

Abstract
Motivation
Systematic identification of molecular targets among known drugs plays an essential role in drug repurposing and understanding of their unexpected side effects. Computational approaches for prediction of drug–target interactions (DTIs) are highly desired in comparison to traditional experimental assays. Furthermore, recent advances of multiomics technologies and systems biology approaches have generated large-scale heterogeneous, biological networks, which offer unexpected opportunities for network-based identification of new molecular targets among known drugs.
Results
In this study, we present a network-based computational framework, termed AOPEDF, an arbitrary-order proximity embedded deep forest approach, for prediction of DTIs. AOPEDF learns a low-dimensional vector representation of features that preserve arbitrary-order proximity from a highly integrated, heterogeneous biological network connecting drugs, targets (proteins) and diseases. In total, we construct a heterogeneous network by uniquely integrating 15 networks covering chemical, genomic, phenotypic and network profiles among drugs, proteins/targets and diseases. Then, we build a cascade deep forest classifier to infer new DTIs. Via systematic performance evaluation, AOPEDF achieves high accuracy in identifying molecular targets among known drugs on two external validation sets collected from DrugCentral [area under the receiver operating characteristic curve (AUROC) = 0.868] and ChEMBL (AUROC = 0.768) databases, outperforming several state-of-the-art methods. In a case study, we showcase that multiple molecular targets predicted by AOPEDF are associated with mechanism-of-action of substance abuse disorder for several marketed drugs (such as aripiprazole, risperidone and haloperidol).
Availability and implementation
Source code and data can be downloaded from https://github.com/ChengF-Lab/AOPEDF.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Identification of drug–target interactions (i.e. interactions between drugs and targets/proteins; DTIs) plays an important role in drug discovery and development. Since experimental determination of DTIs is costly and time-consuming (Haggarty et al., 2003; Kuruvilla et al., 2002), in silico or computational approaches have offered possibilities to identify potential DTIs for accelerating drug development, such as drug repurposing. Several in silico approaches, such as structure-based (Donald, 2011; Morris et al., 2009), ligand-based (Keiser et al., 2007) and machine learning-based methods (Bleakley and Yamanishi, 2009; Wan et al., 2019), have revealed potential in predicting DTIs. However, structure-based methods are limited when 3D structures of proteins are unavailable (Cheng et al., 2007; Rarey et al., 1996) which, unfortunately, is the case for the majority of targets. Ligand-based methods exploit the chemical structures of ligands to make predictions, and their performance is poor when the chemical space of the ligands is out of application domains. A variety of machine learning-based approaches have been developed to predict DTIs (Bleakley and Yamanishi, 2009; He et al., 2010; Mei et al., 2013; Perlman et al., 2011; Xia et al., 2010). These methods fully exploit latent correlations among the related features of drugs and targets and offer moderate accuracy for prediction of DTIs. Altogether, most existing methods for DTI prediction are limited to homogeneous networks or bipartite drug–target networks (Cheng et al., 2012a) and cannot be directly extended to heterogeneous, biological networks (Cheng et al., 2012b).
In comparison to homogeneous networks, heterogeneous networks naturally assemble more objects and complementary information from drugs, targets/proteins and their associated diseases. Several computational approaches have recently been reported to integrate heterogeneous data sources. For example, MSCMF (Zheng et al., 2013) integrates multiple data sources via a weighted averaging scheme and uses the resulting drug and protein similarity matrices to regularize the matrix factorization operation of a given DTI network. However, such data integration may cause substantial losses in network-specific information. HNM (Wang et al., 2014) fuses heterogeneous data by a network diffusion process; however, directly using diffusion states as features or prediction scores may easily suffer from the bias introduced by the noise and high-dimensionality of biological network data. Inspired by the recent surge of deep learning techniques for feature extracting, models with higher predictive capacity have been explored. DeepWalk is a deep learning method by utilizing the similarities from a tripartite, heterogeneous network built from biomedical linked datasets (Zong et al., 2017). NeoDTI integrated neighborhood information of the heterogeneous network and automatically learns topology-preserving representations of drugs and targets (Wan et al., 2019). However, these methods are mainly based on a shallow neural network model with only three layers. Besides, these methods are prone to preserving only the first- or second-order proximity. deepDTnet (Zeng et al., 2020) adopted deep autoencoder to automatically learn high-quality features from heterogeneous networks, and then applied positive-unlabeled (PU)-matrix completion to predict new DTIs. Yet, recent studies have suggested that high-order proximities among diverse types of nodes play crucial roles in capturing the underlying topological structure of the network (Cao et al., 2015; Cui et al., 2019; Perozzi et al., 2014); further, embedding with certain order proximity does not necessarily perform best on all networks. We assorted that incorporating diverse, complementary proximities from different biological networks may improve accuracy further (Zhang et al., 2018).
In this study, we propose arbitrary-order proximity embedded deep forest (AOPEDF), a new computational approach for molecular target identification from known drugs and for target-centered drug repurposing. Specifically, AOPEDF preserves the different order proximity information from 15 networks in a constructed drug–target–disease heterogeneous network. It then utilizes low-dimensional but informative vector representations of features for both drugs and targets/proteins through a cascade deep forest classifier in prediction of DTIs. Theoretically, AOPEDF has the following advantages: (i) AOPEDF integrates diverse information from 15 heterogeneous networks and preserves complementary order proximity information for different networks. Thus, the low-dimensional feature vectors learned by AOPEDF capture rich context information as well as the topological structure of individual networks. (ii) AOPEDF adopts deep forest as a classifier, which achieves high performance in classification but has much fewer hyper-parameters than deep neural networks (DNN) (LeCun et al., 2015). AOPEDF is highly robust to hyper-parameter settings. Importantly, the number of cascade levels can be adaptively determined such that the model complexity can be automatically set. (iii) Tree-based methods implemented in AOPEDF make prediction by inferring decision rules from data, which is more effective in generating interpretable predictions from rich features compared to traditional neural network methods. Via comprehensive evaluation on cross-validation and two external validation sets, we show that AOPEDF achieves higher performance in comparison to several state-of-the-art methods. In summary, AOPEDF offers a powerful tool to predict new DTIs from heterogeneous networks for accelerating target-centered drug repurposing and therapeutic development for understudied diseases.
2 Materials and methods
2.1 Data resource
We collect DTI information from the DrugBank database (v4.3) (Wishart et al., 2018), the therapeutic target database (Yang et al., 2016) and the PharmGKB database (Hernandez-Boussard et al., 2007). Specifically, bioactivity data for drug–target pairs are collected from ChEMBL (v20) (Gaulton et al., 2012), BindingDB (Liu et al., 2007) and IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014). The chemical structure of each drug with SMILES format is extracted from DrugBank (Law et al., 2014). Here, only DTIs meeting the following three criteria are used: (i) the human target is represented by a unique UniProt accession number; (ii) the target is marked as ‘reviewed’ in the UniProt database (Apweiler et al., 2004); and (iii) binding affinities, including Ki, Kd, IC50 or EC50 each ≤10 μM. We used a low binding affinity cutoff of 10 μM as weak-binding drugs play crucial roles in therapeutic development as well (Ohlson, 2008; Wang et al., 2017). In total, a DTI network connecting 732 FDA-approved drugs and 1519 unique human targets (proteins) were used (Supplementary Tables S1 and S2). We randomly selected the matching number of the unknown drug–target pairs (by excluding all known DTIs) as negative samples. The details for building the experimentally validated drug–target network are provided in a recent publication (Cheng et al., 2018). In addition, we construct nine networks for drugs: (i) clinically reported drug–drug interactions, (ii) drug–disease associations, (iii) drug–side effect associations, (iv) chemical similarities, (v) therapeutic similarities derived from the Anatomical Therapeutic Chemical Classification System, (vi) target sequence-derived drug–drug similarities, (vii) Gene Ontology (GO) biological process, (viii) GO cellular component and (ix) GO molecular function, and six networks for proteins: (i) protein–protein interactions, (ii) protein–disease associations, (iii) protein sequence similarities, (iv) GO biological process, (v) GO cellular component and (vi) GO molecular function. The detailed description for building 15 networks is provided in the Supplementary Methods and our recent study (Cheng et al., 2019a,b). For external validation sets, we assembled the newest experimentally validated DTIs from the DrugCentral database (Ursu et al., 2019) and ChEMBL database (Mendez et al., 2019) by excluding overlapping drug–target pairs from the training set. There are partly overlap between two validated sets from DrugCentral and ChEMBL databases, respectively (Supplementary Table S3). There are no any overlap DTIs between the training set and two external validation set.
2.2 Pipeline of AOPEDF
As shown in Figure 1, AOPEDF consists of three steps: (i) data preparation and benchmarking, (ii) arbitrary-order proximity preserved network embedding (AROPE) and (iii) deep forest-based prediction of DTIs. First, we integrate 15 biological networks to construct a complex heterogeneous network which contains diverse information and a multiview perspective in predicting novel DTIs. Then, we preserve arbitrary-order proximity of each network to obtain informative, but low-dimensional vector representations of drugs and targets. Finally, potential DTIs will be predicted using the deep forest classifier.
Fig. 1.
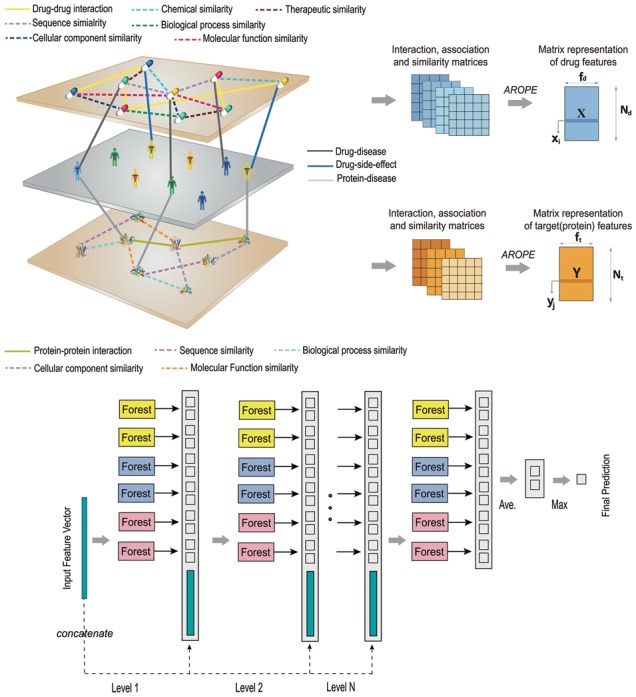
A flowchart of the proposed approach. First, we integrate 15 networks to construct a complicated heterogeneous network which contains diverse chemoinformatics and bioinformatics profiles and a multiview perspective for predicting DTIs. Then, AOPEDF integrates diverse information from the heterogeneous network, and preserves the different order proximity information for different networks through AROPE. Finally, the deep forest classifier is utilized to infer potential DTIs among known drugs
2.3 Arbitrary-order proximity preserved network embedding
We use to denote the adjacency matrix (binary or weighted) of a network with nodes and edges. and stand for its row and column, respectively. is the weight of the edge between nodes and . A is symmetric, denotes the transpose of . Functions are marked by curlicue, e.g. .
Definition 1.
High-order proximity. Given the adjacency matrix of an undirected network, a high-order proximity is defined as a polynomial function of :
(1) where is the order and are the weights. We refer to a proximity of order as the weighted combination of all the orders from the to the , rather than the order alone. We allow if the summation converges. We will assume that for . To preserve the high-order proximity in a low-dimensional vector space, the widely adopted method is matrix factorization, which minimizes the following objective function:
(2) where , are content/context embedding vectors and is the dimensionality of the space. Without loss of generality, we use as the content embedding vectors. From Eckart–Young theorem, the global optimal solution to Equation (2) can be obtained by truncated SVD (Eckart and Young, 1936). Specifically, denote [] as the top- SVD results of , where and each column corresponds to one left/right singular vector, and is a diagonal matrix of singular values in descending order. The embeddings can be obtained by multiplying into U, V:
(3) However, directly calculating and SVD will be both time and space consuming. Besides, since different networks and targets applications usually require the proximities of different orders, how to shift across different orders is also challenging.
Denote the top- eigen-decomposition of as [], where is a diagonal matrix of eigenvalues in descending order of the absolute value, and each column corresponds to an eigenvector. We can refer [], as an eigen-pair. To solve the SVD problem, we can transform it into an eigen-decomposition problem (Strang, 2006).
Theorem 1. For any symmetric matrix , , we have:
(4) where stands for the absolute value function and is the sign function, i.e. if , if and if .
Now, we only need to focus on solving the eigen-decomposition of
As proved in Zhang et al. (2018), to calculate the eigen-decomposition on , we can follow the eigen-decomposition Reweighting theorem:
Theorem 2. Eigen-decomposition Reweighting. If [] is an eigen-pair of , then [] is an eigen-pair of .
The theorem shows that, without performing the eigen-decomposition on , we can obtain the eigen-decomposition results of from the eigen-decomposition results of by replacing with . In fact, the theorem reveals the intrinsic relationship between proximities of different orders. If we regard each eigenvector as a ‘coordinate’ of the nodes in the network and each eigenvalue as a ‘weight’ of the coordinate, then, preserving proximities of different orders is equivalent to reweighting the dimensions.
After the eigen-decomposition reweighting, the order of the eigenvalues may change, including the top- eigen-decomposition of is not necessarily the reweighting of the top- eigen-decomposition of To tackle the problem (Zhang et al., 2018) proved that the top- eigen-decomposition of any is guaranteed to be the reweighting of the top- eigen-decomposition of , where is a function of the network and . Thus, to get the top- eigen-decomposition of any , we need to calculate the top- eigen-decomposition of . Then we can reweight and reorder dimensions and use the top- after reweighting to derive the embedding vectors. Since the top- eigen-decomposition of is shared by arbitrary-order proximities, we can shift between proximities of different orders with a low marginal cost by pre-computing the eigen-decomposition.
2.4 Deep forest algorithm
After learning the low-dimensional vector representation of drugs and proteins, we utilize the deep forest (Zhou and Feng, 2017) for prediction, which provides an alternative approach to DNNs to learn hyper-level representations in low computing cost. Inspired by the layer–layer processing of raw features in DNNs, deep forest employs a cascade structure, each level of cascade receives feature information processed by its preceding level and outputs its processing results to the next level (Fig. 1). Each level is an ensemble of decision tree forest, such as an ensemble of ensembles. Diversity is crucial for ensemble construction. In this study, we used: (i) two random forests (RFs), (ii) two completely random tree forests and (iii) two gradient boosting tree forests. Each forest contains 500 trees and there are 3000 trees in total. For instance, each forest will produce an estimate of class distribution, by counting the percentage of different classes of training examples at the leaf node where the concerned instance falls, and then averaging across all trees in the same forest. The estimated class distribution forms a class vector, which is then concatenated with the original feature vector to be input to the next level of the cascade (Fig. 1). Herein, there are two classes in binary classification, with each of the six forests producing a 2D class vector. In total, the next level of the cascade will receive 12 (=2 × 6) augmented features.
To reduce the risk of overfitting, class vector produced by each forest is generated by k-fold cross-validation (k = 5). In detail, each forest will be used as training data for k−1 times, resulting in k−1 class vectors, which are then averaged to produce the final class vector as augmented features for the next level of cascade. After expanding to a new level, the performance of the whole cascade will be estimated on a validation set, and the training procedure will terminate if there is no significant performance gain. Subsequently, the number of cascade levels is automatically determined. Deep forest employs a multigrained scanning strategy, a sliding window-based approach, to extract local features by scanning raw input to generate a series of local low-dimensional feature vectors. It then trains a series of forests by using those low-dimensional vectors to obtain class distribution of input vectors. More details are provided in previous study (Zhou and Feng, 2017).
3 Results
3.1 Baseline methods
NeoDTI: Neural integration of neighbor information for DTI prediction (Wan et al., 2019) is a nonlinear end-to-end learning model that integrates diverse information from heterogeneous networks and automatically learns topology-preserving representations of drugs and targets for prediction.
deepDTnet: A network-based, deep learning methodology for drug repurposing, which integrates DNN algorithm for network embedding and a PU-matrix completion algorithm for prediction (Zeng et al., 2020).
RLSWNN: Regularized Least Squares with Weighted Nearest Neighbors (van Laarhoven and Marchiori, 2013), which uses a weighted nearest neighbor procedure for inferring a profile for a drug by using interaction profiles of drugs in the training data.
KBMF2K: Kernelized Bayesian matrix Factorization method (Gonen, 2012), which uses a kernelized Bayesian matrix factorization with twin kernels to predict DTIs.
NetLapRLS: An algorithm utilizing the bipartite local model concept (Xia et al., 2010), performs two sets of predictions, including one from the drug side and one from the target side, and then aggregates these predictions to give the final prediction scores for the potential interaction candidates.
DeepWalk: A deep learning method utilizes the similarities within a heterogeneous tripartite network built from biomedical linked datasets (Zong et al., 2017).
3.2 Performance of AOPEDF on the cross-validation
We first evaluated performance of AOPEDF by conducting a 5-fold cross-validation procedure on all positive pairs and a set of matching number of randomly sampled negatives with positive samples (random selection of unknown drug–target pairs by excluding all known DTIs). During each 5-fold cross-validation, we randomly chose a subset of 80% of the known DTI pairs and a matching number of randomly sampled unknown drug–target pairs as the training set, and the remaining 20% known DTI pairs and a matching number of randomly sampled unknown drug–target pairs were held out as the test set. The area under the receiver operating characteristic curve (AUROC) and the area under the precision–recall curve (AUPR) were utilized to evaluate the overall performance of AOPEDF. To reduce the data bias of cross-validation, we performed 10 times of random 5-fold cross-validation and computed the average performance. We found that AOPEDF showed high accuracy (AUROC = 0.985 and AUPR = 0.985) in 5-fold cross-validation, outperforming that of several state-of-the-art methods: NeoDTI (AUROC = 0.971 and AUPR = 0.970), deepDTnet (AUROC = 0.965 and AUPR = 0.969), RLSWNN (AUROC = 0.949 and AUPR = 0.955), KBMF2K (AUROC = 0.936 and AUPR = 0.947) and NetLapRLS (AUROC = 0.923 and AUPR = 0.936) (Supplementary Fig. S1 and Table S4). In addition, AOPEDF outperforms DeepWalk (Zong et al., 2017), a state-of-the-art deep learning approach (Supplementary Fig. S2).
3.3 Performance of AOPEDF on the external validation
Cross-validation on retrospective data probably leads to overoptimistic results. For object performance evaluation, we further collected experimentally validated DTIs from DrugCentral and ChEMBL databases, as two external validation sets (Supplementary Table S3), which can be used to evaluate the generalizable ability of models. The DrugCentral validation set contains 1507 DTIs that were not used in the training set, while the ChEMBL validation set contains 3034 DTIs that were not used in the training set as well. Figure 2a and b illustrates the performance comparison from the DrugCentral validation set. AOPEDF achieves a higher performance over other methods in terms of both AUROC and AUPR. Specifically, AOPEDF achieves AUROC value of 0.868, outperforming that of NeoDTI (0.843), deepDTnet (0.831), RLSWNN (0.772), KBMF2K (0.774) and NetLapRLS (0.738). AOPEDF achieves AUPR value of 0.869, outperforming that of NeoDTI (0.859), deepDTnet (0.861), RLSWNN (0.821), KBMF2K (0.807) and NetLapRLS (0.769) as well. Figure 3a and b shows the performance comparison on the ChEMBL validation set. AOPEDF still yields the best prediction performance in comparison with the other methods. AOPEDF achieves an AUROC value of 0.768 in comparison to NeoDTI (0.744), deepDTnet (0.702), RLSWNN (0.692), KBMF2K (0.648) and NetLapRLS (0.593). AOPEDF achieves an AUPR value of 0.764, outperforming that of NeoDTI (0.745), deepDTnet (0.739), RLSWNN (0.722), KBMF2K (0.684) and NetLapRLS (0.642). However, there are partly overlap between two external validation sets (Supplementary Table S3). The generalizable ability of AOPEDF is warranted to be tested further using more independent validation sets in the future.
Fig. 2.
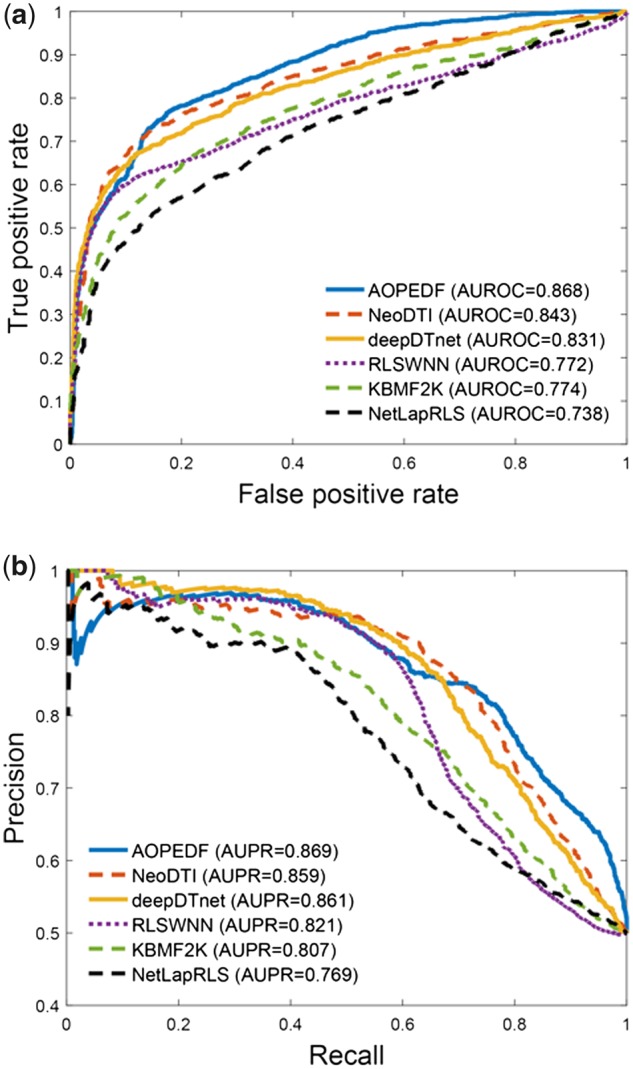
Evaluation of AOPEDF on the external validation set collected from the DrugCentral database (see Section 2). (a) Receiver operating characteristic (ROC) curves of prediction results obtained by applying AOPEDF and five previously published methods. (b) Precision–recall (PR) curves of prediction results obtained by applying AOPEDF and five previously published methods. AUROC, the area under ROC curve; AUPR, the area under PR curve
Fig. 3.
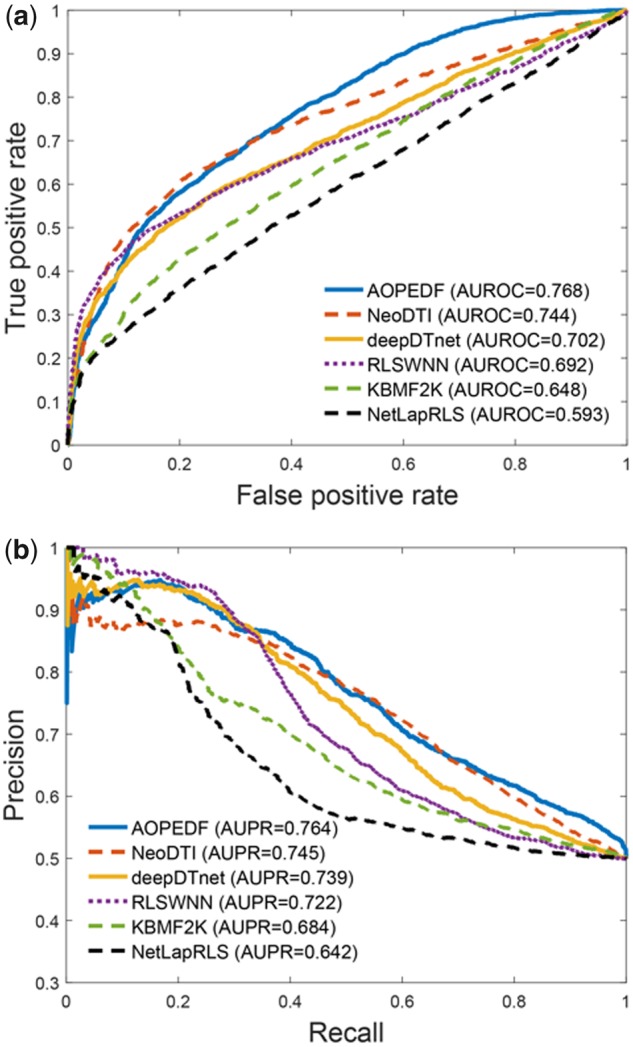
Evaluation of AOPEDF on the external validation set collected from the ChEMBL database (see Section 2). (a) ROC curves of prediction results obtained by applying AOPEDF and five previously published methods. (b) PR curves of prediction results obtained by applying AOPEDF and five previously published methods. AUROC, the area under ROC curve; AUPR, the area under PR curve
3.4 Performance of AOPEDF by ablation analysis
AOPEDF contains two parts, that is, AROPE for feature extraction and deep forest for classification. To examine the contribution of each component, we compared AOPEDF with several combinations. First, we replaced AROPE with LINE (Tang et al., 2015) for feature extraction. Specifically, we integrated 15 networks using AROPE and LINE respectively, and then used the deep forest for prediction. LINE is another network embedding method, which explicitly preserves the first two order proximities, here denoted as , respectively. This operation can inspect the contribution of AROPE. As shown in Supplementary Table S5, we found that AROPE outperformed both . This finding suggested that preserved high-order proximities may provide more effective information for classification. To inspect the contribution of deep forest, we compare deep forest classifier with other traditional classifiers using the same features extracted from AROPE. Specifically, for support vector machine (SVM) (Chang and Lin, 2011), we use a soft margin SVM with linear kernel, which performed better than radial basis function kernel in our experiments. We used a standard RF with 1000 trees, which get the best performance. For DNN (LeCun et al., 2015), we use an MLP having three hidden layers, with 1000, 500 and 200 units, respectively. We use ReLU as activation function and Adam optimizer with learning rate 0.001 to perform gradient descent. The evaluation results of these combinations are reported in Supplementary Table S5. We found that deep forest achieved the best performance. We also evaluated the combination of LINE with traditional classifiers, from which we can further validate the contribution of AROPE and deep forest. In addition, we found that performance of assembling in total 15 networks is much higher than single networks under AOPEDF framework (Supplementary Table S6), indicating power of heterogenous biological network integration. However, when we left each network out, we only found the marginal improvement of in total 15 networks compared to 14 networks (Supplementary Table S7).
3.5 Case study: drug repurposing for substance abuse disorder
Substance abuse disorder, also termed drug addiction, is a serious public health issue and there are no effective treatments available in the clinic (Lo Coco et al., 2019). We next turn to inspect whether AOPEDF could identify molecular targets from marketed drugs in the potential treatment of substance abuse disorder. Here, we centered on 64 G-protein-coupled receptors (GPCRs) which are related to substance abuse disorder (Chen et al., 2019). In total, we computationally identify 648 potential interactions connecting 64 GPCRs and 732 known drugs based on the top 20 predicted candidates by AOPEDF. The network visualization of the top 20 predicted candidates of 64 GPCRs is illustrated in Figure 4. Among the AOPEDF-predicted DTIs, multiple candidates can be supported by previous published literatures. For instance, aripiprazole, an atypical antipsychotic medication, primarily used in the treatment of schizophrenia and bipolar disorder, was predicted by AOPEDF to interact with histamine H3 receptors (HRH3). Such a prediction can be supported by a systematic, unbiased GPCR experimental assay (Lounkine et al., 2012). Besides, we also found that aripiprazole is related to drug abuse (Brunetti et al., 2012). Risperidone, an atypical antipsychotic, is approved to treat schizophrenia, bipolar disorder and irritability associated with autism. Here, we computationally identified that risperidone has potential interactions with HRH3 as well predicted by AOPEDF. This prediction is supported by the previous finding that HRH3 was involved in the pharmacodynamics and clinical efficacy of risperidone (Wei et al., 2012). In addition, risperidone was reported to mediate preclinical efficacy of substance abuse (Machielsen and de Haan, 2009). Haloperidol is a typical antipsychotic medication used in the treatment of schizophrenia, tics in Tourette syndrome, mania in bipolar disorder, nausea and vomiting, delirium, agitation, acute psychosis and hallucinations in alcohol withdrawal. AOPEDF predicts that haloperidol interacts with cholinergic muscarinic 2 receptor, which is also supported by a previous publication (Swathy and Banerjee, 2017). Furthermore, haloperidol was reported to involve in potential treatment of drug abuse as well (Hoffman et al., 1986). In summary, these case studies suggest potential of identifying new molecular targets from marketed drugs in potential treatment of substance abuse disorder. All predictions warrant further preclinical and clinical validations. From a translational perspective, if broadly applied, AOPEDF developed here could help accelerate therapeutic development for multiple understudied diseases as well.
Fig. 4.

An AOPEDF-predicted drug–target network connects 64 substance abuse disorder-related GPCRs and 164 drugs. GPCRs are denoted by gray square nodes. Drug nodes (circle) are labeled by the first-level of the Anatomical Therapeutic Chemical Classification System. Node size represents the degree (connectivity). Several GPCRs and predicted drugs were highlighted and discussed in the main text
4 Discussion and conclusion
In this article, we proposed a deep learning-based computational approach for molecular target identification from known drugs, termed AOPEDF. AOPEDF preserves different order proximity information from 15 networks in a constructed drug–target/protein–disease heterogeneous network. Specifically, AOPEDF formulates DTI prediction as a binary classification task and feeds the low-dimensional but informative vector representations of features for both drugs and proteins into a cascade deep forest classifier. The low-dimensional feature vectors learned by AOPEDF capture rich context information as well as the topological structure of individual networks. In comparison to DNNs approaches, the deep forest classifier of AOPEDF performs excellently in classification but has much fewer hyper-parameters and its performance is quite robust to hyper-parameter settings. Neural network methods often achieve state-of-the-art performance with a ‘black-box’, for which the reasons underlying a prediction cannot be explicitly presented. AOPEDF makes prediction by inferring decision rules from data, which is more effective in generating interpretable predictions from biologically relevant features. We have validated the prediction ability of AOPEDF in terms of 5-fold cross-validation, two external validation sets and a case study. Systematic evaluation demonstrates that AOPEDF achieves state-of-the-art performance for the discovery of DTIs and potential applications of target-centered drug repurposing for substance abuse disorder in the case study. Theoretically, AOPEDF can process various high-dimensional features by utilizing multiple networks with different order features. The deep forest classifier implemented in AOPEDF will utilize the information it needs automatically. In our experiments, for each network we preserve different order proximity, and then choose the best order according to the feature importance and prediction results. Overall, AOPEDF is a scalable framework, which can incorporate more drug and target-related information from various publicly available databases and literatures. Therefore, if broadly applied, we believe that AOPEDF offers a powerful and useful tool to facilitate drug repurposing and therapeutic development in various understudied diseases.
We acknowledged several potential limitations in current study. In this study, we used a low binding affinity value of 10 μM as a threshold to define a physical DTI. Recent studies suggested that weak-binding drugs play crucial roles in therapeutic development as well (Ohlson, 2008; Wang et al., 2017). Our recent studies have successfully applied this low binding affinity cutoff of 10 μM for drug repurposing (Cheng et al., 2018, 2019a, b). However, a stronger binding affinity threshold (e.g. 1 μM) may be a more suitable cutoff in drug discovery although it will generate a small size of drug–target network (Pahikkala et al., 2015). In addition, random selection of unknown drug–target pairs as negative samples may generate possible false positive rate of AOPEDF models. We have repeated the 10 times of random 5-fold cross-validation on each method and added the mean and standard deviation in the Supplementary Table S4. We found that AOPEDF revealed the smallest standard derivation compared to other approaches, suggesting a minor influence of low-quality negative samples on performance. Building regression models to predict the continuous binding affinity (such as Kd, Ki, IC50, EC50) will avoid selecting different biological threshold and avoiding lack of publicly available negative drug–target pairs as well. We found that performance of 5-fold cross-validation (Supplementary Fig. S1) was much higher than performance of two external validation sets (Figs 2 and 3). One possible reason is that experimental assays of DTIs are different between training set and the external validation sets. However, the overfitting risk of AOPEDF is needed to be tested by more independent validation sets in the future. Ablation analysis reveals a marginal improvement of in total 15 networks compared to 14 networks (Supplementary Table S7). Thus, potential risk of information redundancy from multiple networks’ integration is warranted to be tested further. Other feature extraction approaches, such as convolution neural networks (LeCun et al., 2015), could be used to avoid information redundancy in current AOPEDF framework. Finally, the deep forest classifier has much fewer hyper-parameters compared to DNN algorithms, potentially making the proposed method more robust to parameter settings. As shown in the Supplementary Table S8, AOPEDF reveals high robustness by the hyper-parameter settings. Finally, all experimentally validated drug–target networks, including training set and two external validation sets, and codes used in this study are free available at https://github.com/ChengF-Lab/AOPEDF.
Funding
This work was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under Award Number K99HL138272 and R00HL138272 to F.C. This work has been also supported in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract HHSN261200800001E. This research was supported (in part) by the Intramural Research Program of NIH, Frederick National Lab, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government.
Conflict of Interest: none declared.
Supplementary Material
References
- Apweiler R. et al. (2004) UniProt: the universal protein knowledgebase. Nucleic Acids Res., 32, D115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleakley K., Yamanishi Y. (2009) Supervised prediction of drug–target interactions using bipartite local models. Bioinformatics, 25, 2397–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunetti M. et al. (2012) Aripiprazole, alcohol and substance abuse: a review. Eur. Rev. Med. Pharmacol. Sci., 16, 1346–1354. [PubMed] [Google Scholar]
- Cao S. et al. (2015) GraRep: learning graph representations with global structural information. In: Proceedings of the 24th ACM International on Conference on Information and Knowledge ManagementACM, Melbourne, Australia, pp. 891–900.
- Chang C.-C., Lin C.-J. (2011) LIBSVM: a library for support vector machines. ACM Trans. Intell. Syst. Technol., 2, 1–27. [Google Scholar]
- Chen M. et al. (2019) DAKB-GPCRs: an integrated computational platform for drug abuse related GPCRs. J. Chem. Inf. Model., 59, 1283–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A.C. et al. (2007) Structure-based maximal affinity model predicts small-molecule druggability. Nat. Biotechnol., 25, 71–75. [DOI] [PubMed] [Google Scholar]
- Cheng F. et al. (2012a) Prediction of chemical-protein interactions network with weighted network-based inference method. PLoS One, 7, e41064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F. et al. (2012b) Prediction of drug–target interactions and drug repositioning via network-based inference. PLoS Comput. Biol., 8, e1002503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F. et al. (2018) Network-based approach to prediction and population-based validation of in silico drug repurposing. Nat. Commun., 9, 2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F. et al. (2019a) A genome-wide positioning systems network algorithm for in silico drug repurposing. Nat. Commun., 10, 3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F. et al. (2019b) Network-based prediction of drug combinations. Nat. Commun., 10, 1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui P. et al. (2019) A survey on network embedding. IEEE Trans. Knowl. Data Eng., 31, 833–852. [Google Scholar]
- Donald B.R. (2011) Algorithms in Structural Molecular Biology. MIT Press, Cambridge, MA. [Google Scholar]
- Eckart C., Young G. (1936) The approximation of one matrix by another of lower rank. Psychometrika, 1, 211–218. [Google Scholar]
- Gaulton A. et al. (2012) ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res., 40, D1100–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonen M. (2012) Predicting drug–target interactions from chemical and genomic kernels using Bayesian matrix factorization. Bioinformatics, 28, 2304–2310. [DOI] [PubMed] [Google Scholar]
- Haggarty S.J. et al. (2003) Multidimensional chemical genetic analysis of diversity-oriented synthesis-derived deacetylase inhibitors using cell-based assays. Chem. Biol., 10, 383–396. [DOI] [PubMed] [Google Scholar]
- He Z. et al. (2010) Predicting drug–target interaction networks based on functional groups and biological features. PLoS One, 5, e9603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Boussard T. et al. (2007) The pharmacogenetics and pharmacogenomics knowledge base: accentuating the knowledge. Nucleic Acids Res., 36, D913–D918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman A.S. et al. (1986) Catatonic reaction to accidental haloperidol overdose: an unrecognized drug abuse risk. J. Nerv. Ment. Dis., 174, 428–430. [DOI] [PubMed] [Google Scholar]
- Keiser M.J. et al. (2007) Relating protein pharmacology by ligand chemistry. Nat. Biotechnol., 25, 197–206. [DOI] [PubMed] [Google Scholar]
- Kuruvilla F.G. et al. (2002) Dissecting glucose signalling with diversity-oriented synthesis and small-molecule microarrays. Nature, 416, 653–657. [DOI] [PubMed] [Google Scholar]
- Law V. et al. (2014) DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res., 42, D1091–D1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeCun Y. et al. (2015) Deep learning. Nature, 521, 436–444. [DOI] [PubMed] [Google Scholar]
- Liu T. et al. (2007) BindingDB: a web-accessible database of experimentally determined protein–ligand binding affinities. Nucleic Acids Res., 35, D198–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Coco G. et al. (2019) Group treatment for substance use disorder in adults: a systematic review and meta-analysis of randomized-controlled trials. J. Subst. Abuse Treat., 99, 104–116. [DOI] [PubMed] [Google Scholar]
- Lounkine E. et al. (2012) Large-scale prediction and testing of drug activity on side-effect targets. Nature, 486, 361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machielsen M.W., de Haan L. (2009) Differences in efficacy on substance abuse between risperidone and clozapine supports the importance of differential modulation of dopaminergic neurotransmission. Psychopharmacol. Bull., 42, 40–52. [PubMed] [Google Scholar]
- Mei J.P. et al. (2013) Drug–target interaction prediction by learning from local information and neighbors. Bioinformatics, 29, 238–245. [DOI] [PubMed] [Google Scholar]
- Mendez D. et al. (2019) ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res., 47, D930–D940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G.M. et al. (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem., 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlson S. (2008) Designing transient binding drugs: a new concept for drug discovery. Drug Discov. Today, 13, 433–439. [DOI] [PubMed] [Google Scholar]
- Pahikkala T. et al. (2015) Toward more realistic drug–target interaction predictions. Brief. Bioinform., 16, 325–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson A.J. et al. (2014) The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucleic Acids Res., 42, D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman L. et al. (2011) Combining drug and gene similarity measures for drug–target elucidation. J. Comput. Biol., 18, 133–145. [DOI] [PubMed] [Google Scholar]
- Perozzi B. et al. (2014) DeepWalk: online learning of social representations. In: Proceedings of the 20th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining ACM, New York, NY, USA, pp. 701–710.
- Rarey M. et al. (1996) A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol., 261, 470–489. [DOI] [PubMed] [Google Scholar]
- Strang G. (2006) Linear Algebra and Its Applications, 4th edn India Edition. ISBN-10: 9788131501726. [Google Scholar]
- Swathy B., Banerjee M. (2017) Haloperidol induces pharmacoepigenetic response by modulating miRNA expression, global DNA methylation and expression profiles of methylation maintenance genes and genes involved in neurotransmission in neuronal cells. PLoS One, 12, e0184209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J. et al. (2015) LINE: large-scale information network embedding. In: Proceedings of the 24th International Conference on World Wide Web International World Wide Web Conferences Steering Committee, Florence, Italy, pp. 1067–1077.
- Ursu O. et al. (2019) DrugCentral 2018: an update. Nucleic Acids Res., 47, D963–D970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Laarhoven T., Marchiori E. (2013) Predicting drug–target interactions for new drug compounds using a weighted nearest neighbor profile. PLoS One, 8, e66952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan F. et al. (2019) NeoDTI: neural integration of neighbor information from a heterogeneous network for discovering new drug–target interactions. Bioinformatics, 35, 104–111. [DOI] [PubMed] [Google Scholar]
- Wang J. et al. (2017) Weak-binding molecules are not drugs? Toward a systematic strategy for finding effective weak-binding drugs. Brief. Bioinform., 18, 321–332. [DOI] [PubMed] [Google Scholar]
- Wang W. et al. (2014) Drug repositioning by integrating target information through a heterogeneous network model. Bioinformatics, 30, 2923–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Z. et al. (2012) A pharmacogenetic study of risperidone on histamine H3 receptor gene (HRH3) in Chinese Han schizophrenia patients. J. Psychopharmacol., 26, 813–818. [DOI] [PubMed] [Google Scholar]
- Wishart D.S. et al. (2018) DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res., 46, D1074–D1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z. et al. (2010) Semi-supervised drug–protein interaction prediction from heterogeneous biological spaces. BMC Syst. Biol., 4(Suppl. 2), S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H. et al. (2016) Therapeutic target database update 2016: enriched resource for bench to clinical drug target and targeted pathway information. Nucleic Acids Res., 44, D1069–D1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X. et al. (2020) Target identification among known drugs by deep learning from heterogeneous networks. Chem. Sci. In press. DOI: 10.1039/C9SC04336E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z. et al. (2018) Arbitrary-order proximity preserved network embedding. In: Proceedings of the 24th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining ACM, London, UK, pp. 2778–2786.
- Zheng X. et al. (2013) Collaborative matrix factorization with multiple similarities for predicting drug–target interactions. In: Proceedings of the 19th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining ACM, Chicago, IL, USA, pp. 1025–1033.
- Zhou Z.-H., Feng J. (2017) Deep forest: towards an alternative to deep neural networks. In: Proceedings of the Twenty-sixth International Joint Conference on Artificial Intelligence South Wharf, Australia, pp. 3553–3559.
- Zong N. et al. (2017) Deep mining heterogeneous networks of biomedical linked data to predict novel drug–target associations. Bioinformatics, 33, 2337–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.