

Abstract
Purpose:
Monoclonal antibodies (mAb) are used to treat solid and hematological malignancies, and work in part through Fc receptors (FcR) on natural killer cells (NK). However, FcR mediated functions of NK cells from cancer patients are significantly impaired. Identifying the mechanisms of this dysfunction and impaired response to mAb therapy could lead to combination therapies and enhance mAb therapy.
Experimental Design:
Co-cultures of autologous NK cells and MDSC from cancer patients were used to study the effect of MDSC on NK cell FcR mediated functions including antibody dependent cellular cytotoxicity, cytokine production, and signal transduction in vitro. Mouse breast cancer models were utilized to study the effect of MDSC on antibody therapy in vivo and test the efficacy of combination therapies including a mAb and a MDSC targeting agent.
Results:
Cancer patient MDSC were found to significantly inhibit NK cell FcR mediated functions including ADCC, cytokine production, and signal transduction in a contact independent manner. In addition, adoptive transfer of MDSC abolished the efficacy of mAb therapy in a mouse model of pancreatic cancer. Inhibition of iNOS restored NK cell functions and signal transduction. Finally, non-specific elimination of MDSC or inhibition of iNOS in vivo significantly improved the efficacy of mAb therapy in a mouse model of breast cancer.
Conclusions:
MDSC antagonize NK cell FcR mediated function and signal transduction leading to impaired response to mAb therapy in part through nitric oxide production. Thus, elimination of MDSC or inhibition of nitric oxide production offers a strategy to improve mAb therapy.
Keywords: MDSC, NK, Nitric oxide, immunotherapy, Fc receptor
Introduction
Natural killer (NK) cells are large granular lymphocytes that participate in innate immune responses against virus infected and neoplastic cells via receptors for major histocompatibility antigens, markers of cellular stress (NKG2D) and immunoglobulin G (FcR) (1). NK cells are unique in that they constitutively express only a low-affinity, activating FcR (FcγRIIIa or CD16), enabling them to recognize antibody (Ab)–coated targets (1,2). FcR-activated NK cells can kill antibody coated targets and secrete cytokines such as IFN-γ and chemokines (e.g., RANTES and MIP-1α) that inhibit tumor cell proliferation, enhance antigen presentation and promote the chemotaxis of T cells (3). These properties of FcR-activated NK cells are an important component of the response to mAb therapy (4).
NK cells are an important component of immune surveillance against the development of malignancies and the control of established tumors. NK cell infiltration of established tumors is associated with improved disease prognosis (5,6). However, NK cells from cancer patients have reduced cytotoxic function and cytokine production (7). The cause of this dysfunction is not fully understood, but has been attributed to factors secreted by or expressed on the surface of tumor cells (8,9). Only a few studies have examined the role of immune suppressive cells in mediating NK cell dysfunction (10,11) in the setting of cancer.
MDSC are immature myeloid cells with immunosuppressive properties that expand in response to tumor and stroma-derived factors (12). The frequency of circulating MDSC correlates with tumor burden and has prognostic value (13). In mice, MDSC are identified by expression of Gr-1 and CD11b and in humans they are characterized as CD33+/CD11b+/HLA-DRlow/neg (12). MDSC inhibit T-cells by a number of mechanisms including the production of free radicals, expression of amino acid catabolizing enzymes and the secretion of suppressive cytokines (14). Studies in murine models indicate that disruption of MDSC function can improve anti-tumor immune responses and impair tumor growth (15). Given the ability of MDSC to suppress anti-tumor immune responses, they have received significant interest as a potential biomarker and therapeutic target (16).
Studies have shown that MDSC impair NK cell cytokine production and MHC I dependent cytotoxicity through contact dependent mechanisms (e.g. checkpoint ligand expression and reactive oxygen species production) and contact independent mechanisms (arginase and TGF-β production) (10,11,17–21). However, the impact of MDSC on NK cell FcR-mediated function is unknown. Given the prominence of antibody therapy in the treatment of cancer, elucidation of the effects of MDSC on FcR-mediated NK cell function could have important clinical implications. In particular, this approach could identify novel mechanisms of resistance to mAb therapy.
It was hypothesized that MDSC could inhibit FcR-mediated NK cell functions and antagonize monoclonal antibody therapy. Utilizing MDSC and autologous NK cells from cancer patients and murine models of mAb therapy we show that MDSC nitric oxide (NO) production inhibits NK cell FcR-mediated signal transduction and downstream effector functions including ADCC, cytokine production and anti-tumor activity in vivo. To our knowledge, this is the first report to demonstrate that MDSC-derived NO can antagonize NK cell FcR function and blunt the response to antibody therapy.
Results
MDSC Inhibit NK Cell Cytotoxicity and Antibody Dependent Cellular Cytotoxicity
Given the ability of MDSC to interfere with anti-tumor immune responses it was hypothesized that MDSC could inhibit the antibody dependent cellular cytotoxicity (ADCC) function of NK cells. Autologous MDSC inhibited MHC I dependent NK cell cytotoxicity (K562 cytotoxicity assay), which was dose dependent while PBMC had no effect (Figure S1A and S1B). The effect of MDSC on NK cell ADCC was tested next. MDSC isolated from the peripheral blood of patients with melanoma (Figure 1A and S1C), head and neck squamous cell carcinoma (HNSCC, Figure 1B), and breast cancer (Figure 1C) all inhibited NK cell ADCC function. MDSC inhibition of NK cell ADCC reached statistical significance in the study of four melanoma patients while PBMC had no effect (Figure 1A and S1C). Again, the ability of MDSC to inhibit NK cell ADCC was dose dependent (Figure S1D). A phenotype of CD33+, CD11b+, and HLA-DRlow was used to identify MDSC from melanoma patient blood draws. Before isolation the frequency of MDSC was 33.15% amongst total PBMC and after isolation the frequency was 76% representing a 2.3 fold enrichment (Figure S2A and S2B). Isolated MDSC were able to suppress T cell proliferation (Figure S2C).
Figure 1: MDSC inhibit FcR mediated NK cell effector functions and signal transduction.
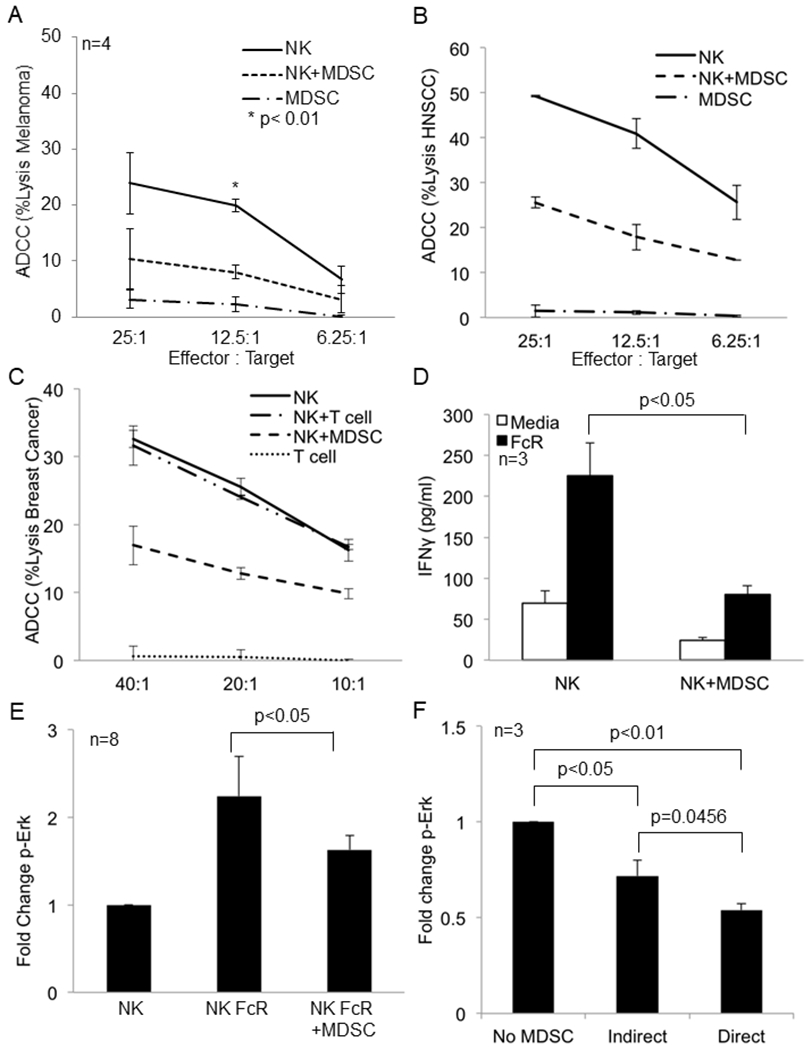
(a) NK cells from the peripheral blood of a melanoma patient were cultured alone or with autologous MDSC overnight and then used in a 51Cr release ADCC assay against cetuximab-coated HT-29 cells. Values represent the mean ± SD from four independent experiments, p<0.01. Significance was determined using the paired t-test. (b) Results from one ADCC assay as conducted in (a) using NK cells and autologous MDSC from a HNSCC patient. (c) Results from one ADCC assay as conducted in (a) using NK cells and autologous MDSC from a breast cancer patient. (d) Autologous NK cells and MDSC from the peripheral blood of melanoma patients were co-cultured at a 1:1 ratio in 96 well plates coated with human IgG (100 μg/ml) or media (control). Supernatants were collected after 48 hrs and cytokine levels measured by ELISA. Quantification of data from three independent experiments, values shown are the mean ± SE, p<0.05. Significance was determined using a paired t-test. (e) Quantification of changes in p-Erk levels in FcR activated CD56+ NK cells measured by flow cytometry in the presence or absence of MDSC. Values are mean ± SE from eight independent experiments, p<0.05. Significance was determined using a paired t-test. (f) NK cells were cultured alone, in direct contact with MDSC (Direct), or physically separated from MDSC by a permeable 0.4 μm Corning Transwell® membrane (Indirect) at a 1:1 ratio overnight. NK cells were then stimulated through the FcR using the anti-CD16 3G8 antibody and F(ab’)2 and levels of p-Erk determined as described above. Values are the mean ± SE from 3 independent experiments. Significance was determined using a paired t-test and Holms method. Representative flow cytometry dot plot for p-Erk is provided in Supplementary Figure 4.
MDSC Inhibit Cytokine Production by FcR-Activated NK Cells
NK cells are an important source of cytokines, such as IFN-γ. Our group has shown in vitro and in phase I clinical trials that co-stimulation of NK cells via the FcγRIIIa and cytokines is a potent stimulus for the production of IFN-γ and chemokines such as RANTES and MIP-1α (22). Therefore, the effect of MDSC on NK cell cytokine production was examined. Co-culture of autologous MDSC and NK cells from melanoma patients significantly inhibited the production of IFN-γ, whereas PBMC did not (Figure 1D, p<0.05 and Figure S3A). This held for FcR-stimulated NK cells cultured with IL-12 (Figure S3B). MDSC inhibition of IFN-γ production was dose dependent, and a time course experiment showed this effect was observable at 24 hours with maximal inhibition at 48 hours (Figure S3C and S3D). Co-culture of NK cells with autologous MDSC also significantly decreased the production of MIP-1α (Figure S3E, p<0.01).
MDSC inhibit FcR Mediated Signal Transduction
Erk activation is critical to NK cell FcR mediated effector functions and natural cytotoxicity (K562 killing). Given the impairment of these NK cell functions in the presence of MDSC it was hypothesized that impaired Erk activation could lead to reduced NK cell FcR-mediated functions following co-culture with MDSC (23). NK cells were stimulated via the FcR using the 3G8 anti-CD16 antibody and a cross-linking F(ab’)2 fragment. Measurement of p-Erk levels in CD56+ NK cells showed that co-culture of melanoma patient NK cells and MDSC resulted in a 40% decrease in p-Erk levels (Figure 1E, p<0.05 and representative dot plot Figure S4). When NK cells were physically separated from MDSC levels of p-Erk in response to FcR stimulation were inhibited by an average of 28.3% (Figure 1F, p<0.05). When these cells were in direct contact, there was a small increase in the level of inhibition in comparison to the contact independent condition (Figure 1F). This result suggests that MDSC inhibition of NK cell FcR-mediated signal transduction relies on diffusible substances with the potential for an additional contact dependent mechanism to play a role.
Inhibition of Nitric Oxide Production Enhances NK cell FcR Mediated Function.
MDSC can promote immune suppression through several contact independent mechanisms including expression of amino acid catabolizing enzymes, immune suppressive cytokines, and production of nitric oxide (NO). To investigate the role of these factors in suppressing FcR-mediated NK cell function, mice bearing 4T1 tumors were treated with neutralizing anti-IL-10 (24) or anti-TGF-β (25) antibodies, or inhibitors targeting 2,3-indolamine dioxygenase (IDO) (26), arginase (27), or inducible nitric oxide synthase (iNOS). NK cells were isolated from the spleen and used in ADCC assays against trastuzumab-coated CT26 cells expressing human HER2. Only inhibition of iNOS and arginase rescued NK cell ADCC activity (Figure 2A–C). Arginase and iNOS both use arginine as a substrate and MDSC express high levels of both enzymes. This suggests that the arginase/iNOS arginine catabolism pathway in MDSC plays an important role in regulating NK cell function, and that manipulation of either pathway could impact NK cell function. The arginase inhibitor produced a reduction in splenic MDSC frequency suggesting that the enhanced NK function in this group could reflect reduced MDSC accumulation (Figure S5A–C). Alternatively, as both arginase and iNOS utilize arginine as a common substrate, and arginine availability has been linked to NK cell function, inhibition of either enzyme could improve NK cell function. If this was the case one could speculate that simultaneous inhibition of both enzymes would dramatically rescue NK cell function. However, when this was tested the inhibition of both enzymes was no more effective at rescuing NK cell function than inhibition of either enzyme alone (Figure S5D). Together, these results suggest that the NO-arginase arginine catabolism pathway plays an important role in the regulation of NK cell FcR-mediated functions and that iNOS is an important mediator of MDSC inhibition of NK cell function. However, since it can be hard to draw firm conclusions from negative data obtained using neutralizing antibodies and chemical inhibitors, we concede that factors in addition to N.O. could still play an important role in the inhibitory effects of MDSC.
Figure 2: iNOS inhibition restores FcR mediated NK cell functions and signal transduction in the presence of MDSC.
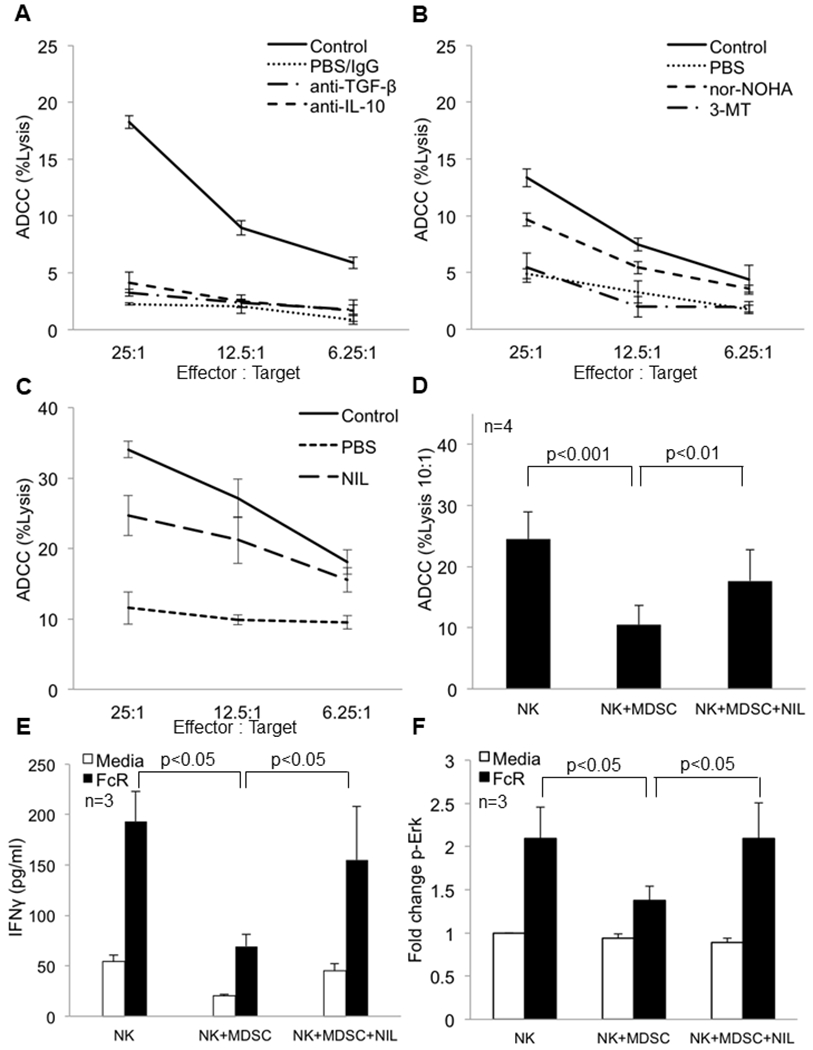
Female Balb/c mice were inoculated with 1x105 4T1 tumor cells or were left un-injected (no tumor-control). Following the establishment of tumors, mice were treated daily with (a) intraperitoneal injections of IgG (250 μg), anti-TGF-β antibody (200 μg), or anti-IL-10 antibody (250 μg) for four consecutive days prior to NK cell isolation for the ADCC assay. The mice were sacrificed 24 hrs after the last treatment. Each group consists of pooled samples from spleens of 4-5 mice. Values represent mean ± SD from one experiment. (b) Mice were treated with PBS (vehicle), arginase inhibitor nor-NOHA (20 mg/kg) i.p or the IDO inhibitor 3-methyltryptophan (MT, 400 mg/kg) via oral gavage prior to NK cell isolation for the ADCC assay. (c) Mice were given intraperitoneal injections of PBS (vehicle) or iNOS inhibitor, L-NIL (20 mg/kg) for one week. NK cells isolated from the spleen were employed in a standard ADCC assay using trastuzumab-coated CT26-HER2 positive tumor cells as targets. (d) NK cells and MDSC from the peripheral blood of melanoma patients were co-cultured overnight at a 1:1 ratio with or without the nitric oxide inhibitor L-NIL (2.5 mM). ADCC function of NK cells displayed as the mean percent lysis of cetuximab-coated HT-29 tumor cells at the 10:1 effector to target ratio. Means ± SE from four independent experiments are shown, p<0.05. Significance was determined using a linear mixed model. Treatment of NK cells with L-NIL alone did not enhance ADCC activity (not shown). (e) Autologous NK cells and MDSC isolated from peripheral blood of melanoma patients were co-cultured in 96 well plates coated with human IgG or media with or without the iNOS inhibitor L-NIL (2.5 mM). Supernatants were harvested after 48 hrs and analyzed for levels of IFNγ by ELISA. Values represent mean ± SE from three independent experiments, p<0.05. Significance was determined using a linear mixed model. (f) p-Erk expression in NK cells co-cultured overnight with MDSC in the presence or absence of L-NIL (2.5 mM). p-Erk levels are expressed as the average fold change ± SE from three independent experiments, p<0.05. Significance was determined using a linear mixed model.
Production of Nitric Oxide by MDSC Is Involved in the Suppression of NK Cell Function
To investigate the role of NO in MDSC inhibition of FcR-mediated NK cell functions, NOS2 expression by MDSC was measured. MDSC expressed high levels of NOS2 compared to NK and T cells (Figure S6A). NO production by MDSC was detected by intracellular flow cytometry using a NO-sensitive dye 4-amino-5-methylamino-2’, 7’-difluorofluorescein diacetate (DAF-FM) (Figure S6B), and the iNOS inhibitor L-NIL significantly inhibited NO production as measured by the Greiss reagent (Figure S6C, p<0.05).
It was hypothesized that L-NIL could rescue FcR-mediated NK cell functions in the presence of MDSC. Melanoma patient NK cells and MDSC were co-cultured in the presence or absence of L-NIL, and L-NIL was found to rescue NK cell ADCC (Figure 2D, p<0.01). These effects were also observed when examining the natural cytotoxicity function of NK cells (Figure S6D). Similarly, L-NIL rescued IFN-γ production and Erk activation in the presence of MDSC (Figures 2E and 2F, p<0.05). When NK cells and MDSC were separated by a permeable membrane L-NIL still rescued the generation of p-Erk supporting a predominant contact independent mechanism (Figure S6E).
Nitric Oxide Is a Negative Regulator of NK Cell Function
To examine the effect of NO on NK cell function, NK cells from normal donors were incubated with a NO donor, S-Nitroso-N-Acetyl–Penicillamine (SNAP, 0.1 mM) or DMSO overnight in the presence or absence of IL-12. SNAP significantly inhibited the ability of NK cells to lyse cetuximab-coated HT-29 colon cancer cells (Figure 3A and 3B, p<0.001). Similarly, SNAP significantly reduced the production of IFN-γ (regardless of the presence of IL-12) (Figure 3C, p<0.05 and Figure S7A). SNAP also inhibited Erk activation in FcR-stimulated NK cells (Figure 3D, p<0.05). Importantly, SNAP was not toxic to NK cells as the viability of SNAP-treated NK cells was > 90% as measured by flow cytometry and trypan blue exclusion (Figure 3E, 3F and Figure S7B). In addition, SNAP did not affect the total number of NK cells remaining after treatment (Figure S7C). Finally, SNAP produced levels of NO that were equivalent to that produced by human MDSC (data not shown). Together, these results suggest that NO can be a negative regulator of NK cell FcR function.
Figure 3: The NO donor SNAP inhibits NK cell function and signal transduction.
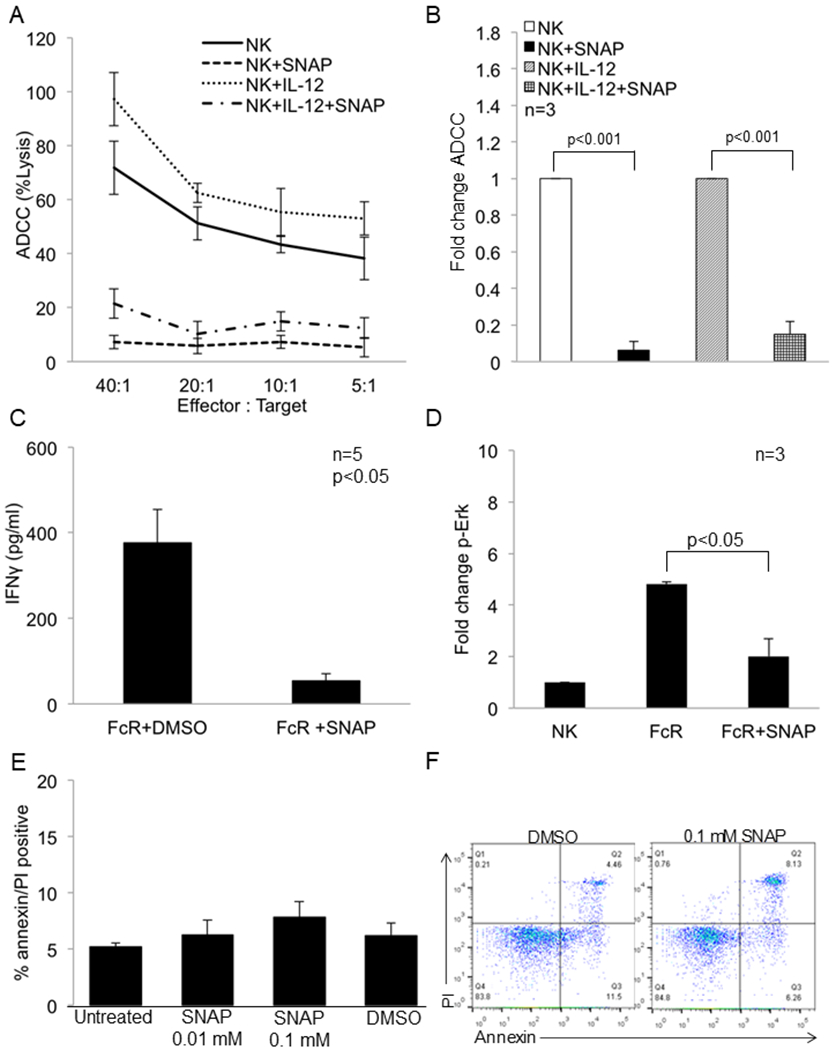
The nitric oxide donor SNAP inhibits NK cell FcR-mediated function and signal transduction. (a) Purified NK cells from normal donors were treated with DMSO or SNAP (0.1 mM) in the presence or absence of IL-12 (10 ng/ml) and then used in a 51Cr release ADCC assay against cetuximab-coated HT-29 cells. Representative results shown are from one of three independent experiments. (b) Fold change in ADCC activity of NK cells (20:1 effector target ratio) treated in a similar fashion as in (a) against cetuximab-coated HT-29 cells. The mean ± SE from three independent experiments are shown, p<0.001. Significance was determined using a t-test. (c) Levels of IFN-γ production after 48 hrs measured by ELISA from healthy donor NK cells treated with DMSO or SNAP (0.1 mM) and activated with immobilized IgG. Values represent mean ± SE from five independent experiments, p<0.05. Significance was determined using a paired t-test. (d) Fold change in the expression of p-Erk in NK cells treated with DMSO or SNAP (0.1 mM) and then stimulated through the FcR via 3G8 antibody. Values represent mean ± SE from three independent experiments, p<0.05. Significance was determined using a paired t-test. (e) NK cells isolated from normal donors were treated with the indicated doses of SNAP or DMSO for 24 hrs. NK cells were then stained with annexin V/PI to determine the percentage of apoptotic cells. Values displayed are from three independent experiments. (f) NK cells isolated from normal donors were treated with the indicated doses of SNAP or DMSO for 24 hrs. NK cells were then stained with annexin V/PI to determine the percentage of apoptotic cells. A representative flow cytometric dot plot is provided for the quantification data in 3E.
Exposure to Nitric Oxide Leads to Nitration of Proteins in NK Cells
Given the ability of MDSC to inhibit Erk activation in NK cells and the ability of L-NIL to rescue this function it was hypothesized that impaired Erk activation could be caused by nitration of signaling molecules downstream of the FcR. To investigate this, NK cells were treated with SNAP or DMSO, probed with anti-nitrotyrosine (NT) and anti-CD16 (FcR) antibodies and analyzed by flow cytometry. SNAP treatment resulted in significant nitration of tyrosine residues on CD16+ NK cells compared to DMSO (Figure 4A, p<0.05). Co-culture of MDSC with NK cells also caused a significant increase in the tyrosine nitration of CD16+ NK cells (Figure 4B, p<0.01). Representative flow cytometry dot plots showing the change in nitrotyrosine positive NK cells in the above two experiments is provided in Figure S8. Treatment of human NK cells with SNAP or co-culture with MDSC had no effect on CD16 expression (data not shown). Next, nitrotyrosine residues were immuno-precipitated from DMSO or SNAP-treated NK cells and probed for Erk. Immunoblot analysis showed that Erk was nitrated in SNAP treated NK cells (Figure 4C). This suggests that MDSC derived NO results in the nitration of tyrosine residues on signaling proteins like Erk as one mechanism by which MDSC inhibit FcR-mediated NK cell functions. With respect to the production of NO by murine MDSC, our group has previously used immunohistochemistry to show that large amounts of NO are produced by MDSC present in the spleens of tumor-bearing mice (28). Also, it has been shown in the literature that murine MDSC produce significant quantities of NO (12,29).
Figure 4: Treatment of NK cells with the nitric oxide donor SNAP or co-culture with MDSC results in nitration of tyrosine residues in NK cells.
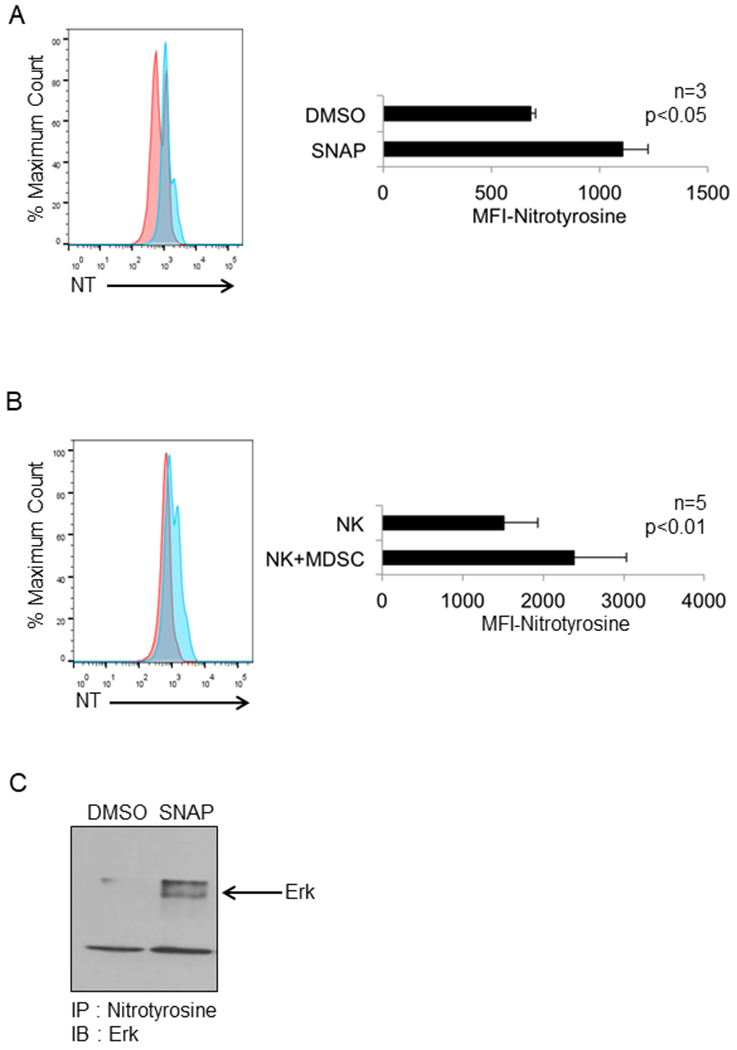
Purified NK cells from normal donors were treated with SNAP (0.01 mM) or DMSO (control). Following permeablization, cells were stained with anti-CD16 and anti-nitrotyrosine antibodies and analyzed by flow cytometry. (a) Representative flow cytometry profile and quantification of nitrotyrosine levels in CD16+ NK cells shown as mean fluorescence intensity (MFI) (right panel) from three independent experiments, p<0.05, significance was determined using a paired t-test. (b) Autologous NK cells and MDSC isolated from peripheral blood of melanoma patients were co-cultured overnight at a ratio of 1:1 and then stained with anti-CD16 and anti-nitrotyrosine antibodies. Representative flow cytometry profile (left) and quantification of nitrotyrosine in CD16+ NK cells as mean fluorescence intensity (MFI) (right) from five independent experiments, p<0.05, significance was determined using a paired t-test. (c) Immunoblot showing the nitration of Erk protein. Purified NK cells from normal donors were treated with SNAP or DMSO (control), immunoprecipitated with anti-nitrotyrosine beads and probed with anti-Erk antibody.
Depletion of MDSC Improves NK Cell ADCC Function in an Animal Model
Chemotherapeutic agents 5-fluorouracil (5-FU) and gemcitabine deplete MDSC in tumor bearing mice (30). Therefore we investigated whether the elimination of MDSC from tumor bearing mice could augment NK cell function. NK cells were isolated from the spleen of mice with 4T1 mammary carcinomas 24 hours after treatment with 5-FU (50 mg/kg), gemcitabine (80 mg/kg) or PBS and used in an ADCC assay against trastuzumab coated CT26-HER2 cells. NK cells from non-tumor bearing mice demonstrated cytotoxic activity in the range of 40% at the 25:1 E:T ratio, whereas NK cells from the PBS group exhibited cytotoxic activity of just 10%. Gemcitabine and 5-FU eliminated MDSC from the spleens of tumor bearing animals and this was associated with NK cell ADCC function in the range of 30% lysis (Figure 5A and 5B and Figure S9A and S9B). In contrast to other reports, treatment with an anti-Gr-1 antibody only modestly reduced MDSC frequency and this correlated with a slight increase in NK cell ADCC function (Figure S9C and S9D). Together, these results demonstrate that MDSC in the spleen can impair NK cell function and their elimination can augment NK cell ADCC.
Figure 5: Depletion of MDSC or inhibition of nitric oxide production in mice augments NK cell-mediated ADCC activity.
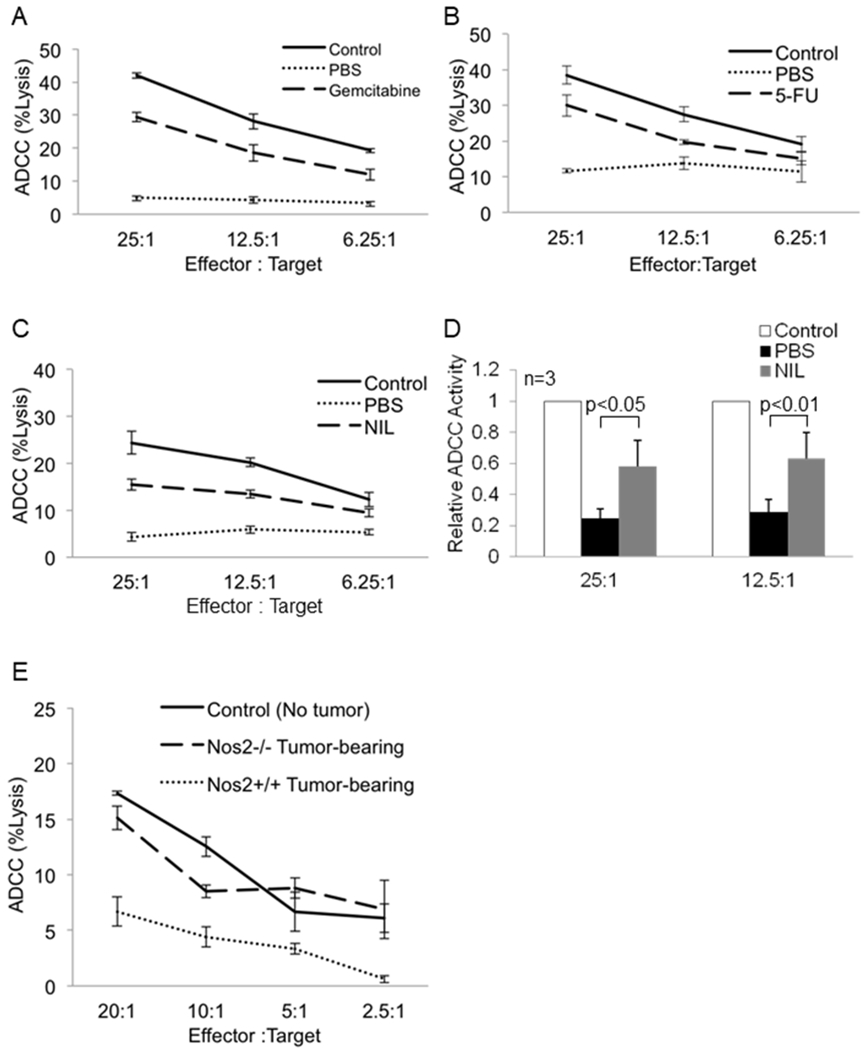
Female Balb/c mice were inoculated with 1x105 4T1 tumor cells or were left un-injected (no tumor-control). The mice were sacrificed 24 hrs after treatment. NK cells isolated from the spleen were used in a standard ADCC assay using trastuzumab-coated CT26-HER2 positive tumor cells as targets. (a) Mice were treated with PBS (vehicle) or gemcitabine (80 mg/kg). Graph displays mean percent lysis by pooled NK cells from 4-5 mice per treatment group. One of two representative experiments is shown. (b) Mice were treated with PBS (vehicle) or 5-fluorouracil (50 mg/kg). Graph displays mean percent lysis by pooled NK cells from 4-5 mice per treatment group. One of two representative experiments is shown. (c) NK cells isolated from mice treated daily for one week with PBS (vehicle) or iNOS inhibitor, L-NIL (20 mg/kg) were used in a standard ADCC assay using trastuzumab-coated CT26-HER2 cells. Each group consists of pooled samples from spleens of 5 mice. (d) Fold change in NK cell ADCC activity from mice treated as in (c). Values displayed are the means ± SE from three independent experiments, p< 0.05 for the 25:1 and 12.5:1 effector target ratios. Data was log-transformed for testing group difference using a linear mixed effect model with random donor effect, and the p-value was calculated using Bonferroni method. (e) Female C57BL/6 (wild type) and Nos2−/− mice were inoculated with B16F10 tumor cells or left un-injected (control). NK cells were isolated from the spleen and used in a standard ADCC assay using trastuzumab coated CT26-HER2 tumor cells. Each group consists of pooled samples from spleens of 3-4 mice. Values represent mean ± SD from one experiment.
Inhibition of NO In Vivo Enhances NK Cell-Mediated ADCC Activity
Next, it was investigated if inhibition of iNOS could augment NK cell function in vivo. Mice bearing 4T1 tumors were treated daily with PBS or the iNOS inhibitor L-NIL (20 mg/kg) for one week (31). NK cells isolated from the spleen were used in an ADCC assay against trastuzumab-coated CT26-HER2 cells. Treatment with L-NIL significantly improved the ADCC function of NK cells (Figure 5C–5D, p<0.05). As seen in figure S9E, there was minimal difference in the frequency of MDSC between the PBS and NIL-treated groups.
Nitric oxide generated by cancer cells has been postulated to dampen anti-tumor immune responses (32,33). To delineate the role of tumor versus MDSC-derived NO, the above experiment was repeated using Nos2−/− deficient mice. NK cells from B16F10 tumor-bearing Nos2−/− mice demonstrated normal ADCC activity compared to NK cells from wild type B16F10 tumor-bearing mice (Figure 5E). These results imply that host-derived and not tumor-derived NO is involved in the regulation of MDSC cell function in this tumor model.
Depletion of MDSC Augments Trastuzumab Therapy In Vivo
Monoclonal antibodies (mAb) like trastuzumab are widely used in the treatment of cancer and there is evidence that NK cells participate in their efficacy (34,35). Therefore, it was hypothesized that MDSC could antagonize mAb therapy. To this end, we employed a well-characterized EMT6 mouse model of mammary carcinoma that overexpresses human HER2 (EMT6-HER2) and responds to Ab therapy in an NK cell-dependent manner (36). Importantly, EMT6-HER2 cells show similar growth kinetics as EMT6 parental cells and drive the expansion of MDSC (unpublished data). Once tumors were palpable (~50 mm3) mice were treated with PBS/IgG, 5-FU (MDSC-depleting agent), trastuzumab or 5-FU followed by trastuzumab. Tumors of mice receiving 5-FU alone or 5-FU in combination with trastuzumab exhibited a significant decrease in MDSC (Figure S10A and S10B). There was only a modest reduction in tumor volume in mice treated with trastuzumab compared to the PBS/IgG-treated control group as might be expected in a model in which the MDSC population is so prominent. Mice treated with 5-FU showed a reduction in tumor volume compared to the PBS/IgG-treated group. Importantly, 5-FU markedly enhanced the effectiveness of trastuzumab compared to either agent alone (Figure 6A, p<0.01 and p<0.001, respectively). It is possible that 5-FU had a direct antitumor effect in this tumor model, but IHC staining for Ki67 and caspase3 showed no difference in proliferation or apoptosis between these treatment groups (Figure S11). These results are consistent with previous studies showing 5-FU did not induce apoptosis of tumor cells or affect the prevalence of immune cells (NK and T cells) within tumors (37). We propose that one major effect of 5-FU on tumor growth is due to its actions on the MDSC compartment.
Figure 6: MDSC antagonize mAb therapy in vivo and MDSC depletion or inhibition of nitric oxide enhances the efficacy of mAb therapy.
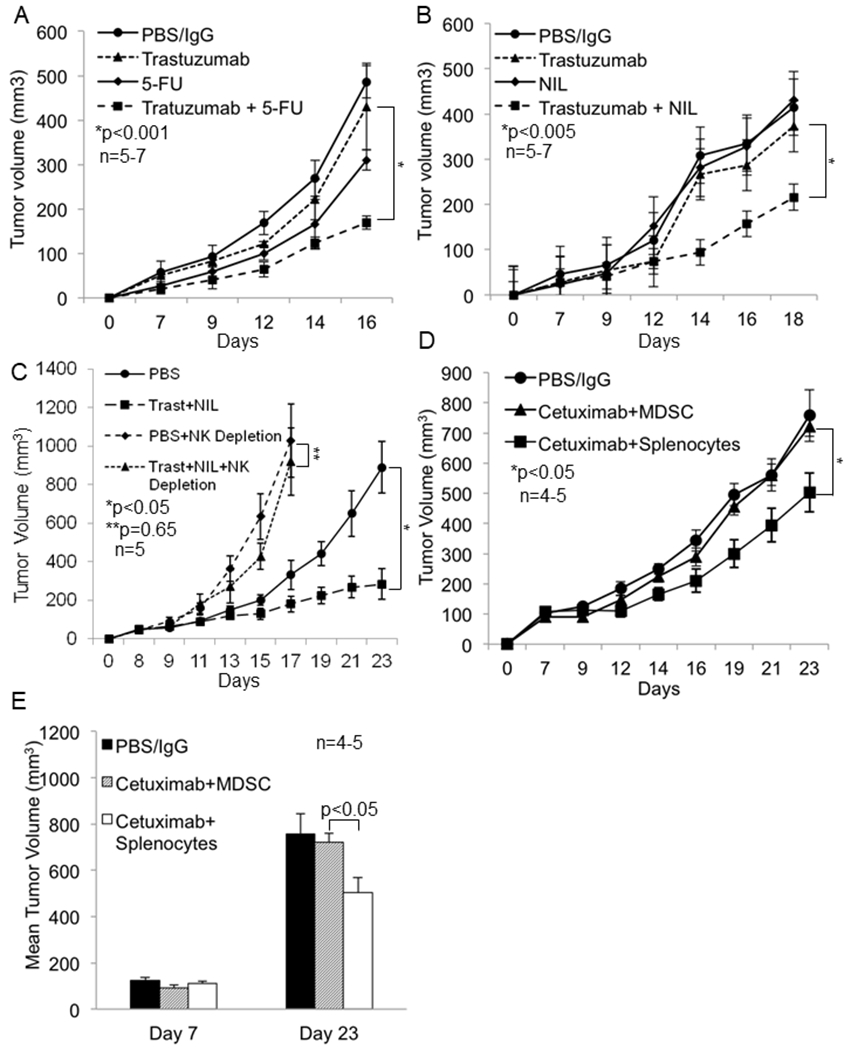
Female Balb/c mice were inoculated with 1x106 EMT6-HER2 tumor cells in the mammary fat pad. Following the establishment of tumors (day 7), mice were treated once a week i.p. with PBS or 5-FU and thrice weekly with IgG or trastuzumab and tumor growth was measured three times a week using digital calipers. (a) Tumor growth in mice treated with PBS and IgG (10 mg/kg), 5-FU (50 mg/kg), trastuzumab (10 mg/kg) or the combination of trastuzumab plus 5-FU. Differences in tumor volumes were tested using a linear mixed model and Student’s t-test. Values represent mean ± SE, p<0.001. (b) Tumor growth in mice treated with PBS and IgG (10 mg/kg), L-NIL (20 mg/kg), trastuzumab (10 mg/kg) or combination of trastuzumab plus L-NIL. Each group consisted of 5-7 mice. Values represent mean ± SE, p<0.005, significance was determined using a linear mixed model and Student’s t-test. (c) Depletion of NK cells with anti-asialo-GM1 abrogates the anti-tumor effect of trastuzumab plus NIL. Balb/c mice were inoculated with 1x106 EMT6-HER2 tumor cells in the mammary fat pad. Three days prior to treatment, mice were administered PBS or 50 µg/mouse of anti asialo GM1 polyclonal antibody to deplete NK cells. On day 8, mice were randomized and treated thrice weekly with PBS and IgG or trastuzumab (10 mg/kg) plus L-NIL (20 mg/kg). NK depleted mice were administered the depleting antibody every 4 days until the end of the study. n=5 for each treatment group. (d) Athymic nude mice were inoculated with 1x106 Panco2-EGFR tumor cells on day zero. After establishment of tumors mice were injected with PBS, splenocytes, or MDSC on day 7 prior to the initiation of treatment with IgG or cetuximab (0.5 mg/kg) on day 9. Mice were re-injected with PBS, splenocytes, or MDSC on day 14 and IgG or cetuximab treatment was continued. (d) Tumor growth curves of mice treated as described above. (e) Mean tumor volume in mice inoculated with Panco2-EGFR cells before (day 7) and after treatment (day 23). Each group consisted of 4-5 mice. Differences in tumor volumes were tested using a linear mixed model and Student’s t-test. Values represent mean ± SE, p<0.001.
Inhibition of MDSC NO Production Enhances Trastuzumab Therapy In Vivo
Next the effect of NO inhibition on the response to trastuzumab was examined. EMT6-HER2 tumor bearing mice were treated with PBS/IgG, L-NIL, trastuzumab or L-NIL followed by trastuzumab. No difference in tumor volume was observed between the PBS/IgG and L-NIL treated groups, and trastuzumab modestly reduced tumor growth (Figure 6B). However, there was a significant decrease in tumor volume when mice received NIL in combination with trastuzumab (p<0.005, Figure 6B). While there was a slight reduction of splenic MDSC in treatment groups compared to the control, the frequency of tumor infiltrating MDSC was similar indicating the observed differences in splenic MDSC frequency were likely driven by group differences in tumor volume (Figure S12). Importantly, the enhanced anti-tumor effect of the combination of trastuzumab and L-NIL was found to be NK cell dependent as depletion of NK cells with anti-asialo GM1 rescued tumor growth even in the setting of combination therapy (Figure 6C and S13A).
Transfer of MDSC Inhibits mAb Therapy
To confirm the ability of MDSC to antagonize mAb therapy of cancer, a MDSC adoptive transfer experiment was performed. Athymic nude mice were inoculated with a pancreatic cancer cell line that overexpresses human HER1 (Panco2-EGFR). The parental and HER1 expressing cell lines grow at identical rates in this mouse strain and there is minimal expansion of MDSC (unpublished data). Following the establishment of tumors, mice were injected with PBS (mock injection), splenocytes from non-tumor bearing mice, or MDSC from syngeneic mice bearing 4T1 mammary carcinoma tumors on day seven prior to the initiation of IgG or cetuximab therapy on day 9. A second round of cell injections was conducted on day 14 (schema shown in Figure S13B). Mice receiving splenocytes and cetuximab showed a significant reduction in tumor volume compared to the control group. In contrast, MDSC ablated the effect of cetuximab therapy (Figure 6D and 6E, p<0.05).
In order to elucidate the role of NO production in the inhibitory effects of transferred MDSC a similar study was conducted using MDSC from nos2−/− deficient mice. As before, injection of wild type MDSC abolished the anti-tumor effect of cetuximab therapy. However, nos2-deficeint MDSC were less antagonistic of cetuximab (Figure S13C). Taken together, these results demonstrate that MDSC can dampen the anti-tumor response of mAb therapies and that nitric oxide production appears to play an important role in mediating this effect.
Discussion
The current study provides evidence for a model in which MDSC inhibit FcR-mediated NK cell actions against antibody-coated tumor cells through the production of NO. The results suggest that nitration of NK cell proteins on tyrosine residues resulted in impaired signal transduction and activation of NK cells following FcR stimulation. Elimination of MDSC from tumor bearing hosts or inhibition of NO production restored NK cell effector functions and improved the anti-tumor effect of mAb therapy in vivo. Finally, transfer of wild-type MDSC significantly antagonized mAb therapy. The results reported provide a mechanism by which MDSC-derived NO leads to impaired FcR mediated NK cell function and reduced efficacy of mAb therapy for cancer.
NK cells play an important role in the response to mAb therapy through their ability to recognize and kill antibody coated tumor cells via engagement of the low affinity activating FcγRIIIa (FcR or CD16) and inhibiting metastatic spread of cancer. As a result NK cells have received attention for their potential therapeutic and prognostic value (38). In both solid and hematological malignancies, alterations in the frequency, maturity, and expression of activating and inhibitory receptors by NK cells have been reported which correspond with impaired NK cell effector functions (39). While the mechanisms of this dysfunction are incompletely understood, reports have begun to explore these observations by analyzing the interaction between NK cells and cells with immune suppressive function including MDSC (10,11,18,19,21,40–42).
The present study provides evidence that MDSC impair NK cell FcR-mediated functions via the production of NO and the nitration of protein tyrosine residues. Our group has previously shown that MDSC-derived NO results in nitration of key tyrosine residues on the STAT1 transcription factor resulting in impaired NK and T cell response to interferon stimulation (28). Other groups have shown that MDSC-derived RNS can cause nitration of the T cell receptor, thus impairing its interaction with the peptide major histocompatibility complex (43). In addition, RNS have been shown to cause post-translational modification of chemokines resulting in impaired T cell trafficking within tumors (44). Finally, tumor-derived NO has also been shown to promote immune dysfunction. Co-culture of PBMCs with melanoma cell lines expressing high levels of NOS1 resulted in tyrosine nitration of T cells and monocytes. The present findings that MDSC-derived NO resulted in the nitration of Erk and impaired FcR-mediated NK cell function are the first to explore the effects of MDSC on the NK cell mediated response to anti-tumor mAb therapy. These results further support the ability of NO to mediate immune suppression and negatively impact mAb-based treatment of cancer.
NK cells also appear to have a role in preventing metastatic spread of cancer and in the control hematologic malignancies. Notably, there are reports showing that immune-suppressive MDSC are increased in patients with leukemia (45). Our results showing that MDSC can inhibit NK cell natural cytotoxicity suggest that MDSC could promote metastasis through impaired NK cell function. Indeed, investigators have shown that MDSC promote metastasis in the setting of breast cancer (46), but to date no report has implicated MDSC inhibition of NK cell function in this process.
Erk is rapidly phosphorylated in primary human NK cells following FcR stimulation and this is critical to NK cell cytokine production and antibody dependent cellular cytotoxicity (23,47). Erk phosphorylation in FcR stimulated NK cells was significantly inhibited by MDSC, but this could be rescued in the presence of an iNOS inhibitor. Treatment of NK cells with the NO donor SNAP resulted in the nitration of Erk on tyrosine residues suggesting that this is one mechanism by which MDSC inhibit Erk activation. Recently, it was identified that MDSC can inhibit p-Erk generation in NK cells through the inhibitory TIGIT receptor on NK cells (17). Analysis of NK cells and MDSC from patients used in this work confirmed expression of these molecules. However, the ability of the iNOS inhibitor L-NIL to rescue p-Erk generation in NK cells cultured in direct contact with MDSC suggests that TIGIT may play an alternative role in this system.
Few studies have investigated the relationship between NO and NK cell function. Earlier studies revealed that autocrine production of NO by NK cells could have a positive effect on NK cell function, and that human NK cells appear to express endothelial nitric oxide synthase but not inducible nitric oxide synthase (48,49). The amount of NO produced by activated NK cells is reported to be in the nanomolar range, which is substantially lower than the 2-20 μM range of NO produced by MDSC. This suggests that exposure to higher levels of NO produced by MDSC is detrimental to NK cell activation and function.
In addition to producing NO, MDSC mediate immune suppression through expression of arginase and 2,3-indoleamine dioxygenase (IDO) and inhibitory cytokines (TGF-β and IL-10) (12). Treatment of tumor bearing mice with neutralizing antibodies against TGF-β and IL-10 or the IDO inhibitor 1-methyl-D-tryptophan did not improve NK cell ADCC. Nevertheless, based on studies in liver and lung cancer models, it is likely that these immune suppressive molecules can contribute to impaired NK cell function in vivo in other murine models and in humans (19). Notably, the arginase inhibitor nor-NOHA modestly improved NK cell ADCC function. This finding is consistent with reports showing that depletion of L-arginine results in impaired NK cell function (50). Arginase and iNOS utilize L-arginine as a substrate and MDSC are known to express high levels of arginase and iNOS (12). As a result, inhibition of either could increase the availability of arginine and decrease NO generation resulting in improved NK cell function.
The presence of immune suppressive cells like MDSC in the tumor microenvironment can be a major impediment to immune-based therapies (51). There are over a dozen mAb approved by the FDA for the treatment of cancer. These agents appear to function in part through the activation of NK cells (4). The finding that MDSC can inhibit FcR-mediated activation and function of NK cells suggests that eliminating MDSC or inhibiting their immune suppressive function could also enhance mAb therapy. Recently sunitinib was found to reduce the frequency of monocytic MDSC and their expression of arginase (52). In addition, imatinib and dasatinib were found to reduce the frequency of suppressive myeloid cells and increase the frequency of NK cells (53). Our group recently demonstrated that ibrutinib, a Bruton’s tyrosine kinase inhibitor, reduced MDSC in tumor bearing mice and MDSC NO production (54). These results suggest that combining antibody therapy with kinase inhibitors active against MDSC could be an effective strategy. In general, strategies to deactivate or deplete MDSC and other immune suppressive cells could enhance the efficacy of immune-based therapies.
In summary, this report provides experimental evidence for an MDSC-mediated mechanism of action for impairment of FcR-medatied NK cell function. MDSC produce NO and are associated with inhibition of NK cell ADCC, cytokine production, FcR signal transduction and response to mAb therapy. These results suggest that mAb therapy in combination with agents targeting MDSC could be a successful therapeutic strategy and provide a rationale for the development of clinical trials to test such combinations.
Material and Methods
Cell lines.
EMT6, 4T1, K562, HT-29, and Raw 264.7 cells were purchased from ATCC and maintained under the recommended cell culture conditions. Panco2-EGFR+ cells were generated as previously described (55). CT26-HER and EMT6-HER2 cells were gifted from the laboratory of Dr. P.T.P. Kaumaya (Ohio State University, Columbus, Ohio, USA). Cells were cultured in RPMI, IMDM, or DMEM with 10% FBS and 1% antibiotic-antimycotic.
NK and MDSC Isolation
Peripheral blood from cancer patients was obtained at the Ohio State University Comprehensive Cancer Center under Institutional Review Board approved protocols (OSU IRB Nos. 1999C0348, 2004C0096 and 2010C0036, respectively). NK cells and MDSC were isolated by 30-minute incubation with the NK or myeloid cell enrichment cocktail (Stem Cell Technologies, Vancouver, BC) followed by Ficoll-Paque centrifugation (GE healthcare). For MDSC, cells were further selected using anti-HLA-DR magnetic beads for 15 minutes at 4oC and isolated using a MS-MACS column (54).
ADCC Assay
Autologous NK cells and MDSC were co-cultured overnight with or without 2.5 mM L-NIL. Healthy donor NK cells were treated with DMSO or 0.1 mM SNAP (Sigma Aldrich) overnight. Target HT-29 tumor cells were labeled with 51Cr and then coated with cetuximab or IgG. Target cells were added to NK cells, and following a 4 hr incubation supernatants were harvested and quantified using a gamma counter. Mean percent cell lysis was determined as previously described (2).
ELISA
96-well flat-bottom plates were coated with 100 μg/mL of polyclonal human IgG in cold PBS overnight at 4°C. NK cells, MDSC, or NK cells plus MDSC were plated at 2 x 105 cells/well as previously described (23). Where indicated cells were treated with 2.5 mM of the iNOS inhibitor L-NIL. Cell-free supernatants were harvested and analyzed for levels of IFN-γ, MIP-1α, or TNF-α (ELISA; R&D Systems, Minneapolis, MN or Ebioscience) (23).
Phospho-Erk Intracellular Staining
Autologous NK cells and MDSC were co-cultured overnight. Where indicated cells were also treated with 2.5 mM of L-NIL. The cells were incubated with the 3G8 anti-CD16 antibody (0.2 mg/mL) on ice for 30 minutes, and then cross-linked with a goat anti-mouse F(ab)2 for 15 minutes. Cells were permeabilized, stained with phospho-Erk and CD56 antibodies and analyzed by FACS (23).
Nitrotyrosine Staining
NK cells were treated with 0.1 mM S-Nitroso-N-acetylpenicillamine (SNAP) (56). Alternatively, autologous NK cells and MDSC were co-cultured overnight at a 1:1 ratio. Cells were washed with cold PBS, incubated in buffer A (Invitrogen) and permeabilized using cold methanol. Cell were stained with anti-CD16 and anti-nitrotyrosine antibodies in buffer B (Invitrogen) and analyzed by FACS.
Immunoprecipitation
NK cells were treated with SNAP as described above. 20 μg of anti-nitrotyrosine beads (Millipore 16310) were added to 50 μg of protein lysate and incubated overnight at 4°C. The beads were washed with cold PBS and re-suspended in 2X Laemelli buffer and boiled. The sample was loaded on a SDS gel for protein expression by immunoblot analysis. The membrane was probed with an anti-Erk antibody (Cell Signaling) (28).
Annexin V/ PI Staining
NK cells isolated from healthy donor leukopaks (American Red Cross) were treated with various concentrations of SNAP or DMSO. NK cells were stained with annexin V and propidium iodide (PI) (BD Pharmingen) (54).
FACS Analysis
Human MDSC were stained with a panel of antibodies recognizing CD11b, CD33, HLA-DR, CD15, and CD14 (Beckman Coulter) as well as TIGIT ligands CD155 and CD112 (Biolegend) (13). NK cells were stained using antibodies against CD16 and CD56 (BD Pharmingen). Mouse MDSC were stained with antibodies against Gr-1 and CD11b (BD Biosciences) (28). Data was acquired using a BD-LSRII and the data was analyzed using the FlowJo.
Nitrite Estimation
MDSC were cultured in 10% human AB (HAB) (Sigma Aldrich) media for 24 hours. Equal amounts of supernatant were mixed with modified Greiss reagent (Sigma Alrdrich). Absorbance at 550 nm was measured using a microplate reader. Nitrite concentrations were determined using a standard curve (54).
DAF-FM Staining
MDSC were stained with 2.5 μM DAFFM (Molecular probes, D-23841) for 30 minutes in serum free media. Cells were then washed three times with serum free media and incubated for an additional 20 minutes in RPMI complete media. Following the incubation, MDSC were washed and analyzed by FACS (57).
Mouse NK Cell Isolation
Balb/c mice received single intraperitoneal injections of gemcitabine (80 mg/kg) or 5-FU (50 mg/kg). MDSC depletion was also accomplished by intraperitoneal injection of anti-Gr-1 (250 μg) daily for 5 days. To inhibit MDSC function Balb/c mice received intraperitoneal injections of the following agents for 5 consecutive days: L-NIL (20 mg/kg), anti-TGF-β Ab (200 μg, BioXcell, clone 1D11.16.8) (25), anti-IL-10 Ab (250 μg, BioXcell, clone JES5-2A5), the arginase inhibitor nor-NOHA (20 mg/kg) (27) or the IDO inhibitor, 1-Methyl-D-tryptophan via oral gavage (400 mg/kg, Sigma Aldrich) (26). Mice were sacrificed 24 hrs after the last treatment. NK cells were isolated from the spleen using a mouse NK cell enrichment kit (Stem Cell Technologies).
Real Time PCR
Following TRIzol extraction (Invitrogen) and RNeasy purification (Qiagen), total RNA was quantitated and reverse transcribed. cDNA was used to measure gene expression by Real-Time PCR using pre-designed primer/probe sets and 2× TaqMan Universal PCR Master Mix with18s rRNA as an internal control (Applied Biosystems) (54).
Tumor Studies
For ex vivo studies of the effect of depletion or inhibition of MDSC on NK cell function, 4-5 week old female Balb/c mice were injected with 1x105 4T1 mammary carcinoma cells in the mammary fat pad. For in vivo studies of the effect of depletion or inhibition of MDSC function on the response to antibody therapy, Balb/c mice were injected with 1x106 EMT6-HER2 mammary carcinoma cells in the mammary fat pad. MDSC transfer studies utilized immune-deficient athymic nude mice (FOXN1−/−) injected subcutaneously with 1x106 Panco2-EGFR+ pancreatic carcinoma cells. For the generation of MDSC in transfer studies, Balb/c mice were injected with 1x105 4T1 mammary carcinoma cells while C57BL/6 or Nos2−/− B6.129P2-Nos2tm1/Lau/J mice were injected with 1x105 B16F10 melanoma cells. MDSC were isolated from the spleen using a myeloid derived suppressor cell isolation kit (Miltenyi). Tumor volume was measured three times weekly using digital calipers. These studies were conducted under a protocol approved by Ohio State University’s Institutional Animal Care and Use Committee (IACUC 2009A0179-R2).
NK Cell Depletion
Three days prior to the start of treatment, a cohort of tumor-bearing mice were injected i.p. with 50 µl anti-asialo GM1 polyclonal antibody (1 mg/ml) to deplete NK cells. NK depleted mice were administered the depleting antibody every 4 days until the end of the study. Depletion of NK cells was confirm via flow cytometry by staining splenocytes from control and anti-asialo GM1 treated mice with an antibody against DX5.
Statistical Analysis
Statistical differences between treatment groups were determined using an ANOVA model and Student’s t-test. For murine tumor studies, a linear mixed model was employed to model longitudinal tumor volume for mice under each treatment. Comparisons were done at each time point and averaged across all time points using t-statistics. The Holm-Bonferroni method was used for adjusting raw p-values for multiple comparisons across treatment groups.
Study Approval
Written informed consent was received from all the human participants prior to inclusion in the experimental studies under Institutional Review Board- approved protocols (OSU IRB Nos. 1999C0348, 2004C0096 and 2010C0036). All mouse studies were conducted under a protocol approved by Ohio State University’s Institutional Animal Care and Use Committee (IACUC 2009A0179-R2).
Supplementary Material
Translational Relevance.
Monoclonal antibodies (mAb) are a mainstay in the current cancer therapeutics landscape with over a dozen FDA approved molecules used to treat solid and hematological malignancies. One of the mechanisms of action of mAbs is the activation of the innate immune system, including NK cells, through engagement of Fc receptors including FcRγIIIA. It is also well appreciated that cancer evolves to evade the immune system in part by promoting the expansion of immune suppressive cells such as myeloid derived suppressor cells (MDSC) which are known to antagonize immune based therapies. Understanding the impact MDSC have on FcR mediated NK cell functions and the response to mAb therapy could lead to the development of novel combination therapies that enhance the efficacy of mAb therapy in multiple disease settings by eliminating or antagonizing the immune suppressive function of MDSC.
Acknowledgement
This work was supported by National Institutes of Health Grants P01 CA95426, K24 CA93670 (W.E.C.), T32 CA90338-27, P30 CA016058, and OSUCCC Translational Therapeutics Award TT142. This work was also supported by the Pelotonia Fellowship Program.
Footnotes
The authors have declared that no conflict of interest exists.
Conflict of interest
Authors declare no conflict of interest.
References
- 1.Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or adaptive immunity? The example of natural killer cells. Science 2011;331:44–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parihar R, Dierksheide J, Hu Y, Carson WE. IL-12 enhances the natural killer cell cytokine response to Ab-coated tumor cells. J Clin Invest 2002;110:983–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roda JM, Parihar R, Magro C, Nuovo GJ, Tridandapani S, Carson WE, 3rd. Natural killer cells produce T cell-recruiting chemokines in response to antibody-coated tumor cells. Cancer research 2006;66:517–26 [DOI] [PubMed] [Google Scholar]
- 4.Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nature reviews Cancer 2012;12:278–87 [DOI] [PubMed] [Google Scholar]
- 5.Ishigami S, Natsugoe S, Tokuda K, Nakajo A, Che X, Iwashige H, et al. Prognostic value of intratumoral natural killer cells in gastric carcinoma. Cancer 2000;88:577–83 [PubMed] [Google Scholar]
- 6.Coca S, Perez-Piqueras J, Martinez D, Colmenarejo A, Saez MA, Vallejo C, et al. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer 1997;79:2320–8 [DOI] [PubMed] [Google Scholar]
- 7.Costello RT, Sivori S, Marcenaro E, Lafage-Pochitaloff M, Mozziconacci MJ, Reviron D, et al. Defective expression and function of natural killer cell-triggering receptors in patients with acute myeloid leukemia. Blood 2002;99:3661–7 [DOI] [PubMed] [Google Scholar]
- 8.Baginska J, Viry E, Paggetti J, Medves S, Berchem G, Moussay E, et al. The critical role of the tumor microenvironment in shaping natural killer cell-mediated anti-tumor immunity. Front Immunol 2013;4:490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ashiru O, Boutet P, Fernandez-Messina L, Aguera-Gonzalez S, Skepper JN, Vales-Gomez M, et al. Natural killer cell cytotoxicity is suppressed by exposure to the human NKG2D ligand MICA*008 that is shed by tumor cells in exosomes. Cancer research 2010;70:481–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cekic C, Day YJ, Sag D, Linden J. Myeloid Expression of Adenosine A2A Receptor Suppresses T and NK Cell Responses in the Solid Tumor Microenvironment. Cancer research 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mao Y, Sarhan D, Steven A, Seliger B, Kiessling R, Lundqvist A. Inhibition of tumor-derived prostaglandin-e2 blocks the induction of myeloid-derived suppressor cells and recovers natural killer cell activity. Clinical cancer research : an official journal of the American Association for Cancer Research 2014;20:4096–106 [DOI] [PubMed] [Google Scholar]
- 12.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009;9:162–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mundy-Bosse BL, Young GS, Bauer T, Binkley E, Bloomston M, Bill MA, et al. Distinct myeloid suppressor cell subsets correlate with plasma IL-6 and IL-10 and reduced interferon-alpha signaling in CD4+ T cells from patients with GI malignancy. Cancer Immunol Immunother 2011;60:1269–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Serafini P, Borrello I, Bronte V. Myeloid suppressor cells in cancer: recruitment, phenotype, properties, and mechanisms of immune suppression. Semin Cancer Biol 2006;16:53–65 [DOI] [PubMed] [Google Scholar]
- 15.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. Journal of immunology (Baltimore, Md : 1950) 2008;181:5791–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schneider T, Sevko A, Heussel CP, Umansky L, Beckhove P, Dienemann H, et al. Serum inflammatory factors and circulating immunosuppressive cells are predictive markers for efficacy of radiofrequency ablation in non-small-cell lung cancer. Clin Exp Immunol 2015;180:467–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sarhan D, Cichocki F, Zhang B, Yingst A, Spellman SR, Cooley S, et al. Adaptive NK Cells with Low TIGIT Expression Are Inherently Resistant to Myeloid-Derived Suppressor Cells. Cancer Res 2016;76:5696–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoechst B, Voigtlaender T, Ormandy L, Gamrekelashvili J, Zhao F, Wedemeyer H, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology (Baltimore, Md) 2009;50:799–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. Journal of immunology (Baltimore, Md : 1950) 2009;182:240–9 [DOI] [PubMed] [Google Scholar]
- 20.Goh CC, Roggerson KM, Lee HC, Golden-Mason L, Rosen HR, Hahn YS. Hepatitis C Virus-Induced Myeloid-Derived Suppressor Cells Suppress NK Cell IFN-gamma Production by Altering Cellular Metabolism via Arginase-1. J Immunol 2016;196:2283–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elkabets M, Ribeiro VS, Dinarello CA, Ostrand-Rosenberg S, Di Santo JP, Apte RN, et al. IL-1beta regulates a novel myeloid-derived suppressor cell subset that impairs NK cell development and function. European journal of immunology 2010;40:3347–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roda JM, Joshi T, Butchar JP, McAlees JW, Lehman A, Tridandapani S, et al. The activation of natural killer cell effector functions by cetuximab-coated, epidermal growth factor receptor positive tumor cells is enhanced by cytokines. Clin Cancer Res 2007;13:6419–28 [DOI] [PubMed] [Google Scholar]
- 23.Kondadasula SV, Roda JM, Parihar R, Yu J, Lehman A, Caligiuri MA, et al. Colocalization of the IL-12 receptor and FcgammaRIIIa to natural killer cell lipid rafts leads to activation of ERK and enhanced production of interferon-gamma. Blood 2008;111:4173–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsukamoto H, Senju S, Matsumura K, Swain SL, Nishimura Y. IL-6-mediated environmental conditioning of defective Th1 differentiation dampens antitumour immune responses in old age. Nat Commun 2015;6:6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manlove LS, Berquam-Vrieze KE, Pauken KE, Williams RT, Jenkins MK, Farrar MA. Adaptive Immunity to Leukemia Is Inhibited by Cross-Reactive Induced Regulatory T Cells. J Immunol 2015;195:4028–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hou DY, Muller AJ, Sharma MD, DuHadaway J, Banerjee T, Johnson M, et al. Inhibition of indoleamine 2,3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res 2007;67:792–801 [DOI] [PubMed] [Google Scholar]
- 27.Narita Y, Kitamura H, Wakita D, Sumida K, Masuko K, Terada S, et al. The key role of IL-6-arginase cascade for inducing dendritic cell-dependent CD4(+) T cell dysfunction in tumor-bearing mice. J Immunol 2013;190:812–20 [DOI] [PubMed] [Google Scholar]
- 28.Mundy-Bosse BL, Lesinski GB, Jaime-Ramirez AC, Benninger K, Khan M, Kuppusamy P, et al. Myeloid-derived suppressor cell inhibition of the IFN response in tumor-bearing mice. Cancer research 2011;71:5101–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raber PL, Thevenot P, Sierra R, Wyczechowska D, Halle D, Ramirez ME, et al. Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. International journal of cancer Journal international du cancer 2014;134:2853–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res 2010;70:3052–61 [DOI] [PubMed] [Google Scholar]
- 31.Schweighofer H, Rummel C, Mayer K, Rosengarten B. Brain function in iNOS knock out or iNOS inhibited (l-NIL) mice under endotoxic shock. Intensive care medicine experimental 2014;2:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jayaraman P, Parikh F, Lopez-Rivera E, Hailemichael Y, Clark A, Ma G, et al. Tumor-expressed inducible nitric oxide synthase controls induction of functional myeloid-derived suppressor cells through modulation of vascular endothelial growth factor release. Journal of immunology (Baltimore, Md: 1950) 2012;188:5365–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Q, Tomei S, Ascierto ML, De Giorgi V, Bedognetti D, Dai C, et al. Melanoma NOS1 expression promotes dysfunctional IFN signaling. J Clin Invest 2014;124:2147–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jaime-Ramirez AC, Mundy-Bosse BL, Kondadasula S, Jones NB, Roda JM, Mani A, et al. IL-12 enhances the antitumor actions of trastuzumab via NK cell IFN-γ production. J Immunol 2011;186:3401–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roda JM, Parihar R, Lehman A, Mani A, Tridandapani S, Carson WE. Interleukin-21 enhances NK cell activation in response to antibody-coated targets. J Immunol 2006;177:120–9 [DOI] [PubMed] [Google Scholar]
- 36.Cho HM, Rosenblatt JD, Tolba K, Shin SJ, Shin DS, Calfa C, et al. Delivery of NKG2D ligand using an anti-HER2 antibody-NKG2D ligand fusion protein results in an enhanced innate and adaptive antitumor response. Cancer research 2010;70:10121–30 [DOI] [PubMed] [Google Scholar]
- 37.Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res 2010;70:3052–61 [DOI] [PubMed] [Google Scholar]
- 38.Cheng M, Chen Y, Xiao W, Sun R, Tian Z. NK cell-based immunotherapy for malignant diseases. Cellular & molecular immunology 2013;10:230–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mamessier E, Sylvain A, Thibult ML, Houvenaeghel G, Jacquemier J, Castellano R, et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. The Journal of clinical investigation 2011;121:3609–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shime H, Kojima A, Maruyama A, Saito Y, Oshiumi H, Matsumoto M, et al. Myeloid-derived suppressor cells confer tumor-suppressive functions on natural killer cells via polyinosinic:polycytidylic acid treatment in mouse tumor models. Journal of innate immunity 2014;6:293–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park YJ, Song B, Kim YS, Kim EK, Lee JM, Lee GE, et al. Tumor microenvironmental conversion of natural killer cells into myeloid-derived suppressor cells. Cancer research 2013;73:5669–81 [DOI] [PubMed] [Google Scholar]
- 42.Husain Z, Huang Y, Seth P, Sukhatme VP. Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. Journal of immunology (Baltimore, Md : 1950) 2013;191:1486–95 [DOI] [PubMed] [Google Scholar]
- 43.Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nature medicine 2007;13:828–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. The Journal of experimental medicine 2011;208:1949–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jitschin R, Braun M, Buttner M, Dettmer-Wilde K, Bricks J, Berger J, et al. CLL-cells induce IDOhi CD14+HLA-DRlo myeloid-derived suppressor cells that inhibit T-cell responses and promote TRegs. Blood 2014;124:750–60 [DOI] [PubMed] [Google Scholar]
- 46.Oh K, Lee OY, Shon SY, Nam O, Ryu PM, Seo MW, et al. A mutual activation loop between breast cancer cells and myeloid-derived suppressor cells facilitates spontaneous metastasis through IL-6 trans-signaling in a murine model. Breast cancer research : BCR 2013;15:R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Djeu JY, Jiang K, Wei S. A view to a kill: signals triggering cytotoxicity. Clinical cancer research : an official journal of the American Association for Cancer Research 2002;8:636–40 [PubMed] [Google Scholar]
- 48.Furuke K, Burd PR, Horvath-Arcidiacono JA, Hori K, Mostowski H, Bloom ET. Human NK cells express endothelial nitric oxide synthase, and nitric oxide protects them from activation-induced cell death by regulating expression of TNF-alpha. Journal of immunology (Baltimore, Md : 1950) 1999;163:1473–80 [PubMed] [Google Scholar]
- 49.Cifone MG, D’Alo S, Parroni R, Millimaggi D, Biordi L, Martinotti S, et al. Interleukin-2-activated rat natural killer cells express inducible nitric oxide synthase that contributes to cytotoxic function and interferon-gamma production. Blood 1999;93:3876–84 [PubMed] [Google Scholar]
- 50.Goh CC, Roggerson KM, Lee HC, Golden-Mason L. Hepatitis C Virus-Induced Myeloid-Derived Suppressor Cells Suppress NK Cell IFN-gamma Production by Altering Cellular Metabolism via Arginase-1. 2016;196:2283–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Science translational medicine 2014;6:237ra67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen HM, Ma G, Gildener-Leapman N, Eisenstein S, Coakley BA, Ozao J, et al. Myeloid derived suppressor cells as an immune parameter in patients with concurrent sunitinib and stereotactic body radiotherapy. Clinical cancer research : an official journal of the American Association for Cancer Research 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Christiansson L, Soderlund S, Mangsbo S, Hjorth-Hansen H, Hoglund M, Markevarn B, et al. The tyrosine kinase inhibitors imatinib and dasatinib reduce myeloid suppressor cells and release effector lymphocyte responses. Molecular cancer therapeutics 2015;14:1181–91 [DOI] [PubMed] [Google Scholar]
- 54.Stiff A, Trikha P, Wesolowski R, Kendra K, Hsu V, Uppati S, et al. Myeloid-Derived Suppressor Cells Express Bruton’s Tyrosine Kinase and Can Be Depleted in Tumor-Bearing Hosts by Ibrutinib Treatment. Cancer research 2016;76:2125–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McMichael EL, Jaime-Ramirez AC, Guenterberg KD, Luedke E, Atwal LS, Campbell A, et al. IL-21 enhances natural killer cell response to cetuximab-coated pancreatic tumor cells. Clin Cancer Res 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lu G, Zhang R, Geng S, Peng L, Jayaraman P, Chen C, et al. Myeloid cell-derived inducible nitric oxide synthase suppresses M1 macrophage polarization. Nat Commun 2015;6:6676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Paul DM, Vilas SP, Kumar JM. A flow-cytometry assisted segregation of responding and non-responding population of endothelial cells for enhanced detection of intracellular nitric oxide production. Nitric Oxide 2011;25:31–40 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.