

SUMMARY
Cancer-associated mutations in genes encoding RNA splicing factors (SFs) commonly occur in leukemias, as well as in a variety of solid tumors, and confer dependence on wild-type splicing. These observations have led to clinical efforts to directly inhibit the spliceosome in patients with refractory leukemias. Here, we identify that inhibiting symmetric or asymmetric dimethylation of arginine, mediated by PRMT5 and type I protein arginine methyltransferases (PRMTs), respectively, reduces splicing fidelity and results in preferential killing of SF-mutant leukemias over wild-type counterparts. These data identify genetic subsets of cancer most likely to respond to PRMT inhibition, synergistic effects of combined PRMT5 and type I PRMT inhibition, and a mechanistic basis for the therapeutic efficacy of PRMT inhibition in cancer.
Graphical Abstract
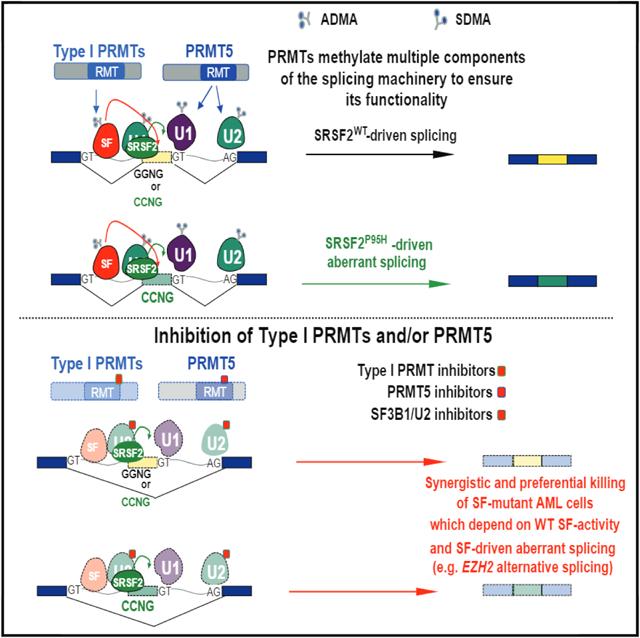
In Brief
Fong et al. show that spliceosomal mutant leukemias are preferentially sensitive to inhibition of protein arginine methyltransferases (PRMTs), that RNA-binding proteins are enriched among substrates of PRMT5 and type I PRMTs, and that combined PRMT5 and type I PRMT inhibition synergistically kill these leukemia cells.
INTRODUCTION
Recent genomic analyses of cancers have identified numerous means by which splicing is altered in cancer (Dvinge et al., 2016; Kahles et al., 2018; Zhang and Manley, 2013). These include change-of-function mutations in RNA splicing factors (SFs) (Harbour et al., 2013; Martin et al., 2013; Wang et al., 2011; Yoshida et al., 2011), mutations that alter splicing in cis (Supek et al., 2014), changes in the expression of splicing regulatory proteins (Anczuków et al., 2012; Kami et al., 2007), and alterations in transcriptional regulators that influence the process of splicing such as c-MYC (Hsu et al., 2015; Koh et al., 2015). In parallel, certain genetic subsets of cancer have been identified to be particularly sensitive to therapeutic inhibition of splicing. These include cells bearing hotspot change-of-function mutations in the RNA SFs SRSF2, SF3B1, and U2AF1 (from hereon collectively referred to as SF-mutants) (Lee et al., 2016; Obeng et al., 2016; Seiler et al., 2018; Shirai et al., 2017), as well as solid tumors and lymphomas driven by MYC amplification and over-expression (Hsu et al., 2015; Koh et al., 2015).
Despite the large numbers of proteins involved in splicing, as well as post-translational modifications of splicing proteins and protein-protein, protein-RNA, and RNA-RNA interactions that regulate splicing, clinical efforts to inhibit splicing have largely utilized drugs inhibiting the interaction of the SF3B complex with RNA (Falco et al., 2011; Kaida et al., 2007; Kotake et al., 2007; Seiler et al., 2018). Currently, however, the safety of direct inhibition of RNA splicing catalysis in patients is unknown. More recently, a series of compounds that result in proteasomal degradation of the RNA SF RBM39 have provided an alternative pharmacologic means to perturb splicing (Han et al., 2017; Uehara et al., 2017). These compounds are effective at degrading RBM39 in vivo and lead to the deregulation of a splicing network required for acute myeloid leukemia (AML) survival (Wang et al., 2019).
PRMT family members regulate the activity of many proteins and their inhibition may affect splicing activities (Koh et al., 2015; Zhang et al., 2015b). With the observation that SF-mutant leukemias are more sensitive to further perturbation of splicing than their wild-type (WT) counterparts, we seek to identify if inhibitors of protein arginine methyltransferases (PRMTs) can preferentially target these leukemias.
RESULTS
Spliceosomal Interacting Proteins Are Targetable Vulnerabilities in SF-Mutant Cells
In an effort to identify additional means to therapeutically impact the process of splicing, we sought to identify proteins with functional relationships to components of the core splicing machinery, which might be druggable with available therapies. We built a network based on protein-protein (protein complexes or enzyme/substrate relationships), protein-DNA (transcriptional), and protein-RNA (post-transcriptional) regulation (Shannon et al., 2003), to obtain a list of 312 genes encoding an extended network of proteins with known or putative interactions with the core spliceosome. We then manually selected all druggable targets and sourced available inhibitors for these proteins (Figure 1A; Table S1). Given that most of the genes in the network (71 %) were transcriptional regulators, and that splicing is a co-transcriptional process influenced by transcription, we also included small molecules inhibiting epigenetic regulatory proteins (chemical probe collection from the SGC, Toronto) as additional controls.
Figure 1. Spliceosomal Interacting Proteins Are Targetable Vulnerabilities in Spliceosomal Mutant Cells.
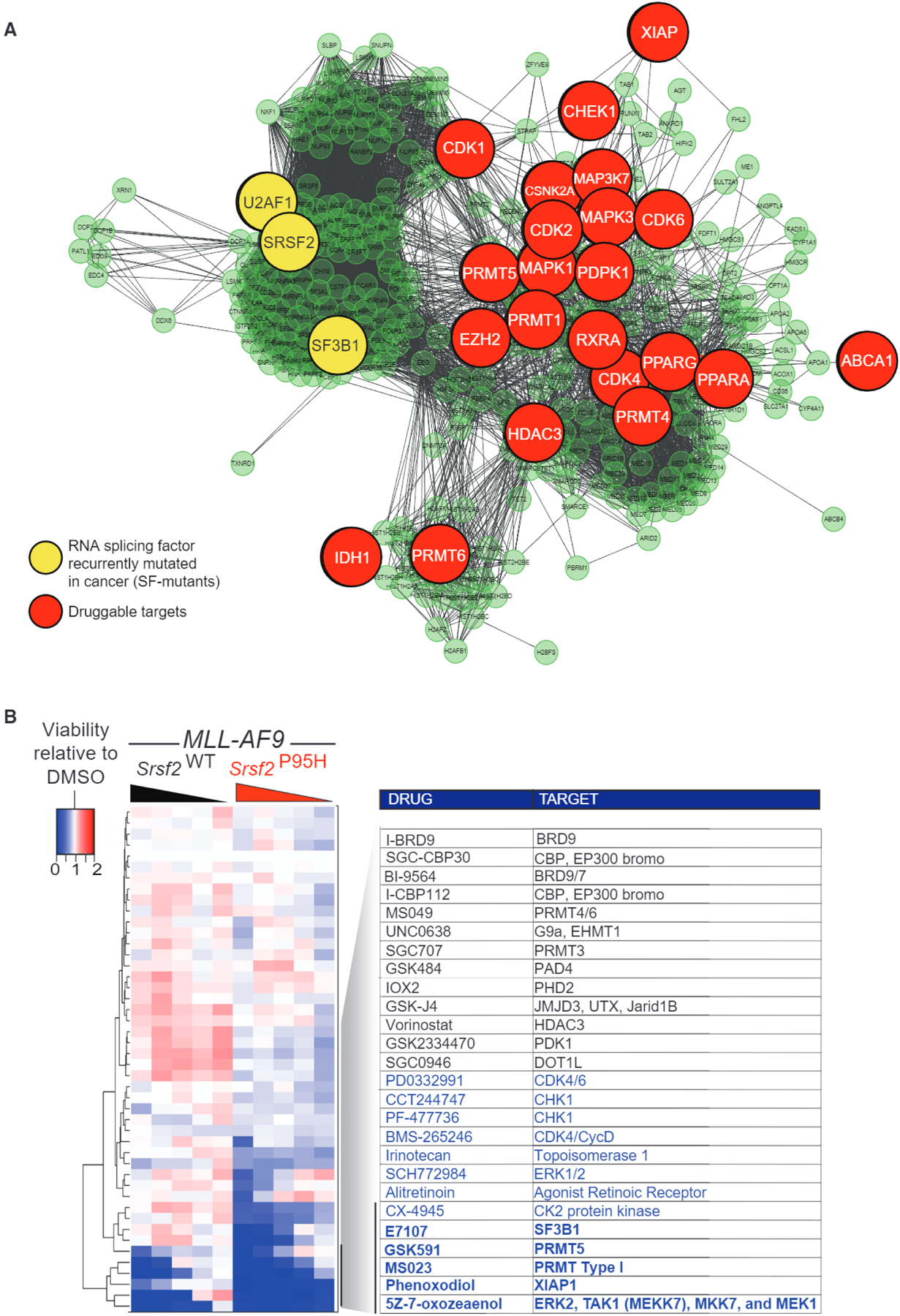
(A) Molecular interaction network generated by Cytoscape 3.4.0 (Shannon et al., 2003) displaying proteins involved in RNA splicing, snRNP assembly, and/or mutated in acute myeloid leukemia (AML) and their nearest neighbors of a given physical entity (e.g., genes or proteins). Genes and proteins are illustrated by nodes, which are connected by lines to nodes based on physical or functional interaction. Nodes that are RNA SFs mutated in cancer are displayed in yellow while those that are druggable targets are indicated in red.
(B) Heatmap of the relative viability of (MLL-AF9/Vav-cre Srsf2WT and MLL-AF9/Vav-cre Srsf2P95H to the indicated compounds following 7 days of growth scored by MTS assay and reported as a ratio to control DMSO-treated cells. Blue indicates a reduction, while red indicates an increase in cell viability, relative to DMSO-treated cells. The experiment was conducted in biological triplicate, and each individual run was repeated in technical triplicate.
See also Table S1.
We assembled a panel of 45 compounds based on the above criteria, and performed a drug screen using a range of five concentrations of each drug for 7 days in isogenic murine AML cells driven by the MLL-AF9 fusion in the absence or presence of mutant Srsf2 (MLL-AF9/Vav-cre Srsf2WT or MLL-AF9/Vav-cre Srsf2P95H cells). The generation of these cells has been described previously in studies identifying the preferential sensitivity of spliceosomal mutant cells to SF3B1 inhibitory compounds, and their growth rate is equivalent both in vitro and in vivo, regardless of Srsf2 genotype (Lee et al., 2016). Moreover, this is a genetically relevant model for human AML as SRSF2 mutations occur in ~10% of adult MLL-rearranged AMLs (Lee et al., 2016). The hallmark global sequence-specific change in RNA splicing characteristic of mutant SRSF2 is evident in the context of both human MLL-rearranged AML and murine MLL/AF9 Srsf2P95H cells (Lee et al., 2016).
We performed an in vitro drug screen and scored cell viability by MTS assay, after 7 days of drug treatment. Values were normalized to DMSO controls. Srsf2-mutant cells were more sensitive than WT counterpart cells to several inhibitors targeting components of the extended splicing network, including the SF3B complex inhibitor E7107 (Figure 1B; Table S1). Moreover, distinct inhibitors of PRMTs resulted in preferential killing of Srsf2-mutant AML cells over WT counterpart cells. These included GSK3203591 (abbreviated GSK591), a selective inhibitor of PRMT5 (Duncan et al., 2016), and MS023, a pan type I PRMTs inhibitor (Eram et al., 2016). Whereas type I PRMTs include PRMT1, 3, 4, and 6, selective inhibitors for PRMT3, PRMT4, or PRMT6 did not impair proliferation of Srsf2-mutant cells, suggesting that PRMT1 was the critical target of MS023 in this context.
PRMT5 and Type I PRMT Inhibition Preferentially Affect SF-Mutant AMLs
We next evaluated the effects of PRMT inhibitors on Srsf2WT versus Srsf2P95H cells over a range of drug concentrations to verify the results of the screen and to determine the half maximal inhibitory concentration for each compound (Figures 2A and 2B). As calculated by CellTiter Glo cell viability assay, Srsf2P95H cells were approximately 10-fold more sensitive than their Srsf2WT isogenic counterpart to GSK591 (120 versus 12 nM in Srsf2WT versus Srsf2P95H-mutant cells, respectively) and MS023 (1, 100 versus 120 nM in Srsf2WT versus Srsf2P95H-mutant cells, respectively). Similar preferential induction of apoptosis was seen for Srsf2P95H-mutant versus WT cells for both GSK591- and MS023-treated cells using a Caspase-Glo 3/7 (Figure S1A). The SF3B1 inhibitor E7107 was used in the same assays as a positive control, given previous data demonstrating enhanced sensitivity of Srsf2P95H cells to E7107 (Lee et al., 2016) (Figure 2C). Overall, these experiments identified that SF-mutant AML cells were >10 times more sensitive to either PRMT5 or type I PRMT inhibition than WT counterparts. The preferential killing of SF-mutant cells was mirrored by dose-dependent reductions of symmetric dimethylarginine and asymmetric dimethylarginine (SOMA and ADMA), respectively (Figures S1B and S1C), thereby also validating the on-target efficacy of these compounds. Moreover, we validated the increased sensitivity of SF-mutant AMLs to PRMT inhibitors across a panel of 32 genetically annotated human AML samples, half of which contained hotspot mutations in SF3B1, SRSF2, or U2AF1 (Figure 2D; Table S2). In this assay, primary AML mononuclear cells were exposed to GSK591 or DMSO for 6 days and evaluated by the number of viable cells at the end of culture. SF-mutant AML cells had a significantly increased sensitivity to GSK591 compared with SF-WT counterparts, an effect seen in SF3B1-, U2AF1-, and SRSF2-mutant samples. Similar effects were additionally observed in drug treatment of nine AML cell lines (four of which had mutations in SRSF2, U2AF1, or SF3B1; Figures S1D and S1E). This latter experiment was performed with the type I PRMT inhibitor GSK3368712, which has very similar potency and selectivity as MS023 (Fedoriw et al., 2019). Importantly these two latter experiments demonstrated that the increased sensitivity of SF-mutant AMLs to PRMT inhibitors extends beyond SRSF2-mutant leukemias to leukemias harboring mutations in SF3B1 and U2AF1 as well.
Figure 2. Preferential Effects of PRMT5 or Type I PRMT Inhibition on SF-Mutant AML over WT Counterparts In Vitro.
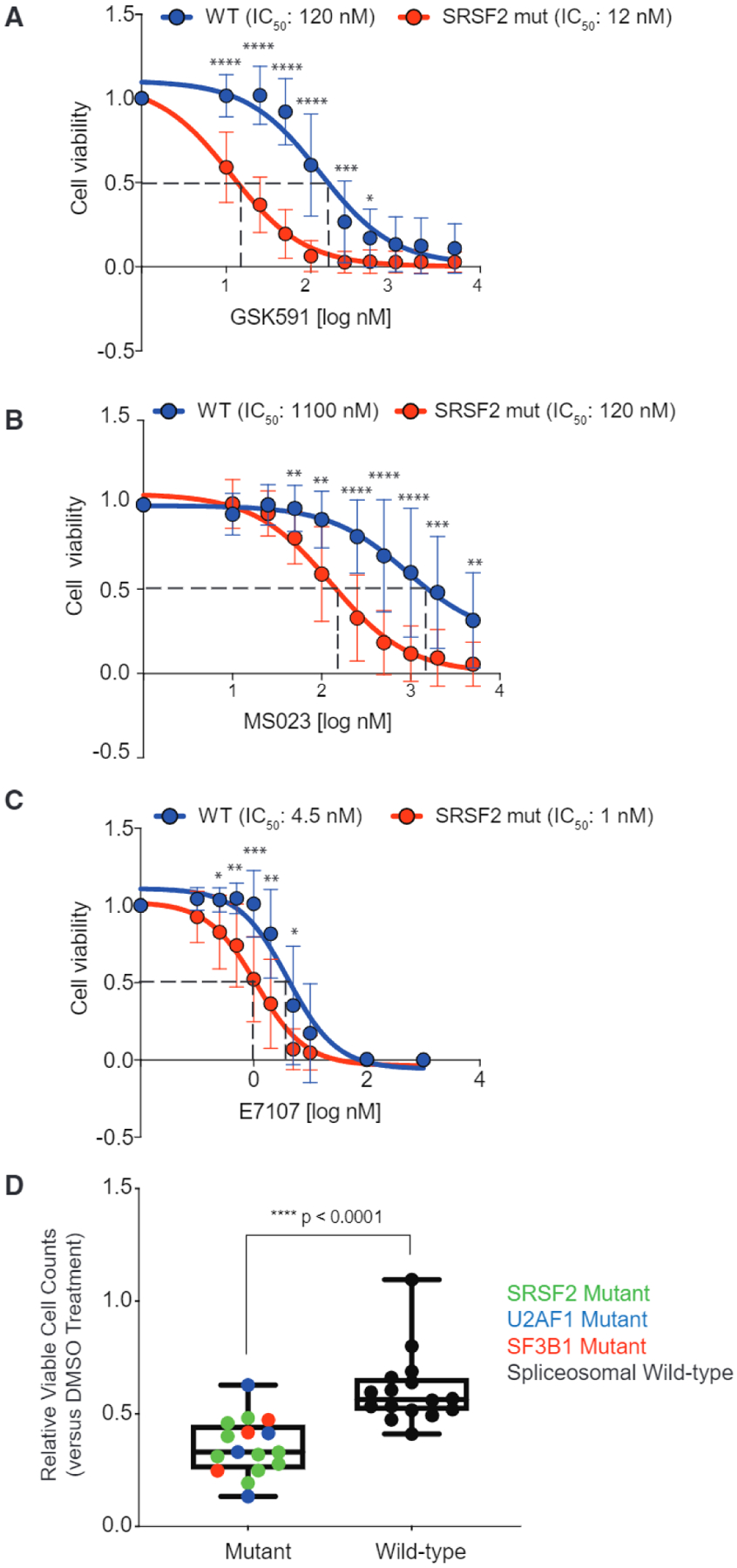
(A–C) Relative cell viability of (MLL-AF9/Vav-cre Srsf2WT and MLL-AF9/Vav-cre Srsf2P95H treated with GSK591 (A), MS023 (B), or E7107 (C), and normalized to control (SGC2096a for PRMT5, MS094 for PRMT1, and DMSO for E7107). Samples were prepared in four to six replicates and averages were calculated, error bars represent SD. Student’s t test was used for statistical analysis.
(D) Relative viable cell counts of AML patient samples to GSK591 based on spliceosomal gene mutation status. Primary AML cells with SF-mutations (n = 16) or WT for SRSF2, U2AF1, and SF3B1 (n = 16) were incubated with DMSO or GSK591 (0.5 mM) for 6 days. Cells were subjected to flow cytometry to detect 7-AAD-negative, YO-PRO1-negative, viable cells. Relative viable cell numbers were compared with Welch’s t test. Boxplot top line of whisker denotes the highest value in dataset and bottom line of whisker denotes the lowest value in dataset, box spans interquartile range and line in box indicates median.
*p = 0.01–0.05, **p = 0.001–0.01, ***P = 0.0001–0.001, ***p < 0.0001. See also Figure S1 and Table S2.
PRMT Inhibition Preferentially Affects SF-Mutant AMLs In Vivo
We next tested the sensitivity of leukemia cells to either PRMT5 or type I PRMT inhibitors based on SF-mutation status in vivo. We used the orally available PRMT5 inhibitor EPZ015666 dosed once a day at 200 mg/kg. Equal numbers of Srsf2WT or Srsf2P95H MLL-AF9-transformed leukemia cells were transplanted into secondary recipient mice and treatment was started at 8 days post-transplant to allow tumor engraftment (Figure S2A). EPZ015666 treatment increased the survival of mice transplanted with Srsf2P95H leukemias but not mice transplanted with Srsf2WT leukemias (Figure 3A). Western blot analysis demonstrated downregulation of global SDMA levels in spleen cells from mice treated with EPZ015666 (Figure 3B).
Figure 3. Preferential Effects of PRMT5 or Type I PRMT Inhibition on SF-Mutant AML over WT Counterparts In Vivo.
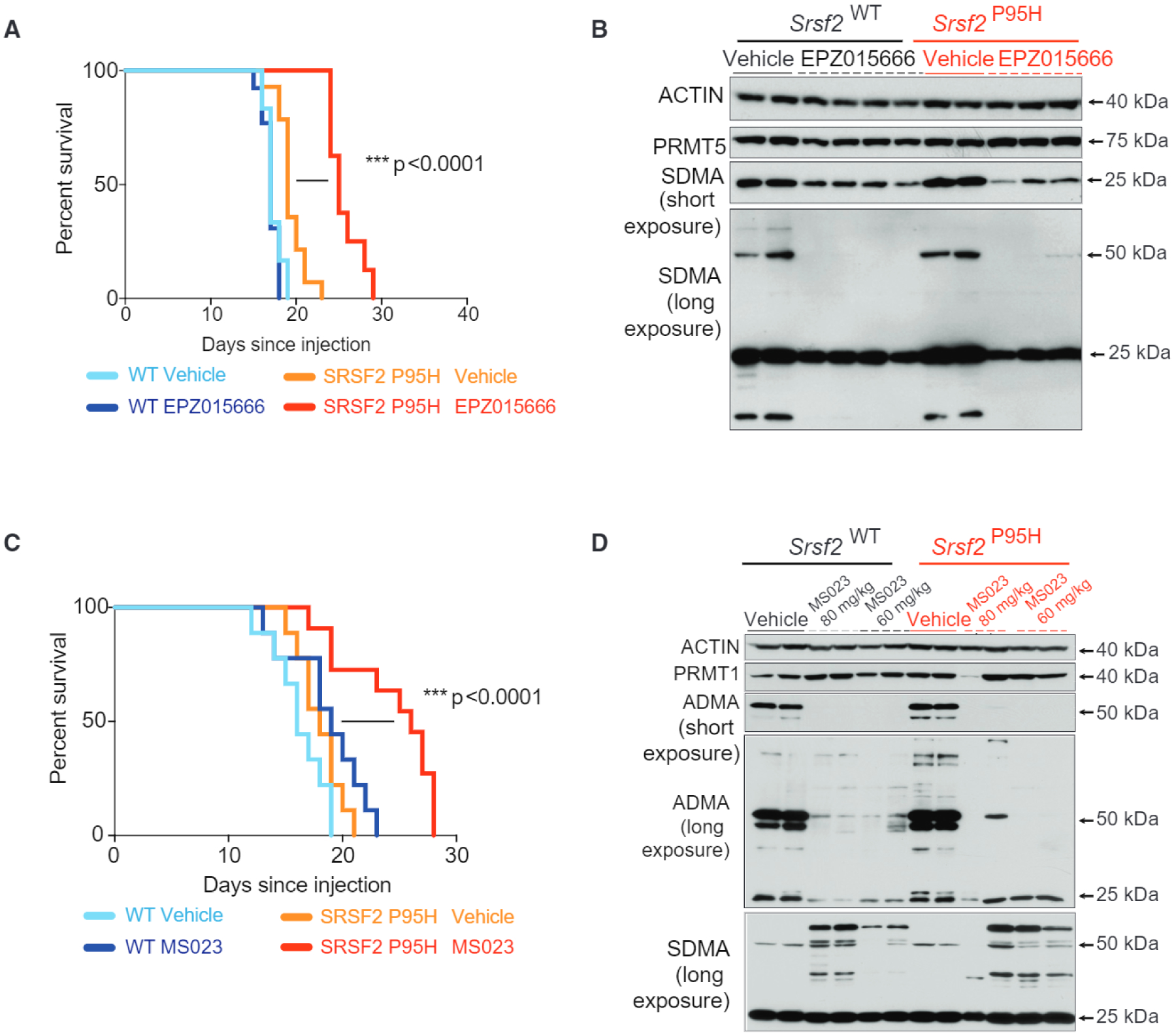
(A and B) Kaplan-Meier survival curve of mice treated with vehicle or EPZ015666. Survival comparison by Mantel-Cox log-ranked test (WT vehicle n = 12; WT drug n = 13, Srsf2P95H vehicle n = 14, Srsf2P95H drug n = 8) (A). Western blot of PRMT5, symmetric dimethyl arginine (SDMA; both a short exposure and a long exposure are shown), and actin in spleens of mice from (A) at time of death. Organs collected were 24 h after the last dose. Each column represents tissue from a distinct individual representative animal (B).
(C and D) Kaplan-Meier survival curve of mice treated with vehicle or MS023. Survival comparison by Mantel-Cox log-ranked test (WT vehicle n = 9, WT drug n = 9, Srsf2P95H vehicle n = 9, Srsf2P95H drug n = 11). (C) Western blot of PRMT1, asymmetric dimethylarginine (ADMA) (both a low exposure and a high exposure are shown), SDMA, and actin in spleens of mice from (C) at time of death. Organs collected were 24 h after the last dose. Each column represents tissue from a distinct individual representative animal (D). *p = 0.01–0.05, **p = 0.001–0.01, ***p = 0.0001–0.001, ****p < 0.0001.
See also Figure S2.
To evaluate the effect of type I PRMT inhibition on leukemias in vivo, we first identified the pharmacokinetic (PK) properties of MS023 (Figure S2B) and established an intraperitoneal dose of 80 mg/kg/day as optimal to downregulate ADMA in most organs (Figure S2C). Similar to that observed upon PRMT5 inhibition, treatment with MS023 delayed disease progression in mice transplanted with Srsf2P95H leukemias but not in the case of Srsf2WT AMLs (Figure 3C). Western blot analysis confirmed downregulation of global ADMA in bone marrow and spleen cells from mice treated with MS023. Concurrent upregulation of SDMA was observed, in accordance with previous reports of a scavenging effect by PRMT5 observed upon type I PRMTs inhibition (Dhar et al., 2013) (Figure 3D). Both EPZ015666 and MS023 were well tolerated in the mouse PK, and in vivo efficacy studies and no apparent toxicity was observed (Chan-Penebre et al., 2015) (and data not shown).
Synergistic Effects of Combined PRMTS, Type I PRMT, and/or SF3B Inhibition
Given the upregulation of SDMA seen with type I PRMT inhibition and the preferential sensitivity of SF-mutant AMLs to inhibition of symmetric as well as asymmetric arginine dimethylation, we next sought to evaluate the effects of simultaneous inhibition of both axes of PRMT function. To evaluate the synergism between the two inhibitors, we calculated their combination index (Cl), which quantitatively defines synergism (Cl < 1), additive effect (Cl = 1) and antagonism (Cl> 1) among two drugs (Chou, 2010). Combined in vitro treatment of Srsf2WT or Srsf2P95H MLL-AF9 leukemia cells with MS023 and GSK591, MS023 and E7107, or GSK591 and E7107, revealed synergistic effects of each combination, regardless of Srsf2 mutational status (Figures 4A–4C). A similar synergistic effect was seen in vivo where combined MS023 (60 mg/kg) and EPZ015666 (150 mg/kg) treatment (given starting at day 8 after engraftment for 10 days) resulted in significantly increased survival in recipient mice engrafted with both Srsf2WT and Srsf2P95H MLL-AF9 leukemia cells (Figure 4D). Consistent with the extended survival seen with combined MS023 and EPZ015666 treatment, PRMT inhibition in vivo resulted in reductions in both ADMA and SDMA levels in splenic tissue (Figure S2D).
Figure 4. Synergistic Effects of Combined PRMT5, Type I PRMT, and/or SF3B Inhibition on Primary Mouse AMLs.
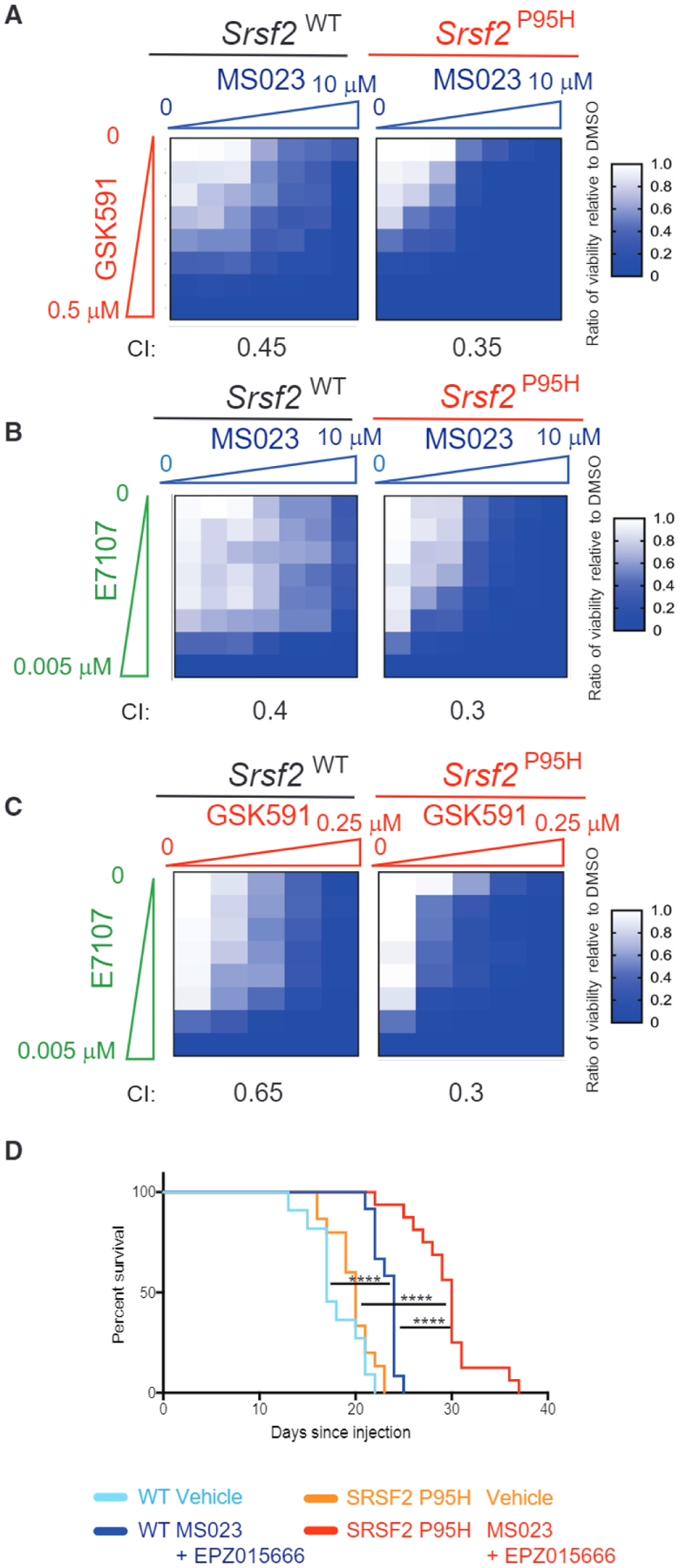
(A–C) Heatmap of half maximal inhibitory concentration values of WT or Srsf2P95H MLL-AF9 cells grown in increasing doses of MS023 plus GSK591 (A), MS023 plus E7107 (B), or GSK591 plus E7107 (C) for 7 days. The combination index (Cl) in each cell type for each drug pair is shown.
(D) Kaplan-Meier curve of mice transplanted with MLL-AF9 cells followed by combined treatment with MS023 plus EPZ015666 in vivo. Survival comparison by Mantel-Cox log-ranked test (WT vehicle n = 11, WT drug n = 12, Srsf2P95H vehicle n = 15, Srsf2P95H drug n = 16). *p = 0.01–0.05, **p = 0.001–0.01, ***p = 0.0001–0.001, ****p < 0.0001.
See also Figure S2.
We then tested the combinatorial effects of PRMT5 and type I PRMT inhibition in additional models. First, we assessed the effect of combined treatment with GSK591 and MS023 in human AML cell lines (Figures 5A–5C). Importantly, the synergy between the two drugs extent beyond MLL-AF9-driven malignancies and was also observed in those with CALM-AF10 (U937) and BCR-ABL (K562) fusions.
Figure 5. Synergistic Effects of Combined PRMT5, Type I PRMT, and/or SF3B Inhibition on Human AML Lines and Patient Samples.
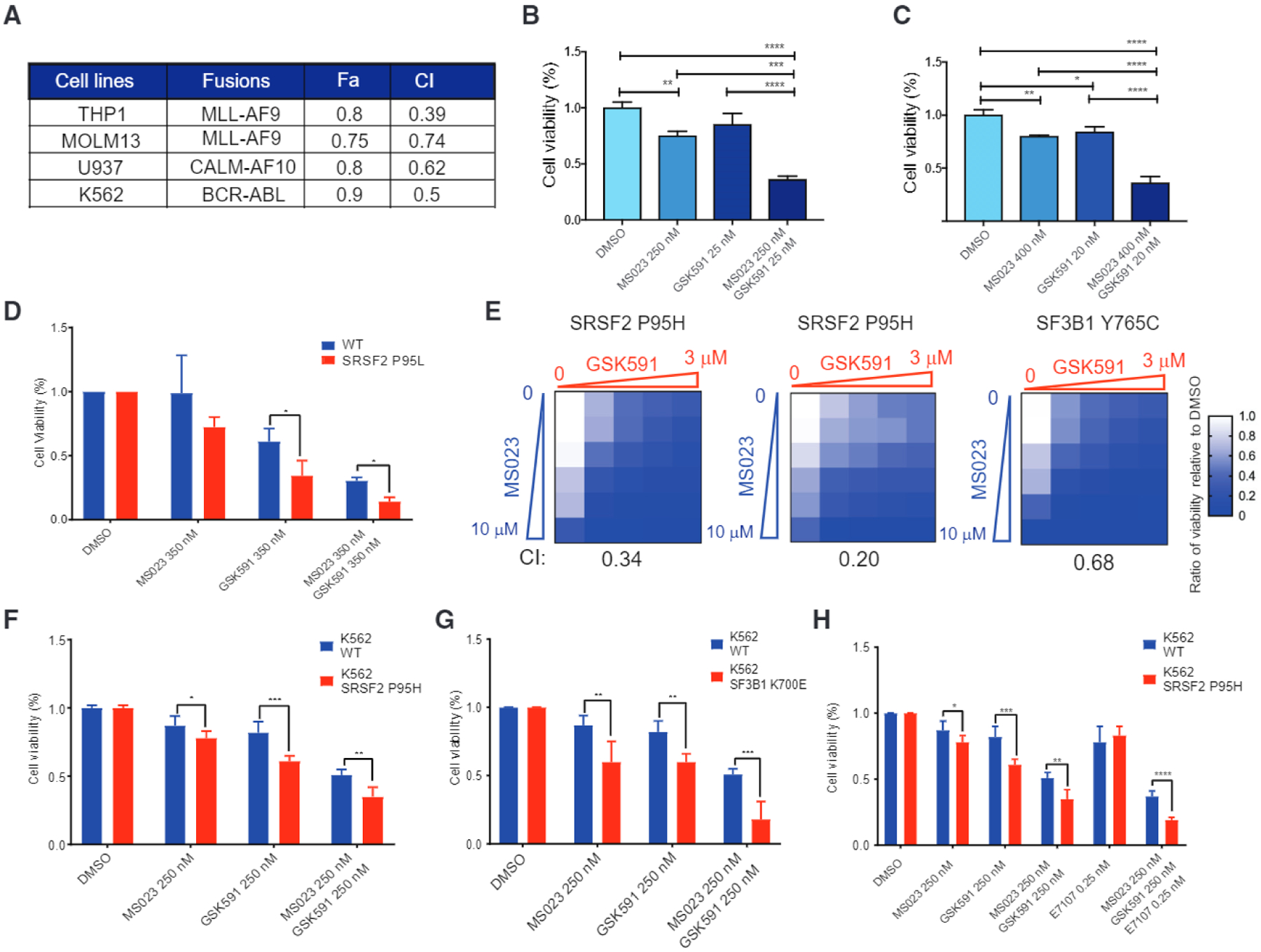
(A) Summary of Cl and fraction affected (Fa) values of human AML cell lines treated with MS023 and GSK591.
(B and C) Percentage of viable THP1 (B) and U937 (C) cells after 8 days exposure to MS023, GSK591, or the combination relative to DMSO in vitro.
(D) Hematopoietic progenitor cells differentiated from the iPSC lines 5–16 Cre20 (SRSF2 P95L) and N-2.12 (isogenic normal) were treated as indicated. Cell viability represents cell counts relative to DMSO-treated cells.
(E) Heatmap of cell viability after 6 days of treatment. Cl at IC50 values of MS023 plus GSK591 is indicated at the bottom.
(F and G) Percentage of viable K562 WT or SRSF2P95H knockin mutant cells (F) and K562 WT or SF3B1K700E knockin mutant cells (G) after 8 days treatment with MS023, GSK591, or the combination relative to DMSO in vitro.
(H) Percentage of viable K562 WT or SRSF2P95H knockin mutant cells after 8 days treatment with MS023, GSK591, E7107 or in combination relative to DMSO in vitro.
All data are representative of three independent experiments, error bars represent SD. *p = 0.01–0.05, **P = 0.001–0.01, ***P = 0.0001–0.001, ****p < 0.0001; one-way ANOVA was used to analyze THP1 and U937, while unpaired t test was used for K562 WT and SRSF2P95H. See also Figures S3 and S4.
Second, we used induced pluripotent stem cells (iPSCs) with a SRSF2P95L mutation knocked in by CRISPR and isogenic normal iPSCs (Chang et al., 2018). We differentiated the iPSCs into hematopoietic progenitor cells and treated them with PRMT inhibitors. The SRSF2-mutant cells were more sensitive than their isogenic WT control (Figure 5D).
Third, we tested three primary patient AML samples, two had Srsf2P95H and one had SF3B1Y765C, in vitro and found that GSK591/MS023 combination treatment resulted in a synergistic effect of reducing cell viability (Figure 5E; Table S3).
Fourth, we tested the efficacy of drug combination on a spliceosomal WT/MLL-rearranged and one SF3B1K700E/EV1-rear-ranged AML PDX models. Treatment with 60 mg/kg MS023 and 150 mg/kg EPZ015666 for 30 days resulted in reduction of human chimerism only in the SF-mutant AML (Figures S3A–S3C).
Fifth, we extended our observation to isogenic K562 human leukemia cell lines with knockin mutations of the endogenous SRSF2 (Srsf2P95H) or SF3B1 (SF3B1K700E) (Figures 5F and 5G). In each case, we observed a strong synergistic effect between GSK591 and MS023, which was more pronounced in SF-mutant isogenic cells. The changes in symmetric and asymmetric dimethylation of arginine in K562 cells, from single and combinatorial use of MS023 and GSK591, were validated by western blot (Figure S4).
Finally, given the reported effects of E7107 (Lee et al., 2016), and the synergy observed upon combining MS023 and GSK591 (Figures 4B and 4C), we tested the effect of a triple-drug combination. As expected, triple combination treatment with MS023, GSK591, and E7107 had a strong effect on cell viability and preferentially killed the SRSF2-mutant line over the WT control (Figure 5H). Altogether these experiments confirm the strong synergy between the drugs impacting different aspects of splicing catalysis (type I PRMTs, PRMT5 and SF3B1 inhibitors), which was accentuated in the presence of an SF mutation.
Global Profiling of PRMT Substrates at Single-Site Resolution by Quantitative Liquid Chromatography-Tandem Mass Spectrometry
We next set out to explore the mechanistic basis for the link between inhibition of arginine methylation and preferential effects on SF-mutant leukemias. Although previous studies have suggested a link between PRMT5 and splicing regulation through arginine methylation of spliceosome-associated proteins (Braun et al., 2017; Koh et al., 2015), PRMT5 has numerous substrates (Guo et al., 2010; Hamard et al., 2018; Jansson et al., 2008; Musiani et al., 2019; Sims et al., 2011; Xu et al., 2001). Similarly, type I PRMTs have a large number of cellular substrates. As a consequence, it is currently unclear which PRMT substrate(s) are most relevant to the observed cellular effects of PRMT inhibition. To address this question, we undertook a proteomic approach to identify PRMT5 and PRMT type I substrates in leukemia. In particular, we employed a stable isotope labeling with amino acids in cell culture (SILAC)-mass spectrometry strategy (Musiani et al., 2019) to characterize the arginine methyl proteome in acute promyelocytic leukemia NB4 cells (Figure 6A; Table S4). In brief, in a forward SILAC experiment, cells cultured in the medium complemented with heavy-labeled arginine (“H”) and lysine were treated with GSK591 or MS023, while cells cultured with light amino acids (“L”) were treated with vehicle. Upon H and L cell harvesting and mixing in equal ratio, protein extraction and in-solution tryptic digestion, peptides bearing mono-methylated arginine (MMA), SDMA, or ADMA, respectively, were enriched using pan-methyl-specific antibodies, before high-resolution nano-liquid chromatography-tandem mass spectrometry analysis for modified peptide identification and quantification (Figure 6A). The SILAC experiments were repeated in biological replicates in SILAC reverse mode, whereby the amino acid labels were swapped among the two functional states (see the STAR Methods). With this strategy, we were able to quantify a total of 391 and 735 A-methyl-peptides in the GSK591 and MS023 experiments, respectively. Specifically, in the GSK591 treatment experiment, we identified 299 peptides bearing MMA, 40 bearing DMA and 52 bearing both modifications; in the MS023 experiment, we found 433 MMA peptides, 200 DMA peptides, and 102 peptides bearing both modifications (Figure S5A). In total, these peptides carried 1, 188 methylation events on arginine, and were distributed on 219 different proteins (Table S4).
Figure 6. Global Profiling of PRMT Substrates at Single-Site Resolution by Quantitative Liquid Chromatography-Tandem Mass Spectrometry.
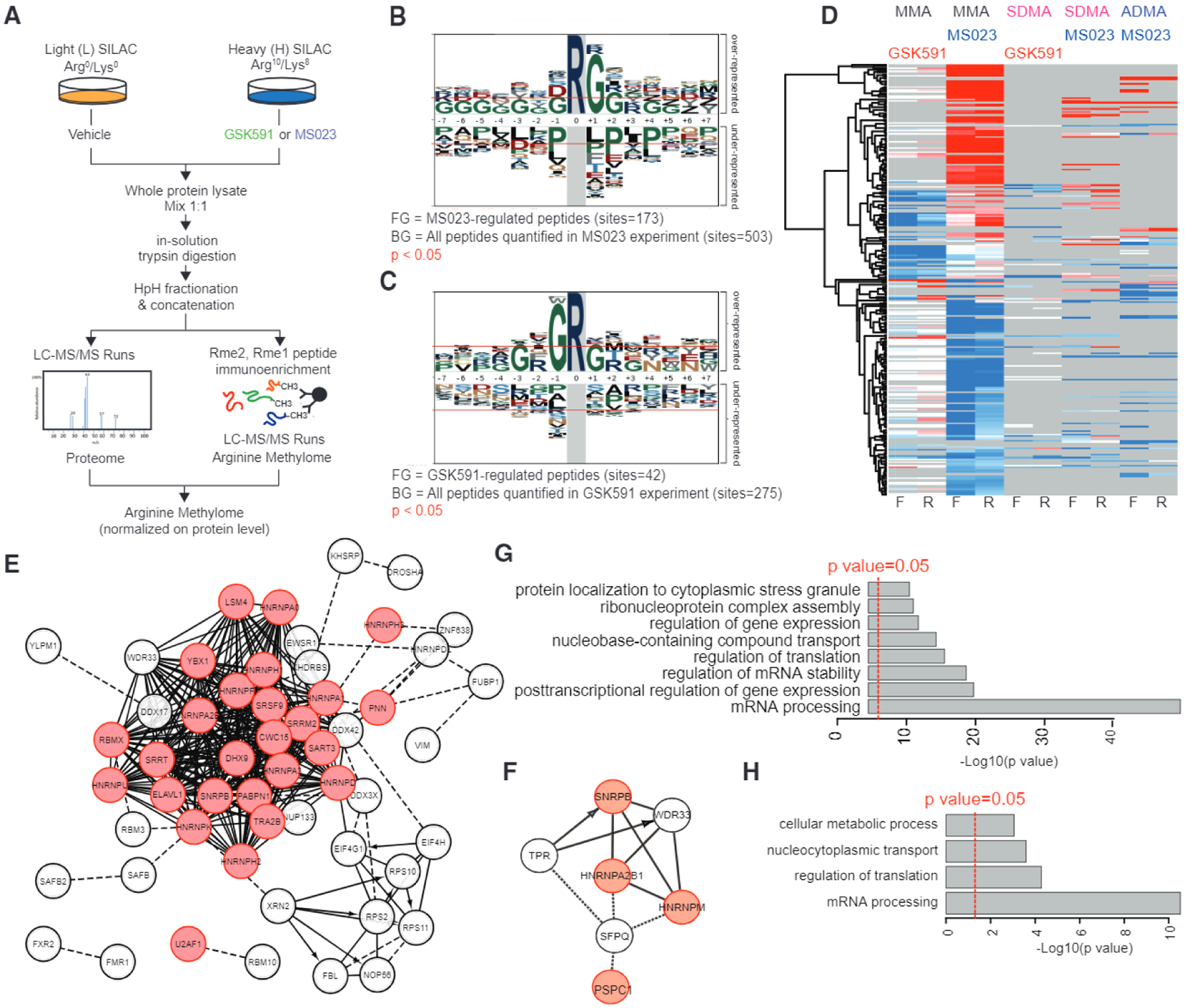
(A) Workflow of the SILAC methyl-R-proteomic experiments carried out to identify mono- and dimethylarginine substrates regulated by GSK591 and MS023 in NB4 leukemia cells.
(B and C) Sequence motif analysis indicates the consensus sequences significantly enriched in the methyl-peptides regulated by MS023 (B) or GSK591 (C).
(D) Heatmap of log2 SILAC ratios of each methyl-peptide identified and quantified from the SILAC experiment diagrammed in (A). For each pharmacological treatment two biological replicates, in forward and reverse SILAC mode, were carried out and different degrees of methylation were enriched and profiled. Only peptides reproducibly regulated in at least one pair of forward-reverse experiment are visualized in the heatmap.
(E and F) Network analysis of the proteins displaying methylation changes upon treatment with MS023 (E) or GSK591 (F). RNA-binding proteins are highlighted in red.
(G and H) Functional analysis of changing methylated proteins highlight the biological process terms significantly enriched among proteins displaying methylation changes upon treatment with MS023 (G) and GSK591 (H).
Analysis of the methyl-peptide SILAC ratios, normalized by the respective protein ratios, revealed that both GSK591 and MS023 caused a prominent downregulation of methyl sites, with 49 (15%) and 135 (16%) methyl-peptides significantly decreased and only 4 (1 %) and 97 (11 %) upregulated, respectively (Figure S5B; Table S4). Log analysis of the significantly regulated methyl-peptides showed specific enrichment for glycine at position + 1 and −1 around the modified arginine in the MS023-and GSK591-treated cells, respectively (Figures 6B and 6C). Interestingly, PRMT5 and type I PRMTs appear to regulate the methylation of distinct and non-overlapping proteins (Figure 6D). We did not detect histones as differentially arginine methylated, rather, proteins whose methylation level changed upon PRMT inhibition were mainly RNA-binding proteins (RBPs) with established roles in RNA export, regulation of RNA stability, and RNA splicing (Figures 6E–6H). In addition, MS023 elicited a specific effect on the methylation of proteins involved in translation (Figures 6E–6G). Most of the methyl-peptides responding to GSK591 (70%) and MS023 (89%) were orthogonally validated through the intersection with a high-quality methyl-proteome dataset annotated through the heavy methyl SILAC labeling strategy (Massignani et al., 2019; Ong et al., 2004) (Figure S5C; Table S4).
Importantly, to exclude a potential bias toward more abundant proteins in our methyl-proteome, we matched the methylated proteins identified with the immuno-enrichment approach to their respective protein abundances, calculated through the Intensity-Based Absolute Quantification (iBAQ) algorithm (Schwanhausser et al., 2011) in the whole proteome dataset, used as input for the methyl-peptide immunoprecipitation (Table S3). This experimental proteome included more than 7,300 proteins, encompassing a dynamic range of abundance of ~ 107 (Figure S5D, black bars). Within this protein abundance distribution, the 219 experimentally identified methyl-proteins covered a dynamic range of abundance of >105, spanning from high (e.g., EEF1A1 and HNRNPs) to low (e.g., KMT2C and SVIL) expressed proteins (Figure S5D, red bars). A similar dynamic range was also observed for proteins directly regulated by PRMT5 or type I PRMT inhibitors (Figure S5E).
This result suggests that, despite being a minor proportion of the detectable cellular proteome, the PRMT-dependent methyl-proteome is not biased toward the high abundant subset; hence, the overrepresentation of RBPs is not a mere reflection of the abundance of these proteins, but linked to the cellular function of PRMTs.
Cell-Cycle Deregulation upon PRMT5 and Type I PRMT Inhibition
To further understand the mechanistic underpinning for the synergistic cytotoxic effects of PRMT inhibition on leukemia cells, regard less of the spliceosomal gene mutational status, we performed RNA sequencing analysis of isogenic Srsf2P95H K562 leukemia cell lines and its WT counterpart, treated with DMSO, GSK591, and MS023, or a combination of the two. We first analyzed the global changes in gene expression caused by drug treatment. Gene ontology (GO) analysis revealed that there is an upregulation of genes involved in mitosis and cell-cycle regulation for both K562 WT and K562 Srsf2P95H cells (Figures 7A and 7B; Table S5). This suggests that drug combination treatment causes cell-cycle deregulation and indeed we could validate this by cell-cycle analysis on cells collected at day 8 of drug treatment. Specifically, inhibition of PRMT5 and type I PRMTs leads to a decrease in proportion of cells in G1 and S phase (Figure 7C). Concomitantly, we observed an increase in an apoptotic sub-G1 population (Figures 7C and 7D). Overall, upon PRMT inhibition, Srsf2P95H-mutant cells exited the cell cycle and induced caspase-3/−7 activity more than their WT counterpart.
Figure 7. Gene Expression Changes and Cell-Cycle Deregulation with PRMT5 and Type I PRMTs Inhibition.
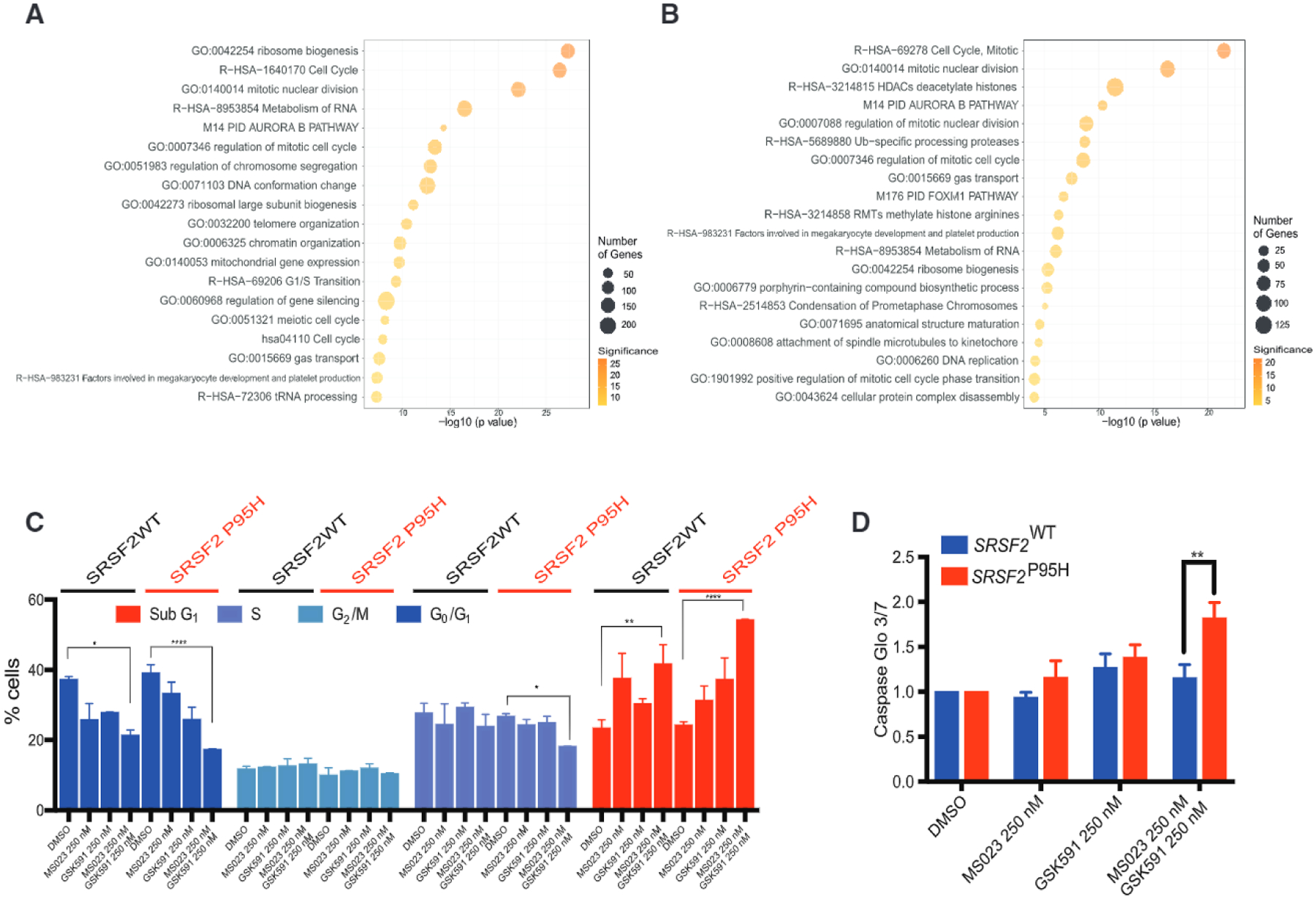
(A and B) GO categories of genes upregulated in K562 WT cells (A) and K562 SRSF2P95H cells (B) upon MS023 and GSK591 combination treatment.
(C) Cell-cycle analysis of K562 WT and SRSF2P95H cells upon MS023 and GSK591 treatment.
(D) Caspase-Glo 3/7 changes following 8 days treatment with MS023 and GSK591 in K562 WT and SRSF2P95H cells.
All data are representative of three independent experiments, error bars represent SD. *p = 0.01–0.05, **p = 0.001–0.01, ***p = 0.0001–0.001, ****p < 0.0001. Student’s t test used for statistical analysis.
PRMT5 and Type I PRMTs Inhibition Leads to Synergistic Changes in Alternative Splicing
We next conducted splicing analysis of the isogenic K562 leukemia cell lines treated with DMSO, GSK591, and MS023, or a combination of the two, to further decipher the possible cause for the overall synergistic killing of PRMT inhibitors, and preferential killing of Srsf2P95H over WT cells. As expected, Srsf2P95H-mutant and WT cells had a distinct transcriptome, typified by unique changes to RNA splicing, as described previously (Kim et al., 2015; Zhang et al., 2015a). First, the analysis revealed an increase in the number of aberrant splicing events upon treatment with both MS023 and GSK591, compared with control DMSO-treated cells. This was true both in WT and SRSF2P95H cells. Interestingly, the number of altered splicing events, particularly in cassette exons, increased upon combined exposure to GSK591 and MS023, both in K562 WT and Srsf2P95H cells (Figures 8A and 8B). In addition, drug combination induced a unique pattern of splicing alteration that is not simply the sum of the cassette exon splicing events caused by the individual drugs (Figure S6A).
Figure 8. PRMT5 and Type I PRMT Inhibition Leads to Synergistic Changes in Alternative Splicing.
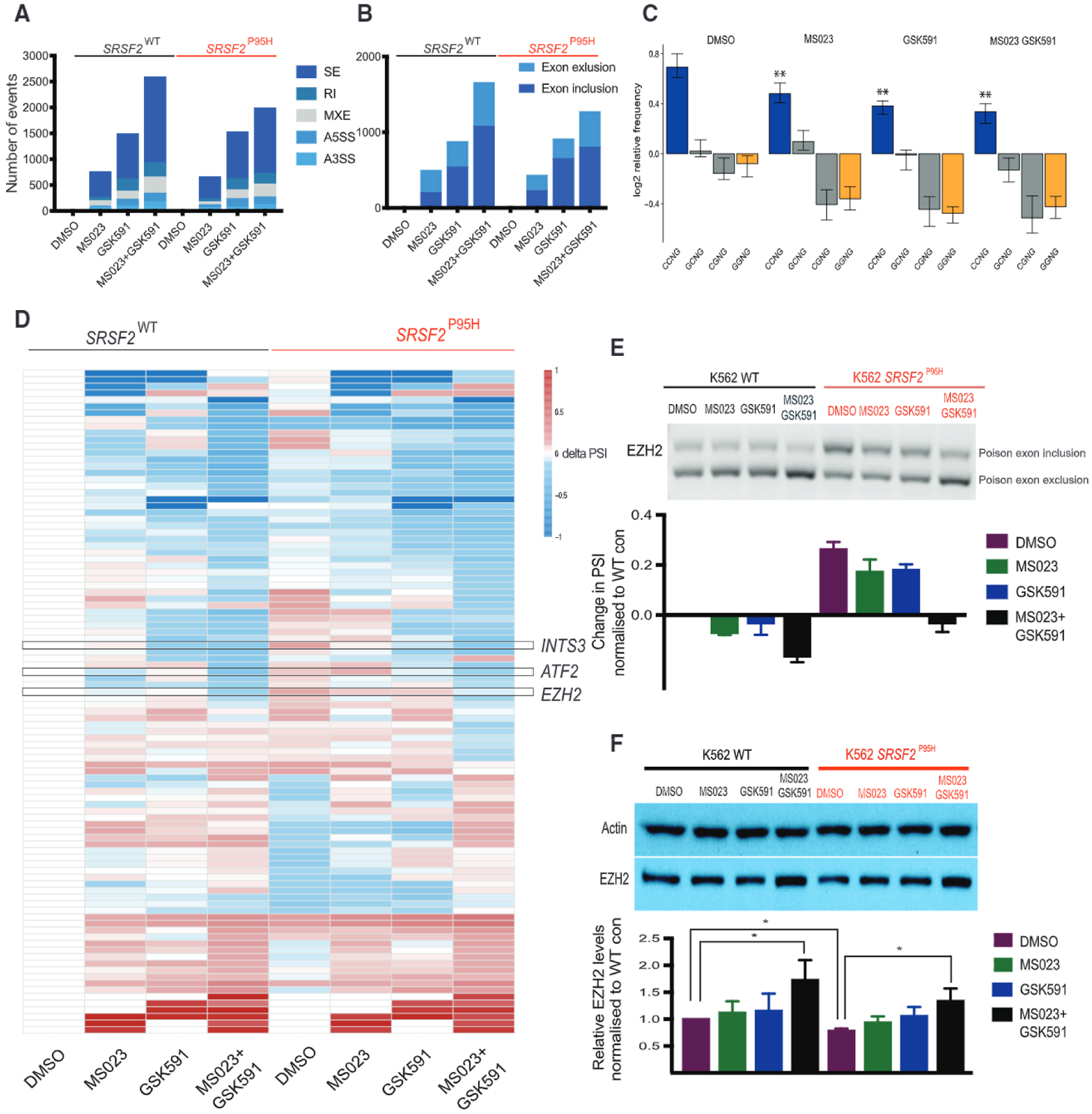
(A) Bar graphs enumerating numbers of significantly differentially spliced events in WT or SRSF2P95H K562 cells treated with each of the compounds listed versus DMSO control. A3SS, alternative 3′ splice site; A5SS, alternative 5′ splice site; MXE, mutually exclusive exon; RI, retained intron; SE, cassette exon.
(B) Bar graphs enumerating numbers of significantly differentially spliced exons in WT or Srsf2P95H K562 cells treated with each of the compounds listed versus DMSO control.
(C) Changes in levels of splicing events with CCNG, GCNG, CGNG, or GGNG motifs upon MS023 and GSK591 treatment in K562 cells.
(D) Heatmap showing change in PSI of exon splicing events normalized to PSI of WT control within the “Cell Cycle” gene ontology category.
(E) RT-PCR data showing changes in PSI levels of EZH2 poison exon inclusion in K562 cells with or without knockin of SRSF2P95H upon MS023 and GSK591 treatment in vitro.
(F) Western blot showing changes in EZH2 protein levels upon MS023 and GSK591 treatment in vitro.
All data are representative of three independent experiments, error bars represent SD. *p = 0.01–0.05, **p = 0.001–0.01, ***p = 0.0001–0.001, ****P < 0.0001. Student’s t test used for statistical analysis. See also Figures S6 and S7 and Table S5.
Second, we compared the overlap of the deregulated events among the two genotypes. Given that Srsf2P95H and WT cells have a different transcriptome to begin with, it was not surprising to note a divergence in the inclusion/exclusion of the affected cassette exons. More specifically, about one-third of the cassette exon events were commonly deregulated upon GSK591 + MS023 treatment, while the rest were uniquely spliced in either WT or Srsf2P95H cells (Figure S6B).
Third, we confirmed that Srsf2P95H-regulated exon inclusion/exclusion events have enrichment for C-rich exonic splicing enhancer sequences over G-rich sequences. Interestingly, treatment with PRMT inhibitors, led to a reduction in events containing a CCNG motif (preferentially regulated by Srsf2P95H) and to an overall depletion of events regulated by WT SRSF2 (containing a GGNG and CGNG motif) (Figure 8C).
We then looked at the GO enrichment of cassette exon events changing upon combination drug treatment in K562 SRSF2P95H cells and focused on the top perturbed GO categories, which included microtubule organization, DNA repair, and cell-cycle regulation (Table S5). We generated heatmaps for events in each GO category to depict the change in percentage spliced in (delta PSI) values upon single- and double-drug treatment in both cell lines, relative to K562 WT (Figures 8D and S6C). The rationale for this approach was to identify those splicing changes that could explain the preferential sensitivity of mutant cells to PRMT inhibition. Within these GO categories, we identified cassette exon splicing events that were changing more in K562 Srsf2P95H than in their WT counterparts and validated some of these events (Figure S6D). As DNA repair genes are broadly deregulated by PRMT inhibition, we also performed western blot of phospho-H2Ax to check for evidence of DNA damage. Indeed, there is a significant upregulation of γH2Ax, especially in the K562 SRSF2P95H cell, upon combination drug treatment (Figure S6E). Overall, although the number of deregulated splicing events is similar between WT and Srsf2P95H-mutant cells, they affect different genes and pathways, ultimately resulting in increased DNA damage and cell-cycle arrest.
Among the splicing events in the cell-cycle regulation GO category, we identified an aberrant splicing event within the mRNA encoding EZH2 (Kim et al., 2015; Lee et al., 2016). This specific event, which is driven by SRSF2 and not by SF3B1 mutations (Pellagatti et al., 2018) (Figures S6F and S6G), leads to the inclusion of a poison exon, to the formation of a premature stop codon, and subsequent nonsense-mediated decay (Kim et al., 2015; Lee et al., 2016). Interestingly, similar to that previously observed for E7107 (Lee et al., 2016), we observed decreased poison exon inclusion upon PRMT inhibition (Figures 8E, S6G, and S7A), leading to increased EZH2 protein abundance (Figure 8F). To functionally test the meaning of this event and assess whether toxicity induced by MS023 and GSK591 was at least in part mediated by EZH2 re-expression, we took two approaches. We first designed an antisense oligonucleotide (AON) to specifically mimic the exon-skipping event induced by PRMT inhibitors. The designed AON was able to reduce the inclusion level of the EZH2 poison exon (Figure S7B), increase EZH2 protein levels (Figure S7C), and reduce cell viability in K562, which was more evident in SRSF2P95H-mutant cells than in WT controls (Figure S7D).
To complement the above AON approach, we also used CRISPR/Cas9 to delete the endogenous EZH2 in K562 cells. We then treated EZH2 null cells or the parental control line with MS023, GSK591, or a combination of the two. EZH2 null cells were significantly more resistant to PRMT inhibition, suggesting that the effect of these small molecules is at least in part mediated by restoration of EZH2 levels in AML cells (Figure S7E).
DISCUSSION
Inhibition of PRMT5 or type I PRMTs has been linked to several downstream effects and proven to impact multiple pathways. Nonetheless, with the exception of the well-described MTAP deletion, which renders cells more susceptible to further depletion of PRMT5 (Kryukov et al., 2016; Marjan et al., 2016; Mavrakis et al., 2016) or inhibition of type I PRMTs (Fedoriw et al., 2019; Gao et al., 2019), there has been no clear indication of additional potential vulnerabilities of cancer cells to PRMT deletion/inhibition.
Overall, our data identify a strong impact of inhibiting protein arginine methylation on RNA splicing. PRMT5 or type I PRMTs inhibition resulted in preferential killing of SF-mutant AML, and overall synergy across genetic backgrounds. Mechanistically, this can be explained through inhibition of arginine post-translational modification (SDMA and ADMA) of a large compendium of RBPs involved in splicing regulation.
Currently, there is no standardized way to predict the effects of arginine methylation on protein function. Even less is known on the interplay between ADMA and SDMA occurring at distinct or overlapping sites. However, despite the current limitations on systematic predictions, there are several examples in the literature describing the effect of individual arginine methylation events on the function of PRMT5 and/or type I PRMTs substrates.
As described here, type I PRMTs and PRMT5 methylate different arginine residues on a common set of RBPs involved in RNA post-transcriptional regulation. Both ADMA and SDMA maintain the positive charge of the arginine side chain, but reduce its hydrogen bonding capabilities. Methylation concurrently increases the hydrophobicity to the arginine side chain, favoring its interaction with aromatic cages. As a consequence, arginine methylation has been shown to alter protein-protein interactions (Erce et al., 2013) and protein-RNA interactions (Denman, 2002; Dolzhanskaya et al., 2006). A well-documented example is the arginine methylation of three of the seven Sm proteins (SNRPB/B’, SNRPD1, and SNRPD3), leading to their binding to the Tudor domain on SMN1 (Bezzi et al., 2013; Friesen et al., 2001; Meister et al., 2001; Meister and Fischer, 2002). This is a critical event in the assembly of spliceosomal snRNPs, and, consistently with previous reports, we detected significant changes in SNRPB methylation in our dataset at arginines 112 and 147, upon PRMT inhibition.
In addition, we identified several arginine methylated residues on SFPQ and PSPC1. Interesting, both proteins are core components of the membrane-less nuclear structures called para-speckles, which play a central role in cancer and stress response (Adriaens et al., 2016). In particular, SFPQR693me1 and PSPC1R507me1 are reduced in the presence of GSK591, while PSPC1R4me1/me2a are reduced in the presence of MS023. Whereas, in the case of SFPQ, arginine methylation is associated with increased RNA binding (Snijders et al., 2015), there is no report describing the role of PSPC1 methylation, a point that warrants future investigation.
Recently, ADMA methylation by PRMT1 has been shown to weaken the cation π interactions between RGG/RG-rich motifs and low complexity regions of several disease-associated prion-like intrinsically disordered proteins (Hofweber et al., 2018; Qamar et al., 2018; Tsai et al., 2016). We detected changes in methylation of several members of this family of proteins (e.g., FUS, G3BP1, hnRNPA2B1, and hnRNPA 1). We hence predict that PRMT5 and PRMT type I inhibitors may have an impact on reversible liquid-liquid phase separation and on the assembly/disassembly of membrane-less organelles, impacting RNA post-transcriptional regulation.
Finally, there are numerous examples of how ADMA/SDMA may regulate localization of proteins including cellular localization of RBPs such as Sam68, Aven, FUS, hnRNPA1, and hnRNPA2 (Cote et al., 2003; Nichols et al., 2000; Thandapani et al., 2015; Tradewell et al., 2012; Wall and Lewis, 2017).
It is well documented that SF genes mutated in cancers are mutually exclusive (Yoshida et al., 2011) and that they have a synthetic lethal relationship when co-mutated in the same cell (Lee et al., 2018). Here we observed a general perturbation of gene expression and, importantly, of alternative splicing (increased number of skipped/included exons and retained intrans) upon PRMT inhibition. Given that SF-mutant cells are preferentially sensitive to genetic or pharmacologic perturbations in splicing, compared with spliceosomal WT counterparts (Lee et al., 2016; Obeng et al., 2016; Seiler et al., 2018; Shirai et al., 2017; Wang et al., 2019), an increase in overall splicing perturbation is predicted to lead to their preferential killing. This has been previously observed upon treatment with SF3B1 inhibitors (Lee et al., 2016; Obeng et al., 2016; Seiler et al., 2018; Shirai et al., 2017; Wang et al., 2019) and RBM39 degrading compounds (Wang et al., 2019). Our results reveal how inhibition of both type I and type II PRMTs can lead to a similar preferential killing of SF-mutant AML and myelodysplastic syndrome cells.
At the same time, it is possible that PRMT inhibition causes the aberrant splicing of oncogenic isoforms required for the survival of SF-mutant cells, leading to their preferential killing over WT counterparts. To test this latter point, we focused our analysis on the aberrant splicing of the EZH2 mRNA. In myeloid malignancies, EZH2 acts as a tumor suppressor, and it is often deleted and/or affected by loss-of-function mutations (Ernst et al., 2010; Nikoloski et al., 2010). Although we acknowledge that multiple genes could mediate the downstream effects of PRMT inhibition, we did observe a partial rescue upon CRISPR-mediated knockout of EZH2.
In human Srsf2P95H-mutant cells, we detected the previously reported aberrant inclusion of a poison exon in the EZH2 transcript, which harbors a premature stop codon and targets it for nonsense-mediated decay (Kim et al., 2015; Lee et al., 2016). Combinatorial use of the PRMT inhibitors caused a reduction in poison exon inclusion and increased EZH2 protein levels. The importance of this splicing event as part of the mechanistic basis for increased sensitivity of SRF2P95H cells to PRMT inhibition was further validated by the increased sensitivity of K562 SRSF2P95H cells to the use of AON that induced skipping of the poison exon, and with CRISPR/Cas9 deletion of EZH2, which reduced sensitivity of K562 cells to PRMT inhibition.
To conclude, the data provided here have important therapeutic implications for patients with spliceosomal gene mutations, given ongoing clinical trials with GSK3326595 (PRMT5 inhibitor from the same chemical series as EPZ015666 and GSK3203591; NCT03614728) and with a type I inhibitor GSK3368715 (Fedoriw et al., 2019; NCT03666988). Specifically, our data provide a strong basis for patient selection in the use of PRMT5 and type I PRMTs inhibitors in ongoing and future clinical trials of these agents based on the presence of genetic alterations impacting RNA splicing. At the same time, our proteomic data identify that PRMT5 and type I PRMTs largely regulate a distinct set of substrates, and, coincident with this, the simultaneous inhibition of both PRMT axes resulted in strong synergistic effects. A notable example is the increased expression of EZH2 protein in SRSF2P95H K562 cells. EZH2 is recurrently deleted or affected by loss-of-function mutations in myeloid malignancies (Ernst et al., 2010), while it is partially inactivated by aberrant splicing in SRSF2-mutant cells. These data extend the therapeutic utility of PRMT inhibitors, identify a combinatorial drug strategy utilizing simultaneous PRMT5 and type I PRMT inhibition, and implicate perturbation of RNA splicing as an important cellular mediator of cell death due to inhibition of PRMTs.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ernesto Guccione (eguccione@imcb.a-star.edu.sg; Ernesto.guccione@mssm.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines and Cell Culture
Primary murine MLL-AF9 leukemia cell lines were generated from bone marrow cells of leukemia-bearing mice and maintained in IMDM/15% FCS supplemented with L-glutamine, murine-SCF (25 ng/mL), murine-lL3 (10 ng/mL) and murine-lL6 (10 ng/mL). K562 isogenic cell lines (engineered to express SRSF2P95H or SF3B1K700E mutations from each respective endogenous locus from Horizon Discovery Inc.) were cultured in IMDM/10% FBS. THP-1, U937, MOLM13, KO52, GDM-1 and HNT34 cell lines were cultured in RPMl/10% FBS. TF1 cells were cultured in RMPl/10% FBS/2 ng/ml recombinant human GM-CSF. OCI-AML5 and F-36p cells were cultured in RPMl/10% FBS/10 ng/ml recombinant human GM-CSF. MonoMac 6 cells were cultured in RPMI/10% FBS/10 μg/ml recombinant human insulin. HEK293T cells were grown in DMEM medium with 10% FBS. The iPSC lines 5–16 Cre20 (SRSF2 P95L) and N-2.12 (isogenic normal) (Chang et al., 2018) were differentiated along the hematopoietic lineage as previously described (Kotini et al., 2017). To induce reprogramming, 10,000–300,000 cells were plated on retronectin-coated 24-well dishes and transduced with the OKMS lentiviral vector CMV-fSV2A (Kotini et al., 2015). The cells were harvested one or two days later and plated on mitotically inactivated MEFs in 6-well plates and centrifuged at 500 rpm for 30 min at RT. The following day and every day thereafter, half of the medium was gently changed to hESC medium with 0.5 mM valproic acid (VPA). In the first 10 days, cells contained in the removed medium were collected by centrifugation and placed back in their original wells. After 3–4 weeks, manually picked colonies with hPSC morphology were expanded. iPSCs were cultured on mitotically inactivated MEFs or in feeder-free conditions on Matrigel with hESC media supplemented with 6 ng/ml FGF2. Primary human AML patient sample cells were grown in lscove’s Modified Dulbecco’s Medium (IMDM), 20% bovine serum albumin, insulin, and transferrin (BIT) 9500 serum substitute, 16.7 μg/ml human low-density lipoproteins, 55 μM beta-mercaptoethanol with recombinant human (rh) G-CSF (20 ng/ml), rhGM-CSF (20 ng/ml), rh IL3 (20 ng/ml), rh IL6 (20 ng/ml), rh FLT3 ligand (50 ng/ml) and rh SCF (50 ng/mL).
Primary Human Samples
Studies were approved by the Institutional Review Boards of the University Health Network, Weill Cornell College of Medicine, and Memorial Sloan Kettering Cancer Center. Studies were conducted in accordance to the Declaration of Helsinki protocol. De-identified primary human AML samples derived from whole peripheral blood or BM mononuclear cells were utilized. The MSK-IMPACT assay was used to perform mutational genotyping of samples, as described previously (Cheng et al., 2015; Zehir et al., 2017). Cord blood was acquired from NY Blood Bank. Informed consent was obtained from all subjects to use the specimens described in this study. Specimens were obtained as part of the Memorial Sloan- Kettering Cancer Center Institutional Review Board approved clinical protocol #06–107 to which all subjects consented. O.A-W is a participating investigator on this protocol.
Animals
6–8 weeks old C57BL/6 mice were purchased from lnVivos. Mice were maintained in individual ventilated cages and fed with auto-claved food and water in the Biological Resource Center, A*STAR Singapore. 10 week-old NSG-SGM3 (NSGS) female mice were purchased from Jackson Laboratory. Mice were maintained in individual ventilated cages and fed with autoclaved food and water in Memorial Sloan Kettering Cancer Center. All animal experiments were conducted in accordance to approved protocols from Institutional Animal Care and Use Committees of Institute of Molecular and Cell Biology (IMCB), A*STAR and Memorial Sloan Kettering Cancer Center. Kaplan-Meier survival curves were compared using the Mantel-Cox Log-rank test via Graphpad Prism 7.
METHOD DETAILS
Identification of Druggable Targets in the Extended Splicing Network
A list of genes belonging to the Gene Ontology (GO) category GO_SPLICEOSOMAL_SNRNP_ASSEMBLY was derived from the Gene Set Enrichment Analysis (GSEA) website (Subramanian et al., 2005). We uploaded this list on the CBIO portal website (Cerami et al., 2012) and selected as a query “Acute Myeloid Leukemia, TCGA” study (Cerami et al., 2012; Gao et al., 2013; Ley et al., 2013). The network tab provided a list of neighboring genes (sif file). This network comprises the nearest neighbors of a given physical entity (e.g. gene, protein or small molecule). The following rules govern the construction of the neighborhood: 1) if A is part of a [complex] (A:B), (A:B) is included in the neighborhood, but none of the interactions involving (A:B) are included. 2) if A is a [CONTROLLER] for a [control] interaction, the reaction that is [CONTROLLED] (and all the participants in that reaction) are included in the neighborhood. 3) if A participates in a [conversion] reaction, and this reaction is [CONTROLLED] by another interaction, the [control] interaction (plus its [CONTROLLER]) are included in the neighborhood. The sif file was then imported into Cytoscape 3.4.0(Shannon et al., 2003) to visualize molecular interaction networks and integrate the data with gene expression profiles and other state data. We next created a network of functional interacting genes using the application ‘Reactome Fl’.(Wu et al., 2014) The list of genes belonging to the extended splicing network was manually curated to identify druggable targets. We used www.drugbank.ca and www.chemicalprobes.org to identify the inhibitors of the druggable genes.
Animal Studies
In leukemia transplant experiments, mice were monitored daily for any sign of distress and leukemia development. The number of mice chosen in each experiment was chosen to give 90% statistical power with a 5% error level given the differences in standard deviation that was observed in the pilot study. Generation and genotyping of the Srsf2P95H/+ conditional knock-in mice as well as MLL-AF9/Vav-cre Srsf2WT/WT (referred to Srsf2WT throughout the text) and MLL-AF9/Vav-cre Srsf2P95H/WT (referred to Srsf2P95H throughout the text), all on C57BL/6 backgrounds, are as previously described (Kim et al., 2015). Briefly, Vav-Cre+Srsf2 WT/WT and Vav-Cre+Srsf2P95H/WT mice were treated with a single dose 5-fluorouracil (150 mg/kg) and bone marrow were harvested 6 days later. The red blood cells from the bone marrow were lysed by ammonium-chloride potassium bicarbonate lysis buffer (ACK lysis buffer). The bone marrow cells were then transduced with viral supernatant containing the murine stem cell virus (MSCV)-driven MLL-AF9 fusion oncogene with an internal ribosomal entry site to express a GFP tag (MSCV-MLL-AF9-IRES-GFP) for 2 days in lscove’s modified Dulbecco’s medium (IMDM) containing 15% fetal bovine serum (FCS) and supplemented with mouse stem cell factor (mSCF) (25 ng/ml), mouse interleukin-6 (10 ng/ml) and mouse interleukin-3 (10 ng/ml). Thereafter, lethally irradiated (9.5 Gy) C57BL6/J mice are injected with ~400,000 cells via tail vein injection to generate primary leukemias. All in vivo drug experiments were conducted as secondary leukemia transplants whereby 6–8 week old C57BL6/J mice are sublethally irradiated (5 Gy) and injected with 250,000 MLL-AF9 leukemia cells collected from moribund mice with leukemia (Lee et al., 2016).
AML PDXs were generated from patient peripheral blood and/or bone marrow mononuclear cells and subsequently transplanted intrafemorally into 10 week-old NSG-SGM3 (NSGS) female mice (Jackson Laboratory). Once human CD45 chimerism was >25% of the bone marrow, NSGS mice were eventually treated with 60 mg/kg MS023 and 150 mg/kg EPZ015666 for 6 weeks.
In Vitro Drug Screen with Murine MLL-AF9 Cells
Both (MLL-AF9/Vav-cre Srsf2WT and MLL-AF9/Vav-cre Srsf2P95H cells were seeded at 900 cells/well in 384 well plate. Cells were treated with indicated drugs for 7 days over various concentrations (see Table S1). Cell viability was measured using MTS or Cell TiterGlo assay (Promega) as per manufacturer’s instructions and values were normalized to DMSO controls.
In Vitro Drug Titration with Murine MLL-AF9 Cells
All cell lines were seeded in white flat-well 96 well plates (Costar) at 1000cells/well unless otherwise indicated. For murine MLL-AF9 cell lines single drug titration experiments, cells were exposed to MS023 from a range of 0–5 μM, to GSK591 from a range of 0–5 μM and to E7107 from a range of 0–1 μM for seven days. For murine MLL-AF9 cell lines combination drug titration experiments, cells were exposed to MS023 from a range of 0–5 μM, to GSK591 from a range of 0–0.5 μM and to E7107 from a range of 0–0.05 μM for seven days. Cell viability read-out was performed using the Cell-titre glo assay (Promega) as per manufacturer’s instructions and normalized to DMSO controls.
In Vitro Drug Treatment with Primary Human AML Patient Samples
Primary AMLcells with SF mutation (n=16) or wild-type for SF (n=16) were incubated with DMSO orGSK591 (0.5 μM) for6 days. Cells were grown in lscove’s Modified Dulbecco’s Medium (IMDM), 20% bovine serum albumin, insulin, and transferrin (BIT) 9500 serum substitute, 16.7 μg/ml human low-density lipoproteins, 55 μM beta-mercaptoethanol with recombinant human (rh) G-CSF (20 ng/ml), rhGM-CSF (20 ng/ml), rh IL3 (20 ng/ml), rh IL6 (20 ng/ml), rh FLT3 ligand (50 ng/ml) and rh SCF (50 ng/mL). Following the 6-day incubation period, cells were subjected to flow cytometry to detect 7-AAD negative, and YO-PRO1 negative, viable cells. Samples were prepared in 4–6 replicates and averages were calculated. Relative viable cell numbers were compared with Welch’s t test.
In Vitro Combination Drug Titration with Primary Human AML Patient Samples
Patient cells derived from either bone marrow or peripheral blood were thawed and allowed to grow for a minimum of three days on OP9 stroma in 6-well tissue culture plates (Greiner) topping up with fresh media and putting cells onto fresh OP9 as necessary. Patient cells were cultured in IMDM media (Gibco) with 2 mM L-glutamine, 10% FBS (Wisent), 55 μM β-mercaptoethanol (Gibco), 100 μg/mL Primocin (lnvivoGen), 100 ng/mL SCF, 50 ng/mL FLT3L, 40 ng/mL THPO, 20 ng/mL IL3, and 20 ng/mL GM-CSF (Shenandoah Biotech or Custom Biologics). Leukemic cells were transferred to GFP labelled OP9 cells in 96-well plates for drug titration at a density of either 25000 or 50000 cells per well in 100 μL media. An additional 100 μL of media with drug was added at day 3–4. Cells were exposed to MS023 from a range of 0–10 μM and to GSK591 from a range of 0–3 μM for six days.
Viable cell number was assessed at day 6. Cells were transferred to 96-well round well suspension plate (Starstedt) along with trypsinized (30uL 0.25% Trypsin-EDTA; Wisent) OP9 stroma and attached leukemic cells. Cells were resuspended in PBS (Wisent) with 2% FBS with 0.2 μM Sytox Blue (Life Technologies) viability dye. Flow cytometry was performed using MACSQuant VYB (Miltenyi), and MACSQuantify software was used to determine viable leukemic cell number (GFP negative, Sytox blue negative).
OP9 mouse stromal cells (ATCC) were grown in α-MEM media without nucleosides that contains GlutaMAX (Gibco), 20% fetal bovine serum (FBS, Wisent), 55 μM β-mercaptoethanol (Gibco) and 100U/100 μg/mL penicillin/streptomycin (Gibco) at 37°C/5% CO2. OP9 cells were transduced with GFP lentivirus and sorted for GFP positive cells to use in assays to test the effect of the drugs on leukemic blasts. GFP positive cells were seeded at 5000–10000 cells/well in 96-well plates (Greiner) and used 1–3 days later for leukemic cell assays.
In Vitro Cell Viability Assays with Human Cell Lines
Cells were treated in duplicated with a 20-point, two-fold dilution series of GSK3203591 (PRMT5 inhibitor) or GSK3368712 (Type I PRMT inhibitor). Cells were treated with drugs for six days and cell growth was measured using Cell-titre glo (Promega).
A plate of untreated cells was read at the time of compound addition to determine the T=0 value representing the starting number of cells. Data were fitted with a four-parameter equation to generate a concentration response curve. Growth inhibition is the percent maximal inhibition and was calculated as 100-((ymin-100)/(ymax-100)*100). Ymin-T0 values were calculated by subtracting the T0 value (100%) from the Ymin value on the curve, and are a measure of net population cell growth or death. Growth Death Index (GDI) is a composite representation of Ymin-T0 and percent maximal inhibition. If Ymin-T0 values are negative, then GDI equals Ymin-T0; otherwise, GDI represents the fraction of cells remaining relative to DMSO control (ymax) and (ymin): (ymin-100)/(ymax-100)*100).
Treatment of iPSC-derived Hematopoietic Progenitor Cells with PRMT Inhibitors
The iPSC lines 5–16 Cre20 (SRSF2 P95L) and N-2.12 (isogenic normal) (Chang et al., 2018) were differentiated along the hematopoietic lineage as previously described (Kotini et al., 2017). 250.000 single cells from day 12 of hematopoietic differentiation were plated in 24-well ultra-low attachment plates. MS023 and GSK591 inhibitors or DMSO were added on days 12 and 17 of hematopoietic differentiation culture at a concentration of 350 nM. Live cells were quantified by counting in a hemocytometer 10 days after the beginning of treatment (day 22 of differentiation culture).
Pharmacokinetic Studies of MS023 In Vivo
Male Swiss Albino mice at Sai Life Sciences were administered a single 80 mg/kg intraperitoneal (IP) injection (n=3) or a single oral 150 mg/kg dose of MS023 (n=3). Plasma concentrations of MS023 were then evaluated at 0.083, 0.25, 0.5, 1, 2, 4, 8, and 12 hr post dosing.
Administration of PRMT Inhibitors In Vivo
For in vivo drug sensitivity studies, 8 mg (80 mg/kg dose) or 6 mg (60 mg/kg dose) of MS023 was dissolved in 50 μL N-Methyl-2-pyrrolidone, 200 μL Captisol, 200 μL polyethylene glycol 400 and 550 μL PBS and administered via IP injection once per day. 20 mg/ml (200 mg/kg dose) or 15 mg/ml (150 mg/kg dose) of EPZ015666 was reconstituted in 0.5% methylcellulose and administered to each mouse via oral gavage route once per day. To generate leukemia in C57BL6 mice (6–10 weeks old), the mice were sublethally irradiated at 5 Gy total body γ irradiation, and tail vein injected with 250,000 primary MLL-AF9 leukemic cells. (Lee et al., 2016). In single drug administration experiment, 80 mg/kg MS023 or 200 mg/kg EPZ015666 was administered from Day 8 of experiment till mice are moribund. In combinatorial drug administration experiment, 60 mg/kg MS023 and 150 mg/kg EPZ015666 were administered daily to the mice from Day 8 of experiment for 10 days.
Western Blotting
Protein concentrations were determined using RC DC protein assay kit from Bio-Rad. Proteins were run on 8–15% gels and separated with SOS-PAGE. The membranes were blotted with the primary antibodies, anti-actin (Santa Cruz) at 1:1000, anti-PRMT5 (Abeam) at 1:1000, anti-PRMT1 (Cell signaling) at 1:1000, anti-SD MA (Cell signaling) at 1:1000, anti-ADMA (Cell signaling) at 1:1000, anti-MMA (Cell signaling) at 1:1000, anti-EZH2 (Cell signaling) at 1:1000 and anti-γH2Ax (Cell signaling) at 1:1000. Blots were incubated with primary antibodies overnight at 4°C. The next day, blots were washed in TBST, incubated with HAP-conjugated secondary antibodies for 1 hr and visualised on X-ray films with West Pico Chemiluminescent Substrate.
RNA-seq Sample Preparation
RNA was extracted from all human and mouse cell samples using RNeasy mini kit (Qiagen) as per manufacturers instructions. Poly(A)-selected, unstranded lllumina libraries were prepared with a modified TruSeq protocol. 0.5X AMPure XP beads were added to the sample library to select for fragments <400 bp, followed by 1 X beads to select for fragments > 100 bp. These fragments were then amplified with PCR (15 cycles) and separated by gel electrophoresis (2% agarose). 300 bp DNA fragments were isolated and sequenced on the lllumina HiSeq 2000 (~100 million 101 bp reads per sample).
Arginine Methyl-Peptides Separation and Enrichment Prior to LC-MS/MS Analysis
NB4 cells were treated for 3 days with MS023 (3 μM) or GSK591 (1 μM). Equal numbers of Light and Heavy-labelled NB4 cells differentially treated were mixed in a 1:1 ratio, pelleted and washed twice with PBS. Cell pellets were lysed in urea lysis buffer (9 M urea, 20 mM HEPES pH 8.0), supplemented with 1X Roche proteases and phosphatases inhibitors, sonicated and cleared by ultracentrifugation (20.000 × g for 15 min at 15°C). For in-solution digestion, 50 mg of proteins were reduced by adding 4.5 mM DTT (Sigma-Aldrich) for 30 min at 55°C, alkylated with 5.5 mM iodoacetamide (IAA: 10% v/v for 15 min at room temperature in the dark, Sigma Aldrich) and digested overnight with sequencing-grade trypsin (1:100 w/w, Promega), after a fourfold dilution in 25 mM ammonium bicarbonate solution. Protease digestion was terminated with the addition of trifluoroacetic acid (TFA) to adjust pH < 3. Precipitated material was removed by centrifugation for 15 min at 1780 × g at room temperature. Soluble peptides were purified using reversed-phase Sep-Pak C18 cartridges (Waters, Milford, MA) and eluted off the Sep-Pak with 40% can, followed by a step of acetonitrile removal and concentration through 48 hours of lyophilization. Lyophilized peptides were dissolved in 25 mM ammonium hydroxide (NH4OH) and subsequently off-line fractionated by High-pH (HpH) reversed-phased chromatographic separation using a Phenomenex Jupiter® C12 4 μm Proteo 90 Å, LC column 250 × 4.6 mm, on an ÄKTA-FPLC (fast protein liquid chromatography) system (GE Healthcare) operating at 1 ml/min. Buffer A was 25 mM NH4OH and Buffer B was 25 mM NH4OH in 90% ACN. Fractions were collected using a collector in a 96-deep well plate at 1-min intervals. Samples were initially loaded onto the column at 1 ml/min for 3 min, after which the fractionation gradient was as follows: 5% B to 30% B in 60 min, 30% B to 60% in 2 min and ramped to 70% B for 3 min. Then, fraction collection was halted and the gradient was held at 100% B for 5 min, before being ramped back to 5% B, when the column was washed. The 60 fractions collected were concatenated to 14 throughout each experiment. After lyophilisation, each concatenated fraction was dissolved in 250 μl of 1x immuno-Affinity Purification Buffer (IAP buffer, #9993, Cell Signaling Technologies, CST) and subjected to two consecutive steps of methyl-R-peptides enrichment using the SDMA antibody-conjugated beads (PTMScan [sdme-R] Kit #13563, Cell Signaling Technologies) and the MMA antibody-conjugated beads (PTMScan Mono-Methyl Arginine Motif [mme-RG] Kit #12235, Cell Signaling Technologies), following the manufacturer’s instructions. For the MS023 experiments, ADMA-bearing peptides were also enriched using the specific PTMScan kit #13474, starting from the same amount of desalted and lyophilized peptides. After peptides incubation with the antibody conjugated beads for 2 hours at 4°C, the immuno-precipitates were washed twice in ice-cold IAP buffer, followed by three washes in water; then, the bound methyl-peptides were eluted with two consecutive runs of elution in 50 μI 0.15% TFA. Peptide eluates were desalted on reversed-phase C18 Stage Tips, as described previously (Rappsilber et al., 2007) and subjected to a second round of trypsin digestion prior to nano-LC-MS/MS analysis.
Nano-LC-MS/MS Analysis
Peptide mixtures were analysed by on line nano-flow liquid chromatography tandem mass spectrometry using an EASY-nLC™ 1000 (Thermo Fisher Scientific, Odense, Denmark) connected to a Q-Exactive instrument (Thermo Fisher Scientific) through a nano-electrospray ion source. The nano-LC system was operated in one column set-up with a 50 cm analytical column (75 μm inner diameter, 350 μm outer diameter) packed with C18 resin (EasySpray PEPMAP RSLC C18 2M 50 cm × 75 M, Thermo Fisher Scientific) configuration. Solvent A was 0.1 % formic acid (FA) and solvent B was 0.1 % FA in 80% ACN. Samples were injected in an aqueous 0.1 % TFA solution at a flow rate of 500 nL/min. SILAC immuno-enriched methyl-peptides were separated with a gradient of 5–40% solvent B over 90 min followed by a gradient of 40–60% for 10 min and 60–80% over 5 min at a flow rate of 250 nL/min in the EASY-nLC 1000 system.
The Q-Exactive was operated in the data-dependent mode (DDA) to automatically switch between full scan MS and MSMS acquisition. Survey full scan MS spectra (from m/z 300–1150) were analysed in the Orbitrap detector with resolution R=35,000 at m/z 400. The ten most intense peptide ions with charge states ≥2 were sequentially isolated to a target value of 3e6 and fragmented by Higher Energy Collision Dissociation (HCD) with a normalized collision energy setting of 25%. The maximum allowed ion accumulation times were 20 ms for full scans and 50ms for MSMS and the target value for MSMS was set to 1 e6. The dynamic exclusion time was set to 20 s.
Validation of RNAseq
Primers were designed to flank the exon of interest for validation of cassette exon events. cDNA is synthesized using the Maxima first strand cDNA synthesis kit (ThermoFisher Scientific). The PCR cycling conditions to amplify the alternatively spliced transcripts are as follows: 95°C for 5 min, 28 cycles of 95°C for 45 s, 58°C for 30 s and 72°C for 1 min, followed by 72°C for 5 min.
The primers used to validate the splicing events are as follows:
EZH2
Forward primer: TTTCATGCAACACCCAACACT
Reverse primer: CCCTGCTTCCCTATCACTGT
ATF2
Forward primer: AGTT ACATGTGAA TTCTGCCAGG
Reverse primer: CTCAAATGGACTCGCCAACTC
INTS3
Forward primer: ATGCCAAGCTGGCTTTGTTTT
Reverse primer: TCCGACATATGGTTGTCCATCTC
HDAC7
Forward primer: GGAAGAATCCACTGCTCCGA
Reverse primer: GACTGGGCAAAGTGGAAGGG
TRPT1
Forward primer: GGCCAACCAGGGCCATT
Reverse primer: ATCACCAGCCAAGGAAAGGG
LEF1
Forward primer: CCACCCATCCCGAGAACATC
Reverse primer: AGGCTTCACGTGCATTAGGT
AON Electroporation
K562 cells were transiently transfected with antisense oligonucleotides using the BTX Gemini X2 electroporation system. Cells were resuspended in cytoporation medium T (BTX #47–0002) at a density of 2×107 cells/ml with 10 μM of AON. Thereafter, cells were electroporated at 250 V × 10 ms × 1 pulse and plated onto fresh medium (3 ml medium per 100 μL electroporated cells). At 48 hours after electroporation, cells were collected for RNA, protein and cell viability readout using Cell-titre Glo (Promega).
AON sequence:
SCR: CGGUGUGUGUAUCAUUCUCUAGUGU
EZH2 (1042): UGAAUCUUCUGUCCAAAAUCCAACAGGCAAUAUA
CRISPR/Cas9 Knockout of EZH2 in K562 Cells
Guide RNA (gRNA) sequences were cloned into a lentiCRISPR V2 vector (Addgene) at the BsmBI restriction site.
Target sequences are
gRNA1:TTATCAGAAGGAAATTTCCG
gRNA2: TTATGATGGGAAAGTACACG
To generate lentivirus, 293T cells were transfected using Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturer’s protocol. Virus was collected at 48 and 72 hours post transfection, concentrated with Lenti-X concentrator (Takara), and titered using Lenti-X GoStix (Takara). 1×106 K562 cells were spinoculated with virus (MOI of 3 and 5 μg/ml polybrene) for 1 hour at 2400 rpm and 37°C in non-tissue culture treated plates. Two days post-infection, cells were selected with 5 μg/ml puromycin for four days before starting experiments.
QUANTIFICATION AND STATISTICAL ANALYSIS
Computation of the Combination Index
The presence of synergistic or additive effects was determined following the theorem of Chou-Talalay (Chou, 2010). The resulting combination index (Cl) offers quantitative definition for additive effect (Cl= 1), synergism (Cl < 1), and antagonism (Cl > 1) in drug combinations.
RNA-Sequencing and Bioinformatics Analysis
Library preparation was performed following the TruSeq RNA Sample preparation v2 guide (lllumina). In brief, the sequenced reads were mapped to mm9 build of the mouse genome or hg19 build of the human genome using STAR version 2.4.2a. Differential expression analysis was performed using the edgeR package in R. Enriched Gene Ontology terms and KEGG pathways were identified using Metascape. Heatmaps of gene expressions (FPKM) were generated with in-house scripts with R. Alternative splicing analysis was done using rMATS version 4.0.1 with annotation versions Ensembl.NCBIM37v65 for mouse and Ensembl.GRCh37v72 for human. Significant alternative splicing events were defined at FDR=0.05 and inclusion level difference of 10%.
Replicates
RNA-Seq was conducted with 3–5 biological replicates from each group. Genetic phenotyping experiments were replicated three times independently. For in vivo experiments, the number of animals was chosen to ensure 90% power with 5% error based on observed standard deviation. Flow cytometric experiments were replicated independently two-three times. Pilot studies were conducted with drug studies and results were replicated in a larger study to achieve enough statistical power. In vitro experiments were replicated two-three times, with viability experiments being completed in triplicate.
Data Analysis of SILAC Arginine Methyl-Peptides
Acquired raw data were analysed using the integrated MaxQuant software v1.3.0.5 or v.1.5.2.8, using the Andromeda search engine (Cox and Mann, 2008). In MaxQuant, the estimated false discovery rate (FDR) of all peptide identifications was set to a maximum of 1 %. The main search was performed with a mass tolerance of 7 ppm. Enzyme specificity was set to Trypsin/P. A maximum of 3 missed cleavages was permitted, and the minimum peptide length was fixed at 7 amino acids. Carbamidomethylation of Cysteine was set as a fixed modification. The January 2016 version of the Uniprot sequence was used for peptide identification.
To assign and quantify SILAC methyl-peptides, each raw file was analysed with the following set of variable modifications: N-terminal acetylation, Methionine oxidation, mono-methyl-K/R and di-methyl-K/R. The MaxQuant evidence.txt output file was then filtered as follows: potential contaminants and reverse sequences were removed; methyl-peptides not fulfilling the quality criteria of Andromeda score ≥ 25 and PTM localization probability ≥ 0.50 were also removed. For the methyl-peptides quantified more than once, the median SILAC ratio was calculated. Finally, methyl-peptide SILAC ratios were normalised on the respective protein SILAC ratios extracted from the proteinGroups.txt MaxQuant output file. These were calculated using unmodified peptides in the “input” experiment. To define significantly up- or down-regulated methyl-peptides by GSK591, we used mean(μ) and standard deviation (σ), based on the distribution of the unmodified peptide SILAC ratios calculated separately in the forward and reverse experiments and we applied a μ ± 3σ cut-off to the distributions of the modified peptides of the respective replicate (see Table S4).
HmSEEKER: A Perl-Based Pipeline for High-Confidence Assignment of Methyl-Peptides from hmSILAC Data
To assign hmSILAC peptide sequences, we defined new modifications in MaxQuant with the mass increment and residue specificities corresponding to heavy mono-methylation (mono-methyl4-K/R) and di-methylation (di-methyl4-K/R). Additionally, we defined new modifications for heavy methionine (Met4) and oxidized heavy methionine (OxMet4). To reduce the search complexity, raw data were analysed twice with the following sets of variable modifications: (ii) N-terminal acetylation, Met4, OxMet4, oxidation, mono-methyl-K/R, mono-methyl4-K/R; (ii) N-terminal acetylation, Met4, OxMet4, oxidation, di-methyl-K/R, di-methyl4-K/R.
Identification of high confidence methyl-sites was carried out with an in-house developed, Perl-based pipeline, named hmSEEKER, which identifies doublets of heavy and light hmSILAC peptides from MaxQuant output tables (Massignani et al., 2019). hmSEEKER performs the following steps: methyl-peptides identified in the msms file are first filtered to remove: (i) all contaminants and decoy peptides, (ii) all peptides with single charge and (iii) all peptides bearing simultaneous heavy and light modifications. Then each peptide is associated to its corresponding MS1 peak in the allPeptides file. Finally, the H or L counterpart of each peak is searched among other peaks detected in the same raw data file. Because the pair is searched in msmsScans, hmSEEKER enables the identification of peptide doublets even when one of the two counterparts has not been MS/MS sequenced, thus not appearing in the msms file. We used hmSEEKER to automatically filter the MaxQuant msms.txt file and remove contaminants and reverse sequences, as well as peptides carrying simultaneously light and heavy modifications. To increase the confidence of our findings, remaining peptides were further filtered to remove any peptide with Andromeda score < 25 or Andromeda delta score < 12 and any methylation with a PTM localization probability < 0.75. Heavy and light methyl-peptide pairs were accepted when the difference between calculated and expected mass shift was < 2 ppm and the difference between their retention times was < 30 s.
Use of hmLINKER to Intersect the SILAC Methyl-proteome with the hmSILAC Dataset
Validation of the methylated peptide identified in the SILAC experiments through the hmSILAC identifications was achieved by using hmLINKER, another in-house developed bioinformatic tool that compares the sequences of the peptides in the SILAC dataset to those in the hmSILAC repository (hmSEEKER output). If a match is not found at the sequence level, the peptide is not immediately discarded, but a second round of match-attempt is performed using a 31 amino acids sequence window, centered on each modification site
Motif Analysis of R Methyl-Peptides
Motif analysis of R methyl-sites was performed using the plogo web application (O’Shea et al., 2013), which allows the visualization of significant enrichment variations between the set of GSK591-regulated sequences and the unchanging methyl-peptides used as a background set. p value threshold was set to 0.05.
Bioinformatic Analysis of R Methyl-Peptides
Motif analysis of changing methyl-sites was performed using the plogo web application (O’Shea et al., 2013), which allows the visualization of significant enrichment variations between the set of drug-regulated sequences and the unchanging methyl-peptides used as a background set. P value threshold was set to 0.05. Networks on regulated methyl-proteins were built with the Reactome application of Cytoscape (Fabregat et al., 2018; Shannon et al., 2003). The Gene Ontology analysis was carried out with GOrilla (Eden et al., 2009) and Revigo (Supek et al., 2011).
Calculation of the Protein Abundance Index in the Annotated Proteome
We matched the methylated proteins identified via the immuno-enrichment approach with their respective protein, annotated in the whole proteome, which was used ad INPUT for the methyl-peptides IP. To calculate the abundance index of each protein in this proteome, we employed the iBAQ algorithm (Intensity-Based Absolute Quantification (Schwanhausser et al., 2011), embedded in the MaxQuant suit.
Statistical Analyses
Statistical significance was determined by unpaired Student’s t-test after testing for normal distribution unless indicated otherwise. The Mantel-Cox log-rank test was used to compare survival curves. P values of < 0.05 were considered statistically significant. Data were plotted using GraphPad Prism 7 software as mean values and error bars represent standard deviation. Asterisks indicate * p = 0.01–0.05; ** p = 0.001–0.01, *** p = 0.0001–0.001, **** p <0.0001.
DATA AND CODE AVAILABILITY
The accession number for the RNA-seq data reported in this paper is GEO:GSE123774.
The accession number for the MS-proteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE (Vizcaino et al., 2016) partner repository with the dataset identifier PRIDE: PXD012007.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Actin | Santa Cruz | sc-147778; RRID: AB_626632 |
| Anti-PRMT5 | Abeam | Abeam ab109451; RRID: AB_10863428 |
| Anti-SDMA | Cell signaling | #13222; RRID: AB_2714013 |
| Anti-ADMA | Cell signaling | #13522; RRID: AB_2665370 |
| Anti-PRMT1 | Cell signaling | CST #2449; RRID: AB_2237696 |
| Anti-MMA | Cell signaling | #8015; RRID: AB_10891776 |
| Anti-EZH2 | Cell signaling | CST #5246; RRID: AB_10694683 |
| Anti- γH2Ax | Cell signalling | CST #9718; RRID: AB_2118009 |
| Biological Samples | ||
| AML primary patient samples | Princess Margaret Hospital, Memorial Sloan Kettering Cancer Center, & Weill Cornell College of Medicine | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| GSK3203591 | SGC Toronto | https://thesgc.org/chemical-probes/GSK591 |
| MS023 | SGC Toronto | https://www.thesgc.org/chemical-probes/MS023 |
| EPZO15666 | Tocris | Cat. No. 6516 |
| GSK3368712 | GSK | N/A |
| E7107 | H3 Biomedicine Inc. | N/A |
| I-BRD9 | SGC Toronto | https://www.thesgc.org/chemical-probes/l-BRD9 |
| SGC-CBP30 | SGC Toronto | https://www.thesgc.org/chemical-probes/CBP30 |
| BI-9564 | SGC Toronto | https://www.thesgc.org/chemical-probes/BI-9564 |
| I-CBP112 | SGC Toronto | https://www.thesgc.org/chemical-probes/l-CBP112 |
| MS049 | SGC Toronto | https://www.thesgc.org/chemical-probes/MS049 |
| UNC0638 | SGC Toronto | https://thesgc.org/chemical-probes/UNC0638 |
| SGC707 | SGC Toronto | https://www.thesgc.org/chemical-probes/SGC707 |
| GSK484 | SGC Toronto | https://www.thesgc.org/chemical-probes/GSK484 |
| 10X2 | SGC Toronto | https://www.thesgc.org/chemical-probes/IOX2 |
| GSK-J4 | SGC Toronto | https://www.thesgc.org/chemical-probes/gskj1 |
| Vorinostat | Cayman chemicals | CAS 149647-78-9 |
| GSK2 334470 | Cayman chemicals | CAS 1227911-45-6 |
| SGC0946 | SGC Toronto | https://www.thesgc.org/chemical-probes/SGC0946 |
| PD0332991 | Cayman chemicals | CAS 827022-32-2 |
| CCT244747 | Cayman chemicals | CAS 1404095-34-6 |
| PF-477736 | Cayman chemicals | CAS 952021-60-2 |
| BMS-265246 | Cayman chemicals | CAS 582315-72-8 |
| Irinotecan | Cayman chemicals | CAS 100286-90-6 |
| SCH772984 | Cayman chemicals | CAS 942183-80-4 |
| Alitretinoin | Cayman chemicals | CAS 5300-03-8 |
| CX-4945 | Cayman chemicals | CAS 1009820-21-6 |
| Phenoxodiol | Sigma | CAS 81267-65-4 |
| 5Z-7-oxozeaenol | Cayman chemicals | CAS 253863-19-3 |
| Critical Commercial Assays | ||
| Caspase-Glo. 3/7 luminescence assay | Promega | G8090 |
| Cell titer Glo | Promega | G7570 |
| MTS assay | Promega | G3582 |
| PTMScan® Asymmetric Di-Methyl Arginine Motif [adme-R] Kit | Cell Signaling Technologies | Kit #13474 |
| PTMScan® Symmetric Di-Methyl Arginine Motif [sdme-RG] Kit | Cell Signaling Technologies | Kit #13563 |
| PTMScan Mono-Methyl Arginine 13 Motif [mme-RG] Kit | Cell Signaling Technologies | Kit #12235 |
| Deposited Data | ||
| RNA-seq data | This paper | GEO:GSE123774 |
| MS-proteomics data | This paper | PRIDE: PXD012007 |
| Experimental Models: Cell Lines | ||
| Human: K562 cells | ATCC | CLL-243 |
| Human: THP1 cells | ATCC | TIB-202 |
| Human: M0LM13 cells | DSMZ | ACC 554 |
| Human: U937 cells | ATCC | CRL-1593.2 |
| Human: K562 SRSF2P95H/WT knockin cells | Horizon Discovery | N/A |
| Human: K562 SF3B1K700E/WT knockin cells | Horizon Discovery | N/A |
| Human: TF1 cells | ATCC | CRL-2003 |
| Human: 0CI-AML5 cells | DSMZ | ACC 247 |
| Human: F-36P cells | DSMZ | ACC 543 |
| Human: GDM-1 cells | ATCC | CRL-2627 |
| Human: KO52 cells | JCRB | CVCL-1321 |
| Human: HNT34 cells | DSMZ | ACC 600 |
| Human: MonoMac6 cells | DSMZ | ACC 124 |
| Human: 5–16 Cre20 (SRSF2 P95L) | (Chang et al., 2018) | N/A |
| Human: N-2.12 (isogenic normal) | (Chang et al., 2018) | N/A |
| Experimental Models: Organisms/Strains | ||
| Mice: Srsf2P95H/+ | (Kim et al., 2015). | N/A |
| Mice: MLL-AF9/Vav-cre Srsf2WT/WT | (Kim et al., 2015). | N/A |
| Mice: MLL-AF9/Vav-cre Srsf2P95H/WT | (Kim et al., 2015). | N/A |
| Mice: Vav-cre transgenic mice (B6.Cg-Tg(Vav1-icre)A2Kio/J) | The Jackson Laboratory | JAX: 008610 |
| Oligonucleotides | ||
| Human EZH2 F | (Lee et al., 2016) | TTTCATG CAACACCC AACACT |
| Human EZH2 R | (Lee et al., 2016) | CCCTGCTTCCCTATCACTGT |
| Human ATF2 F | This paper | AGTTACATGTGAATTCTG CCAG G |
| Human ATF2 R | This paper | CTCAAATG G ACTCG CCAACTC |
| Human INTS3 F | This paper | ATGCCAAG CTGGCTTTGTTTT |
| Human INTS3 R | This paper | TCCGACATATGGTTGTCCATCTC |
| Human TRPT1 F | This paper | GG CCAACCAG G GCCATT |
| Human TRPT1 R | This paper | ATCACCAG CCAAG GAAAG GG |
| Human HDAC7 F | This paper | GG AAG AATCCACTG CTCCGA |
| Human HDAC7 R | This paper | GACTGGG CAAAGTGG AAG GG |
| Human LEF1 F | This paper | CCACCCATCCCGAGAACATC |
| Human LEF1 R | This paper | AGG CTTCACGTG CATTAGGT |
| Anti-sense oligonucleotide SCR | This paper | CGGUGUGUGUAUCAUUCUCUAGUGU |
| Anti-sense oligonucleotide EZH2 | This paper | U GAAU C U U CU G U CCAAAAU CCAACAGGCAAU AUA |
| sgRNA 1 sequence for EZH2 | This paper | TTATCAG AAG G AAATTTCC G |
| sgRNA 2 sequence for EZH2 | This paper | TTATG ATG G GAAAGTACACG |
| Recombinant DNA | ||
| LentiCRISPR v2 | N/A | Addgene: 52961 |
| Software and Algorithms | ||
| Cytoscape 3.4.0 | (Shannon et al., 2003) | https://cytoscape.org/ |
| Reactome Fl | (Wuetal., 2014) | N/A |
| plogo web application | (O’Shea et al., 2013) | N/A |
| hmLINKER | (Massignani et al., 2019) | N/A |
| hmSEEKER | (Massignani et al., 2019) | N/A |
| Prism 7.0 | Graphpad | https://graphpad.com/ |
| lmageJ | NIH | https://irnagej-nih-gov.libproxy1.nus.edu.sg/ij/index.html |
| STAR | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| HTSeq | (Anders et al., 2(15) | N/A |
| R | R Core Team | https://www.r-project.org/ |
| Bioconductor | (Huber et al., 2015) | N/A |
| DESeq2 | (Love et al., 2014) | 10.18129/B9.bioc.DESeq2 |
| rMATS | (Shen et al., 2014) | http://maseq-mats.sourceforge.net/ |
| ggsashimi | (Garrido-Martin et al., 2018) | https://github.com/guigolab/ggsashimi |
Highlights.
Inhibition of SDMA or ADMA preferentially kills splicing factor (SF)-mutant cells
Combined inhibition of PRMT5 and type I PRMTs has synergistic effects
RNA-binding proteins are the most enriched cellular substrates of PRMTs
Inhibition of RNA splicing underlies the cytotoxic effects of PRMT inhibition
Significance.
Pharmacologic suppression of PRMT5 and type I PRMTs is being pursued as a cancer treatment approach, and numerous mechanisms have been proposed for the efficacy of PRMT inhibition. Here we identify that spliceosomal mutant leukemias are preferentially sensitive to PRMT inhibition and that RNA-binding proteins are the most enriched substrates of PRMT5 and/or type I PRMTs. Accordingly, combined PRMT5 and type I PRMT inhibition resulted in synergistic cell killing and pronounced effects on splicing compared with inhibiting either enzymatic activity alone. These data provide a rational combinatorial strategy and a strong basis for ongoing and future clinical trials based on the presence of genetic alterations impacting RNA splicing.
ACKNOWLEDGMENTS
We thank H3 Biomedicine for providing E7107. We thank the BRC Shared facilities for technical support. We thank F. Raimondi (IEO) for technical support w ith the proteome to transcriptome data integration analysis. We are grateful to the GIS Genome Sequencing Team for help with the Solexa high-throughput sequencing; to the entire EG lab for critical discussion. This work was supported by AGS fellowship to J.Y.F., AGA-SINGA (Singapore Graduate Award) fellowships to P.A.G. and L.P. E.G. is supported by NMRC/OFI RG/0032/2017 and NRF-CRP17-2017-06 grants. This research was additionally supported by the RNA Biology Center at the Cancer Science Institute of Singapore, NUS, as part of funding under the Singapore Ministry of Education’s AcRF Tier 3 grants, grant no. MOE2014-T3-1-006. S.C.-W.L. and O.A.W. are supported by the Leukemia and Lymphoma Society. O.A.W. is supported by grants from NIH/NHLBI (R01 HL 128239), NIH/NCI (1 R01 CA201247-01A1), the US Department of Defense Bone Marrow Failure Research Program (W81XWH-12-1-0041), Cycle For Survival, the Edward P. Evans Foundation, the Henry & Marilyn Taub Foundation, the Pershing Square Sohn Foundation, and Starr Foundation grants I8-A8-075 and I9-A9-059. C.H.A. and G.M.L. were supported by the Leukemia and Lymphoma Society of Canada. The Structural Genomics Consortium is a registered charity (1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer lngelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) (ULTRADD grant no. 115766), Janssen, Merck, Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, Sao Paulo Research Foundation FAPESP, Takeda, and the Wellcome Trust. Research in the T.B. group was supported by grants from the Italian Ministry of Health (GR-2011-02347880), the Epigen Flagship project (CNR), and the Italian Association for Cancer Research (IG-201 8-21834). D.M. was supported by the Fondazione Umberto Veronesi (FUV) fellowship and the Fondazione lstituto Europeo di Oncologia (FIEO) fellowship. A.R. acknowledges support through the NIH (award no. T32 CA078207). J.J. acknowledges support by grant R01GM122749 from the US NIH.
DECLARATION OF INTERESTS
O.A.-W. has received research funding from H3 Biomedicine, has served as a consultant for H3 Biomedicine and Merck, and has served on advisory board for Foundation Medicine, H3 Biomedicine, and Janssen. E.G. has received research funding from Eli-Lilly and Prelude Therapeutics, has served as a consultant for Prelude Therapeutics, SKBP, and has served on the advisory board for LION TCR and Janssen, he is a co-founder of lmmuNOA Pie. Ltd.. C.M.K. is currently an employee at Mechanistic Biology and Profiling, Discovery Sciences, IMED Biotech Unit, AstraZeneca Pharmaceuticals LP. 35 Gate-house Dr, Waltham, MA 02451, USA. C.T. and O.B. are Employees and stake-holders of GSK. M.M. receives honorariums from Amgen, Pfizer, and Astellas, and funding from Pfizer, Notable Labs. All SGC Affiliated authors receive funding from the nine SGC-funding companies. C.H.A. and G.M.L. were supported by the Leukemia Lymphoma Society of Canada. The Structural Genomics Consortium is a registered charity (1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer lngelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) (ULTRADD grant no. 115766), Janssen, Merck, Novartis Pharma, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation FAPESP, Takeda, and the Wellcome Trust.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.ccell.2019.07.003.
REFERENCES
- Adriaens C, Standaert L, Barra J, Latil M, Verfaillie A, Kalev P, Boeckx B, Wijnhoven PW, Radaelli E, Vermi W, et al. (2016). p53 induces formation of NEAT1 lncRNA-containing paraspeckles that modulate replication stress response and chemosensitivity. Nat. Med 22, 861–868. [DOI] [PubMed] [Google Scholar]
- Anczuków O, Rosenberg AZ, Akerman M, Das S, Zhan L, Kami R, Muthuswamy SK, and Krainer AR (2012). The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat. Struct. Mol. Biol 19, 220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezzi M, Teo SX, Muller J, Mok WC, Sahu SK, Vardy LA, Sonday ZQ, and Guccione E (2013). Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes Dev. 27, 1903–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun CJ, Stanciu M, Bautz PL, Patterson JC, Calligaris D, Higuchi F, Neupane R, Fenoglio S, Cahill DP, Wakimoto H, et al. (2017). Coordinated splicing of regulatory detained intrans within oncogenic transcripts creates an exploitable vulnerability in malignant glioma. Cancer Cell 32, 411–426.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan-Penebre E, Kuplast KG, Majer CR, Boriack-Sjodin PA, Wigle TJ, Johnston LD, Rioux N, Munchhof MJ, Jin L, Jacques SL, et al. (2015). A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat. Chem. Biol 11, 432–437. [DOI] [PubMed] [Google Scholar]
- Chang CJ, Kotini AG, Olszewska M, Georgomanoli M, Teruya-Feldstein J, Sperber H, Sanchez R, DeVita R, Martins TJ, Abdel-Wahab O, et al. (2018). Dissecting the contributions of cooperating gene mutations to cancer phenotypes and drug responses with patient-derived iPSCs. Stem Cell Reports 10, 1610–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, Chandramohan R, Liu ZY, Won HH, Scott SN, et al. (2015). Memorial Sloan Kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J. Mol. Diagn 17, 251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TC (2010). Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 70, 440–446. [DOI] [PubMed] [Google Scholar]
- Cote J, Boisvert FM, Boulanger MC, Bedford MT, and Richard S (2003). Sam68 RNA binding protein is an in vivo substrate for protein arginine N-methyltransferase 1. Mol. Biol. Cell 14, 274–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Denman RB (2002). Methylation of the arginine-glycine-rich region in the fragile X mental retardation protein FMRP differentially affects RNA binding. Cell. Mol. Biol. Lett 7, 877–883. [PubMed] [Google Scholar]
- Dhar S, Vemulapalli V, Patananan AN, Huang GL, Di Lorenzo A, Richard S, Comb MJ, Guo A, Clarke SG, and Bedford MT (2013). Loss of the major type I arginine methyltransferase PRMT1 causes substrate scavenging by other PRMTs. Sci. Rep 3, 1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolzhanskaya N, Merz G, Aletta JM, and Denman RB (2006). Methylation regulates the intracellular protein-protein and protein-RNA interactions of FMRP. J. Cell Sci 119, 1933–1946. [DOI] [PubMed] [Google Scholar]
- Duncan KW, Rioux N, Boriack-Sjodin PA, Munchhof MJ, Reiter LA, Majer CR, Jin L, Johnston LD, Chan-Penebre E, Kuplast KG, et al. (2016). Structure and property guided design in the identification of PRMT5 tool compound EPZ015666. ACS Med. Chem. Lett 7, 162–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvinge H, Kim E, Abdel-Wahab O, and Bradley RK (2016). RNA splicing factors as oncoproteins and tumour suppressors. Nat. Rev. Cancer 16, 413–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Navan R, Steinfeld I, Lipson D, and Yakhini Z (2009). GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eram MS, Shen Y, Szewczyk M, Wu H, Senisterra G, Li F, Butler KV, Kaniskan HU, Speed BA, Dela Sena C, et al. (2016). A potent, selective, and cell-active inhibitor of human type I protein arginine methyltransferases. ACS Chem. Biol 11, 772–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erce MA, Abeygunawardena D, Low JK, Hart-Smith G, and Wilkins MR (2013). Interactions affected by arginine methylation in the yeast protein-protein interaction network. Mol. Cell Proteomics 12, 3184–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, Waghom K, Zoi K, Ross FM, Reiter A, et al. (2010). Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat. Genet 42, 722–726. [DOI] [PubMed] [Google Scholar]
- Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, Haw R, Jassal B, Kominger F, May B, et al. (2018). The reactome pathway knowledgebase. Nucleic Acids Res. 46, D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedoriw A, Rajapurkar SR, O’Brien S, Gerhart SV, Mitchell LH, Adams ND, Rioux N, Lingaraj T, Ribich SA, Pappalardi MB, et al. (2019). Anti-tumor activity of the type I PRMT inhibitor, GSK3368715 with PRMT5 inhibition through MTAP loss. Cancer Cell 10.1016/j.ccell.2019.05.014. [DOI] [PubMed] [Google Scholar]
- Falco EG, Coil KE, and Reed R (2011). The anti-tumor drug E7107 reveals an essential role for SF3b in remodeling U2 snRNP to expose the branch point-binding region. Genes Dev. 25, 440–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen WJ, Massenet S, Paushkin S, Wyce A, and Dreyfuss G (2001). SMN, the product of the spinal muscular atrophy gene, binds preferentially to dimethylarginine-containing protein targets. Mol. Cell 7, 1111–1117. [DOI] [PubMed] [Google Scholar]
- Gao G, Zhang L, Villarreal OD, He W, Su D, Bedford E, Moh P, Shen J, Shi X, Bedford MT, et al. (2019). PRMT1 loss sensitizes cells to PRMT5 inhibition. Nucleic Acids Res. 47, 5038–5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, p|1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido-Martín D, Palumbo E, Guigó R, and Breschi A (2018). ggsashimi: Sashimi plot revised for browser- and annotation-independent splicing visualization. PLoS Comput. Biol 14, e1006360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Zheng L, Xu H, Dai H, Zhou M, Pascua MR, Chen QM, and Shen B (2010). Methylation of FEN1 suppresses nearby phosphorylation and facilitates PCNA binding. Nat. Chem. Biol 6, 766–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamard PJ, Santiago GE, Liu F, Karl DL, Martinez C, Man N, Mookhtiar AK, Duffort S, Greenblatt S, Verdun RE, et al. (2018). PRMT5 regulates DNA repair by controlling the alternative splicing of his-tone-modifying enzymes. Cell Rep. 24, 2643–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han T, Goralski M, Gaskill N, Capata E, Kim J, Ting TC, Xie Y, Williams NS, and Nijhawan D (2017). Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 356, 10.1126/science.aal3755. [DOI] [PubMed] [Google Scholar]
- Harbour JW, Roberson ED, Anbunathan H, Onken MD, Worley LA, and Bowcock AM (2013). Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat. Genet 45, 133–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofweber M, Hutten S, Bourgeois B, Spreitzer E, Niedner-Boblenz A, Schifferer M, Ruepp MD, Simons M, Niessing D, Madi T, et al. (2018). Phase separation of FUS is suppressed by its nuclear import receptor and arginine methylation. Cell 173, 706–719.e13. [DOI] [PubMed] [Google Scholar]
- Hsu TY, Simon LM, Neill NJ, Marcotte R, Sayad A, Bland CS, Echeverria GV, Sun T, Kurley SJ, Tyagi S, et al. (2015). The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature 525, 384–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, et al. (2015). Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 12, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson M, Durant ST, Cho EG, Sheahan S, Edelmann M, Kessler B, and La Thangue NB (2008). Arginine methylation regulates the p53 response. Nat. Cell Biol 10, 1431–1439. [DOI] [PubMed] [Google Scholar]
- Kahles A, Lehmann KV, Toussaint NC, Huser M, Stark SG, Sachsenberg T, Stegle O, Kohlbacher O, Sander C, Cancer Genome Atlas Research, N., et al. (2018). Comprehensive analysis of alternative splicing across tumors from 8,705 patients. Cancer Cell 34, 211–224.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaida D, Matayoshi H, Tashiro E, Nojima T, Hagiwara M, lshigami K, Watanabe H, Kitahara T, Yoshida T, Nakajima H, et al. (2007). Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat. Chem. Biol 3, 576–583. [DOI] [PubMed] [Google Scholar]
- Kami R, de Stanchina E, Lowe SW, Sinha R, Mu D, and Krainer AR (2007). The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat. Struct. Mal. Biol 14, 185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, Li Y, Chung YR, Micol JB, Murphy ME, et al. (2015). SRSF2 mutations contribute to myelodysplasia by mutant-specific effects on exon recognition. Cancer Cell 27, 617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh CM, Bezzi M, Low DH, Ang WX, Teo SX, Gay FP, AI-Haddawi M, Tan SY, Osato M, Sabo A, et al. (2015). MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature 523, 96–100. [DOI] [PubMed] [Google Scholar]
- Kotake Y, Sagane K, Owa T, Mimori-Kiyosue Y, Shimizu H, Uesugi M, lshihama Y, Iwata M, and Mizui Y (2007). Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat. Chem. Biol 3, 570–575. [DOI] [PubMed] [Google Scholar]
- Kotini AG, Chang CJ, Boussaad I, Delrow JJ, Dolezal EK, Nagulapally AB, Perna F, Fishbein GA, Klimek VM, Hawkins RD, et al. (2015). Functional analysis of a chromosomal deletion associated with myelodysplastic syndromes using isogenic human induced pluripotent stem cells. Nat. Biotechnol 33, 646–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotini AG, Chang CJ, Chow A, Yuan H, Ho TC, Wang T, Vora S, Solovyov A, Husser C, Olszewska M, et al. (2017). Stage-specific human induced pluripotent stem cells map the progression of myeloid transformation to transplantable leukemia. Cell Stem Cell 20, 315–328.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryukov GV, Wilson FH, Ruth JR, Paulk J, Tsherniak A, Marlow SE, Vazquez F, Weir BA, Fitzgerald ME, Tanaka M, et al. (2016). MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 351, 1214–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, Dvinge H, Kim E, Cho H, Micol JB, Chung YR, Durham BH, Yoshimi A, Kim YJ, Thomas M, et al. (2016). Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat. Med 22, 672–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, North K, Kim E, Jang E, Obeng E, Lu SX, Liu B, Inoue D, Yoshimi A, Ki M, et al. (2018). Synthetic lethal and convergent biological effects of cancer-associated spliceosomal gene mutations. Cancer Cell 34, 225–241.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Hoadley K, Triche TJ Jr., Laird PW, Baty JD, et al. (2013). Genomic and epigenomic landscapes of adult de nova acute myeloid leukemia. N. Engl. J. Med 368, 2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marjan K, Cameron MJ, Quang P, Clasquin MF, Mandley E, Kunii K, McVay M, Choe S, Kernytsky A, Gross S, et al. (2016). MTAP deletions in cancer create vulnerability to targeting of the MAT2NPRMT5/RIOK1 axis. Cell Rep. 15, 574–587. [DOI] [PubMed] [Google Scholar]
- Martin M, Masshofer L, Temming P, Rahmann S, Metz C, Bornfeld N, van de Nes J, Klein-Hitpass L, Hinnebusch AG, Horsthemke B, et al. (2013). Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat. Genet 45, 933–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massignani E, Cuomo A, Musiani D, Jammula S, Pavesi G, and Bonaldi T (2019). hmSEEKER: identification of hmSILAC doublets in maxquant output data. Proteomics 19, e1800300. [DOI] [PubMed] [Google Scholar]
- Mavrakis KJ, McDonald ER 3rd, Schlabach MR, Billy E, Hoffman GR, deWeck A, Ruddy DA, Venkatesan K, Yu J, McAllister G, et al. (2016). Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science 351, 1208–1213. [DOI] [PubMed] [Google Scholar]
- Meister G, Eggert C, Buhler D, Brahms H, Kambach C, and Fischer U (2001). Methylation of Sm proteins by a complex containing PRMT5 and the putative U snRNP assembly factor plCln. Curr. Biol 11, 1990–1994. [DOI] [PubMed] [Google Scholar]
- Meister G, and Fischer U (2002). Assisted RNP assembly: SMN and PRMT5 complexes cooperate in the formation of spliceosomal UsnRNPs. EMBO J. 21, 5853–5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musiani D, Bok J, Massignani E, Wu L, Tabaglio T, Ippolito MR, Cuomo A, Ozbek U, Zorgati H, Ghoshdastider U, et al. (2019). Proteomics profiling of arginine methylation defines PRMT5 substrate specificity. Sci. Signal 12, 10.1126/scisignal.aat8388. [DOI] [PubMed] [Google Scholar]
- Nichols RC, Wang XW, Tang J, Hamilton BJ, High FA, Herschman HR, and Rigby WF (2000). The RGG domain in hnRNP A2 affects subcellular localization. Exp. Cell Res 256, 522–532. [DOI] [PubMed] [Google Scholar]
- Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tonnissen ER, van der Heijden A, Scheele TN, Vandenberghe P, de Witte T, et al. (2010). Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat. Genet 42, 665–667. [DOI] [PubMed] [Google Scholar]
- O’Shea JP, Chou MF, Quader SA, Ryan JK, Church GM, and Schwartz D (2013). pLogo: a probabilistic approach to visualizing sequence motifs. Nat. Methods 10, 1211–1212. [DOI] [PubMed] [Google Scholar]
- Obeng EA, Chappell RJ, Seiler M, Chen MC, Campagna DR, Schmidt PJ, Schneider RK, Lord AM, Wang L, Gambe RG, et al. (2016). Physiologic expression of Sf3b1 (K700E) causes impaired erythropoiesis, aberrant splicing, and sensitivity to therapeutic spliceosome modulation. Cancer Cell 30, 404–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong SE, Mittler G, and Mann M (2004). Identifying and quantifying in vivo methylation sites by heavy methyl SILAC. Nat. Methods 1, 119–126. [DOI] [PubMed] [Google Scholar]
- Pellagatti A, Armstrong RN, Steeples V, Sharma E, Repapi E, Singh S, Sanchi A, Radujkovic A, Horn P, Dolatshad H, et al. (2018). Impact of spliceosome mutations on RNA splicing in myelodysplasia: dysregulated genes/pathways and clinical associations. Blood 132, 1225–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qamar S, Wang G, Randle SJ, Ruggeri FS, Varela JA, Lin JQ, Phillips EG, Miyashita A, Williams D, Strohl F, et al. (2018). FUS phase separation is modulated by a molecular chaperone and methylation of arginine cation-pi interactions. Cell 173, 720–734.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Mann M, and lshihama Y (2007). Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc 2, 1896–1906. [DOI] [PubMed] [Google Scholar]
- Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, and Selbach M (2011). Global quantification of mammalian gene expression control. Nature 473, 337–342. [DOI] [PubMed] [Google Scholar]
- Seiler M, Yoshimi A, Darman R, Chan B, Keaney G, Thomas M, Agrawal AA, Caleb B, Csibi A, Sean E, et al. (2018). H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat. Med 24, 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and ldeker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, Zhou Q, and Xing Y (2014). rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. U S A 111, E5593–E5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai CL, White BS, Tripathi M, Tapia R, Ley JN, Ndonwi M, Kim S, Shao J, Carver A, Saez B, et al. (2017). Mutant U2AF1-expressing cells are sensitive to pharmacological modulation of the spliceosome. Nat. Commun 8, 14060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims RJ 3rd, Rojas LA, Beck DB, Bonasio R, Schuller R, Drury WJ 3rd, Eick D, and Reinberg D (2011). The C-terminal domain of RNA polymer-ase II is modified by site-specific methylation. Science 332, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijders AP, Hautbergue GM, Bloom A, Williamson JC, Minshull TC, Phillips HL, Mihaylov SR, Gjerde DT, Hornby DP, Wilson SA, et al. (2015). Arginine methylation and citrullination of splicing factor praline- and glutamine-rich (SFPQ/PSF) regulates its association with mRNA. RNA 21, 347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Bosnjak M, Skunca N, and Smuc T (2011). REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6, e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Minana B, Valcarcel J, Gabaldon T, and Lehner B (2014). Synonymous mutations frequently act as driver mutations in human cancers. Cell 156, 1324–1335. [DOI] [PubMed] [Google Scholar]
- Thandapani P, Song J, Gandin V, Cai Y, Rouleau SG, Garant JM, Boisvert FM, Yu Z, Perreault JP, Topisirovic I, et al. (2015). Aven recognition of RNA G-quadruplexes regulates translation of the mixed lineage leukemia protooncogenes. Elife 4, 10.7554/elife.06234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tradewell ML, Yu Z, Tibshirani M, Boulanger MC, Durham HD, and Richard S (2012). Arginine methylation by PRMT1 regulates nuclear-cytoplasmic localization and toxicity of FUS/TLS harbouring ALS-linked mutations. Hum. Mal. Genet 21, 136–149. [DOI] [PubMed] [Google Scholar]
- Tsai WC, Gayatri S, Reineke LC, Sbardella G, Bedford MT, and Lloyd RE (2016). Arginine demethylation of G3BP1 promotes stress granule assembly. J. Biol. Chem 291, 22671–22685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara T, Minoshima Y, Sagane K, Sugi NH, Mitsuhashi KO, Yamamoto N, Kamiyama H, Takahashi K, Kotake Y, Uesugi M, et al. (2017). Selective degradation of splicing factor CAPERalpha by anticancer sulfonamides. Nat. Chem. Biol 13, 675–680. [DOI] [PubMed] [Google Scholar]
- Vizcaino JA, Csordas A, del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, et al. (2016). 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, 0447–0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall ML, and Lewis SM (2017). Methylarginines within the RGG-motif region of hnRNP A1 affect its IRES trans-acting factor activity and are required for hnRNP A1 stress granule localization and formation. J. Mal. Biol 429, 295–307. [DOI] [PubMed] [Google Scholar]
- Wang E, Lu SX, Pastore A, Chen X, Imig J, Chun-Wei Lee S, Hockemeyer K, Ghebrechristos YE, Yoshimi A, Inoue D, et al. (2019). Targeting an RNA-binding protein network in acute myeloid leukemia. Cancer Cell 35, 369–384.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, Werner L, Sivachenko A, Deluca DS, Zhang L, et al. (2011). SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N. Engl. J. Med 365, 2497–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Dawson E, Duong A, Haw R, and Stein L (2014). ReactomeFIViz: a Cytoscape app for pathway and network-based data analysis. F1000Res 3, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Chen H, Du K, Asahara H, Tini M, Emerson BM, Montminy M, and Evans RM (2001). A transcriptional switch mediated by cofactor methylation. Science 294, 2507–2511. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. (2011). Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478, 64–69. [DOI] [PubMed] [Google Scholar]
- Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, Srinivasan P, Gao J, Chakravarty D, Devlin SM, et al. (2017). Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med 23 (6), 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Lieu YK, Ali AM, Penson A, Reggio KS, Rabadan R, Raza A, Mukherjee S, and Manley JL (2015a). Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc. Natl. Acad. Sci. USA 112, E4726–E4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Tran NT, Su H, Wang R, Lu Y, Tang H, Aoyagi S, Guo A, Khodadadi-Jamayran A, Zhou D, et al. (2015b). Cross-talk between PRMT1-mediated methylation and ubiquitylation on RBM15 controls RNA splicing. Elife 4, e07938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, and Manley JL (2013). Misregulation of pre-mRNA alternative splicing in cancer. Cancer Discov. 3, 1228–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-seq data reported in this paper is GEO:GSE123774.
The accession number for the MS-proteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE (Vizcaino et al., 2016) partner repository with the dataset identifier PRIDE: PXD012007.