

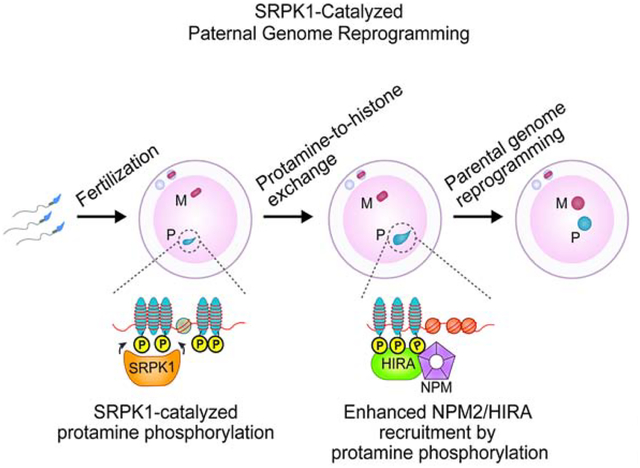
SUMMARY
The paternal genome undergoes massive exchange of histone with protamine for compaction into sperm during spermiogenesis. Upon fertilization, this process is potently reversed, which is essential for parental genome reprogramming and subsequent activation; however, it remains poorly understood how this fundamental process is initiated and regulated. Here, we report that the previously characterized splicing kinase SRPK1 initiates this life-beginning event by catalyzing site-specific phosphorylation of protamine, thereby triggering protamine-to-histone exchange in the fertilized oocyte. Interestingly, protamine undergoes a DNA-dependent phase transition to gel-like condensates and SRPK1-mediated phosphorylation likely helps open up such structures to enhance protamine dismissal by nucleoplasmin (NPM2) and enable the recruitment of HIRA for H3.3 deposition. Remarkably, genome-wide ATAC-seq analysis reveals that selective chromatin accessibility in both sperm and MII oocytes is largely erased in early pronuclei in a protamine phosphorylation-dependent manner, suggesting that SRPK1-catalyzed phosphorylation initiates a highly synchronized reorganization program in both parental genomes.
Graphical Abstract
In Brief
Upon fertilization, maternal splicing kinase SRPK1 phosphorylates protamine to induce paternal chromatin decondensation (protamine-to-histone replacement) and parental genome reprogramming.
eTOC Blurb
In this issue of Cell, Gou et al. reports a key role of the splicing kinase SRPK1 in catalyzing site-specific phosphorylation in protamines, which resemble SR proteins. This proves to be critical for initiating protamine-to-histone exchange in fertilized mouse oocyte. SRPK1-catalyzed protamine phosphorylation opens up its condensates with DNA and enables efficient recruitment of nucleoplasmin for protamine removal and the histone chaperone HIRA for histone deposition. Accompanying with paternal genome reprogramming, maternal genome also undergoes reprogramming in a synchronized fashion before the two pronuclei merge.
INTRODUCTION
Histones have been long known to be gradually replaced by protamines during spermiogenesis (histone-to-protamine exchange) in order to tightly package paternal DNA into the sperm head, resulting in a 10 to 20-fold reduction in nuclear volume to ensure transmission of genetic information to offspring with both high fidelity and efficiency (Balhorn, 2007; Oliva, 2006). After fertilization, the paternal genome undergoes a programmed reversal of histone-to-protamine exchange in the egg (Adenot et al., 1991; Lee et al., 2014), which is essential for paternal genome reprogramming, including the recruitment of the DNA dioxygenase TET3 to induce active DNA demethylation (Gu et al., 2011), re-establishing the 3D genome in both gametes (Du et al., 2017), and subsequent zygotic genome activation (ZGA) (Zhou and Dean, 2015). Despite the rapid progress in delineating these pre-implantation developmental events, especially by recent genomic profiling of different stages during embryonic development (Du et al., 2017; Lu et al., 2016; Wu et al., 2016; Wu et al., 2018; Xia et al., 2019), our understanding remains primitive as to how the whole process is initiated shortly after the sperm enters the egg.
Protamines are a class of small, highly arginine (Arg)-enriched nuclear proteins, which are thought to evolve from specialized histones through protamine-like proteins to true protamines (Balhorn, 2007). In Xenopus, for example, the sperm contains several basic proteins that resemble protamine plus classic H3 and H4, and during post-fertilization chromatin remodeling, those protamine-like proteins are replaced by maternal H2A and H2B (Prado et al., 2004). In mammals, protamine 1 (P1) is primarily responsible for packaging sperm DNA, although a second protamine, P2, is additionally involved in primates, some rodents, and a subset of placental mammals (Balhorn, 1989; Lee and Cho, 1999). During spermiogenesis in mouse testis, typical histones are first replaced by transition proteins and then P1 and P2 (Oliva, 2006). According to the Balhorn model proposed about three decades ago (Balhorn, 1982), which obviously needs refinement with direct structural evidence, the Arg-rich core of protamine binds the phosphate backbone of genomic DNA, which is then stacked on one another to form a sheet-like structure locked by inter-protamine disulfide bridges. Remarkably, such tightly packed chromatin is rapidly decondensed in the fertilized egg, which is facilitated by released proteins from the nucleus of the oocyte upon germinal vesicle breakdown (GVBD). Fundamental, but still unsolved, questions are which protein(s) catalyzes the protamine replacement with histones and how the process might be regulated.
Multiple acidic protein factors have been implicated in mediating protamine-to-histone exchange in the fertilized egg. The primary candidate is nucleoplasmin (NPM), which is the most abundant protein in matured oocytes. Immunodepletion of NPM from Xenopus oocyte extracts blocked, while purified NPM induced, sperm DNA decondensation in vitro (Inoue et al., 2011; Philpott and Leno, 1992; Philpott et al., 1991; Shintomi et al., 2017; Shintomi et al., 2015). However, sperm DNA decondensation still occurred when the major NPM gene (NPM2) was genetically ablated in mice (Burns et al., 2003). This might be due to redundant functions of the other two relatively minor NPM family members, NPM1 and NPM3, expressed in mouse oocytes (Okuwaki et al., 2012). Additionally, template-activating factor 1β (TAF1β) also exhibited a similar sperm DNA decondensation activity in vitro (Matsumoto et al., 1999). Therefore, multiple proteins may be involved in protamine removal independently or cooperatively. Accompanying with protamine removal, histones are incorporated for de novo assembly of nucleosomes and the H3.3 chaperone HIRA is thought to play a major role in this process (Inoue and Zhang, 2014; Lin et al., 2014; Loppin et al., 2005), perhaps in synergy with the chromatin remodeler CHD1 (Konev et al., 2007). Interestingly, sperm DNA was still able to undergo decondensation without HIRA or CHD1 (Inoue and Zhang, 2014; Konev et al., 2007; Lin et al., 2014), indicating that protamine removal and histone deposition may be coordinated, but not necessarily tightly coupled.
Compared to specific factors involved in the protamine-to-histone exchange, even less is known about how the process is regulated. Both protamine and NPM are known to be phosphorylated and NPM phosphorylation appears to enhance its ability to decondense sperm DNA in vitro (Banuelos et al., 2007; Leno et al., 1996). Mammalian protamines contain several potential phosphorylation sites (Chirat et al., 1993; Pirhonen et al., 1993), one of which in mouse P2 appears to play a regulatory role during spermiogenesis (Itoh et al., 2019). Multiple kinases have also been implicated in phosphorylating protamines (Papoutsopoulou et al., 1999; Pirhonen et al., 1994; Wu et al., 2000), but all studies to date were based on in vitro phosphorylation assays with purified kinases, thus lacking direct evidence for the physiological kinase-substrate relationship, which has to be established by both loss- and gain-of-function studies with candidate kinase and defined phosphorylation site(s) in the substrate.
In this study, we demonstrate that protamines are extended members of the Ser/Arg-rich (SR) family of proteins, which are best known for their functions in pre-mRNA splicing (Fu, 1995; Lin and Fu, 2007; Zhong et al., 2009). SR proteins are well known to be regulated from nuclear import to spliceosome assembly by evolutionarily conserved SR protein-specific kinases (SRPKs) (Giannakouros et al. 2011; Zhou and Fu, 2013). Mammalian genomes encode for three SRPKs: SRPK1 is ubiquitously expressed with the highest expression in germ cells (Papoutsopoulou et al., 1999), whereas SRPK2 and SRPK3 are largely restricted to neurons and muscles, respectively (Nakagawa et al., 2005; Wang et al., 1998). We now show that SRPK1 and two SRPK1-catalyzed phosphorylation sites in P1 in mice are vital for initiating protamine-to-histone exchange in the fertilized egg by facilitating direct protamine interactions with NPM2 for protamine removal and HIRA for H3.3 deposition to drive synchronized reprogramming of both paternal and maternal genomes. These findings establish a central, splicing-independent function of SRPK1 in catalyzing one of the earliest life events in mammals.
RESULTS
Requirement of Maternal SRPK1 for Paternal Genome Decondensation
Both mammalian P1 and P2 are highly enriched with Ser/Arg dipeptides characteristic of SR proteins, which are uniquely recognized by SRPKs (Gui et al., 1994a; 1994b; Colwill et al., 1996; Wang et al., 1998). To begin to test the hypothesis that SRPK1 might play a key role in zygotic development by phosphorylating protamines to trigger protamine-to-histone exchange, we performed immunostaining on both germline vesicle (GV) and Metaphase II (MII) oocytes with a specific antibody against SRPK1. By confocal microscopy, we found that SRPK1 was diffusely distributed throughout the GV oocyte in which interchromatin granules (also known as nuclear speckles) were detectable within GV by anti-SC35; in contrast, nuclear speckles are “melted” by excessive SRPK1 as a consequence of GVBD in the MII oocyte (Figures 1A and S1A), as seen in somatic cells during mitosis (Gui et al., 1994a; Ding et al., 2006). As expected, SRPK2 was largely undetectable (Figure S1B). Notably, SRPK1 became associated with some patches in the MII oocyte, which likely resulted from remaining interchromatin granules released from the broken GV, which has also been reported in somatic cells during mitosis (Spector et al., 1991).
Figure 1. Inhibition of SRPK1 arrested fertilized oocytes at the 1-cell stage.
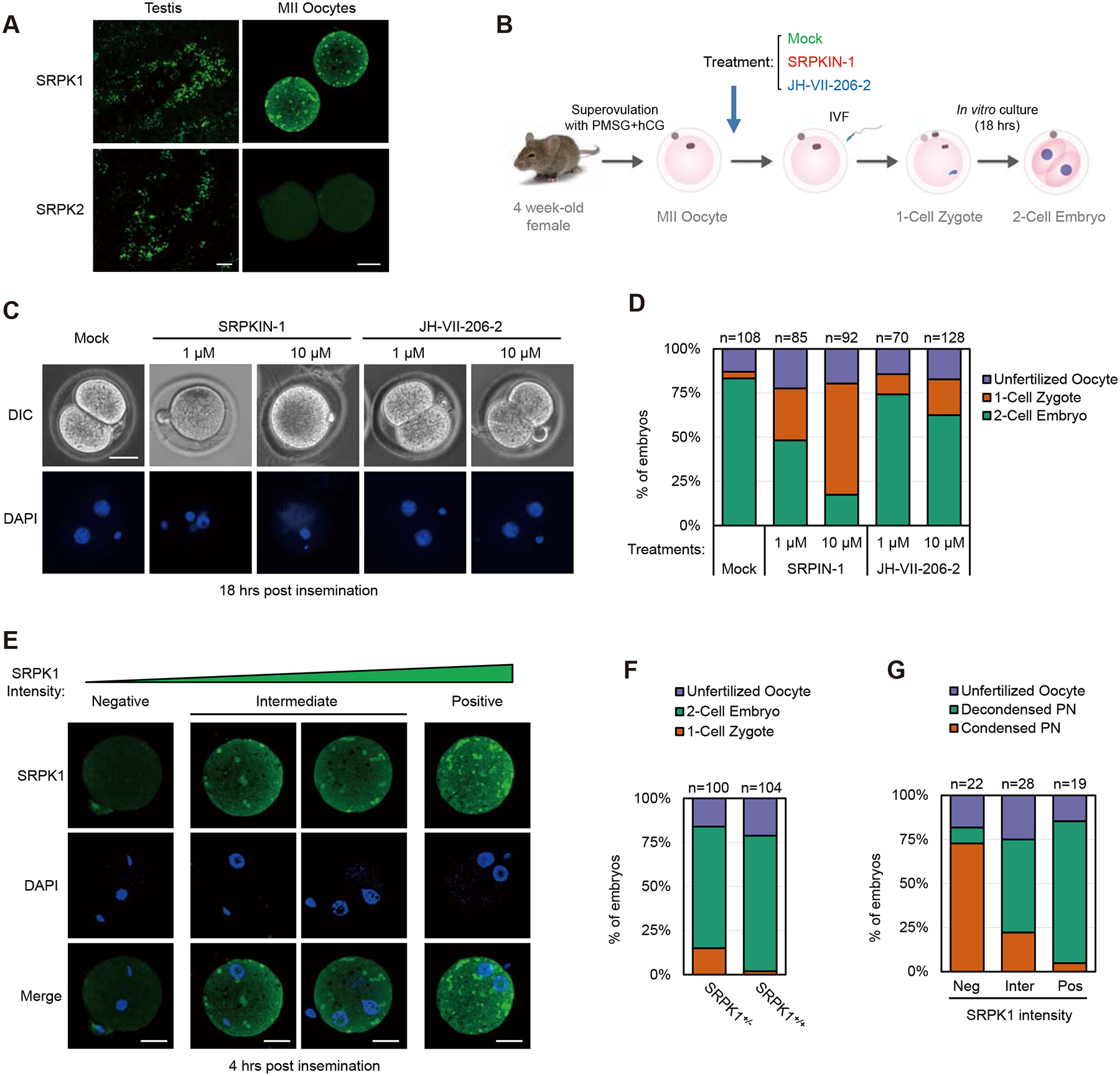
(A) Immunostaining of mouse testis and MII oocyte for SRPK1 and SRPK2. Scale bar, 20 μm. See Figure S1A for negative staining with anti-SRPK2 in comparison without primary antibody.
(B) Scheme for treatment with an SRPK1 inhibitor during in vitro fertilization (IVF). SRPKIN-1 is a specific SRPK1 inhibitor and JH-VII-206–2 is a compound with a related structure to SRPKIN-1 but without effect on SRPK1 kinase activity. See Figure S1B for compound structures.
(C) Representative images of chemically treated oocytes examined by differential interference contrast (DIC) microscopy (upper panel) or DAPI-stained DNA (lower panel). Maternal (M) and paternal (P) genomes are labeled.
(D) Quantification of unfertilized oocyte, 1-cell zygotes, and 2-cell embryos in response to treatment with the SRPK1 inhibitor or control compound. The numbers of zygotes quantified from three independent experiments are indicated above the bars.
(E) Representative images of zygotes with low, intermediate, and high levels of residual SRPK1 (quantified with ImageJ), correlated to different degrees of paternal genome decondensation. Scale bar, 20 μm.
(F and G) Quantification of unfertilized oocyte, 1-cell zygotes, and 2-cell embryos from fertilized oocytes in response to SRPK1 depletion (F) and percentages of the paternal genome that remained condensed in 1-cell zygotes relative to the remaining levels of SRPK1 (G). The numbers of zygotes quantified from three independent experiments are indicated above the bars. See also Figure S1C–F for additional details.
To determine if SRPK1 is required for preimplantation embryonic development upon in vitro fertilization (IVF) of mouse eggs (Figure 1B), we first took advantage of a newly developed SRPK1 inhibitor SRPKIN-1 (Figure S1C), which forms a specific covalent bond with a tyrosine phenol group uniquely present in the ATP-binding pocket of the kinase (Hatcher et al., 2018). SRPKIN-1 treatment had no effect on fertilization, based on the presence of both gametes detected by DAPI staining, but blocked the transition of fertilized eggs from 1-cell to 2-cell embryos in a dosage-dependent manner, as indicated by the reduction of 2-cell embryos from 83.3% of mock (DMSO) treated eggs to 48.2% and 17.4% of eggs respectively treated with 1 μM and 10 μM of SRPKIN-1 (Figures 1C, 1D, and S1D). A control compound JH-VII-206–2, which has a structure similar to SRPKIN-1, but cannot bind SRPK1 (Figure S1C), showed little effect (74.3% at 1 μM and 62.5% at 10 μM) on zygotic development (Figures 1C, 1D, and S1D). These dosage-dependent effects mirrored paternal DNA that remained in the condensed state, showing a significant population of zygotes with condensed paternal DNA in SRPKIN-1 treated, but not control compound treated, eggs (Figure S1E). These observations strongly suggest a key role of maternal SRPK1 in driving paternal genome decondensation in fertilized eggs.
Given the possibility that chemical inhibitors might have some general toxic effects, we next took a genetic approach to further establish the functional requirement of SRPK1 in early development. To this end, we utilized heterozygous SRPK1+/− mice we previously generated (Wang et al., 2014), taking advantage of differential transcriptional silencing in late stage GV oocytes before reaching the MII stage (Bouniol-Baly et al., 1999). We reasoned that, if the wild-type copy of SRPK1 in heterozygous SRPK1+/− oocytes became silenced earlier before ovulation, such oocytes would have a degree of reduced SRPK1 mRNA and protein. Indeed, we found that 12.5% to 21.7% of in vitro fertilized eggs from SRPK1+/− females were arrested at the 1-cell stage, compared to ~2.5% of inseminated oocytes from SRPK1+/+ females (Figures 1E, 1F, and S1F). This partial effect likely resulted from incomplete depletion of SRPK1 protein, and if so, we would predict a negative correlation between remaining SRPK1 levels and developmental arrest. We therefore divided the fertilized eggs into three categories based on the immunostaining intensity of SRPK1 (negative, intermediate, positive according to ImageJ) and found that ~4-hour post insemination, the majority of SRPK1-negative zygotes (60 to 80%) failed to decondense their paternal genome; many fewer zygotes (20 to 30%) with intermediate SRPK1 staining showed this phenotype; and most SRPK1-positive zygotes developed normally (Figures 1G and S1G). This correlative evidence strongly suggests a central role of SRPK1 in zygotic development by promoting paternal genome decondensation.
Functional Rescue of Genome Decondensation and Protamine Removal by Active SRPK1
Because SRPK1 protein was only partially depleted in oocytes from SRPK1+/− females, we took a third approach to further demonstrate the requirement of SRPK1 for paternal genome decondensation. To this end, we followed a previously established microinjection protocol to deplete maternal mRNA during oocyte growth, which also allows complementation with exogenous mRNA (Inoue et al., 2014; Inoue and Zhang, 2014). We isolated growing oocytes from young females and injected them with an established siRNA against SRPK1 (see Star*Methods), and after in vitro culture to allow oocyte growth, ~70% of injected oocytes were able to progress to the MII stage by Day 13 (Figure S2A), and these were used for IVF (Figure 2A). Using this strategy, we successfully depleted maternal SRPK1, as evidenced by negative staining in resulting zygotes after IVF, and found that SRPK1 depletion largely prevented paternal genome decondensation (Figure 2B and 2D–F, panels I and II), leading to a potent blockage of zygotic development, as only 13.8% of siSRPK1-treated zygotes progressed to the 2-cell stage, whereas 79.6% of scrambled (Scr) siRNA-treated zygotes developed normally (Figures 2C and S2B).
Figure 2. Kinase activity of SRPK1 is required for protamine removal.
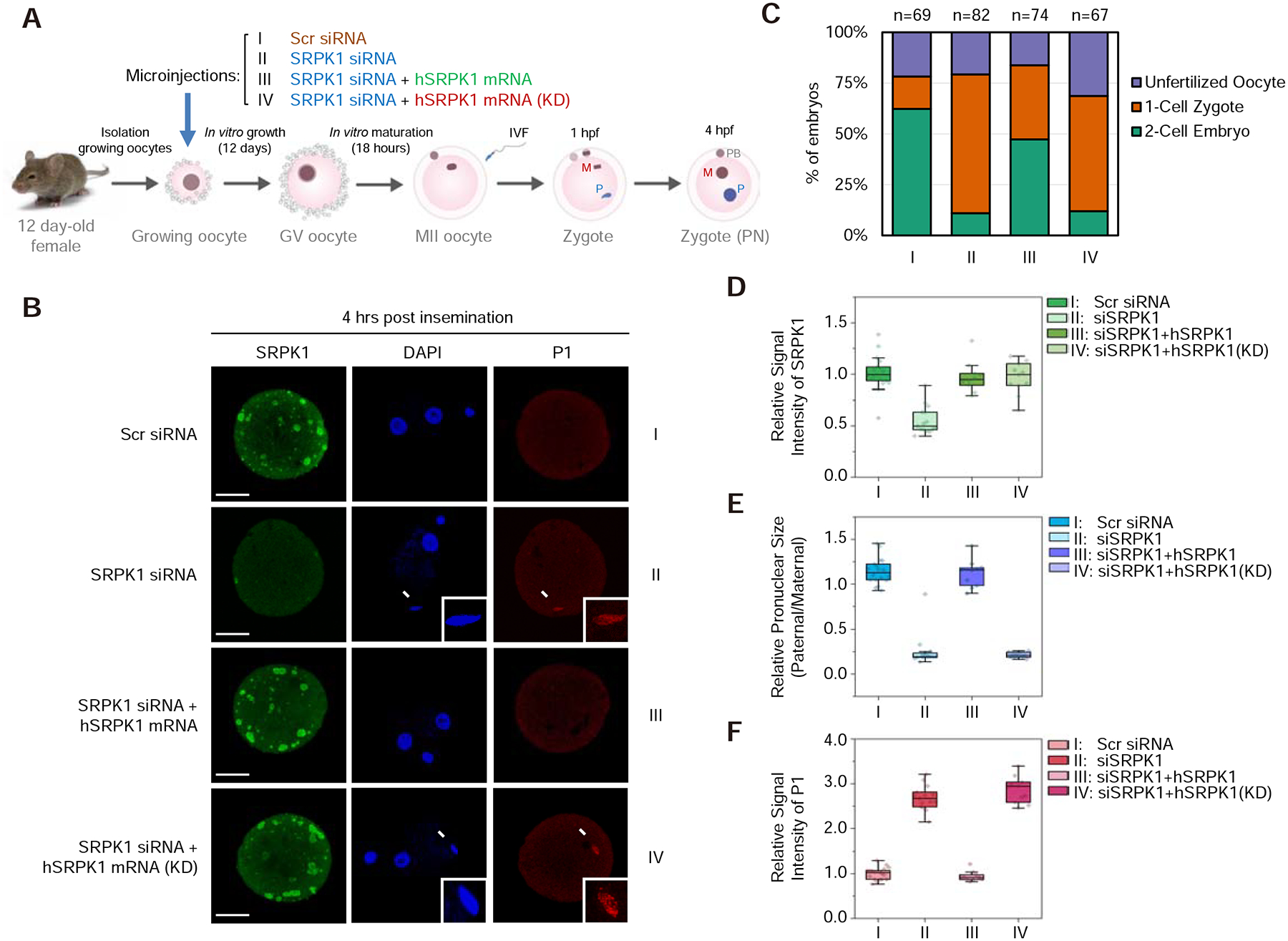
(A) Scheme for siRNA microinjection into growing oocytes to deplete maternal SRPK1 and functional rescue with siRNA-resistant SRPK1 mRNA. See Figure S2A for characterization of the growing oocytes.
(B) Representative images of fertilized eggs stained with anti-SRPK1 (green), anti-P1 (red), and DAPI (blue) 4 hours post insemination. Panel I to IV respectively show oocytes injected with scrambled (Scr) siRNA (I), SRPK1 siRNA (II), SRPK1 siRNA plus human SRPK1 mRNA (III) and SRPK1 siRNA plus human kinase-dead SRPK1 mRNA (KD) (IV). Arrows indicate paternal DNA with a zoomed image in the insert. P, paternal DNA; M, maternal DNA; PB, polar body. Scale bar, 20 μm.
(C) Quantified impact on zygotic development in response to different treatments as in B. The numbers of zygotes quantified from three independent experiments are indicated above the bars. See Figure S2B for additional details.
(D,E,F) Relative SRPK1 staining signals (D), relative values of pronuclear size (paternal/maternal) (E), and relative P1 staining signals (F) in fertilized eggs. The values of zygotes treated with Scr siRNA were set as 1.0 in each case. **P < 0.01 by two-tailed Student’s t-test; error bars, mean±SEM.
Importantly, the defect could be functionally rescued by co-injecting a siRNA-resistant human SRPK1 mRNA (note that human SRPK1 is nearly identical to mouse SRPK1) (Figure 2B and 2D–F, panel III), as indicated by the restoration of 2-cell embryos from 13.8% to 56.5% (Figures 2C and S2B). Consistent with our hypothesis that SRPK1 may catalyze protamine dismissal, we found that P1 remained on condensed paternal chromatin in SRPK1-depleted zygotes, but was removed in the presence of exogenous SRPK1 (Figure 2B and 2D–F). These results demonstrated that SRPK1 is necessary to facilitate paternal genome decondensation and protamine removal, which is required for zygotic development beyond the 1-cell stage. This functional complementation system also enabled us to ask whether the kinase activity of SRPK1 is essential for early zygotic development. We thus co-injected developing oocytes with a kinase-dead version of human SRPK1 (KD). As predicted, although SRPK1 expression was fully restored, the paternal genome still remained condensed along with P1 (Figure 2B–F, panel IV). Considering together the three lines of evidence from chemical, genetic, and functional complementation studies, we conclude that kinase-active, maternal SRPK1 is required for catalyzing protamine eviction to allow paternal genome decondensation.
Determining SRPK1 Phosphorylation Sites in Protamines in vitro and in vivo
SRPK1 is a Ser-specific kinase and exclusively requires an adjacent Arg for signaling site-specific phosphorylation (Gui et al., 1994b). Inspection of P1 and P2 indicates 4 and 6 potential phosphorylation sites in them where all 4 Ser residues in P1 and 4 out of 6 Ser residues in P2 are flanked by an Arg residue (Figures 3A and S3A). To begin to determine which of these potential sites are phosphorylated by SRPK1, we first performed in vitro phosphorylation with purified kinase on candidate substrates. Because P1 is small (51 aa) and P2 is relatively larger (107 aa), we chemically synthesized full-length P1 and 3 fragments of P2 as well as various Ala substitution mutations in candidate Ser residues, either singly or in combination (Figures 3A and S3A), and then tested their phosphorylation with purified SRPK1 in vitro. Quantitative analysis of incorporated phosphate into wild-type and mutant substrates suggested that SRPK1 was able to phosphorylate P1 at Ser9 and Ser43 (Figures 3B and S3B, S3C) and P2 at Ser8, 10, 56 and 69 (Figure S3D). We noted that one of these mapped SRPK1 phosphorylation sites (Ser56) in P2 corresponds to a recently characterized phosphorylation event important for functional interaction with a testis-specific phosphatase (Itoh et al., 2019), consistent with a potential role of SRPK1-regulated protamine phosphorylation in sperm production during spermiogenesis.
Figure 3. Maternal SRPK1 catalyzes site-specific phosphorylation of Protamine 1.
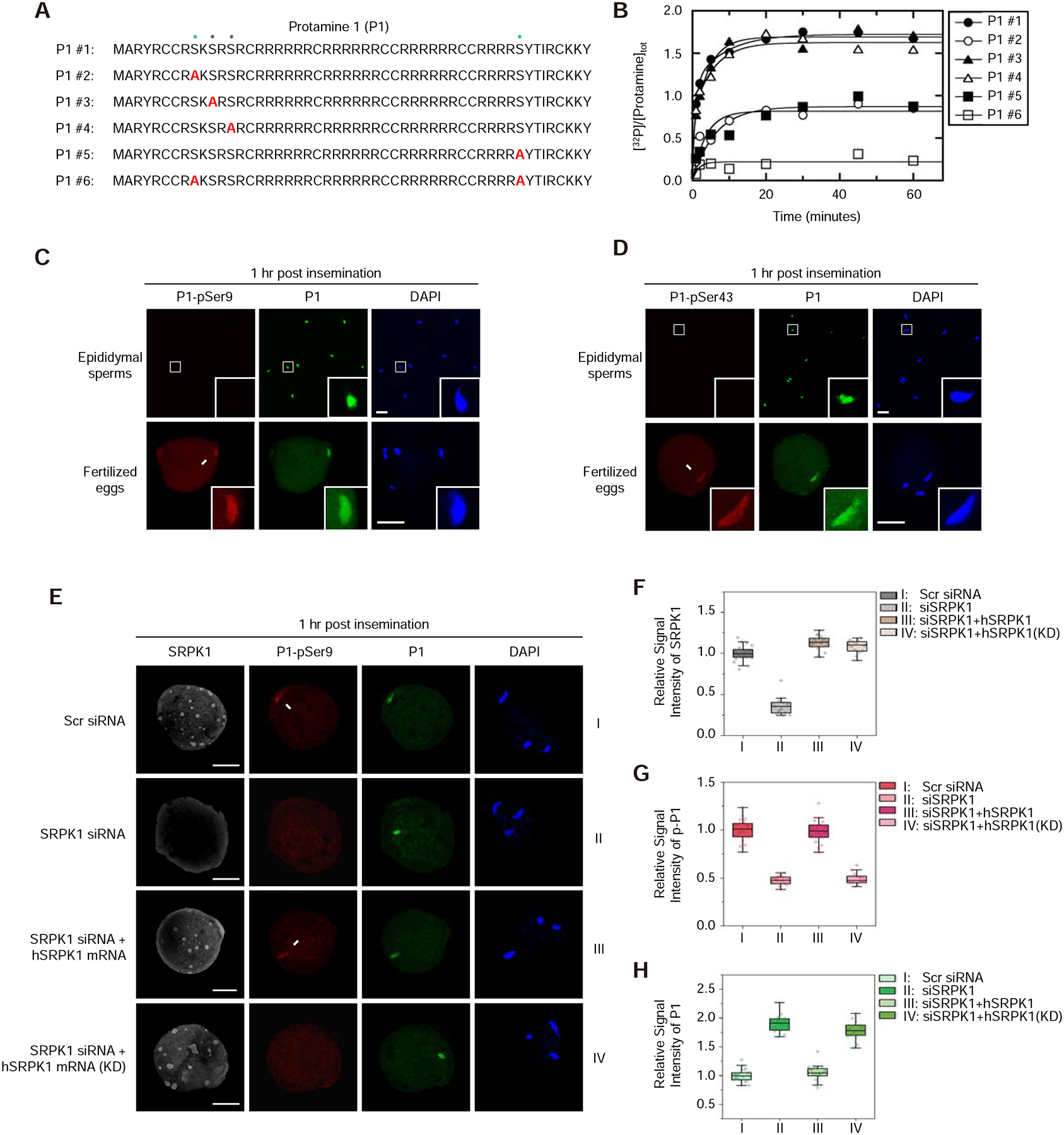
(A) The peptide sequence of mouse P1 (#1) and a series (#2 to #6) of peptides containing specific Ser-to-Ala mutations (red). See Figure S3A for 3 separately synthesized P2 peptides and mapped phosphorylation sites by SRPK1 in vitro.
(B) Time-dependent 32P-phosphate transfer to P1 by SRPK1. Data were fit to either single or double exponential function to obtain total phosphorylation content per peptide at reaction endpoints. See Figure S3B and C for the phosphoryl content transferred to P1 peptides by SRPK1 and the velocity of each phosphorylation reaction; see Figure S3D for SRPK1-catalyzed phosphorylation of individual P2 peptides.
(C, D) Immunostaining of total P1 (green) and phosphorylated P1 (red) at Ser9 (C) or Ser43 (D) in mouse epididymal sperms and zygotes 1-hour post insemination. Arrows indicate P1 on paternal DNA, each with a zoomed image in the insert. P, paternal DNA; M, maternal DNA; PB, polar body. Scale bar, 20 μm. See Figure S3E and F for characterizing the specificity of individual anti-P1 phospho-specific antibodies.
(E) Representative images of fertilized eggs treated with siSRPK1 or rescued with SRPK1 mRNA as in Figure 2B. See also Figure S3G for a time course experiment with anti-pSer43-specific antibody and Figure S3H for negative staining with anti-pSer11 and anti-pSer13 antibodies.
(F,G,H) Relative staining signals with anti-SRPK1 (F), anti-pSer9 (G) and anti-P1 (H) in fertilized eggs. The values of control zygote treated with Scr siRNA were set as 1.0 in each case. **P < 0.01 by two-tailed Student’s t-test; error bars, mean±SEM. See Figure S3I for a similar set of experiments with anti-pSer43.
Because P1 is universally involved in paternal genome packaging in mammals and P2 is species-specific, we focused on establishing in vivo phosphorylation events in P1 and their functional significance in subsequent studies. To this end, we synthesized phosphorylated P1 at individual candidate sites and used these phospho-peptides to raise polyclonal antibodies in rabbits (Figure S3E). Affinity-purified antibodies showed the expected specificity in dot blotting, as individual antibodies reacted with their prospective phospho-peptide antigens without cross-reactions with others (Figure S3F). We next used these reagents to determine if SRPK1 is responsible for protamine phosphorylation in vivo. By immunostaining on both epididymal sperm and zygote 1-hour post IVF, an optimal time window for us to detect P1 phosphorylation before its dismissal from the paternal genome (Figure S3G), we detected clear fluorescence signals on paternal chromatin with both anti-P1-pSer9 and anti-P1-pSer43 antibodies in fertilized eggs (Figure 3C and 3D), but not with anti-P1-pSer11 and anti-P1-pSer13 antibodies (Figure S3H). In contrast, both anti-P1-pSer9 and anti-P1-pSer43 antibodies stained negatively on epididymal sperm, and given positive staining with total P1, these data indicate that P1 is unphosphorylated at these two sites in mature sperm (Figure 3C and 3D). These data establish site-specific phosphorylation of P1 at Ser9 and Ser43 residues in fertilized eggs.
To determine if P1-Ser9 and P1-Ser43 were indeed phosphorylated by SRPK1, we again utilized the functional rescue system (see Figure 2). We found that the P1-pSer9 signal was readily detectable in control siRNA-treated oocyte 1-hour post IVF, but such signal vanished in SRPK1-depleted oocyte, and significantly, exogenous active SRPK1, but not the kinase-dead version of the kinase, restored the signal (Figure 3E). Importantly, at this critical window when P1 still remained on paternal chromatin, quantitative analysis revealed that the immunostaining intensities of total P1 were inversely correlated with its phosphorylation levels in the presence of active SRPK1 (Figure 3F–H). We obtained the same results with P1-pSer43 (Figure S3I). These data further established the specificity of the phospho-specific antibodies we raised and demonstrated P1 phosphorylation at Ser9 and Ser43 by maternal SRPK1 immediately after the sperm enters the egg to facilitate P1 dismissal from the paternal genome.
Requirement of SRPK1-catalyzed Protamine 1 Phosphorylation for Male Fertility
To demonstrate the importance of SRPK1-catalyzed P1 phosphorylation in the physiological setting, we sought to introduce specific mutations in P1 knock-in mice. For this purpose, we utilized the CRISPR/Cas9 technology to introduce Ser-to-Ala mutations at Ser9 and Ser43 either singly or in combination (Figures 4A and S4A). We obtained homozygous mice and confirmed the intended mutations by Sanger sequencing (Figure S4B). We also selected a set of genomic regions based on sequence similarity to our guide RNAs to examine potential off-target effects, and by PCR and Sanger sequencing, we detected no mutation in any of the regions we examined (Figure S4C). We next determined male fertility from these knock-in strains by crossing them with wild-type females and found that mice with single mutations (P1S9A or P1S43A) were largely fertile, comparable to wild-type, but the fertility of double P1S9S43A mutant mice was severely compromised, as indicated by the drastic reduction in litter size (Figure 4B). This phenotype correlated to the developmental potential of eggs fertilized by sperm of different genotypes: Only a small fraction (~17%) of eggs fertilized by double mutant sperm was able to progress to the 2-cell stage 18-hour post IVF, which was in sharp contrast to the majority (~80%) of eggs fertilized with both single mutants, although we observed a slight reduction in both cases compared to wild-type sperm (Figure 4C).
Figure 4. Knock-in mutations in Protamine 1 blocked paternal genome decondensation.
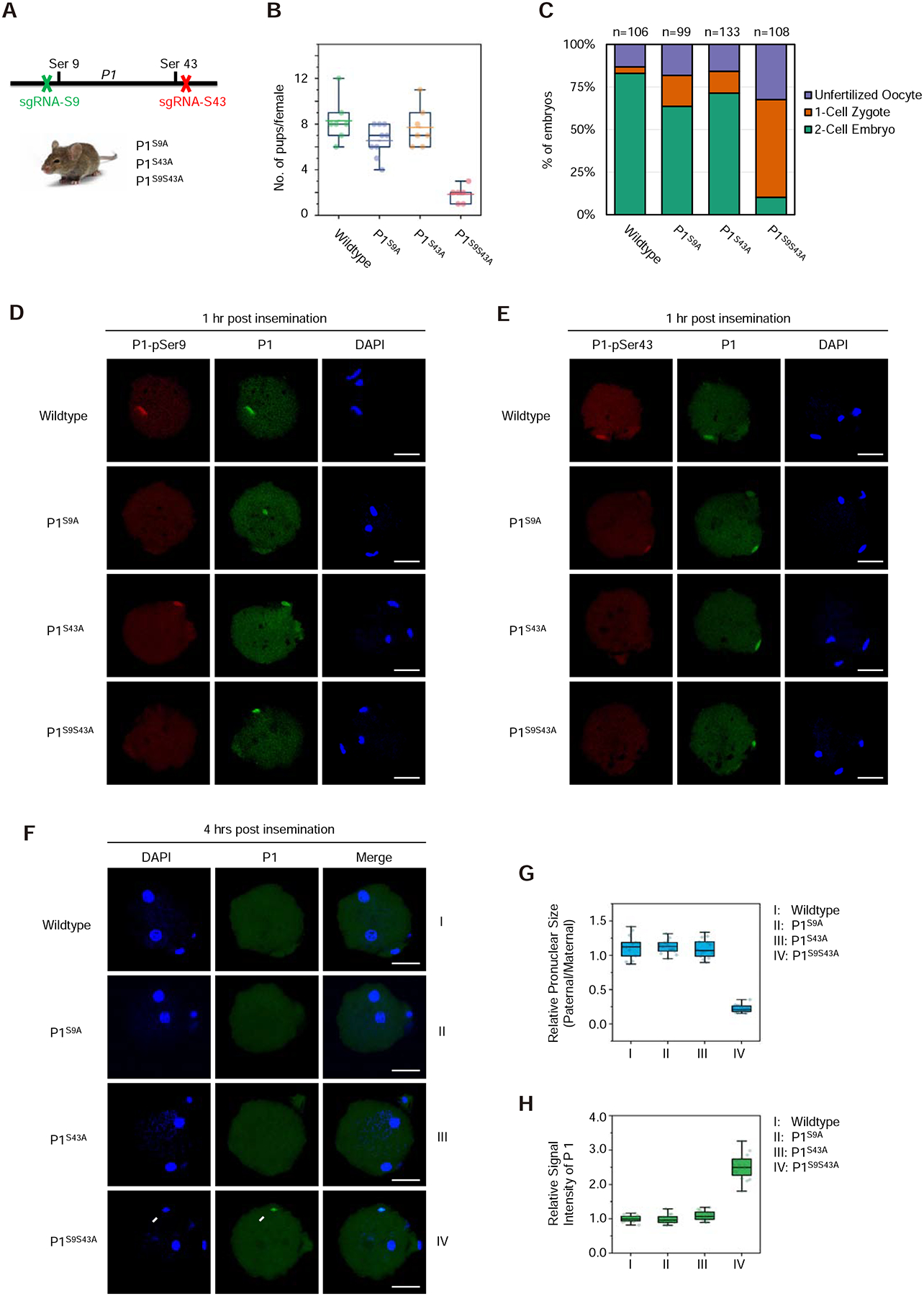
(A) Generation of knock-in P1 mutant mice with CRISPR/Cas9. See Figure S4A–C for gRNA design, confirmation of mutations by Sanger sequencing, and lack of mutations on potential off-target loci examined.
(B) Quantification of litter size of wild-type and mutant males after mating with wild-type females. See Figure S4D–F for sperm counts from wild-type and mutant males as well as DNA compaction examined by aniline blue staining and transmission electron microscopic images of cauda epididymal sperm.
(C) Quantification of developmental potential of zygotes after fertilization with wild-type, single and double P1 mutant sperm. The numbers of zygotes quantified from three independent experiments are indicated above the bars.
(D,E) Representative images of fertilized eggs stained with anti-pSer9 (D) or anti-pSer43 (E), total P1 (green) and DAPI (blue) 1-hour post insemination when protamine still remained on paternal chromatin. P, paternal DNA; M, maternal DNA; PB, polar body. Scale bar, 20 μm.
(F) Representative images of fertilized eggs stained for total P1 (green) and DAPI (blue) 4-hour post insemination when protamine was largely displaced from the paternal genome and disbursed throughout the ooplasm. Arrow indicates P1 staining signal remained on the paternal chromatin only with the double mutant. P, paternal DNA; M, maternal DNA; PB, polar body. Scale bar, 20 μm.
(G,H) Relative values of pronuclear size (paternal/maternal) (G) and relative P1 staining signals on paternal chromatin (H) in fertilized eggs. **P < 0.01 by two-tailed Student’s t-test; error bars, mean±SEM.
To further differentiate the contribution of P1 phosphorylation defects to male infertility during spermiogenesis and/or zygotic development, we examined sperm counts from mutant males and found normal sperm count from both wild-type and single mutant males, but a slight reduction in sperm count from double mutant males (Figure S4D). To examine DNA compaction in double mutant sperm, we employed aniline blue staining (Gou et al., 2017) and found that mutant sperm were similar to wild-type sperm, both not efficiently stained with aniline blue (Figure S4E). Transmission electron microscopy (TEM) further revealed well-condensed sperm head/nucleus in both wild-type and double mutant epididymal sections (Figure S4F). These data strongly suggest appropriately compacted sperm genome from double mutant males.
To directly demonstrate that the functional defects largely resulted from defects in zygotic development, likely due to failed protamine-to-histone exchange in fertilized eggs, we performed immunostaining of fertilized egg 1-hour post IVF with our phospho-specific antibodies. We detected P1 phosphorylation at Ser43, but not Ser9, on Ser9-mutated paternal genome; conversely, P1 phosphorylation at Ser9, but not Ser43, in Ser43-mutated paternal genome; and negative staining with both antibodies on double mutant-fertilized eggs (Figure 4D and 4E). As P1 still remained associated with paternal chromatin at this stage, we next examined zygotes 4-hour post IVF and found that P1 was dismissed in eggs fertilized with wild-type or single mutant sperm, but remained on paternal chromatin in eggs fertilized with double mutant sperm (Figure 4F). Quantitative analysis further showed that the immunostaining signals of P1 were inversely correlated with the sizes of the paternal pronucleus (Figure 4G and 4H). As double mutant-fertilized eggs were literally blocked at the 1-cell stage (see Figure 4C), we conclude that SRPK1-catalyzed phosphorylation is required for protamine removal, which is a prerequisite for paternal genome decondensation in the fertilized egg and subsequent zygotic development.
DNA-induced Phase Transition of Protamine 1 to a Gel-like Structure
Because the Arg-rich domain in a SR-like splicing factor (U2AF65) has been implicated in promoting RNA-RNA interactions during spliceosome assembly (Valcarcel et al., 1996), we asked how the interaction of P1 with DNA might be modulated by SRPK1-catalyzed phosphorylation. We thus performed the gel mobility shift assay to determine how P1 might bind DNA, and as expected, P1 bound double-stranded DNA in a dosage-dependent manner, and in the presence of active SRPK1, the affinity was only slightly reduced (Figure S5A). During this analysis, we noted that the P1/DNA complex hardly entered the gel, indicating a DNA-dependent phase transition, consistent with a predicted intrinsically disordered region in P1 (Figure 5A). To pursue this intriguing property of P1, we incubated P1 with genomic DNA and observed the formation of turbid solution with increasing P1 concentrations, even without the need to reduce temperature or include a crowding reagent (Figure S5B). Visualizing this turbid material under a microscope revealed small particles with low P1 concentrations and large mesh-like networks with higher P1 concentrations (Figure 5B). Interestingly, these different degrees of aggregation were independent of DNA length, indicating that such mesh-like structures were not due to smaller particles arrayed on the DNA string (Figure S5C). By spiking in a small fraction of fluorescence-labeled P1 or DNA in the reaction, we performed fluorescence recovery after photobleaching (FRAP) and found that neither P1 nor DNA fluorescence could be recovered within the time frame of the experiment (Figure 5C), indicating the formation of a gel-like structure. We suspect that this DNA-induced phase separation might be related to DNA packaging into the sperm head in late spermiogenesis.
Figure 5. DNA-induced phase transition of Protamine 1 is partially reversed by SRPK1.
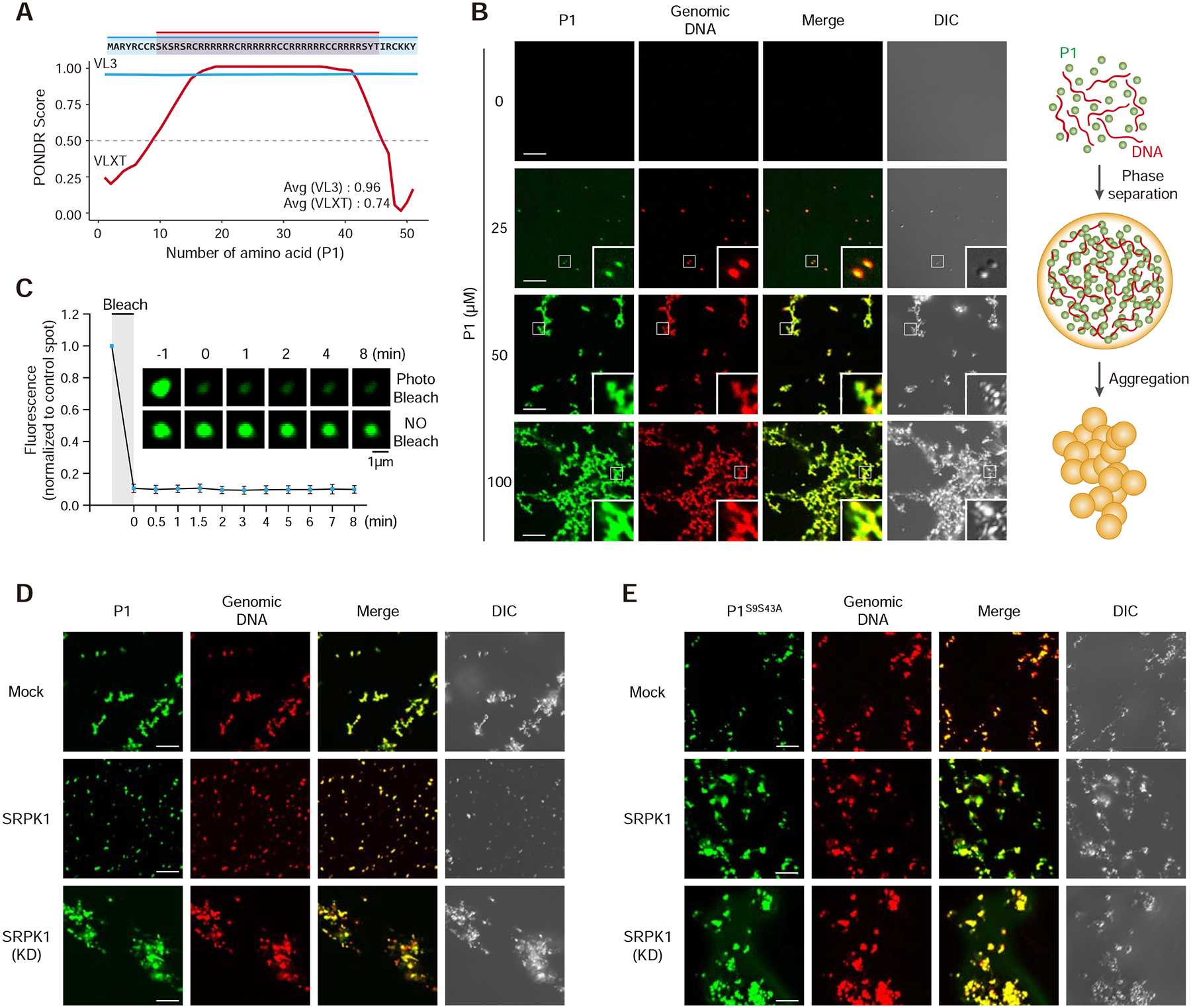
(A) Prediction of intrinsically disordered region (IDR) in P1. Note that the mapped SRPK1 phosphorylation sites are right on both edges of the IDR.
(B) P1 phase separation in the presence of DNA with increasing concentrations of P1 protein. 10% P1 was labeled with Alexa 488 and 1% DNA was labeled with Alexa 594. Insert: amplified images. Scale bar, 10 μm. See Figure S5A–C for protein concentration-dependent, but DNA length-independent P1 phase transition.
(C) Fluorescent recovery after photobleaching (FRAP) analysis on P1/DNA gel-like particles. Bleaching was performed at the indicated time points. Quantified data are based on 3 independent experiments. Error bars, mean±SEM.
(D,E) Representative images of wild-type (D) and double mutant (E) P1 phase separation in the presence of active or kinase dead SRPK1. See Figure S5D,E for similar analysis with single mutants in P1 phosphorylation sites.
We next asked how SRPK1-catalyzed P1 phosphorylation might affect such phase transition. We first tested incubation of P1 with genomic DNA in a buffer we used for the in vitro kinase reaction and observed mesh-like structures (Figure 5D). Note that our buffer contains DTT, which would prevent the formation of intra- and inter-steric disulfide bonds in protamine molecules, suggesting that the mesh-like structure is not mediated by crosslinking between protamine-containing particles. After incubation at 37°C for 30 min with active SRPK1 (conditions that allow efficient P1 phosphorylation), but not kinase-dead SRPK1, we detected only gel-like particles, not mesh-like networks (Figure 5D). We next tested single and double phosphorylation mutant P1 and found that both single mutants behaved similar to wild-type P1 in an active SRPK1-dependent manner (Figure S5D and S5E) and only the double mutant was able to transit to mesh-like networks even in the presence of active SRPK1 (Figure 5E). Together, these results suggest that small gel-like particles may form through some form of stacking interactions among P1/DNA complexes, which is independent of the phosphorylation state of P1, while the transition to mesh-like networks is blocked by P1 phosphorylation. This suggests that SRPK1-mediated P1 phosphorylation may facilitate opening of the highly compacted paternal genome for specific chaperones to act upon, rather than directly causing protamine dismissal.
SRPK1-mediated Protamine Phosphorylation Enhances the Function of NPM
Previous studies have reported that purified NPM2 was sufficient to induce protamine removal from sperm after removing its membrane with Streptolysin O (SLO) and disruption of disulfide bonds with DTT (Inoue et al., 2011; Shintomi et al., 2017; Shintomi et al., 2015; Yamaguchi et al., 2018). However, this assay normally takes 2 to 24 hours to observe detectable effects, indicating that purified NPM2 may be sufficient to slowly extract protamine from DNA during prolonged incubation. Following the same protocol (Figure S6A), we repeated the assay with purified recombinant human NPM2 (Figure S6B) and observed partial sperm decondensation from 1 to 2 hours (Figure S6C). Under the same conditions, we tested the effect of SRPK1 and found that, while purified SRPK1 alone is insufficient to decondense the sperm genome (Figure S6D), SRPK1 treatment greatly accelerated NPM2-mediated sperm decondensation as early as 15 min, while the kinase-dead SRPK1 failed to show this effect (Figure 6A, upper panel). These observations are well in line with slow paternal DNA decondensation, as seen in SRPK1-depleted oocytes ~10-hour post IVF (Figure S6E). We also tested sperm from double mutant males, and as expected, those double mutant sperm no longer responded to SRPK1 in accelerating NPM2-mediated sperm chromatin decondensation (Figure 6A, lower panel), again in line with slow decondensation of double mutant paternal chromatin in the presence of maternal SRPK1 (Figure S6F). We tested and detected no effect of purified NPM2 in the absence or presence of active SRPK1 on in vitro assembled P1/DNA gel-like particles (Figure S6G), indicating that NPM2 may not be able to fully strip off protamines from the paternal genome.
Figure 6. Phosphorylation-dependent interactions of Protamine 1 with NPM and HIRA.
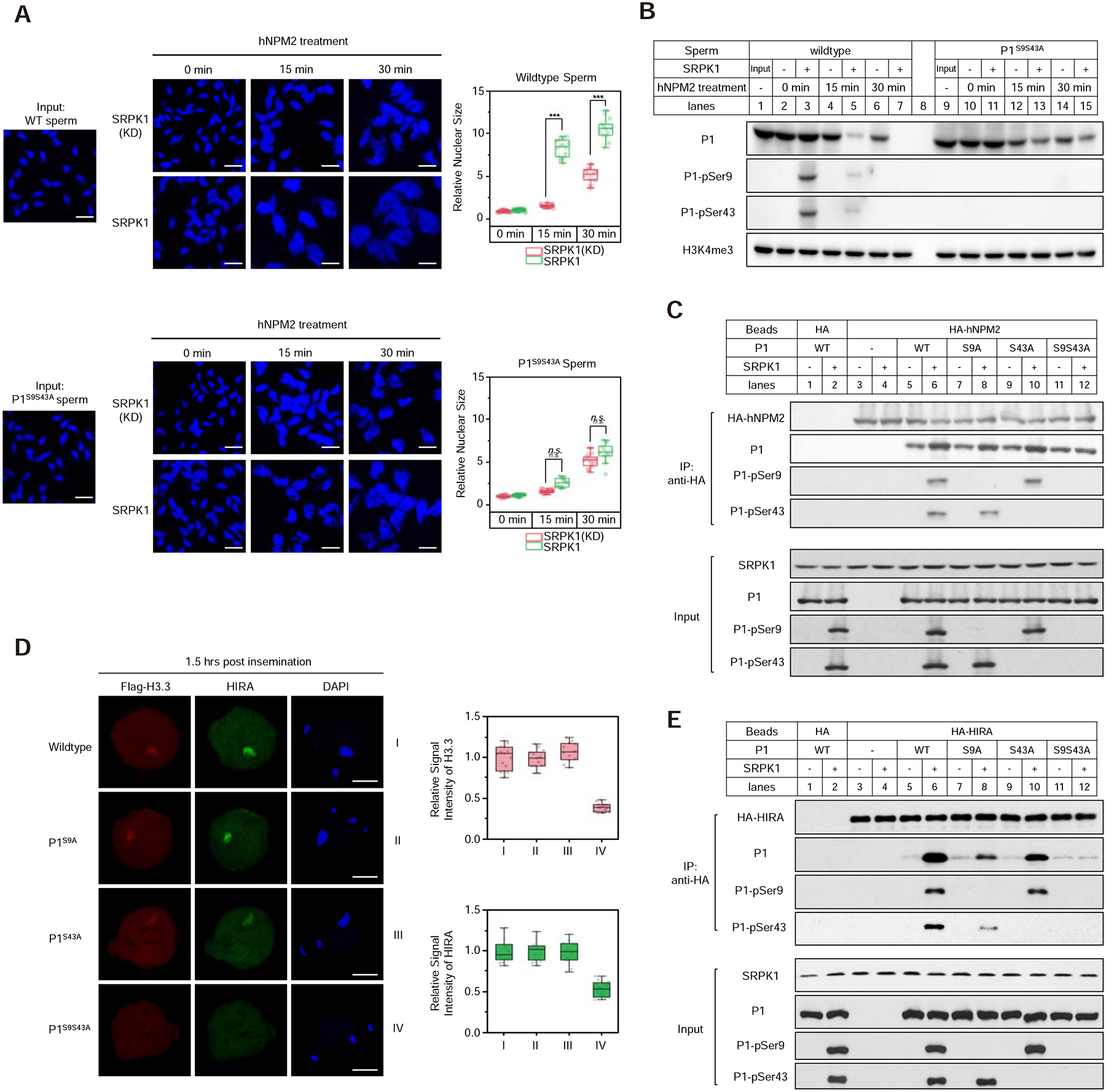
(A) SRPK1 phosphorylation-dependent enhancement of sperm DNA decondensation with purified NPM2. Isolated wild-type (upper panels) and double mutant (lower panels) sperm were treated with purified NPM2 for different time points in the presence of kinase-dead or active SRPK1. Sperm volumes were quantified, as shown on the right. ***P < 0.001 by two-tailed Student’s t-test; error bars, mean±SEM. See Figure S6A for the in vitro sperm DNA decondensation assay.
(B) Western blot of retained P1 on sperm chromatin from the experiments as in A. Total and specifically phosphorylated P1 were blotted with specific antibodies. See Figure S6B for purification of recombinant NPM2, Figure S6C and S6D for the levels of retained P1 upon longer NPM2 or SRPK1 treatment, Figure S6E and S6F for in vivo results on paternal DNA decondensation in SRPK1-depleted and P1 double mutant oocytes ~10-hour post insemination, and Figure S6G for the lack of effect of NPM2 on in vitro assembled P1/DNA particles.
(C) Pulldown assay to determine the interaction between HA-tagged human NPM2 and P1 in the presence or absence of SRPK1. Anti-HA pulldowns were blotted with individual antibodies as indicated.
(D) Representative images of fertilized eggs stained for FLAG-H3.3 (red), HIRA (green) and DNA (blue) 1.5-hour post insemination with wild-type, single and double mutant sperm. P, paternal DNA; M, maternal DNA; PB, polar body. Scale bar, 20 μm. Fluorescence intensity was quantified by ImageJ, as shown on the right for FLAG-H3.3 and HIRA.
(E) Pulldown assay to determine the interaction between HA-tagged human HIRA and P1 in the presence or absence of SRPK1. Anti-HA pulldowns were blotted with individual antibodies as indicated.
Because chromatin decondensation may reflect loosen structures, not necessarily protamine removal, we next wished to directly evaluate P1 that remained on sperm chromatin by Western blotting and ask whether P1 removal might be induced by SRPK1-catalyzed phosphorylation. We first incubated SLO/DTT-pretreated sperm with SRPK1 for 30 min and then proceeded with the DNA decondensation assay. We found that SRPK1 phosphorylated P1 at Ser9 and Ser43, and importantly, while NPM2 alone slowly removed chromatin-bound P1, SRPK1 greatly enhanced NPM2-dependent P1 removal, and as expected, the double mutant P1 no longer responded to SRPK1 treatment (Figure 6B). We next asked how SRPK1 might affect the interaction between P1 and NPM2 by using HA-tagged NPM2 purified from HEK293T cells to pull down wild-type and mutant P1 with or without SRPK1. We found that HA-NPM2 was able to bind P1, which was enhanced by SRPK1-mediated phosphorylation on wild-type and single mutants, but not double mutant (Figure 6C). These data suggest a mechanism for SRPK1-enhanced, NPM2-mediated protamine removal from sperm chromatin.
Enhanced Recruitment of the H3.3 Chaperone HIRA to Phosphorylated Protamine
It has been well established that the histone H3.3 chaperone HIRA is selectively recruited to the paternal pronucleus for chromatin remodeling (Inoue and Zhang, 2014; Lin et al., 2014). However, it has been unclear how such selectivity is achieved and how HIRA recruitment to the paternal genome may be regulated in the fertilized egg. We hypothesized that SRPK1-mediated protamine phosphorylation might facilitate coordinated protamine removal and histone H3.3 deposition. To test this hypothesis, we again took advantage of single and double P1 mutant sperm to perform IVF and stained for H3.3 and HIRA 1.5-hour post IVF. Interestingly, we detected efficient HIRA recruitment to and H3.3 deposition on the paternal pronucleus with wild-type and single P1 mutant sperm, but little HIRA signal on the double mutant paternal chromatin (Figure 6D). Thus, HIRA recruitment to the paternal genome appears to be largely mediated by phosphorylated P1. The similar timing (1 to 1.5 hours) between P1 removal and H3.3 deposition suggests that the two processes are synchronous and occur shortly after fertilization.
To determine whether HIRA recruitment was directly mediated by protamine in a SRPK1 phosphorylation-dependent manner, we performed the in vitro pull down assay in the presence or absence of active SRPK1 using purified HA-tagged HIRA from HEK293T cells. We found that HIRA bound wild-type P1 in a strictly SRPK1-dependent manner (Figure 6E). Both single mutant P1 reduced the efficiency whereas the double mutant P1 abolished the interaction with HIRA (Figure 6E). These data are fully in line with the in vivo recruitment results, together, suggesting that SRPK1-catalyzed protamine phosphorylation also facilitates the selective recruitment of HIRA to the paternal genome to coordinate with the function of NPM proteins during paternal genome reprogramming.
Coordinated Chromatin Remodeling in Both Paternal and Maternal Pronuclei
It is striking to note that the defect in male fertility caused by the double mutant P1 is more severe than that caused by deleting any of the factors implicated in protamine-to-histone exchange reported to date, indicating that SRPK1-catalyzed protamine phosphorylation is the first step to potentiate the action of various chaperones. Given that all potential chaperones are present in the MII oocytes and the ability of purified NPM2 to slowly decondense sperm in vitro, we wondered whether paternal DNA still underwent partial decondensation without P1 phosphorylation, thus altering the accessibility of sperm chromatin that could be captured by ATAC-seq. We thus characterized the chromatin state of parental genomes by performing low input ATAC-seq (with ~100 fertilized eggs), as established earlier (Wu et al., 2016), to capture early genome reprogramming events ~5-hour post IVF, which corresponds to late pronucleus (PN) stage 2 and early stage 3 (Wossidlo et al., 2010), a developmental window too early to physically isolate paternal and maternal pronuclei for genomic profiling. In order to differentiate between paternal and maternal genomes, we took advantage of extensive (~20 million) single nucleotide polymorphisms (SNPs) between C57BL/6 and BALB/cJ mice (Link et al., 2018). As our P1 knock-in mice were constructed in the C57BL/6 strain, we utilized sperm from this strain to fertilize MII oocytes from the BALB/cJ strain (Figure 7A).
Figure 7. Coordinated parental genome reprogramming in early pronuclei.
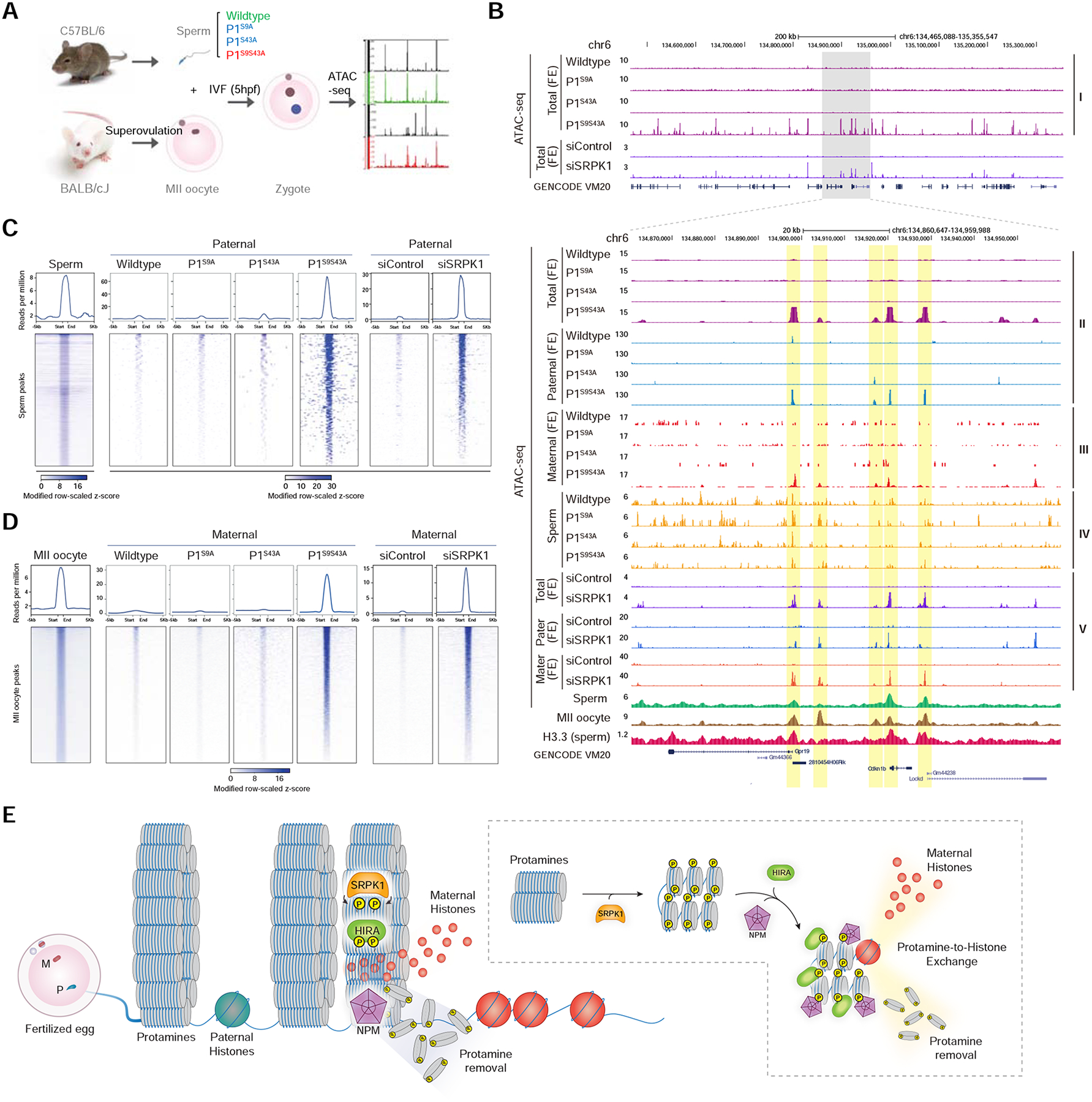
(A) Scheme for using C57BL/6 sperm to fertilize BALB/cJ MII oocytes followed by ATAC-seq. See Figure S7A–C for statistics of the ATAC-seq libraries generated and comparison with the public ATAC-seq data from sperm.
(B) The UCSC browser view of a representative genomic region for ATAC-seq signals in fertilized eggs (FE) by wild-type, single and double mutant sperm (Layer I), ATAC-seq signals uniquely mapped to the paternal (Layer II) or maternal (Layer III) genome, ATAC-seq signals on isolated sperm of different genotypes (Layer IV), and ATAC-seq signals in fertilized eggs (FE) derived from SRPK1-depleted and control siRNA-treated oocytes (Layer V). Public ATAC-seq data on wild-type sperm (GSE79230) and MII oocyte (GSE116854), and ChIP-seq signals for H3.3 on sperm DNA (GSE42629) are displayed at bottom. See Figure S7D and S7E for mapping rates of ATAC-seq reads on the paternal and maternal genomes. All data were normalized to 1 million total counts, and the same scales in y-axes were used for displaying comparable data for comparison.
(C, D) Mega-gene analysis on paternal genome-specific (C) and maternal genome-specific (D) ATAC-seq signals in eggs fertilized with wild-type, single and double P1 mutant sperm, and in control siRNA-treated or siSRPK1-treated eggs fertilized with wild-type sperm. Upper panels: accumulated ATAC-seq signals aligned on the centers of ATAC-seq peaks from wild-type sperm (C) or from MII oocyte (D); Low panels: Distribution of ATAC-seq signals on individual loci based on scaled z-scores. Different z-score scales were used to display sperm ATAC-seq profile versus paternal signals in fertilized eggs (C) because of the higher background detected in sperm, likely due to more compacted paternal genome in sperm relative to fertilized eggs where inter-protamine disulfide bonds are removed to enable more robust ATAC-seq profiling. See Figure S7F and S7G for the accumulation of previously mapped H3.3 ChIP-seq signals on the ATAC-seq peaks we identified and their enrichment at TSSs.
(E) Proposed model for phosphorylation regulation of the protamine-to-histone exchange in fertilized oocyte by SRPK1.
We performed ATAC-seq in duplicate and obtained 10–28 million uniquely mapped reads to the mouse genome from individual libraries (Figure S7A). The ATAC-seq data were of high quality, as indicated by high reproducibility between individual duplicates from fertilized eggs (Figure S7B) as well as between wild-type and different mutant sperm in comparison with the public sperm ATAC-seq data (Jung et al., 2017; Figure S7C). Surprisingly, despite similar read densities, we detected broadly distributed ATAC-seq reads across the mouse genome even before deconvoluting the reads to paternal and maternal gametes formed with wild-type and single mutant sperm, and in contrast, we saw individual peaks with zygotes formed with the double mutant sperm, as illustrated on a representative genomic region (Figure 7B, layer I). Treatment with siSRPK1, but not control siRNA, caused similar effects (Figure 7B). We next assigned ATAC-seq reads to the paternal versus maternal genome based on SNPs, as reported earlier (Link et al., 2018; Xie et al., 2012). From a total of 478,021 and 110,608 SNP-containing reads mapped to the maternal and paternal genome, respectively, cross-genome analysis showed little cross-genome assignment (Figure S7D and E). This is also the case with both single and double mutant ATAC-seq data (Figure S7D). We noted that, while 81.2% of SNP-containing reads were assigned to the maternal genome, 18.8% of SNP-containing reads were assignable to the paternal genome in eggs fertilized by wild-type sperm, which progressively decreased with single and double mutants (14.2%, 10.8%, 8.0%) (Figure S7F). This likely reflects more compaction of the paternal genome compared to the maternal genome.
After deconvoluting paternal and maternal ATAC-seq reads and normalizing all libraries to an equal density (counts per million or CPM), we found that only the double mutant showed significant ATAC-seq peaks (Figure 7B, layer II), despite much reduced read density even among top 10,000 ATAC-seq reads-containing genomic bins (Figure S7G). This was also the case with the ATAC-seq reads uniquely mapped to the maternal genome (Figure 7B, layer III). Using the same data treatment strategy to focus on SNP-containing ATAC-seq reads, we found that wild-type, single, and double mutant sperm all showed similar ATAC-seq peak distributions, which also resembled the published ATAC-seq profile on wild-type sperm (Figure 7B, layer IV), indicating that the paternal genome was similarly packaged during spermiogenesis. Aligning on these sperm ATAC-seq peak centers, we found that the paternal genome in eggs fertilized with the double mutant sperm largely retained sperm-specific ATAC-seq peaks (Figure 7C), which were also overlapped with the published H3.3 ChIP-seq signals on wild-type sperm (Figure S7H), mostly corresponding to transcription start sites (TSSs) (Figure S7I). We noted that the background for ATAC-seq signals on isolated sperm was much higher than that in eggs fertilized with the double mutant sperm, as indicated by the lower z-score scale with the ATAC-seq signals from sperm compared to fertilized eggs, which is likely due to the more compacted paternal genome in sperm relative to fertilized egg where extensive inter-protamine disulfide bonds are likely removed.
Strikingly, the ATAC-seq peaks on the maternal genome from eggs fertilized with the double mutant P1 sperm also largely resembled those in the MII oocyte (Jung et al., 2019), despite the ability of the double mutant-fertilized oocyte to form the maternal pronucleus (indicative of unaffected progression of fertilized MII oocyte beyond metaphase), which is in sharp contrast to vanished ATAC-seq peaks on the maternal genome in eggs fertilized with wild-type and single mutant P1 sperm (Figure 7D). Note that this comparison was made with the normalized data using the same scale. This might explain the inability to detect ATAC-seq peaks at the 1-cell stage, which was thought to result from “failed” ATAC-seq profiling experiments in a previous report (Wu et al., 2016). siSRPK1 treatment mirrored the effects of the double mutant P1 sperm on both paternal and maternal genome reprogramming (Figure 7B–D). Based on these data, we draw two important conclusions: (i) dramatic chromatin remodeling takes place in a highly coordinated fashion in both paternal and maternal pronuclei before they merge, and (ii) SRPK1-catalyzed protamine phosphorylation initiates such synchronized parental genome reprogramming.
DISCUSSION
Based on the evidence presented in this study, we propose a model for a cascade of events in chromatin remodeling shortly after fertilization (Figure 7E). The highly compacted sperm DNA first needs to be opened up by removing inter-protamine disulfide bonds (Emelyanov and Fyodorov, 2016), which exposes protamines for phosphorylation by SRPK1. Protamine phosphorylation may weaken interactions between protamine molecules, as reflected by the reduction of large aggregates to smaller condensates in the presence of SRPK1, and subsequently promote their interactions with various chaperones to mediate the protamine-to-histone exchange on the paternal genome. Importantly, this process appears to be highly synchronized with chromatin remodeling in the maternal genome before fusion of the two gametes.
Protamines as Part of the SR Superfamily and a Splicing-independent Function of SRPK1
The superfamily of SR proteins is characterized by a domain(s) containing multiple Arg/Ser dipeptides (RS domain) flanked by one or two RNA binding domains, which primarily function in constitutive and regulated pre-mRNA splicing (Fu, 1995). All RS domain-containing proteins studied to date are specially recognized by SR protein-specific kinases (SRPKs) to promote their nuclear import, protein-protein interactions, and protein-RNA interactions to facilitate spliceosome assembly (Zhou and Fu, 2013). SRPKs specifically require Arg to catalyze Ser phosphorylation in RS domains (Gui et al., 1994b). Both P1 and P2 contain several RS dipeptides and P1 has been shown to be an excellent substrate for SRPK1 in the in vitro kinase reaction (Papoutsopoulou et al., 1999); however, given the fact that various basic proteins, such as histone H1, often serve as surrogate substrates for kinases, it is our current work that demonstrates that SRPK1-catalyzed protamine phosphorylation is a critical physiological event in early embryonic development. Protamines may thus be considered new members of the SR superfamily of proteins.
Interestingly, SRPK1 is conserved from budding yeast to humans and protamine is also highly conversed among vertebrates, suggesting that protamine and SRPK1-catalyzed protamine phosphorylation may be a universal mechanism for facilitating protamine-to-histone exchange within fertilized eggs in animal kingdom. As a matter of fact, SR proteins have been implicated in multiple cellular activities beyond RNA processing from transcription to RNA export to translational control (Zhong et al., 2009). Our current finding of SRPK1-facilitated chromatin remodeling on the paternal genome points to a potential function of SRPKs at the chromatin level in somatic cells by modulating the regulatory activities of classic SR proteins on chromatin during transcription (Ji et al., 2013; Lin et al., 2008; Mo et al., 2013; Xiao et al., 2019).
Regulation of Protamine-to-Histone Exchange during Spermiogenesis and Fertilization
SRPK1 is highly expressed in testis and oocyte, consistent with its vital roles in both spermiogenesis and preimplantation development of fertilized egg. It is interesting to note that a S56A mutation in P2 has been reported to bypass the requirement for a phosphatase regulator (Hspa4I) to produce functional sperm during spermiogenesis (Itoh et al., 2019). It is well known that histones are sequentially replaced by transition proteins and then by protamines in spermatids (Oliva, 2006), indicating that SRPK1-catalyzed protamine phosphorylation may ensure such sequential packaging of the paternal DNA. However, protamine has to be largely dephosphorylated in the final stage of spermiogenesis, consistent with the lack of immunostaining signals on isolated wild-type sperm with our phospho-specific antibodies, which may enable DNA-dependent phase transition, as we now observed in vitro. According to this likely scenario, we predict that hyper-phosphorylation of protamine, such as those mimicked by Ser-to-Asp mutations in SRPK1 phosphorylation sites, may prevent the production of functional sperm during spermatogenesis, a hypothesis we are actively pursuing.
If dephosphorylation is required for histone-to-protamine exchange in spermatids, our current work demonstrates that this process is potently reversed by SRPK1-catalyzed phosphorylation shortly after fertilization to initiate paternal genome reprogramming. This process requires a dual phosphorylation event in P1. Interestingly, we note that the N-terminus of P1 contains 3 potential phosphorylation sites, only one of which (Ser9) is phosphorylated. Importantly, this phosphorylation site is highly conserved in vertebrates, and in contrast, the C-terminal phosphorylation site (Ser43) is present only in a subset of mammals, but not in humans. We show that phosphorylation at either one of these two sites in mouse P1 is sufficient for promoting the protamine-to-histone exchange. Thus, in the absence of a C-terminal site, the conserved N-terminal phosphorylation site may provide the regulatory function in most species.
It is important to point out that a small number of eggs fertilized by double mutant P1 sperm were still able to develop into 2-cell embryos, which is consistent with the observation that P1S9S43A males could occasionally give live birth. This phenotype likely results from SRPK1-catalyzed P2 phosphorylation and/or the ability of NPM2 to slowly extract protamine from sperm, thus enabling some double mutant sperm to eventually make through the process.
Coordinated, but not Necessarily Coupled, Protamine-to-Histone Exchange
The protamine-to-histone exchange may be a much more complex process than it appears. In the literature, NPM2 has been thought to play a major role in extracting protamine from sperm DNA, but the paternal genome still undergoes efficient decondensation in fertilized oocytes of NPM2 KO mice (Burns et al., 2003), which is likely due to related functions of NPM1 and NPM3 (Okuwaki et al., 2012) and/or TAF1β (Matsumoto et al., 1999). Likewise, the histone chaperone HIRA is responsible for deposing the histone 3 variant H3.3 onto paternal DNA, but this process does not have to be coupled with protamine dismissal, as sperm DNA still undergoes efficient decondensation in HIRA-depleted oocytes (Inoue and Zhang, 2014; Lin et al., 2014).
Interestingly, while purified NPM2 is sufficient to drive protamine removal from sperm chromatin, which is accelerated by SRPK1-catalyzed protamine phosphorylation, we were unable to detect this effect on in vitro assembled P1/DNA particles. This suggests that the P1/DNA condensates may simply be in vitro artifacts, and thus, do not reassemble the conformation of native sperm chromatin. A more interesting alternative scenario is that NPM2 may initiate protamine removal on chromatin regions that harbor retained H3.3 residues, rather than randomly from anywhere. As H3.3 bound chromatin regions correspond to ATAC-seq peaks, suggesting that those accessible regions might provide docking sites for NPM2 to act on, given the ability of NPM2 to interact with both protamines and histones (Ellard et al., 2016). As H3.3 is entirely missing from the in vitro assembled P1/DNA particles, NPM2 may not be accessible to these particles even in the presence of SRPK1. Therefore, as illustrated in Figure 7E, we propose a working model where NPMs may begin to extract protamine from H3.3-containing regions, thus emphasizing a functional requirement for a small fraction of retained histone. SRPK1 not only enhances the interaction of NPMs with sperm chromatin, but also enables phosphorylated protamine to recruit HIRA to deposit H3.3 onto vacated DNA regions. This process may thus be orchestrated to drive protamine-to-histone exchange in a coordinated fashion.
Synchronized Chromatin Remodeling of Both Gametes in Fertilized Egg
One of the most striking findings presented in this work is the coordinated reprogramming between paternal and maternal genomes upon fertilization even though both gametes’ genomes still reside in separate pronuclei. Recent genomic profiling for chromatin state by DNase I hypersensitivity assay (Lu et al., 2016) and ATAC-seq (Wu et al., 2016) revealed a gradual gain of chromatin accessibility during pre-implantation zygotic development, which is associated with progressive formation of topologically associating domains (TADs) (Du et al., 2017) and redistribution of the chromatin mark H3K4me3 (Dahl et al., 2016; Zhang et al., 2016). Interestingly, although the paternal DNA is predominantly packed with protamine whereas the maternal genome still with histones, both genomes show extensively overlapped ATAC-seq peaks, predominantly at TSSs (Jung et al., 2019), indicating that both genomes are poised for activation.
Because PN3 is the earliest stage profiled prior to our work, it remains unclear whether the detected DNase-seq peaks at the PN3 stage are those inherited from sperm and MII oocyte or newly established after fertilization. By performing ATAC-seq profiling right after fertilization between late PN2 and early PN3 stages, we unexpectedly found that both paternal and maternal genomes simultaneously lost ATAC-seq peaks, indicating that both parental genomes undergo a rather complete reprogramming after fertilization. Importantly, by using the double mutant P1 sperm or in response to SRPK1 depletion, we found that ATAC-seq peaks on both paternal and maternal genomes remained, indicative of dynamic chromatin remodeling in both genomes. The sperm-induced maternal genome reprogramming might be related to a curious phenomenon, called hybridogenesis, where fertilization occurs with the sperm from a closely related species, but the paternal DNA is not passed on to the developing embryo, as seen in various frogs and salamander species (Lavanchy and Schwander, 2019), indicating that the incoming sperm may provide a signal to activate the maternal genome. Our current findings have thus laid a new foundation for future studies to understand how the paternal genome communicates with the maternal genome in fertilized egg.
STAR*METHODs
LEAD CONTACT AND MATERIALS AVAILABILITY
Please direct any requests for further information and resources to the Lead Contact, Xiang-Dong Fu (xdfu@ucsd.edu). All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Female heterozygous SRPK1+/− (Wang et al., 2014) and control mice at 3–4 weeks of age were used in this study. P1S9A/+, P1S43A/+, and P1S9S43A/+ knock-in mice were generated by using a CRISPR/Cas9-mediated genome editing approach (Paquet et al., 2016). Specifically, chemically synthesized tracrRNA, crRNA, and single-stranded donor oligonucleotides were co-injected with Cas9 protein (NEB) into the pronuclei of fertilized eggs by the UCSD Transgenic Mouse Core. After in vitro culture for 2 hours, embryos were transferred into the oviducts of pseudo-pregnant ICR females. Offspring genomic DNA was extracted and subjected to PCR and Sanger sequencing for genotyping. Potential off-target sites were selected according to CRISPR Design (http://crispr.mit.edu) and PCR-amplified for sequencing. Male P1 knock-in and control mice at 10–12 weeks of age were used in these analyses. All mouse models were generated in the C57BL/6 background.
All animal work was performed under the guidelines of Institutional Animal Care and Use Committee (IACUC), with protocols (S99116) approved by University of California San Diego, consistent with the Guide for Care and Use of Laboratory Animals, National Research Council, 1996 (institutional animal welfare assurance no. A-3125–01).
Antibodies
Rabbit polyclonal antibodies against P1 phosphorylation at Ser9, Ser11, Ser13, and Ser43 were generated by immunizing rabbits with chemically synthesized P1 peptides with phosphorylation (P9: MARYRCCRSpKSRSRCR; P11: MARYRCCRSKSpRSRCR; P13: YRCCRSKSRSpRCRRRRR; P43: RCCRRRRSpYTIRCKKY; Figure S3E), followed by affinity purification with antigens. Other antibodies used are provided in the Key Resource Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal antibody anti-SRPKl | BD Biosciences | 611072 |
| Mouse monoclonal antibody anti-SRPK2 | BD Biosciences | 611118 |
| Mouse monoclonal antibody anti-Protamine 1 | Briar Patch Biosciences | Hup1N |
| Mouse monoclonal antibody anti-HIRA | Active Motif | 39557 |
| Mouse monoclonal antibody anti-SC35 | Gui et al., 1994a | N/A |
| Goat polyclonal antibody anti-Protamine 1 | Santa Cruz | SC-23107 |
| Rabbit polyclonal antibody anti-Flag | Sigma-Aldrich | F7425 |
| Rabbit polyclonal antibody anti-HA | Cell Signaling | 3724 |
| Rabbit polyclonal antibody anti-SRPK1 | ABclonal | A5854 |
| Rabbit polyclonal anti-P1-pSer9 | This paper | N/A |
| Rabbit polyclonal anti-P1-pSer11 | This paper | N/A |
| Rabbit polyclonal anti-P1-pSer13 | This paper | N/A |
| Rabbit polyclonal anti-P1-pSer43 | This paper | N/A |
| Bacterial and Virus Strains | ||
| BL21 (DE3) competent E.coli | BioLabs | C2527 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Gonadotropin Pregnant Mare Serum (PMSG) | Millipore | 367222 |
| Chorionic Gonadotropin Human (hCG) | Millipore | C1063 |
| EmbryoMax HTF | Millipore | MR-070-D |
| EmbryoMax M2 | Millipore | MR-015-D |
| EmbryoMax M2 with Hyaluronidase | Millipore | MR-051-F |
| EmbryoMax Acidic Tyrode’s Solution | Millipore | MR-004-D |
| EmbryoMax KSOM | Millipore | MR-121-D |
| Aniline Blue solution | Sigma-Aldrich | B8563 |
| Insulin-transferrin-sodium selenite media supplement | Sigma-Aldrich | I1884 |
| Recombinant Human EGF | R&D Systems | 236-EG |
| Chemically synthesized peptides, see Table S1 | This paper | See Table S1 |
| EnGen™ Cas9 NLS | BioLabs | M0646T |
| Critical Commercial Assays | ||
| Nextera DNA Library Prep Kit | Illumina | 15028212 |
| SPRI Beads | Beckman Coulter | B23319 |
| DNA Clean & Concentrator −5 kit | Zymo Research | D4013 |
| Amicon Ultra-0.5 Centrifugal Filter devices | Millipore | UFC500324 |
| Atto 488 protein labeling kit | Sigma-Aldrich | 38371 |
| ULYSIS Nucleic acid labeling kits | Molecular Probes | U21654 |
| Micro Bio-spin columns P-30, Tris | Bio-Rad | 732–6250 |
| Mouse Testis genomic DNA | ZYAGEN | MG-401 |
| HiTrap Columns, CM Sepharose FF (1 mL) | GE Healthcare | 95056–086 |
| HiTrap Columns, DEAE Sepharose FF (1 mL) | GE Healthcare | 95056–084 |
| Dynabeads™ Protein G | ThermoFisher | 10009D |
| Deposited Data | ||
| ATAC-seq data | This paper | GSE136403 |
| Experimental Models: Cell Lines | ||
| Human HEK293T | ATCC | CRL-3216 |
| Oligonucleotides | ||
| ON-TARGET plus Mouse Srpk1 siRNA | Dharmacon | J-061647–08 |
| crRNAs, see Table S2 | This paper | N/A |
| tracrRNA, see Table S2 | This paper | N/A |
| Donor DNA, see Table S2 | This paper | N/A |
| 25, 50, 100 nt Cy3 labelled sense oligos, Table S2 | This paper | N/A |
| 25, 50, 100 nt Antisense oligos, see Table S2 | This paper | N/A |
| Recombinant DNA | ||
| pRSETb-His-SRPK1 | Aubol et al., 2012 | N/A |
| pRSETb-His-SRPK1 (KD) | Aubol et al., 2012 | N/A |
| pET23-hNpm2 | Platonova et al., 2011 | N/A |
| pcDNA3-HA | This paper | N/A |
| pcDNA3-HA-hNpm2 | This paper | N/A |
| pcDNA3-HA-HIRA | Hall et al., 2001 | N/A |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.sov/ij/ |
METHOD DETAILS
In Vitro Fertilization
IVF was performed as described (Sztein et al., 2000). Specifically, sperm were collected from caudal epididymides of 8 to 12-week old male C57BL/6 mice and then capacitated for 1 hour in human tubal fluid (HTF) medium (Millipore) at 37°C in 5% CO2 incubator. Sperm were incubated with intact cumulus-oocyte complexes or MII oocytes for 1 to 4 hours. After removing excess sperm and cumulus lysate, presumptive fertilized eggs were transferred to KSOM medium (Millipore) for further culture and analysis.
For inhibitor treatment, isolated cumulus-oocyte complexes were incubated with M2 medium (Millipore) containing Hyaluronidase (Millipore) to release MII oocytes. After washing, the oocytes were transferred to HTF medium (Millipore) containing SRPKIN-1 or JH-VII-206–2 (Hatcher et al., 2018) suspended in DMSO at indicated concentrations for 1 hour before insemination.
Microinjection of Growing Oocytes and In Vitro Growth
Injection and culture of growing oocytes were performed according to Inoue et al., 2014; Inoue and Zhang, 2014. Specifically, oocyte-granulosa cell complexes were isolated from ovaries of 12-day old C57BL/6 female mice and transferred into α-MEM with GlutaMax (Life Technologies, 32571–036) containing 5% FBS, 100 IU/L PMSG, 5 μg/ml insulin (Sigma-Aldrich), 5 μg/ml transferrin (Sigma-Aldrich), and 5 ng/ml sodium selenite (Sigma-Aldrich). After 1-hour incubation, the complexes were switched to M2 medium (Millipore) for microinjection. 5~10 pL siSRPK1 (20 μM) or control siRNA (both from Dharmacon) was injected into the cytoplasm of growing oocytes with Femtojet (Eppendorf), evident by movement of cytoplasmic granules within the oocyte. For rescue experiments, 100 ng/μl of T7 in vitro transcribed human SRPK1 mRNA was co-injected with mouse siSRPK1. After microinjection, oocytes were transferred to M2 medium containing 2% PVP (MW 360,000, Sigma-Aldrich). Medium was changed every other day (Figure S2). After 12-day in vitro culture, fully grown oocytes surrounded by cumulus cells were transferred to α-MEM supplemented with 5% FBS and 10 ng/ml EGF for meiotic maturation overnight. MII oocytes were transferred to HTF medium (Millipore) and inseminated with capacitated sperm. To detect H3.3 deposition during maternal chromatin remodeling, isolated MII oocytes were injected with 100 ng/μl Flag-H3.3 mRNA T7 in vitro transcribed from linearized pcDNA3.1-Flag-H3.3 construct and purified by lithium chloride precipitation followed by suspension in nuclease-free water.
Immunofluorescence, Aniline Blue Staining, and Electron Microscopy
Zygotes were washed with M2 medium (Millipore) and treated with acidic Tyrode’s solution (Sigma-Aldrich) to remove zona pellucida. Samples were fixed in 4% paraformaldehyde (Sigma-Aldrich) in PBS for 15 min at room temperature. After permeabilization with 0.2% Triton X-100 for 20 min at room temperature (for protamine staining, sperm or zygotes were additionally treated with Tris-HCl pH 9.5 and 25 mM DTT for 10 min after permeabilization), samples were washed in PBS containing 0.1% BSA, blocked in PBS containing 1% BSA and 0.05% Tween-20 for 1 hour, and incubated with the following primary antibodies overnight at 4°C: mouse anti-SRPK1 (1:500, BD Biosciences), rabbit anti-SRPK1 (1:100, ABclonal), mouse anti-SRPK2 (1:250, BD Biosciences), mouse anti-protamine 1 (1:100, Briar Patch Biosciences), goat anti-protamine 1 (1:50, Santa Cruz), rabbit antisera against phosphorylated protamine 1 (1:100, custom raised in this study), mouse anti-HIRA (1:100, Active Motif), rabbit anti-Flag (1:200, Sigma-Aldrich). After washing with PBS containing 0.05% Tween-20 and 0.1% BSA, samples were incubated with second antibodies conjugated to Alexa Fluor 488, Alexa 594 or Alexa 647 (Molecular Probes) for 1 hour at room temperature, washed and stained with DAPI for 5 min. After an additional wash with PBS, Fluoromount-G mounting media (SouthernBiotech) was applied onto glass slides. Images were acquired with Olympus microscope. Staining signal intensity and pronuclear size were quantified by using ImageJ (NIH). For SRPK1 intensity quantification, all the examined zygotes showed a Mean Intensity distribution from 10 to 60, the zygotes were divided into 3 categories based on their Mean Intensity (Negative: 10~25; Intermediate: 25~45; Positive: 45~60).
Aniline blue staining and transmission electron microscopy (TEM) were performed to determine the chromatin compaction of sperm nuclei (Gou et al., 2017). Specifically, sperm from cauda epididymis were harvested, spread on slides and air-dried. Samples were fixed in 4% paraformaldehyde (Sigma-Aldrich) for 30 min and washed with PBS, then treated with 0.2% Triton X-100 for 15 min. Slides were stained using acidic aniline blue staining (5% methyl blue in 100 ml 4% acetic acid, Sigma-Aldrich) for 10 min. TEM was performed at the UCSD EM core: cauda epididymis sections were fixed in 2% paraformaldehyde containing 0.05% glutaraldehyde in 0.15M sodium cacodylate buffer (pH 7.4), and then fixed in 1% osmium tetroxide. Dehydration was carried out in ethanol and the samples were embedded in Durcupan. Ultrathin sections were counterstained with uranyl acetate and lead citrate, and images were captured on JEOL 1400 plus TEM at 80KV with Gatan 4k×4k camera.
SRPK1 Purification and In Vitro Phosphorylation Reactions
Wild-type and kinase-dead SRPK1 were expressed from the pRSETb vector containing a 6×HisTag at the N-terminus and purified (Aubol et al., 2012). Protamine phosphorylation (Supplemental Table S1) with 10 μM purified SRPK1 in vitro was carried out in reaction buffer (100 mM Mops-pH 7.4, 2 mM DTT, 10 mM MgCl, and 50 μM 32P-ATP) at 37°C for 1 hour. All reactions were carried out in a total reaction volume of 10 μL and then were quenched with 90 μL 30% Acetic Acid. Phosphorylated protamine (50 μL) was separated from unreacted 32P-ATP by spotting onto a charged paper filter dish (Whatman grade P81), gently washed three times with 0.5% o-phosphoric acid (Fisher Scientific), rinsed with acetone, dried, and counted on the 32P channel in liquid scintillation counter. The total amount of phosphate incorporated into the substrate was determined by considering the specific activity (cpm/min) of the reaction mixture and the background retention of 32P-ATP in the absence of SRPK1.
Preparation of Fluorescence-Labeled DNA and Protein
Mouse testis genomic DNA (ZYAGEN) was labeled with Alexa Fluor 594 using ULYSIS Nucleic Acid Labeling Kits (Molecular Probes), according to manufacturer’s protocol. Specifically, 1 μg of sperm DNA was incubated with Alexa Fluor 594 in labeling buffer (5 mM Tris-Cl pH 8.0 and 1 mM EDTA pH 8.0) for 15 min at 80°C without a denaturation step. Labeled sperm DNA was purified using Micro Bio-Spin P-30 Gel column (Bio-Rad). 25nt and 50nt Cy3-labeled and non-labeled DNA oligonucleotides (Integrated DNA Technologies), and 100nt Cy3-labeled DNA oligonucleotide (Sigma-Aldrich) were chemically synthesized (Supplemental Table S2). To generate double-strand DNA, sense and antisense single-stranded DNA oligonucleotide were annealed for 5 min at 95°C in annealing buffer (20 mM Tris-Cl pH 7.5 and 50 mM NaCl) followed by ramping down from 95°C to 25°C at 0.1C/sec.
Protamine peptides were fluorescently labeled using Atto 488 Protein Labeling Kit (Sigma-Aldrich), according to manufacturer’s protocol. Specifically, 0.35 μg of chemically synthesized peptide was incubated with Atto 488 reactive dye in sodium bicarbonate buffer for 5 hours at 4°C. During the reaction, the mixture was gently rotated and shielded from light. Excess dye was removed by multiple rounds of washing with wash buffer (20 mM Tris-Cl pH 7.5 and 150 mM NaCl) with the 3K Amicon Centrifugal Filter (Millipore).
In Vitro Phase Separation Assay and FRAP Assay
Phase separation of Protamine 1 (10% Atto 488-labeled) with sperm DNA (1% Alexa Fluor 594-labeled) or DNA in different lengths (1% Cy3-labeled) was performed in phase separation buffer (20 mM Tris-Cl pH 7.5 and 75 mM NaCl). After incubation for 10 min, gel-like particles were imaged under Olympus microscope. For SRPK1 treatment, gel-like particles were generated by mixing 50 μM of Protamine 1 (10% Atto 488-labeled) and 350 ng/μl of sperm DNA (1% Alexa Fluor 594-labeled) in buffer (2 mM DTT, 20 mM Tris-Cl pH 7.5, 10 mM MgCl2, 100 μM ATP, and 30 mM NaCl). After incubation for 5 min, 10 μM wild-type or kinase-dead SRPK1 was added to the mixture followed by further incubation for 30 min at 37°C.
Fluorescence recovery after photobleaching (FRAP) was performed on Olympus microscope equipped with 488 nm laser. Photobleaching was performed for 10 sec using 30% laser power and images were captured every 30 sec. Fluorescence intensity was measured by ImageJ. Background signal was subtracted and the intensity of bleached spot was normalized to non-bleached control spot at each time point. The normalized intensity of bleached spot was compared with the intensity of pre-bleaching time point.
NPM Purification and Treatment of Sperm Nuclei
The human NPM2 expression unit was from the Akey lab (Platonova et al., 2011). Recombinant NPM2 was expressed in E. coli BL21 cells and purified. Specifically, NPM2 expressing BL21 (DE3) cells were cultured in 400 ml 2×YT medium supplemented with 0.5% glycerol, 0.2% lactose and 2 mM MgCl2 for 20 hours at 37°C with shaking at 200 rpm. After harvesting cells at 4°C, the pellet was lysed for 1 hour on ice in lysis buffer (50 mM Tris-HCl pH 8.0, 100 mM NaCl, 5 mM EDTA, 5 mM 2-mercaptoethanol, 1 mM PMSF, 1.3 mg/ml lysozyme and 1.3 mg/ml deoxycholic acid). After removing insoluble components by centrifugation (23,000g) for 15 min, the supernatant was incubated on ice for 8 hours. Lysis buffer was added to the supernatant up to 37 ml and then centrifuged at 10,000g for 30 min. The supernatant was transferred to a new 50 ml tube, incubated at 80°C for 10 min, and then immediately placed on ice for 20 min. After centrifugation (10,000g) for 30 min to remove denatured insoluble components, the supernatant was concentrated and desalted by centrifugation (4°C, 7,500g) for 40 min.
Sperm nuclei were extracted and treated with purified NPM2, as described previously (Shintomi et al., 2017; Yamaguchi et al., 2018). Specifically, sperm tails were removed by sonication for 10 cycles (on: 0.5s; off: 5.0s). After washing with PBS, sperm were permeabilized with streptolysin-O (10 units/mL, Sigma-Aldrich) and disulfide bond reduced with DTT (2 mM). About 107 sperm were incubated with the phosphorylation reaction buffer (2 mM DTT, 100 mM Tris-Cl pH 7.5, 10 mM MgCl2, 50 μM ATP) with 10 μM wild-type or kinase-dead SRPK1 at 37°C for 30 min. After washing with PBS, sperm were resuspended with 1 mL treatment buffer (2.5 mM MgCl2, 5 mM sodium butyrate, 20 mM HEPES-KOH pH 7.7, 100 mM KCl, 10 mM DTT, Complete, and EDTA free) containing 100 to 1,000 μM NPM2 and incubated for 15 to 120 min at 37°C on a ThermoMixer (Eppendorf). After incubation, NPM2-treated sperm were pelleted at 20,400g for 5 min at 4°C for microscopic analysis.
In Vitro Binding Assay
HA-tagged human NPM2 or HIRA proteins were pulled down from transfected 293T cell lysates with anti-HA antibody-conjugated Protein G beads (ThermoFisher). Protamine peptides were incubated with phosphorylation reaction buffer (2 mM DTT, 100 mM Tris-Cl pH 7.5, 10 mM MgCl2, 50 μM ATP) with 10 μM wild-type or kinase-dead SRPK1 at 37°C for 30 min. After quenching the reaction with 5% acetic acid, protamine peptides were incubated with beads at room temperature for 30 min. After washing the beads with washing buffer (50 mM Tris-HCl (pH 7.4), 0.1% Triton X-100, 500 mM NaCl, 5 mM EDTA, proteinase inhibitor cocktail), the samples were analyzed by SDS-PAGE for NPM or HIRA. Protamine samples were dissolved in buffer (0.9N acetic acid, 0.5M 2-mercaptoethanol, 20% sucrose) and separated by electrophoresis in Acid Urea Gel containing 2.5M urea (Waterborg, 2002).
ATAC-seq Library Generation and Sequencing
Sperm were harvested from caudal epididymides of wild-type or knock-in male mice in the C57BL/6 background. MII oocytes for insemination were isolated from superovulated BALB/cJ females. Five hours post insemination, fertilized eggs (confirmed by pronucleus formation under microscope) were harvested for ATAC-seq. Generation of ATAC-seq libraries was according to omniATAC-seq (Corces et al., 2017). Specifically, fertilized zygote or sperm were incubated in cold resuspension buffer (10 mM Tris-Cl pH 7.4, 10 mM NaCl, and 3 mM MgCl2) containing 0.1% NP-40, 0.1% Tween-20, and 0.01% digitonin on ice for 3 min and then washed with cold resuspension buffer containing 0.1% Tween-20. Tagmentation was next performed with Tn5 transposon enzyme (Illumina) in tagmentation buffer (20 mM Tris-Cl pH 7.4, 10 mM MgCl2, 20% dimethyl formamide, 0.1% Tween-20, and 0.01% digitonin) for 30 min at 37°C on a thermomixer with shaking at 1,000 rpm. Tagmentated DNA was purified using DNA Clean & Concentrator-5 Kit (Zymo Research). PCR was performed to amplify each library for 14 cycles. After purification using DNA Clean & Concentrator-5 Kit (Zymo Research), DNA fragments between 200- to 700-bp in length were selected by double-selection (0.55X and 1.5X) on SPRI beads (Beckman Coulter) for constructing ATAC-seq libraries followed by pair-end sequencing on HiSeq4000 (Illumina) in the UCSD Genomics Core.
Quantifications and Statistical Analysis
Statistical parameters were reported either in individual figures or corresponding figure legends. Quantified data were in general presented as bar/line plots, with the error bar corresponding to mean±SEM or boxplot, showing the median (middle line), first and third quartiles (box boundaries), and furthest observation or 1.5 times of the interquartile (end of whisker). All statistical analyses were done in R. Whenever asterisks are used to indicate statistical significance, *stands for P <0.05; ** for P <0.01, and *** for P <0.001.
Data and Code Availability
The raw data FASTQ files and processed BigWig files for all sequencing data have been deposited in NCBI GEO under GSE136403.
Supplementary Material
Figure S1. Chemical and genetic evidence for the requirement of SRPK1 to catalyze paternal genome decondensation, Related to Figure 1
(A) Staining of SRPK1 (green) and SC35 (red) in GV oocyte in comparison with MII oocyte. Scale bar, 20 μm.
(B) Positive staining of SRPK1 and negative staining of SRPK2 in MII oocytes. Stained oocytes with only secondary antibody served as negative control.
(C)Chemistry structures of the SRPK1 inhibitor SRPKIN-1 and a related compound lacking detectable inhibitory activity against SRPK1.
(D) Dosage-dependent effect of SRPKIN-1 on early zygotic development from 3 independent experiments.
(E) Dosage-dependent effect of SRPKIN-1 on paternal genome decondensation in fertilized eggs from 3 independent experiments. Right: Quantified data displayed by bar graph.
(F) Impact of genetic ablation of SRPK1 on early zygotic development from 3 independent experiments. Right: Quantified data displayed by bar graph with indicated p-value.
(G) Correlation between remaining SRPK1 immunostaining signals and degree of paternal genome decondensation in SRPK1-ablated oocytes. Right: Quantified data displayed by bar graph with indicated p-value.
Figure S2. In vitro growth of isolated oocytes for siRNA-mediated SRPK1 deletion and functional rescue with siRNA-resistant SRPK1 mRNA, Related to Figure 2
(A) Representative images of growing oocytes after in vitro culture for different days. Bottom panels: matured oocyte at the GV and MII stages.
(B) Impact of SRPK1 depletion on zygotic development and functional rescue with SRPK1 mRNA.
Figure S3. Mapping SRPK1 phosphorylation in P1 and P2 and evidence for SRPK1-mediated phosphorylated P1 in vivo, Related to Figure 3
(A) Sequence of P2 and three chemically synthetic fragments and their mutant versions for in vitro phosphorylation with SRPK1. Green stars denote the mapped phosphorylation sites.
(B,C) Phosphorylation contents (B) and reaction velocity (C) of SRPK1-catalyzed phosphorylation on wild-type and mutant P1.
(D) Kinetics of SRPK1-catalyzed phosphorylation on wild-type and mutant P2 fragments.
(E,F) P1 peptides used to raise phospho-specific antibodies in rabbits (E) and immunoblot analysis of affinity-purified anti-phosphoantibodies on individual Protamine peptides (F).
(G) Time-course analysis of P1 phosphorylation at Ser43 after in vitro fertilization.
(H) Lack of evidence for P1 phosphorylation at Ser11 and Ser13 in vivo.
(I) Rescue of P1 phosphorylation at Ser43 with active SRPK1, but not the kinase-dead mutant, in siSRPK1-treated oocyte.
Figure S4. Engineering knock-in mutations of SRPK1 phosphorylation sites in P1 and functional impact on sperm production, Related Figure 4
(A,B) Design of specific gRNAs to target Ser9 and Ser43 in P1 (A) and confirmation of specific mutations introduced in mouse germ line by Sanger sequencing (B).
(C) Potential off-targets PCR-amplified and sequenced to show the lack of detectable off-target effects.
(D) Sperm counts from males of wild-type, single, and double P1 mutant.
(E) Aniline blue staining of cauda epididymal sperm from dpp 12-week mice. Arrows indicate adjacent somatic cells that are efficiently stained by aniline blue, which served as positive control. Scale bar, 10 μm.
(F) Representative images of transmission electron micrographs of wild-type and double mutant sperm heads from dpp 12-week mice. Scale bar, 1 μm
Figure S5. P1 phase separation in the presence of DNA and impact of SRPK1-catalyzed phosphorylation on the formation of mesh-like networks, Related to Figure 5
(A,B) Gel mobility shift assay for P1 interaction with dsDNA (A) and formation of insoluble material (B).
(C) Analysis of DNA concentration-dependent (left) and DNA length-dependent effect on P1 phase transition. Different degrees of phase separation are indicated by the keys on the right.
(D,E) SRPK1 still prevented phase transition of single mutant at Ser9 (D) or Ser43 (E) from small particles to mesh-like structures.
Figure S6. In vitro sperm decondensation with purified NPM2, Related to Figure 6
(A) Scheme for the in vitro sperm decondensation assay. Isolated sperm were permeabilized with SLO and disulfide bonds removed with DTT for the decondensation assay with purified human NPM2. Retained P1 on paternal chromatin was quantified by Western blotting.
(B) Purification of recombinant human NPM2 expressed in bacteria.
(C) Human NPM2-mediated sperm decondensation and P1 release over an extended period of treatment. Retained P1 on sperm DNA was quantified at bottom. The H3K4me3 signal on paternal chromatin served as control.
(D) Representative images of sperm DNA (DAPI staining) (left panel) and western blot of retained P1 on sperm chromatin (right panel) at the indicated SRPK1 treatment time points Scale bar, 10 μm. Total and specifically phosphorylated P1 were blotted with specific antibodies.
(E) Left, representative images of fertilized eggs stained with anti-SRPK1 (green) and DAPI (blue) ~10 hours post insemination. Top and bottom panels show oocytes injected with scrambled (Scr) siRNA and siSRPK1, respectively. P, paternal DNA; M, maternal DNA; PBs, polar bodies. Scale bar, 20 μm. Right, relative SRPK1 staining signals (top) and relative values of pronuclear size (paternal/maternal) (bottom) in fertilized eggs.
(F) Left, representative images of fertilized eggs stained with anti-P1 (red) and DAPI (blue) ~10 hours post insemination. Top and bottom panels show oocytes fertilized with wild-type and double P1 mutant sperm, respectively. P, paternal DNA; M, maternal DNA; PB, polar body. Scale bar, 20 μm. Right, relative P1 staining signals (top) and relative values of pronuclear size (paternal/maternal) (bottom) in fertilized eggs.
(G) Alexa 488-labeled P1 and Alexa 594-labled DNA were mixed to form condensates in vitro followed by testing the potential effect of purified NPM2 on these condensates in the presence of active or kinase-dead SRPK1. The data indicate that the condensates remain a gel-like structure even in the presence of NPM2 and active SRPK1.
Figure S7. Global analysis of chromatin accessibility by ATAC-seq on sperm and fertilized egg, Related to Figure 7
(A) ATAC-seq libraries generated. Shown are total mapped reads, reads after removing those mapped to mitochondrial DNA (-ChrM), and reads uniquely mapped to the paternal and maternal genomes based on SNPs.
(B,C) Reproducibility of duplicated ATAC-seq libraries (B) and comparison between our libraries and with the public ATAC-seq data on wild-type sperm (C).
(D,E) Cross-assignment of maternal (BALB/cJ) and paternal (C57BL/6) reads to C57BL/6 and BALB/cJ genomes, respectively.
(F,G) Mapping rates of ATAC-seq reads to the paternal and maternal genomes (F) and relative reads density (Paternal over Maternal) distribution among top 10,000 DNA bins (G), showing significantly reduced ATAC-seq signals on the paternal genome relative to the maternal genome with double mutant-fertilized eggs.
(H,I) Overlap of our ATAC-seq peaks on wild-type, single and double mutant sperm with the public H3.3 ChIP-seq signals on wild-type sperm (H) and the distribution of ATAC-seq signals at TSS regions (I).
Highlights.
SRPK1 phosphorylates protamine to initiate paternal genome reprogramming
Phosphorylation site mutations in protamine inhibit protamine-to-histone exchange
Phosphorylated protamine permits efficient recruitment of NPM2 and HIRA
Paternal and maternal genomes undergo synchronized reprogramming upon fertilization
ACKNOWLEDGMENTS
This project was supported by grants from NIH (GM052872 and HG004659) to X-D.F. The authors are grateful to members of the Fu lab for cooperation, reagent sharing, and insightful discussion. We thank Dr. Christopher Akey of Boston University for the gift of NPM2 vectors; Dr. Peter Adams of Sanford Burnham Prebys Medical Discovery Institute for the gift of the HA-HIRA vector; Dr. Bang-An Wang of the Salk Institute, Dr. Man Zhang of GRMH-GDL, Dr. Chen Chu of Harvard Medical School, Drs. Ian Huggins and Marten Hoeksema of UC San Diego for helpful comments on manuscript and experimental assistance. J.Z. and P.L.M. were partially supported by P30 DK063491, P30 CA023100, and P42 ES010337. B.E.A and J.A.A were supported by GM067969. L-T.G. and this research were supported by NICHD under award number K99HD094901.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing financial interests.
REFERENCES
- Adenot PG, Szollosi MS, Geze M, Renard JP, and Debey P (1991). Dynamics of Paternal Chromatin Changes in Live One-Cell Mouse Embryo after Natural Fertilization. Mol Reprod Dev 28, 23–34. [DOI] [PubMed] [Google Scholar]
- Aubol BE, Plocinik RM, McGlone ML, and Adams JA (2012). Nucleotide Release Sequences in the Protein Kinase SRPK1 Accelerate Substrate Phosphorylation. Biochemistry-Us 51, 6584–6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balhorn R (1982). A Model for the Structure of Chromatin in Mammalian Sperm. J Cell Biol 93, 298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balhorn R (1989) Mammalian Protamines: Structure and Molecular Interactions In: Adolph KW (eds) Molecular Biology of Chromosome Function. Springer, New York, NY [Google Scholar]
- Balhorn R (2007). The protamine family of sperm nuclear proteins. Genome Biol 8, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banuelos S, Omaetxebarria MJ, Ramos I, Larsen MR, Arregi I, Jensen ON, Arizmendi JM, Prado A, and Muga A (2007). Phosphorylation of both nucleoplasmin domains is required for activation of its chromatin decondensation activity. J Biol Chem 282, 21213–21221. [DOI] [PubMed] [Google Scholar]
- Bouniol-Baly C, Hamraoui L, Guibert J, Beaujean N, Szollosi MS, and Debey P (1999). Differential transcriptional activity associated with chromatin configuration in fully grown mouse germinal vesicle oocytes. Biol Reprod 60, 580–587. [DOI] [PubMed] [Google Scholar]
- Burns KH, Viveiros MM, Ren YS, Wang P, DeMayo FJ, Frail DE, Eppig JJ, and Matzuk MM (2003). Roles of NPM2 in chromatin and nucleolar organization in oocytes and embryos. Science 300, 633–636. [DOI] [PubMed] [Google Scholar]
- Chirat F, Arkhis A, Martinage A, Jaquinod M, Chevaillier P, and Sautiere P (1993). Phosphorylation of Human Sperm Protamines Hp1 and Hp2 - Identification of Phosphorylation Sites. Biochim Biophys Acta 1203, 109–114. [DOI] [PubMed] [Google Scholar]
- Colwill K, Feng LL, Yeakley JM, Gish GD, Caceres JF, Pawson T, and Fu XD (1996). SRPK1 and Clk/Sty protein kinases show distinct substrate specificities for serine/arginine-rich splicing factors. J Biol Chem 271, 24569–24575. [DOI] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl JA, Jung I, Aanes H, Greggains GD, Manaf A, Lerdrup M, Li G, Kuan S, Li B, Lee AY, et al. (2016). Broad histone H3K4me3 domains in mouse oocytes modulate maternal-to-zygotic transition. Nature 537, 548–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding JH, Zhong XY, Hagopian JC, Cruz MM, Ghosh G, Feramisco J, Adams JA, and Fu XD (2006). Regulated cellular partitioning of SR protein-specific kinases in mammalian cells. Mol Biol Cell 17, 876–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, Zheng H, Huang B, Ma R, Wu J, Zhang X, He J, Xiang Y, Wang Q, Li Y, et al. (2017). Allelic reprogramming of 3D chromatin architecture during early mammalian development. Nature 547, 232–235. [DOI] [PubMed] [Google Scholar]
- Ellard K, Serpa JJ, Petrotchenko EV, Borchers CH, and Ausio J (2016). Expression and purification of the full murine NPM2 and study of its interaction with protamines and histones. Biochem Biophys Rep 6, 165–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emelyanov AV, and Fyodorov DV (2016). Thioredoxin-dependent disulfide bond reduction is required for protamine eviction from sperm chromatin. Gene Dev 30, 2651–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu XD (1995). The Superfamily of Arginine Serine-Rich Splicing Factors. Rna 1, 663–680. [PMC free article] [PubMed] [Google Scholar]
- Giannakouros T, Nikolakaki E, Mylonis I, and Georgatsou E (2011). Serine-arginine protein kinases: a small protein kinase family with a large cellular presence. Febs J 278, 570–586. [DOI] [PubMed] [Google Scholar]
- Gou LT, Kang JY, Dai P, Wang X, Li F, Zhao S, Zhang M, Hua MM, Lu Y, Zhu Y, et al. (2017). Ubiquitination-Deficient Mutations in Human Piwi Cause Male Infertility by Impairing Histone-to-Protamine Exchange during Spermiogenesis. Cell 169, 1090–1104 e1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu TP, Guo F, Yang H, Wu HP, Xu GF, Liu W, Xie ZG, Shi LY, He XY, Jin SG, et al. (2011). The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 477, 606–U136. [DOI] [PubMed] [Google Scholar]
- Gui JF, Lane WS, and Fu XD (1994a). A Serine Kinase Regulates Intracellular-Localization of Splicing Factors in the Cell Cycle. Nature 369, 678–682. [DOI] [PubMed] [Google Scholar]
- Gui JF, Tronchere H, Chandler SD, and Fu XD (1994b). Purification and Characterization of a Kinase Specific for the Serine-Rich and Arginine-Rich Pre-Messenger-Rna Splicing Factors. Proc Natl Acad Sci USA 91, 10824–10828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall C, Nelson DM, Ye X, Baker K, DeCaprio JA, Seeholzer S, Lipinski M, and Adams PD (2001). HIRA, the human homologue of yeast Hir1p and Hir2p, is a novel cyclin-cdk2 substrate whose expression blocks S-phase progression. Mol Cell Biol 21, 1854–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatcher JM, Wu G, Zeng C, Zhu J, Meng F, Patel S, Wang W, Ficarro SB, Leggett AL, Powell CE, et al. (2018). SRPKIN-1: A Covalent SRPK1/2 Inhibitor that Potently Converts VEGF from Pro-angiogenic to Anti-angiogenic Isoform. Cell Chem Biol 25, 460–470 e466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Ogushi S, Saitou M, Suzuki MG, and Aoki F (2011). Involvement of Mouse Nucleoplasmin 2 in the Decondensation of Sperm Chromatin after Fertilization. Biol Reprod 85, 70–77. [DOI] [PubMed] [Google Scholar]
- Inoue A, Sunaga K, Aoki F, and Zhang Y (2014). siRNA-Mediated Depletion of Stable Proteins in Mouse Oocytes. Protocol Exchange (July 21). [Google Scholar]
- Inoue A, and Zhang Y (2014). Nucleosome assembly is required for nuclear pore complex assembly in mouse zygotes. Nat Struct Mol Biol 21, 609–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Kondoh G, Miyachi H, Sugai M, Kaneko Y, Kitano S, Watanabe H, Maeda R, Imura A, Liu Y, et al. (2019). Dephosphorylation of protamine 2 at serine 56 is crucial for murine sperm maturation in vivo. Sci Signal 12, eaao7232. [DOI] [PubMed] [Google Scholar]
- Ji X, Zhou Y, Pandit S, Huang JE, Li HR, Lin CY, Xiao R, Burge CB, and Fu XD (2013). SR Proteins Collaborate with 7SK and Promoter-Associated Nascent RNA to Release Paused Polymerase. Cell 153, 855–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung YH, Kremsky I, Gold HB, Rowley MJ, Punyawai K, Buonanotte A, Lyu X, Bixler BJ, Chan AWS, and Corces VG (2019). Maintenance of CTCF- and Transcription Factor-Mediated Interactions from the Gametes to the Early Mouse Embryo. Mol Cell 75, 154–171 e155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung YH, Sauria MEG, Lyu XW, Cheema MS, Ausio J, Taylor J, and Corces VG (2017). Chromatin States in Mouse Sperm Correlate with Embryonic and Adult Regulatory Landscapes. Cell Rep 18, 1366–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konev AY, Tribus M, Park SY, Podhraski V, Lim CY, Emelyanov AV, Vershilova E, Pirrotta V, Kadonaga JT, Lusser A, et al. (2007). CHD1 motor protein is required for deposition of histone variant h3.3 into chromatin in vivo. Science 317, 1087–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavanchy G, and Schwander T (2019). Hybridogenesis. Curr Biol 29, R9–R11. [DOI] [PubMed] [Google Scholar]
- Lee CH, and Cho YH (1999). Aspects of mammalian spermatogenesis: Electrophoretical analysis of protamines in mammalian species. Mol Cells 9, 556–559. [PubMed] [Google Scholar]
- Lee MT, Bonneau AR, and Giraldez AJ (2014). Zygotic Genome Activation During the Maternal-to-Zygotic Transition. Annu Rev Cell Dev Bi 30, 581–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leno GH, Mills AD, Philpott A, and Laskey RA (1996). Hyperphosphorylation of nucleoplasmin facilitates Xenopus sperm decondensation at fertilization. J Biol Chem 271, 7253–7256. [DOI] [PubMed] [Google Scholar]
- Lin CJ, Koh FM, Wong P, Conti M, and Ramalho-Santos M (2014). Hira-Mediated H3.3 Incorporation Is Required for DNA Replication and Ribosomal RNA Transcription in the Mouse Zygote. Dev Cell 30, 268–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SR, Coutinho-Mansfield G, Wang D, Pandit S, and Fu XD (2008). The splicing factor SC35 has an active role in transcriptional elongation. Nat Struct Mol Biol 15, 819–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SR, and Fu XD (2007). SR proteins and related factors in alternative splicing. Adv Exp Med Biol 623, 107–122. [DOI] [PubMed] [Google Scholar]
- Link VM, Duttke SH, Chun HB, Holtman IR, Westin E, Hoeksema MA, Abe Y, Skola D, Romanoski CE, Tao J, et al. (2018). Analysis of Genetically Diverse Macrophages Reveals Local and Domain-wide Mechanisms that Control Transcription Factor Binding and Function. Cell 173, 1796–1809 e1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loppin B, Bonnefoy E, Anselme C, Laurencon A, Karr TL, and Couble P (2005). The histone H3.3 chaperone HIRA is essential for chromatin assembly in the male pronucleus. Nature 437, 1386–1390. [DOI] [PubMed] [Google Scholar]
- Lu FL, Liu YT, Inoue A, Suzuki T, Zhao KJ, and Zhang Y (2016). Establishing Chromatin Regulatory Landscape during Mouse Preimplantation Development. Cell 165, 1375–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto K, Nagata K, Miyaji-Yamaguchi M, Kikuchi A, and Tsujimoto M (1999). Sperm chromatin decondensation by template activating factor I through direct interaction with basic proteins. Mol Cell Biol 19, 6940–6952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo S, Ji X, and Fu XD (2013). Unique role of SRSF2 in transcription activation and diverse functions of the SR and hnRNP proteins in gene expression regulation. Transcription 4, 251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa O, Arnold M, Nakagawa M, Hamada H, Shelton JM, Kusano H, Harris TM, Childs G, Campbell KP, Richardson JA, et al. (2005). Centronuclear myopathy in mice lacking a novel muscle-specific protein kinase transcriptionally regulated by MEF2. Gene Dev 19, 2066–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuwaki M, Sumi A, Hisaoka M, Saotome-Nakamura A, Akashi S, Nishimura Y, and Nagata K (2012). Function of homo- and hetero-oligomers of human nucleoplasmin/nucleophosmin family proteins NPM1, NPM2 and NPM3 during sperm chromatin remodeling. Nucleic Acids Res 40, 4861–4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva R (2006). Protamines and male infertility. Hum Reprod Update 12, 417–435. [DOI] [PubMed] [Google Scholar]
- Papoutsopoulou S, Nikolakaki E, Chalepakis G, Kruft V, Chevaillier P, and Giannakouros T (1999). SR protein-specific kinase 1 is highly expressed in testis and phosphorylates protamine 1. Nucleic Acids Res 27, 2972–2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquet D, Kwart D, Chen A, Sproul A, Jacob S, Teo S, Olsen KM, Gregg A, Noggle S, and Tessier-Lavigne M (2016). Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 533, 125–129. [DOI] [PubMed] [Google Scholar]
- Philpott A, and Leno GH (1992). Nucleoplasmin Remodels Sperm Chromatin in Xenopus Egg Extracts. Cell 69, 759–767. [DOI] [PubMed] [Google Scholar]
- Philpott A, Leno GH, and Laskey RA (1991). Sperm Decondensation in Xenopus Egg Cytoplasm Is Mediated by Nucleoplasmin. Cell 65, 569–578. [DOI] [PubMed] [Google Scholar]
- Pirhonen A, Linnalakankkunen A, and Maenpaa PH (1994). P2 Protamines Are Phosphorylated in-Vitro by Protein-Kinase-C, Whereas P1 Protamines Prefer Camp-Dependent Protein-Kinase - a Comparative-Study of 5 Mammalian-Species. Eur J Biochem 223, 165–169. [DOI] [PubMed] [Google Scholar]
- Pirhonen A, Valtonen P, Linnalakankkunen A, and Maenpaa PH (1993). Invitro Phosphorylation Sites of Stallion and Bull P1-Protamines for Cyclic Adenosine 3’,5’-Monophosphate-Dependent Protein-Kinase and Protein-Kinase-C. Biol Reprod 48, 821–827. [DOI] [PubMed] [Google Scholar]
- Platonova O, Akey IV, Head JF, and Akey CW (2011). Crystal Structure and Function of Human Nucleoplasmin (Npm2): A Histone Chaperone in Oocytes and Embryos. Biochemistry-Us 50, 8078–8089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado A, Ramos I, Frehlick LJ, Muga A, and Ausio J (2004). Nucleoplasmin: a nuclear chaperone. Biochem Cell Biol 82, 437–445. [DOI] [PubMed] [Google Scholar]
- Shintomi K, Inoue F, Watanabe H, Ohsumi K, Ohsugi M, and Hirano T (2017). Mitotic chromosome assembly despite nucleosome depletion in Xenopus egg extracts. Science 356, 1284–1287. [DOI] [PubMed] [Google Scholar]
- Shintomi K, Takahashi TS, and Hirano T (2015). Reconstitution of mitotic chromatids with a minimum set of purified factors. Nat Cell Biol 17, 1014–U1336. [DOI] [PubMed] [Google Scholar]
- Spector DL, Fu XD, and Maniatis T (1991). Associations between Distinct Pre-Messenger-Rna Splicing Components and the Cell-Nucleus. Embo J 10, 3467–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztein JM, Farley JS, and Mobraaten LE (2000). In vitro fertilization with cryopreserved inbred mouse sperm. Biol Reprod 63, 1774–1780. [DOI] [PubMed] [Google Scholar]
- Valcarcel J, Gaur RK, Singh R, and Green MR (1996). Interaction of U2AF65 RS region with pre-mRNA branch point and promotion of base pairing with U2 snRNA [corrected]. Science 273, 1706–1709. [DOI] [PubMed] [Google Scholar]
- Wang HY, Lin W, Dyck JA, Yeakley JM, Zhou SY, Cantley LC, and Fu XD (1998). SRPK2: A differentially expressed SR protein-specific kinase involved in mediating the interaction and localization of pre-mRNA splicing factors in mammalian cells. J Cell Biol 140, 737–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang PP, Zhou ZH, Hu AC, de Albuquerque CP, Zhou Y, Hong LX, Sierecki E, Ajiro M, Kruhlak M, Harris C, et al. (2014). Both Decreased and Increased SRPK1 Levels Promote Cancer by Interfering with PHLPP- Mediated Dephosphorylation of Akt. Mol Cell 54, 378–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterborg JH (2002). Acid-urea-triton polyacrylamide gel electrophoresis of histones In The Protein Protocols Handbook, Walker JM, ed. (Totowa, NJ: Humana Press; ), pp. 113–123. [Google Scholar]
- Wossidlo M, Arand J, Sebastiano V, Lepikhov K, Boiani M, Reinhardt R, Scholer H, and Walter J (2010). Dynamic link of DNA demethylation, DNA strand breaks and repair in mouse zygotes. Embo J 29, 1877–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Huang B, Chen H, Yin Q, Liu Y, Xiang Y, Zhang B, Liu B, Wang Q, Xia W, et al. (2016). The landscape of accessible chromatin in mammalian preimplantation embryos. Nature 534, 652–657. [DOI] [PubMed] [Google Scholar]
- Wu JY, Ribar TJ, Cummings DE, Burton KA, McKnight GS, and Means AR (2000). Spermiogenesis and exchange of basic nuclear proteins are impaired in male germ cells lacking Camk4. Nat Genet 25, 448–452. [DOI] [PubMed] [Google Scholar]
- Wu J, Xu J, Liu B, Yao G, Wang P, Lin Z, Huang B, Wang X, Li T, Shi S, et al. (2018). Chromatin analysis in human early development reveals epigenetic transition during ZGA. Nature 557, 256–260. [DOI] [PubMed] [Google Scholar]
- Xia W, Xu J, Yu G, Yao G, Xu K, Ma X, Zhang N, Liu B, Li T, Lin Z, et al. (2019). Resetting histone modifications during human parental-to-zygotic transition. Science 365, 353–360. [DOI] [PubMed] [Google Scholar]
- Xiao R, Chen JY, Liang Z, Luo D, Chen G, Lu ZJ, Chen Y, Zhou B, Li H, Du X, et al. (2019). Pervasive Chromatin-RNA Binding Protein Interactions Enable RNA-Based Regulation of Transcription. Cell 178, 107–121 e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Barr CL, Kim A, Yue F, Lee AY, Eubanks J, Dempster EL, and Ren B (2012). Base-Resolution Analyses of Sequence and Parent-of-Origin Dependent DNA Methylation in the Mouse Genome. Cell 148, 816–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi K, Hada M, Fukuda Y, Inoue E, Makino Y, Katou Y, Shirahige K, and Okada Y (2018). Re-evaluating the Localization of Sperm-Retained Histones Revealed the Modification-Dependent Accumulation in Specific Genome Regions. Cell Rep 23, 3920–3932. [DOI] [PubMed] [Google Scholar]
- Zhang B, Zheng H, Huang B, Li W, Xiang Y, Peng X, Ming J, Wu X, Zhang Y, Xu Q, et al. (2016). Allelic reprogramming of the histone modification H3K4me3 in early mammalian development. Nature 537, 553–557. [DOI] [PubMed] [Google Scholar]
- Zhong XY, Wang PP, Han JH, Rosenfeld MG, and Fu XD (2009). SR Proteins in Vertical Integration of Gene Expression from Transcription to RNA Processing to Translation. Mol Cell 35, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou LQ, and Dean J (2015). Reprogramming the genome to totipotency in mouse embryos. Trends Cell Biol 25, 82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou ZH, and Fu XD (2013). Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma 122, 191–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Chemical and genetic evidence for the requirement of SRPK1 to catalyze paternal genome decondensation, Related to Figure 1
(A) Staining of SRPK1 (green) and SC35 (red) in GV oocyte in comparison with MII oocyte. Scale bar, 20 μm.
(B) Positive staining of SRPK1 and negative staining of SRPK2 in MII oocytes. Stained oocytes with only secondary antibody served as negative control.
(C)Chemistry structures of the SRPK1 inhibitor SRPKIN-1 and a related compound lacking detectable inhibitory activity against SRPK1.
(D) Dosage-dependent effect of SRPKIN-1 on early zygotic development from 3 independent experiments.
(E) Dosage-dependent effect of SRPKIN-1 on paternal genome decondensation in fertilized eggs from 3 independent experiments. Right: Quantified data displayed by bar graph.
(F) Impact of genetic ablation of SRPK1 on early zygotic development from 3 independent experiments. Right: Quantified data displayed by bar graph with indicated p-value.
(G) Correlation between remaining SRPK1 immunostaining signals and degree of paternal genome decondensation in SRPK1-ablated oocytes. Right: Quantified data displayed by bar graph with indicated p-value.
Figure S2. In vitro growth of isolated oocytes for siRNA-mediated SRPK1 deletion and functional rescue with siRNA-resistant SRPK1 mRNA, Related to Figure 2
(A) Representative images of growing oocytes after in vitro culture for different days. Bottom panels: matured oocyte at the GV and MII stages.
(B) Impact of SRPK1 depletion on zygotic development and functional rescue with SRPK1 mRNA.
Figure S3. Mapping SRPK1 phosphorylation in P1 and P2 and evidence for SRPK1-mediated phosphorylated P1 in vivo, Related to Figure 3
(A) Sequence of P2 and three chemically synthetic fragments and their mutant versions for in vitro phosphorylation with SRPK1. Green stars denote the mapped phosphorylation sites.
(B,C) Phosphorylation contents (B) and reaction velocity (C) of SRPK1-catalyzed phosphorylation on wild-type and mutant P1.
(D) Kinetics of SRPK1-catalyzed phosphorylation on wild-type and mutant P2 fragments.
(E,F) P1 peptides used to raise phospho-specific antibodies in rabbits (E) and immunoblot analysis of affinity-purified anti-phosphoantibodies on individual Protamine peptides (F).
(G) Time-course analysis of P1 phosphorylation at Ser43 after in vitro fertilization.
(H) Lack of evidence for P1 phosphorylation at Ser11 and Ser13 in vivo.
(I) Rescue of P1 phosphorylation at Ser43 with active SRPK1, but not the kinase-dead mutant, in siSRPK1-treated oocyte.
Figure S4. Engineering knock-in mutations of SRPK1 phosphorylation sites in P1 and functional impact on sperm production, Related Figure 4
(A,B) Design of specific gRNAs to target Ser9 and Ser43 in P1 (A) and confirmation of specific mutations introduced in mouse germ line by Sanger sequencing (B).
(C) Potential off-targets PCR-amplified and sequenced to show the lack of detectable off-target effects.
(D) Sperm counts from males of wild-type, single, and double P1 mutant.
(E) Aniline blue staining of cauda epididymal sperm from dpp 12-week mice. Arrows indicate adjacent somatic cells that are efficiently stained by aniline blue, which served as positive control. Scale bar, 10 μm.
(F) Representative images of transmission electron micrographs of wild-type and double mutant sperm heads from dpp 12-week mice. Scale bar, 1 μm
Figure S5. P1 phase separation in the presence of DNA and impact of SRPK1-catalyzed phosphorylation on the formation of mesh-like networks, Related to Figure 5
(A,B) Gel mobility shift assay for P1 interaction with dsDNA (A) and formation of insoluble material (B).
(C) Analysis of DNA concentration-dependent (left) and DNA length-dependent effect on P1 phase transition. Different degrees of phase separation are indicated by the keys on the right.
(D,E) SRPK1 still prevented phase transition of single mutant at Ser9 (D) or Ser43 (E) from small particles to mesh-like structures.
Figure S6. In vitro sperm decondensation with purified NPM2, Related to Figure 6
(A) Scheme for the in vitro sperm decondensation assay. Isolated sperm were permeabilized with SLO and disulfide bonds removed with DTT for the decondensation assay with purified human NPM2. Retained P1 on paternal chromatin was quantified by Western blotting.
(B) Purification of recombinant human NPM2 expressed in bacteria.
(C) Human NPM2-mediated sperm decondensation and P1 release over an extended period of treatment. Retained P1 on sperm DNA was quantified at bottom. The H3K4me3 signal on paternal chromatin served as control.
(D) Representative images of sperm DNA (DAPI staining) (left panel) and western blot of retained P1 on sperm chromatin (right panel) at the indicated SRPK1 treatment time points Scale bar, 10 μm. Total and specifically phosphorylated P1 were blotted with specific antibodies.
(E) Left, representative images of fertilized eggs stained with anti-SRPK1 (green) and DAPI (blue) ~10 hours post insemination. Top and bottom panels show oocytes injected with scrambled (Scr) siRNA and siSRPK1, respectively. P, paternal DNA; M, maternal DNA; PBs, polar bodies. Scale bar, 20 μm. Right, relative SRPK1 staining signals (top) and relative values of pronuclear size (paternal/maternal) (bottom) in fertilized eggs.
(F) Left, representative images of fertilized eggs stained with anti-P1 (red) and DAPI (blue) ~10 hours post insemination. Top and bottom panels show oocytes fertilized with wild-type and double P1 mutant sperm, respectively. P, paternal DNA; M, maternal DNA; PB, polar body. Scale bar, 20 μm. Right, relative P1 staining signals (top) and relative values of pronuclear size (paternal/maternal) (bottom) in fertilized eggs.
(G) Alexa 488-labeled P1 and Alexa 594-labled DNA were mixed to form condensates in vitro followed by testing the potential effect of purified NPM2 on these condensates in the presence of active or kinase-dead SRPK1. The data indicate that the condensates remain a gel-like structure even in the presence of NPM2 and active SRPK1.
Figure S7. Global analysis of chromatin accessibility by ATAC-seq on sperm and fertilized egg, Related to Figure 7
(A) ATAC-seq libraries generated. Shown are total mapped reads, reads after removing those mapped to mitochondrial DNA (-ChrM), and reads uniquely mapped to the paternal and maternal genomes based on SNPs.
(B,C) Reproducibility of duplicated ATAC-seq libraries (B) and comparison between our libraries and with the public ATAC-seq data on wild-type sperm (C).
(D,E) Cross-assignment of maternal (BALB/cJ) and paternal (C57BL/6) reads to C57BL/6 and BALB/cJ genomes, respectively.
(F,G) Mapping rates of ATAC-seq reads to the paternal and maternal genomes (F) and relative reads density (Paternal over Maternal) distribution among top 10,000 DNA bins (G), showing significantly reduced ATAC-seq signals on the paternal genome relative to the maternal genome with double mutant-fertilized eggs.
(H,I) Overlap of our ATAC-seq peaks on wild-type, single and double mutant sperm with the public H3.3 ChIP-seq signals on wild-type sperm (H) and the distribution of ATAC-seq signals at TSS regions (I).
Data Availability Statement
The raw data FASTQ files and processed BigWig files for all sequencing data have been deposited in NCBI GEO under GSE136403.