

Abstract
Background
Unexpected allergic reactions to peanut are the most common cause of fatal food-related anaphylaxis. Mechanisms underlying the variable severity of peanut allergic reactions remain unclear.
Objectives
To expand mechanistic understanding of reaction severity in peanut allergy.
Methods
We performed an integrated transcriptomic and epigenomic study of peanut allergic children as they reacted in vivo during double-blind, placebo-controlled peanut challenges. We integrated whole blood transcriptome and CD4+ T-cell epigenome profiles to identify molecular signatures of reaction severity. Through linear mixed effects modeling, network construction, and causal mediation analysis, we identified genes, CpGs, and their interactions that mediate reaction severity. Findings were replicated in an independent cohort.
Results
We identified 318 genes with changes in expression during the course of reaction associated with reaction severity, and 203 CpG sites with differential DNA methylation associated with reaction severity. After replicating these findings in an independent cohort, we constructed interaction networks with the identified peanut severity genes and CpGs. These analyses and leukocyte deconvolution highlighted neutrophil-mediated immunity. We identified NFKBIA and ARG1 as hubs in the networks and three groups of interacting key node CpGs and peanut severity genes encompassing immune response, chemotaxis, and regulation of macroautophagy. Additionally, we found that gene expression of PHACTR1 and ZNF121 causally mediate the association between methylation at corresponding CpGs and reaction severity, suggesting that methylation may serve as an anchor upon which gene expression modulates reaction severity.
Conclusions
Our findings enhance current mechanistic understanding of the genetic and epigenetic architecture of reaction severity in peanut allergy.
Keywords: food allergy, peanut allergy, reaction severity, transcriptome, epigenome, integrated network, causal mediation
Capsule Summary
This integrated transcriptomic and epigenomic study of peanut allergic children reacting to peanut in vivo identified peripheral blood gene expression and methylation signatures, and interactions between them, associated with reaction severity.
Graphical Abstarct
Introduction
Peanut allergy is a clinical and public health problem affecting 2 to 5% of children1 and 1% of the overall population in the United States.2 Unexpected allergic reactions to peanut are the most common cause of fatal food-related anaphylaxis.3 Peanut allergic individuals are at daily risk for potentially life-threatening angioedema, respiratory difficulty, cardiovascular compromise, and/or anaphylaxis following ingestion of peanut.4 There is no cure for peanut allergy and our understanding of its causation and pathobiology remains incomplete.4
Mechanisms underlying the variable severity of peanut allergic reactions remain unclear.5 Exposure to equal amounts of peanut may cause anaphylaxis in one individual but only minor throat itching in another, despite similar profiles on clinical assays such as serum specific IgE (sIgE) and allergy skin prick test (SPT). Mechanisms underlying this heterogeneity in reaction severity are not known.5 The limitations of currently available assays and our understanding of peanut allergy have far-reaching implications on health and lifestyle, as many peanut allergic individuals constantly worry about peanut exposure and the possibility of severe reactions.6 Elucidating mechanisms underlying reaction severity in peanut allergy could enable effective treatments to mitigate or possibly prevent severe reactions.
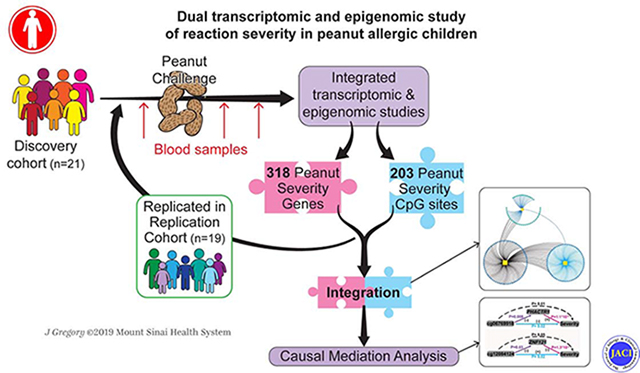
In this study, we performed an integrated transcriptomic and epigenomic study of peripheral blood longitudinally collected from peanut allergic children as they reacted in vivo to peanut during double-blind, placebo-controlled oral peanut challenge to identify gene expression changes, CpGs, biological processes, and interactions associated with reaction severity (Fig 1). After replicating results in an independent cohort of peanut allergic children, we integrated our gene expression and epigenetic findings to characterize interactions between CpG sites and gene expression. We identified severity-associated methylation loci that were also associated with severity-associated gene expression changes (i.e. peanut severity eCpGs). Causal mediation analysis then enabled us to identify eCpGs responsible for reaction severity.
Fig. 1. Study design and analytical flow.

Schematic outlining the clinical sample collections, transcriptome and epigenome profiling, and main analyses. A discovery cohort consisting of 21 peanut allergic children underwent physician-supervised peanut challenge. For each subject, peripheral blood samples were drawn at baseline immediately before the peanut challenge, and at two hours and four hours after start of the peanut challenge, resulting in 3 samples per subject. To identify genes with expression changes over time associated with reaction severity (peanut severity genes), whole blood RNA sequencing for all samples and linear mixed-effects models were employed (pink box). To identify DNA methylation sites associated with reaction severity (peanut severity CpGs), CD4+ lymphocytes were isolated from each baseline peripheral blood sample, DNA methylation profiling was performed, and linear regression models were constructed (blue box). An independent cohort of an additional 19 peanut allergic children were then similarly profiled and used to replicate the peanut severity genes and peanut severity CpGs identified (green box). Integration of the transcriptome and epigenome results (purple box) was next pursued by building a combined network, identifying expression CpGs (eCpGs, i.e. CpGs associated with expression of peanut severity genes), and causal mediation analysis.
Our data-driven approach to studying reaction severity in peanut allergic children through in vivo transcriptomic profiling and integrated epigenome-wide study represents a systems-wide approach for elucidating novel mechanisms underlying reaction severity in peanut allergy.
Methods
Please see Supplementary Methods for full details.
Study design
The discovery cohort included 21 peanut allergic children who reacted to peanut challenge during randomized, double-blind, placebo-controlled food challenges.7 Peripheral blood samples were collected immediately before food challenge (baseline), 2 hours from food challenge start (during challenge), and 4 hours from food challenge start (after challenge) (Fig. 1). The replication cohort included 19 independent peanut allergic children following the same protocol as the replication cohort. Written informed consent was obtained from all participants.
To measure reaction severity in response to peanut challenge, a threshold-weighted reaction severity score was calculated for each subject as the product of symptom grade (Table E1) and dose factor to capture both symptom(s) experienced during peanut challenge and the eliciting dose. This scoring approach has been used in prior studies of peanut allergy severity.8, 9
For transcriptome profiling, RNA isolated from peanut challenge peripheral blood samples was used to construct sequencing libraries10 and RNA sequencing (RNAseq) was performed on the Illumina HiSeq 2500 System with a per-sample target of 40–50 million 100 bp paired-end reads. Epigenome profiles were generated using DNA isolated from peripheral blood CD4+ cells of baseline samples and Illumina Infinium HumanMethylation450 arrays.
Analyses
To identify genes associated with reaction severity, peripheral blood transcriptome profiles at baseline, 2 hours, and 4 hours from the peanut challenges were used together to construct linear mixed-effects models for each gene. The model used for each gene was as follows: Gene expression ~ Severity + time + severity*time + age + study site + (1|subject). Genes significantly associated with reaction severity in the discovery cohort (FDR≤0.05) and replicated in the replication cohort (P≤0.05 and with consistent direction of effects between the two cohorts) were defined as “peanut severity” genes. A functional interaction network was built for the top 10 peanut severity genes (ranked by P-value) using ConsensusPathDB.11 To further investigate the biological context and relationships of all peanut severity genes, gene coexpression networks were constructed using Weighted Gene Co-expression Network Analysis (WGCNA).12 Changes in the distribution of leukocytes cell fractions were estimated from the transcriptomic profiles using CIBERSORT13 and tested for associations with reaction severity.
To identify CpG sites associated with reaction severity, epigenome profiles at baseline were used to construct linear regression models for each gene-associated CpG with the following model: Methylation β value ~ Severity + Age + Study site. Similar to peanut severity genes, CpGs significantly associated with reaction severity in the discovery cohort and replicated in the replication cohort were defined as “peanut severity” CpGs. The peanut severity genes and CpGs identified were then used to construct integrated networks using xMWAS.14 CpGs identified in the integrated networks were assessed for the degree to which their DNA methylation variation at baseline affects gene expression over the course of peanut challenge. CpG sites significantly associated with gene expression changes (i.e. eCpGs) were then included in causal mediation analysis to assess the degree to which the effects of CpG on reaction severity were causally mediated by gene expression.
Results
Clinical characteristics of participants
The clinical characteristics of this discovery cohort are shown in Table 1. Children ranged in age from 7 to 17 years. Their peanut sIgE levels ranged from 0.41 to 213.0 kUA/L, and SPT wheal size from 3.0 to 32.0 mm. All participants reacted within the first two hours of peanut challenge start, and none reacted to placebo. The median cumulative dose at first objective symptom was the fourth dose (30 mg dose, for a cumulative intake of 44 mg peanut protein). The most common symptoms experienced during peanut challenge were distress, followed by angioedema, throat tightness, and urticaria. Threshold-weighted severity scores for reaction severity that incorporated both symptoms experienced during peanut challenge and eliciting dose (see Supplementary Methods for details), and henceforth referred to as “reaction severity” for ease, ranged from 3.0 to 70.0, with interquartile range 21.0. Reaction severity did not correlate with clinical variables including age, presence of asthma, SPT size, or sIgE (each P>0.05).
Table 1.
Characteristics of the peanut allergic discovery cohort (n=21) and replication cohort (n=19)
| Discovery Cohort (n=21) | Replication Cohort (n=19) | P-value* | |
|---|---|---|---|
| Sex- Female | 7 (33.3%) | 7 (36.8%) | 0.99 |
| Age (years) | 11.0 (5.0) | 12.0 (4.0) | 0.33 |
| Parental allergy | 21 (100.0%) | 16 (84.2%) | 0.10 |
| Peanut sIgE (kUa/L) | 87.4 (140.5) | 68.0 (82.8) | 0.50 |
| Peanut skin prick test (mm) | 13.0 (11.0) | 12.0 (6.0) | 0.32 |
| Cumulative dose at first objective symptom (g peanut protein) | 0.044 (0.14) | 0.014 (0.44) | 0.59 |
| Cumulative successfully consumed dose (g peanut protein) | 0.144 (0.40) | 0.144 (0.48) | 0.80 |
| Reaction severity score | 21.0 (21.0) | 12.0 (13.5) | 0.02 |
| Symptoms experienced during peanut challenge | |||
| Distress | 14 (66.7%) | 13 (68.4%) | 0.99 |
| Throat tightness | 6 (28.6%) | 9 (47.4%) | 0.33 |
| Rhinorrhea | 2 (9.5%) | 3 (15.8%) | 0.65 |
| Rash | 4 (19.0%) | 1 (5.3%) | 0.35 |
| Rubbing of eyes, nose or scratching | 3 (14.3%) | 2 (10.5%) | 0.98 |
| Urticaria: 1–2 lesions | 6 (28.6%) | 3 (15.8%) | 0.46 |
| Urticaria: >3 lesions | 5 (23.8%) | 2 (10.5%) | 0.41 |
| Angioedema | 9 (42.9%) | 4 (21.1%) | 0.19 |
| Vomit: single | 2 (9.5%) | 2 (10.5%) | 0.99 |
| Vomit: multiple times | 0 (0.0%) | 1 (5.3%) | 0.48 |
| Abdominal pain: severe | 5 (23.8%) | 2 (10.5%) | 0.41 |
| Diarrhea | 1 (4.8%) | 0 (0.0%) | 0.99 |
| Wheezing | 1 (4.8%) | 0 (0.0%) | 0.99 |
| Stridor | 0 (0.0%) | 1 (5.3%) | 0.48 |
| Hypotension | 0 (0.0%) | 0 (0.0%) | 1.00 |
Number (percent) or Median (IQR) are shown.
Fisher’s Exact test for categorical variables and Wilcoxon signed rank test for continuous variables.
Peanut severity genes: genes with changes in expression associated with reaction severity
To identify genes with changes in expression over the course of peanut challenge associated with reaction severity, we profiled peripheral blood samples obtained from each subject undergoing peanut challenge at baseline, 2 hours, and 4 hours from challenge start using RNAseq (Fig 1). No outlier samples were detected following data processing, normalization, and quality control. Our previous15 and current work (Fig E1) has shown that gene expression levels during placebo challenge are similar to levels at baseline, so placebo RNAseq profiles were not incorporated into this study. Using the peanut challenge RNAseq data from all time points, we constructed linear-mixed effects models to test for the association between gene transcript level change over the course of peanut challenge and reaction severity. This enabled us to use each individual’s baseline sample as their own control, improving precision. After controlling inflation,16 we identified 3,090 genes for which change in expression over time during peanut challenge was associated with reaction severity (FDR≤0.05).
To assess the robustness of the genes identified, we recruited an independent replication cohort of 19 peanut allergic children. The subjects in this replication cohort were similar to those in the discovery cohort in terms of age and symptoms experienced during peanut challenge (P>0.05) (Table 1). However, the median reaction severity score in the replication cohort was lower compared to the discovery cohort (P=0.02), supporting that as a group, these subjects had less severe reactions to peanut challenge (Fig E2).
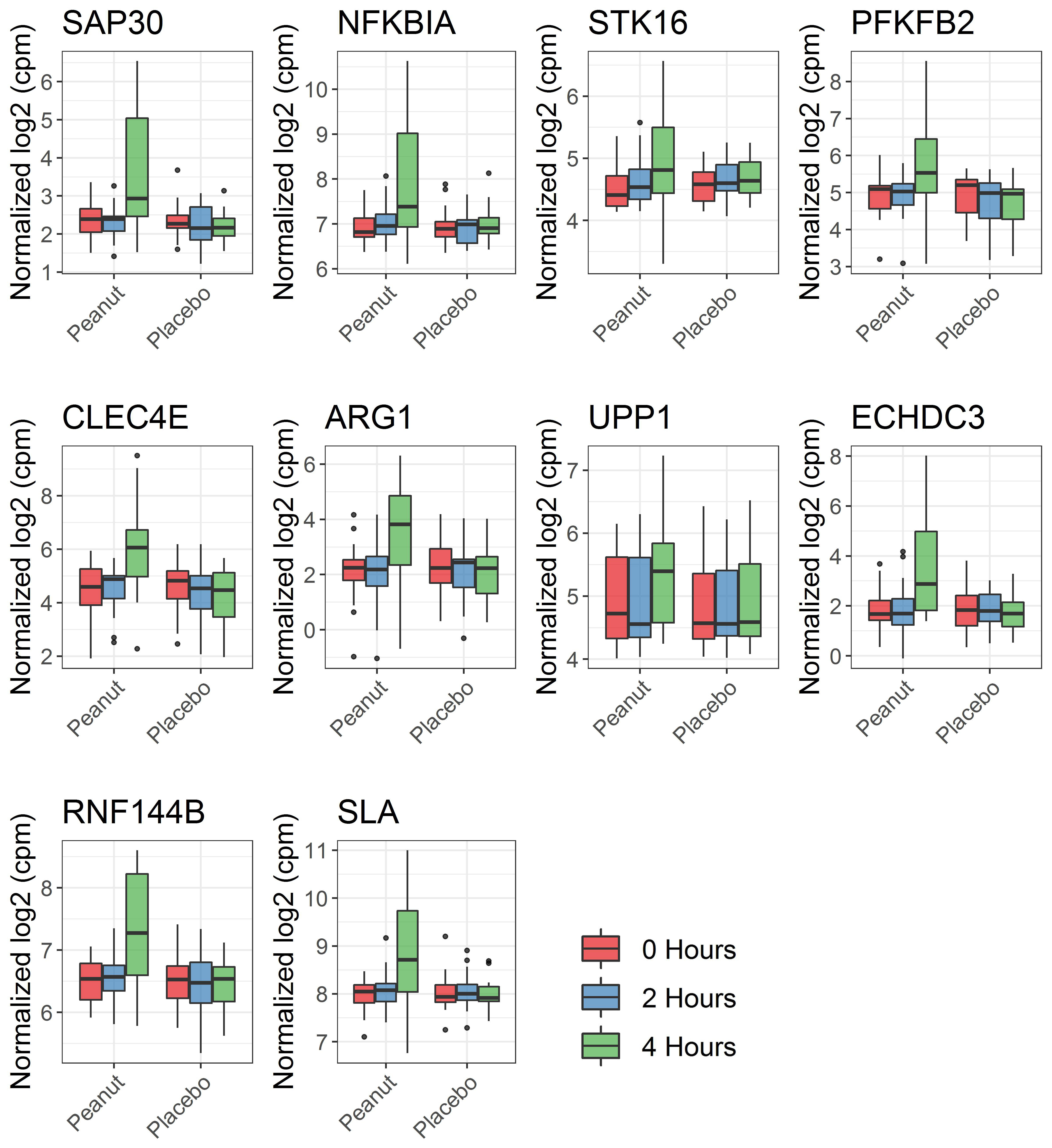
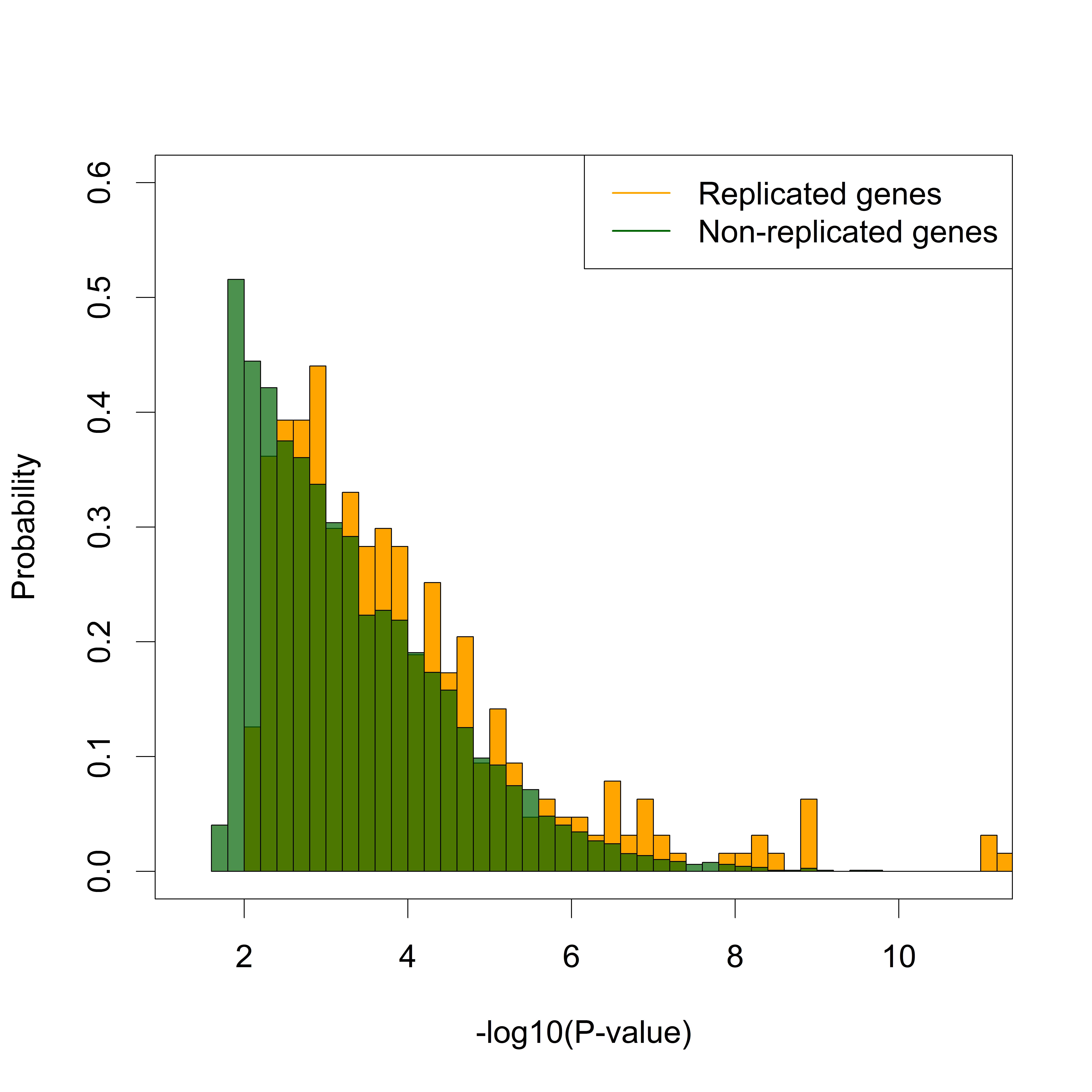
Among genes significantly associated with reaction severity in the discovery cohort, 318 were replicated in the replication cohort at P≤0.05 and with consistent direction of effect (Table E2), representing a significant enrichment over that expected by chance (fold enrichment=2.3, Fisher’s exact test P=2.8×10−53). The discovery cohort P-values for replicated genes had a significantly lower P-value distribution (Kolmogorov-Smirnov statistic = 0.54, P=2.0×10−16) than genes significant in the discovery cohort only (Fig E3). We henceforth refer to these replicated genes as “peanut severity genes.” Results for peanut severity genes in the discovery and replication cohorts are shown in Table E2. Across all peanut severity genes, we observed a greater magnitude of gene expression change between baseline and four hours compared to baseline and two hours, as shown in Fig E1 for ten selected genes. Because effect size estimates from linear mixed effects models can be challenging to interpret, changes in gene expression between baseline and four hours, and between baseline and two hours, are shownin Table E2 in addition to the P values and FDRs from linear mixed effects models for each gene. The relationship between change in gene expression and reaction severity for the top ten (ranked by P-value) peanut severity genes are shown in Fig 2.
Fig. 2. Change in expression of peanut severity genes over the course of reaction is associated with reaction severity.

Linear mixed effects modeling showed that change in gene expression over the course of reaction was associated with reaction severity for peanut severity genes, including these top 10 genes (ranked by P-value) shown here. Because effect sizes from linear mixed effects models are less intuitive to visualize, change in gene expression from baseline to four hours after peanut challenge start (blue points) and change in gene expression from baseline to two hours after peanut challenge start (pink points) are each plotted against reaction severity score for the top 10 peanut severity genes identified in the discovery cohort. Shaded areas show 95% confidence intervals for each association. P-values for the interaction between reaction severity and time from the linear mixed effects models are shown for each gene. These associations were all replicated in the replication cohort. cpm: counts per million.
Biological context and network of peanut severity genes
To identify biological processes associated with the 318 peanut severity genes, we performed Gene Ontology (GO) analysis. The top 20 GO biological process terms associated with peanut severity genes at FDR≤0.05 and absolute fold enrichment ≥2.0, ranked by fold enrichment, are shown in Fig 3A. Regulation of nitric oxide process (fold enrichment=23.0; FDR=0.02), macrophage activation (fold enrichment=11.9; FDR=0.003), and neutrophil activation (fold enrichment=4.9; FDR=6.9× 10−6) were among the GO biological processes associated with peanut severity genes with greatest fold enrichment. Fig 3B shows clustering of the identified GO biological process terms using REVIGO17.
Fig. 3. Gene ontology processes for peanut severity genes encompass major immunemediated pathways.

(A) Top twenty gene ontology (GO) enrichment terms associated with peanut severity genes (FDR≤0.05), ranked by fold-enrichment. (B) Clustering of GO terms associated with peanut severity genes (FDR≤0.05) by REVIGO17, with box size inversely corresponding to P-value for association. Bold-faced terms indicate higher order GO processes. (C) Top twenty GO terms for peanut severity genes (ranked by fold-enrichment) that were upregulated with increasing reaction severity (FDR≤0.05). (D) Clustering of GO terms for upregulated peanut severity genes (FDR≤0.05) by REVIGO17, with box size inversely corresponding to P-value of association. Bold-faced terms are for higher order GO processes.
To gain better insight into biological processes associated with up- and down-regulated genes among the 318 peanut severity genes, we also performed stratified analyses. Fig 3C shows the top 20 biological processes associated with the 245 (77%) peanut severity genes that were upregulated with increasing peanut reaction severity at FDR≤0.05 and fold enrichment ≥ 2, ranked by fold enrichment. The GO biological terms associated with upregulated peanut severity genes centered around neutrophil-related functions, including phagocytosis (fold enrichment=6.6, FDR=0.04), neutrophil activation (fold enrichment=4.9; FDR=1.1×10−7) and neutrophil degranulation (fold enrichment=4.5; FDR=1.8×10−6). REVIGO17 identified leukocyte activation and defense response as higher-level representative terms for the upregulated peanut severity genes (Fig 3D). Stratified GO analysis of the 73 downregulated peanut severity genes yielded no significant biological process terms.
To identify potential interactions between the top ten peanut severity genes (FDR<7.4×10−6), we next constructed an interaction network using ConsensusPathDB11. The resulting network included protein products from six of the top ten genes (based on P-values) as nodes, among other gene protein products (Fig 4). The majority of nodes in this network have recognized roles in inflammation, including NFKBIA and ARG1. The hub node of this network is NFKBIA, which is involved in both innate and adaptive immunity through regulation of cytokines, chemokines, and cell adhesion molecules of immune cells.18 ARG1, a member of the innate immune system connects to NFKBIA via CCAAT/Enhancer Binding Protein Beta (CEBPB), a well-known transcription factor of the immune system.19
Fig. 4. Interaction network analysis demonstrates interaction between six of the top ten peanut severity genes, with NFKBIA serving as a hub node.

Protein products from six of the top ten peanut severity genes were found to interact by ConsensusPathDB11 and are shown in black. The four genes without protein product interactions are not shown. Orange edges denote protein-protein interactions. Blue edges denote directional interactions via transcriptional activation. The hub node of this network is NFKBIA, which is involved in both innate and adaptive immunity through regulation of cytokines, chemokines, and cell adhesion molecules of immune cells. Through UBE2L3, NFKBIA interacts with pro-inflammatory ubiquitin ligase RNF144B to promote macrophage apoptosis. NFKBIA also interacts with STK16, a membrane kinase regulating the TGF-β response, via TID1. Another contributor to innate immunity, ARG1, interacts with NFKBIA through CEBPB and NCOR2. ARG1 activates macrophages via a type 2 cytokine-dependent manner and regulates T lymphocyte functions. NFKBIA interacts with SAP30 histone deacetylase to via PCBD1 or NCOR2 to promote chromatin compaction. This has downstream effects on glycolytic PFKFB2, which encodes a protein for energy generation by glycolysis.
Toward a broader context of gene expression beyond the single gene level, we constructed transcriptome-wide gene networks in the discovery cohort across all individuals and time-points using WGCNA.12 We identified 25 groups (i.e. modules) of highly interconnected genes (Table E3). A single coexpression module was significantly enriched for peanut severity genes. Associated with the GO term “neutrophil activation”, this module included 208 peanut severity genes (of 5694 genes in the module), equivalent to a 2.4-fold enrichment over that expected by chance (Fisher’s exact test FDR=2.3×10−43) (Table E3). Additional GO terms associated with this module are shown in Table E4.
Inferred cellular changes associated with reaction severity
We used a leukocyte deconvolution algorithm to infer the proportions of 19 leukocyte populations from the RNAseq expression profiles of each sample at baseline, 2 hours, and 4 hours (Fig 5A). This deconvolution enabled us to obtain a discovery-oriented and data-driven survey of 19 leukocyte subsets over time, including some low-frequency cellular subsets. Changes in the proportions of three specific cell types were associated with reaction severity, including naïve B cell (FDR=7×10−5), neutrophils (FDR=0.01), and naïve CD4+ T cells (FDR=0.02) (Fig 5B). Naïve B and CD4+ T cell proportions were negatively associated with reaction severity during peanut challenge, while neutrophil proportions were positively associated.
Fig. 5. Leukocyte compositional estimates during peanut challenge and their association with reaction severity.

(A) Cell fractions of 19 leukocyte subsets estimated from transcriptome wide RNA-seq gene expression signatures are shown at baseline, 2 hours, and 4 hours, witheach column representing an individual, and individuals ordered by increasing reaction severity from left to right. Cell types estimated by the analysis are indicated in the legend. (B) Three cell subsets were significantly associated with reaction severity. Change in leukocyte composition from baseline to four hours after peanut challenge start is plotted against reaction severity score for B cells (naïve), neutrophils, and CD4+ T cells (naïve). Shaded areas show 95% confidence intervals for each association. P-values for the interaction between reaction severity and time are shown.
Peanut severity CpGs: CpG sites associated with reaction severity
A secondary goal of this study was to examine the potential role of epigenetics, namely methylation, in reaction severity. While we recognized limited power to detect small-to-medium effect sizes between CpG sites and reaction severity associations, we viewed this CpG survey as an important exploratory study, given the limited number of studies published to date on methylation in rigorously phenotyped food allergic subjects. To this end, we performed methylation profiling of peripheral blood CD4+ lymphocytes from discovery cohort subjects at baseline. We chose to focus on CD4+ lymphocytes in particular given known cell-specific variability in methylation that can be obscured by whole blood methylation profiling,20 and the recognized role of CD4+ lymphocytes in allergic inflammation.21
Following data processing, normalization, and quality control filtering, 370,051 CpG sites remained for analysis. All CpGs mapped to a gene within 10Mb, based on gene coordinates available in the UCSC gene annotation set (GRCh37/hg19). Using linear regression models and after control for deflation16, we identified 11,152 CpG sites associated with reaction severity at FDR≤0.05 in the discovery cohort. Analogous to our RNAseq analytic flow, we also conducted the same CpG methylation analysis in the replication cohort of peanut allergic children (Table 1), observing replication of 203 CpGs with P≤0.05 and same direction of effect between the two cohorts. The discovery cohort P-values for replicated CpGs had a significantly lower P-value distribution (Kolmogorov-Smirnov statistic = 0.59, P = 1.0 × 10−16) than CpG sites that were significant in the discovery cohort only. We refer to these replicated CpG sites henceforth as “peanut severity CpGs.” Detailed results for peanut severity CpGs in the discovery and replication cohorts are provided in Table E5. The peanut severity CpGs mapped to 197 unique genes, with all CpGs mapping to at least one gene. The relationship between four example peanut severity CpGs and reaction severity score in the discovery (Fig E4) and replication (Fig E5) cohorts are shown; these four peanut severity CpGs map to peanut severity genes and are highlighted because they feature prominently in downstream analyses (see below).
Integrated analyses of peanut severity genes and peanut severity CpGs
First, we assessed the overlap between the 203 peanut severity CpGs and 318 peanut severity genes identified, finding that 4 CpGs resided within 10Mb of a peanut severity gene. Using xMWAS14, we then used methylation levels at these 4 CpGs and changes in gene expression for the 318 peanut severity genes to construct groups of CpGs and genes that exhibited high correlation coefficients between members. We identified three groups, collectively consisting of 363 strong correlations (R2≥0.7 and P≤0.05) between 4 key node CpGs and 273 peanut severity genes (Fig 6). The lists of genes and CpGs for the three groups, as well as their pairwise correlations, are provided in Table E6. Gene ontology analysis for these three groups revealed enrichment for terms all consistent with those for peanut severity genes, including immune response, chemotaxis, and regulation of macroautophagy (Fig 6). The majority of the observed correlations between CpG methylation and gene expression were negative (62%) (i.e. higher CpG methylation at baseline was associated with decreased gene expression from baseline to four hours). Notably, all top ten peanut severity genes were correlated with at least one of the key node CpGs identified (Fig 6): PFKFB2 was negatively correlated with cg06769918 (PHACTR1) in the immune response group; SAP30, NFKBIA, STK16, ECHDC3, and UPP1 were positively correlated with cg12084124 (ZNF121) in the regulation of macroautophagy group; and ARG1, CLEC4E, RNF144B, and SLA were negatively correlated with cg17545300 (CHST15) in the chemotaxis group (Fig 6).
Fig. 6. Integrated network of top interactions (R2≥0.7 and P≤0.05) between peanut severity genes and peanut severity CpGs.

Orange square nodes denote CpGs. Circular nodes denote genes. Blue edges illustrate negative correlations between DNA methylation level and gene expression change from baseline to four hours. Orange edges represent positive correlations between DNA methylation level and gene expression change from baseline to four hours. Circular nodes are colored by correlated group. Node size positively corresponds with the number of edges connected to that node. Bolder edges reflect higher correlation coefficients. The top ten peanut severity genes are shown with gene names.
Peanut severity eCpGs: CpGs associated with reaction severity and gene expression
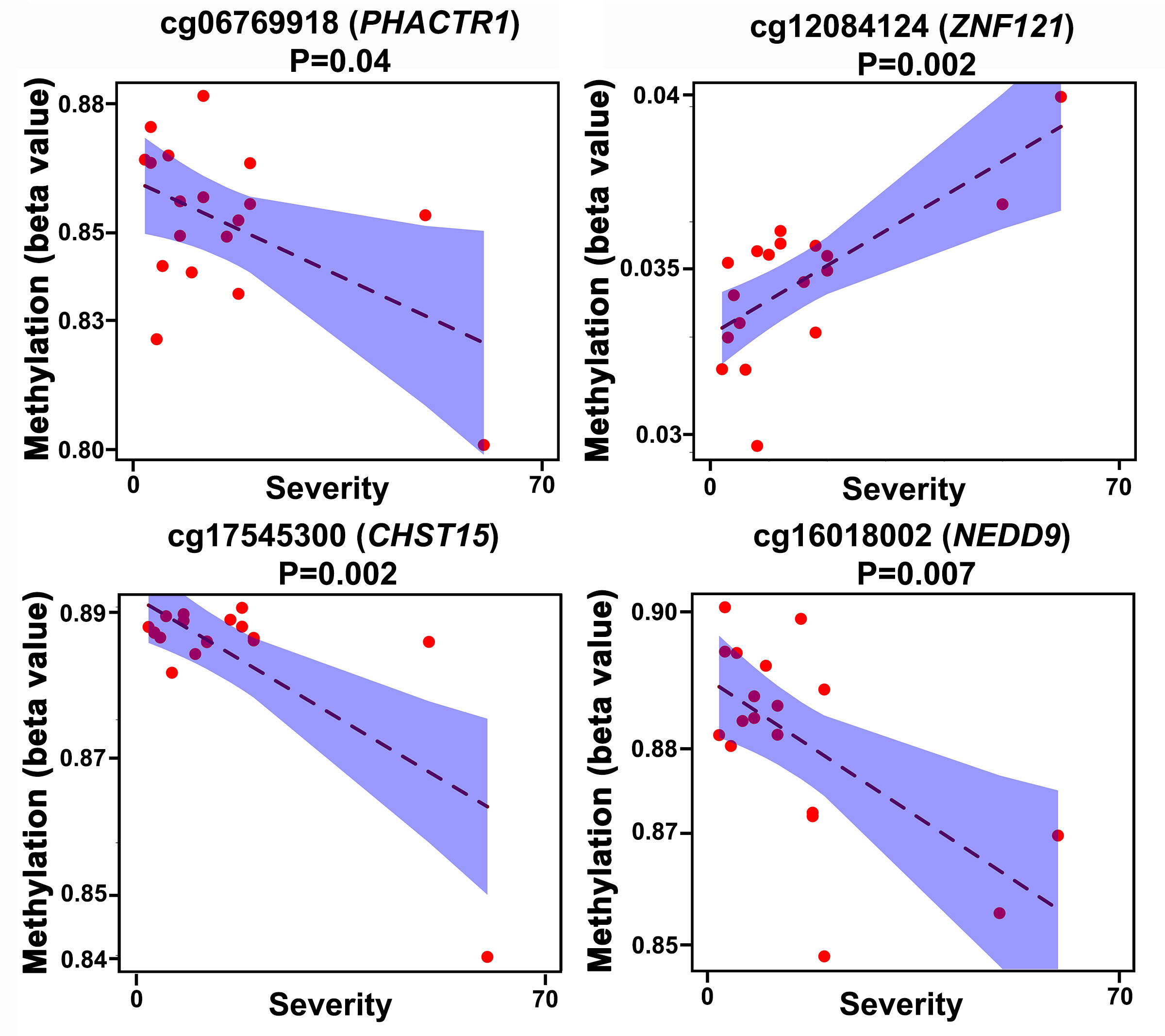
To further investigate the 4 key node CpGs identified above (Fig 6), we performed eCpG analysis. In Fig E6, we show the relationship between methylation levels and gene expression changes for these 4 eCpGs. We found that 2 of 4 key node CpGs were significantly associated (FDR≤0.05) with the expression of a peanut severity gene located within 10 Mb (Table E7). We will refer to these 2 CpG sites associated with both reaction severity and gene expression as peanut severity eCpGs. Both eCpGs (cg06769918 (PHACTR1) and cg12084124 (ZNF121) ) exhibited negative associations between baseline methylation levels and gene expression changes that occurred during peanut challenge.
For the 2 eCpGs identified, we next investigated the extent to which the association between CpG methylation and reaction severity was mediated by gene expression. While CpG methylation is unlikely to change acutely during peanut challenge, CpG methylation may serve as an anchor upon which gene expression changes to modulate reaction severity. Causal mediation analysis showed that the associations between the two eCpGs (cg06769918 (PHACTR1) and cg12084124 (ZNF121)) and reaction severity were each causally mediated by gene expression changes of their respective gene. Specifically, the association between lower methylation at cg06769918 and increased reaction severity was causally mediated by increased gene expression of PHACTR1 (P=0.01, FDR = 0.02), and the association between lower methylation at cg12084124 and increased reaction severity was causally mediated by increased gene expression of ZNF121 (P=0.05, FDR = 0.05) (Fig 7).
Fig. 7. Causal mediation analysis for peanut severity CpGs, peanut severity genes, and reaction severity.

Associations between (1) CpGs and reaction severity are shown in blue; (2) CpGs and peanut severity genes are shown in purple; (3) peanut severity genes and reaction severity are shown in pink. Positive (+) and negative (−) associations are as indicated. Dashed arrows indicate mediation of the association between CpG and reaction severity by peanut severity gene expression. P-values from this mediation analysis are shown in black.
Discussion
In this integrated transcriptomic and epigenomic study of peanut allergic children reacting to peanut in vivo, we identified and replicated peripheral blood gene expression and methylation signatures associated with reaction severity (Fig 1). This study fills an important niche in food allergy research where mechanisms driving variable severity of reactions remain poorly understood. Food allergic individuals must live with the uncertainty of potentially severe reactions,22 with no cures nor clinical assays to reliably predict reaction severity.23 The identification of genes and molecular processes driving reaction severity could lead to prognostic biomarkers as well as therapeutic interventions. While clinical variables such as age,24 allergen dose,25 asthma status,24 SPT size,26 sIgE level,27 and basophil activity28 have been associated with reaction severity, these factors neither reliably predict reaction severity24 nor elucidate mechanisms driving severity. Given that food allergic reactions can quickly manifest across multiple organ systems, we hypothesized that transcriptomic analysis of peripheral blood during in vivo reactions would identify novel genes and biological processes that actively change with reaction severity. Further, given known genetic risk factors29, 30 and the role of epigenetics in modulating gene expression,31 we also hypothesized that differentially methylated CpG sites at baseline could be associated with reaction severity. We focused on CD4+ lymphocytes given their recognized role in food allergy32 and the cell-specificity of methylation changes.20
We identified and replicated peanut severity genes exhibiting changes in expression during peanut challenge that were significantly associated with reaction severity (Fig 2, Table E2). The biological processes encompassed by peanut severity genes as a group included regulation of nitric oxide processes and activation of neutrophils and macrophages (Fig 3). Nitric oxide is known to regulate the functional activity, growth, and death of many immune and inflammatory cell types including neutrophils and macrophages.33 Both neutrophils and macrophages hold primary roles in inflammation and allergic disease.34,35, 36 For example, depletion of macrophages and neutrophils in several mouse models modulates symptoms of peanut-induced anaphylaxis.35, 37 Genes specifically upregulated with increasing severity were associated with neutrophil activation and neutrophil-mediated immunity (Fig 3). These results were consistent with the significant positive association between neutrophil fraction and reaction severity, based on our leukocyte deconvolution analyses (Fig 5). Our results were also consistent with previous studies showing increased expression of several markers of neutrophil activation and trafficking in patients with anaphylaxis.38, 39 Further, neutrophil activation occurs early during reactions, independent of mast cell activation.39 The results from this study and prior findings by us15 and others collectively support that targeting macrophage and neutrophil activity may be a high-yield approach for further understanding reaction severity in food allergy, and for identifying biomarkers and therapy for severe allergic reactions.
Among the top ten peanut severity genes (Fig 2), we had previously identified eight (SAP30, NFKBIA, PFKFB2, CLEC4E, ARG1, ECHDC3, RNF144B, SLA) as genes associated with reaction to peanut but not placebo, including ECHDC3, which had also been identified as one of six key drivers causally regulating networks associated with peanut allergic reactions.15 IL1R2, another key driver of peanut allergic reactions we reported previously,15 was also among the 318 peanut severity genes identified here. Additionally, 59.4% (189/318) of peanut severity genes found in this study overlap with peanut response genes previously reported,15 suggesting crosstalk between genes involved in the occurrence of peanut allergic reactions overall and those that modulate reaction severity. However, the significant non-overlap suggests that there are also distinct biological processes that underlie reaction state and reaction severity.
The interaction network built with the top ten peanut severity genes (Fig 4) highlighted NFKBIA as a central hub. NFKBIA is a well-known regulator of NF-κB signaling involved in both innate and adaptive immunity through modulation of cytokines, chemokines, and cell adhesion molecules in immune cells.18 We previously identified upregulation of NF-kB signaling in our study of acute allergic reactions to peanut vs. placebo.15 Others have also reported associations of NF-kB with risk for asthma and allergy-related phenotypes.40 Another hub and peanut severity gene in our interaction network was ARG1, which regulates group 2 innate lymphoid cell proliferative capacity and pro-inflammatory functions that promote type 2 inflammation.19 ARG1 also modulates T lymphocyte function,19 and dysregulation of ARG1 has been associated with asthma.41 ARG1 interacts with NFKBIA via CEBPB, a transcription factor that facilitates differentiation along the Th2 lineage and regulates acute-phase reactions. RNF144B, another peanut severity gene in the network, promotes lipopolysaccharide-induced inflammation and apoptosis in human macrophages.42 Three other peanut severity genes in our network (STK16, SAP30, PFKFB2) are not known to be associated with allergy or immune-related function, but our interaction network suggests that they contribute to reaction severity in peanut allergy via indirect interactions with NFKBIA.
Our integrated analyses of peanut severity genes and peanut severity CpGs identified four interconnected CpG-gene groups enriched for immune response, chemotaxis, and regulation of macroautophagy (Fig 6). These groups consisted of genes specifically involved in neutrophil and macrophage activity whose functions have been well documented in allergy and anaphylaxis as discussed above.43, 44 Our approach also allowed for the discovery of novel causal links between gene expression and methylation signatures of PHACTR1 and ZNF121 in the context of reaction severity. Specifically, we found that baseline methylation levels of cg06769918 were inversely and causally associated with PHACTR1 expression and reaction severity. The same causal relationship was found for cg12084124, ZNF121, and reaction severity. In addition, our interaction network (Figure 6) showed that cg06769919 (PHACTR1) and cg12084124 (ZNF121) each directly or indirectly interact with all top ten peanut severity genes. PHACTR1 is a phosphatase and actin regulator expressed in macrophages and lymphocytes that has been studied thus far in the context of cardiovascular inflammation and pneumonia.45 ZNF121 is a zinc finger transcription factor that has not been well studied thus far, but is thought to regulate cell proliferation and differentiation.46 To our knowledge, our study is the first to link PHACTR1 and ZNF121 to food allergy.
A major strength of this study is our use of double-blind, placebo-controlled peanut challenges to characterize peanut allergy and capture reaction severity in all subjects. These food challenges are vastly more accurate than self-reported allergy or severity, sIgE level, or SPT as markers of food allergy.19 Second, the collection of longitudinal samples from each subject over the course of the peanut challenge enabled us to use each subject as their own control. This study design provides improved precision, precisely captures acute peanut reactions and transient changes of gene expression, as well as augments power compared to case-control designs used in previous gene expression and epigenetic studies of children with food allergy.32, 47–50 Finally, because our study employed parallel transcriptomic and epigenomic profiling in the same subjects, followed by network-based and mediation analysis, we were able to identify not only novel genes, but also biological processes and interactions that contribute to reaction severity in peanut allergy.
Our study has some limitations. First, our sample size was limited, although comparable to prior gene-expression based studies of food allergy.32, 47–50 However, with the subjects studied, each underwent rigorous clinical phenotyping and dual transcriptomic and epigenomic profiling to yield unprecedented, novel parallel data in well-characterized peanut allergic subjects. Further, we also replicated our findings in an independent cohort of peanut allergic children. Second, while it would have been interesting to also directly measure (e.g. by FACS sorting) the 19 cellular subsets that we inferred by leukocyte deconvolution, this would have required considerable effort and the limited blood volumes may have been precluded measurement of low frequency cell types. While leukocyte deconvolution enabled us to infer the proportions of 19 cellular subsets during the course of reaction in an unbiased, and data-driven fashion, the study does not define if changes in cellular proportions precede or follow transcriptomic changes. Last, we recognized that our examination of methylation changes and reaction severity could be limited by the high dimensionality of CpG profiling (i.e. 450K CpG sites) that would amplify the statistical challenges of high-dimensional data (i.e. p>>n),51 leading to limited findings by traditional approaches. We took a conservative approach to identifying peanut severity CpGs that required replication and incorporated causal mediation analysis to link our methylation findings to our more robust transcriptome results. We consider our methylation findings an exploratory dimension that can be further studied with these initial results in mind.
The genes, biological processes, and interactions identified by this study represent potential therapeutic targets for mitigating reaction severity in peanut allergy. For example, NFKBIA was a central hub in the interaction network from this study. The NF-κB signaling pathway is a well-known target of glucocorticoids, which are commonly administered to individuals presenting with acute food allergic reaction.52 The consistency of our network-based finding with current practice provides reassurance regarding the clinical applicability of our findings. A second hub in this network, ARG1, is blocked by arginase inhibitors to inhibit airway hyper-responsiveness in asthmatic mice. Arginase inhibitors have been proposed for treatment of airway inflammation and asthma in human beings.53 However, the potential use of arginase inhibitors in food allergy has not been explored. Next steps motivated by this study include interventional investigations of the gene, biological pathway, and interaction targets identified by our study as potential modulators of reaction severity in peanut and other allergies.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Key Messages.
In this integrated transcriptomic and epigenomic study of peanut allergic children reacting to peanut in vivo, we identified and replicated peripheral blood gene expression and methylation signatures associated with reaction severity.
The biological processes encompassed by the peanut severity genes identified included neutrophil and macrophage activation, and genes specifically upregulated with increasing severity were associated with neutrophil degranulation and neutrophil-mediated immunity. NFKBIA and ARG1 were identified as hubs in interaction networks.
Integrated analyses of peanut severity genes and peanut severity CpGs identified four interconnected CpG-gene groups enriched for immune response, chemotaxis, and regulation of macroautophagy.
Acknowledgments
We thank the families who participated, the staff at each institution, and the Statistical and Clinical Coordinating Center.
Funding: This study was supported by the National Institutes of Health (R01AI118833, U19AI136053, U19AI066738 and U01AI066560) and the Mindich Child Health and Development Institute at Mount Sinai. The project was also supported by UL1 TR- 002535 (National Jewish), UL1 TR-000067 (Mount Sinai), UL1 TR-000039 (Arkansas), UL1 TR-000083 (U North Carolina) and UL1 TR-000424 (Johns Hopkins) from the National Center for Research Resources (NCRR), a component of the National Institutes of Health. The funding bodies had no role in the collection, analysis, interpretation of data, or writing of the manuscript.
Abbreviations
- sIgE
specific IgE
- SPT
skin prick test
- RNAseq
RNA sequencing
- GO
Gene Ontology
- WGCNA
Weighted Gene Co-expression Network Analysis
- cpm
counts per million
- Th2
T helper type 2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bunyavanich S, Rifas-Shiman SL, Platts-Mills TA, Workman L, Sordillo JE, Gillman MW, et al. Peanut allergy prevalence among school-age children in a US cohort not selected for any disease. Journal of Allergy and Clinical Immunology 2014; 134:2011–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McGowan EC, Keet CA. Prevalence of self-reported food allergy in the National Health and Nutrition Examination Survey (NHANES) 2007–2010. J Allergy Clin Immunol 2013; 132:1216–9 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Food Allergy: Quick Facts. National Institute of Allergy and Infectious Diseases 2010:https://web.archive.org/web/20100407195412/http://www.niaid.nih.gov/topics/foodAllergy/understanding/Pages/quickFacts.aspx, downloaded 3/25/2017.
- 4.Boyce JA, Assa’ad A, Burks AW, Jones SM, Sampson HA, Wood RA, et al. Guidelines for the diagnosis and management of food allergy in the United States: report of the NIAID-sponsored expert panel. J Allergy Clin Immunol 2010; 126:S1–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flinn A, Hourihane JO. Allergic reaction to peanuts: can we predict reaction severity in the wild? Curr Allergy Asthma Rep 2013; 13:645–50. [DOI] [PubMed] [Google Scholar]
- 6.Polloni L, DunnGalvin A, Ferruzza E, Bonaguro R, Lazzarotto F, Toniolo A, et al. Coping strategies, alexithymia and anxiety in young patients with food allergy. Allergy 2016. [DOI] [PubMed] [Google Scholar]
- 7.Sampson HA, Gerth van Wijk R, Bindslev-Jensen C, Sicherer S, Teuber SS, Burks AW, et al. Standardizing double-blind, placebo-controlled oral food challenges: American Academy of Allergy, Asthma & Immunology-European Academy of Allergy and Clinical Immunology PRACTALL consensus report. J Allergy Clin Immunol 2012; 130:1260–74. [DOI] [PubMed] [Google Scholar]
- 8.Flinterman AE, Knol EF, Lencer DA, Bardina L, den Hartog Jager CF, Lin J, et al. Peanut epitopes for IgE and IgG4 in peanut-sensitized children in relation to severity of peanut allergy. J Allergy Clin Immunol 2008; 121:737–43 e10. [DOI] [PubMed] [Google Scholar]
- 9.Lewis SA, Grimshaw KE, Warner JO, Hourihane JO. The promiscuity of immunoglobulin E binding to peanut allergens, as determined by Western blotting, correlates with the severity of clinical symptoms. Clin Exp Allergy 2005; 35:767–73. [DOI] [PubMed] [Google Scholar]
- 10.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010; 26:139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herwig R, Hardt C, Lienhard M, Kamburov A. Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat Protoc 2016; 11:1889–907. [DOI] [PubMed] [Google Scholar]
- 12.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008; 9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015; 12:453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uppal K, Ma C, Go YM, Jones DP, Wren J. xMWAS: a data-driven integration and differential network analysis tool. Bioinformatics 2018; 34:701–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watson CT, Cohain AT, Griffin RS, Chun Y, Grishin A, Hacyznska H, et al. Integrative transcriptomic analysis reveals key drivers of acute peanut allergic reactions. Nat Commun 2017; 8:1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Iterson M, van Zwet EW, Consortium B, Heijmans BT. Controlling bias and inflation in epigenome- and transcriptome-wide association studies using the empirical null distribution. Genome Biol 2017; 18:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Supek F, Bosnjak M, Skunca N, Smuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 2011; 6:e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lawrence T The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol 2009; 1:a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Monticelli LA, Buck MD, Flamar AL, Saenz SA, Tait Wojno ED, Yudanin NA, et al. Arginase 1 is an innate lymphoid-cell-intrinsic metabolic checkpoint controlling type 2 inflammation. Nat Immunol 2016; 17:656–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol 2014; 15:R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ling MF, Luster AD. Allergen-Specific CD4(+) T Cells in Human Asthma. Ann Am Thorac Soc 2016; 13 Suppl 1:S25–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shaker MS, Schwartz J, Ferguson M. An update on the impact of food allergy on anxiety and quality of life. Curr Opin Pediatr 2017; 29:497–502. [DOI] [PubMed] [Google Scholar]
- 23.Sicherer SH, Sampson HA. Food allergy: A review and update on epidemiology, pathogenesis, diagnosis, prevention, and management. J Allergy Clin Immunol 2018; 141:41–58. [DOI] [PubMed] [Google Scholar]
- 24.Turner PJ, Baumert JL, Beyer K, Boyle RJ, Chan CH, Clark AT, et al. Can we identify patients at risk of life-threatening allergic reactions to food? Allergy 2016; 71:1241–55. [DOI] [PubMed] [Google Scholar]
- 25.Pettersson ME, Koppelman GH, Flokstra-de Blok BMJ, Kollen BJ, Dubois AEJ. Prediction of the severity of allergic reactions to foods. Allergy 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wainstein BK, Studdert J, Ziegler M, Ziegler JB. Prediction of anaphylaxis during peanut food challenge: usefulness of the peanut skin prick test (SPT) and specific IgE level. Pediatr Allergy Immunol 2010; 21:603–11. [DOI] [PubMed] [Google Scholar]
- 27.Rolinck-Werninghaus C, Niggemann B, Grabenhenrich L, Wahn U, Beyer K. Outcome of oral food challenges in children in relation to symptom-eliciting allergen dose and allergen-specific IgE. Allergy 2012; 67:951–7. [DOI] [PubMed] [Google Scholar]
- 28.Song Y, Wang J, Leung N, Wang LX, Lisann L, Sicherer SH, et al. Correlations between basophil activation, allergen-specific IgE with outcome and severity of oral food challenges. Ann Allergy Asthma Immunol 2015; 114:319–26. [DOI] [PubMed] [Google Scholar]
- 29.Sicherer SH, Furlong TJ, Maes HH, Desnick RJ, Sampson HA, Gelb BD. Genetics of peanut allergy: a twin study. J Allergy Clin Immunol 2000; 106:53–6. [DOI] [PubMed] [Google Scholar]
- 30.Hong X, Hao K, Ladd-Acosta C, Hansen KD, Tsai HJ, Liu X, et al. Genome-wide association study identifies peanut allergy-specific loci and evidence of epigenetic mediation in US children. Nat Commun 2015; 6:6304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nature Reviews Genetics 2012; 13:484–92. [DOI] [PubMed] [Google Scholar]
- 32.Martino D, Joo JE, Sexton-Oates A, Dang T, Allen K, Saffery R, et al. Epigenome-wide association study reveals longitudinally stable DNA methylation differences in CD4+ T cells from children with IgE-mediated food allergy. Epigenetics 2014; 9:998–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bogdan C Nitric oxide synthase in innate and adaptive immunity: an update. Trends Immunol 2015; 36:161–78. [DOI] [PubMed] [Google Scholar]
- 34.Munoz-Cano R, Picado C, Valero A, Bartra J. Mechanisms of Anaphylaxis Beyond IgE. J Investig Allergol Clin Immunol 2016; 26:73–82; quiz 2p following 3. [DOI] [PubMed] [Google Scholar]
- 35.Arias K, Chu DK, Flader K, Botelho F, Walker T, Arias N, et al. Distinct immune effector pathways contribute to the full expression of peanut-induced anaphylactic reactions in mice. Journal of Allergy and Clinical Immunology 2011; 127:1552–61.e1. [DOI] [PubMed] [Google Scholar]
- 36.Jönsson F, Mancardi Da, Kita Y, Karasuyama H, Iannascoli B, Rooijen NV, et al. Mouse and human neutrophils induce anaphlaxis. 2011; 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reber LL, Marichal T, Mukai K, Kita Y, Tokuoka SM, Roers A, et al. Selective ablation of mast cells or basophils reduces peanut-induced anaphylaxis in mice. J Allergy Clin Immunol 2013; 132:881–8 e1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Munoz-Cano R, Pascal M, Bartra J, Picado C, Valero A, Kim DK, et al. Distinct transcriptome profiles differentiate nonsteroidal anti-inflammatory drug-dependent from nonsteroidal anti-inflammatory drug-independent food-induced anaphylaxis. J Allergy Clin Immunol 2016; 137:137–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Asai Y, Eslami A, van Ginkel CD, Akhabir L, Wan M, Ellis G, et al. Genome-wide association study and meta-analysis in multiple populations identifies new loci for peanut allergy and establishes c11orf30/EMSY as a genetic risk factor for food allergy. J Allergy Clin Immunol 2017. [DOI] [PubMed] [Google Scholar]
- 40.Martino DJ, Bosco A, McKenna KL, Hollams E, Mok D, Holt PG, et al. T-cell activation genes differentially expressed at birth in CD4+ T-cells from children who develop IgE food allergy. Allergy 2012; 67:191–200. [DOI] [PubMed] [Google Scholar]
- 41.Litonjua AA, Lasky-Su J, Schneiter K, Tantisira KG, Lazarus R, Klanderman B, et al. ARG1 is a novel bronchodilator response gene: screening and replication in four asthma cohorts. Am J Respir Crit Care Med 2008; 178:688–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ariffin JK, Kapetanovic R, Schaale K, Gatica-Andrades M, Blumenthal A, Schroder K, et al. The E3 ubiquitin ligase RNF144B is LPS-inducible in human, but not mouse, macrophages and promotes inducible IL-1beta expression. J Leukoc Biol 2016; 100:15561. [DOI] [PubMed] [Google Scholar]
- 43.Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol 2008; 8:34–47. [DOI] [PubMed] [Google Scholar]
- 44.van Egmond M, Vidarsson G, Bakema JE. Cross-talk between pathogen recognizing Toll-like receptors and immunoglobulin Fc receptors in immunity. Immunol Rev 2015; 268:311–27. [DOI] [PubMed] [Google Scholar]
- 45.Reschen ME, Lin D, Chalisey A, Soilleux EJ, O’Callaghan CA. Genetic and environmental risk factors for atherosclerosis regulate transcription of phosphatase and actin regulating gene PHACTR1. Atherosclerosis 2016; 250:95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luo A, Zhang K, Zhao Y, Zhu Z, Fu L, Dong JT. ZNF121 interacts with ZBRK1 and BRCA1 to regulate their target genes in mammary epithelial cells. FEBS Open Bio 2018; 8:1943–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Syed A, Garcia MA, Lyu SC, Bucayu R, Kohli A, Ishida S, et al. Peanut oral immunotherapy results in increased antigen-induced regulatory T-cell function and hypomethylation of forkhead box protein 3 (FOXP3). J Allergy Clin Immunol 2014; 133:500–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paparo L, Nocerino R, Cosenza L, Aitoro R, D’Argenio V, Del Monaco V, et al. Epigenetic features of FoxP3 in children with cow’s milk allergy. Clin Epigenetics 2016; 8:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kosoy R, Agashe C, Grishin A, Leung DY, Wood RA, Sicherer SH, et al. Transcriptional Profiling of Egg Allergy and Relationship to Disease Phenotype. PLoS One 2016; 11:e0163831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saulnier N, Nucera E, Altomonte G, Rizzi A, Pecora V, Aruanno A, et al. Gene expression profiling of patients with latex and/or vegetable food allergy. Eur Rev Med Pharmacol Sci 2012; 16:1197–210. [PubMed] [Google Scholar]
- 51.Johnstone IM, Titterington DM. Statistical challenges of high-dimensional data. Philos Trans A Math Phys Eng Sci 2009; 367:4237–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schuliga M NF-kappaB Signaling in Chronic Inflammatory Airway Disease. Biomolecules 2015; 5:1266–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ogino K, Kubo M, Takahashi H, Zhang R, Zou Y, Fujikura Y. Anti-inflammatory effect of arginase inhibitor and corticosteroid on airway allergic reactions in a Dermatophogoides farinae-induced NC/Nga mouse model. Inflammation 2013; 36:141–51. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.