation of binding affinity between the N protein and 3′‐terminal sequences with efficiency of the (−)‐strand RNA synthesis. (A) RNA probes with poly(A) tails of various lengths or deletion of 3′‐terminal 55 nt. (B–F) Determination of Kd between N and RNA probes illustrated in (A). Upper panel: EMSA showing binding experiments using a fixed concentration of 0.2 nm 32P‐labeled RNA probe with increasing amounts of N protein. Lower panel: plot of a fraction of bound RNA against protein concentration based on the EMSA results shown in the upper panel, which was used for the determination of Kd using the Hill equation. Lower panel: Kd values for the RNA probe and the N protein. (G) Values of Kd based on the results shown in (B–F). The values in (B–G) represent the mean ± SD (n = 3) of three independent experiments. b, protein‐bound RNA; ub, unbound RNA.
Züst et al. 19 proposed that two helical stem structures (designated S3 and S4, Fig. 5A) form at the end of the MHV 3′ UTR, and S3 is predicted to be important for initiating (−)‐strand RNA synthesis. Using reverse‐genetic approaches, Liu et al. 20 demonstrated that mutation in the 3′‐side of S3 (designated construct E in that study) to disrupt the stem structure produced a lethal phenotype; however, the data regarding the relative level of (−)‐strand RNA synthesis between the mutant (construct E) and wild‐type (wt) virus are not available. Conversely, mutation in the 3′‐side of S4 to disrupt the stem structure resulted in a viable virus. To experimentally determine the effect of the two structures on (−)‐strand RNA synthesis, sequence substitution based on the aforementioned study by Liu et al. 20 at the 3′‐side of S3 and S4 to disrupt the stem structures was performed using DI RNA BM25A; these constructs were designated BMmtS3 and BMmtS4, respectively (Fig. 5B). As shown in Fig. 5C, the efficiency of (−)‐strand RNA synthesis from BMmtS3 was significantly impaired, whereas the efficiency of (−)‐strand RNA synthesis from BMmtS4 was not altered in comparison with that from BM25A. Note that no recombination occurred between the helper virus BCoV and the three DI RNA constructs, and no sequence in these DI RNA constructs was altered (data not shown). The results, therefore, suggest that the S3 structure is critical for (−)‐strand RNA synthesis. Accordingly, the results are in agreement with those of the study by Liu et al. 20 in terms of virus viability and also support the model proposed by Züst et al. 19. Next, we performed EMSA to determine the binding affinity between the N protein and the two helical stems, S3 and S4, and to evaluate whether binding affinity is also linked to the efficiency of (−)‐strand RNA synthesis. For this aim, RNA probes (Fig. 5D) predicted by the Mfold algorithm and containing intact S3 and S4 structures (wtS3S4), disrupted S3 and intact S4 structures (mtS3, by mutation at the 3′‐side of S3) and intact S3 and disrupted S4 structures (mtS4, by mutation at the 3′‐side of S4) were generated, and EMSA was performed to determine Kd. As shown in Fig. 5E,G, the binding affinity between the N protein and wtS3S4 (Kd: 70.3 nm) was similar to that between N protein and mtS4 (Kd: 66.3 nm). Conversely, the Kd value for the N protein and mtS3 could not be calculated (Fig. 5F), suggesting that the value is higher than 500 nm and that binding affinity is very low. Because the higher binding affinity (wtS3S4 and mtS4 with N) corresponds to more efficient (−)‐strand RNA synthesis (BM25A and BMmtS4) and lower binding affinity (mtS3 with N protein) corresponds to less efficient (−)‐strand RNA synthesis (BMmtS3) (Figs. 5C,H), the binding affinity of N protein to S3 structure is linked to the efficiency of (−)‐strand RNA synthesis. Therefore, the results suggest the involvement of N protein and S3 structure in (−)‐strand RNA synthesis and thus support our hypothesis and the model proposed by Züst et al. 19.
Figure 5.
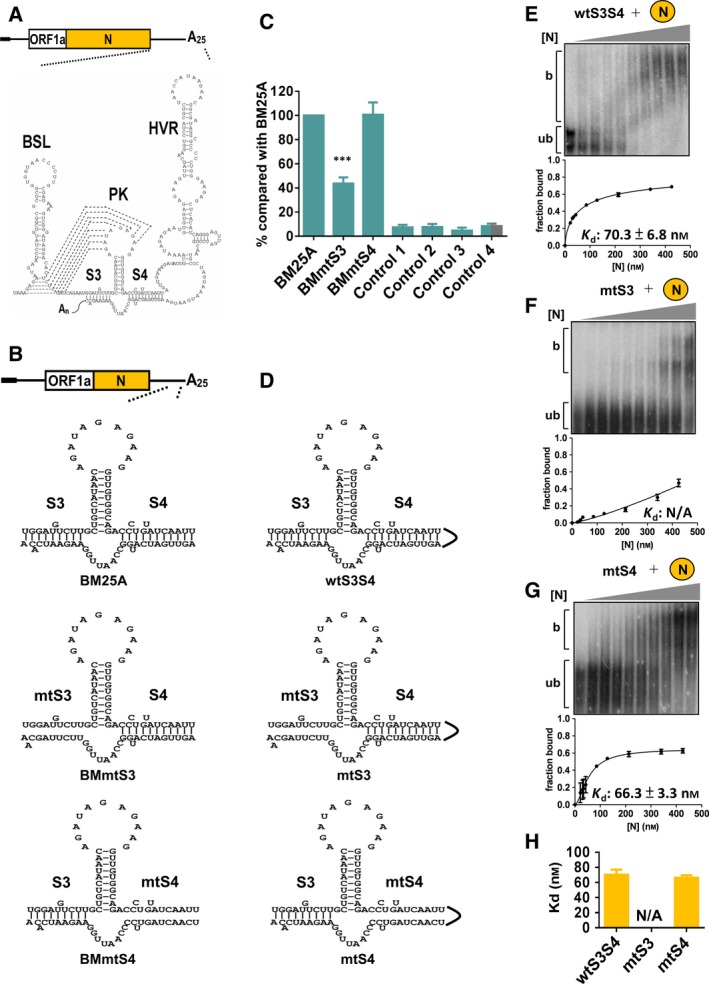
Correlation of binding affinity between the N protein and 3′‐terminal structures S3 and S4 with efficiency of the (−)‐strand RNA synthesis. (A) Schematic diagram depicting RNA structures in the 3′‐UTR of MHV including the bulged stem‐loop, BSL; PK, HVR and two helical stems, S3 and S4. (B) BM‐DI RNA constructs with mutations to disrupt the stem structure of S3 (BMmtS3) or S4 (BMmtS4). (C) Relative level of (−)‐strand RNA synthesis from BM‐DI RNA constructs illustrated in (B), as measured by RT‐qPCR. Control 1 – total cellular RNA from mock‐infected cells. Control 2 – total cellular RNA from BCoV‐infected cells. Control 3 – total cellular RNA from BM25A‐transfected mock‐infected cells. Control 4 – a mixture of BCoV‐infected cellular RNA extracted at 8 hpt and 200 ng of BM25A transcript. (D) RNA probes with an intact structure of S3 and S4 (wtS3S4) or a disrupted structure of S3 (mtS3) or S4 (mtS4). (E–G) Determination of Kd between N and RNA probes illustrated in (D). Upper panel: EMSA showing binding experiments using a fixed concentration of 0.2 nm 32P‐labeled RNA probe with increasing amounts of N protein. Lower panel: plot of a fraction of bound RNA against protein concentration based on the EMSA results shown in the upper panel, which was used for the determination of Kd using the Hill equation. (H) Values of Kd based on the results shown in (E–G). The values in (C) and (E–H) represent the mean ± SD (n = 3) of three independent experiments. b, protein‐bound RNA; ub, unbound RNA; N/A, not available. The statistical significance was evaluated using a t‐test versus BM25A: ***P < 0.001.
Binding of the N protein to the 5′‐terminal sequence is involved in (−)‐strand RNA synthesis
It has been shown that (−)‐strand N sgmRNA can be synthesized using (+)‐strand N sgmRNA as a template 39, though the efficiency is decreased in comparison with that of DI RNA 10, a surrogate for the CoV genome. The discrepancy between the structure of N sgmRNA and DI RNA lies in the 5′‐terminal sequence: DI RNA contains the 5′‐terminal 498 nt of the CoV genome, but N sgmRNA contains only the 5′‐terminal 77 nt of the genome. It was, therefore, speculated that such a structural difference is the main factor leading to various efficiencies of (−)‐strand RNA synthesis. In the current study, as the interaction between N protein and 3′‐terminal sequence was correlated with (−)‐strand RNA synthesis (Figs. 1, 2, 3, 4, 5), we hypothesized that the binding affinity of N protein with the 5′‐terminal structures of N sgmRNA and DI RNA may also explain the discrepancy in the efficiency of (−)‐strand RNA synthesis between the two viral RNA species. To test the hypothesis, we first repeated the experiment by detecting (−)‐strand RNA synthesis from N sgmRNA (sBM25A) and DI RNA (BM25A) 10. Note that, similar to BM25A, sBM25A was also engineered to contain the MHV 3′ UTR for subsequent analysis of (−)‐strand RNA detection. As shown in Fig. 6A, the efficiency of (−)‐strand RNA synthesis from sBM25A was decreased by ~ 50% when compared with that from DI RNA BM25A. In addition, no recombination was found between the helper virus BCoV and these DI RNA constructs and no sequence in these DI RNA constructs was altered (data not shown). Thus, the results are consistent with a previously published study 10. To evaluate whether such a decrease in (−)‐strand RNA synthesis correlates with the binding affinity between the N protein and the 5′‐terminal sequence, 120 nt of RNA probes BM5′‐120 nts and sBM5′‐120 nts (Fig. 6B), which are derived from the 5′‐terminus of BM25A and sBM25A, respectively, and thus have different structures, were subjected to EMSA to determine the binding affinities with N protein. The Kd for BM5′‐120 nt was 66.1 nm; however, that for sBM5′‐120 nt could not be calculated (Figs. 6C,D), suggesting that the N protein binds to the 5′‐terminal sequence of DI RNA BM25A with higher affinity than that of N sgmRNA. Because the efficiency of (−)‐strand RNA synthesis from DI RNA is better than that from N sgmRNA and because N binds to the 5′‐terminal sequence of DI RNA BM25A with better efficiency than that of N sgmRNA, the results also suggest the involvement of N protein in (−)‐strand RNA synthesis and provide further evidence explaining how the discrepancy between 5′ terminal structure of DI RNA and N sgmRNA affects the efficiency of (−)‐strand synthesis in the previously published study 10.
Figure 6.
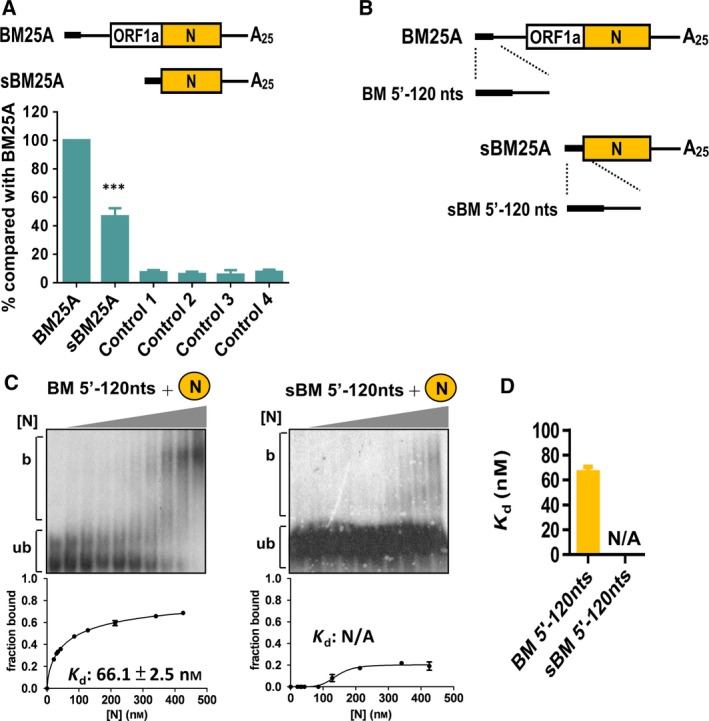
Correlation of binding affinity between the N protein and 5′‐terminal structures with efficiency of (−)‐strand RNA synthesis. (A) Upper panel: BM‐DI RNA construct containing the 5′‐terminal 498 nt of the CoV genome (BM25A) or only the 5′‐terminal 77 nt of the genome (sBM25A). Lower panel: relative level of (−)‐strand RNA between BM25A and sBM25A, as measured by RT‐qPCR. (B) Schematic diagram depicting the RNA probe containing the 5′‐terminal 120 nt of the genome (BM 5′‐120 nts) or N subgenome (sBM 5′‐120 nts). (C) Upper panel: EMSA showing binding experiments using a fixed concentration of 0.2 nm 32P‐labeled RNA probe with increasing amount of N protein. Lower panel: plot of a fraction of bound RNA against protein concentration based on the EMSA results shown in the upper panel, which was used for Kd determination using the Hill equation. (D) Kd values based on the results shown in (C). Controls in (A): Control 1 – total cellular RNA from mock‐infected cells. Control 2 – total cellular RNA from BCoV‐infected cells. Control 3 – total cellular RNA from BM25A‐transfected mock‐infected cells. Control 4 – a mixture of BCoV‐infected cellular RNA extracted at 8 hpt and 200 ng of BM25A transcript. The values in (A) (C) and (D) represent the mean ± SD (n = 3) of three independent experiments. b, protein‐bound RNA; ub, unbound RNA; N/A, not available. The statistical significance was evaluated using a t‐test versus BM25A: ***P < 0.001.
Interaction between the 5′‐ and 3′‐ends of the coronavirus genome via the N protein as a bridge
Our results showed that the N protein is able to bind to the 5′‐ and 3′‐terminal sequences of the CoV genome (Figs 2, 4, 5, 6). In addition, N protein can interact with other N monomers via its C‐terminal domain 40, 41, 42. It was therefore hypothesized that the N protein mediates interaction between the 5′‐ and 3′‐ends of the CoV genome, circularizing the genome. To examine this possibility, RNAs containing the DI RNA BM25A 5′‐terminal 120 nt (BM5′‐120 nts) and the 3′‐terminal 55 nt plus the poly(A) tail (3′‐55 ntsA25) (Fig. 7A), which are terminal RNA elements of CoV genome important for (−)‐strand RNA synthesis (Figs 4,5 and 6), were labeled with biotin and [α‐32P]ATP, respectively. The labeled RNAs (biotin‐BM5′‐120 nts and 32P‐3′‐55 ntsA25) were then incubated with the N protein followed by a streptavidin pull‐down assay, RNA extraction and denaturing gel electrophoresis to detect the signal of 32P‐3′‐55 ntsA25 (Fig. 7B). As shown in Fig. 7C, lane 3, the 32P‐labeled RNA (32P‐3′‐55 ntsA25) was detected (indicated by an arrow). However, the same 32P‐labeled RNA was not observed when biotin‐BM5′‐120 nts and 32P‐3′‐55 ntsA25 were incubated with Glutathione S‐transferase (lane 5). The results suggest that N protein is able to mediate the interaction between the 5′‐ and 3′‐terminal sequences of the CoV genome, circularizing the genome. Similar results were also found when biotin‐sBM5′‐120 nts, an RNA element which is derived from the 5′‐terminal 120 nt of N sgmRNA (sBM25A) (Fig. 7A) and has inefficient binding affinity with N protein (Fig. 6), and 32P‐3′‐55 ntsA25 were incubated with the N protein although the signal of 32P‐3′‐55 ntsA25 was weaker (Fig. 7C, lane 4). The amount of 32P‐3′‐55 ntsA25 in lane 4 was ~ 6% of that in lane 3. Together, the results suggest that the N protein can act as a bridge to facilitate interaction between the 5′‐ and 3′‐terminal sequences of the CoV genome and subgenome, leading to their circularization. In addition, because the (−)‐strand RNA synthesis (Fig. 6) and genome circularization from DI RNA (BM25A) are more efficient than those from N sgmRNA (sBM25A) (Fig. 7C), the results may also suggest that the circularization efficiency is involved in the (−)‐strand RNA synthesis.
Figure 7.
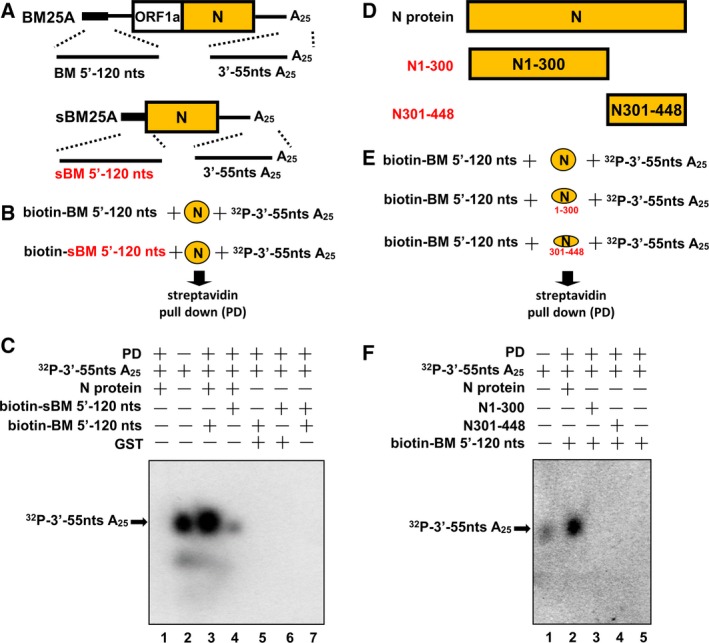
Interaction between the 5′‐ and 3′‐ends of the genome mediated by the N protein. (A) Schematic diagram depicting the sequences located at the 5′‐ and 3′‐termini of the CoV DI RNA genome (BM25A) and N subgenome (sBM25A) used for evaluating crosstalk between the two ends mediated by the N protein. (B) Diagram showing the strategy for assessing interaction between the 5′‐ and 3′‐ends of the genome and N subgenome. A biotin‐labeled 5′‐terminal RNA was incubated with N protein and 3′‐terminal 32P‐labeled RNA followed by a streptavidin pull‐down assay and RNA extraction. (C) Detection of 32P‐labeled RNA (indicated by arrow) by 6% denaturing gel electrophoresis to determine interaction between the two ends of the genome and N subgenome. (D) Schematic diagrams depicting the structure of BCoV N protein and its mutants N1–300 (N protein amino acids 1–300) and N301–448 (N protein amino acids 301–448). (E) Diagram showing the strategy for assessing the domains in N protein responsible for the interaction between the 5′‐ and 3′‐ends of the genome. (F) Detection of 32P‐labeled RNA (indicated by arrow) by 6% denaturing gel electrophoresis to identify the domain in N protein involved in the interaction between the 5′‐ and 3′‐ends of the genome. PD, pull‐down.
It has been shown that human CoV OC43 (HCoV‐OC43) N protein shares 97% amino acid sequence identity with BCoV N protein 43. In addition, it has been identified that in HCoV‐OC43 N protein, the N‐terminal region contains RNA‐binding domain (amino acids 1–300) and C‐terminal region is mainly involved in the oligomerization of the N protein (amino acids 301–448) 42. Therefore, to determine the domains of BCoV N protein involved in the genome circularization and thus to further verify the 5′‐ and 3′‐terminal interaction event of the genome, the truncated forms of the BCoV N protein were constructed based on the aforementioned conclusion from HCoV‐OC43 42: one contained only the N‐terminal domain (N1–300) and the other contained only the C‐terminal domain (N301–448) (Fig. 7D), followed by a streptavidin pull‐down assay (Fig. 7E). As shown in Fig. 7F, lane 2, when the full‐length N protein was used in the assay, the 32P‐labeled RNA (32P‐3′‐55 ntsA25) was detected (indicated by an arrow). However, the same 32P‐labeled RNA was not observed when the truncated N proteins (N1–300 and N301–448) were used (lanes 3 and 4). The results suggest that both the N‐ and C‐terminal domains of N protein are required for the genome circularization. That is, the 5′‐ and 3′‐terminal sequences of the genome may bind to N‐terminal domain of the individual N protein monomer and then the two N protein monomers can interact with each other via its C‐terminal domain, circularizing genome. Thus, the results further determine the involvement of the N protein in the interaction between the 5′‐ and 3′‐terminal sequences of the CoV genome.
Discussion
In this study, analysis of whether poly(A) tail length and sequence are involved in the efficient (−)‐strand RNA synthesis led us to discover that interaction between the N protein and 5′‐ and 3′‐terminal cis‐acting RNA elements are associated with (−)‐strand RNA synthesis. In addition, we for the first time demonstrate that interaction between the 5′‐ and 3′‐end of the CoV genome can be achieved via the N protein as a bridge, resulting in circularization of the genome. The biological relevance of the current findings is discussed below.
The minimum size of a poly(A) tail to initiate efficient (−)‐strand RNA synthesis for Sindbis virus and poliovirus is ~ 12 A residues 44, 45. For both viruses, the efficiency of (−)‐strand RNA synthesis does not increase as a linear function of poly(A) tail length. These studies indicate that a certain length of poly(A) tail may be sufficient to serve as a mediator to recruit replication‐associated factors for (−)‐strand synthesis 35, 44, 45, 46. In the current study, it was found that DI RNA with a 15‐nt poly(A) tail is sufficient for efficient (−)‐strand RNA synthesis and such synthesis does not increase as a linear function of poly(A) tail length in DI RNA ranging from 0 to 25 nt. In addition, the efficiency of (−)‐strand RNA synthesis is similar between DI RNA with a poly(U), poly(C) or poly(G) tail (Fig. 1C). These results support the speculation based on findings for Sindbis virus and poliovirus 44, 45 that factors other than the poly(A) tail that interact with both the poly(A) tail and adjacent 3′‐terminal sequence may also be involved in (−)‐strand RNA synthesis of CoV. Indeed, this argument is reinforced by the results presented in Figs 2, 4 and 5, whereby binding of N protein to the 3′‐terminal sequence along with the poly(A) tail appears to be associated with (−)‐strand synthesis. Because N protein in CoVs has been suggested to interact with replication complex 27, 28, 29, 47, the argument may also indicate that the binding of N protein with 3′‐terminal sequence plus the 15‐nt poly(A) tail is sufficient to form a stable replication complex for initiating (−)‐strand synthesis. Consequently, such argument in turn can explain why (−)‐strand synthesis does not increase as a linear function of poly(A) tail length in CoV DI RNA as well as Sindbis virus and poliovirus 44, 45.
In the current study, RNA‐protein binding assays and analysis of (−)‐strand RNA synthesis revealed the connection between the N protein and (−)‐strand RNA synthesis, whereby the binding affinity between N, but not PABP, and the 5′‐ and 3′‐terminal sequence or structure nearly corresponded to the efficiency of (−)‐strand synthesis. Based on the results, it is speculated that such binding of the N protein to the 5′‐ and 3′‐terminal sequence or structure has an advantage in initiating (−)‐strand RNA synthesis because this region of DI RNA bound by N rather than by PABP may prevent the binding of PABP to the poly(A) tail as well as subsequent translation factors 25. Under this circumstance, the N protein is able to efficiently interact with replication‐associated replicase proteins 27, 28, 29, 47 and initiate (−)‐strand synthesis. Accordingly, the CoV N protein appears to have a more important role than PABP in (−)‐strand RNA synthesis in terms of binding to the CoV 3′‐terminal sequence including the poly(A) tail. As PABP does have a positive effect on replication 18, 48, we still cannot rule out its importance in other steps of the CoV life cycle, for example, translation, which may affect subsequent RNA synthesis.
In addition to the 3′‐terminal 55 nt, a structure designated S3, which was formed by base pairing between the 3′‐most nt and loop 1 of the PK stem (Fig. 5A), is proposed by Züst et al. 19 to have an important function in the initiation of (−)‐strand RNA synthesis. Although the subsequent study by Liu et al. 20 showed that mutation to disrupt the stem structure of S3 generates a lethal virus, the role of the S3 in (−)‐strand RNA synthesis is still not known. In the current study, by using the same strategy to disrupt the structure of S3 but with DI RNA (BMmtS3, Fig. 5B), we found the efficiency of (−)‐strand RNA synthesis from the BMmtS3 mutant to be significantly impaired (Fig. 5C), experimentally demonstrating the important role of the S3 structure in (−)‐strand RNA synthesis. The results, therefore, strongly support the (−)‐strand initiation model proposed by Züst et al. 19 and may explain in part why disruption of S3 produced a lethal phenotype in the study by Liu et al. 20. More importantly, we extend the findings to associate decreased efficiency in (−)‐strand synthesis with the poor binding affinity between the N protein and the disrupted S3 structure (Fig. 5), further highlighting the involvement of N protein in (−)‐strand RNA synthesis and expanding the model proposed by Züst et al. 19. Indeed, the result that opening the S4 structure does not impair the efficiency of (−)‐strand synthesis also supports the model and is consistent with that by Liu et al. 20. In addition, our findings also can explain why deletion of the 3′‐terminal 55 nt, which also removes the S3 structure, from DI RNA severely impairs (−)‐strand RNA synthesis in the current (Fig. 5C) and previous studies 16, 17. Based on our findings and those of others 19, 20, we extend the Züst model and propose that stable binding of the N protein to the poly(A) tail and S3 structure is a critical step in the initiation of (−)‐strand RNA synthesis in CoVs.
Several studies have suggested that cis‐acting RNA elements located at the 5′‐terminus of the genome are also critical for (−)‐strand RNA synthesis 8, 10, 49, 50. In the current study, we found that in addition to the 3′‐terminal sequence or structure, the binding affinity of N protein to the 5′‐terminal sequence also correlates with (−)‐strand RNA synthesis. As (−)‐strand RNA synthesis is initiated from the 3′‐terminus of the (+)‐strand genome, the question remains as to how binding of the N protein to 5′‐terminal RNA elements in the (+)‐strand genome affects (−)‐strand RNA synthesis. We argue that genome circularization may be an important mechanism to address the question above. Indeed, a link between the 5′‐ and 3′‐termini of the viral genome via RNA‐RNA or RNA‐protein interaction has been suggested to initiate RNA synthesis in RNA viruses 9, 33, 35, 51, 52, 53, 54, 55, 56, 57. As the N protein can bind to both the 5′‐ and 3′‐terminal sequences of the CoV genome (Figs 2, 4, 5, 6) and N protein can interact with other N protein monomers, it is reasonable that the 5′‐terminus can interact with the 3′‐terminus via the N protein, circularizing the genome, as evidenced by the results shown in Fig. 7. In addition, alternations in the 5′‐terminal sequence impair the efficiency of the interaction (Fig. 7C, lane 4) further explain the previously unsolved question of how 5′‐terminal RNA elements in the (+)‐strand genome affect (−)‐strand RNA synthesis. Because the CoV N protein has been suggested to interact with CoV replicase proteins and cellular proteins, it is postulated that the interaction between the 5′‐and 3′‐terminal sequences and N protein may function as a platform to recruit replication‐associated proteins to constitute a stable replication complex, thereby initiating (−)‐strand RNA synthesis. In this sense, mutations in the 5′‐ or 3′‐terminal sequence, which decrease the binding affinity between N protein and both terminal sequences and thus the stability of the replication complex, lead to decreased (−)‐strand RNA synthesis. Consequently, without N protein to act as a bridge to form the replication complex, (−)‐strand RNA cannot be initiated efficiently, impairing subsequent (+)‐strand RNA synthesis. This may explain why CoV recovery in many CoV reverse‐genetic systems can be significantly increased with addition of the N protein 30, 31, 47. Although RNA‐RNA or RNA‐protein interaction facilitating interaction between the 5′‐and 3′‐termini of the CoV genome has been suggested to be involved in RNA synthesis 6, 9, 36, 58, such interaction achieved via the N protein as a bridge has not been previously documented in CoVs.
Based on the data presented herein, and the results of others, we link the N protein and genome circularization to (−)‐strand RNA synthesis and extend Züst's model 19 as follows (Fig. 8). Based on Züst's model, the S3 structure is formed by interaction between the 3′‐terminal sequence and loop 1 of the PK. Binding of N protein to the (−) strand RNA synthesis‐associated 5′‐ and 3′‐terminal sequences or structures (e.g., S3) then leads to circularization of the CoV genome, which serves as a platform to recruit cellular proteins and viral replicase proteins, initiating (−)‐strand synthesis using the poly(A) tail as a template. The proposed model, in turn, is able to explain the results presented in this study. In conclusion, we show the involvement of the N protein and genome circularization in (−)‐strand RNA synthesis. The findings support and extend Züst's model for (−)‐strand RNA synthesis.
Figure 8.
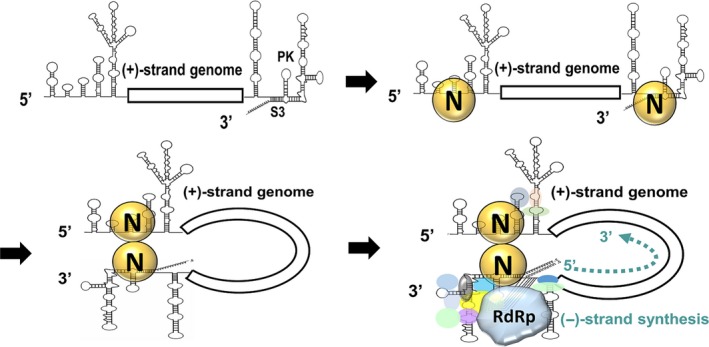
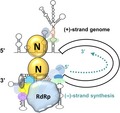
Extended model for initiation of CoV (−)‐strand RNA synthesis. The S3 structure is formed by interaction between the 3′‐terminal sequence and the PK loop 1. In the initial stage of (−)‐strand RNA synthesis, the N protein binds to both the 5′‐ and 3′‐terminal sequences. This interaction leads to crosstalk between the 5′‐ and 3′‐ends of the genome, as bridged by the N protein, recruiting replicase proteins and initiating (−)‐strand RNA synthesis. RdRp, RNA‐dependent RNA polymerase.
Materials and methods
Viruses and cells
Bovine coronavirus Mebus strain (GenBank accession no. U00735), which was obtained from David A. Brian (University of Tennessee, TN, USA), was grown in HRT‐18 cells and plaque‐purified 59, 60. HRT‐18 cell line was also obtained from David A. Brian and maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS (HyClone Laboratories, Inc., Logan, UT, USA) at 37 °C with 5% CO2 61.
Plasmid constructs
Constructs pBM0A, pBM5A, pBM15A, pBM25U, pBM25C, pBM25G, pBMΔ55, pBMmst3 and pBMmst4 were generated as described previously 10, 16, 62; briefly, pBM25A DNA was used as the template with appropriate oligonucleotides containing various mutated 3′‐terminal sequences corresponding to the constructs for PCR. The resulting PCR product was digested with SpeI and MluI and cloned into SpeI‐ and MluI‐linearized pBM‐25A to create the aforementioned constructs. For RNA probes, PCR was performed using the oligonucleotides to generate DNA fragments, which were then used as templates for synthesizing RNA probes 3′‐55ntsA25, 3′‐55ntsU25, 3′‐55ntsC25, 3′‐55ntsG25, 3′‐55ntsA0, mtS3, mtS4 and A25. The DNA fragments for producing the RNA probes BM 5′‐120nts and sBM‐120nt were also generated by the PCR, but with pBM25A and psBM25A as the template, respectively 10. Note that each RNA probe contains additional three guanosine residues at its 5′ end after in vitro transcription.
Expression of recombinant proteins
pET32aN carrying the BCoV N protein gene, pET32aN1‐300 carrying the BCoV N protein gene 1‐900, pET32aN300‐448 carrying the BCoV N protein gene 901–1344, pET32aMHVN carrying the MHV N protein gene (GenBank accession no. AY700211.1) and pET28aPABP carrying human cytoplasmic PABP1 (PABPC1) gene (GenBank accession no. NM_002568) were constructed as described previously 25. Expression of His‐tagged N protein and PABP was also performed as described previously 25. Briefly, to express MHV N protein, BCoV N protein and its mutants, Escherichia coli BL21 (DE3) cells containing pET32aMHVN, pET32aN, pET32aN1‐300 or pET32aN301‐448 were inoculated into LB medium supplemented with 50 μg·mL−1 ampicillin, induced with isopropyl thio‐β‐D‐galactoside, collected by centrifugation and resuspended in PBS. The resuspended cells were sonicated and the supernatant was collected by removing cell debris with centrifugation. The recombinant protein in collected supernatant was purified through the 6xHis tag by immobilized metal ion affinity chromatography with EDTA‐resistant Ni Sepharose excel resin (GE Healthcare, Houston, TX, USA) and loaded on a nickel‐chelating column (GE Healthcare). Fractions which contain recombinant N protein were dialyzed and collected. To obtain PABP, pET28aPABP‐containing E. coli BL21 (DE3) pLysS cells were inoculated into LB medium supplemented with 50 μg·mL−1 kanamycin. The cells were also induced with isopropyl thio‐β‐D‐galactoside, centrifuged and resuspended in 20 mm Tris–HCl (pH 7.6) buffer. The resuspended cells were sonicated and centrifuged, and the supernatant was collected. The supernatant containing the recombinant PABP was loaded onto a Ni SepharoseTM 6 Fast Flow column (GE Healthcare) followed by washing with buffer containing 20 mm Tris–HCl, 0.5 m NaCl, pH 7.4 and 40, 100, 200, 300 and 500 mm imidazole. Fractions containing recombinant PABP were pooled and dialyzed. The purified recombinant N protein and PABP were stored in buffer (50 mm Tris‐Glycine, pH 7.4, 0.15 m NaCl, 30% Glycerol, and 0.01% NaN3) at −20 °C until use.
Evaluation of (−)‐strand RNA synthesis
To evaluate the efficiency of (−)‐strand RNA synthesis 8, 10, 16, MluI‐linearized plasmid DNA construct was transcribed in vitro with mMessage mMachine T7 transcription kit (Thermo Fisher Scientific, Waltham, MA, USA) at 37 °C for 90 min according to the manufacturer's instructions and then chromatographed through a Biospin 6 column (Bio‐Rad, Hercules, CA, USA). HRT‐18 cells were infected with BCoV and after 2 h of infection, 3 μg of DI RNA transcript was transfected into BCoV‐infected HRT‐18 cells. RNA was extracted with TRIzol (Thermo Fisher Scientific) at 8 h post‐transfection (hpt) and 6 μg of RNA was treated with tobacco acid pyrophosphatase (TAP; Epicentre, Madison, WI, USA) to generate monophosphate RNA. Head‐to‐tail ligation of RNA was performed using T4 RNA ligase I (New England Biolabs, Ipswich, MA, USA) followed by phenol‐chloroform extraction and ethanol precipitation. For RT reaction, 1 μg of TAP‐treated and ligated RNA was used with oligonucleotide MHV3′UTR3(−) and SuperScript III reverse transcriptase (Invitrogen) 8, 10, 16. To detect the (−)‐strand RNA without head‐to‐tail ligation, RT‐PCR was performed with 1 μg of RNA with oligonucleotides MHV3′UTR‐DR(−) (for RT) and MHV3UTR‐DR(+) 10. Real‐time PCR amplification was performed using TagMan® Universal PCR Master Mix (Applied Biosystems, Carlsbad, CA, USA) according to the manufacturer's recommendations with a LightCycler® 480 instrument (Roche Applied Science, Penzberg, Germany) with the primers MHV3′UTR6(−) and BCV23‐40(+) 8, 10, 16. Dilutions of plasmids containing the same sequence as the detected RT‐PCR product of (−) –strand DI RNA were run in parallel with quantitated cDNA for use as standard curves (dilutions ranged from 108 to 10 copies of each plasmid). In addition, the levels of 18S rRNA, (+)‐strand reporter‐containing DI RNA or sgmRNA and helper virus M sgmRNA were applied as internal controls for normalization of (−)‐strand DI RNA synthesis 8, 10, 16. The reactions were conducted with an initial preincubation at 95 °C for 5 min, followed by 35 amplification cycles as follows: 95 °C for 15 s and 60 °C for 60 s.
EMSA and Kd
Electrophoretic mobility shift assays were performed essentially as described previously 18, 25, 63, 64. Briefly, 32P‐labeled RNA was generated as specified by the manufacturer (Promega, Fitchburg, WI, USA) and separated on 6% sequencing gels. Passive elution was performed at 4 °C followed by phenol‐chloroform extraction. To determine binding affinity, a fixed concentration of 0.2 nm 32P‐labeled RNA was titrated with the protein of interest in binding buffer [20 mm HEPES (pH 7.5), 6 mm MgCl2, 1.5 μm EGTA, 22.5 mm NaCl, 330 mm KCl, 36% glycerol, 3.6 mm DTT, 82.5 μg·mL−1 BSA] for 15 min at 37 °C with 1 U·mL−1 RNasin (Promega). The bound RNA‐protein complexes were separated from unbound RNA by 8% native polyacrylamide gel electrophoresis with crosslink ratio 2.7%, dried, and analyzed by autoradiography. Free and bound RNAs were quantitated and fit to the Hill equation: , where b is the upper binding limit, [P] is the protein concentration, n is the Hill coefficient. graphpad prism (GraphPad Software, Inc., La Jolla, CA, USA) was then used to derive Kd, and the Kd values presented were determined based on at least three independent experiments.
Assay for interaction between the 5′‐ and 3′‐ends of the genome bridged by the N protein
The assay was based on the methods of Huang et al. 36, with some modifications. DNA templates pBM25A and psBM25A were used for in vitro transcription with T7 polymerase (Promega) in the presence of a biotin‐UTP labeling NTP mixture (Roche Applied Science), as recommended by the manufacturer, to synthesize biotinylated RNA with 5′‐terminal 120 nt of genome or N subgenome, respectively. After purification, the biotinylated RNA was incubated with recombinant N protein and 32P‐labeled RNA containing 3′‐terminal 55 nt plus the poly(A) tail in a reaction buffer containing 5 U of RNasin (Promega), 25 mm KCl, 5 mm HEPES (pH 7.6), 2 mm MgCl2, 0.1 mm EDTA, 0.2% glycerol, and 2 mm DTT. After incubation at room temperature for 30 min, a streptavidin suspension (MagQu, Taipei, Taiwan) was added to the mixture and incubated for 30 min at room temperature followed by three washes with binding buffer. RNAs bound to streptavidin agarose beads were eluted by a buffer [7 m urea, 350 mm NaCl, 10 mm Tris‐HCl (pH 8.0), 10 mm EDTA, and 1% SDS] followed by phenol‐chloroform extraction and ethanol precipitation. The samples were resolved on a 6% polyacrylamide gel.
Statistical analysis
Data were analyzed with Student's unpaired t‐test using prism 6.0 software (GraphPad Software, Inc.). The values in the study are presented as the mean ± SD (n = 3); *P < 0.05, **P < 0.01 and ***P < 0.001.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
HYW and CYL conceived and designed the experiments. CYL, TLT and CHL performed all the experiments. HYW, CYL, TLT, CNL, and CHL contributed to data analysis and discussion. HYW and CYL wrote the manuscript.
Acknowledgements
We thank Dr Wei‐Li Hsu and Chih‐Jung Kuo at National Chung Hsing University, Taiwan, for the assistance of protein preparation and RNA‐protein interaction experiment. This work was supported by grants 105‐2320‐B‐005‐004‐ and 106‐2313‐B‐005‐046‐MY3 from the Ministry of Science and Technology (MOST), R.O.C. (https://www.most.gov.tw/?l=ch).
References
- 1. International Committee on Taxonomy of Viruses & King AM (2012) Virus Taxonomy: Classification and Nomenclature of Viruses : Ninth Report of the International Committee on Taxonomy of Viruses. Academic Press, London: Waltham, MA. [Google Scholar]
- 2. Brian DA & Baric RS (2005) Coronavirus genome structure and replication. Curr Top Microbiol Immunol 287, 1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lee S & Lee C (2014) Complete genome characterization of Korean Porcine deltacoronavirus strain KOR/KNU14‐04/2014. Genome Announc 2, e01191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Masters PS (2006) The molecular biology of coronaviruses. Adv Virus Res 66, 193–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Madhugiri R, Fricke M, Marz M & Ziebuhr J (2016) Coronavirus cis‐acting RNA elements. Adv Virus Res 96, 127–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sola I, Mateos‐Gomez PA, Almazan F, Zuniga S & Enjuanes L (2011) RNA‐RNA and RNA‐protein interactions in coronavirus replication and transcription. RNA Biol 8, 237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang D & Leibowitz JL (2015) The structure and functions of coronavirus genomic 3′ and 5′ ends. Virus Res 206, 120–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ke TY, Liao WY & Wu HY (2013) A leaderless genome identified during persistent bovine coronavirus infection is associated with attenuation of gene expression. PLoS ONE 8, e82176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li L, Kang H, Liu P, Makkinje N, Williamson ST, Leibowitz JL & Giedroc DP (2008) Structural lability in stem‐loop 1 drives a 5′ UTR‐3′ UTR interaction in coronavirus replication. J Mol Biol 377, 790–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yeh PY & Wu HY (2014) Identification of cis‐acting elements on positive‐strand subgenomic mRNA required for the synthesis of negative‐strand counterpart in bovine coronavirus. Viruses 6, 2938–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hsue B, Hartshorne T & Masters PS (2000) Characterization of an essential RNA secondary structure in the 3′ untranslated region of the murine coronavirus genome. J Virol 74, 6911–6921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hsue B & Masters PS (1997) A bulged stem‐loop structure in the 3′ untranslated region of the genome of the coronavirus mouse hepatitis virus is essential for replication. J Virol 71, 7567–7578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Williams GD, Chang RY & Brian DA (1999) A phylogenetically conserved hairpin‐type 3′ untranslated region pseudoknot functions in coronavirus RNA replication. J Virol 73, 8349–8355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Goebel SJ, Miller TB, Bennett CJ, Bernard KA & Masters PS (2007) A hypervariable region within the 3′ cis‐acting element of the murine coronavirus genome is nonessential for RNA synthesis but affects pathogenesis. J Virol 81, 1274–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu Q, Johnson RF & Leibowitz JL (2001) Secondary structural elements within the 3′ untranslated region of mouse hepatitis virus strain JHM genomic RNA. J Virol 75, 12105–12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liao WY, Ke TY & Wu HY (2014) The 3′‐terminal 55 nucleotides of bovine coronavirus defective interfering RNA harbor cis‐acting elements required for both negative‐ and positive‐strand RNA synthesis. PLoS ONE 9, e98422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lin YJ, Liao CL & Lai MM (1994) Identification of the cis‐acting signal for minus‐strand RNA synthesis of a murine coronavirus: implications for the role of minus‐strand RNA in RNA replication and transcription. J Virol 68, 8131–8140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Spagnolo JF & Hogue BG (2000) Host protein interactions with the 3′ end of bovine coronavirus RNA and the requirement of the poly(A) tail for coronavirus defective genome replication. J Virol 74, 5053–5065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zust R, Miller TB, Goebel SJ, Thiel V & Masters PS (2008) Genetic interactions between an essential 3′ cis‐acting RNA pseudoknot, replicase gene products, and the extreme 3′ end of the mouse coronavirus genome. J Virol 82, 1214–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu P, Yang D, Carter K, Masud F & Leibowitz JL (2013) Functional analysis of the stem loop S3 and S4 structures in the coronavirus 3′UTR. Virology 443, 40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen H, Gill A, Dove BK, Emmett SR, Kemp CF, Ritchie MA, Dee M & Hiscox JA (2005) Mass spectroscopic characterization of the coronavirus infectious bronchitis virus nucleoprotein and elucidation of the role of phosphorylation in RNA binding by using surface plasmon resonance. J Virol 79, 1164–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nelson GW & Stohlman SA (1993) Localization of the RNA‐binding domain of mouse hepatitis virus nucleocapsid protein. J Gen Virol 74 , 1975–1979. [DOI] [PubMed] [Google Scholar]
- 23. Cologna R, Spagnolo JF & Hogue BG (2000) Identification of nucleocapsid binding sites within coronavirus‐defective genomes. Virology 277, 235–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grossoehme NE, Li LC, Keane SC, Liu PH, Dann CE, Leibowitz JL & Giedroc DP (2009) Coronavirus N Protein N‐Terminal Domain (NTD) specifically binds the transcriptional regulatory sequence (TRS) and melts TRS‐cTRS RNA duplexes. J Mol Biol 394, 544–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tsai T‐L, Lin C‐H, Lin C‐N, Lo C‐Y & Wu H‐Y (2018) Interplay between the poly(A) tail, poly(A)‐binding protein and coronavirus nucleocapsid protein regulates gene expression of the coronavirus and host cell. J Virol 92, e01162–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bost AG, Prentice E & Denison MR (2001) Mouse hepatitis virus replicase protein complexes are translocated to sites of M protein accumulation in the ERGIC at late times of infection. Virology 285, 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Verheije MH, Hagemeijer MC, Ulasli M, Reggiori F, Rottier PJ, Masters PS & de Haan CA (2010) The coronavirus nucleocapsid protein is dynamically associated with the replication‐transcription complexes. J Virol 84, 11575–11579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hurst KR, Koetzner CA & Masters PS (2013) Characterization of a critical interaction between the coronavirus nucleocapsid protein and nonstructural protein 3 of the viral replicase‐transcriptase complex. J Virol 87, 9159–9172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hurst KR, Ye R, Goebel SJ, Jayaraman P & Masters PS (2010) An interaction between the nucleocapsid protein and a component of the replicase‐transcriptase complex is crucial for the infectivity of coronavirus genomic RNA. J Virol 84, 10276–10288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Almazan F, Galan C & Enjuanes L (2004) The nucleoprotein is required for efficient coronavirus genome replication. J Virol 78, 12683–12688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schelle B, Karl N, Ludewig B, Siddell SG & Thiel V (2005) Selective replication of coronavirus genomes that express nucleocapsid protein. J Virol 79, 6620–6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zuniga S, Cruz JL, Sola I, Mateos‐Gomez PA, Palacio L & Enjuanes L (2010) Coronavirus nucleocapsid protein facilitates template switching and is required for efficient transcription. J Virol 84, 2169–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Villordo SM & Gamarnik AV (2009) Genome cyclization as strategy for flavivirus RNA replication. Virus Res 139, 230–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Romero‐Lopez C & Berzal‐Herranz A (2009) A long‐range RNA‐RNA interaction between the 5′ and 3′ ends of the HCV genome. RNA 15, 1740–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Herold J & Andino R (2001) Poliovirus RNA replication requires genome circularization through a protein‐protein bridge. Mol Cell 7, 581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huang P & Lai MM (2001) Heterogeneous nuclear ribonucleoprotein a1 binds to the 3′‐untranslated region and mediates potential 5′‐3′‐end cross talks of mouse hepatitis virus RNA. J Virol 75, 5009–5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hofmann MA & Brian DA (1991) The 5′ end of coronavirus minus‐strand RNAs contains a short poly(U) tract. J Virol 65, 6331–6333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wu HY, Ke TY, Liao WY & Chang NY (2013) Regulation of coronaviral Poly(A) tail length during infection. PLoS ONE 8, e70548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wu HY & Brian DA (2010) Subgenomic messenger RNA amplification in coronaviruses. Proc Natl Acad Sci USA 107, 12257–12262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lo YS, Lin SY, Wang SM, Wang CT, Chiu YL, Huang TH & Hou MH (2013) Oligomerization of the carboxyl terminal domain of the human coronavirus 229E nucleocapsid protein. FEBS Lett 587, 120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yu IM, Gustafson CL, Diao J, Burgner JW 2nd, Li Z, Zhang J & Chen J (2005) Recombinant severe acute respiratory syndrome (SARS) coronavirus nucleocapsid protein forms a dimer through its C‐terminal domain. J Biol Chem 280, 23280–23286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Huang CY, Hsu YL, Chiang WL & Hou MH (2009) Elucidation of the stability and functional regions of the human coronavirus OC43 nucleocapsid protein. Protein Sci 18, 2209–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hogue BG, King B & Brain DA (1984) Antigenic relationships among proteins of bovine coronavirus, human respiratory coronavirus‐Oc43, and mouse hepatitis coronavirus‐A59. J Virol 51, 384–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hardy RW & Rice CM (2005) Requirements at the 3′ end of the sindbis virus genome for efficient synthesis of minus‐strand RNA. J Virol 79, 4630–4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Silvestri LS, Parilla JM, Morasco BJ, Ogram SA & Flanegan JB (2006) Relationship between poliovirus negative‐strand RNA synthesis and the length of the 3′ poly(A) tail. Virology 345, 509–519. [DOI] [PubMed] [Google Scholar]
- 46. Barton DJ, O'Donnell BJ & Flanegan JB (2001) 5 ‘cloverleaf in poliovirus RNA is a cis‐acting replication element required for negative‐strand synthesis. EMBO J 20, 1439–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yount B, Denison MR, Weiss SR & Baric RS (2002) Systematic assembly of a full‐length infectious cDNA of mouse hepatitis virus strain A59. J Virol 76, 11065–11078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Galan C, Sola I, Nogales A, Thomas B, Akoulitchev A, Enjuanes L & Almazan F (2009) Host cell proteins interacting with the 3′ end of TGEV coronavirus genome influence virus replication. Virology 391, 304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu P, Li L, Keane SC, Yang D, Leibowitz JL & Giedroc DP (2009) Mouse hepatitis virus stem‐loop 2 adopts a uYNMG(U)a‐like tetraloop structure that is highly functionally tolerant of base substitutions. J Virol 83, 12084–12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu P, Li L, Millership JJ, Kang H, Leibowitz JL & Giedroc DP (2007) A U‐turn motif‐containing stem‐loop in the coronavirus 5′ untranslated region plays a functional role in replication. RNA 13, 763–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Alvarez DE, Filomatori CV & Gamarnik AV (2008) Functional analysis of dengue virus cyclization sequences located at the 5′ and 3′UTRs. Virology 375, 223–235. [DOI] [PubMed] [Google Scholar]
- 52. Alvarez DE, Lodeiro MF, Luduena SJ, Pietrasanta LI & Gamarnik AV (2005) Long‐range RNA‐RNA interactions circularize the dengue virus genome. J Virol 79, 6631–6643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Filomatori CV, Lodeiro MF, Alvarez DE, Samsa MM, Pietrasanta L & Gamarnik AV (2006) A 5′ RNA element promotes dengue virus RNA synthesis on a circular genome. Genes Dev 20, 2238–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ooms M, Abbink TE, Pham C & Berkhout B (2007) Circularization of the HIV‐1 RNA genome. Nucleic Acids Res 35, 5253–5261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sola I, Moreno JL, Zuniga S, Alonso S & Enjuanes L (2005) Role of nucleotides immediately flanking the transcription‐regulating sequence core in coronavirus subgenomic mRNA synthesis. J Virol 79, 2506–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zuniga S, Sola I, Alonso S & Enjuanes L (2004) Sequence motifs involved in the regulation of discontinuous coronavirus subgenomic RNA synthesis. J Virol 78, 980–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Enjuanes L, Almazan F, Sola I & Zuniga S (2006) Biochemical aspects of coronavirus replication and virus‐host interaction. Annu Rev Microbiol 60, 211–230. [DOI] [PubMed] [Google Scholar]
- 58. Moreno JL, Zuniga S, Enjuanes L & Sola I (2008) Identification of a coronavirus transcription enhancer. J Virol 82, 3882–3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. King B & Brian DA (1982) Bovine coronavirus structural proteins. J Virol 42, 700–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lapps W, Hogue BG & Brian DA (1987) Sequence analysis of the bovine coronavirus nucleocapsid and matrix protein genes. Virology 157, 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tompkins WA, Watrach AM, Schmale JD, Schultz RM & Harris JA (1974) Cultural and antigenic properties of newly established cell strains derived from adenocarcinomas of the human colon and rectum. J Natl Cancer Inst 52, 1101–1110. [DOI] [PubMed] [Google Scholar]
- 62. Peng YH, Lin CH, Lin CN, Lo CY, Tsai TL & Wu HY (2016) Characterization of the Role of Hexamer AGUAAA and Poly(A) Tail in Coronavirus Polyadenylation. PLoS ONE 11, e0165077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Raman S, Bouma P, Williams GD & Brian DA (2003) Stem‐loop III in the 5′ untranslated region is a cis‐acting element in bovine coronavirus defective interfering RNA replication. J Virol 77, 6720–6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Raman S & Brian DA (2005) Stem‐loop IV in the 5′ untranslated region is a cis‐acting element in bovine coronavirus defective interfering RNA replication. J Virol 79, 12434–12446. [DOI] [PMC free article] [PubMed] [Google Scholar]