

Abstract
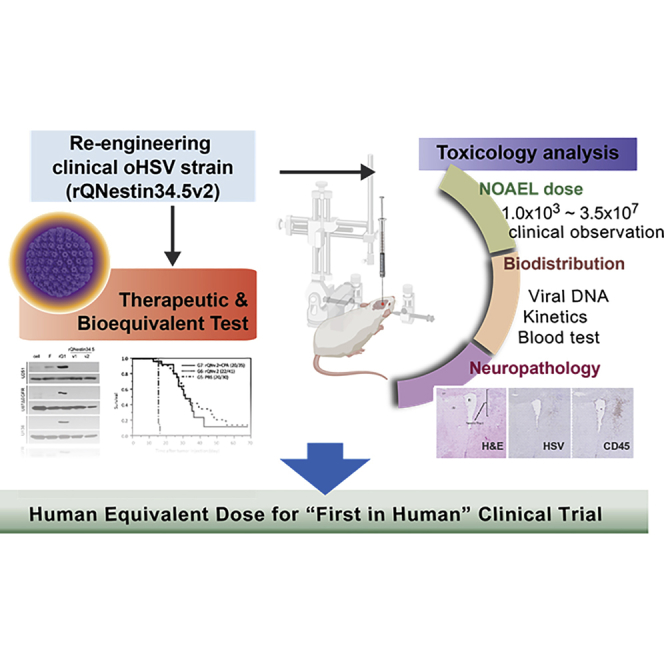
rQNestin34.5v.2 is an oncolytic herpes simplex virus 1 (oHSV) that retains expression of the neurovirulent ICP34.5 gene under glioma-selective transcriptional regulation. To prepare an investigational new drug (IND) application, we performed toxicology and efficacy studies of rQNestin34.5v.2 in mice in the presence or absence of the immunomodulating drug cyclophosphamide (CPA). ICP34.5 allows HSV1 to survive interferon and improves viral replication by dephosphorylation of the eIF-2α translation factor. rQNestin34.5v.2 dephosphorylated eIF-2α in human glioma cells, but not in human normal cells, resulting in significantly higher cytotoxicity and viral replication in the former compared to the latter. In vivo toxicity of rQNestin34.5v.2 was compared with that of wild-type F strain in immunocompetent BALB/c mice and athymic mice by multiple routes of administration in the presence or absence of CPA. A likely no observed adverse effect level (NOAEL) dose for intracranial rQNestin34.5v.2 was estimated, justifying a phase 1 clinical trial in recurrent glioma patients (ClinicalTrials.gov: NCT03152318), after successful submission of an IND.
Keywords: herpes simplex virus; ICP34.5; nestin, oncolytic virus, toxicology, glioblastoma, brain tumor, preclinical study
Graphical Abstract
Introduction
Glioblastoma multiforme (GBM) (World Health Organization [WHO] grade III–IV) is one of the most aggressive types of cancer, with a median survival of approximately 15 months.1,2 When newly diagnosed, the first line of treatment consists of surgery followed by radiotherapy with concomitant temozolomide chemotherapy and then additional adjuvant temozolomide therapy.3,4 Additional adjuvant treatments such as tumor-treating fields, intratumoral chemotherapy wafers, and bevacizumab are also US Food and Drug Administration (FDA) approved.5 However, GBM almost always recurs within months. The absence of treatments that lead to reliable durable anticancer responses mandates the search for more effective agents. Oncolytic viruses (OVs) act via a multimodal mechanism of action: (1) they cause direct cytotoxicity by infecting tumor cells; (2) they biodistribute within the tumor mass, leading to more cytotoxicity; and (3) they evoke an immune response against tumor and viral antigens, which is thought to lead to lasting adaptive immunity.6, 7, 8, 9 Oncolytic herpes simplex virus type 1 (oHSV) has been one of the most widely studied OVs: one type of oHSV has been FDA approved for the treatment of melanoma.10 The HSV1 peptide ICP34.5 has pleiotropic functions, but importantly it has been shown to be HSV1’s major neurovirulence factor in mice.11, 12, 13, 14 Because of this safety concern, most oHSVs in clinical trials have been engineered to lack ICP34.5 function.13,15, 16, 17, 18, 19, 20, 21 However, some functions of the ICP34.5 protein benefit the viral life cycle, including evasion from interferon action and counteracting the protein kinase R (PKR) host defense mechanism that leads to translational shut-off by phosphorylation of eiF-2α.22, 23, 24, 25, 26 In fact, lack of ICP34.5 severely attenuates viral replicative ability.27,28 To counteract this effect, several oHSVs have been engineered with second-site mutations that complement this viral defect:29 both the FDA-approved oHSV, talimogene laherparepvec (Imlygic),30 and a second oHSV in clinical trials in Japan15,31 possess defects in the ICP47 gene shown to revert some of the ICP34.5 mutant phenotypes.
We have utilized a different strategy restoring one copy of ICP34.5 under the transcriptional control of the nestin promoter/enhancer element.27,32 Nestin is overexpressed in a variety of cancers, including GBM. This oHSV was named rQNestin34.5.32 After mouse GBM efficacy studies showed the therapeutic anticancer potency of rQNestin34.5, we approached the FDA to discuss investigational new drug (IND)-enabling studies. From these discussions a new oHSV based on the rQNestin34.5 concept was engineered: rQNestin34.5v.2. The primary genotypic and phenotypic differences consist of a deletion of a fusion transcript made by sequences encoding for green fluorescent protein (GFP) linked to the carboxyl terminus of the ICP6 gene. In addition, we have shown that the immunomodulating drug cyclophosphamide (CPA) significantly enhanced oHSV replication and survival in tumors by limiting initial innate antiviral responses.33, 34, 35 The primary objective of the study was to find a dose of rQNestin34.5v.2 (with and without CPA) that led to no animal lethality and no or minimal neuropathologic findings. This would then define the human equivalent dose (HED) to start a phase 1 clinical trial. Herein, we report efficacy, toxicology, and biodistribution studies of rQNestin34.5v.2 with and without CPA in naive mice and mice bearing GBM that enabled us to define a dose of the agent that led to lethality and/or neuropathologic findings, thus allowing definition of a HED as the starting dose for a phase 1 human clinical trial in recurrent GBM.
Results
Engineering rQNestin34.5v.2
Although we had initially reported on the anticancer efficacy of rQNestin34.5,32 pre-IND discussion with the FDA resulted in modifying this oHSV to remove a fusion ICP6-GFP transcript. This transcript encoded for the enhanced GFP linked to the carboxyl terminus of ICP6. This carboxyl terminus has been linked to neuronal necroptosis and toxicity.36,37 To engineer rQNestin34.5 lacking this transcript, we had to restart from completely new reagents (Figure S1). The generated rQnestin34.5v.2 was initially characterized by Southern blotting with the desired genetic identity (data not shown) and then sequenced for confirmation (data not shown).
rQNestin34.5v.2 Infection Suppressed Phosphorylation of eIF-2α in a Glioma-Specific Manner
One of the host-immune evasion mechanisms used by HSV1 is the suppression of eIF-2α phosphorylation by ICP34.5.26,38 We sought to determine whether the ICP34.5-positive rQNestin34.5v.2 could suppress phosphorylation of eIF-2α in a glioma-selective fashion. Four human glioma cell lines (U251, U87ΔEGFR, U138, Gli36ΔEGFR), primary human glioma cells (OG02), and two primary human normal cells (human astrocytes [HAs], human umbilical vein endothelial cells [HUVECs]) were infected with rQNestin34.5v.2, rQNestin34.5v.1, the ICP34.5 null oncolytic strain rHsvQ1, and wild-type strain F at an MOI of 1. rQNestin34.5 v.2 and rQNestin34.5 suppressed phosphorylation of eIF-2α in glioma cell lines and primary glioma cells, but not in normal cells (Figure S2). The positive control (strain F) suppressed eIF-2α phosphorylation in all cell lines, whereas the negative control (ICP34.5 null mutant rHsvQ1) induced phosphorylation of eIF-2α in all cell lines tested. These results demonstrated that the reinserted gene encoding for ICP34.5 in rQNestin34.5v.2 (as well as in rQNestin34.5v.1) suppressed phosphorylation of eIF-2α in a glioma-specific manner.
rQNestin34.5v.2 Exhibits Selective Cytotoxicity and Replication toward Human Glioma Cells Compared to Normal Cells
We sought to determine whether rQNestin34.5v.2 exhibited glioma-specific cytotoxicity and replication. First, three human glioma cell lines (U87, U87ΔEGFR, U251), one patient-derived glioma cell (OG02), four primary human normal cells (astrocytes, pulmonary fibroblasts, smooth muscle cells, skeletal muscle cells), and primary mouse astrocytes were infected with rQNestin34.5v.2 at an MOI of 0.1. rQNestin34.5v.2 was cytotoxic for all glioma cells, with reduced cell survival to less than 20% of control (Figure S3A). Alternatively, no significant cytotoxicity was observed for human and mouse normal cells, with more than 80% of cells surviving at the end of a 72-h incubation period. We then tested the replication of rQNestin34.5v.2 against four established glioma cell lines (U251, Gli36, T98G, U87ΔEGFR), three patient-derived glioma cells (G97, OG02, X12), and four primary human normal cells (astrocytes, pulmonary fibroblasts, smooth muscle cells, skeletal muscle cells). Whereas wild-type strain F showed high replication in all cell lines tested, rQNestin34.5v.2 and rHsvQ1 replicated in glioma cells with almost no replication in normal cells (Figure S3B). The maximal viral yields of rQNestin34.5v.2 in glioma cells were higher than those in normal cells by at least 2 orders of magnitude (≥103 plaque-forming units [PFU]/mL versus ≤101 PFU/mL). rQNestin34.5v.2 replicated to higher yields than did the ICP34.5 null control rHsvQ1 in glioma cells. These data thus showed that rQNestin34.5v.2 replicated to much higher levels and was more cytotoxic in glioma cells compared to normal cells.
Preliminary In Vivo Toxicity Studies of rQNestin34.5v.2 in Athymic and Immunocompetent Mice with or without CPA
To determine doses of rQNestin34.5v.2 to employ for a planned GLP (good laboratory practice) toxicology and biodistribution study (required for IND filing), we first performed a non-GLP dose-escalation study of intracerebral (i.c.) administration of rQNestin34.5v.2 into the brains of athymic mice at 8 weeks of age. In parallel, a second dose-escalation study was performed with pre-administered intraperitoneal CPA at doses of 300 mg/kg or 200 mg/kg. Intracranial injections in athymic mice brains at doses of rQNestin34.5v.2 of 1.2 × 104 PFU and up to 1.2 × 105 PFU were not associated with lethality throughout the 60-day observation (Table S1). At higher doses, lethality was encountered. Wild-type F strain injections were lethal in 100% of athymic mice at a dose of 104 PFU. Based on these preliminary findings, we thus selected a dose that was intermediate between 1.2 × 104 and 1.2 × 105 PFU (i.e., 4.2 × 104 PFU) for the GLP-grade toxicology studies.
When one dose of intraperitoneal CPA (300 mg/kg) was pre-administered, 103 PFU was the highest dose of intracerebrally administered rQNestin34.5v.2 that was non-lethal. A dose of 3 × 103 PFU led to 1/17 lethality, 4 days after agent injection. Neuropathologic analysis of this brain did not reveal evidence of HSV-induced encephalitis or neurotoxicity, and the reason for death remained unexplained (data not shown). A lower dose of CPA (200 mg/kg) allowed increase in the intracerebral virus dose to 104 PFU without lethality, while this same dose of virus with 300 mg/kg CPA led to lethality in 3/25 mice. Because the 200 mg/kg dose of CPA was found to be ineffective in improving oHSV survival in tumors (data not shown), it was not pursued further. Based on this, we focused on a dose of intracerebral 3 × 103 PFU to test in CPA-pretreated (300 mg/kg) athymic mice for a GLP toxicology and biodistribution study.
We also wanted to determine doses of rQNestin34.5v.2 that did not cause lethality in immunocompetent mice. We employed HSV-susceptible BALB/c mice. In these studies, we also tested lethality utilizing various routes of administration. Intracranial injections of rQNestin34.5v.2 at a dose of 107 PFU were carried out in 28 mice at 8 weeks of age. There was one death that occurred about 3 days after injection, but the remaining 27 mice remained clinically well, until they were euthanized 60 days later as per the protocol to harvest brains and organs for histology and distribution studies. Unfortunately, it was not possible to obtain tissues from the single dead mouse, since it occurred unexpectedly (i.e., the animal appeared well without loss of weight or neurologic issues but was found dead the next morning in the cage with signs of having been cannibalized by other mice). In contrast, 3/5 and 19/22 BALB/c mice injected intracerebrally with wild-type F strain at doses of 104 and 105 PFU, respectively, perished, usually within the first 10 days from injection with neurologic signs. Intrathecal (i.t.) injections were tolerated by 5/5 8-week-old mice for the rQNestin34.5v.2 group at 106 PFU and 10/11 at 107 PFU, but only 1/5 mice for the F strain group at 105 PFU. In older mice (>6 months), 9/9 mice tolerated intrathecally 107 PFU of rQNestin34.5v.2, but 2/5 mice tolerated 105 PFU of wild-type F. For intrahepatic (i.h.) and intravenous (i.v.) injections, all mice tolerated 107 PFU or more of rQNestin34.5v.2.
As per the experiments in athymic mice, two doses of CPA were tested (300 and 200 mg/kg) in 8-week-old BALB/c mice, where 106 PFU of intracranial rQNestin34.5v.2 led to 0/18 mortalities with pre-administration of high-dose CPA. Higher dose of virus (3 × 106 and 107 PFU) led to 3/27 and 4/28 deaths, respectively. With low-dose CPA, there was no mortality even with 107 PFU of intracranial virus. Intrathecal injections of virus were tolerated without lethality at 107 PFU with both high and low doses of CPA. Intrahepatic and intravenous administrations of 107 PFU were tolerated by 8/10 and 9/10 animals receiving high-dose CPA, respectively, and by 12/12 and 12/12 animals with low-dose CPA.
Taken together, for all four injection routes tested, rQNestin34.5 v.2 led to greatly reduced in vivo toxicity compared to that of wild-type F strain, by at least 2 orders of magnitude. CPA pretreatment increased mortality in immune-competent BALB/c mice, presumably due to its known side effects. However, when the dose of CPA was reduced to 200 mg/kg, no lethality was observed for rQNestin34.5v.2 at the dose of 1 × 107 PFU. The non-lethal dose of rQNestin34.5v.2 alone was estimated to be 2 × 107 PFU or more (higher doses were not tested) for intravenous and intrahepatic administration, and less than 1 × 107 PFU for intracerebral and intrathecal administration. In combination with CPA (300 mg/kg) pretreatment, the non-lethal dose of rQNestin34.5 v.2 was estimated at 1 × 106 PFU for intracerebral and intrathecal administration in young mice, and at 1 × 107 PFU for intrathecal administration in old mice. We did not test lower non-lethal doses for the other routes of administration.
Ultimately, for the GLP biodistribution and toxicology study, after meeting with the FDA, we decided that the intracerebral route in athymic mice was the most appropriate model to test because (1) it showed higher dose-related toxicity than did the immunocompetent model, and (2) the proposed clinical trial would utilize intracerebral administration.
GLP Toxicology and Biodistribution Study of rQNestin34.5v.2 Intracerebral Administration in Athymic Mice
Based on discussions with the FDA and the experiments shown in Table S1, athymic mice were assigned to one of four groups (both sexes, n = 18 per group): group 1, control; group 2, CPA pre-administration only (300 mg/kg); group 3, intracerebral injection with 4.2 × 104 PFU; or group 4, pre-administration with CPA followed by intracerebral injection with 3 × 103 PFU (see Table 5 in Materials and Methods). Six mice (three per sex) from each group at the 4-, 31-, or 61-day time point after oHSV injection were scheduled for euthanasia. Endpoints used to evaluate potential toxicities of rQNestin34.5v.2 were mortality, clinical observations, body weights, food consumption, clinical pathology, gross necropsy (including organ weights), histopathology (from selected mice), and HSV and immune cell immunohistochemistry (IHC) of the brains of selected mice. Biodistribution of rQNestin34.5v.2 was evaluated by quantitative polymerase chain reaction (qPCR) of blood and of the following tissues, when available: brain (with tumor), spinal cord, bone marrow, kidney, lungs, gonads, heart, liver, spleen, lymph node, and any skin lesions.
Table 5.
Experimental Groups (see Materials and Methods)
| Group | Target Human U87ΔEGFR Glioma Cells (2 × 105) | Target CPA (mg/kg) | Target rQNestin34.5v.2 (PFU) | Total No. of Mice | Target No. of Animals at Each Necropsy |
||
|---|---|---|---|---|---|---|---|
| Day 4 (M/F) | Day 31 (M/F) | Day 61 (M/F) | |||||
| 1 | 0 | 0 | 0a | 36 | 6/6 | 6/6 | 6/6 |
| 2 | 0 | 300 | 0a | 36 | 6/6 | 6/6 | 6/6 |
| 3 | 0 | 0 | 4.2 × 104 | 36 | 6/6 | 6/6 | 6/6 |
| 4 | 0 | 300 | 3 × 103 | 36 | 6/6 | 6/6 | 6/6 |
| 5 | L | 0 | 0a | 30 | 0/10 | 0/10 | 0/10 |
| 6 | L | 0 | 3.5 × 107 | 41 | 0/10 | 0/10 | 0/10 |
| 7 | L | 300 | 3.5 × 107 | 35 | 0/10 | 0/10 | 0/10 |
M, male; F, female.
Dulbecco’s phosphate-buffered saline.
Clinical Observations and Premature Lethality
There were 12 early deaths distributed within the CPA group (group 2), the 4.2 × 104 PFU virus group (group 3), and the 3 × 103 PFU virus + CPA group (group 4) (Table S2). Following histopathology evaluation (hematoxylin and eosin staining) of collected tissues from mice in the CPA (group 2) and 3 × 103 PFU virus + CPA groups (group 4), there was no apparent cause noted for these premature deaths that could be attributed to CPA and/or virus administration with one exception: one female mouse in the 4.2 × 104 PFU virus group (group 3) died at 54 days post-infection and was noted to have a positive viral infection in the brain via IHC evaluation (see Table 2 later). All of the other mice administered rQNestin34.5v.2 and all of the mice in the control and CPA groups remained clinically normal throughout the in-life period.
Table 2.
Histopathology and Immunohistochemistry Evaluation
| Animal ID | Day of Death | CD45 Immunoreactive | Interpretation | ||
|---|---|---|---|---|---|
| Group 1 | |||||
| Control males, scheduled deaths | 101 | 4 | no significant microscopic lesions | ||
| 103 | 4 | ||||
| 105 | 4 | ||||
| 108 | 31 | ||||
| 110 | 31 | ||||
| 112 | 31 | ||||
| 113 | 61 | ||||
| 116 | 61 | ||||
| 117 | 61 | ||||
| Control females, scheduled deaths | 151 | 4 | no significant microscopic lesions | ||
| 153 | 4 | ||||
| 155 | 4 | ||||
| 157 | 31 | ||||
| 160 | 31 | ||||
| 162 | 31 | ||||
| 163 | 61 | ||||
| 165 | 61 | ||||
| 167 | 61 | ||||
| Group 2 | |||||
| CPA males, scheduled deaths | 202 | 4 | no significant microscopic lesions | ||
| 204 | 4 | ||||
| 206 | 4 | ||||
| 208 | 31 | ||||
| 210 | 31 | ||||
| 211 | 31 | ||||
| 214 | 61 | ||||
| CPA males, early deaths | 215 | 52 (FD) | cause of death not apparent | ||
| 218 | 55 (FD) | cause of death not apparent | |||
| CPA females, scheduled deaths | 252 | 4 | no significant microscopic lesions | ||
| 254 | 4 | ||||
| 256 | 4 | ||||
| 257 | 31 | ||||
| 259 | 31 | ||||
| 262 | 31 | ||||
| 267 | 61 | ||||
| CPA females, early deaths | 264 | 59 (FD) | cause of death not apparent | ||
| 265 | 54 (FD) | cause of death not apparent | |||
| Group 3 | |||||
| 4.2 × 104 PFU virus, male scheduled deaths | 302 | 4 | minimal HSV infection of nervous tissue in the absence of cytopathic effect or inflammatory change | ||
| 304 | 4 | no significant microscopic lesions | |||
| 306 | 4 | √ | minimal HSV infection of nervous tissue in the absence of cytopathic effect or inflammatory change | ||
| 307 | 31 | no significant microscopic lesions | |||
| 312 | 31 | no significant microscopic lesions | |||
| 315 | 31 | no significant microscopic lesions | |||
| 314 | 61 | no significant microscopic lesions | |||
| 317 | 61 | no significant microscopic lesions | |||
| 4.2 × 104 PFU virus, female scheduled deaths | 352 | 4 | no significant microscopic lesions | ||
| 354 | 4 | √ | minimal viral infection with no cytopathic effect and a minimal inflammatory response | ||
| 356 | 4 | √ | minimal viral infection of ependyma and periventricular tissues in the absence of cytopathic effect and a minimal inflammatory response | ||
| 358 | 31 | no significant microscopic lesions | |||
| 359 | 31 | no significant microscopic lesions | |||
| 361 | 31 | no significant microscopic lesions | |||
| 367 | 61 | no significant microscopic lesions | |||
| 4.2 × 104 PFU virus, male early deaths | 309 | 6 (FD) | cause of death not apparent | ||
| 316 | 53 (FD) | cause of death not apparent | |||
| 4.2 × 104 PFU virus, female early deaths | 365 | 54 (FD) | cause of death not apparent | ||
| 364 | 55 (FD) | √ | death attributed to viral infection in brain | ||
| Group 4 | |||||
| 3 × 103 PFU virus + CPA, male scheduled deaths | 402 | 4 | no significant microscopic lesions | ||
| 404 | 4 | no significant microscopic lesions | |||
| 406 | 4 | there was a single HSV antigen-positive cell that was not associated with cytopathic effect or an inflammatory response | |||
| 408 | 31 | no significant microscopic lesions | |||
| 410 | 31 | no significant microscopic lesions | |||
| 411 | 31 | no significant microscopic lesions | |||
| 415 | 61 | no significant microscopic lesions | |||
| 3 × 103 PFU virus + CPA, female scheduled deaths | 452 | 4 | no significant microscopic lesions | ||
| 454 | 4 | no significant microscopic lesions | |||
| 456 | 4 | no significant microscopic lesions | |||
| 458 | 31 | minimal focal viral infection in the absence of inflammatory responses or cytopathic effect | |||
| 459 | 31 | no significant microscopic lesions | |||
| 461 | 31 | no significant microscopic lesions | |||
| 464 | 61 | no significant microscopic lesions | |||
| 466 | 61 | no significant microscopic lesions | |||
| 3 × 103 PFU virus + CPA, male early deaths | 418 | 53 (FD) | cause of death not apparent | ||
| 413 | 54 (FD) | cause of death not apparent | |||
| 3 × 103 PFU virus + CPA, female early death | 467 | 53 (FD) | cause of death not apparent | ||
FD, found dead.
Body Weights
The male CPA group (group 2) had significantly lower group mean body weights when compared to the control and 4.2 × 104 PFU virus groups on days 1, 4, 8, 22, 29, 43, 50, and 57 (data not shown). Group mean body weights also were significantly lower for the CPA group females (group 2) when compared to the control and 4.2 × 104 PFU virus groups (groups 1 and 3, respectively) on days 1, 4, and 31. The lower group mean body weights in the CPA-treated groups was due to a slower rate of body weight gain, rather than body weight loss. Both the male and female CPA groups had similar group mean body weights when compared to the respective 3 × 103 PFU virus + CPA group, suggesting that the administration of CPA had an adverse effect on body weights (affecting male mice more often than female mice). The administration of rQNestin34.5v.2 did not affect body weights in these groups. Group mean body weights for male mice administered 4.2 × 104 PFU virus were lower, with statistical significance on days 1 and 8 when compared to the control group.
Food Consumption
Male mice in the CPA group had significantly lower mean feed consumed per day values, when compared with the mean daily feed consumed per day values of the control group male mice for the period of days 36–43, days 43–50, and days 50–57. For all other days and groups, group mean feed consumed per day values for male and female mice in the toxicity assessment groups were similar to the respective comparison groups (control group for the CPA and 4.2 × 104 PFU virus groups and the CPA group for the 3 × 103 PFU virus + CPA group).
qPCR
Biodistribution of rQNestin34.5v.2 was assayed by qPCR analyses of injected brains in six mice per group at days 4, 31, and 61 post-injection of oHSV (Table 1). On day 4, rQNestin34.5v.2 was detected in the brains of mice in the 4.2 × 104 PFU virus group (group 3) at 8.6 × 103 to 6.0 × 104 copies/μg DNA and in the brains of all 3 × 103 PFU virus + CPA group 4 mice at 8.4 × 102 to 8.0 × 103 copies/μg DNA. Additionally, the kidney for mouse 351 (female, 4.2 × 104 PFU virus group) and the heart for mouse 353 (female, 4.2 × 104 PFU virus group) were positive for rQNestin34.5v.2 with 5.3 × 101 and 2.9 × 102 copies/μg DNA, respectively. rQNestin34.5v.2 was not detected from other tissues collected at the day 4 necropsy.
Table 1.
Days 4, 31, and 61 rQNestin34.5v.2-Positive Results in Brain: Toxicity Assessment of Groups Administered rQNestin34.5v.2
| Group | Day 4 |
Day 31 |
Day 61 |
|||
|---|---|---|---|---|---|---|
| Animal ID | Copies/μg DNA | Animal ID | Copies/μg DNA | Animal ID | Copies/μg DNA | |
| 4.2 × 104 PFU virus(group 3) | 301 | 8.8 × 103 | 308 | 5.9 × 103 | 313 | 1.2 × 102 |
| 303 | 8.6 × 103 | 310 | 1.3 × 103 | 318 | 8.5 × 102 | |
| 305 | 1.2 × 104 | 311 | 1.9 × 103 | |||
| 351a | 9.2 × 103 | 357 | 2.2 × 103 | 363 | 4.2 × 102 | |
| 353b | 6.0 × 104 | 360 | 1.9 × 103 | 366 | 8.4 × 101 | |
| 355 | 4.4 × 104 | 362 | 1.5 × 103 | 368 | 5.2 × 102 | |
| 3 × 103 PFU virus + CPA (group 4) |
401 | 8.4 × 102 | 407 | 4.4 × 101 | 414 | 3.3 × 103 |
| 403 | 4.6 × 103 | 409 | 1.8 × 102 | 416 | not detected | |
| 405 | 8.0 × 103 | 412 | not detected | 417 | 9.6 × 101 | |
| 451 | 1.2 × 103 | 457 | 5.8 × 102 | 463 | not detected | |
| 453 | 1.7 × 103 | 460 | 1.1 × 102 | 465 | not detected | |
| 455 | 2.1 × 103 | 462 | 1.4 × 102 | 468 | not detected | |
rQNestin34.5v.2 was also detected in the kidney.
rQNestin34.5v.2 was also detected in the heart.
On day 31, rQNestin34.5v.2 was detected in the brains of the 4.2 × 104 PFU virus group mice with a viral load of 1.3 × 103 to 5.9 × 103 copies/μg DNA. On day 31, rQNestin34.5v.2 was detected in the spinal cord of animal 459 (3 × 103 PFU virus + CPA group) with 9.7 × 101 copies/μg DNA; the brain for this animal was sent for IHC evaluation, and rQNestin34.5v.2 was not observed in the brain of this animal. The brains of 5/6 animals designated for qPCR evaluation at day 31 were positive for rQNestin34.5v.2 (4.4 × 101 to 5.8 × 102 copies/μg DNA), although the average concentration was approximately 15-fold less than the average concentration from the brains on day 4. No other tissues from the day 31 necropsy were positive for rQNestin34.5v.2.
On day 61, rQNestin34.5v.2 was detected only in the brains of the 4.2 × 104 PFU virus group mice with a viral load of 8.4 × 101 to 8.5 × 102 copies/μg DNA: the average values were approximately 60- and 6-fold less than the average concentration seen on days 4 and 31, respectively. rQNestin34.5v.2 was not detected in any other tissue on day 61. On day 61, the brains of two of the three male mice submitted for qPCR (414 and 417) were positive for rQNestin34.5v.2 at concentrations of to 3.3 × 103 to 9.6 × 101copies/μg DNA; the average value was approximately 8-fold higher than the average concentration seen in the day 31 male brains. rQNestin34.5v.2 was not detected in the brains of the three female mice (463, 465, and 468) submitted for qPCR on day 61, or in any of the spinal cords analyzed.
Hematology and Serum Chemistries
None of the hematology results indicated effects due to rQNestin34.5v.2 administration (data not shown). Changes noted in hematology parameters included decreases in reticulocyte counts (absolute and percent) on day 4 for male and female mice in the CPA-only group when compared with male and female mice in the control group. These changes were an expected result of CPA administration and were not noted on day 31, indicating recovery from the effects of CPA. There were no differences in reticulocyte values when the CPA-only and the 3 × 103 PFU virus + CPA groups were compared. Decreases in red blood cell count, hemoglobin, and hematocrit values were noted in the CPA-only group when compared to the control group. These changes were suspected to be a result of CPA administration. None of the serum chemistry results indicated any effects due to rQNestin34.5v.2 administration.
Organ Weights
Statistically significant decreases noted in absolute spleen weight, spleen-to-body weight, and spleen-to-brain weight ratios for the CPA group males when compared to the control group males on day 4 were likely due to an effect of CPA administration on splenic hematopoiesis (data not shown). Clinical pathology findings indicate decreased hematopoiesis at day 4 in this group likely because of CPA administration. These decreases were not seen on day 31, indicating recovery from this effect. There were statistically significant reductions in the CPA groups for terminal body weight and for the weight of other organs at various time points when compared to other groups. These were all likely due to the effect of CPA. There were no rQNestin34.5v.2 effects on body or organ weights.
Histopathology and IHC
Neuropathologic H&E (hematoxylin and eosin) and immunohistochemical evaluation for HSV antigen and CD45 immunoreactive cells were carried out for mice that were scheduled for euthanasia and for those that encountered early unscheduled premature deaths (Table 2). Of relevance, in the four early-death mice in the 4.2 × 104 PFU virus group (males 309 and 316 and females 365 and 364) only female mouse 364 (found dead on day 55) was noted with rQNestin34.5v.2 in the brain, and the early death was attributed to viral infection in the brain. For the mice that underwent scheduled euthanasia in the 4.2 × 104 PFU virus group (males 302 and 306 and females 354 and 356), minimal viral infection was detected with no cytopathic effect or inflammatory response in day 4 brain tissue from four of the six mice examined. No rQNestin34.5v.2 was detected by IHC in day 31 or day 61 brains. For the scheduled euthanasia for mice that received CPA in addition to rQNestin34.5v.2, evidence of viral presence was only detected by IHC in a single day 4 brain tissue and in a single day 31 brain tissue. Male 406 (day 4 scheduled necropsy) was noted with a single viral antigen-positive cell that was not associated with cytopathic effect or an inflammatory response, and female 458 (day 31 scheduled necropsy) was noted with minimal focal viral infection in the absence of inflammatory responses or cytopathic effect. Evidence of rQNestin34.5v.2 in the brain was not detectable by IHC in any other brains from mice treated with rQNestin34.5v.2 plus CPA at scheduled or unscheduled necropsies. There were no gross or microscopic findings in any of the early-death mice examined from the CPA group (215, 216, and 218 [males] and 264 and 265 [females]) or in the 3 × 103 PFU virus + CPA group (413 and 418 [males] and 467 [female]). As expected, there was no rQNestin34.5v.2 detected by IHC in the brains of male or female mice in the control or CPA group at any of the scheduled, or unscheduled, necropsies. In conclusion, there was no lethality or neuropathologic findings for a rQNestin34.5v.2 dose of 3 × 103 PFU (with CPA). This dose was also not associated with significant abnormalities and minimal evidence of long-term biodistribution outside of the injected area.
GLP Toxicology and Biodistribution of rQNestin34.5v.2 after Administration in Human GBM Cells Implanted In Athymic Mice Brains
The previous studies were conducted in athymic mice without tumors. However, after discussion with the FDA, we wanted to know whether the toxicology and biodistribution of rQNestin34.5v.2 changed when inoculated in human GBM cells established in the brains of athymic mice. Therefore, a GLP experiment was conducted where human U87DEGFR GBM cells where injected into the brains of athymic mice. At the 7-day time period, 3.5 × 107 PFU of the oHSV (group 6) or vehicle (group 5) were stereotactically injected in tumor. One group (group 7) was pretreated with one dose of CPA 2 days before oHSV administration. Mouse mortality was followed for 61 days after oHSV injection, and attempts to assign cause of lethality to tumor or oHSV were made. Inoculation with rQNestin34.5v.2 significantly improved survival of tumor-bearing mice (Figure 1), but this improvement was not affected by the addition of CPA. This was likely due to our published observations that CPA’s enhancement or oHSV-mediated survival occurs at low viral doses and diminishes as oHSV dose increases.35 These survival findings reproduced the significant anti-glioma effect of the agent, in an experiment performed under GLP conditions, in a randomized blinded fashion, by an individual entity independent from the study principal investigator (PI).
Figure 1.

Kaplan-Meier Survival Curves
Comparison of survival curves for athymic mice with an orthotopic human glioma treated with PBS (group 5), 3.5 × 107 PFU of rQNestin34.5v.2 (group 6), or 3.5 × 107 PFU of rQNestin34.5v.2 with CPA pre-administration, 2 days before virus injection. Animals scheduled to undergo scheduled necropsy at day 4 or 31 are not included. In group 7, there were 10 mice that underwent scheduled necropsy at day 4 and 4 that underwent scheduled necropsy at day 31. One mouse was still alive at the study termination and underwent scheduled necropsy at this time (included in graph). For group 6, there were 10 mice that underwent scheduled necropsy at days 4 and 7 that underwent scheduled necropsy at day 31. Two mice were still alive at study termination and underwent scheduled necropsy at this time (included in graph). For group 5, there were 10 mice that underwent scheduled necropsy at day 4, but none was alive for the day 31 and thereafter scheduled necropsies. There was a statistically significant increase in survival of animals from group 6 and 7 when compared to those from group 5 (p < 0.0001, log-rank test).
Except for mice that were necropsied as scheduled on day 4, all tumor-bearing mice, inoculated with PBS control, were found dead or euthanized due to moribund condition by day 9 after PBS administration (Figure 1; Table S3). In contrast, 23/41 mice in the tumor + virus group and 20/35 mice in the tumor + virus + CPA group were found dead or were humanely euthanized due to moribund condition prior to scheduled necropsy, and these deaths occurred through day 48. The remaining mice survived until they were scheduled for termination.
Neuropathology of Mice with Brain Tumors Treated with rQNestin3.5v.2 Alone
The brains from mice with brain tumors treated with rQNestin34.5v.2 that were either moribund or died before scheduled necropsy dates were analyzed histologically and immunohistochemically to determine whether the growing tumor versus viral encephalitis/meningitis was the cause of death. Table 3 lists the animal identification (ID) for each scheduled necropsy and premature death as well as the neuropathological findings and, when possible, an interpretation for the probable cause of death. In 17/21 premature deaths, the volume, location, and extent of tumor in brain was the likely cause of early mouse demise. When viral antigen was present in brain outside of tumor, its limited amounts were not deemed sufficient to lead to animal demise. In 3/21 mice the cause of death was unknown: for mouse nos. 673, 676, and 678 there was evidence of extensive tumor necrosis with some virus antigen present in brain, but neither was judged to be extensive enough to lead to animal demise. In 1/12 (mouse 684), the likely cause of early animal demise was a result of viral encephalitis and meningitis. In this mouse, there was a very small tumor located rostrally with virus disseminated in areas outside of tumor. The needle tract appeared to be separate from tumor (Figure 2). This suggested that the initial rQNestin34.5v.2 injection was incorrectly directed into brain rather than into tumor probably because tumor also had not grown well, due to its small size by this time. There were also several instances of selectivity for viral antigen and CPE within tumor (Figures S4 and S5).
Table 3.
Neuropathologic Findings and HSV Immunohistochemistry for Brains of Athymic Mice Harboring Human Gliomas Injected with rQNestin34.5v.2 (3.5 × 10E7 PFUs)
| Braina |
Gliomab |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Mouse No. | Datec | Antigend | CPEe | CD45f | Necrosisg | Antigend | CD45e | Volumeh | Interpretation |
| 653 | 4 | 0 | 0 | 0 | 4 | 2 | 3 | 6% | tumor in striatum; 80% of tumor is HSV antigen+ and necrotic |
| 654 | 4 | 3 | 0 | 3 | 2–3 | 2 | 3 | 5% | extension of HSV from tumor to ependymal with spread through ventricular system (ventricular spread); 50% of tumor is necrotic and HSV antigen+ |
| 655 | 4 | 3 | 2 (adjacent to tumor) | 3 | 4 | 2 | 3 | 6% | extension of HSV from tumor to ependymal with ventricular spread; 80% of tumor is necrotic and HSV antigen+ |
| 659 | 5i | 3 | 3 | 3 | 4 | 2 | 3 | 4% | extension of HSV from tumor to ependymal cells with ventricular spread; 100% of tumor is necrotic and HSV antigen+ |
| 660 | 5i | 3 | 0 | 3 | 4 | 2 | 3 | 1.5% | extension of HSV from tumor to ependymal with ventricular spread; two tumor nodules: one is 100% necrotic and HIV antigen+ |
| 661 | 19j | 3 | 0 | 2 | 0 | 0 | 0 | 10% | mild periventricular distribution of viral antigen; Tumor in brainstem is large and likely cause of death |
| 662 | 31 | 0 | 0 | 0 | 4 | 2 | 1 | 1% | single necrotic tumor in striatum that is HSV antigen+ |
| 663 | 28j | 0 | 0 | 0 | 2 | 2 | 0 | 25% | several nodules with two that are 100% necrotic and HSV+; cause of death due to tumor edema/hemorrhage |
| 664 | 24j | 0 | 0 | 0 | 1 | 2 | 2 | 25% | one tumor nodule is 100% necrotic and HSV antigen+; others exhibit partial necrosis but are HSV antigen−; death attributed to white matter edema associated with tumor |
| 665 | 23j | 0 | 0 | 0 | 2 | 2 | 2 | 55% | multiple tumor nodules with variable necrosis, and necrotic foci are both HSV antigen+ HSV antigen−; death is attributed to high tumor burden and associated hemorrhage |
| 666 | 21j | 0 | 0 | 0 | 2 | 2 | 3 | 45% | extensive tumor necrosis (45%) that is viral antigen+; death is attributed to the high tumor burden |
| 669 | 18jj | 0 | 0 | 0 | 0 | 0 | 0 | 50% | death is attributed to the high tumor burden |
| 670 | 23j | 0 | 0 | 0 | 2 | 2 | 2 | 30% | multiple tumor foci, 45% necrosis with regions that are HSV antigen− and HSV antigen+; death is attributed to the high tumor burden |
| 671 | 31 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | there is evidence of an inoculation track in striatum but no tumor or HSV |
| 673 | 24j | 2 | 2 | 2 | 1 | 2 | 2 | 15% | two tumor nodules with necrosis associated with HSV antigen (one complete, the other10%); forebrain with focus HSV+ cells, CPE, and inflammation, but evidence of extensive viral dissemination is lacking; cause of death is not apparent |
| 675 | 19j | 0 | 0 | 0 | 0 | 0 | 0 | 25% | death is attributed to the high tumor burden in the brainstem |
| 676 | 14j | 1 | – | 2 | 1 | 2 | 2 | 5% | tumor necrosis that is HSV antigen+, with limited viral spread into adjacent brain; cause of death not apparent |
| 677 | 31 | 0 | 0 | 0 | 1 | 2 | 3 | 26% | large tumor nodule 100% necrotic and HSV antigen+ |
| 678 | 16j | 0 | 0 | 0 | 3 | 2 | 2 | 30% | one large and one small tumor nodule (50% and 100% necrosis, respectively) that are HSV antigen+; cause of death not apparent |
| 679 | 61 | 0 | 0 | 0 | 4 | 2 | 3 | <1% | small necrotic tumor that is weakly HSV antigen+ and associated with intense CD45+ immunoreactivity |
| 680 | 7j | 3 | 1 | 2 | 1 | 2 | 2 | <10% | multiple tumor nodules in brainstem that are HSV antigen+ and associated CD45+ infiltrates; some HSV antigen in periventricular areas; death attributed to tumor-associated edema and hemorrhage |
| 682 | 41j | 0 | 0 | 0 | 1 | 2 | 0 | 50% | large tumor masses, where necrosis is associated with HSV antigen but no CD45+ infiltrates; death attributed to effects of tumor burden |
| 683 | 32j | 0 | 0 | 0 | 0 | 0 | 0 | 20% | death attributed to effects of tumor burden |
| 684 | 10j | 3 | 2 | 3 | 0 | 0 | 0 | low | low tumor volume in brainstem; evidence of ventricular dissemination of HSV with CPE and inflammation, particularly in brainstem; death attributed to virus infection |
| 685 | 22j | 2 | 0 | 0 | 1 | 0 | 1 | 50% | large tumors associated with edema and hemorrhage that are the cause of death |
| 686 | 48j | 0 | 0 | 0 | 1 | 0 | 1 | 30% | death attributed to effects of tumor burden |
| 687 | 20j | 0 | 0 | 0 | 1 | 0 | 1 | 55% | death attributed to tumor-associated edema, hemorrhage, and necrosis of parenchyma (infarction) |
| 689 | 23j | 0 | 0 | 0 | 1 | 2 | 1 | 25% | one nodule of tumor is necrotic and HSV antigen+; death attributed to tumor-associated edema and hemorrhage |
| 690 | 39j | 0 | 0 | 0 | 0 | 0 | 1 | 25% | death attributed to tumor-associated edema and hemorrhage |
| 691 | 26j | 0 | 0 | 0 | 0 | 0 | 0 | 15% | death attributed to brainstem localization of tumor and associated edema |
All analyses were conducted by Dr. M. Oglesbee (Ohio State University College of Veterinary Medicine).
Brain pathology.
Glioma pathology.
Date of scheduled euthanasia or unexpected death from time of rQNestin34.5v.2 injection.
HSV antigen (scored 0–3) (see Materials and Methods for explanation of scores).
Viral cytopathic effect (scored 0–3).
CD45+ cells, suggestive of activated microglia/inflammation (scored 0–3).
Necrosis (scored 0–3).
Estimated percent of brain occupied by glioma.
Scheduled necropsies conducted on day 5 rather than day 4.
Premature death or euthanasia for moribund state.
Figure 2.

Brain Histology and Immunohistochemistry for Mouse 684
H&E (A, D, and G), HSV IHC (B, E, and H), and CD45 IHC (C, F, I) for brain from mouse 684. (A)–(F) are from the frontal area of brain located 1.32 mm anterior to bregma and where the needle tract with a band of pannecrosis and microgliosis was observed, but this tract was not located near tumor (not shown here). (D)–(F) are high-power microphotographs of (A)–(C). (G)–(I) are from a more caudal area, located 1.62 posterior to bregma, where periventricular HSV and CD45-positive cells were seen. CC, corpus callosum; LV, lateral ventricle; LSI, lateral septal nucleus; Hi, hippocampus; CP, choroid plexus; 3V, 3rd ventricle; MHb, medial habenular nucleus.
Neuropathology of CPA-Pretreated Mice with Tumors, Inoculated with rQNestin34.5v.2
The brains from mice that were either moribund or died before scheduled necropsy dates were analyzed histologically and immunohistochemically to determine whether the growing tumor or viral encephalitis/meningitis was the cause of death. Table 4 lists the animal ID for each premature death as well as the neuropathological interpretation for cause of death. In 13/19 premature deaths, the volume, location, and extent of tumor in the brain was the likely cause of early mouse demise. In most cases where there was viral antigen outside of the tumor, the level of antigen was low and of limited extent, not associated with cytopathic effect or with significant CD45+ infiltrates. Presence of viral antigen outside of the tumors may reflect mild nestin immunohistochemical staining that was detected in reactive astrocytes and ependymal cells in mouse brains (Figure 3). In 2/19 premature deaths (mouse nos. 774 and 779), there were small or no tumors and no virus antigen. Therefore, a cause of death could not be determined. In 3/19, the cause of death was possibly due to tumor, but the small size rendered it more equivocal (mouse nos. 781 and 763). In 1/19 (mouse 770), the cause of early animal demise (day 27 after virus injection/34 days after tumor injection) was likely a result of disseminated viral encephalitis and meningitis, although tumors were also detected in the brainstem, which could contribute to the clinical outcome (Figure S6). This suggested that the initial rQNestin34.5v.2 injection was incorrectly directed into brain rather than into tumor probably because tumor had grown in the brainstem and not in the cortex were rQNestin34.5v.2 was injected.
Table 4.
Neuropathologic Findings and HSV Immunohistochemistry for Brains of Group 7 Athymic mice Harboring Human Gliomas Injected with rQNestin34.5v.2 (3.5 × 107 PFU) after CPA Pre-administration
| Braina |
Gliomab |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Mouse No. | Datec | Antigend | CPEe | CD45f | Necrosisg | Antigend | CD45f | Volumeh | Interpretation |
| 754 | 4 | 2 | 2 | 2 | 4 | 2 | 3 | 3% | tumor in striatum is necrotic, HSV, and CD45 antigen+; adjacent brain and injection tract is HSV+ and CD45+; 2nd focus of HSV antigen in brainstem |
| 755 | 4 | 1 | 0 | 1 | 4 | 2 | 2 | 3% | two tumors in striatum that are 90% HSV+; single focus of HSV+ neurons in mesencephalon |
| 756 | 5i | 3 | 1 | 2 | 0 | 0 | 0 | 0 | there is no tumor; injection tract in striatum with HSV+ cells; ventricular spread of virus based upon HSV+ cells in ependyma/subependyma |
| 757 | 5i | 3 | 0 | 0 | 4 | 2 | 3 | 2% | tumor in injection tract is 100% necrotic with strong HSV+/CD45+ signal; HSV+ staining extends from tumor into surrounding neurons, ependymal cells, and subependymal cells |
| 758 | 5i | 3 | 0 | 2 | 4 | 2 | 3 | 1% | tumor in injection tract with 80% necrotic and HSV+/CD45+; HSV+ staining extends from tumor to surrounding neurons with spread along the ependymal cells and subependyma |
| 761 | 17j | 0 | 0 | 0 | 1 | 2 | 2 | 10% | tumor in forebrain is necrotic, HSV+, CD45+; there are other large tumors that are HSV−; cause of death not apparent |
| 762 | 20j | 1 | 0 | 1 | 0 | 0 | 0 | 5% | one limited focus of HSV+ cells adjacent to lateral ventricle; death attributed to multiple HSV− tumors in brainstem |
| 763 | 20j | 3 | 0 | 0 | 1 | 2 | 2 | 30% | tumor in forebrain is 100% necrotic, HSV+, CD45+; other large tumors are HSV−; limited HSV+ cells in brainstem/cortex; cause of death not apparent |
| 766 | 17j | 3 | 0 | 1 | 0 | 0 | 1 | 5% | sporadic HSV+ cells in periventricular areas; death attributed to multiple HSV− tumors in brainstem, meninges |
| 767 | 10j | 3 | 0 | 0 | 1 | 2 | 2 | <5% | HSV+ cells in rostral periventricular areas; rostral necrotic tumor is HSV+, CD45+; death attributed to tumors in the brainstem |
| 768 | 28j | 1 | 0 | 1 | 1 | 0 | 1 | 55% | one focus of HSV+ cells in cortex; death is attributed to large tumor burden |
| 769 | 23j | 0 | 0 | 0 | 1 | 0 | 1 | 50% | death attributed to large tumor burden |
| 770 | 20j | 3 | 1 | 1 | 0 | 0 | 1 | 5% | disseminated HSV+ cells with low CPE and CD45+ infiltrates; death was attributed to virus infection, although cannot exclude tumor localization in brainstem as a cause |
| 771 | 31 | 0 | 0 | 0 | 4 | 2 | 2 | <1% | small tumor in striatum with 100% necrosis and HSV+ staining, with peripheral CD45+ infiltrates |
| 772 | 31 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | no tumor or virus |
| 773 | 29j | 0 | 0 | 0 | 1 | 0 | 1 | 60% | cause of death attributed to large tumor burden and tumor-associated hemorrhage |
| 774 | 19j | 0 | 0 | 0 | 0 | 0 | 0 | 10% | cause of death not apparent |
| 775 | 23j | 2 | 0 | 1 | 1 | 2 | 2 | 35% | large tumor that is necrotic, HSV+ with CD45+ infiltrates; HSV+ cells in brain around tumor; death is attributed to the large tumor burden |
| 776 | 27j | 1 | 0 | 0 | 2 | 2 | 2 | 40% | Tumor with 15% necrosis that is HSV+; two HSV+ neurons in cortex; death attributed to large tumor burden |
| 777 | 35j | 0 | 0 | 0 | 2 | 2 | 1 | <5% | tumor in striatum is necrotic, HSV+; death attributed to multiple brainstem tumors |
| 779 | 29j | 0 | 0 | 0 | 0 | 0 | 0 | 8% | cause of death not apparent |
| 780 | 27j | 0 | 0 | 0 | 1 | 2 | 2 | 50% | large tumors in forebrain with margins that are necrotic, HSV+, CD45+; death is attributed to large tumor burden |
| 781 | 17j | 0 | 0 | 0 | 0 | 0 | 0 | 25% | cause of death not apparent |
| 782 | 24j | 0 | 0 | 0 | 1 | 2 | 2 | 30% | focal tumor is 100% necrotic, HSV+, CD45+; cause of death attributed to tumor-associated edema and hemorrhage |
| 783 | 28j | 0 | 0 | 0 | 1 | 2 | 2 | 55% | one small focus of tumor is necrotic, HSV+; death is attributed to multiple large tumors associated with hemorrhage |
| 785 | 22j | 0 | 0 | 0 | 1 | 0 | 1 | 50% | death is attributed to the high tumor burden. |
All analyses were conducted by Dr. M. Oglesbee (Ohio State University College of Veterinary Medicine).
Brain pathology.
Glioma pathology.
Date of scheduled euthanasia or unexpected death from time of rQNestin34.5v.2 injection.
HSV antigen (scored 0–3) (see Materials and Methods for explanation of scores).
Viral cytopathic effect (scored 0–3).
CD45+ cells, suggestive of activated microglia/inflammation (scored 0–3).
Necrosis (scored 0–3).
Estimated percent of brain occupied by glioma.
Scheduled necropsies conducted on day 5 rather than day 4.
Premature death or euthanasia for moribund state.
Figure 3.

Brain Histology and Immunohistochemistry for Mouse 758
(A) H&E-stained section of brain from mouse 758. Arrow points to needle inoculation site. Immediately past the needle tract is a tumor showing extensive necrosis that is attributed to rQNestin34.5v.2 injection. (B) HSV immunohistochemistry (IHC) reveals extensive tumor infection with extension of the infection toward the ventricle (arrow). (C) High-power microphotograph of HSV IHC, showing cells at the ependymal surface and subependymal region that are HSV antigen-positive. (D) CD45 IHC showing several inflammatory cells present along the ependyma. (E) IHC for nestin in reactive astrocytes adjacent to tumor. Arrow points to a mitotic astrocyte. (F) Ependyma showing mild, yet extensive, nestin immunohistochemical staining in the same areas where HSV and CD45 cells are observed.
qPCR Analyses
In mice whose brain tumors were injected with rQNestin34.5v.2, there was rQNestin34.5v.2 DNA in the brain/brain tumor at day 4 with an average of 1.3 × 107 (range, 6.6 × 105 to 4.2 × 107) copies/mg (Figure 4A). This had decreased by day 31 to an average of 9.8 × 105 (range, 1.5 × 105 to 3.1 × 106) copies/mg and even further by day 61 to 8.4 × 104 copies/mg, albeit in the only mouse available for analysis. In CPA-pretreated mice whose brain tumors were injected with rQNestin34.5v.2, there was rQNestin34.5v.2 DNA in the brain/tumor in this group at day 4 with an average of 3.2 × 106 (range, 2.9 × 105 to 7.4 × 106) copies/mg at day 4 that increased to 7.4 × 107 copies/mg at day 31 (Figure 4B). By day 61, the only mouse available for analysis revealed a decrease in viral DNA in brain/tumor to 3.7 × 104 copies/mg.
Figure 4.

Longitudinal Persistence of oHSV Genomes in Brain
(A) qPCR for rQNestin34.5v.2 genomes in brains, 4, 31, and 61 days after injection into brains with tumors. (B) qPCR for rQNestin34.5v.2 genomes in brains, 4, 31, and 61 days after injection into brains with tumors, in mice pretreated with CPA.
qPCR data for mice that were injected with rQNestin34.5v.2 was obtained from bone marrow (n = 11), whole blood (n = 5), brain (n = 6), heart (n = 2), kidney (n = 11), testes/ovaries (n = 0), liver (n = 9), lung (n = 11), spinal cord (n = 2), spleen (n = 2), and lymph nodes (n = 0) at the day 4 or day 5 time point. Additional qPCR was also performed at the day 31 time point for bone marrow (n = 7), whole blood (n = 3), brain (n = 4), heart (n = 7), kidney (n = 7), testes/ovaries (n = 7), liver (n = 7), lung (n = 7), spinal cord (n = 7), spleen (n = 7), and lymph nodes (n = 7). There was rQNestin34.5v.2 DNA detected in several organs at days 4 and 5. At the day 31 time point only mouse 668 liver where 9.3 copies/mg DNA were detected, mouse 672 liver where 10 copies/mg DNA were detected, and mouse 677 where 26 copies/mg agent were measured in liver and 14,000 copies/mg were measured in tumor exuding from the burr hole underneath the scalp. There was also viral DNA measured in spinal cord at the day 31 time point for mice 672 (89 copies/mg) and 674 (10 copies/mg) out of seven analyzed. This prompted us to analyze liver and spinal cord DNA at the day 61 time point, and these were both negative in the remaining two mice left for analyses (Table S4).
For the CPA-pretreated mice whose tumors were injected with rQNestin34.5v.2, qPCR data for rQNestin34.5v.2 were obtained from bone marrow (n = 11), whole blood (n = 5), brain (n = 6), heart (n = 3), kidney (n = 11), testes/ovaries (n = 0), liver (n = 9), lung (n = 11), spinal cord (n = 0), spleen (n = 0), and lymph nodes (n = 0) at the day 4 or day 5 time point. Additional qPCR was also performed at the day 31 time point for bone marrow (n = 4), whole blood (n = 2), brain (n = 2), heart (n = 4), kidney (n = 4), testes/ovaries (n = 4), liver (n = 4), lung (n = 4), spinal cord (n = 4), spleen (n = 4), and lymph nodes (n = 4). There was rQNestin34.5v.2 DNA detected in some of the other organs at day 4 and in some of the mice that did not survive to the day 31 scheduled necropsy. Minimal viral DNA was seen at the day 31 time point for mouse 772 in liver (16 copies/mg), spinal cord (15 copies/mg), and lymph node (14 copies/mg), which became undetectable by day 61 (mouse 784) (Table S5). Of note, in blood there was no evidence of viral genomes after the day 4 or 5 time points.
Blood Analyses
Serum chemistries, hematology, and liver function tests for mice with brain tumors treated with rQNestin3.5v.2 alone were mostly normal, except for the only mouse analyzed at day 61, who appeared to have a very high white blood cell (WBC) and neutrophil counts in serum, of uncertain significance (data not shown).
Body Weight Analyses
Body weights of all animals with brain tumors treated with rQNestin34.5v.2 alone from day 1 until scheduled euthanasia at day 4, 31, or 61 or unscheduled death were recorded. The average change in weight at scheduled sacrifice compared to their maximum achieved body weight was −16.8%, while the average change in weight at unscheduled death compared to maximum achieved body weight was −33.26% (data not shown). This was highly statistically significant (p < 1 × 10−4, two-tailed t test). When comparing the average loss of body weight between all scheduled euthanasia of these mice and those of mice with tumors treated with control PBS or unscheduled deaths between these two groups, the change was not significant, suggesting that weight loss was due to tumor growth rather than to an effect of rQNestin34.5v.2 injection.
Discussion
The aim of this study was to establish a dose of rQNestin34.5v.2 that was non-lethal and nonpathogenic upon intracerebral injection in the brain of athymic mice. Establishing this dose (the no observed adverse effect level [NOAEL] dose) in mice would then justify a starting dose for a first-in-human phase 1 trial in patients with recurrent GBM. We were able to show that (1) a dose of 3 × 103 PFU injected in the brains of athymic mice was non-lethal and showed minimal to no evidence of inflammatory or cytopathic changes in brain during a 60-day study time; (2) this dose was tolerated clinically by mice without laboratory abnormalities; (3) this dose was not associated with systemic biodistribution of viral genomes, and in brain there was evidence of rapid clearance of viral DNA; (4) higher doses were associated with increased toxicity; (5) doses of 3 × 107 PFU injected in athymic mice with human GBM xenografts increased animal survival from tumor-induced death, but there were also instances of virus-induced inflammatory changes (with or without cytopathic effects) in brain presumably from misplaced injections in brain rather than tumor; and (6) these higher doses in the GBM model also led to increases in viral genomes in tumors and occasionally in other organs over time. The sum of these experiments thus led to a successful IND application proposing a starting dose of 106 PFU in humans, based on a conversion factor of 3,750 (3 × 103 PFU × 37,500 = 1.125 × 107 PFU) between mouse and human brain with a further reduction by one log, based on FDA recommendations.39
OVs based on HSV1 are among the most widely studied biotherapies for cancer. Most oHSVs in clinical trials (except for one)40, 41, 42 have been engineered with defective and/or deleted expression of the late gene γ134.5 that encodes for ICP34.5. This gene was initially described to be the major determinant of herpes neurovirulence and encephalitis in mice.14 The likely mechanism for this neurovirulence relates to binding of ICP34.5 to neuronal beclin-1. This is thought to prevent autophagy-mediated major histocompatibility complex (MHC) class II presentation, leading to reduced viral clearance by the adaptive immune system.11,12 However, ICP34.5 possesses additional roles that benefit viral kinetics and replication: (1) it counteracts an infected host cell mechanism that utilizes PKR to phosphorylate the eiF-2α translation factor, shutting off translation;22,25,26 (2) it also counteracts interferon-mediated signaling,24,43,44 including inhibition of STING;45,46 and (3) it is a structural component of the viral capsid, interacting with the infected cell’s cytoskeleton for intracellular trafficking.23 Lack of ICP34.5 attenuates oHSV replication, and thus avenues to circumvent this have been sought out. Our approach has been to re-express one copy of its gene under transcriptional control of tumor-specific promoters,27,47 such as the nestin transcriptional element.32,35 The first version of this engineered oHSV, however, maintained expression of a fusion GFP-ICP6 transcript with a hybrid gene product that would express the carboxyl terminus of ICP6 that has been linked to inhibiting infected cell necroptosis, allowing for increased viral propagation and virulence.36,37,48, 49, 50 After discussion with the FDA, we thus re-engineered the first version of rQNestin34.5 to remove this GFP-ICP6 fusion transcript and this second version (rQNestin34.5v.2) was then exploited for the toxicology and biodistribution studies reported herein. Other approaches to circumvent the ICP34.5 defect have included a second site recombinant in the viral gene Us1115,31 that supplements some of ICP34.5’s functions,26,29 deletion of ICP34.5’s beclin 1 binding domain,28 or expression of ortholog genes with functions that allow for translation without virulence, such as the cytomegalovirus (CMV) gene IRS151, 52, 53 or the human gene GADD34.54
Once rQNestin34.5v.2 was engineered we had to demonstrate that it remained as efficacious as the original rQNestin34.5. In fact, in vitro it reversed eiF-2α phosphorylation just as well as version 1 (v.1), and its cytotoxicity profile against a panel of tumor and normal cells was also very similar (Figures S1 and S2). In vivo, GLP analyses of survival also showed that rQNestin34.5v.2 significantly improved survival of mice with brain tumors (Figure 1). A comparative analysis of rQNestin34.5v.2 versus v.1 also showed superior therapeutic effects of the former (data not shown). Therefore, rQNestin34.5v.2 was at least therapeutically bioequivalent to v.1.
Next, for clinical trial development, we had to determine a starting dose for the phase 1 clinical trial, since an ICP34.5-expressing oHSV had never been inoculated in human brains. We approached this problem by first performing a dose-escalation assay of rQNestin34.5v.2 compared to the parental wild-type F strain with and without CPA in both immunocompetent BALB/c mice and athymic mice via intracerebral, intrathecal, intravenous, and intrahepatic routes (Table S1). These pilot data established doses of rQNestin34.5v.2 that led to lethality by all four routes. Not unexpectedly, immunocompetent mice were more resistant to dose-dependent lethality than were athymic mice. Intrathecal/intravenous/intrahepatic routes of administration were also more resistant to dose-dependent lethality than were intracerebral routes. As expected, CPA increased lethality. These data provided the basis for pre-IND discussions with the FDA about the final toxicology and biodistribution study. It was decided that for a first-in-human study we should replicate as much as possible the proposed phase 1 clinical trial: this would consist of a single injection in brains. Therefore, the GLP study would also consist of an intracerebral route administration in the most susceptible mouse strain, i.e., athymic mice. The selected dosing that led to no lethality in athymic mice brains was 4.2 × 104 PFU for mice without CPA pretreatment and 3 × 103 PFU for mice with CPA pretreatment. In addition, it was also decided to study the biodistribution and toxicology in mice brains with tumors treated with the highest possible dose of the agent available, i.e., 3.5 × 107 PFU.
This independently conducted GLP study established doses of rQNestin34.5v.2 that were not lethal and pathogenic in mice. There was one death in a female mouse (no. 364) 55 days after intracerebral injection (4.2 × 104 PFU) that was clearly attributable to HSV encephalitis. This death led to a selection of the next lowest dose tested (3 × 103 PFU) as the dose considered for the clinical trial. Except for the one premature death, all other mice did well without deaths attributable to HSV encephalitis or evidence of HSV-mediated cytopathic effects. CPA was associated with expected CPA-mediated toxicity but with the absence of neuropathology. Viral genomes did not persist in mouse brains after injection and were essentially cleared by a month after injection. There was minimal if any presence of viral genomes outside the CNS. These results thus validated the relative non-pathogenicity of this low dose of rQNestin34.5v.2 in athymic mouse brains.
In comparison, there was much more neuropathology in the experiments conducted with mice with GBM injected with a dose of 3.5 × 107 PFU. This is a combination of the high dose but also of the relative amplification of progeny viruses. The neuropathology of ICP34.5+ HSV1 in mice is well established. This neuropathogenicity was evident in inflammatory and cytopathic effects in brains. We noted that there were areas of nestin immunopositivity, particularly in tanycytes along the subventricular zone (SVZ). The neuropathologic findings appear to support a mechanism of action where when the agent was injected primarily in tumor, intensive HSV antigen expression (likely replication) and spread with an intense inflammatory response (monocytes and neutrophils) occurred rapidly (mouse nos. 653, 654, 655, 659, and 660). There was some spillage of HSV into surrounding brain and also into CSF spaces and ventricles. Over time, tumors underwent necrosis, but in multiple instances tumors escaped the viral effect, grew, and killed the mouse (most mice in group 6). In some mice, there appeared to be complete regression of tumors from the agent (mouse nos. 662 and 679). In one mouse (no. 684) there was minimal tumor in the brainstem but also HSV burden in brain areas that may have been the reason for animal demise. The data appear to suggest that safe administration requires injection of the agent within the tumor with caution to avoid spillage into CSF spaces.
Viral genomes in these GBM groups were initially higher but also rapidly decreased. This was either due to tumor regression, which would expectedly lead to reduced replication, or inability of oHSV to replicate efficiently in growing tumors. Interestingly, CPA did seem to increase the extent of viral genomes within tumors up to a month later, with mouse 764 expressing 1.8 × 107 and mouse 765 1.3 × 108 copies/mg viral DNA. Since both values were higher than any of the six mice analyzed at day 4, the data suggest that there may be continuous persistence and replication of the agent at day 31. Since this was not visualized with group 6 (oHSV alone) animals, this appears to agree with our published data related to CPA effects on enhancement of OV replication.33,35 Although we did not attempt to determine infectious oHSV yields in this study, we have previously shown the temporal kinetics of infectious oHSV after injection in human brain tumors and the effects of CPA or other pharmacologic modulators on oHSV titers (see, for example, Fulci et al.55 and Otsuki et al.56).
Nestin is an intermediate filament protein and is mostly expressed in neural cells during the early stage of development in mice and downregulated in adulthood.57,58 Our data, however, showed expression of nestin in adult mice in cells surrounding ventricle.59,60 In humans, nestin is expressed in gliomas, particularly in the glioma stem cell compartment.61, 62, 63, 64, 65 In human adults with gliomas, there is extensive literature showing nestin expression in tumor and not brain. Nestin immunopositivity correlates with tumor grade.66 Zhang et al.65 estimated in 69 high-grade gliomas that 58% of tumors had between 30% and 60% of cells staining positive, and 33% of tumors had more than 60% of cells staining positive. Although nestin represents a marker of neural stem cells (NSCs) found in the ependymal/subependymal layer of humans,67,68 these studies have been carried out in children or very young adults. However, there is very little evidence, if any, in the literature of nestin expression in the NSC niche of older human adults, who represent the predominant population that is affected by Glioblastoma (GBM). In fact, there was a study that extensively studied the SVZ of adult humans harvested during surgery.69 Although several markers (e.g., glial fibrillary acidic protein [GFAP], tubulin beta-3 chain [TuJI]) were utilized to detect potential NSCs, interestingly there was no use or mention of nestin. Very recently, a group has published a study with four human adult brains.70 They report nestin-immunopositive neurons in a few brain regions of the basal forebrain and midline, but not in white matter, where gliomas arise. Independently, a group has published a study reviewing nestin expression in adult brains afflicted by gliomas versus other CNS disease.66 They report no nestin immunopositivity in brain.66 Our mouse data, however, were still concerning for possible nestin expression in the brains of adult humans. Independently, we sought to identify whether there were nestin-positive cells in a brain surrounding a resected GBM and in a brain with resected GBMs that had undergone surgery, radiation, and chemotherapy. In neither case did we find nestin-immunopositive cells (unpublished data). This suggests that mouse toxicity and lethality from rQNestin34.5 may arise from ICP34.5 expression in nestin-positive cells along the ventricles, but in human adults this toxicity is less likely due to absence of nestin-expressing cells. In support of this, the current ongoing phase 1 clinical trial in humans with recurrent GBM treated with rQNestin34.5v.2 (ClinicalTrials.gov: NCT03152318) has not shown evidence of viral-mediated toxicity or encephalitis (unpublished data). This indicates that athymic mice likely overestimated the toxicity of intracerebral rQNestin34.5v.2.
CPA has been used extensively by us and others to improve oncolytic virus replication and survival in injected tumors both preclinically and clinically.33, 34, 35,67,71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89 CPA may function by limiting initial innate host responses against the oncolytic virus (consisting of microglia, macrophages, and natural killer [NK] cells33, 34, 35,55,90) and by reducing the T regulatory cell population.72,80 In the GBM tumor model, CPA led to increased persistence of viral genomes, but it did not increase efficacy or toxicity. This is likely due to the high dose of rQNestin34.5v.2 used in the experiment, since we previously published that the CPA effect on oHSV efficacy was observed only with a low and not high dose of oHSV.35 However, in the ongoing clinical trial, an arm with pre-administration of CPA before rQNestin34.5v.2 administration is planned, since several of the cohorts would entail viral doses that have been shown to benefit from CPA pre-administration.
HSV1 is endemic in humans, with a majority exhibiting pre-existent HSV1 antibodies, which could be reasonably expected to modulate the effect of the virotherapy, but in the reported phase 3 clinical trial of the oHSV, Imlygic, for melanoma the results did not show a significant difference in responses as a function of pre-existent antibodies.17
In conclusion, the extensive preclinical data presented were submitted as part of a new IND application to the FDA in 2015 and again in 2016, leading to enrollment of the first human subject with a recurrent GBM treated with an intratumoral dose of 106 PFU in September 2017.
Materials and Methods
GLP Animal Studies
GLP animal studies were performed by a contract research organization (Battelle, West Jefferson, OH, USA). These studies were approved by Institutional Animal Care and Use Committee (IACUC) of Battelle and performed in accordance with the guidelines and regulations of Battelle. Two hundred fifty nu/nu mice (athymic) were initially assigned to one of seven dose groups as presented in Table 5. Groups 1–4, considered as the toxicity assessment group, were comprised of 36 mice (18 per sex per group) with a target of 6 per sex assigned to each scheduled necropsy. Groups 5–7, considered as the efficacy group, were comprised of all female mice. Due to expected mortality in the tumor-bearing groups, 41 and 35 mice were assigned to groups 6 and 7, respectively, in order to allow for a target of 10 mice per group at the day 61 scheduled necropsy. Pre-GLP animal studies were approved by IACUC of the Ohio State University (OSU) and performed in accordance with the guidelines and regulations of OSU.
Endpoints used to evaluate the potential toxicity of rQNestin34.5v.2 were mortality, clinical observations, body weights, food consumption, clinical pathology, gross necropsy (including organ weights), histopathology (selected tissues), and IHC (of the brains of selected mice). From selected mice, biodistribution of rQNestin34.5v.2 was evaluated in blood, brain (with tumor), spinal cord, bone marrow, kidney, lungs, gonads, heart, liver, spleen, and lymph node by qPCR. A staggered start was used in order to accommodate the surgery schedule for all of the mice.
Viruses, Cells, and Media
Preclinical lots of rqNestin34.5v.2 were prepared in a GLP manner. The oHSV was manufactured by Meridian Life Sciences (Memphis, TN, USA). The lot used on this study (02190074) was manufactured on March 2, 2013 and supplied by PeriphaGen (Pittsburgh, PA). Eleven vials containing 110 μL per vial with a reported titer of 1.2 × 107 PFU/μL were received by Battelle on July 30, 2013 and stored in a freezer unit set to maintain −85°C to −60°C. While in use, the test article was stored on wet ice. Stability of rQNestin34.5v.2 was evaluated at −65°C or below for up to 12 months. Stability of a similar viral construct was evaluated at 2°C–8°C for up to 8 h. Certificates of analyses are available (data not shown). Human U87ΔEGFR glioma cells were sent from the Brigham and Women’s Hospital and to Battelle and received on July 19, 2013 at a concentration of 1 × 107 cells/mL. Cells were prepared in Bambanker cryopreservation medium (Lymphotec). Human U87ΔEGFR glioma cells were stored at the Battelle Biomedical Research Center (BRC) test site (West Jefferson, OH, USA). CPA was received from Crosby Drugs on June 6, 2013. Lot no. 2I778A (Baxter) was stored at room temperature with an expiration date of September 2015. CPA was received as a dry powder. Dulbecco’s phosphate-buffered saline (DPBS), lot RNBC5905 (Sigma), was the vehicle for rQNestin34.5v.2. DPBS was stored per instructions on the bottle (15°C–30°C), not per protocol (2°C–8°C), and used prior to the expiration date of October, 2014. Hanks’ balanced salt solution (HBSS), lot no. 1300251 (Gibco brand, Life Technologies), was the vehicle for the human U87ΔEGFR glioma cells. HBSS was stored at 15°C–30°C and used prior to the expiration date of March 30, 2016. 0.9% sodium chloride for injection (sterile saline), lot no. C899575 (Baxter), was the vehicle for the CPA and was used prior to the September 2014 expiration date.
Preparation of Cells, oHSV, Media, and CPA
A stock of rQNestin34.5v.2 was provided at 1.2 × 107 PFU/μL. Two secondary stocks were prepared at 1.2 × 105 PFU/5 μL (dilution A) and 1.2 × 104 PFU/5 μL (dilution B) by diluting the stock with DPBS 1:100 and 1:10, respectively. Dilution A was further diluted 14:186 with DPBS to achieve the 4.2 × 104 PFU/5 μL concentration used for the 4.2 × 104 PFU virus group mice. Dilution B was further diluted 10:190 with DPBS to achieve the 3.5 × 103 PFU/5 μL concentration used for the 3 × 103 PFU virus group mice. The stock concentration of rQNestin34.5v.2 (1.2 × 107 PFU/μL) was also diluted 233:167 with DPBS to achieve the 3.5 × 107 PFU/5 μL concentration used for the tumor + virus group mice and the tumor + virus + CPA group mice. rQNestin34.5v.2 was prepared within a laminar flow hood once for each day of use. The number of vials needed were thawed on ice prior to dilution. Dilutions were maintained on wet ice and used (injected) within 8 h of the initial thawing of the stock. Human U87ΔEGFR glioma cells were seeded, maintained, and expanded as needed. The culturing of the human U87ΔEGFR glioma cells was initiated by seeding approximately 1 × 105 cells per flask containing growth medium (Dulbecco’s modified Eagle’s medium [DMEM] with 10% heat-inactivated fetal bovine serum [FBS], 100 U/mL penicillin, and 100 μg/mL streptomycin). Cells were then re-fed the following day by completely exchanging the growth medium. Once the cells had reached approximately 65%–80% confluence, they were washed with PBS and removed from the flask surface using 0.05% trypsin/ethylenediaminetetraacetic acid (EDTA) followed by the addition of passaging medium (DMEM with 2% FBS) to inactivate the trypsin. An aliquot of the cell suspension was then counted using a hemocytometer and trypan blue to stain non-viable cells. The human U87ΔEGFR glioma cells were then passaged by seeding an appropriate volume of the cell suspension into flasks containing growth medium. In order to prepare the cells for injection, three to eight flasks were harvested using trypsin/EDTA followed by passaging medium and then counted as described above. Afterward, the cells were pelleted by centrifugation (5 min, 500 RCF [relative centrifugal force], 20°C) and resuspended in HBSS at a target of 4 × 104 cells per μL, such that each mouse would receive a target of 2 × 105 cells in 5 μL. Once prepared for injection, the human U87ΔEGFR glioma cells were stored in a refrigerator unit set to maintain 2°C–8°C or on wet ice for up to 3 h 46 min prior to injection. For 12 mice (no. 561 [tumor group], nos. 654, 662, 676, 680, 684, 687, and 688 [tumor + virus group], and nos. 753, 766, 768, and 769 [tumor + virus + CPA group]), the human U87ΔEGFR glioma cells were injected up 46 min past the 3-h time point. CPA was prepared within a laminar flow hood once for the study by adding 50 mL of 0.9% sodium chloride for injection (sterile saline) to the contents of one bottle of CPA, yielding a 20 mg/mL concentration. The reconstituted CPA was stored at 2°C–8°C.
Mice
Ninety male and 286 female nu/nu mice (athymic) Crl:NU-Foxn1nu strain code 088 (homozygous) were received at Battelle on July 23, 2013 from Charles River Laboratories (Wilmington, MA, USA). Mice were 5 weeks of age at receipt, and males weighed 20.9–29.5 g on day 1 and females weighed 18.1–25.9 g on day 1. All mice were quarantined per testing facility standard operating procedure (SOP). The housing and animal care practices met current Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC) standards and current requirements stated in the Guide for the Care and Use of Laboratory Animals (National Research Council [NRC], 2011). Mice were housed in a positive pressure high-efficiency particulate air (HEPA)-filtered containment area (bioBUBBLE, Fort Collins, CO, USA). Mice were individually housed in plastic cages with filter-media lids and irradiated corn cob bedding. All animal housing and environmental conditions followed testing facility SOP. The environmental conditions of the animal study room conformed to testing facility SOP. The light/dark cycle was controlled by electronic timers set to maintain 12 h of light and 12 h of dark each day using fluorescent lighting set to start at 6:00 am each day. The room temperature and relative humidity controls were set to maintain ranges of 68°F–79°F and 30%–70%, respectively. Room temperature and humidity were monitored for conformance per testing facility SOP. Additionally, the temperature and humidity within the bioBUBBLE were monitored and recorded at least twice daily using a calibrated thermometer and calibrated hygrometer, respectively. Fresh air was set to provide a minimum of 10 changes of room air per hour. Cages and feeders were changed and sanitized at least weekly according to testing facility SOP. All mice had ad libitum access to irradiated NTP-2000 wafer feed (Zeigler Brothers, Gardners, PA, USA) ad libitum according to testing facility SOP. Water was provided by Hydropacs. Each animal received, at a minimum, one Hydropac per week, which was changed out at a minimum of weekly intervals. Documentation of the preparation of the Hydropacs, lot/batch numbers, and results of analyses was provided by the supplier. The city water conformed to the current US Environmental Protection Agency (EPA) drinking water standards. There were no known or reported contaminants in either the water or feed that would have any impact on study results or interpretations. Analytical reports of water analysis and each lot of certified feed used are maintained under the direction of Battelle.
Mouse Group Assignment and Randomization
Prior to group assignment, mice were identified by cage card. Mice were assigned to dose groups by body weight during week −2 (control, CPA, 4.2 × 104 PFU virus, and 3 × 103 PFU virus + CPA groups) and week −1 (tumor, tumor + virus, and tumor + virus + CPA groups) following injection of human U87ΔEGFR glioma cells and prior to CPA injection using a computer program (Provantis, Instem), which ensured similar group mean body weights. This assigned number (unique within the study) was considered the animal ID number, and all electronic data were recorded by this ID (Tables 6 and 7). Following group assignment, a second, unique number was given to each mouse that was considered the treatment ID number. Mice were identified by tail mark (dorsal side of tail) with the treatment ID number and a temporary cage card. The temporary cage card contained the study number and treatment ID number, at a minimum, but did not contain information related to group assignment. A cross-reference containing the animal ID number and the treatment ID number list was maintained. The treatment ID was used to blind staff performing stereotaxic injections to the assigned groups. Upon completion of the dose administration period, the animal ID number allocated at group assignment was added to the ventral side of the tail via tail mark and a new cage card was generated. Each new cage card contained information including study number, group assignment, and animal ID number.
Table 6.
Target Animal Identification Numbers (see Materials and Methods)
| Group | Description | Identification Numbers |
|
|---|---|---|---|
| Males | Females | ||
| 1 | control | 101–118 | 151–168 |
| 2 | CPA | 201–218 | 251–268 |
| 3 | 4.2 × 104 PFU virus | 301–318 | 351–368 |
| 4 | 3 × 103 PFU virus + CPA | 401–418 | 451–468 |
| 5 | tumor | – | 551–580 |
| 6 | tumor + virus | – | 651–69a |
| 7 | tumor + virus + CPA | – | 751–785a |
Due to exhaustion of formulated CPA, one animal designated for group 7 was reassigned to group 6.
Table 7.
Target Animal Identification Numbers per Scheduled Necropsy Day
| Group | Color Code | Day 4 |
Day 31 |
Day 61 |
|||
|---|---|---|---|---|---|---|---|
| Male | Female | Male | Female | Male | Female | ||
| 1 | white | 101–106 | 151–156 | 107–112 | 157–162 | 113–118 | 163–168 |
| 2 | lilac | 201–206 | 251–256 | 207–212 | 257–262 | 213–218 | 263–268 |
| 3 | blue | 301–306 | 351–356 | 307–312 | 357–362 | 313–318 | 363–368 |
| 4 | green | 401–406 | 451–456 | 407–412 | 457–462 | 413–418 | 463–468 |
| 5 | yellow | – | 551–560 | – | 561–570 | – | 571–580 |
| 6 | orange | – | 651–660 | – | 661–670 | – | 671–680 |
| 7 | red | – | 751–760 | – | 761–770 | – | 771–780 |
In order to maintain a sufficient number of animals available for each day of necropsy, animals were reassigned to another day of necropsy, as needed.
Surgical Procedure
On day −7, mice were anesthetized with ketamine (100 mg/kg), xylazine (10 mg/kg), and acepromazine (3 mg/kg). During the surgery, the animals’ vital signs and level of anesthesia were monitored by observing respiration, color, and toe pinch reflexes. Once anesthetized, each animal’s head was secured to the stereotactic apparatus. Then, the skin was cleaned by wiping with a surgical scrub cotton swab followed by an alcohol-soaked cotton swab. A 1-cm linear incision was made in the skull midline using a disposable sterile scalpel (no. 10). After the bregma was identified, a small burr hole was drilled into the skull (using a sterile drill bit) at stereotactic coordinates: 1 mm frontal from bregma/2 mm right lateral from bregma (for intraparenchymal administration). A sterile Hamilton syringe with a 27-gauge needle was loaded with 5 μL of tumor cell suspension (day −7, groups 5, 6, and 7), 5 μL of the appropriate concentration of rQNestin34.5v.2 solution (day 1, groups 3, 4, 6, and 7), or 5 μL of DPBS (day 1, groups 1, 2, and 5) and secured to the stereotaxic frame. The needle was slowly lowered into the burr hole to puncture the dura and then introduced to a final depth of 3 mm into the brain. The cells, virus suspension, or DPBS was slowly injected during a period of 5–6 min, and the needle was slowly retracted during an additional 2–6 min. Following the injection and retraction of the needle, the burr hole was covered with sterile bone wax, and the skin incision was closed with 4.0 nylon suture and wiped with a clean cotton swab soaked in surgical scrub solution. The stereotaxic apparatus was wiped with alcohol between surgeries, and the Hamilton syringes were cleaned internally with three cycles of PBS. Post-operative analgesics were buprenorphine (0.1 mg/kg, administered via subcutaneous [s.c.] injection once immediately after regaining consciousness) and meloxicam (0.2 mg/kg, once daily for 3 days following surgery).
CPA Administration
On day −2, mice in the CPA-designated groups received an intraperitoneal injection of CPA at a volume of 15 mL/kg delivering a dose of 300 mg/kg CPA. Animal 786 (tumor + virus + CPA group) was reassigned as animal 691 (tumor + virus group) because there was insufficient CPA to dose all assigned animals.
Clinical Observations, Body Weights, Food Consumption, and Serum Collection
Observations for morbidity/moribund status and mortality were performed on all mice in the study room according to testing facility SOP. A detailed clinical observation was also performed on potential study mice at the time of body weights for group assignment, on mice receiving a stereotaxic injection of glioma cells on day −7 (prior to injection), on mice receiving an intraperitoneal injection of CPA or saline on day −2 (prior to injection), on mice receiving a stereotaxic injection of rQNestin34.5v.2 or vehicle on day 1 (prior to injection), and at a target of weekly intervals throughout the study on surviving study animals. Beginning on day −2 for mice in the control, CPA, 4.2 × 104 PFU virus, and 3 × 103 PFU virus + CPA non-tumor bearing groups and on day −7 for mice in the tumor groups, cage-side clinical observations were performed once daily (with the exception of days with scheduled detailed clinical observations) throughout the in-life period. Additional observations were recorded as needed. On the days of scheduled necropsy, a detailed clinical observation was performed on mice scheduled for necropsy.
Body weights were recorded from animals pre-study (week −2) for group assignment. Individual animal body weights were recorded on days −7, −2, and 1 and a target of weekly thereafter (to coincide with detailed clinical observations) for surviving animals. Fasted body weights were recorded for mice selected for necropsy on the day of each animal’s scheduled necropsy.
Feed consumed was quantitatively assessed during the study on a weekly basis to coincide with weekly body weights. Feed consumed was not performed for several early-death animals.
On each day of scheduled necropsy, blood was collected from all surviving mice scheduled for necropsy. Specimens were analyzed at the Battelle King Avenue Test Site and not at the testing facility, as specified in the protocol; however, this had no impact on the study, as the specimens were analyzed on appropriate equipment by trained staff. Mice were anesthetized with CO2/O2, and whole-blood specimens were collected via cardiac puncture. Blood was utilized for qPCR evaluation or for analysis of hematology and/or clinical chemistry parameters following a randomized selection, as follows:
From a target of half the mice scheduled for necropsy (five mice per group in the tumor-bearing groups and a target of three mice per sex per group for the toxicity groups), whole-blood specimens were collected into tubes containing sodium citrate as the anticoagulant. The whole-blood/citrate tubes were placed immediately on dry ice prior to storage in a freezer unit set to maintain at −85°C to −60°C until shipment for qPCR evaluation.
From the remaining mice scheduled for necropsy (a target of five mice per group in tumor-bearing groups and a target of three mice per sex per group for the toxicity groups), whole-blood specimens (as much as possible) were collected for the evaluation of hematology and serum chemistry parameters. Blood specimens for serum chemistry parameters measurement were placed into tubes containing serum separator gel. Blood specimens for hematology parameters measurement were placed into tubes with tripotassium ethylenediaminetetraacetate (K3 EDTA).
Blood was not collected from mice found dead.
Serum Chemistry, and Hematology
At a minimum, serum chemistry parameters indicated with an asterisk were evaluated: alanine aminotransferase,∗ albumin/globulin ratio,∗ albumin,∗ alkaline phosphatase,∗ aspartate aminotransferase,∗ cholesterol, creatinine, chloride,∗ potassium, triglycerides,∗ sodium,∗ γ-glutamyl transferase, globulin, glucose,∗ protein, total,∗ urea nitrogen. Hematological parameters measured are listed as follows: cell morphology, erythrocyte count, hematocrit, hemoglobin, leukocyte count, total, leukocyte differential, absolute, mean corpuscular hemoglobin, mean corpuscular hemoglobin concentration, mean corpuscular volume, nucleated red blood cell count (nRBC), platelet count, reticulocyte count (%).
qPCR
DNA was extracted from collected tissues and blood using a QIAGEN DNeasy tissue kit according to the vendor’s suggested protocol. The yield and quality of DNA from each sample were determined by spectrophotometry. qPCR was performed on each specimen in duplicate using a primer and probe set targeting the HSV1 gene ICP22. The qPCR method was partially qualified. Each qPCR test included a standard curve consisting of 105, 104, 103, 102, 0.5 × 102, 10, and 0.5 copies per reaction of pSG25 reference plasmid, buffer control (BC), and a non-template control. ICP22 copy number in each tissue was assessed by comparison to the standard curve. Tissues were processed according to SOP BESTC BIO IV-312 and BESTC BIO V-061, and qPCR for rQNestin34.5v.2 vector (ICP22 target) was completed according to BESTC BIO IV-313. The limit of detection and quantitation for the assay was five copies of ICP22 per 5 μL of purified DNA. Final reportable values were copies/μg DNA. Each tissue extraction batch included a process or BC to monitor cross-contamination. For all tissues except the brain, a fraction of the tissue (unless the entire tissue was ≤25 mg [≤10 mg for spleen], in which case the entire tissue was used) was processed for qPCR analysis. For brains, the viral load in the entire brain was wanted, which meant processing the entire brain and removing a portion for qPCR analysis. qPCR was performed on selected tissues from early-death animals in the toxicity groups and on spinal cords from early-death animals in the tumor-bearing groups for which cause of death was undetermined or possibly due to rQNestin34.5v.2 based on the IHC evaluation. Finally, qPCR data from all tissues were obtained for mouse nos. 668 (tumor + virus group), 688 (tumor + virus group), and 778 (tumor + virus + CPA group).
Necropsies and Tissue Processing
Complete necropsies were performed on all study mice that died or were terminated at an unscheduled time. Moribund mice were humanely euthanized per testing facility SOP. An attempt was made to collect blood specimens for hematology and serum chemistry parameters assessment or for qPCR. Tissues, when present, were collected as listed for scheduled necropsies (Table 8). The brains (whole with tumor intact) from a target of one-half of early-death mice in the control, CPA, and tumor-only groups were processed for shipment for IHC evaluation, and the other half were processed for qPCR. Remaining tissues were processed as for scheduled necropsies. Blood specimens were processed with target of one-half of the mice in each group designated for hematology and serum chemistry analysis and a target of one-half of the mice in each group designated for qPCR. For scheduled necropsies, a target of 12 mice (six per sex per group) in the toxicity groups and a target of 10 mice per group in the tumor-bearing groups were scheduled for necropsy on days 4 (except for mouse nos. 656–660 (tumor + virus group) and 756–758 (tumor + virus + CPA group) that were necropsied on day 5), 31, and 61. Animals were fasted overnight, weighed, and humanely terminated per testing facility SOP in conjunction with blood collection for the assessment of clinical pathology parameters. All scheduled necropsies proceeded with a board-certified veterinary pathologist available for consultation. Each necropsy included examination of the external surface of the body and all orifices; the cranial, thoracic, abdominal, and pelvic cavities and their contents; and collection of tissues. Listed tissues, when present at scheduled necropsy or moribund termination, were collected and weighed from all animals. Paired organs were weighed together. For each animal, instruments were cleaned in diluted bleach and rinsed in 10% ethanol prior to the collection of each tissue.
Table 8.
List of Tissues for Necropsy
| Tissue | Processing at Necropsy |
|---|---|
| Tumor with brain | whole brain with tumor intact—tumor encompassed injection site; from target half of mice (random selection), whole brains (with tumor) flash-frozen in liquid nitrogen for qPCR; from target half of mice (random selection), whole brains (with tumor) placed into 10% NBF |
| Lymph nodes (LNs) | collected mandibular/cervical LNs as a single entity (as many as possible); left flash-frozen in liquid nitrogen for qPCR; right side placed into 10% NBF and stored for possible processing and histopathology |
| Spinal cord | cervical and thoracolumbar—(no bone in section) for qPCR; cervical and thoracolumbar—fix in situ, decalcified later for histopathology evaluation |
| Bone marrow (femur) | left femur—bone marrow flushed with phosphate-buffered saline and flash-frozen in liquid nitrogen for qPCR; right femur placed into 10% NBF and stored for processing and histopathology |
| Kidney, lungs, ovaries/testes | left side flash frozen in liquid nitrogen for qPCR; right side placed into 10% NBF and stored for processing and histopathology, with the exception of testes, which were preserved in modified Davidson’s fixative and subsequently transferred to 10% NBF according to testing facility SOP |
| Heart, liver, spleen | sample (~5 mm3) flash-frozen in liquid nitrogen for qPCR; remainder of tissue placed into 10% NBF and stored for processing and histopathology |
| Animal identification | collected but not processed for evaluation |
| Gross lesions (i.e., skin ulceration) | collected and processed as heart, liver, spleen, and retained at testing facility for possible qPCR and histopathology evaluation |
NBF, neutral buffered formalin.
Specimens collected at necropsy for qPCR analysis were flash frozen in liquid nitrogen (with the exception of blood) and stored in a freezer unit set to maintain −85°C to −60°C.
Tissue Histopathology and IHC
Tissues were received by the Comparative Pathology and Mouse Phenotyping Shared Resource (CPMPSR) in the Department of Veterinary Biosciences, Ohio State University. Four standard coronal sections of brain were made, representing telencephalon, diencephalon, mesencephalon, and metencephalon, and placed in cassettes for dehydration and paraffin embedding by the Histology Laboratory of the CPMPSR. Five- to 7-μm sections were placed on glass slides for routine H&E staining, and immunohistochemical staining for HSV antigen (Dako, Agilent Technologies, Santa Clara, CA, USA), CD45 (Abcam, Cambridge, MA, USA), and nestin (Acris, Origene Technologies, Rockville, MD, USA). Slides were mounted with glass coverslips and evaluated. Tissues([bone marrow, ovaries/testes, heart, liver, lungs, lymph nodes (mandibular/cervical), kidney, spinal cord (cervical and thoracolumbar), and spleen from animals 215, 216, 218, 264, and 265 [CPA group] and 413, 418, and 467 [3 × 103 PFU virus + CPA group]) were processed to slides and stained with H&E. Tissues from early-death mice in the 4.2 × 104 PFU virus group were not examined, as the proposed clinical starting dose is the human bio-equivalent of the 3 × 103 PFU virus for mice and not the 4.2 × 104 PFU virus. Slides were examined microscopically by Dr. M. Oglesbee, a board-certified veterinary pathologist.
Statistical Analyses
Statistical comparisons were made as per groups listed in Table 9. All appropriate quantitative in-life data collected at Battelle using the Provantis system were analyzed for test article effects by parametric or nonparametric approaches. For all data, normality was determined by the Shapiro-Wilk test, and homogeneity of variances was tested by Levene’s test. Data were log-transformed to achieve normality when assumptions were not met. For parametric data determined to be normally distributed and homogeneous among groups, an analysis of variance (ANOVA) model was used to test for differences among the group means. Ad hoc pairwise comparisons were adjusted using Dunnett’s test.. For nonparametric data that were not normally distributed and/or nonhomogeneous, a Kruskal-Wallis test was used to determine whether there were differences among the group means. Ad hoc pairwise comparisons were done by Wilcoxon tests and adjusted for multiplicity by the Bonferroni-Holm method. All statistical tests were performed at the 0.05 level of significance (p < 0.05), after accounting for multiple comparisons where indicated. All appropriate quantitative clinical pathology and post-mortem data collected at Battelle were analyzed statistically when n ≥ 3. All data were analyzed for test article effects by analysis of variance. For homogeneous data, as determined by Bartlett’s test for homogeneity at the 0.05 level, tests for differences between the control and comparison groups were made using Dunnett’s test. For nonhomogeneous data, as determined by Bartlett’s test for homogeneity at the 0.05 level, tests for pairwise differences between the control and each of the comparison groups were made using Cochran and Cox’s modified two-sample t test. Statistical significance for each comparison was reported at the 0.05 level.
Table 9.
Groups Analyzed Statistically
| Group | Compared to Which Group |
|---|---|
| Control | – |
| CPA | control |
| 4.2 × 104 PFU virus | control |
| 3 × 103 PFU virus + CPA | CPA |
| Tumor | – |
| Tumor + virus | tumor |
| Tumor + virus + CPA | tumor and tumor + virus |
The random selection of mice for clinical pathology-qPCR versus IHH/histopathology-qPCR assignment was accomplished through the use of a computer-generated list of random numbers produced by Microsoft Excel. Briefly, the random number generation capability was selected with the following options: number of variables = 1; number of random numbers = the total number of entries (N); distribution = uniform; parameters = “0” and “1.” The animal IDs were entered into a column (A) in numerical order. The random numbers generated by Excel were placed into a second column (B). The columns were sorted by the random number list in a lowest-to-highest order; the spreadsheet retained the original entries, and the sorted values were placed into columns D and E. The final sample processing assignment was designated in another column (F). This procedure was repeated to define a separate brain processing assignment. As such, the selection for blood and brain dispositions was completed independently. By following this process, the animals assigned for blood qPCR were not necessarily the same animals assigned for brain qPCR. This process was continued for all groups and all termination days (4, 31, and 61, and early termination of group 5) until protocol amendment 2 was effective and defined the exception that the brains of all early-death mice in virus groups (with or without injections of tumor cells) would go only for IHC/histopathology at Ohio State University (all other mice were subjected to the above-defined randomization process).
Author Contributions
E.A.C., H.N., and K.K. were responsible for the design, analyses, interpretation, and preparation of the manuscript. H.N. and K.K. were responsible for the conduct of experiments. S.A.F. was responsible for the biostatistical analyses. M.O. was responsible for the neuropathological analyses and preparation of the manuscript.
Conflicts of Interest
E.A.C. and H.N. are named inventors on patents related to rQNestin34.5v.2. E.A.C. is a consultant to Advantagene, Inc., who has an option to license rQNestin34.5v.2. E.A.C. is currently an advisor to Alcyone Biosciences, Insightec, Inc., Voyager Therapeutics, Sangamo Therapeutics, Seneca Therapeutics, Immunomic Therapeutics, and DNAtrix, Inc., and has equity interests in Immunomic Therapeutics, Seneca Therapeutics, and DNAtrix, Inc.; he has also advised Oncorus, Merck, Tocagen, Ziopharm, Stemgen, NanoTx, Ziopharm Oncology, Cerebral Therapeutics, Genenta, Janssen, Karcinolysis, Shanaghai Biotech, and Sigilon Therapeutics. He has received research support from NIH, US Department of Defense, American Brain Tumor Association, National Brain Tumor Society, Alliance for Cancer Gene Therapy, Neurosurgical Research Education Foundation, Advantagene, NewLink Genetics, and Amgen. The remaining authors declare no competing interests.
Acknowledgments
This work was supported by NIH grants U01NS061811, P01CA163205, R01NS110942, and P01CA236749 and by a grant from the Alliance for Cancer Gene Therapy (Greenwich, CT) (to E.A.C.). The technical expertise of Fang Liu, Samantha Kerr, and Jennifer Dumman is acknowledged.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.03.028.
Supplemental Information
References
- 1.Ostrom Q.T., Gittleman H., Kruchko C., Louis D.N., Brat D.J., Gilbert M.R., Petkov V.I., Barnholtz-Sloan J.S. Completeness of required site-specific factors for brain and CNS tumors in the Surveillance, Epidemiology and End Results (SEER) 18 database (2004–2012, varying) J. Neurooncol. 2016;130:31–42. doi: 10.1007/s11060-016-2217-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang A.S., Ostrom Q.T., Kruchko C., Rogers L., Peereboom D.M., Barnholtz-Sloan J.S. Complete prevalence of malignant primary brain tumors registry data in the United States compared with other common cancers, 2010. Neuro-oncol. 2017;19:726–735. doi: 10.1093/neuonc/now252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hegi M.E., Diserens A.C., Gorlia T., Hamou M.F., de Tribolet N., Weller M., Kros J.M., Hainfellner J.A., Mason W., Mariani L. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 4.Stupp R., Mason W.P., van den Bent M.J., Weller M., Fisher B., Taphoorn M.J., Belanger K., Brandes A.A., Marosi C., Bogdahn U., European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups. National Cancer Institute of Canada Clinical Trials Group Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 5.Wen P.Y., Reardon D.A. Neuro-oncology in 2015: progress in glioma diagnosis, classification and treatment. Nat. Rev. Neurol. 2016;12:69–70. doi: 10.1038/nrneurol.2015.242. [DOI] [PubMed] [Google Scholar]
- 6.Chiocca E.A., Nassiri F., Wang J., Peruzzi P., Zadeh G. Viral and other therapies for recurrent glioblastoma: is a 24-month durable response unusual? Neuro-oncol. 2019;21:14–25. doi: 10.1093/neuonc/noy170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iorgulescu J.B., Reardon D.A., Chiocca E.A., Wu C.J. Immunotherapy for glioblastoma: going viral. Nat. Med. 2018;24:1094–1096. doi: 10.1038/s41591-018-0142-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lawler S.E., Speranza M.C., Cho C.F., Chiocca E.A. Oncolytic viruses in cancer treatment: a review. JAMA Oncol. 2017;3:841–849. doi: 10.1001/jamaoncol.2016.2064. [DOI] [PubMed] [Google Scholar]
- 9.Peruzzi P., Chiocca E.A. Viruses in cancer therapy—from benchwarmers to quarterbacks. Nat. Rev. Clin. Oncol. 2018;15:657–658. doi: 10.1038/s41571-018-0077-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poh A. First oncolytic viral therapy for melanoma. Cancer Discov. 2016;6:6. doi: 10.1158/2159-8290.CD-NB2015-158. [DOI] [PubMed] [Google Scholar]
- 11.Leib D.A., Alexander D.E., Cox D., Yin J., Ferguson T.A. Interaction of ICP34.5 with Beclin 1 modulates herpes simplex virus type 1 pathogenesis through control of CD4+ T-cell responses. J. Virol. 2009;83:12164–12171. doi: 10.1128/JVI.01676-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Orvedahl A., Alexander D., Tallóczy Z., Sun Q., Wei Y., Zhang W., Burns D., Leib D.A., Levine B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1:23–35. doi: 10.1016/j.chom.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 13.Andreansky S., Soroceanu L., Flotte E.R., Chou J., Markert J.M., Gillespie G.Y., Roizman B., Whitley R.J. Evaluation of genetically engineered herpes simplex viruses as oncolytic agents for human malignant brain tumors. Cancer Res. 1997;57:1502–1509. [PubMed] [Google Scholar]
- 14.Chou J., Kern E.R., Whitley R.J., Roizman B. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science. 1990;250:1262–1266. doi: 10.1126/science.2173860. [DOI] [PubMed] [Google Scholar]
- 15.Todo T., Martuza R.L., Rabkin S.D., Johnson P.A. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc. Natl. Acad. Sci. USA. 2001;98:6396–6401. doi: 10.1073/pnas.101136398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thomas S., Kuncheria L., Roulstone V., Kyula J.N., Mansfield D., Bommareddy P.K., Smith H., Kaufman H.L., Harrington K.J., Coffin R.S. Development of a new fusion-enhanced oncolytic immunotherapy platform based on herpes simplex virus type 1. J. Immunother. Cancer. 2019;7:214. doi: 10.1186/s40425-019-0682-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andtbacka R.H., Kaufman H.L., Collichio F., Amatruda T., Senzer N., Chesney J., Delman K.A., Spitler L.E., Puzanov I., Agarwala S.S. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015;33:2780–2788. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 18.Markert J.M., Medlock M.D., Rabkin S.D., Gillespie G.Y., Todo T., Hunter W.D., Palmer C.A., Feigenbaum F., Tornatore C., Tufaro F., Martuza R.L. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000;7:867–874. doi: 10.1038/sj.gt.3301205. [DOI] [PubMed] [Google Scholar]
- 19.Patel D.M., Foreman P.M., Nabors L.B., Riley K.O., Gillespie G.Y., Markert J.M. Design of a phase I clinical trial to evaluate M032, a genetically engineered HSV-1 expressing IL-12, in patients with recurrent/progressive glioblastoma multiforme, anaplastic astrocytoma, or gliosarcoma. Hum. Gene Ther. Clin. Dev. 2016;27:69–78. doi: 10.1089/humc.2016.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Papanastassiou V., Rampling R., Fraser M., Petty R., Hadley D., Nicoll J., Harland J., Mabbs R., Brown M. The potential for efficacy of the modified (ICP 34.5−) herpes simplex virus HSV1716 following intratumoural injection into human malignant glioma: a proof of principle study. Gene Ther. 2002;9:398–406. doi: 10.1038/sj.gt.3301664. [DOI] [PubMed] [Google Scholar]
- 21.Rampling R., Cruickshank G., Papanastassiou V., Nicoll J., Hadley D., Brennan D., Petty R., MacLean A., Harland J., McKie E. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther. 2000;7:859–866. doi: 10.1038/sj.gt.3301184. [DOI] [PubMed] [Google Scholar]
- 22.Zhang C., Tang J., Xie J., Zhang H., Li Y., Zhang J., Verpooten D., He B., Cao Y. A conserved domain of herpes simplex virus ICP34.5 regulates protein phosphatase complex in mammalian cells. FEBS Lett. 2008;582:171–176. doi: 10.1016/j.febslet.2007.11.082. [DOI] [PubMed] [Google Scholar]
- 23.Radtke K., Kieneke D., Wolfstein A., Michael K., Steffen W., Scholz T., Karger A., Sodeik B. Plus- and minus-end directed microtubule motors bind simultaneously to herpes simplex virus capsids using different inner tegument structures. PLoS Pathog. 2010;6:e1000991. doi: 10.1371/journal.ppat.1000991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manivanh R., Mehrbach J., Knipe D.M., Leib D.A. Role of herpes simplex virus 1 γ34.5 in the regulation of IRF3 signaling. J. Virol. 2017;91:e01156-17. doi: 10.1128/JVI.01156-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y., Zhang C., Chen X., Yu J., Wang Y., Yang Y., Du M., Jin H., Ma Y., He B., Cao Y. ICP34.5 protein of herpes simplex virus facilitates the initiation of protein translation by bridging eukaryotic initiation factor 2α (eIF2α) and protein phosphatase 1. J. Biol. Chem. 2011;286:24785–24792. doi: 10.1074/jbc.M111.232439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He B., Gross M., Roizman B. The γ134.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the α subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. USA. 1997;94:843–848. doi: 10.1073/pnas.94.3.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chung R.Y., Saeki Y., Chiocca E.A. B-myb promoter retargeting of herpes simplex virus γ34.5 gene-mediated virulence toward tumor and cycling cells. J. Virol. 1999;73:7556–7564. doi: 10.1128/jvi.73.9.7556-7564.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanai R., Zaupa C., Sgubin D., Antoszczyk S.J., Martuza R.L., Wakimoto H., Rabkin S.D. Effect of γ34.5 deletions on oncolytic herpes simplex virus activity in brain tumors. J. Virol. 2012;86:4420–4431. doi: 10.1128/JVI.00017-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He B., Chou J., Brandimarti R., Mohr I., Gluzman Y., Roizman B. Suppression of the phenotype of gamma(1)34.5- herpes simplex virus 1: failure of activated RNA-dependent protein kinase to shut off protein synthesis is associated with a deletion in the domain of the alpha47 gene. J. Virol. 1997;71:6049–6054. doi: 10.1128/jvi.71.8.6049-6054.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu B.L., Robinson M., Han Z.Q., Branston R.H., English C., Reay P., McGrath Y., Thomas S.K., Thornton M., Bullock P. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003;10:292–303. doi: 10.1038/sj.gt.3301885. [DOI] [PubMed] [Google Scholar]
- 31.Taguchi S., Fukuhara H., Todo T. Oncolytic virus therapy in Japan: progress in clinical trials and future perspectives. Jpn. J. Clin. Oncol. 2019;49:201–209. doi: 10.1093/jjco/hyy170. [DOI] [PubMed] [Google Scholar]
- 32.Kambara H., Okano H., Chiocca E.A., Saeki Y. An oncolytic HSV-1 mutant expressing ICP34.5 under control of a nestin promoter increases survival of animals even when symptomatic from a brain tumor. Cancer Res. 2005;65:2832–2839. doi: 10.1158/0008-5472.CAN-04-3227. [DOI] [PubMed] [Google Scholar]
- 33.Fulci G., Breymann L., Gianni D., Kurozomi K., Rhee S.S., Yu J., Kaur B., Louis D.N., Weissleder R., Caligiuri M.A., Chiocca E.A. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc. Natl. Acad. Sci. USA. 2006;103:12873–12878. doi: 10.1073/pnas.0605496103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ikeda K., Ichikawa T., Wakimoto H., Silver J.S., Deisboeck T.S., Finkelstein D., Harsh G.R., 4th, Louis D.N., Bartus R.T., Hochberg F.H., Chiocca E.A. Oncolytic virus therapy of multiple tumors in the brain requires suppression of innate and elicited antiviral responses. Nat. Med. 1999;5:881–887. doi: 10.1038/11320. [DOI] [PubMed] [Google Scholar]
- 35.Kambara H., Saeki Y., Chiocca E.A. Cyclophosphamide allows for in vivo dose reduction of a potent oncolytic virus. Cancer Res. 2005;65:11255–11258. doi: 10.1158/0008-5472.CAN-05-2278. [DOI] [PubMed] [Google Scholar]
- 36.Guo H., Omoto S., Harris P.A., Finger J.N., Bertin J., Gough P.J., Kaiser W.J., Mocarski E.S. Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe. 2015;17:243–251. doi: 10.1016/j.chom.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang Z., Wu S.Q., Liang Y., Zhou X., Chen W., Li L., Wu J., Zhuang Q., Chen C., Li J. RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe. 2015;17:229–242. doi: 10.1016/j.chom.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 38.Chou J., Chen J.J., Gross M., Roizman B. Association of a M(r) 90,000 phosphoprotein with protein kinase PKR in cells exhibiting enhanced phosphorylation of translation initiation factor eIF-2 alpha and premature shutoff of protein synthesis after infection with gamma 134.5- mutants of herpes simplex virus 1. Proc. Natl. Acad. Sci. USA. 1995;92:10516–10520. doi: 10.1073/pnas.92.23.10516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.US Food and Drug Administration . 2005. Guidance for industry: estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy volunteers.https://www.fda.gov/media/72309/download [Google Scholar]
- 40.Hirooka Y., Kasuya H., Ishikawa T., Kawashima H., Ohno E., Villalobos I.B., Naoe Y., Ichinose T., Koyama N., Tanaka M. A phase I clinical trial of EUS-guided intratumoral injection of the oncolytic virus, HF10 for unresectable locally advanced pancreatic cancer. BMC Cancer. 2018;18:596. doi: 10.1186/s12885-018-4453-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kasuya H., Kodera Y., Nakao A., Yamamura K., Gewen T., Zhiwen W., Hotta Y., Yamada S., Fujii T., Fukuda S. Phase I dose-escalation clinical trial of HF10 oncolytic herpes virus in 17 Japanese patients with advanced cancer. Hepatogastroenterology. 2014;61:599–605. [PubMed] [Google Scholar]
- 42.Takakuwa H., Goshima F., Nozawa N., Yoshikawa T., Kimata H., Nakao A., Nawa A., Kurata T., Sata T., Nishiyama Y. Oncolytic viral therapy using a spontaneously generated herpes simplex virus type 1 variant for disseminated peritoneal tumor in immunocompetent mice. Arch. Virol. 2003;148:813–825. doi: 10.1007/s00705-002-0944-x. [DOI] [PubMed] [Google Scholar]
- 43.Verpooten D., Ma Y., Hou S., Yan Z., He B. Control of TANK-binding kinase 1-mediated signaling by the γ134.5 protein of herpes simplex virus 1. J. Biol. Chem. 2009;284:1097–1105. doi: 10.1074/jbc.M805905200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma Y., Jin H., Valyi-Nagy T., Cao Y., Yan Z., He B. Inhibition of TANK binding kinase 1 by herpes simplex virus 1 facilitates productive infection. J. Virol. 2012;86:2188–2196. doi: 10.1128/JVI.05376-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ishikawa H., Barber G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pan S., Liu X., Ma Y., Cao Y., He B. Herpes simplex virus 1 γ134.5 protein inhibits STING activation that restricts viral replication. J. Virol. 2018;92:e01015-18. doi: 10.1128/JVI.01015-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakamura H., Kasuya H., Mullen J.T., Yoon S.S., Pawlik T.M., Chandrasekhar S., Donahue J.M., Chiocca E.A., Chung R.Y., Tanabe K.K. Regulation of herpes simplex virus γ134.5 expression and oncolysis of diffuse liver metastases by Myb34.5. J. Clin. Invest. 2002;109:871–882. doi: 10.1172/JCI10623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ali M., Roback L., Mocarski E.S. Herpes simplex virus 1 ICP6 impedes TNF receptor 1-induced necrosome assembly during compartmentalization to detergent-resistant membrane vesicles. J. Biol. Chem. 2019;294:991–1004. doi: 10.1074/jbc.RA118.004651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guo H., Gilley R.P., Fisher A., Lane R., Landsteiner V.J., Ragan K.B., Dovey C.M., Carette J.E., Upton J.W., Mocarski E.S., Kaiser W.J. Species-independent contribution of ZBP1/DAI/DLM-1-triggered necroptosis in host defense against HSV1. Cell Death Dis. 2018;9:816. doi: 10.1038/s41419-018-0868-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mocarski E.S., Guo H., Kaiser W.J. Necroptosis: the Trojan horse in cell autonomous antiviral host defense. Virology. 2015;479–480:160–166. doi: 10.1016/j.virol.2015.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cassady K.A. Human cytomegalovirus TRS1 and IRS1 gene products block the double-stranded-RNA-activated host protein shutoff response induced by herpes simplex virus type 1 infection. J. Virol. 2005;79:8707–8715. doi: 10.1128/JVI.79.14.8707-8715.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cassady K.A., Bauer D.F., Roth J., Chambers M.R., Shoeb T., Coleman J., Prichard M., Gillespie G.Y., Markert J.M. Pre-clinical assessment of C134, a chimeric oncolytic herpes simplex virus, in mice and non-human primates. Mol. Ther. Oncolytics. 2017;5:1–10. doi: 10.1016/j.omto.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Friedman G.K., Nan L., Haas M.C., Kelly V.M., Moore B.P., Langford C.P. γ134.5-deleted HSV-1-expressing human cytomegalovirus IRS1 gene kills human glioblastoma cells as efficiently as wild-type HSV-1 in normoxia or hypoxia. Gene Ther. 2015;22:348–355. doi: 10.1038/gt.2014.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakashima H., Nguyen T., Kasai K., Passaro C., Ito H., Goins W.F., Shaikh I., Erdelyi R., Nishihara R., Nakano I. Toxicity and efficacy of a novel GADD34-expressing oncolytic HSV-1 for the treatment of experimental glioblastoma. Clin. Cancer Res. 2018;24:2574–2584. doi: 10.1158/1078-0432.CCR-17-2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fulci G., Dmitrieva N., Gianni D., Fontana E.J., Pan X., Lu Y., Kaufman C.S., Kaur B., Lawler S.E., Lee R.J. Depletion of peripheral macrophages and brain microglia increases brain tumor titers of oncolytic viruses. Cancer Res. 2007;67:9398–9406. doi: 10.1158/0008-5472.CAN-07-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Otsuki A., Patel A., Kasai K., Suzuki M., Kurozumi K., Chiocca E.A., Saeki Y. Histone deacetylase inhibitors augment antitumor efficacy of herpes-based oncolytic viruses. Mol. Ther. 2008;16:1546–1555. doi: 10.1038/mt.2008.155. [DOI] [PubMed] [Google Scholar]
- 57.Fisher L.J. Neural precursor cells: applications for the study and repair of the central nervous system. Neurobiol. Dis. 1997;4:1–22. doi: 10.1006/nbdi.1997.0137. [DOI] [PubMed] [Google Scholar]
- 58.Kawaguchi A., Miyata T., Sawamoto K., Takashita N., Murayama A., Akamatsu W., Ogawa M., Okabe M., Tano Y., Goldman S.A., Okano H. Nestin-EGFP transgenic mice: visualization of the self-renewal and multipotency of CNS stem cells. Mol. Cell. Neurosci. 2001;17:259–273. doi: 10.1006/mcne.2000.0925. [DOI] [PubMed] [Google Scholar]
- 59.Cho J.M., Shin Y.J., Park J.M., Kim J., Lee M.Y. Characterization of nestin expression in astrocytes in the rat hippocampal CA1 region following transient forebrain ischemia. Anat. Cell Biol. 2013;46:131–140. doi: 10.5115/acb.2013.46.2.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krishnasamy S., Weng Y.C., Thammisetty S.S., Phaneuf D., Lalancette-Hebert M., Kriz J. Molecular imaging of nestin in neuroinflammatory conditions reveals marked signal induction in activated microglia. J. Neuroinflammation. 2017;14:45. doi: 10.1186/s12974-017-0816-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jin X., Jin X., Jung J.E., Beck S., Kim H. Cell surface Nestin is a biomarker for glioma stem cells. Biochem. Biophys. Res. Commun. 2013;433:496–501. doi: 10.1016/j.bbrc.2013.03.021. [DOI] [PubMed] [Google Scholar]
- 62.Lv D., Lu L., Hu Z., Fei Z., Liu M., Wei L., Xu J. Nestin expression is associated with poor clinicopathological features and prognosis in glioma patients: an association study and meta-analysis. Mol. Neurobiol. 2017;54:727–735. doi: 10.1007/s12035-016-9689-5. [DOI] [PubMed] [Google Scholar]
- 63.Strojnik T., Rosland G.V., Sakariassen P.O., Kavalar R., Lah T. Neural stem cell markers, nestin and musashi proteins, in the progression of human glioma: correlation of nestin with prognosis of patient survival. Surg. Neurol. 2007;68:133–143. doi: 10.1016/j.surneu.2006.10.050. [DOI] [PubMed] [Google Scholar]
- 64.Tomita T., Akimoto J., Haraoka J., Kudo M. Clinicopathological significance of expression of nestin, a neural stem/progenitor cell marker, in human glioma tissue. Brain Tumor Pathol. 2014;31:162–171. doi: 10.1007/s10014-013-0169-6. [DOI] [PubMed] [Google Scholar]
- 65.Zhang M., Song T., Yang L., Chen R., Wu L., Yang Z., Fang J. Nestin and CD133: valuable stem cell-specific markers for determining clinical outcome of glioma patients. J. Exp. Clin. Cancer Res. 2008;27:85. doi: 10.1186/1756-9966-27-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kitai R., Horita R., Sato K., Yoshida K., Arishima H., Higashino Y., Hashimoto N., Takeuchi H., Kubota T., Kikuta K. Nestin expression in astrocytic tumors delineates tumor infiltration. Brain Tumor Pathol. 2010;27:17–21. doi: 10.1007/s10014-009-0261-0. [DOI] [PubMed] [Google Scholar]
- 67.Peng K.W., Myers R., Greenslade A., Mader E., Greiner S., Federspiel M.J., Dispenzieri A., Russell S.J. Using clinically approved cyclophosphamide regimens to control the humoral immune response to oncolytic viruses. Gene Ther. 2013;20:255–261. doi: 10.1038/gt.2012.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Roy N.S., Wang S., Jiang L., Kang J., Benraiss A., Harrison-Restelli C., Fraser R.A., Couldwell W.T., Kawaguchi A., Okano H. In vitro neurogenesis by progenitor cells isolated from the adult human hippocampus. Nat. Med. 2000;6:271–277. doi: 10.1038/73119. [DOI] [PubMed] [Google Scholar]
- 69.Sanai N., Tramontin A.D., Quiñones-Hinojosa A., Barbaro N.M., Gupta N., Kunwar S., Lawton M.T., McDermott M.W., Parsa A.T., Manuel-García Verdugo J. Unique astrocyte ribbon in adult human brain contains neural stem cells but lacks chain migration. Nature. 2004;427:740–744. doi: 10.1038/nature02301. [DOI] [PubMed] [Google Scholar]
- 70.Hendrickson M.L., Rao A.J., Demerdash O.N., Kalil R.E. Expression of nestin by neural cells in the adult rat and human brain. PLoS ONE. 2011;6:e18535. doi: 10.1371/journal.pone.0018535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Burke M.J., Ahern C., Weigel B.J., Poirier J.T., Rudin C.M., Chen Y., Cripe T.P., Bernhardt M.B., Blaney S.M. Phase I trial of Seneca Valley virus (NTX-010) in children with relapsed/refractory solid tumors: a report of the Children’s Oncology Group. Pediatr. Blood Cancer. 2015;62:743–750. doi: 10.1002/pbc.25269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cerullo V., Diaconu I., Kangasniemi L., Rajecki M., Escutenaire S., Koski A., Romano V., Rouvinen N., Tuuminen T., Laasonen L. Immunological effects of low-dose cyclophosphamide in cancer patients treated with oncolytic adenovirus. Mol. Ther. 2011;19:1737–1746. doi: 10.1038/mt.2011.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Currier M.A., Gillespie R.A., Sawtell N.M., Mahller Y.Y., Stroup G., Collins M.H., Kambara H., Chiocca E.A., Cripe T.P. Efficacy and safety of the oncolytic herpes simplex virus rRp450 alone and combined with cyclophosphamide. Mol. Ther. 2008;16:879–885. doi: 10.1038/mt.2008.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dhar D., Toth K., Wold W.S. Cycles of transient high-dose cyclophosphamide administration and intratumoral oncolytic adenovirus vector injection for long-term tumor suppression in Syrian hamsters. Cancer Gene Ther. 2014;21:171–178. doi: 10.1038/cgt.2014.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dispenzieri A., Tong C., LaPlant B., Lacy M.Q., Laumann K., Dingli D., Zhou Y., Federspiel M.J., Gertz M.A., Hayman S. Phase I trial of systemic administration of Edmonston strain of measles virus genetically engineered to express the sodium iodide symporter in patients with recurrent or refractory multiple myeloma. Leukemia. 2017;31:2791–2798. doi: 10.1038/leu.2017.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hasegawa N., Abei M., Yokoyama K.K., Fukuda K., Seo E., Kawashima R., Nakano Y., Yamada T., Nakade K., Hamada H. Cyclophosphamide enhances antitumor efficacy of oncolytic adenovirus expressing uracil phosphoribosyltransferase (UPRT) in immunocompetent Syrian hamsters. Int. J. Cancer. 2013;133:1479–1488. doi: 10.1002/ijc.28132. [DOI] [PubMed] [Google Scholar]
- 77.Hofmann E., Weibel S., Szalay A.A. Combination treatment with oncolytic Vaccinia virus and cyclophosphamide results in synergistic antitumor effects in human lung adenocarcinoma bearing mice. J. Transl. Med. 2014;12:197. doi: 10.1186/1479-5876-12-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ichikawa T., Petros W.P., Ludeman S.M., Fangmeier J., Hochberg F.H., Colvin O.M., Chiocca E.A. Intraneoplastic polymer-based delivery of cyclophosphamide for intratumoral bioconversion by a replicating oncolytic viral vector. Cancer Res. 2001;61:864–868. [PubMed] [Google Scholar]
- 79.Ikeda K., Wakimoto H., Ichikawa T., Jhung S., Hochberg F.H., Louis D.N., Chiocca E.A. Complement depletion facilitates the infection of multiple brain tumors by an intravascular, replication-conditional herpes simplex virus mutant. J. Virol. 2000;74:4765–4775. doi: 10.1128/jvi.74.10.4765-4775.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kottke T., Thompson J., Diaz R.M., Pulido J., Willmon C., Coffey M., Selby P., Melcher A., Harrington K., Vile R.G. Improved systemic delivery of oncolytic reovirus to established tumors using preconditioning with cyclophosphamide-mediated Treg modulation and interleukin-2. Clin. Cancer Res. 2009;15:561–569. doi: 10.1158/1078-0432.CCR-08-1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li H., Zeng Z., Fu X., Zhang X. Coadministration of a herpes simplex virus-2 based oncolytic virus and cyclophosphamide produces a synergistic antitumor effect and enhances tumor-specific immune responses. Cancer Res. 2007;67:7850–7855. doi: 10.1158/0008-5472.CAN-07-1087. [DOI] [PubMed] [Google Scholar]
- 82.Lun X.Q., Jang J.H., Tang N., Deng H., Head R., Bell J.C., Stojdl D.F., Nutt C.L., Senger D.L., Forsyth P.A., McCart J.A. Efficacy of systemically administered oncolytic vaccinia virotherapy for malignant gliomas is enhanced by combination therapy with rapamycin or cyclophosphamide. Clin. Cancer Res. 2009;15:2777–2788. doi: 10.1158/1078-0432.CCR-08-2342. [DOI] [PubMed] [Google Scholar]
- 83.Myers R.M., Greiner S.M., Harvey M.E., Griesmann G., Kuffel M.J., Buhrow S.A., Reid J.M., Federspiel M., Ames M.M., Dingli D. Preclinical pharmacology and toxicology of intravenous MV-NIS, an oncolytic measles virus administered with or without cyclophosphamide. Clin. Pharmacol. Ther. 2007;82:700–710. doi: 10.1038/sj.clpt.6100409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Qiao J., Wang H., Kottke T., White C., Twigger K., Diaz R.M., Thompson J., Selby P., de Bono J., Melcher A. Cyclophosphamide facilitates antitumor efficacy against subcutaneous tumors following intravenous delivery of reovirus. Clin. Cancer Res. 2008;14:259–269. doi: 10.1158/1078-0432.CCR-07-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Roulstone V., Khan K., Pandha H.S., Rudman S., Coffey M., Gill G.M., Melcher A.A., Vile R., Harrington K.J., de Bono J., Spicer J. Phase I trial of cyclophosphamide as an immune modulator for optimizing oncolytic reovirus delivery to solid tumors. Clin. Cancer Res. 2015;21:1305–1312. doi: 10.1158/1078-0432.CCR-14-1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Studebaker A.W., Hutzen B.J., Pierson C.R., Haworth K.B., Cripe T.P., Jackson E.M., Leonard J.R. Oncolytic herpes virus rRp450 shows efficacy in orthotopic xenograft group 3/4 medulloblastomas and atypical teratoid/rhabdoid tumors. Mol. Ther. Oncolytics. 2017;6:22–30. doi: 10.1016/j.omto.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Thomas M.A., Spencer J.F., Toth K., Sagartz J.E., Phillips N.J., Wold W.S. Immunosuppression enhances oncolytic adenovirus replication and antitumor efficacy in the Syrian hamster model. Mol. Ther. 2008;16:1665–1673. doi: 10.1038/mt.2008.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ungerechts G., Frenzke M.E., Yaiw K.C., Miest T., Johnston P.B., Cattaneo R. Mantle cell lymphoma salvage regimen: synergy between a reprogrammed oncolytic virus and two chemotherapeutics. Gene Ther. 2010;17:1506–1516. doi: 10.1038/gt.2010.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Young B.A., Spencer J.F., Ying B., Tollefson A.E., Toth K., Wold W.S. The role of cyclophosphamide in enhancing antitumor efficacy of an adenovirus oncolytic vector in subcutaneous Syrian hamster tumors. Cancer Gene Ther. 2013;20:521–530. doi: 10.1038/cgt.2013.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Alvarez-Breckenridge C.A., Yu J., Price R., Wojton J., Pradarelli J., Mao H., Wei M., Wang Y., He S., Hardcastle J. NK cells impede glioblastoma virotherapy through NKp30 and NKp46 natural cytotoxicity receptors. Nat. Med. 2012;18:1827–1834. doi: 10.1038/nm.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.