

Abstract
Despite good responses to first-line treatment with platinum-based combination chemotherapy, most ovarian cancer patients will relapse and eventually develop a platinum-resistant disease with a poor overall prognosis. The molecular events leading to the cisplatin resistance of ovarian cancer cells are not fully understood. Here, we performed a proteomic analysis to identify protein candidates deregulated in a cisplatin-resistant ovarian cancer cell line (A2780CP20) in comparison to their sensitive counterpart (A2780). Forty-eight proteins were differentially abundant in A2780CP20, as compared with A2780, cells. Enolase-1 (ENO1) was significantly decreased in cisplatin-resistant ovarian cancer cells. Western blots and RT-PCR confirmed our findings. Ectopic ENO1 expression increased the sensitivity of ovarian cancer cells to cisplatin treatment. In contrast, small-interfering (siRNA)-based ENO1 silencing in A2780 cells reduced the sensitivity of these cells to cisplatin treatment. Whereas glucose consumption was lower, intracellular levels were higher in cisplatin-resistant ovarian cancer cells as compared with their cisplatin-sensitive counterparts. Senescence-associated β-galactosidase (β-Gal) levels were higher in cisplatin-resistant ovarian cancer cells as compared with cisplatin-sensitive ovarian cancer cells. β-Gal levels were decreased in ENO1 overexpressed clones. Protein levels of the cell cycle regulators and senescence markers p21 and p53 showed opposite expression patterns in cisplatin-resistant compared with cisplatin sensitive cells. Our studies suggest that decreased expression of ENO1 promotes glucose accumulation, induces senescence, and leads to cisplatin resistance of ovarian cancer cells.
Keywords: Enolase, ENO1, senescence, p21, p53, glucose, cisplatin resistance, ovarian cancer, beta-Gal
Introduction
Cytoreductive surgery and platinum/taxane combination chemotherapy are the most common treatments for patients with ovarian cancer [1]. Although the majority of ovarian cancer patients respond to front-line platinum combination chemotherapy, relapse occurs in over 60% of treated patients, resulting in chemoresistant fatal disease [1]. Several molecular mechanisms of cisplatin chemoresistance have been postulated, including decreased cisplatin accumulation inside cells, increased sulfur-cisplatin complexes and DNA repair mechanisms, the activation of anti-apoptotic signals and inactivation of pro-apoptotic pathways, and dysregulation of oncogenes, tumor suppressor genes, and non-coding RNAs [2,3]. However, the key molecular pathways responsible for the cisplatin resistance of ovarian cancer cells have not been completely elucidated.
Evidence indicates that posttranscriptional and posttranslational regulation of gene expression governs the function of several proteins associated with cancer initiation, progression, and drug resistance. Recent advances in proteomics have made possible the simultaneous quantitative comparison of the proteome profile in drug-sensitive and drug-resistant cancer cells [4]. For example, Bruce and co-workers identified 374 proteins with significantly altered expression levels in cisplatin-resistant cells as compared to cisplatin-sensitive HeLa cells [5]. Many of these proteins had not been previously associated with cisplatin resistance. Similarly, changes in protein abundance associated with the mitochondrial proteomes that promote evasion of apoptosis, tumor invasiveness, and metastasis of cisplatin-resistant epithelial ovarian cancer cells were reported [6]. Moreover, studies in ovarian cancer have also identified differential protein abundance in chemoresistant metastatic tissue and cell lines [4,7]. This evidence indicates that proteomic approaches are useful in identifying key molecules in molecular pathways that contribute to cisplatin resistance in ovarian cancer.
Aiming to identify key proteins associated with cisplatin resistance in ovarian cancer, we performed a proteomic analysis in cisplatin-sensitive (A2780) and cisplatin-resistant (A2780CP20) ovarian cancer cells. Results showed low levels of Enolase-1 (ENO1) in A2780CP20 compared to A2780 cells. Western blot and real-time PCR studies confirmed that ENO1 is significantly reduced in a panel of cisplatin-resistant ovarian cancer cells as compared with cisplatin-sensitive ovarian cancer cells. Because ENO1 is an enzyme involved in glucose metabolism, we measured the glucose consumption and the intracellular glucose levels and we observed lower glucose consumption in cisplatin-resistant compared with cisplatin-sensitive ovarian cancer cells. However, the intracellular glucose levels were higher in cisplatin-resistant cells than in cisplatin-sensitive cells. We further investigated how ENO1 silencing, or its overexpression, correlated with the intracellular glucose levels and with the sensitivity of these cells to cisplatin treatment. Finally, we measured beta-galactosidase (β-Gal) levels in cisplatin-resistant and cisplatin-sensitive ovarian cancer cells and we noted that β-Gal levels were lower in cisplatin-resistant ovarian cancer cells. β-Gal levels were also correlated with ENO1 expression levels. Western blot analysis showed that the levels of the cell cycle regulators and senescence markers p21 and p53 showed opposite expression patterns in cisplatin-resistant compared with cisplatin sensitive cells. Our studies suggest that the decreased expression of ENO1 promotes glucose accumulation, induces senescence, and increases cisplatin resistance in ovarian cancer cells.
Materials and methods
Cell lines and culture conditions
The human ovarian epithelial cancer cell lines A2780 and A2780CIS were purchased from the European Collection of Cell Cultures (ECACC). OV-90 and OVCAR3 cells were purchased from the American Type Culture Collection (ATCC). OV-90CIS and OVCAR3CIS were generated by exposing the parental cells to increasing concentrations of cisplatin. A2780CP20, HEYA8, and HEYA8-MDR were provided by Dr. Anil K. Sood (MD Anderson Cancer Center) and have been described elsewhere [8-11]. Each cell line was screened using Mycoplasma removal agent, as described by the manufacturer (AbD Serotec). For in vitro propagation, cells were maintained in RPMI-1640 medium (Thermo Scientific) alone (A2780, A2780CP20, A2780CIS, HEYA8, and HEYA8-MDR), or containing 0.01 mg/mL insulin (Sigma-Aldrich; OVCAR3 and OVCAR3CIS) or 0.5 mg/mL G418 (Sigma-Aldrich; Empty Vector and ENO1-overexpressing clones). OV-90 and OV-90CIS cells were maintained in M199 (Gibco, Life Technologies)/MCDB-105 (Sigma-Aldrich) medium. In all cases, the medium was supplemented with 10% fetal bovine serum (FBS; Thermo Scientific) and 0.1% antibiotic/antimycotic solution (Thermo Scientific). Cell lines and clones were grown at 37°C in 5% CO2 with 95% air. In vitro experiments were performed at 70%-75% cell confluence. The concentration of cisplatin inhibiting 50% of cell growth (IC50) was calculated by the Alamar Blue method 72 hours after the incubation of each cell line with cisplatin [11,12].
Proteomics analysis
2-D Fluorescence Difference Gel Electrophoresis (2D-DIGE) analysis
The first and second dimension separation was performed as published by Ciborowski and coworkers with some modifications [13]. Briefly, first dimension separation was carried out with an IPGphor III apparatus (GE Healthcare). Protein cell extracts (500 µg of A2780 and 500 µg of A2780CP20) were loaded onto Immobiline DryStrips gels (24 cm long) with linear immobilized pH gradient 3-11 non-linear gradient (NL) and rehydrated overnight. Isoelectric focusing was performed at a constant temperature of 20°C with a total of 45 kVh. Strips were then incubated with an equilibration solution (50 mM Tris-HCL pH 8.8, 6 M urea, 30% glycerol, 2% SDS, 0.01% bromophenol blue) containing 100 mM DTT for 15 minutes. Gel strips were also incubated with 100 mM iodoacetamide in equilibration solution for 15 minutes to alkylate proteins. Then, the strips were loaded onto 12% polyacrylamide gels and fixed with 0.5% agarose. The second dimension separation was performed using an Ettan Dalttwelve Electrophoresis System (GE Healthcare) at 20°C. For visualization of protein spots, signals were collected at excitation wavelength for Cy2-, Cy3-, and Cy5-labeled samples at 488, 520, and 620 nm, respectively, using Ettan DIGE Imager (GE Healthcare). Gels were analyzed using DeCyder 2D 6.5 software (GE Healthcare). Spot normalization was done relative to the total volume of all spots in the whole gel. The protein spots with differences of 1.2-fold between A2780 and A2780CP20 cells (p-values < 0.05) were considered for MS identification. Protein spots selected for protein identification after DeCyder analysis were picked from preparative gels (loaded with 125 µg of protein extracts) using an automatic Ettan Spot Picker (GE Healthcare) with a 2.0 mm diameter picking head.
Preparation of peptide digests for identification by tandem mass spectrometry
Protein spots collected from preparative gels were washed at room temperature with 50% acetonitrile (ACN)/50 mM NH4HCO3 for 1 hour. Gel pieces were then dried in a speed vac and incubated with trypsin (Promega, Madison, WI) in 50 mM NH4HCO3 overnight at 37°C. Digested peptides were then extracted with 60% ACN and 0.1% trifluoroacetic acid (TFA), dried on a speed vac and resuspended in 0.5% TFA. All samples were purified using C18 ZipTips (Millipore) according to the manufacturer’s recommendations and resuspended in 2% ACN with 0.1% formic acid prior to LC-MS/MS analysis.
Mass spectrometry and protein identification
Peptides were fractionated on a microcapillary RP-C18 column (NewObjectives) followed by fragmentation using ESI-LC-MS/MS system (ProteomeX System with LTQ, ThermoElectron, Inc.) in a nano-spray configuration. The spectra obtained from mass spectrometric analyses were searched using Sequest™ search engine (BioWorks 3.2 software from ThermoElectron Inc). In the TurboSEQUEST search parameters, 10,000 thresholds and 1.4 precursor mass tolerance for Dta generation were used. For Dta Search, we used a peptide tolerance of 1.5 and fragment ions tolerance of 0.02 with charge state set on “Auto”. An indexed human.fasta.idx database with the following five keywords: Homo, sapiens, human, man, primate was created from nr.fasta retrieved from http.ncbi.nih.gov. Keratins and cytokeratins were excluded from our human.fasta.idx database. The protein identity was accepted only if the probability was a significant threshold level with P≤0.05, and at least two peptides matched.
Western blot analysis
Each cell line was detached with trypsin (0.25%) at 37°C, washed with Phosphate Buffer Saline (PBS), harvested, and stored at -80°C until processed. Cells were lysed with ice-cold lysis buffer (1% Triton X, 150 mM NaCl, 25 mM Tris HCl, 0.4 mM NaVO4, 0.4 mM NaF, and protease inhibitors) and incubated on ice for 30 min. Whole-cell lysates were centrifuged, supernatants were collected, and protein concentration was determined using Bio-Rad Protein Reagents. In all cases, protein lysates (30-50 µg) were separated by SDS-PAGE, blotted onto nitrocellulose membranes, and probed with the appropriate dilution of AL7A1 (ALDH7) (Abcam ab106815), PRDX6 (Abcam ab 59585), RL27 (RPL27) (Abcam ab94537), DOPD (DDT) (Abcam ab150338), ENOA (ENO1) (Sigma, AV3476), ILKAP (Abcam ab11857), CYTB (Sigma HPA017380), p21 (Cell Signaling 2974), and p53 (cell Signaling 2527) primary antibodies. Membranes were rinsed and incubated with horseradish peroxidase-conjugated secondary antibodies. Bound antibodies were detected using enhanced chemiluminescence (GE Healthcare) followed by autoradiography in a FluorChemTM 8900 (Alpha Innotech Corporation). Densitometry analysis of band intensities for each protein, including β-actin, was performed. Fold changes in protein levels were calculated, first relative to the β-actin (Sigma A5441), and then relative to the protein levels in A2780 cells that were taken as 1 in each replicate.
RNA isolation, cDNA synthesis, and SYBR-I-based real-time PCR analysis
Total RNA was isolated using the GenElute Mammalian Total RNA Miniprep kit from Sigma Aldrich. RNA was converted into complementary DNA (cDNA) with the Enhanced Avian RT first strand synthesis kit from Sigma-Aldrich. In brief, total RNA (1 µg), 500 mM dNTP, 2.5 mM random nonamers, and nuclease-free water were mixed up to 10 mL total volume. The mixture was centrifuged and heated at 70°C for 10 minutes and combined with 1 mL of enhanced avian RT, 2 mL 10X buffer, 1 mL RNase inhibitor, and nuclease-free water up to 20 mL total volume. Samples were then preincubated at 25°C for 15 minutes, followed by incubation at 45°C for 50 minutes. SYBR-I-based Real-Time PCR (qPCR) was performed in a StepOne plus Real-Time PCR System (Thermo Scientific). In brief, 10 mL of Power SYBR Green PCR master mix reagent (Applied Biosystems), 0.5 mL of forward primer, 0.5 mL reverse primer (0.4 mM final concentration each), 2 mL of cDNA product, and nuclease-free water up to 20 mL final volume. Primers: ENO1 forward: GATCTCTTCACCTCAAAAGC, ENO1 reverse: TTCCATCCATCTCGATCATC. Beta-actin (β-actin) was used as an endogenous control. Primers: β-actin forward: CCCTTTTTGTCCCCCAAC, β-actin reverse: CTGGTCTCAAGTCAGTGTACAGGT. Cycling conditions: one cycle of 10 minutes at 95°C, and 40 cycles of 15 seconds at 95°C, 30 seconds at 60°C and 30 seconds at 72°C. Melt curve analysis was performed at the end of the PCR reaction. Relative ENO1 expression was calculated with the ΔΔCt-method [14,15].
ENO1 stable transfection
Ectopic ENO1 expression was performed in A2780CP20 cells. ENO1 (ORF expression clone for ENO1, catalog # EX-C0061-M02, accession number: NM_001428) and Empty Vector (pReceiver-M02, catalog # EX-NEG-M02) were purchased from GeneCopoeia. In brief, A2780CP20 cells were seeded onto 6-well plates at a concentration of 3.5 × 104 cells/mL and incubated at 37°C, 5% CO2. The next day, 2 µg of ENO1 or Empty Vector in combination with MegaTran 1.0 Transfection reagent (ratio 1:1.5 w/v) (OriGene, catalog # TT200002) was added to each well. Twenty-four hours later, the culture media was replaced by RPMI-1640-containing G418 (0.5 mg/mL) to select stable transfected clones. Individual clones were picked up and grown in independent flasks. ENO1 expression levels in each clone were measured by Western blot analysis.
Transient transfection of small-interfering RNA (siRNA)
Since the ENO1 gene is shared between two isoforms, ENO1 (NM_001428.5) and Myc binding protein 1 (MBP-1) (NM_001201483.3) [16], two different siRNA sequences were designed: siENO1(1) targets the ENO1 region 5’-GGTGCTTCAACTGGTATCT-3’; while siENO1(2) targeted the ENO1 region 5’-AGATACCAGTTGAAGCACC-3’, which is common for both ENO1 and MBP-1 isoforms. ENO1 siRNAs and negative control, siRNA (NC-siRNA), were purchased from Sigma. Prior to transfection, 1.5 × 105 cells/mL of A2780 were plated into 10 cm Petri dishes. Twenty-four hours later, siRNAs were mixed with HiPerfect transfection reagent (Qiagen) at a 1:2 ratio (siRNA: transfection reagent, vol/vol) and incubated at room temperature (RT) for 20 minutes. The mix was added drop by drop to the cells and allowed to be transfected at 37°C, 5% CO2, and collected 24 hours later for Western blot analysis.
Cell viability and colony formation assays
For cell viability, A2780CP20-ENO1 or Empty Vector clones (2 × 104 cells/mL) were seeded into 96-well plates. The next day, cells were treated with different concentrations of cisplatin and incubated for 48 hours. For cell viability upon siRNA transfection, cells (3.5 × 104 cells/mL) were seeded into 96-well plates and the next day cells were transfected with 100 nM of siRNAs as described above. Twenty-four hours later, 2 mM cisplatin was added for an additional 48 hours. At the end of the treatment, the medium was removed and 95 µl of Alamar blue (Invitrogen) dye was added. OD values were obtained spectrophotometrically in a plate reader (BioRad) after a maximum of 4 hours of dye incubation. In all cases, percentages of cell viability were obtained after blank OD subtraction, taking the values of the untreated cells as a normalization control.
For colony formation assays, clones (3.5 × 104 cells/mL) were plated into 6-well plates, and the next day 2 mM cisplatin was added. Eight hours later, the media was removed and 1000 cells were seeded into 10 cm Petri dishes. Ten days later, colonies were stained with 0.5% crystal violet in methanol. Colonies of at least 50 cells were counted in five random fields (10X) using the Nikon Eclipse TS100 microscope. The percentage of colonies was calculated relative to the number of colonies in the Empty Vector plate, which was considered as 100%.
Glucose consumption and intracellular glucose measurements
Glucose consumption was measured using a Gluc Cell glucose monitoring system (Thermo Fisher Scientific). Cells (3 × 104 cells/ml) were plated in 6-well plates, and the next day the culture media was replaced by fresh media. Two µl of the media was taken directly from the cell culture supernatants, placed in the Test Strips, and inserted in the monitoring system. This procedure was done at 0, 12, 24, 48 and 72 h after adding fresh media. Immediately after each measurement, cells were collected and counted by the trypan blue method. Glucose levels were expressed in mg/dL and normalized to 100,000 cells. The intracellular glucose levels were measured with the Glucose Assay Kit from Abcam (catalog # AB65333) as per the manufacturer specifications. In brief, cells were washed with 1X PBS, detached with 0.25% of Trypsin, collected, and resuspended in cold 1X PBS at 1 × 106 cells/mL. Two million cells were lysed with Assay Buffer and deproteinized with a Deproteinizing Sample Preparation Kit (Abcam catalog # AB204708), according to the manufacturer specifications. Processed samples were analyzed by colorimetric measurements using a microplate reader (BioRad) at an OD of 570 nm. The amount of glucose in each sample was extrapolated from a glucose standard curve prepared for each experiment with known concentrations of glucose.
Senescence-associated β-galactosidase activity
Senescence was measured in a panel of ovarian cancer cells and A2780CP20 ENO1 clones with a beta-galactosidase (β-Gal) Detection Kit from Abcam (catalog # AB176721). This kit uses the fluorogenic fluorescein digalactoside (FDG) galactosidase substrate, which, upon cleavage by β-Gal, generates a fluorescent product that can be measured. In brief, cells were collected, lysed with protein lysis buffer (included in the kit), and diluted at 1 µg/mL protein concentration. Protein aliquots of each sample were incubated with FDG for 4 hours. After this period of time, a stop buffer was added, and the fluorescence in each sample was quantified with a Thermo Scientific Varioskan Flash spectral reader at 490 nm excitation and 525 nm emission. β-Gal levels in each sample were calculated using a β-galactosidase standard curve prepared for each experiment.
Statistical analysis
For in vitro and in vivo experiments, statistical analysis was performed using Student’s t-test. P-values of <0.05 were considered statistically significant. GraphPad Prism software was used for graphing and statistical analysis.
Results
Proteomic analysis revealed several proteins differentially abundant in cisplatin-resistant and cisplatin-sensitive ovarian cancer cells
Following 2-DIGE protein separation and DeCyder analysis, protein spots with >1.2-fold changes and p-values ≤0.05 were selected for protein identification by mass spectroscopy (MS). The MS data was analyzed and filtered using TurboSEQUEST with the following parameters: DelCn of 0.1, XCorr of 1.5 and 70% of protein coverage. By using these parameters, 147 proteins were identified (Supplementary Table 1). Forty-eight out of the 147 proteins were differentially abundant in cisplatin-resistant (A2780CP20), as compared with cisplatin-sensitive (A2780), cells (Supplementary Table 2). Based on the human.fasta.idx index, fold change (higher than 2-fold), and their biological roles, seven differentially abundant proteins, including ENOA (ENO1), ILKAP, RL27, PRDX6, CYTB, DOPD and AL7A1 (Table 1), were selected for further validation by Western blots.
Table 1.
Candidate proteins from the proteomics studies selected for further validation
| Protein Symbol | Fold Change A2780CP20 vs A2780 | Biological Role |
|---|---|---|
| ENOA | -2.69 | Functions as a glycolytic enzyme. ENOA is also a multifunctional enzyme involved in growth control, cellular stress, parasitic infections, autoantigen activities, and cancer. |
| ILKAP | -2.52 | Protein phosphatase that may play a role in regulation of cell cycle progression via dephosphorylation of its substrates. |
| RL27 | +4.34 | Part of the 60S subunit: DNA replication, transcription and repair, RNA splicing and modification. |
| PRDX6 | +2.78 | Mitochondrial protein Involved in redox regulation of cells; protects against oxidative injuries. It can reduce H2O2, short-chain organic, fatty acid, and phospholipid hydroperoxides. |
| CYTB | -2.67 | Intracellular thiol proteinase inhibitor. Tightly binding reversible inhibitor of cathepsins L, H, and B. |
| DOPD | +5.07 | Enzyme: Tautomerization of D-dopachrome with decarboxylation to give 5,6-dihydroxindole (DHI). |
| AL7A1 | +2.74 | Play a major role in the detoxification of aldehydes generated by alcohol metabolism and lipid peroxidation. |
Western blots and densitometric analysis of the band intensities showed non-significant differences in protein abundance between cisplatin-sensitive (A2780) and cisplatin-resistant (A2780CP20) ovarian cancer cells for RL27, CYTB, DOPD or AL7A1 (Figure 1A, 1B). The protein levels of PRDX6 showed the opposite tendency in the Western blots and the proteomic studies (Figure 1A, 1B). On the other hand, ILKAP and ENOA (ENO1) protein levels showed the same tendency in the Western blots and the proteomic studies (Figure 1A, 1B). ILKAP is a protein phosphatase that plays a role in the regulation of cell cycle progression via dephosphorylation of its substrates, primarily ILK [17-21]. The role of ILKAP and ILK in ovarian cancer has been studied elsewhere [20,22-24]. However, the biological consequences of ENO1 downregulation in ovarian cancer cells and its association with cisplatin resistance have not been investigated.
Figure 1.
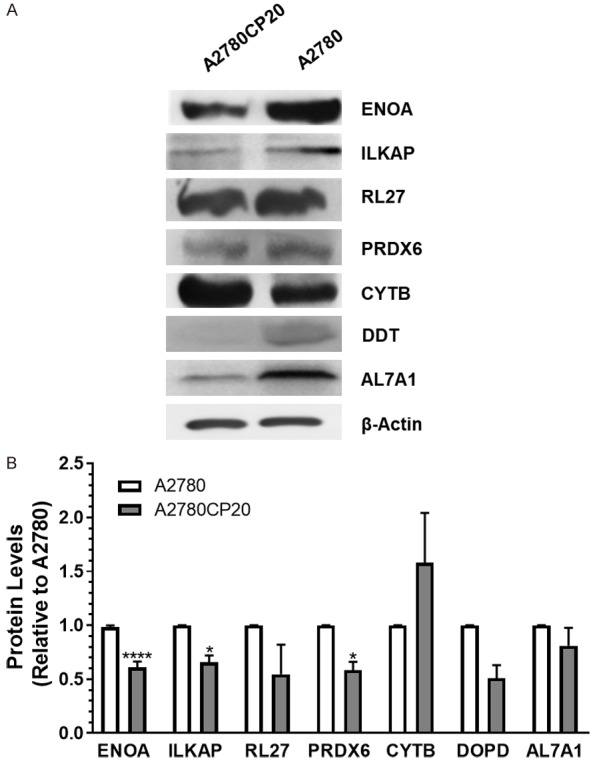
Western blot validation of the proteomic results. (A) Western blot analysis was performed using 30-50 µg of protein extracts. Beta-Actin (β-actin) was used as a loading control. (B) Densitometry analysis of band intensities shown in (A). Fold changes in protein levels were calculated relative to A2780 cells. Averages ± SEM are shown for three independent experiments. *P<0.05, ****P<0.0001.
ENO1 protein and mRNA levels are lower in cisplatin-resistant ovarian cancer cells as compared with cisplatin-sensitive ovarian cancer cells
To determine if the decreased expression of ENO1 also occurred in other cisplatin-resistant ovarian cancer cells, we performed Western blots and SYBR-I-based real-time PCR. Supplementary Table 3 shows the cisplatin IC50 values for the panel of ovarian cancer cells used in this study. Our results confirmed that ENO1 protein levels were decreased in cisplatin-resistant ovarian cancer cell lines (A2780CP20, A2780CIS, OV-90CIS, and OVCAR3CIS) when compared with their cisplatin-sensitive counterparts (A2780, OV-90, and OVCAR3) (Figure 2A). OV-90 and OVCAR3 are high-grade serous ovarian cancer (HGSOC) cell lines [25,26]. HGSOC is the most common and lethal of the ovarian cancer types [25,26]. The densitometric analysis of the Western blot bands showed that the decreased ENO1 levels were significantly lower in cisplatin-resistant ovarian cancer cells than in cisplatin-sensitive ovarian cancer cells (Figure 2B). The MBP-1 (Myc binding protein 1) band intensity in all cell lines is negligible compared with ENO1 levels (Supplementary Figure 1A). MBP-1 is a nuclear isoform of ENO1 [27]. Quantitative PCR results showed that the mRNA levels of ENO1 also were significantly lower in cisplatin-resistant cells as compared with cisplatin-sensitive ovarian cancer cells (Figure 2C). These results suggest that ENO1 levels in cisplatin-resistant ovarian cancer cells are altered also at the transcriptional level.
Figure 2.
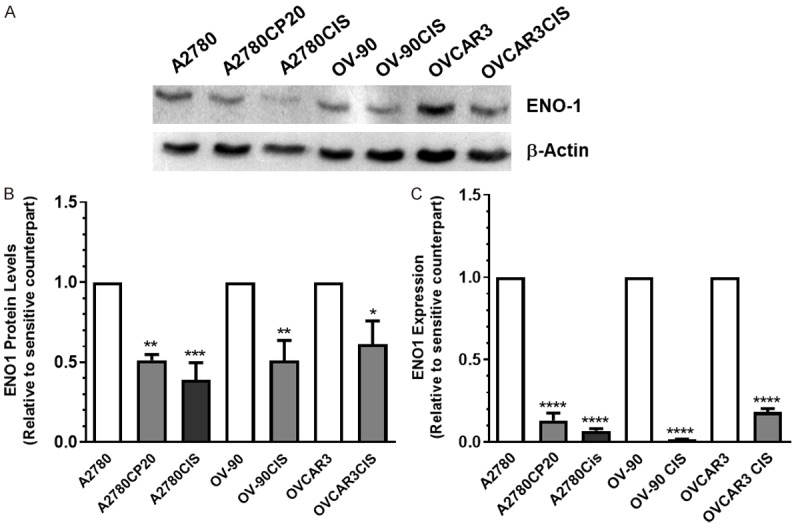
ENO1 protein and mRNA levels in a panel of ovarian cancer cells. (A) Western blot analysis was performed using protein extracts (30-50 µg) of cisplatin-sensitive (A2780, OV-90, and OVCAR3) and cisplatin-resistant (A280CP20, A2780CIS, OV-90CIS, and OVCAR3CIS) ovarian cancer cells. (B) Densitometry analysis of band intensities, shown in (A). Fold changes in protein levels were calculated relative to the cisplatin sensitive pair. Averages ± SEM are shown for three independent experiments (C) ENO1 mRNA expression levels were assessed by qPCR. β-actin was used as a PCR internal control. Fold changes in mRNA levels were calculated relative to the cisplatin sensitive pair. Averages ± SEM are shown for three independent experiments. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
ENO1 expression levels correlate with the sensitivity of ovarian cancer cells to cisplatin treatment
We evaluated the hypothesis that ENO1 expression levels are associated with the sensitivity of ovarian cancer cells to cisplatin treatment. ENO1 was stably transfected into A2780CP20 cells. Figure 3A is a Western blot to assess ENO1 protein levels in Empty Vector (EV) and ENO1-stable transfected clones. Ectopic ENO1 expression did not affect MBP-1 expression levels (Supplementary Figure 1B). Figure 3B is a cell viability experiment showing that the ENO1-clone-5 was more sensitive (IC50 8.1 mM) to cisplatin treatment compared with the EV clone (IC50 11.4 mM). In a colony formation assay, we observed a reduced number of colonies in the ENO1-clone-5 compared with the EV clone (Figure 3C). Importantly, cisplatin treatment reduced the number of colonies in almost 70% (****P<0.0001) in the ENO1-clone-5 compared to the EV clone (Figure 3D). Images of the whole Petri dishes for the colony formation assay are shown in Supplementary Figure 2.
Figure 3.
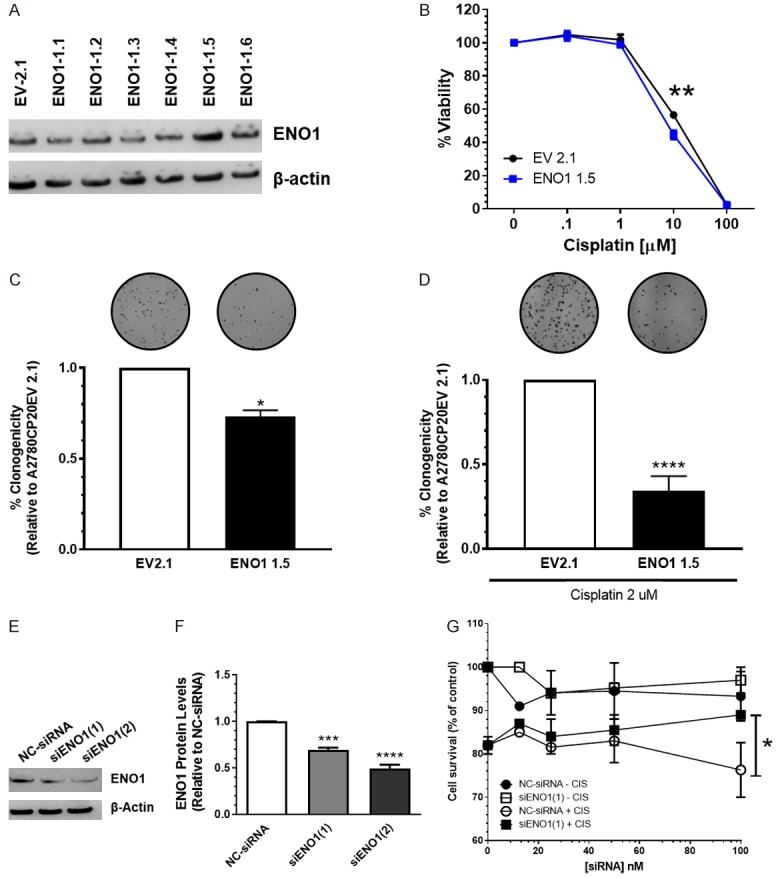
In vitro effect of ENO1 overexpression or silencing on cell growth and proliferation. (A) A2780CP20 cells were stably transfected with an empty vector (EV) or with an ENO1-containing vector. Western blot analysis was performed with 50 µg of protein extracts. (B) EV and ENO1 clones (3 × 104 cell/ml) were exposed to different concentrations of cisplatin for 72 h. Cell viability values were calculated relative to untreated cells. (C, D) Percentages of clonogenicity were calculated relative to EV cells. (E) A2780 cells (3 × 104 cells/ml) were transiently transfected with a negative control siRNA (NC-siRNA) or the two ENO1-targeted siRNAs. Western blot was performed with 50 µg of protein extracts. (F) Densitometry analysis of band intensities from (E). Protein levels were calculated relative to NT cells. (G) A2780 cells (3 × 104 cells/ml) cells were plated in 96-wells and the next day cells were transfected with different concentrations of siRNAs, as described in (E) Twenty-four hours after siRNA transfection, cisplatin (2 mM, final concentration) was added to the cells. Forty-eight hours later, cell viability was measured. Averages ± SEM are shown for three independent experiments. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Next, we transiently transfected A2780 cells with ENO1-targeted siRNAs. Since the ENO1 gene is shared between the two isoforms [16], ENO1 and MBP-1, two different siRNA sequences were designed: siENO1(1) targets the mRNA of ENO1 only; while siENO1(2) targets both the ENO1 and MBP-1 isoforms. Figure 3E is a Western blot showing that both siRNAs decreased ENO1 protein levels. Figure 3F shows the densitometric analysis of the band intensities of Figure 3E. We observed that both siRNAs were able to reduce the ENO1 protein levels [siENO1(1) ***P<0.001 and siENO1(2) ****P<0.0001] as compared with NC-siRNA-transfected cells. Furthermore, compared with the NC-siRNA, the siENO1(1) reduced the sensitivity of A2780 cells to cisplatin treatment (2 mM final concentration) (Figure 3G). For example, at 100 nM of siRNA, the cell viability was 93% with the NC-siRNA and 97% with siENO1(1). When cells were exposed to cisplatin, the cell viability was 76% for NC-siRNA and 89% for siENO1(1) (Figure 3G). Transient transfection of the siRNA targeting both ENO1 and MBP-1 [siRNA(2)] did not induce significant changes in the sensitivity of A2780 cells to cisplatin treatment as compared with the NC-siRNA transfected cells (Supplementary Figure 3). Together, these results suggest that ENO1 levels correlate with the sensitivity of A2780 cells to cisplatin treatment.
Intracellular glucose levels are higher in cisplatin-resistant ovarian cancer cells than in cisplatin-sensitive ovarian cancer cells
ENO1 is a glycolytic enzyme responsible for catalyzing the conversion of 2-phosphoglycerate to phosphoenolpyruvate [27,28]. Lack of the ENO1 enzyme promotes the intracellular accumulation of glucose [27,29]. Therefore, we assessed if ENO1 levels correlated with the intracellular glucose levels in cisplatin-sensitive cells and cisplatin-resistant ovarian cancer cells. Figure 4A-C show that the intracellular glucose levels were significantly higher in cisplatin-resistant than in cisplatin-sensitive cells. In addition, the intracellular glucose levels were significantly lower (***P<0.001) in the ENO1-overexpressing clone as compared with the EV clone (Figure 4D). Moreover, the intracellular glucose levels were significantly higher (***P<0.001) in siRNA-mediated ENO1 silenced cells as compared with NC-siRNA transfected cells (Figure 4E).
Figure 4.
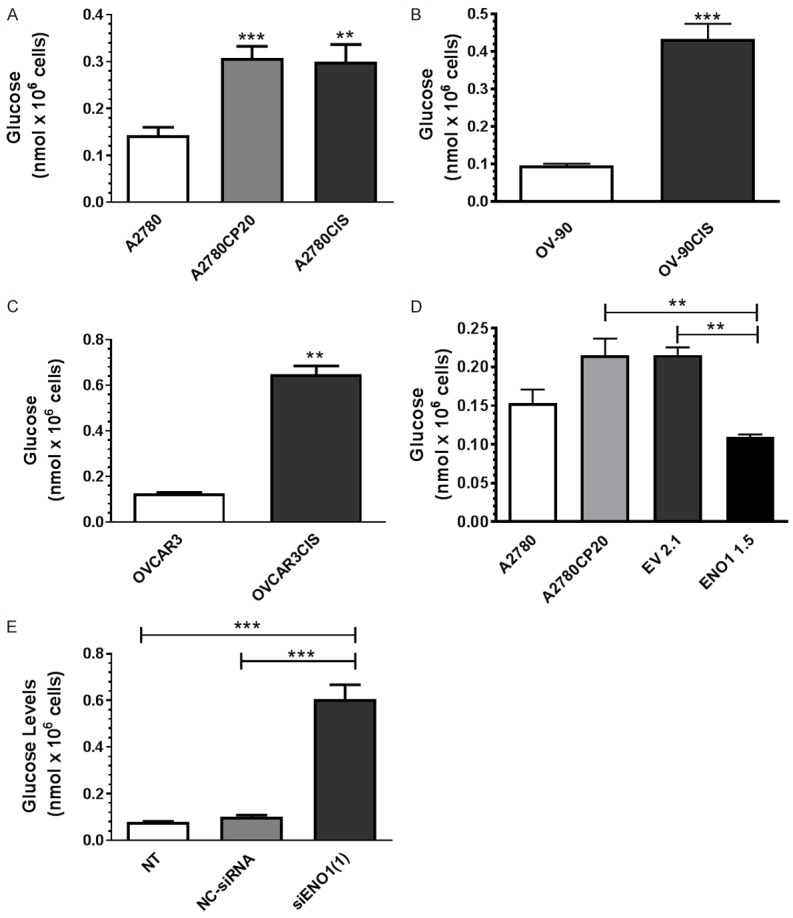
Intracellular glucose measurements. (A-D) Cells (1 × 104 cells/ml) were plated in 6-well plates. Twenty-four hours later, cells were rinsed with PBS, and 2 million cells of each cell line were used to assess the intracellular glucose levels. Averages ± SEM are shown for five independent experiments. (E) A2780 (1 × 104 cells/ml) cells were transfected with siRNA. Twenty-four hours later, cells were collected for glucose content assessment, as described in (A-D). Averages ± SEM are shown for five independent experiments. *P<0.05, **P<0.01, ***P<0.001.
Cisplatin resistant ovarian cancer cells consume less glucose than cisplatin sensitive ovarian cancer cells
As higher intracellular glucose levels in cisplatin-resistant cells may be caused by higher glucose consumption, we measured glucose consumption in the panel of ovarian cancer cells and the ENO1 clones. Figure 5 shows the glucose levels measured in the cultured media at different time points. In all cell lines tested, the glucose levels decreased in a time-dependent manner. However, cisplatin-resistant cells consumed a smaller glucose amount (higher glucose levels in the culture media) than their cisplatin sensitive counterparts at all time-points tested (Figure 5A-C). Although we observed statistically significant differences in the glucose levels between the EV and ENO1 clones (Figure 5D), these differences were minimal compared with the changes observed between the cisplatin-sensitive and the cisplatin-resistant ovarian cancer cells. Together, results showed in Figures 4 and 5 indicate that, although cisplatin-resistant ovarian cancer cells consume less glucose, they accumulate higher amounts of it compared with cisplatin-sensitive cells.
Figure 5.
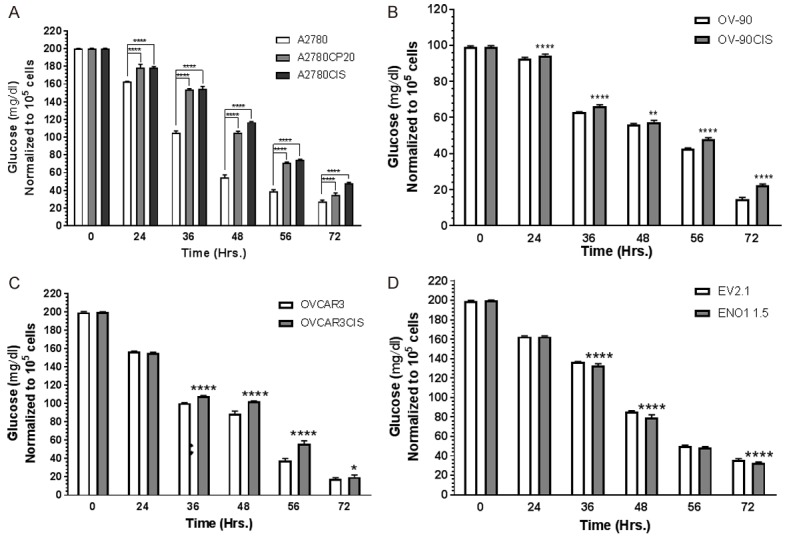
Glucose consumption measurements. A-D. Cells (1 × 104 cells/ml) were plated in 6-well plates. Glucose was measured in the cultured media. Averages ± SEM are shown for three independent experiments. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Senescence-associated markers in ovarian cancer cells
Previous studies have shown that the acquisition of drug resistance is accompanied by a senescence phenotype of cancer cells [30-32]. Evidence also indicates that decreased expression of ENO1 and high glucose intracellular levels promote cellular senescence [30]. Thus, we measured the senescence-associated beta-galactosidase (β-Gal) levels in cisplatin-resistant and cisplatin-sensitive ovarian cancer cells and the ENO1-overexpressing clone. Figure 6A shows that the β-Gal levels were significantly higher in the cisplatin-resistant cells compared with their cisplatin-sensitive counterparts. Importantly, the β-Gal levels decreased in the ENO1-overexpressing clone as compared with the EV clone (Figure 6B). Finally, we measured the levels of p21 and p53, cell cycle progression regulators and senescence-associated markers [31]. The Western blot image showed in Figure 6C indicates that the p21 protein levels were almost absent in cisplatin-resistant as compared with their cisplatin sensitive counterparts, where the p21 levels were prominent. Opposite tendency was observed for p53, as the levels of this protein was increased in cisplatin resistant as compared with cisplatin sensitive cells. Original Western blot images were included in the Supplementary Figure 4. Of note, OV-90 cells exhibited low p21 and high p53 protein levels (Figure 6C). Together, these results suggest that decreased expression of ENO1 promotes senescence of cisplatin-resistant ovarian cancer cells by regulating p21 and p53 protein levels.
Figure 6.
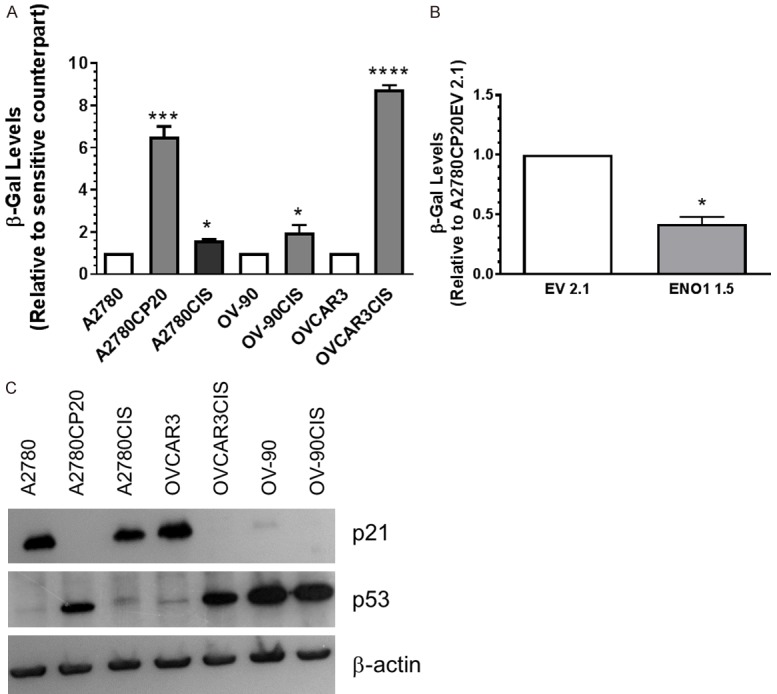
Senescence-associated β-galactosidase and senescence-associated markers. Cells (1 × 104 cells/ml) were plated in 6-well plates. Twenty-four hours later, cells were rinsed with PBS, and protein extracts were prepared and diluted at a 1 µg/mL protein concentration. Senescence-associated β-galactosidase activity (SA-β-gal) was assessed by fluorescence. β-galactosidase levels were calculated relative to each cisplatin-sensitive cell pair (A) or relative to the EV clone (B). Averages ± SEM are shown for three independent experiments. *P<0.05, ***P<0.001, ****P<0.0001. (C) Western blot analysis was performed using 50 µg of protein extracts.
Discussion
By using a proteomic approach, we identified Enolase-1 (ENO1) as significantly decreased in cisplatin-resistant as compared with cisplatin-sensitive ovarian cancer cells. Our results were confirmed in HGSOC cells by using Western blot and qPCR. Although our proteomic analysis identified several proteins differentially abundant in cisplatin-sensitive and cisplatin-resistant ovarian cancer cells, Western blot analysis only confirmed the proteomic expression patterns for ILKAP and enolase-α (ENOA or ENO1). The protein expression levels of the additional five proteins assessed using Western blots did not correlate with the proteomic results. One possible explanation for this observation is that the protein assessed by immunoblots was an isoform or a paralog of the protein identified by MS [33]. Another possibility is a misidentification of candidate proteins with the parameters used for protein searching and identification with the TurboSequest program [34]. For example, as we used only 70% of ion coverage, some spectra cannot be matched to the correct sequence and they could be erroneously assigned to a different peptide in the database [34]. Additionally, as many peptides in proteomic studies still have amino acid modifications, spectra derived from modified peptides are commonly assigned to the wrong amino acid sequences [35]. This problem commonly leads to false protein identifications and errors in protein quantification [35]. Furthermore, poor protein digestion, highly hydrophilic or very small peptides that are not retained in a column or highly large/hydrophobic peptides that are stuck in gel or are too large for mass spectrometer analysis, also could contribute to inaccuracies in our proteomic data [35]. In any case, future studies could explore the role of other differentially abundant proteins (Supplementary Table 2) in cisplatin-resistant and cisplatin-sensitive ovarian cancer cells.
Enolases are metalloenzymes responsible for the conversion of 2-phosphoglycerate (2-PG) to phosphoenolpyruvate (PEP) during glycolysis [27,28]. ENO1 (ENO-α) is one of three enolase isoforms, the other two being ENO2 (ENO-γ) and ENO3 (ENO-β) [28]. Besides glycolysis, ENO1 is a multifunctional enzyme involved in growth control, cellular stress, parasitic infections, autoantigen activities, cancer metastasis and drug resistance [28]. ENO1 is also present on the surface of several cell types, such as leukocytes and neurons where it serves as a receptor and activator of plasminogen [28]. In our study, decreased ENO1 levels were associated with intracellular glucose accumulation and senescence of cisplatin-resistant ovarian cancer cells. These results are in agreement with information stating that, besides metabolism control, ENO1 is a key enzyme involved in cell growth, survival and drug resistance, and that all of these events are intimately associated with each other [28].
The reduced protein levels of ENO1 in cisplatin-resistant cells were confirmed using HGSOC cell (OVCAR3/OVCAR3CIS and OV-90/OV-90CIS) pairs. As HGSOC is the most common and malignant of the gynecological cancers [1], our findings suggest that reduced ENO1 protein levels could be used as a predictor of drug response in women with HGSOC. This hypothesis should be confirmed at the protein level by immunohistochemical (IHC) experiments with HGSOC patient samples. When we interrogated the KM Plotter database (www.kmplot.com) which includes close to 1600 ovarian cancer samples we observed that ovarian cancer patients with low ENO1 mRNA levels recur faster (PFS) than patients with high ENO1 mRNA levels (data not shown). However, this correlation was not statistically significant. Our findings that mRNA levels are reduced in cisplatin-resistant cells suggest the absence/repression of transcription factors responsible for ENO1 expression. Oncogenes such as RAS, MYC, and HIF-1α have been reported to increase ENO1 expression [16,30,36]. However, we and others have shown that these transcription factors are overexpressed/mutated in cisplatin-resistant ovarian cancer cells [16,36,37]. It could be speculated that a repressor protein is responsible for the reduced expression of ENO1 in cisplatin-resistant ovarian cancer cells. The identification of this protein is crucial to fully understand how ENO1 is regulated in ovarian cancer cells. Interestingly, in a proteomic study, Kotz et al. found that ENO1 is associated with the fluorescent carboxy-fluorescein-diacetate-labelled cisplatin analog (CFDA-cisplatin) in A2780 and A2780CIS cells [38]. This interaction might decrease the net intracellular cisplatin levels and contribute to the drug resistance of ovarian cancer cells. This hypothesis should be further investigated.
Alternative splicing of the ENO1 mRNA transcript generates a cytoplasmic protein (ENO1) or a nuclear protein referred to as Myc-binding protein-1 (MBP-1) [16,39]. Subramanian and Miller reported that MBP-1 binds to the P2 promoter DNA region of c-MYC and down-regulates c-MYC expression [39]. As c-MYC levels are highly abundant in cisplatin-resistant ovarian cancer cells [37], the negligible expression levels of MBP-1 in ovarian cancer cells could partially contribute to the high c-MYC levels observed in those cells [37]. In fact, when we used the siRNA to reduce the expression of ENO1 only, cells become less sensitive to cisplatin treatment. However, when we used the siRNA targeting, both ENO1 and MBP-1, changes in cisplatin sensitivity were not observed. These results could be due to the elimination of the c-MYC repression by MBP-1 [16]. Because designing siRNAs targeting only MBP-1 is particularly difficult (as the mRNA sequence of MBP-1 spans the same mRNA region of ENO1), the role of MBP-1 in ovarian cancer should be investigated with other, different approaches. Nevertheless, when we ectopically expressed ENO1 in cisplatin-resistant ovarian cancer cells, these cells became more sensitive to cisplatin treatment. Together, these results indicate that the decreased expression of ENO1 contributes to the cisplatin resistance of ovarian cancer cells.
The role of ENO1 in cancer is controversial. Liu and co-workers found that ENO1 overexpression positively correlated with clinical stage, lymph node metastasis, and poor prognosis of patients with pancreatic cancer [40]. Qi and co-workers reported that the mRNA and protein levels of ENO1 were upregulated in glioma tissues compared to normal brains [41]. ShRNA-mediated ENO1 knocking-down decreased cell proliferation, inhibited cell migration and invasion; and reduced in vivo tumorigenesis of glioma cells [41]. Elevated expression of ENO1 has also been observed in cell lines and tissue samples of breast, lung, prostate, and pancreatic cancers, as well as in neuroendocrine tumors, neuroblastoma, cholangiocarcinoma, thyroid carcinoma, hepatocellular carcinoma, and Burkitt lymphoma [27]. In contrast, a proteomic study performed by Auer et al. in endometrioid endometrial cancer samples found that ENO1 was decreased as compared with normal controls [42]. By using Western blotting and IHC analysis, Mao and co-workers reported that ENO1 is frequently down-regulated in non-small cell lung cancer (NSCLC) tissue patients, and its down-regulation correlated with aggressive tumor behavior [43]. An explanation for the discrepancy regarding ENO1 expression is not currently available. Changes in ENO1 levels by cellular stress [27], the occurrence of the MBP-1 isoform and ENO1 pseudogenes, the abundance of microRNAs (miRNAs) post-transcriptionally controlling ENO1 protein expression [29], and the presence of heterogeneous cell populations in a tumor might contribute to the opposing ENO1 levels reported in the literature. Particularly, Jin and co-workers found that reduced expression of miR-22 increases ENO1 expression levels, stimulates glycolysis, and promotes cisplatin resistance in gastric cancer cells [29]. If cisplatin-resistant ovarian cancer cells express higher levels of miR-22 than their cisplatin sensitive counterparts, this could explain the opposite findings of Jin and co-workers and our findings. In fact, while, in some cancers, miR-22 acts as a tumor suppressor gene, in other tumors it exhibits oncogenic properties [44]. Thus, studies to clarify if ENO1 is post-transcriptionally regulated by non-coding RNAs in ovarian cancer should be performed. Nevertheless, our findings that ENO1 was decreased in cisplatin-resistant ovarian cancer cells compared with cisplatin-sensitive ovarian cancer cells are in agreement with reports that lower levels of ENO1 lead to intracellular glucose accumulation, senescence, and the selection of certain cell populations which are more resistant to chemotherapy [30,45,46].
Another important finding of our study was that cisplatin-resistant cells consume less glucose than their cisplatin-sensitive counterparts. Possible explanations for this finding include the decreased levels of glucose transporters that reduce the influx of cisplatin to the inside of cells. Two types of glucose transport have been described: sodium-glucose linked transporters (SGLTs) and facilitated diffusion glucose transporters (GLUT) [47]. Six members of the SGLT and twelve members of the GLUT transports have been reported [47]. Ishiko and co-workers assessed the expression of GLUT1, GLUT3, and GLUT4 in human ovarian tumor samples using immunohistochemical analysis, and observed that the expressions of GLUT1 and GLUT4 correlated with the tumor stage, and the expressions of GLUT1, GLUT3 and GLUT4 correlated positively with VEGF expression [48]. More recently, Fang and co-workers used a GLUT1-specific inhibitor and observed in vitro inhibition of cell proliferation and in vivo tumor reduction with ovarian cancer cell line- and patient-derived xenograft mouse models [49]. Further studies should measure the expression levels of all of these receptors in cisplatin-sensitive and cisplatin-resistant ovarian cancer cells. The differences in glucose consumption rates were higher in A2780CP20/A2780CIS/A2780 than in OV-90CIS/OV-90 or OVCAR3CIS/OVCAR3 pairs of cells. Although A2780 is classified as an endometrial ovarian cancer (EOC) cell line [25] and would not present a good model of HGSOC [25,50], these cells are still used to study molecular mechanisms of cisplatin resistance [37]. Yet, differences in gene expression between EOC and HGSOC could account for the observed differences in glucose consumption rates. Additionally, A2780CP20 and A2780CIS are ~50 and ~8 times more resistant, respectively, to cisplatin than A2780 cells. However, OV-90CIS and OVCAR3CIS are only 3-times and 4.5-times more resistant than OV-90 and OVCAR3, respectively, to cisplatin treatment (Supplementary Table 3) which could also account for the differences in glucose consumption rates we observed. Our findings that cisplatin-resistant cells also contain higher intracellular glucose levels than their sensitive counterparts support our hypothesis that the reduced expression of ENO1 avoids the glucose consumption in cisplatin-resistant cells. Importantly, Avril and co-workers found that, opposite to cisplatin sensitive cells, cisplatin-resistant ovarian cancer cells rely on glutamine more than glucose as their main energy source [51]. Future experiments could address how glutamine-dependent metabolism leads to the senescence of cisplatin-resistant ovarian cancer cells.
Novelli and co-workers studied the role of ENO1 in the Warburg effect [30]. The Warburg effect, also known as aerobic glycolysis, is a characteristic of most solid tumors, increasing the glycolysis rate both in hypoxic conditions and in the presence of normal oxygen level, independent of mitochondrial status [52,53]. Novelli and co-workers used shRNA-mediated ENO1 depleted cells and observed increases in reactive oxygen species (ROS) which were generated through the sorbitol and NADPH oxidase pathways [30]. Interestingly, ENO1 silenced cells are forced to use glucose through the pentose phosphate and the polyol pathways, with a consequent decrease in lactate levels [30]. ENO1 silencing leads to cell cycle arrest in the G2/M phase and induced senescence [30]. In agreement with these results, we observed higher levels of intracellular glucose and senescence-associated β-Gal in cisplatin-resistant compared with cisplatin-sensitive cells that correlated with ENO1 expression levels. In fact, the effects of overexpressing ENO1 were more dramatic on clonogenic than in cell viability assays. The clonogenic assays measure long-term effects on cell proliferation, which include changes in gene expression, a step required for the acquisition of a senescence phenotype, as we observed in the cisplatin-resistant ovarian cancer cells. Together, our results suggest that the decreased expression of ENO1 in cisplatin-resistant ovarian cancer cells is part of their metabolic rewiring, a hallmark of cancer cells that includes re-programming for the optimal use of molecular precursors of proteins, lipids, and nucleotides required to maintain the enhanced growth, proliferation and metastatic potential of those cells [54].
Pillai and co-workers working with breast cancer cells and breast tumors obtained after neoadjuvant chemotherapy observed that oxidative stress caused by chemotherapeutic agents generated senescent-like colonies with an aggressive tumor stem cell-like phenotype [46]. These cells contained low levels of reactive oxygen species (ROS), high levels of antioxidant enzymes, and displayed higher CD133 and Oct-4 expression. Our findings are in agreement with the hypothesis of Pillai and co-workers [46] and Chen and co-workers that, although therapy-induced senescence has short-term benefits, the response also causes the reprogramming of gene expression which leads to the selection of highly drug-resistant phenotype clones [45]. Although increased p21 and decreased p53 protein levels are associated with a senescence phenotype [31] we observed opposite tendency, as the p21 protein levels were reduced while the p53 levels were increased in cisplatin-resistant as compared with cisplatin sensitive cells. Downregulation of p21 and upregulation of p53 could promote a senescent drug-resistant phenotype in cancer cells. These hypotheses should be further investigated.
In conclusion, using quantitative proteomics and Western blot analysis, we demonstrated that ENO1 was reduced in cisplatin-resistant ovarian cancer cells as compared with cisplatin-sensitive ovarian cancer cells. ENO1 overexpression sensitized cells to cisplatin treatment, decreased the glucose intracellular levels, and reduced the senescence of cisplatin-resistant ovarian cancer cells. Molecules in pathways leading to the restoration of ENO1 levels could be considered as targets to design specific therapies to overcome the cisplatin resistance of ovarian cancer cells.
Acknowledgements
This research was funded by RCMI grant U54 MD007600 (National Institute on Minority Health and Health Disparities) from the National Institutes of Health, institutional seed funds from the University of Puerto Rico Comprehensive Cancer Center (PEVM), and the NIGMS-RISE Grant Number R25-GM061838 (NGR, BIQD, GSS, ELLD, IMEV). We would like to thank Mary Helen Mays, Ph.D. for critical reading and scientific editing of the manuscript and Juliana Perez-Laspiur, Ph.D. for running proteomic samples.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Matulonis UA, Sood AK, Fallowfield L, Howitt BE, Sehouli J, Karlan BY. Ovarian cancer. Nat Rev Dis Primers. 2016;2:16061. doi: 10.1038/nrdp.2016.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shen DW, Pouliot LM, Hall MD, Gottesman MM. Cisplatin resistance: a cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol Rev. 2012;64:706–721. doi: 10.1124/pr.111.005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Damia G, Broggini M. Platinum resistance in ovarian cancer: role of DNA repair. Cancers (Basel) 2019;11 doi: 10.3390/cancers11010119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cruz IN, Coley HM, Kramer HB, Madhuri TK, Safuwan NA, Angelino AR, Yang M. Proteomics analysis of ovarian cancer cell lines and tissues reveals drug resistance-associated proteins. Cancer Genomics Proteomics. 2017;14:35–51. doi: 10.21873/cgp.20017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chavez JD, Hoopmann MR, Weisbrod CR, Takara K, Bruce JE. Quantitative proteomic and interaction network analysis of cisplatin resistance in HeLa cells. PLoS One. 2011;6:e19892. doi: 10.1371/journal.pone.0019892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chappell NP, Teng PN, Hood BL, Wang G, Darcy KM, Hamilton CA, Maxwell GL, Conrads TP. Mitochondrial proteomic analysis of cisplatin resistance in ovarian cancer. J Proteome Res. 2012;11:4605–4614. doi: 10.1021/pr300403d. [DOI] [PubMed] [Google Scholar]
- 7.Coscia F, Watters KM, Curtis M, Eckert MA, Chiang CY, Tyanova S, Montag A, Lastra RR, Lengyel E, Mann M. Integrative proteomic profiling of ovarian cancer cell lines reveals precursor cell associated proteins and functional status. Nat Commun. 2016;7:12645. doi: 10.1038/ncomms12645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Behrens BC, Hamilton TC, Masuda H, Grotzinger KR, Whang-Peng J, Louie KG, Knutsen T, McKoy WM, Young RC, Ozols RF. Characterization of a cis-diamminedichloroplatinum(II)-resistant human ovarian cancer cell line and its use in evaluation of platinum analogues. Cancer Res. 1987;47:414–418. [PubMed] [Google Scholar]
- 9.Landen CN Jr, Goodman B, Katre AA, Steg AD, Nick AM, Stone RL, Miller LD, Mejia PV, Jennings NB, Gershenson DM, Bast RC Jr, Coleman RL, Lopez-Berestein G, Sood AK. Targeting aldehyde dehydrogenase cancer stem cells in ovarian cancer. Mol Cancer Ther. 2010;9:3186–3199. doi: 10.1158/1535-7163.MCT-10-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vivas-Mejia P, Benito JM, Fernandez A, Han HD, Mangala L, Rodriguez-Aguayo C, Chavez-Reyes A, Lin YG, Carey MS, Nick AM, Stone RL, Kim HS, Claret FX, Bornmann W, Hennessy BT, Sanguino A, Peng Z, Sood AK, Lopez-Berestein G. c-Jun-NH2-kinase-1 inhibition leads to antitumor activity in ovarian cancer. Clin Cancer Res. 2010;16:184–194. doi: 10.1158/1078-0432.CCR-09-1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vivas-Mejia PE, Rodriguez-Aguayo C, Han HD, Shahzad MM, Valiyeva F, Shibayama M, Chavez-Reyes A, Sood AK, Lopez-Berestein G. Silencing survivin splice variant 2B leads to antitumor activity in taxane--resistant ovarian cancer. Clin Cancer Res. 2011;17:3716–3726. doi: 10.1158/1078-0432.CCR-11-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larson EM, Doughman DJ, Gregerson DS, Obritsch WF. A new, simple, nonradioactive, nontoxic in vitro assay to monitor corneal endothelial cell viability. Invest Ophthalmol Vis Sci. 1997;38:1929–1933. [PubMed] [Google Scholar]
- 13.Rozek W, Ricardo-Dukelow M, Holloway S, Gendelman HE, Wojna V, Melendez LM, Ciborowski P. Cerebrospinal fluid proteomic profiling of HIV-1-infected patients with cognitive impairment. J Proteome Res. 2007;6:4189–4199. doi: 10.1021/pr070220c. [DOI] [PubMed] [Google Scholar]
- 14.Vargas IM, Vivas-Mejia PE. Assessment of mRNA splice variants by qRT-PCR. Methods Mol Biol. 2013;1049:171–186. doi: 10.1007/978-1-62703-547-7_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 16.Feo S, Arcuri D, Piddini E, Passantino R, Giallongo A. ENO1 gene product binds to the c-myc promoter and acts as a transcriptional repressor: relationship with Myc promoter-binding protein 1 (MBP-1) FEBS Lett. 2000;473:47–52. doi: 10.1016/s0014-5793(00)01494-0. [DOI] [PubMed] [Google Scholar]
- 17.Lorenzato A, Torchiaro E, Olivero M, Di Renzo MF. The integrin-linked kinase-associated phosphatase (ILKAP) is a regulatory hub of ovarian cancer cell susceptibility to platinum drugs. Eur J Cancer. 2016;60:59–68. doi: 10.1016/j.ejca.2016.02.022. [DOI] [PubMed] [Google Scholar]
- 18.Hausmann C, Temme A, Cordes N, Eke I. ILKAP, ILK and PINCH1 control cell survival of p53-wildtype glioblastoma cells after irradiation. Oncotarget. 2015;6:34592–34605. doi: 10.18632/oncotarget.5423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakrieko KA, Vespa A, Mason D, Irvine TS, D’Souza SJ, Dagnino L. Modulation of integrin-linked kinase nucleo-cytoplasmic shuttling by ILKAP and CRM1. Cell Cycle. 2008;7:2157–2166. doi: 10.4161/cc.7.14.6241. [DOI] [PubMed] [Google Scholar]
- 20.Kumar AS, Naruszewicz I, Wang P, Leung-Hagesteijn C, Hannigan GE. ILKAP regulates ILK signaling and inhibits anchorage-independent growth. Oncogene. 2004;23:3454–3461. doi: 10.1038/sj.onc.1207473. [DOI] [PubMed] [Google Scholar]
- 21.Leung-Hagesteijn C, Mahendra A, Naruszewicz I, Hannigan GE. Modulation of integrin signal transduction by ILKAP, a protein phosphatase 2C associating with the integrin-linked kinase, ILK1. EMBO J. 2001;20:2160–2170. doi: 10.1093/emboj/20.9.2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghatak S, Morgner J, Wickstrom SA. ILK: a pseudokinase with a unique function in the integrin-actin linkage. Biochem Soc Trans. 2013;41:995–1001. doi: 10.1042/BST20130062. [DOI] [PubMed] [Google Scholar]
- 23.Ahmed N, Riley C, Oliva K, Stutt E, Rice GE, Quinn MA. Integrin-linked kinase expression increases with ovarian tumour grade and is sustained by peritoneal tumour fluid. J Pathol. 2003;201:229–237. doi: 10.1002/path.1441. [DOI] [PubMed] [Google Scholar]
- 24.Hannigan G, Troussard AA, Dedhar S. Integrin-linked kinase: a cancer therapeutic target unique among its ILK. Nat Rev Cancer. 2005;5:51–63. doi: 10.1038/nrc1524. [DOI] [PubMed] [Google Scholar]
- 25.Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun. 2013;4:2126. doi: 10.1038/ncomms3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hernandez L, Kim MK, Lyle LT, Bunch KP, House CD, Ning F, Noonan AM, Annunziata CM. Characterization of ovarian cancer cell lines as in vivo models for preclinical studies. Gynecol Oncol. 2016;142:332–340. doi: 10.1016/j.ygyno.2016.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ji H, Wang J, Guo J, Li Y, Lian S, Guo W, Yang H, Kong F, Zhen L, Guo L, Liu Y. Progress in the biological function of alpha-enolase. Anim Nutr. 2016;2:12–17. doi: 10.1016/j.aninu.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diaz-Ramos A, Roig-Borrellas A, Garcia-Melero A, Lopez-Alemany R. alpha-Enolase, a multifunctional protein: its role on pathophysiological situations. J Biomed Biotechnol. 2012;2012:156795. doi: 10.1155/2012/156795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qian X, Xu W, Xu J, Shi Q, Li J, Weng Y, Jiang Z, Feng L, Wang X, Zhou J, Jin H. Enolase 1 stimulates glycolysis to promote chemoresistance in gastric cancer. Oncotarget. 2017;8:47691–47708. doi: 10.18632/oncotarget.17868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Capello M, Ferri-Borgogno S, Riganti C, Chattaragada MS, Principe M, Roux C, Zhou W, Petricoin EF, Cappello P, Novelli F. Targeting the Warburg effect in cancer cells through ENO1 knockdown rescues oxidative phosphorylation and induces growth arrest. Oncotarget. 2016;7:5598–5612. doi: 10.18632/oncotarget.6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gordon RR, Nelson PS. Cellular senescence and cancer chemotherapy resistance. Drug Resist Updat. 2012;15:123–131. doi: 10.1016/j.drup.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rebbaa A. Targeting senescence pathways to reverse drug resistance in cancer. Cancer Lett. 2005;219:1–13. doi: 10.1016/j.canlet.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 33.Chandramouli K, Qian PY. Proteomics: challenges, techniques and possibilities to overcome biological sample complexity. Hum Genomics Proteomics. 2009;2009 doi: 10.4061/2009/239204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim MS, Zhong J, Pandey A. Common errors in mass spectrometry-based analysis of post-translational modifications. Proteomics. 2016;16:700–714. doi: 10.1002/pmic.201500355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bogdanow B, Zauber H, Selbach M. Systematic errors in peptide and protein identification and quantification by modified peptides. Mol Cell Proteomics. 2016;15:2791–2801. doi: 10.1074/mcp.M115.055103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, Yan H, Wang W, Chen S, Viale A, Zheng H, Paik JH, Lim C, Guimaraes AR, Martin ES, Chang J, Hezel AF, Perry SR, Hu J, Gan B, Xiao Y, Asara JM, Weissleder R, Wang YA, Chin L, Cantley LC, DePinho RA. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–670. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reyes-Gonzalez JM, Armaiz-Pena GN, Mangala LS, Valiyeva F, Ivan C, Pradeep S, Echevarria-Vargas IM, Rivera-Reyes A, Sood AK, Vivas-Mejia PE. Targeting c-MYC in platinum-resistant ovarian cancer. Mol Cancer Ther. 2015;14:2260–2269. doi: 10.1158/1535-7163.MCT-14-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kotz S, Kullmann M, Kalayda GV, Dyballa-Rukes N, Jaehde U, Metzger S. Optimized two-dimensional gel electrophoresis in an alkaline pH range improves the identification of intracellular CFDA-cisplatin-protein adducts in ovarian cancer cells. Electrophoresis. 2018;39:1488–1496. doi: 10.1002/elps.201700377. [DOI] [PubMed] [Google Scholar]
- 39.Subramanian A, Miller DM. Structural analysis of alpha-enolase. Mapping the functional domains involved in down-regulation of the c-myc protooncogene. J Biol Chem. 2000;275:5958–5965. doi: 10.1074/jbc.275.8.5958. [DOI] [PubMed] [Google Scholar]
- 40.Yin H, Wang L, Liu HL. ENO1 overexpression in pancreatic cancer patients and its clinical and diagnostic significance. Gastroenterol Res Pract. 2018;2018:3842198. doi: 10.1155/2018/3842198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Song Y, Luo Q, Long H, Hu Z, Que T, Zhang X, Li Z, Wang G, Yi L, Liu Z, Fang W, Qi S. Alpha-enolase as a potential cancer prognostic marker promotes cell growth, migration, and invasion in glioma. Mol Cancer. 2014;13:65. doi: 10.1186/1476-4598-13-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lomnytska MI, Becker S, Gemoll T, Lundgren C, Habermann J, Olsson A, Bodin I, Engström U, Hellman U, Hellman K, Hellström AC, Andersson S, Mints M, Auer G. Impact of genomic stability on protein expression in endometrioid endometrial cancer. Br J Cancer. 2012;106:1297–1305. doi: 10.1038/bjc.2012.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang YS, Wu W, Walsh G, Hong WK, Mao L. Enolase-alpha is frequently down-regulated in non-small cell lung cancer and predicts aggressive biological behavior. Clin Cancer Res. 2003;9:3641–3644. [PubMed] [Google Scholar]
- 44.Wang J, Li Y, Ding M, Zhang H, Xu X, Tang J. Molecular mechanisms and clinical applications of miR-22 in regulating malignant progression in human cancer (Review) Int J Oncol. 2017;50:345–355. doi: 10.3892/ijo.2016.3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang L, Fang J, Chen J. Tumor cell senescence response produces aggressive variants. Cell Death Discov. 2017;3:17049. doi: 10.1038/cddiscovery.2017.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Achuthan S, Santhoshkumar TR, Prabhakar J, Nair SA, Pillai MR. Drug-induced senescence generates chemoresistant stemlike cells with low reactive oxygen species. J Biol Chem. 2011;286:37813–37829. doi: 10.1074/jbc.M110.200675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Navale AM, Paranjape AN. Glucose transporters: physiological and pathological roles. Biophys Rev. 2016;8:5–9. doi: 10.1007/s12551-015-0186-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tsukioka M, Matsumoto Y, Noriyuki M, Yoshida C, Nobeyama H, Yoshida H, Yasui T, Sumi T, Honda K, Ishiko O. Expression of glucose transporters in epithelial ovarian carcinoma: correlation with clinical characteristics and tumor angiogenesis. Oncol Rep. 2007;18:361–367. [PubMed] [Google Scholar]
- 49.Ma Y, Wang W, Idowu MO, Oh U, Wang XY, Temkin SM, Fang X. Ovarian cancer relies on glucose transporter 1 to fuel glycolysis and growth: anti-tumor activity of BAY-876. Cancers (Basel) 2018;11 doi: 10.3390/cancers11010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Viscarra T, Buchegger K, Jofre I, Riquelme I, Zanella L, Abanto M, Parker AC, Piccolo SR, Roa JC, Ili C, Brebi P. Functional and transcriptomic characterization of carboplatin-resistant A2780 ovarian cancer cell line. Biol Res. 2019;52:13. doi: 10.1186/s40659-019-0220-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hudson CD, Savadelis A, Nagaraj AB, Joseph P, Avril S, DiFeo A, Avril N. Altered glutamine metabolism in platinum resistant ovarian cancer. Oncotarget. 2016;7:41637–41649. doi: 10.18632/oncotarget.9317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8:519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci. 2016;41:211–218. doi: 10.1016/j.tibs.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fouad YA, Aanei C. Revisiting the hallmarks of cancer. Am J Cancer Res. 2017;7:1016–1036. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.