

Abstract
Background and Purpose –
Our recent study demonstrated that release of peroxiredoxin 2 (Prx2) from red blood cells (RBC) is involved in the inflammatory response and brain injury after intracerebral hemorrhage. The current study investigated the role of extracellular Prx2 in hydrocephalus development after experimental intraventricular hemorrhage.
Methods –
There were four parts in this study. First, Sprague-Dawley rats received an intraventricular injection of lysed RBC or saline and were euthanized at one hour for Prx2 measurements. Second, rats received an intraventricular injection of Prx2, deactivated Prx2 or saline. Third, lysed-RBC was co-injected with conoidin A, a Prx2 inhibitor, or vehicle. Fourth, rats received Prx2 injection and were treated with minocycline or saline (i.p.). The effects of Prx2 and the inhibitors were examined using magnetic resonance imaging assessing ventriculomegaly, histology assessing ventricular wall damage, and immunohistochemistry to assess inflammation, particularly at the choroid plexus,
Results –
Intraventricular injection of lysed-RBC resulted in increased brain Prx2 and hydrocephalus. Intraventricular injection of Prx2 alone caused hydrocephalus, ventricle wall damage, activation of choroid plexus epiplexus cells (macrophages) and an accumulation of neutrophils. Conoidin A attenuated lysed-RBC induced injury. Systemic minocycline treatment reduced the epiplexus cell activation and hydrocephalus induced by Prx2.
Conclusions –
Prx2 contributed to the intraventricular hemorrhage-induced hydrocephalus, probably by inducing inflammatory responses in choroid plexus and ventricular wall damage.
Keywords: hydrocephalus, intraventricular hemorrhage, minocycline, peroxiredoxin 2
Graphical Abstract
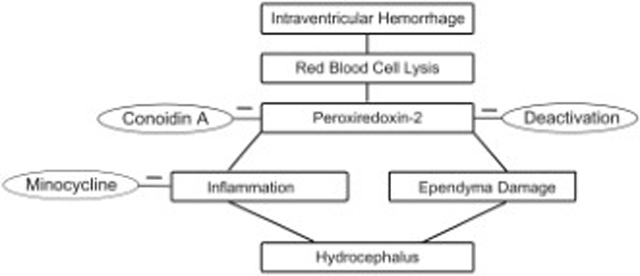
After intraventricular hemorrhage, lysis of red blood cells results in peroxiredoxin 2 release and causes hydrocephalus, which is reduced by conoidin A, an inhibitor of peroxiredoxin 2. Intraventricular peroxiredoxin 2-induced hydrocephalus is associated with inflammation in the choroid plexus and ventricular wall damage, and can be attenuated by minocycline, a non-specific inhibitor of macrophages/microglia. These data suggest that peroxiredoxin 2 may be a potential therapeutic target for hydrocephalus following intraventricular hemorrhage
Introduction:
Many patients with intracerebral hemorrhage (ICH) or subarachnoid hemorrhage (SAH) also have intraventricular hemorrhage (IVH) and it is a significant cause of morbidity and mortality.1 Hydrocephalus develops in up to 55% of IVH patients and is recognized as a critical determinant of prognosis2-4 and recent studies indicate that both IVH and hydrocephalus are predictors of poor outcome after ICH.5-7 However, the underlying mechanisms of hydrocephalus development after IVH are still not fully understood.8, 9 Erythrocyte lysis and the release of intracellular components may play an important role IVH-induced hydrocephalus.10-14
Peroxiredoxins are a group of proteins that regulate redox signaling playing an essential role in cell metabolism by catalyzing peroxide reduction to balance cellular hydrogen peroxide levels.15 Peroxiredoxin 2 (Prx2), one of the six members of peroxiredoxin family, is the third most abundant protein in red blood cells (RBC).16 Evidence suggests that the release of Prx2 to the extracellular space is involved in the progression of brain injury after ICH,17 SAH,18 traumatic brain injury (TBI)19 and ischemic brain injury20 by initiating inflammation. However, the role of Prx2 in hydrocephalus development after IVH and its possible mechanisms have not been studied.
Conoidin A is a cell-permeable inhibitor of the two mammalian peroxiredoxin homologues (Prx1 and Prx2).21, 22 In this study, we use conoidin A as a Prx2 inhibitor23 and explored its effects in rat IVH model. Minocycline, a tetracycline derivative with anti-inflammatory effects, has been shown to reduce brain injury after ICH24, TBI25 or reperfusion injury in experimental and human ischemic stroke by reducing microglia/macrophage activation.26, 27 Our recent studies found that macrophage activation in the choroid plexus is associated with hydrocephalus development28, 29. Minocycline reduces epiplexus macrophage activation and hydrocephalus in spontaneous hypertensive rats28.
The current study examined: (1) the role of Prx2 in the development of hydrocephalus after experimental IVH, as well as its impact on the ventricular wall and choroid plexus (CP) inflammatory events; (2) whether co-injection of a Prx2 inhibitor, conoidin A, could attenuate the injury caused by intraventricular injection of lysed-RBC; and (3) the effects of systemic minocycline treatment on Prx2-induced hydrocephalus.
Materials and Methods:
The authors declare that all supporting data are available within the article.
Animal Preparation and Intraventricular Injection
Animal protocols were approved by the University of Michigan Committee on the Use and Care of Animals. The University of Michigan has an Animal Welfare Assurance on file with the Office for Protection from Research Risks and is fully accredited by the American Association for the Accreditation of Laboratory Animal Care. The studies followed the Guide for The Care and Use of Laboratory Animals (National Research Council) and comply with the ARRIVE guidelines for reporting in vivo experiments.
A total of 99 Sprague-Dawley rats were used in this study, 87 males and 12 females that were 3 to 4 months old and weighed 250 to 350 g (Charles River Laboratories, Portage, MI, USA). Animals were anesthetized with pentobarbital (50 mg/kg, i.p.), the right femoral artery was catheterized for blood collection and blood pressure monitoring. Core body temperature was maintained at 37.5°C with a feedback-controlled heating pad. Rats were then positioned in a stereotaxic frame (Kopf Instruments, Tujunga, CA, USA). A cranial burr hole (1 mm) was drilled 0.6 mm posterior, 4.5 mm ventral, and 1.7 mm lateral to the bregma and a 26-gauge needle was inserted perpendicularly through the burr hole into the right lateral ventricle.30
Lysed-RBCs were prepared as previously described.31 Briefly, autologous blood was obtained and washed with saline for three times. Packed-RBCs (hematocrit level about 87%) were obtained by centrifugation of the washed autologous blood with supernatant and buffy coat discarded. Lysed-RBCs were prepared by freezing the packed-RBCs in liquid nitrogen (3 minutes) followed by thawing at 37°C (5 minutes) for a total of three times.
Prx2 solution was prepared with recombinant rat peroxiredoxin 2 protein (1mg/ml; Novus Biological cop.; NBP2–52150, 25 μg), diluted with equal volume of saline. Deactivated Prx2 (Prx2(heat)) solution was obtained by heating the Prx2 solution at 56°C for 30 minutes32 one day before injection.
Conoidin A, a Prx2 inhibitor (Cayman Chemical; 15605), was diluted in dimethyl sulfoxide (DMSO) at concentration of 5mM and then co-injected with lysed-RBC (1:100 dilution) at a final concentration of 50μM. That dose of Conoidin A could reduce brain injury caused by lysed RBCs in an ICH model17. The same volume of DMSO was co-injected with lysed-RBC (1:100 dilution) as vehicle control. 30μl of lysed-RBC, saline, lysed-RBC+conoidin A or lysed-RBC+vehicle was injected into the right lateral ventricle over 6 minutes (5μl/min) by a micro infusion pump (Harvard Apparatus Inc.). Fifty μl of Prx2 solution, deactivated Prx2 solution or saline was injected over 7 minutes (~7 μl/min). The needle was removed 1 minute after injection, burr holes filled by bone wax and skin incisions sutured closed.
Experimental Groups
There were four parts to this study. First, 12 male rats randomly received an intraventricular injection of either lysed-RBC or saline (30μl, n=6 each group). All rats were euthanized 1 hour after injection and the brain removed for either Western blot analysis (n=4 each group) or histological examination. Second, 18 male and 12 female rats were randomly divided into Prx2, Prx2(heat), saline, Prx2-female and saline-female groups (n=6 each group) and received an 50μl intraventricular injection of either Prx2 (25 μg) solution, deactivated Prx2 (25 μg) solution or saline. All of them received an MRI scan one day later and were then euthanized. In addition, 12 male rats were randomly divided into Prx2 day-3 group and saline day-3 group (n=6 each group). Each rat received an intraventricular injection of Prx2 solution or saline as described before. Rats were euthanized 3 days later after MRI scans and brains used for histology. Third, 28 male rats were randomly divided into 2 groups and had a 30μl intraventricular injection of lysed-RBC+conoidin A or lysed-RBC+vehicle (DMSO). Eight rats from each group were euthanized at one day and 6 rats from each group were euthanized at 3 days after injection, all rats had MRI scans just before euthanasia. The brains were used for histological examination. Fourth, 17 male rats received intraventricular injection of Prx2 solution (50μl, 25 μg) as described above. Rats were divided into minocycline group (n=9) and control group (n=8) and treated with intraperitoneal injection of minocycline (45 mg/kg, one hour and 12 hours after Prx2 injection at day 0 and 22.5 mg/kg at day 1) or saline (equal volume) starting right after intraventricular injection.24 Rats were euthanized at day 1 after MRI scans and the brains were used for histology. Randomization was carried out using odd/even numbers for the treatment groups. Dead animals were excluded from this study.
Magnetic Resonance Imaging and Ventricular Volume Measurement
MRIs were performed with rats anesthetized by 2% isoflurane using a 9.4-T Varian MR scanner (Varian, Palo Alto, CA, USA) with a T2 fast spin-echo sequence. A total of 25 coronal slices were obtained for each scan with a view field of 35 mm×35 mm and a slice thickness of 0.5 mm. Ventricular volumes were measured as previously described.10 Bilateral ventricles were outlined in each slice and measured by another person blinded to the experiments. Ventricular volume was calculated by multiplying the area measured in each slice with the slice thickness and then summing over all slices.
Western Blot Analysis
Western blot analysis was performed as previously described.11, 12 Briefly, rats were overdosed with pentobarbital (390 mg/kg, intraperitoneally) and underwent transcardiac perfusion with 0.1 mol/L phosphate-buffered saline (pH 7.4). Periventricular brain tissue was obtained after perfusion, immersed in western sample buffer and sonicated. Protein concentration was measured with Bio-Rad (Hercules, CA, USA) protein assay kit and 50μg protein from each sample was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a Hybond-C pure nitrocellulose membrane (Amersham, Pittsburgh, PA, USA).
Membranes were probed with the following primary antibodies: rabbit anti rat monoclonal peroxiredoxin 2 antibody (1:5000 dilution; Novus Biological cop.; NBP2-67887) and mouse anti rat β-actin (1:40000 dilution; Cell Signaling cop.; 4970S). The secondary antibodies were goat anti rabbit IgG (Bio-Rad, 1:5000) and goat anti-mouse IgG (Bio-Rad, 1:5000). Antigen-antibody complexes were visualized with the ECL chemiluminescence system (Amersham) and exposed to a Kodak X-OMAT film (Rochester, NY, USA). The relative densities of bands against β-actin were analyzed with NIH ImageJ program.
Immunohistochemistry
After an overdose of pentobarbital (390 mg/kg, intraperitoneally), rats underwent transcardiac perfusion with 4% paraformaldehyde in 0.1 mol/L phosphate-buffered saline (pH 7.4). Brains were removed, kept in 4% paraformaldehyde for 24 hours, and then immersed in 30% sucrose for 2 to 3 days at 4°C. Brains were embedded in optimal cutting temperature compound (Sakura Finetek, Torrance, CA, USA) and sectioned at 18 μm using a cryostat. Immunohistochemical staining was performed using the avidin–biotin complex technique as previously described.10
The primary antibodies used in this study were: goat anti albumin goat anti Iba-1 (1:400 dilution; Abcam, Cambridge, MA, USA; ab5076), rabbit anti myeloperoxidase (MPO; 1:200 dilution; Invitrogen; PA5–16672), and mouse anti CD68 (1:200 dilution; Abcam, Cambridge, MA, USA; ab31630). Hematoxylin was used as a counterstain. Negative controls omitted the primary antibody.
Ventricle wall damage and other measurements
Statistical analyses of ventricle wall damage were performed as previously described.33 Hematoxylin and eosin (H&E) stained brain sections were obtained for each rat. The length of the ependyma that was disrupted or detached from the periventricular parenchyma was measured, as well as the total bilateral ventricle wall length. The degree of ventricle wall damage was calculated as a percentage by dividing the length of disruption over the total ventricular surface perimeter. In order to analyze inflammatory cell changes at the CP, the number of positive cells was calculated as a percentage of the total cell count of the bilateral CPs for each rat. Morphological changes in positive cells were evaluated by the mean value of soma size measured in six different views (×40 magnification) taken from bilateral CPs for each rat. Three views were taken from both the left and right lateral ventricle CP to maximally cover the whole CP. All analyses were performed using Image J software by a blinded observer.
Statistical Analysis
Values are shown as mean ± SD. Student’s t-test and one-way ANOVA test with Tukey post hoc test were used for data analyses. Differences were considered as significant with p<0.05. The sample size of minocycline treatment was determined based on the data of part 2 (ventricular volumes at day 1 after Prx2 injection: 31.3 ± 4.7 mm3). An n=8 per group would give 90% power for detecting a 20% decrease in ventricular volume with minocycline.
Results:
Mortality within 24 hours was 13% (5 of 39) after lysed-RBC injection, 2% (1 of 58) after Prx2 protein injection and zero in all other groups. All dead rats were excluded from analyses for failing to accomplish the entire course of the experiment.
Prx2 protein levels were significantly increased in the periventricular zone 1 hour after lysed-RBC injection (Prx2/β-actin ratio: 0.86 ± 0.58 vs. 0.21 ± 0.12 in saline, p <0.05, Fig. 1A). Injection of exogenous Prx2 caused significant ventricle dilation in both male and female rats at 24 hours (Fig. 1B). However, the ventricular dilation in females was less severe than in males (p<0.01, Fig. 1B) and all the following experiments were performed in males.
Fig. 1.

(A) Prx2 protein levels in the ipsilateral periventricular zone one hour after lysed-RBC or saline injection. Values are mean ± SD, n=4, *p<0.05 vs. saline group; (B) Examples of T2 imaging and quantification of ventricle volumes in male and female rats one day after intraventricular injection of saline or Prx2. Values are mean ± SD, n=6, #p<0.01 vs. saline-male group, ##p<0.01 vs. Prx2-female group.
T2 MRIs showed bilateral ventricle dilation rats injected with either deactivated Prx2 (18.6 ± 6.3 mm3) or Prx2 (31.3 ± 4.7 mm3) at day 1 compared to saline injection (10.7 ± 1.6 mm3, p<0.01, Fig.2A). However, ventricular volumes were much greater with Prx2 than deactivated Prx2 (p<0.01, Fig.2A). In addition, there were significant ventricle wall damage and disruption of the ependymal surface with Prx2 injection group compared with saline injection group (50 ± 4 % vs. 12 ± 3 % in saline, p<0.01, Fig. 2B). In contrast, there was no significant change of ventricle wall damage and disruption of the ependymal surface in the deactivated Prx2 injection group (14 ± 3%, p > 0.05, Fig. 2B) compared to saline injection. The Prx2-induced damage was especially noticeable in the frontal and lateral horns of the ventricles.
Fig. 2.

(A) Examples of T2 imaging and quantification of ventricular volume in male rats one day after injection of 50μl saline, deactivated Prx2 (Prx2(heat)) or Prx2 (25 μg) into the right lateral ventricle. Values are mean ± SD, n=10 in saline, n=6 in Prx2(heat) and n=13 in Prx2 group, #p<0.01 vs. saline group, ##p<0.01 vs. Prx2(heat) group; (B) Examples of H&E stained sections showing ventricle wall damage in male rats one day after intraventricular injection of saline, Prx2(heat) or Prx2 injection that are quantified in the bar graph. Values are mean ± SD, n=6, #p<0.01 vs. the other groups. Scale bar=50μm.
An accumulation of inflammatory cells was seen in the ventricles. Thus, the numbers of MPO (7.7 ± 4.8 % vs. 0.4 ± 0.1 % in saline, p<0.01) and Iba-1 (14.6 ± 4.1 % vs. 4.7 ± 1.9 % in saline, p<0.01) positive cells at the CP were significantly increased by Prx2 injection. In contrast, there was no significant change in the number of MPO (2.7 ± 1.2, p>0.05) or Iba-1 (8.8 ± 1.6, p<0.01) positive cells at the CP after deactivated Prx2 injection (Fig.3). The soma size of Iba-1 positive cells at the CP was significantly increased in the Prx2 injection group (36.6 ± 2.1 vs. 11.0 ± 1.9 μm2 in saline, p<0.01, Fig.3), indicating macrophage activation. The soma size of Iba1 labeled cells was also significantly increased in the deactivated Prx2 injection group (26.5 ± 5.6, p<0.01, Fig.3), but to a lesser extent than with Prx2 (p<0.01, Fig. 3).
Fig. 3.
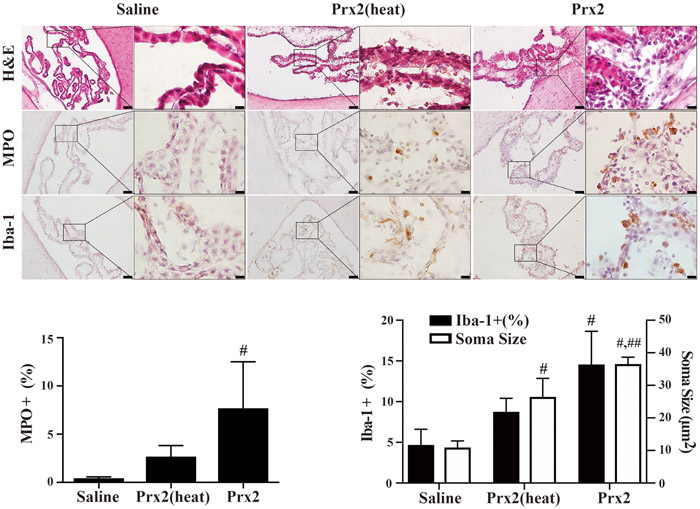
Examples of H&E stained sections myeloperoxidase (MPO; neutrophil marker) and Iba-1 (macrophage marker) immunohistochemistry at the choroid plexus one day after intraventricular saline, Prx2(heat) and Prx2 injection. The numbers of MPO and Iba-1 positive cells were quantified. Values are mean ± SD, n=6, #p<0.01 vs. saline group, ##p<0.01 vs. Prx2(heat) group. Scale bar = 50μm at low magnification, =10μm at high magnification.
Co-injection of a Prx2 inhibitor, conoidin A, significantly reduced the ventricular dilation induced by lysed-RBC (43.3 ± 6.1 mm3) compared with the vehicle co-injection group (55.8 ± 8.1 mm3) at 24 hours (p<0.01, Fig.4A). A similar tendency was also found at 3 days after injection, although that was not statistically significant (data not shown). Comparing with rats in vehicle group, a significant reduction of ventricle wall damage was found in rats with lysed-RBC+ conoidin A injection (52 ± 8 vs. 64 ± 7 % in lysed-RBC + vehicle, p<0.01, Fig.4B). Histological analysis of the accumulated inflammatory cells showed a tendency towards a decrease in Iba-1 positive cells at the CP, but this didn’t reach significance (Fig.4C). However, the difference in soma size between groups was significant (36.5 ± 3.0 vs. 62.9 ± 6.6 μm2 in lysed-RBC + vehicle, p<0.01, Fig.4C).
Fig. 4.

Effects of conoidin A on lysed RBC-induced hydrocephalus, ventricle wall damage and choroid plexus inflammation. (A) Examples of T2 MRIs one day after intraventricular injection of 30μl lysed-RBC with vehicle or conoidin A and quantification of ventricle volume. (B) Examples of H&E staining in the same two groups showing ventricular wall damage and quantification of that damage. (C) Examples of Iba-1 immunohistochemistry at the choroid plexus and corresponding H&E sections one day after intraventricular injection of 30μl lysed-RBC with vehicle or conoidin A. The number Iba-1 positive cells and soma size were quantified. Values are mean ± SD, n=8, #p<0.01 vs. lysed-RBC + vehicle group. Scale bar= 50μm at low magnification, =10μm at high magnification.
Large numbers of inflammatory cells were found gathered in and around the site of the lysed-RBC injection at 24 hours (Fig.5A). Co-injection of conoidin A significantly attenuated the accumulation of MPO (24 ± 7 vs. 37 ± 9 % in lysed-RBC + vehicle, p<0.01, Fig.5B), Iba-1 (26 ± 6 vs. 44 ± 13 % in lysed-RBC + vehicle, p<0.01, Fig. 5C) and CD68 (23 ± 3 vs. 39 ± 11 in lysed-RBC + vehicle, p<0.01, Fig.5D) positive cells in the lysed-RBC mass within the ventricle.
Fig. 5.
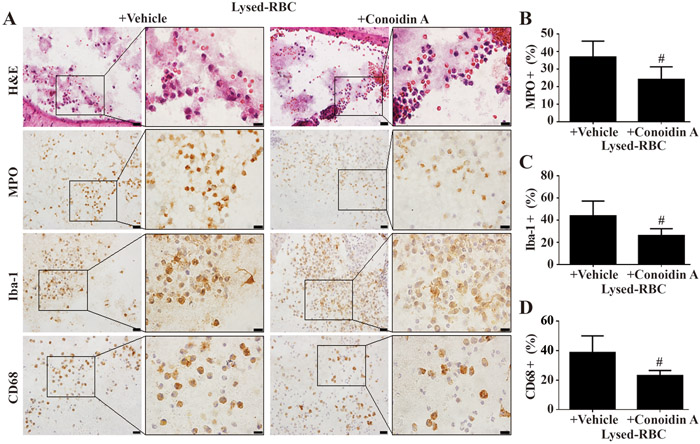
Effects of conoidin A on ventricular inflammation one day after intracerebroventricular injection of lysed-RBC. (A) H&E staining showed material from the lysed RBC within the ventricle that was associated with leukocytes. Immunohistohemistry for myeloperoxidase (MPO), Iba-1 and CD68 demonstrated the presence of both neutrophils (MPO positive) and macrophages (Iba-1 and CD68 positive cells). Conoidin A reduced the percentage of MPO (B), Iba-1 (C) and CD68 (D) positive cells associated with this material compared to vehicle treatment. Values are mean ± SD, n=8, #p<0.01 vs. lysed-RBC + vehicle group. Scale bar= 20μm at low magnification, =10μm at high magnification.
Systemic treatment with minocycline significantly reduced the ventricular dilation induced by Prx2 injection (15.6 ± 4.4 vs. 28.7 ± 6.3 mm3 in control, p<0.01, Fig.6A) at one day after injection. Ventricle wall damage was also significantly reduced with minocycline (42 ± 6%) compared to vehicle controls (51 ± 7 %, p<0.01; Fig.6B). While the number of Iba-1 positive cells at the CP was not significantly decreased, they were significantly smaller in size (28.9 ± 3.8 vs. 50.7 ± 3.7 μm2 in control, p<0.01, Fig.6C) after minocycline treatment. Also, the number of CD68 positive cells at the CP was significantly reduced in minocycline group (2.9 ± 1.4 vs. 7.5 ± 2.7 % in control, p<0.01, Fig.6C) at day 1 after Prx2 injection.
Fig. 6.

Effects of minocycline (Mino) on Prx2-induced hydrocephalus and choroid plexus inflammation. (A) Examples of T2 MRI one day after intraventricular Prx2 (25μg) injection with control or minocycline treatment. The MRIs were used to quantify ventricular volume with and without minocycline treatment. (B) Examples of H&E staining of different areas of the lateral ventricle in relation with and without minocycline treatment. The location of the areas in relation to MRIs in the same animals is also shown. The H&E stained sections were used to quantify ventricle wall damage with different treatment (bar graph). (C) Examples of H&E staining and immunohistochemistry of the choroid plexus after Prx2 injection with and without minocycline treatment. These were used to determine the effect of minocycline on the number of Iba-1 and CD68 positive cells at the choroid plexus as well as soma size for the Iba-1 positive cells (bar graphs). Values are mean ± SD, n=8 in control and n=9 in Mino group, #p<0.01vs. Control group. Scale bar= 50μm in (A) and low magnification of (B) and (C), =10μm in high magnification of (B) and (C).
Discussion:
The major findings the current study were: (1) intraventricular injection of lysed-RBC caused an increase in periventricular Prx2; (2) intraventricular injection of Prx2 resulted in hydrocephalus, ventricle wall damage and significant inflammation at the CP. However, deactivated Prx2 caused slight ventricle dilation and there are glycerol and dithiothreitol in Prx2 solution, suggesting that intraventricular glycerol, dithiothreitol and deactivated Prx2 protein may cause moderate ventricle dilation; (3) co-injection of a Prx2 inhibitor, conoidin A, attenuated lysed-RBC induced hydrocephalus, ventricle wall damage and inflammatory responses both at the CP and in the ventricle around the lysed-RBC mass; (4) minocycline treatment attenuated Prx2-induced hydrocephalus and ventricle wall damage, as well as macrophage activation at the CP.
RBC lysis and hydrocephalus development after IVH have been studied in the recent years.10, 11, 13, 14 In the current study, lysed RBCs resulted in more severe hydrocephalus. We have demonstrated that hemoglobin and its degradation products (e.g. iron) can cause hydrocephalus.11 Prx2 is one of the major components in RBCs and may be released extracellularly initiating a series of destructive inflammatory responses, such as neutrophil accumulation, macrophage activation, and release of pro-inflammatory factors.34-37 Considering the abundance of Prx2 in the RBC cytoplasm (5.6 mg/ml),38, 39 its contribution to IVH-induced hydrocephalus after RBC lysis may be of great importance, which has not yet been examined.
The current study confirmed the hydrocephalus-inducing effect of intraventricular lysed-RBC injection, which is in accordance with a previous study.11 There was a significant elevation of Prx2 level in the periventricular tissue one hour after lysed RBC injection. Although lysed RBC mass was removed during brain tissue sampling, some lysed RBCs might penetrate into the periventricular zone. Therefore, Prx2 protein measured by Western blotting represents the total Prx2 levels, which could come from release of lysed RBCs, brain cells and some RBCs penetrating into the periventricular zone. To explore the role of extracellular Prx2, intracerebroventricular injections were employed. Similar and significant impacts were observed in the intraventricular Prx2 injection model as with lysed-RBC injection, including ventricle dilation, ventricle wall damage, neutrophil accumulation and macrophage activation at the CP. Furthermore, co-injection of a Prx2 inhibitor, conoidin A, significantly attenuated the hydrocephalus, ventricle wall damage and epiplexus cell activation caused by lysed-RBC. Conoidin A inhibits peroxiredoxins by binding to the catalytic site on the enzyme21. The IC50 of Conoidin A is 23 μM.22 All of these findings pinpoints that Prx2 can play a major role in the development of IVH-induced hydrocephalus. Interestingly, while intracerebroventricular injection of Prx2 caused ventriculomegaly in both male and female rats, the effect was significantly greater in males. The underlying mechanism for this difference merits further investigation.
The underlying mechanisms associated with this Prx2-induced ventricular dilation have not been studied. Extracellular Prx2, along with its reduced form and substrates, can act as damage-associated molecular patterns (DAMPs) that induce microglial/macrophage activation, inflammatory responses and neuronal death in models of hemorrhagic and ischemic stroke.15, 17, 34, 35, 37, 40, 41 The current study found marked increase in the accumulation of inflammatory cells in the CP and ependymal cell destruction along the ventricle wall after both Prx2 and lysed-RBC intraventricular injection, with the latter being reduced by the Prx2 inhibitor conoidin A. This confirms a similar inflammation-related, damage-inducing effect of extracellular Prx2 on the ventricular system.
Morphological changes were observed in the Iba-1 positive cells (macrophages) at the CP after both lysed-RBC and Prx2 injections. They became round rather than ramified in shape, and had a significantly upregulated soma size indicative of macrophage activation that might contribute to hydrocephalus development. Using the number of CD68 positive cells as an indicator for macrophage activation,27 we tested this hypothesis and showed that both Prx2 inhibition and minocycline treatment downregulated macrophage activation and were able to attenuate the ventricle dilation and related damage caused by lysed-RBC and Prx2 injection. These findings further support the role of macrophage activation in the hydrocephalus induced by Prx2 protein after IVH.
The material from the RBC lysate caused a significant influx of leukocytes (neutrophils and macrophages) into the ventricle. The source of those ventricular leukocytes, across the CP or the ventricular wall, is still uncertain. The triggering signal and specific pathway for Prx2 to activate these cells and their recruitment route into the ventricle need to be determined.
The current study used lysed-RBC and Prx2 injection models to analyze the effect of extracellular Prx2 in hydrocephalus development after IVH. The deactivation and degradation of Prx2, which was already completely released when injected and exposed to various enzymes in the cerebrospinal fluid, may impact the degree of hydrocephalus development. The reduced ventricle dilation with denatured Prx2 supports this hypothesis. Further studies with a whole-blood injection model and longer follow-up are essential to better resemble the physiological release and potential degradation of Prx2 after IVH.
There are several other limitations to this study. 1) Only the acute effects of Prx2 on ventriculomegaly and underlying mechanisms were examined. While those effects were detrimental, it is possible that it may have beneficial effects later after IVH (e.g. in tissue repair). 2) Conoidin A is an inhibitor of peroxiredoxin protein family and could affect the function of the several members of this family. Thus, further studies with a more specific approach to inhibiting Prx2 are necessary in order to rule out possible effects due to other Prx members. 3) While Prx2 release from RBC may be an initial trigger, the release of other Prx members from other cell types may further potentiate injury. 4) Although the effects of Prx2 on hydrocephalus was also examined in adult females, the estrous cycle of female rats was not monitored. 5) The activity of Prx2 was not measured.
In conclusion, the present study revealed that Prx2 released from RBC into the extracellular space contributes to IVH-induced hydrocephalus and related damage, including triggering downstream inflammatory responses. Prx2 may be a potential therapeutic target for hydrocephalus after IVH.
Supplementary Material
Acknowledgments
Sources of Funding
YH, RFK and GX are supported by grants NS-091545, NS-090925, NS-096917, NS-106746 and NS-112394 from the National Institutes of Health (NIH).
Footnotes
Disclosures
None.
References:
- 1.Nieuwkamp DJ, de Gans K, Rinkel GJ, Algra A. Treatment and outcome of severe intraventricular extension in patients with subarachnoid or intracerebral hemorrhage: A systematic review of the literature. J Neurol. 2000;247:117–121 [DOI] [PubMed] [Google Scholar]
- 2.Bhattathiri PS, Gregson B, Prasad KS, Mendelow AD, Investigators S. Intraventricular hemorrhage and hydrocephalus after spontaneous intracerebral hemorrhage: Results from the stich trial. Acta Neurochir Suppl. 2006;96:65–68 [DOI] [PubMed] [Google Scholar]
- 3.Passero S, Ulivelli M, Reale F. Primary intraventricular haemorrhage in adults. Acta Neurol Scand. 2002;105:115–119 [DOI] [PubMed] [Google Scholar]
- 4.Lee SH, Park KJ, Park DH, Kang SH, Park JY, Chung YG. Factors associated with clinical outcomes in patients with primary intraventricular hemorrhage. Med Sci Monit. 2017;23:1401–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanley DF. Intraventricular hemorrhage: Severity factor and treatment target in spontaneous intracerebral hemorrhage. Stroke. 2009;40:1533–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mustanoja S, Satopaa J, Meretoja A, Putaala J, Strbian D, Curtze S, et al. Extent of secondary intraventricular hemorrhage is an independent predictor of outcomes in intracerebral hemorrhage: Data from the helsinki ich study. Int J Stroke. 2015;10:576–581 [DOI] [PubMed] [Google Scholar]
- 7.Trifan G, Arshi B, Testai FD. Intraventricular hemorrhage severity as a predictor of outcome in intracerebral hemorrhage. Front Neurol. 2019;10:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Strahle J, Garton HJ, Maher CO, Muraszko KM, Keep RF, Xi G. Mechanisms of hydrocephalus after neonatal and adult intraventricular hemorrhage. Transl Stroke Res. 2012;3:25–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karimy JK, Zhang J, Kurland DB, Theriault BC, Duran D, Stokum JA, et al. Inflammation-dependent cerebrospinal fluid hypersecretion by the choroid plexus epithelium in posthemorrhagic hydrocephalus. Nat Med. 2017;23:997–1003 [DOI] [PubMed] [Google Scholar]
- 10.Chen Z, Gao C, Hua Y, Keep RF, Muraszko K, Xi G. Role of iron in brain injury after intraventricular hemorrhage. Stroke. 2011;42:465–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao C, Du H, Hua Y, Keep RF, Strahle J, Xi G. Role of red blood cell lysis and iron in hydrocephalus after intraventricular hemorrhage. J Cereb Blood Flow Metab. 2014;34:1070–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao F, Liu F, Chen Z, Hua Y, Keep RF, Xi G. Hydrocephalus after intraventricular hemorrhage: The role of thrombin. J Cereb Blood Flow Metab. 2014;34:489–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garton T, Hua Y, Xiang J, Xi G, Keep RF. Challenges for intraventricular hemorrhage research and emerging therapeutic targets. Expert Opin Ther Targets. 2017;21:1111–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Q, Feng Z, Tan Q, Guo J, Tang J, Tan L, et al. Post-hemorrhagic hydrocephalus: Recent advances and new therapeutic insights. J Neurol Sci. 2017;375:220–230 [DOI] [PubMed] [Google Scholar]
- 15.Park MH, Jo M, Kim YR, Lee CK, Hong JT. Roles of peroxiredoxins in cancer, neurodegenerative diseases and inflammatory diseases. Pharmacol Ther. 2016;163:1–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sarafian TA, Verity MA, Vinters HV, Shih CC, Shi L, Ji XD, et al. Differential expression of peroxiredoxin subtypes in human brain cell types. J Neurosci Res. 1999;56:206–212 [PubMed] [Google Scholar]
- 17.Bian L, Zhang J, Wang M, Keep RF, Xi G, Hua Y. Intracerebral hemorrhage-induced brain injury in rats: The role of extracellular peroxiredoxin 2. [published online July 4, 2019]. Transl Stroke Res. 2019. https://link.springer.com/article/10.1007/s12975-019-00714-x. Accessed July 5, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang ZH, Han YL, Wang CX, Zhou CH, Wu LY, Zhang HS, et al. The effect of subarachnoid erythrocyte lysate on brain injury: A preliminary study. Biosci Rep. 2016;36:e00359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Connor DE Jr., Chaitanya GV, Chittiboina P, McCarthy P, Scott LK, Schrott L, et al. Variations in the cerebrospinal fluid proteome following traumatic brain injury and subarachnoid hemorrhage. Pathophysiology. 2017;24:169–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shichita T, Hasegawa E, Kimura A, Morita R, Sakaguchi R, Takada I, et al. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat Med. 2012;18:911–917 [DOI] [PubMed] [Google Scholar]
- 21.Haraldsen JD, Liu G, Botting CH, Walton JG, Storm J, Phalen TJ, et al. Identification of conoidin a as a covalent inhibitor of peroxiredoxin ii. Org Biomol Chem. 2009;7:3040–3048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu G, Botting CH, Evans KM, Walton JA, Xu G, Slawin AM, et al. Optimisation of conoidin a, a peroxiredoxin inhibitor. ChemMedChem. 2010;5:41–45 [DOI] [PubMed] [Google Scholar]
- 23.Ryu DY, Kim KU, Kwon WS, Rahman MS, Khatun A, Pang MG. Peroxiredoxin activity is a major landmark of male fertility. Sci Rep. 2017;7:17174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dai S, Hua Y, Keep RF, Novakovic N, Fei Z, Xi G. Minocycline attenuates brain injury and iron overload after intracerebral hemorrhage in aged female rats. Neurobiol Dis. 2019;126:76–84 [DOI] [PubMed] [Google Scholar]
- 25.Hanlon LA, Huh JW, Raghupathi R. Minocycline transiently reduces microglia/macrophage activation but exacerbates cognitive deficits following repetitive traumatic brain injury in the neonatal rat. J Neuropathol Exp Neurol. 2016;75:214–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Y, Salayandia VM, Thompson JF, Yang LY, Estrada EY, Yang Y. Attenuation of acute stroke injury in rat brain by minocycline promotes blood-brain barrier remodeling and alternative microglia/macrophage activation during recovery. J Neuroinflammation. 2015;12:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yew WP, Djukic ND, Jayaseelan JSP, Walker FR, Roos KAA, Chataway TK, et al. Early treatment with minocycline following stroke in rats improves functional recovery and differentially modifies responses of peri-infarct microglia and astrocytes. J Neuroinflammation. 2019;16:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gu C, Hao X, Li J, Hua Y, Keep RF, Xi G. Effects of minocycline on epiplexus macrophage activation, choroid plexus injury and hydrocephalus development in spontaneous hypertensive rats. J Cereb Blood Flow Metab. 2019;39:1963–1948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wan Y, Hua Y, Garton HJ, Novakovic N, Keep RF, Xi G. Activation of epiplexus macrophages in hydrocephalus caused by subarachnoid hemorrhage and thrombin. CNS Neuroscience & Therapeutics. 2019;25:1134–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lodhia KR, Shakui P, Keep RF. Hydrocephalus in a rat model of intraventricular hemorrhage. Acta Neurochir Suppl. 2006;96:207–211 [DOI] [PubMed] [Google Scholar]
- 31.Xi G, Keep RF, Hoff JT. Erythrocytes and delayed brain edema formation following intracerebral hemorrhage in rats. J Neurosurg. 1998;89:991–996 [DOI] [PubMed] [Google Scholar]
- 32.Simon J, Muller J, Ghazaryan A, Morsbach S, Mailander V, Landfester K. Protein denaturation caused by heat inactivation detrimentally affects biomolecular corona formation and cellular uptake. Nanoscale. 2018;10:21096–21105 [DOI] [PubMed] [Google Scholar]
- 33.Okubo S, Strahle J, Keep RF, Hua Y, Xi G. Subarachnoid hemorrhage-induced hydrocephalus in rats. Stroke. 2013;44:547–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garcia-Bonilla L, Iadecola C. Peroxiredoxin sets the brain on fire after stroke. Nat Med. 2012;18:858–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salzano S, Checconi P, Hanschmann EM, Lillig CH, Bowler LD, Chan P, et al. Linkage of inflammation and oxidative stress via release of glutathionylated peroxiredoxin-2, which acts as a danger signal. Proc Natl Acad Sci U S A. 2014;111:12157–12162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knoops B, Argyropoulou V, Becker S, Ferte L, Kuznetsova O. Multiple roles of peroxiredoxins in inflammation. Mol Cells. 2016;39:60–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu Y, Zhang XS, Zhang ZH, Zhou XM, Gao YY, Liu GJ, et al. Peroxiredoxin 2 activates microglia by interacting with toll-like receptor 4 after subarachnoid hemorrhage. J Neuroinflammation. 2018;15:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Low FM, Hampton MB, Peskin AV, Winterbourn CC. Peroxiredoxin 2 functions as a noncatalytic scavenger of low-level hydrogen peroxide in the erythrocyte. Blood. 2007;109:2611–2617 [DOI] [PubMed] [Google Scholar]
- 39.Low FM, Hampton MB, Winterbourn CC. Peroxiredoxin 2 and peroxide metabolism in the erythrocyte. Antioxid Redox Signal. 2008;10:1621–1630 [DOI] [PubMed] [Google Scholar]
- 40.Zhong Q, Zhou K, Liang QL, Lin S, Wang YC, Xiong XY, et al. Interleukin-23 secreted by activated macrophages drives gammadeltat cell production of interleukin-17 to aggravate secondary injury after intracerebral hemorrhage. J Am Heart Assoc. 2016;5:e004340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gulke E, Gelderblom M, Magnus T. Danger signals in stroke and their role on microglia activation after ischemia. Ther Adv Neurol Disord. 2018;11:1756286418774254. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.