

Abstract
The CRISPR/Cas9 nucleases have been widely applied for genome editing in various organisms. Cas9 nucleases complexed with a guide RNA (Cas9–gRNA) find their targets by scanning and interrogating the genomic DNA for sequences complementary to the gRNA. Recognition of the DNA target sequence requires a short protospacer adjacent motif (PAM) located outside this sequence. Given that the efficiency of target location may depend on the strength of interactions that promote target recognition, here we sought to compare affinities of different Cas9 nucleases for their cognate PAM sequences. To this end, we measured affinities of Cas9 nucleases from Streptococcus pyogenes, Staphylococcus aureus, and Francisella novicida complexed with guide RNAs (gRNAs) (SpCas9–gRNA, SaCas9–gRNA, and FnCas9–gRNA, respectively) and of three engineered SpCas9–gRNA variants with altered PAM specificities for short, PAM-containing DNA probes. We used a “beacon” assay that measures the relative affinities of DNA probes by determining their ability to competitively affect the rate of Cas9–gRNA binding to fluorescently labeled target DNA derivatives called “Cas9 beacons.” We observed significant differences in the affinities for cognate PAM sequences among the studied Cas9 enzymes. The relative affinities of SpCas9–gRNA and its engineered variants for canonical and suboptimal PAMs correlated with previous findings on the efficiency of these PAM sequences in genome editing. These findings suggest that high affinity of a Cas9 nuclease for its cognate PAM promotes higher genome-editing efficiency.
Keywords: DNA-binding protein, DNA–protein interaction, CRISPR/Cas, fluorescence, RNA binding protein, Cas9, fluorescent Cas9 beacon, genome editing
Introduction
The type II CRISPR–Cas3 bacterial adaptive immune systems that use a single Cas9 protein programmed with a guide RNA (gRNA) to identify and degrade target DNA have been harnessed for a vast range of genome editing applications (1–6). The type IIA Cas9 nucleases from Streptococcus pyogenes (SpCas9) and Staphylococcus aureus (SaCas9) are commonly used to introduce desired mutations in eukaryotic cells. SaCas9 is smaller than SpCas9, which makes its delivery to cells using viral vectors more convenient (7, 8). In contrast to SpCas9 and SaCas9, the type IIB Cas9 from Francisella novicida (FnCas9) cleaves the target DNA in a staggered pattern to leave 4-nucleotide 5′-overhangs (9). It was also reported that FnCas9 exhibits significantly higher cleavage specificity as compared with SpCas9 (10). Cas9 derivatives that lack endonuclease activity but can bind to targets specified by gRNA (dCas9) are used for transcriptome modulation, base-specific genome editing, and visualization of genomic loci in live cells (11–13).
Both recognition and cleavage of target DNA by Cas9–gRNA complexes require a short protospacer adjacent motif (PAM) located near the targeted sequence (14). The PAM sequences are diverse among orthologous Cas9 nucleases. The requirement for PAM sequences constrains the choice of sites amenable for genome editing. To address this issue, engineered Cas9 mutants with altered PAM specificities have been developed, but many often show editing activities lower than that of the WT protagonists (15–19). Crystal structures of SpCas9 and SaCas9 bound to target DNAs have revealed that these enzymes employ a major-groove PAM recognition mechanism involving direct and water-mediated hydrogen-bonding interactions with cognate canonical PAMs (5′-NGG and 5′-NNGRRT for SpCas9 and SaCas9, respectively) (20, 21). Genome editing experiments have showed that in vivo, PAM specificities of SpCas9 and its engineered variants are not quite strict. One well-known example is SpCas9 recognition of NAG and NGA sequences as PAM sites that can support cleavage, although significantly less efficiently than the consensus NGG PAM (22, 23). The recognition of noncanonical PAM sequences should be taken into account when designing target sequences to minimize off-target effects (22). Factors that determine efficiencies of various suboptimal PAMs are not fully understood.
The Cas9–gRNA effector complexes locate a target site by scanning and interrogating the genomic DNA (24–28). The Cas9 target search in eukaryotic cells is a slow process that may limit the efficiency of genome editing, particularly when the concentration of Cas9–gRNA complexes is low to ensure specificity of target recognition (27, 28). Single-molecule and bulk experiments have shown that the DNA interrogation process is triggered by Cas9–gRNA binding to PAM that leads to destabilization of the DNA duplex close to PAM and allows initial pairing of crRNA with complementary DNA bases (24, 29). The initial Cas9–PAM interaction is often a target for anti-CRISPR proteins and synthetic small inhibitors (30, 31). Because the rate of a target location by DNA-binding proteins may depend on the strength of interactions that initiate target recognition (32), the affinity of a CRISPR–Cas effector for PAM might be one of the factors determining the efficiency of genome editing. Given the diversity of DNA-targeting CRISPR–Cas systems, it is thus of interest to compare affinities of both native and engineered CRISPR–Cas effectors for cognate PAM sequences. Here, we report a comparison of affinities of SpCas9–gRNA, SaCas9–gRNA, FnCas9–gRNA, and three engineered SpCas9–gRNA variants with altered PAM specificities (Cas9–VQR, xCas9, and Cas9–NG) for various PAMs in the context of short nontarget DNA substrates bearing cognate PAM sequences. We find that affinities of the studied Cas9 effector complexes for their optimal PAM sequences differ significantly, with the highest affinity observed for SaCas9. The results show that the relative affinities of SpCas9–gRNA and its engineered variants for consensus and suboptimal PAM sequences correlate with reported effectiveness of these PAMs in genome editing, suggesting that strong binding to PAM may lead to higher genome editing efficiency. The data also suggest a difference between modes of FnCas9 and SpCas9 interaction with PAM-proximal DNA during target location.
Results
Experimental setup
The affinity of SpCas9–gRNA for canonical NGG PAM has been previously assessed by measuring SpCas9–gRNA binding to ∼50-bp-long model DNA substrates containing no protospacer matching the gRNA spacer but bearing multiple NGG sites (24, 29, 31). As expected, in contrast to target DNA sequences, such nontarget substrates bound to Cas9–gRNA relatively weakly (24, 29, 31). We previously found that nontarget DNA substrates bearing short duplex segments noncomplementary to gRNA upstream of PAM bind to SpCas9–gRNA significantly stronger than similar substrates bearing longer upstream segments (29). The difference in the binding strength was suggested to result from obstructing the entry of duplex DNA beyond a certain threshold distance from PAM into the Cas9–sgRNA interior by an element of the effector complex. In this work, we used ∼20-bp-long DNA substrates (probes) that contained a single PAM sequence and a short upstream segment that was either noncomplementary or complementary to the gRNA spacer as a tool to compare affinity of Cas9–gRNA complexes for various PAM sequences. We expected that comparatively high affinity of such probes for Cas9–gRNA may facilitate detection of interactions of the Cas9 effectors with suboptimal PAM sequences. The relative affinities of DNA probes were determined by measuring their ability to competitively affect the rate of Cas9–gRNA binding to “Cas9 beacons,” fluorescently labeled target DNA derivatives containing a protospacer complementary to spacer part of gRNA and a functional PAM (33, 34). Schematic representation of the beacon assay is shown in Fig. 1. Beacon 1 consists of two fully complementary oligonucleotides, whereas beacon 2 consists of three oligonucleotides: its nontarget strand contains a discontinuity. Cas9–gRNA binding to a beacon results in a readily measurable increase in fluorescence intensity. Beacons composed of three oligonucleotides show the same kinetics of fluorescence increase upon binding to Cas9–gRNA and dCas9–gRNA, but the kinetics of fluorescence increases observed upon Cas9–gRNA and dCas9–gRNA binding to beacons composed of two oligonucleotides may differ (33, 34). The rate of beacon binding and maximal increase in fluorescence intensity depend on the beacon sequence and structure, which can be varied easily (29, 35). The advantages of the beacon assay are that it reports only on functional effector complexes capable of specific binding to the beacon and allows detection of low-affinity binding of a competitor to the effector complex (34). The assay also allows one to calculate the dissociation constant of Cas9–gRNA with a DNA competitor probe, provided that nearly complete beacon binding to Cas9–gRNA in the presence of competitor is reached on a suitable time scale (minutes to a few hours) (29). In practice this condition is fulfilled when the competitor concentration is not too high and its affinity for Cas9–gRNA is not too strong. Beacons used in this work were designed to have binding rates in the absence of competitors on the scale of several minutes. Sequences of competitors were adjusted to avoid formation of hairpin structures. Structures of all beacons and competitor probes are shown in Figs. S2 and S3. Competitors were preincubated with Cas9–gRNA complexes for 15 min prior to the addition of beacon.
Figure 1.
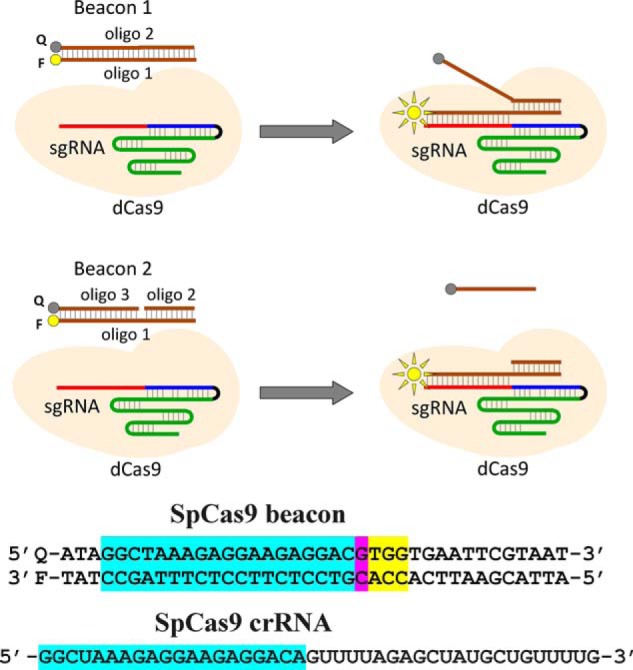
Principle of the Cas beacon assay and structure of SpCas9 beacon. Beacon 1 consists of two fully complementary oligonucleotides (oligo), whereas beacon 2 has a discontinuity in its nontarget strand. The PAM-distal ends of the beacon target and nontarget strands are labeled with a fluorophore (F) and a fluorescence quencher (Q), respectively. The baseline fluorescence intensity of beacon is low because of proximity of the fluorophore with the quencher. The Cas9–gRNA effector complex binds to beacon in a way that mimics their binding to target DNA, which leads to separation of the fluorophore and the quencher and a readily detectable increase in fluorescence intensity. The sequence of SpCas9 crRNA and beacon used in experiments with SpCas9 and are shown below the panel. The crRNA spacer and beacon protospacer bases complementary to the spacer sequence are highlighted in blue, the mismatched protospacer bases are highlighted in pink, and the PAM sequence is highlighted in yellow.
Comparison of affinities of SpCas9 and its engineered variants with altered PAM specificities for optimal and suboptimal PAMs
The reported PAM preferences of SpCas9 and its engineered variants may correlate with their affinities for corresponding PAMs. To test this conjecture, we measured the affinities of dSpCas9, dCas9–VQR, dxCas9, and dCas9–NG effector complexes for short DNA probes bearing PAM sequences of interest using the competition Cas9 beacon assay. The engineered Cas9–VQR variant recognizes the NGAN sequence as PAM (15). The base in the fourth position affects the efficiency of Cas9–VQR genome editing. In human and plant cells the NGAG > NGAT ≈ NGAA > NGAC relative PAM preferences have been found (15, 36). The preference for the NGAG PAM is consistent with base-specific recognition of a guanine base in the fourth position (37). The relative affinities of dCas9–VQR–gRNA for the NGAN PAMs were compared in the context of competitor probes 1.1–1.8 (Fig. 2A). The probes contained the TGAG, TGAT, TGAA, or TGAC PAM variants and 2-bp segments upstream of PAM, which were either noncomplementary (probes 1.1–1.4) or complementary (probes 1.5–1.8) to the gRNA spacer. As expected, probes bearing the complementary 2-bp segments upstream of PAM were more effective competitors than corresponding fully noncomplementary probes. Probes 1.1 and 1.5 bearing the TGAG PAM were significantly stronger competitors of beacon binding and thus had higher affinities for Cas9–VQR–gRNA than probes bearing other TGAN PAM variants (Fig. 2, B and C, and Fig. S4). The calculated Kd for probe 1.1 was 6.4 ± 1.3 nm, whereas Kd for probes 1.2–1.4 were ∼200 nm. Although we did not observe a difference between competitor strengths of probes 1.2–1.4 (Fig. 2B), experiments with partially complementary probes showed that probes 1.6 and 1.7 containing the TGAA and TGAT PAM sequences, respectively, are stronger competitors than probe 1.8, which bears the TGAC PAM (Fig. 2C). The data thus show that the Cas9–VQR PAM variants known to be more efficient in genome editing have higher affinities for Cas9–VQR–gRNA, suggesting that strong binding to PAM favors Cas9–VQR genome editing.
Figure 2.
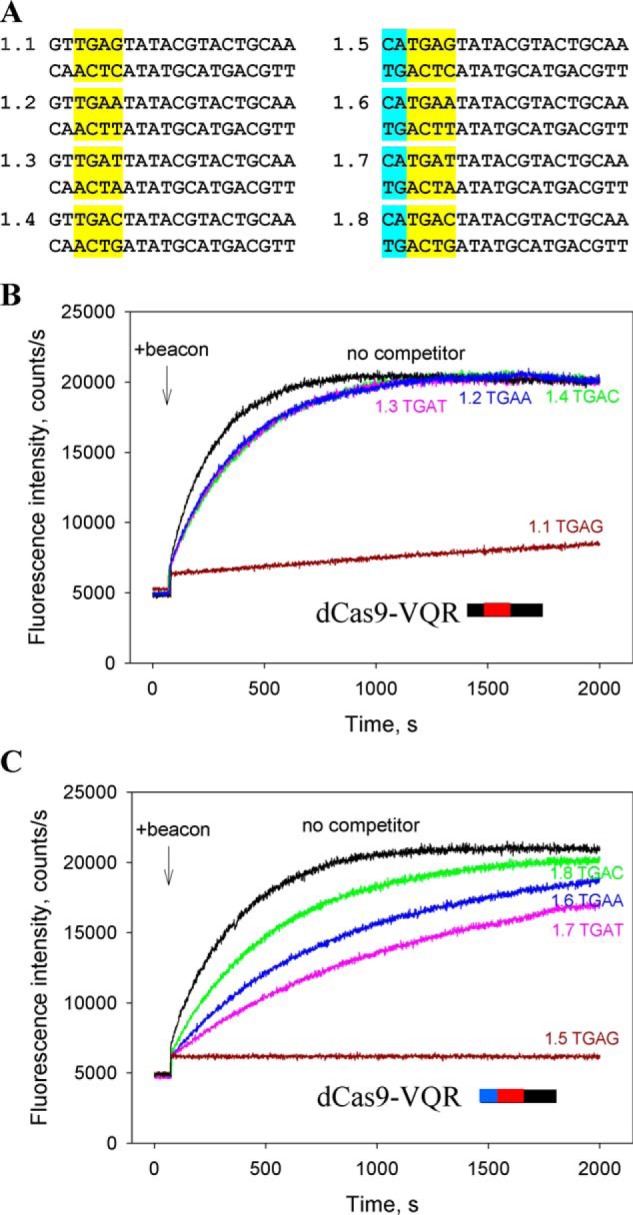
Competition assays with dCas9–VQR. A, structures of competitor DNA probes. The probes bore 2-bp segments upstream of PAM sequences that were either noncomplementary (probes 1.1–1.4) or complementary (probes 1.5–1.8) to gRNA. The PAM sequences are highlighted in yellow, and 2-bp segments matching the spacer sequence are highlighted in blue. B and C, effect of competitor probes 1.1–1.4 (B) and 1.5–1.8 (C) that bore indicated PAM sequences on the kinetics of dCas9–VQR–gRNA binding to beacon. The concentrations of competitors were 200 and 40 nm in experiments shown in B and C, respectively.
The relative affinities of dSpCas9–gRNA for various PAM sequences were compared in the context of parent probe 2.1 bearing the canonical TGG PAM and its derivatives 2.2–2.8 in which the TGG PAM sequence was substituted for either suboptimal TGA/TAG or nonfunctional TGT, TGC, TTG, TCG, and TAA sequences (Fig. S3). The probes bore 2-bp segments complementary to gRNA spacer immediately upstream of PAM to increase the strength and specificity of the binding. Probe 2.1 bearing the TGG PAM was a much stronger competitor than all other probes (Fig. 3). The beacon-binding rate dropped down by ∼400-fold in the presence of 200 nm of probe 2.1, whereas the addition of probes 2.2 and 2.3 with suboptimal TGA and TAG PAMs, correspondingly, caused a decrease in the binding rate by only 1.7-fold. Probes 2.4–2.8 bearing nonfunctional PAM sequences were the weakest competitors and caused less than 25% decrease in the beacon-binding rate (Fig. 3).
Figure 3.
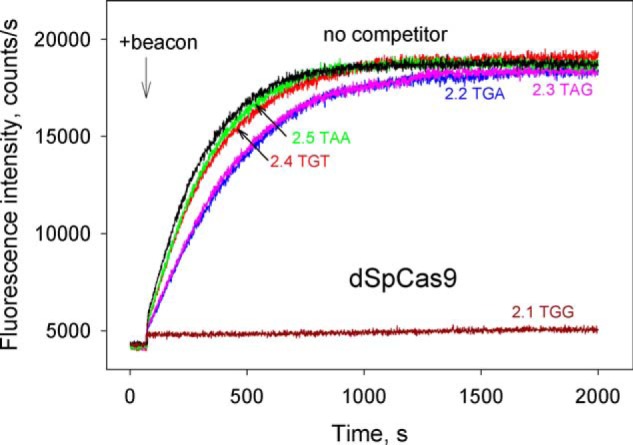
Competition assays with dSpCas9 using DNA probes bearing canonical, suboptimal, or nonfunctional PAM sequences. Shown are the effects of competitor DNA probes 2.1–2.5 (shown in Fig. S3) bearing indicated PAM sequences on the kinetics of dCas9–gRNA binding to beacon. The concentration of all competitor probes was 200 nm.
The xCas9 and Cas9–NG variants of SpCas9 recognize the relaxed NGN sequence as PAM (although SpCas9–NG was found to be less active at NGC relative to NGD PAMs in human cells) (17, 18). In addition to its broader PAM compatibility, xCas9 has considerably reduced off-target activity compared with WT SpCas9 (17, 38, 41). The available data also show that in many cases Cas9–NG is more efficient than xCas9 in inducing mutations at target sites with NGN PAMs (18, 38–41). We compared affinities of dxCas9–gRNA and dCas9–NG–gRNA for probes 3.1–3.5 containing AGG, AGA, AGT, AGC, and ATG PAM variants and 2-bp segments complementary to the gRNA spacer upstream of PAM (shown in Fig. 4A) using the same beacon. In the absence of competitors, dCas9–NG–gRNA bound beacon ∼6-fold faster than dxCas9–gRNA (Fig. 4, B and C), consistent with the higher rate of Cas9–NG–induced DNA cleavage observed in in vitro assays of Cas9–NG and xCas9 activities (18). In the presence of 200 nm of probe 3.1 containing the AGG PAM, the rate of beacon binding to dxCas9–gRNA decreased 7-fold, whereas other probes caused ∼2-fold decreases in the beacon-binding rate (Fig. 4B). Much higher inhibitory effects were observed in similar experiments with dCas9–NG. The rate of beacon binding to dCas9–NG–gRNA decreased ∼600- and 120-fold in the presence of 200 and 40 nm of probe 3.1, respectively. The calculated Kd for probe 3.1 binding to dxCas9–gRNA was ∼80-fold higher than for dCas9–NG–gRNA (Kd = 30 ± 6 nm and Kd = 0.35 ± 0.11 nm, respectively). We compared the effects caused by probes 3.1–3.5 on beacon binding to Cas9–NG–gRNA at 40 nm probe concentrations. As can be seen from Fig. 4C and Fig. S5, the AGG PAM containing probe 3.1 was a slightly stronger inhibitor than probes 3.2 and 3.3 bearing, respectively, the AGT and AGA PAM sequences, whereas the inhibition effect caused by probe 3.4 (AGC PAM) was weaker than that of probe 3.1 by 4.8-fold. The latter observation is in agreement with lower Cas9–NG editing activity at NGC sites (18). Control probe 3.5 bearing a T at the second PAM position was a weaker inhibitor than probes 3.1–3.4 (Fig. 4C). The data thus show that dCas9–NG binds short DNA probes bearing the relaxed NGN PAM stronger than dxCas9, which suggests that the observed difference between xCas9 and Cas9–NG cleavage efficiencies (18, 38–41) may be, at least in part, accounted for by stronger interaction of Cas9–NG with its PAM. Overall, the above data show that there is a correspondence between the affinities of the studied Cas9 effectors for PAM sequences and their reported in vivo activities.
Figure 4.
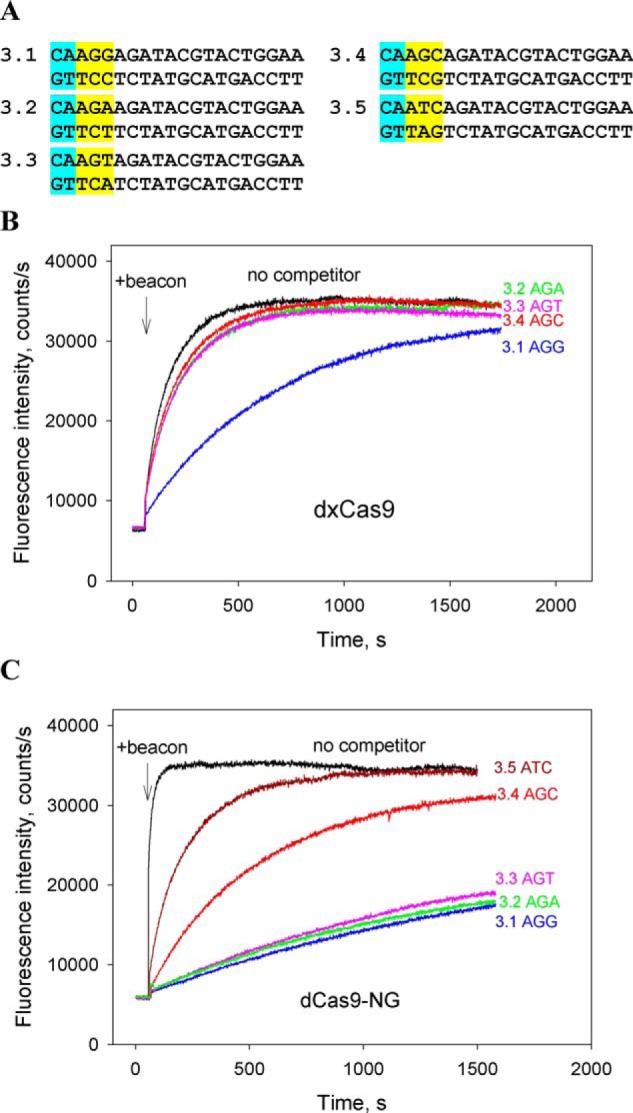
Competition assays with dxCas9 and dCas9–NG. A, structures of competitor DNA probes. The probes bore 2-bp segments upstream of PAM sequences (highlighted in yellow) that were complementary to gRNA. B and C, effect of competitor probes bearing indicated PAM sequences on the kinetics of dxCas9–gRNA (B) and dCas9–NG–gRNA (C) binding to beacon. The concentrations of competitor DNA probes were 200 and 40 nm in experiments shown in B and C, respectively.
Affinity of SaCas9 for its canonical PAM is significantly higher than that of SpCas9
We compared affinities of dSpCas9 and dSaCas9 for their PAMs in the context of noncomplementary to sgRNA probes 4.1–4.7 that had the same downstream boundary but different upstream edges at positions +1, −2, −3, −5, and −7 (Fig. 5A). The probes bore overlapping NGG and NNGRRG canonical PAM sequences and thus could be used in competition beacon assay experiments with both SaCas9 and SpCas9. The probes 4.1–4.3 bearing, respectively, no nucleotides and a 1- or 2-bp segment upstream of PAM very strongly inhibited dSaCas9–gRNA binding to beacon at 40 nm probe concentration, whereas further extensions of the upstream edge to −3, −5, and −7 positions in, respectively, probes 4.4, 4.5, and 4.6 led to gradual decrease of the inhibitory effect (Fig. 5B). The substitution of a G for a T at the third PAM position of probe 4.3 almost eliminated the competition effect of resulting probe 4.7, proving that probe 4.3 binding was PAM-dependent (Fig. S6). As can be seen in Fig. 5C, similar dependence of relative probe competition strength on upstream probe boundary was found in experiments with dSpCas9, in agreement with our previous measurements performed under somewhat different conditions (29). However, the inhibition effects caused by the probes on beacon binding to dSaCas9 were much stronger than those observed with dSpCas9 (Fig. 5, B and C). Consistently, the Kd for binding of probe 4.5 to dSaCas9–gRNA was 26-fold lower than for binding to dSpCas9–gRNA (3.2 ± 0.5 and 85 ± 21 nm, respectively). The Kd values for probe 4.3 binding to dSpCas9–gRNA and dSaCas9–gRNA were 6.7 ± 0.7 and <0.4 nm, respectively.
Figure 5.
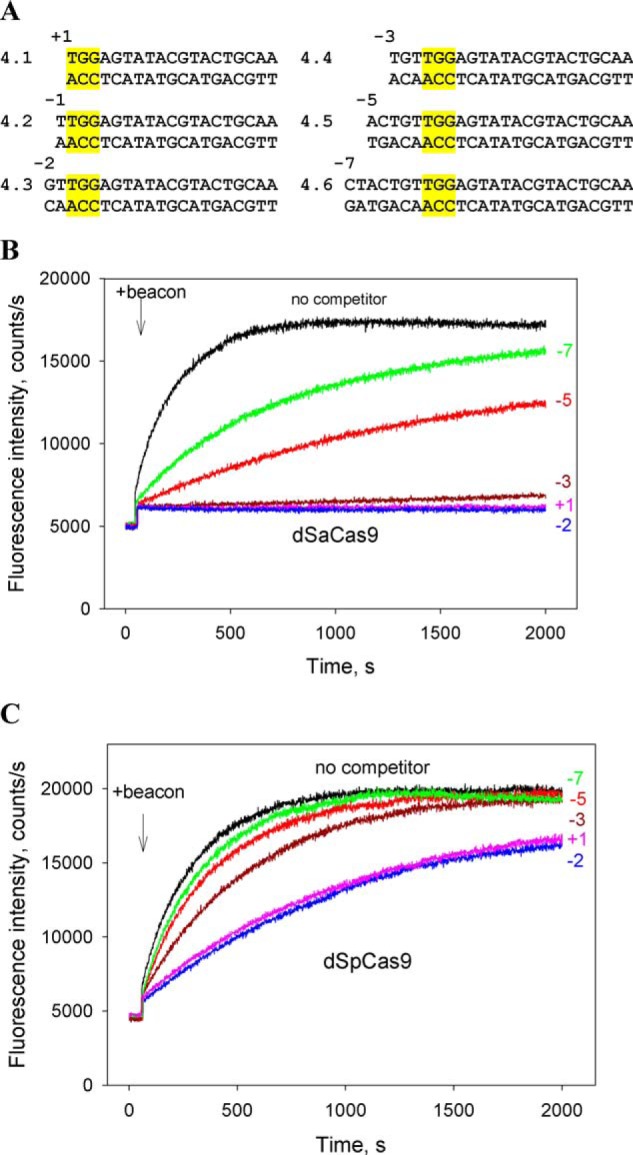
Competition assays with dSpCas9 and dSaCas9 using DNA probes with upstream edges at different positions. A, structures of competitor DNA probes. All probes bore no gRNA guide sequence complementarity. The SpCas9 PAM sequence is highlighted in yellow. B and C, effect of probes 4.1–4.6 on the kinetics of dSaCas9–gRNA (B) and dSpCas9–gRNA (C) binding to beacons. Positions of upstream edges of the probes are indicated in the panels. The concentration of all competitor probes was 40 nm.
Next, we conducted competition beacon assay measurements using as a nontarget competitor the pUC19 plasmid, which contains multiple (330 NGG and 78 NNGRRT) PAMs for both enzymes in various contexts. Consistent with data obtained with short probes, the dSaCas9–gRNA binding to the beacon was strongly competed by 3 nm of pUC19, whereas only ∼2-fold decrease in the rate of dSpCas9–gRNA binding to the beacon was observed (Fig. 6, A and B). We also measured the inhibition effects caused by probe 4.8 bearing a truncated PAM lacking the nonconserved upstream base pair in the 5′-TGGAGT PAM segment. As can be seen in Fig. S7 (A and B), probe 4.8 was a significantly weaker inhibitor than probe 4.1 bearing the full PAM sequence. This observation suggests that non–sequence-specific contacts of SaCas9 and SpCas9 with upstream nucleotides of their PAMs seen in crystal structures contribute noticeably to affinities toward PAMs (20, 21, 42). To verify the results of beacon assays, the binding of probe bearing a 2-bp segment upstream of PAM and control probe bearing mutated PAM sequences to dSaCas9–gRNA and dSpCas9–gRNA was measured by the electrophoretic mobility shift assay. As can be seen from Fig. S8, the probe affinity for dSaCas9–sgRNA is considerably higher than for dSpCas9–sgRNA. Overall, the above data show that dSaCas9–gRNA binds short PAM-containing substrates much stronger than does dSpCas9–gRNA, suggesting that SaCas9 affinity for PAM is significantly higher than that of SpCas9.
Figure 6.
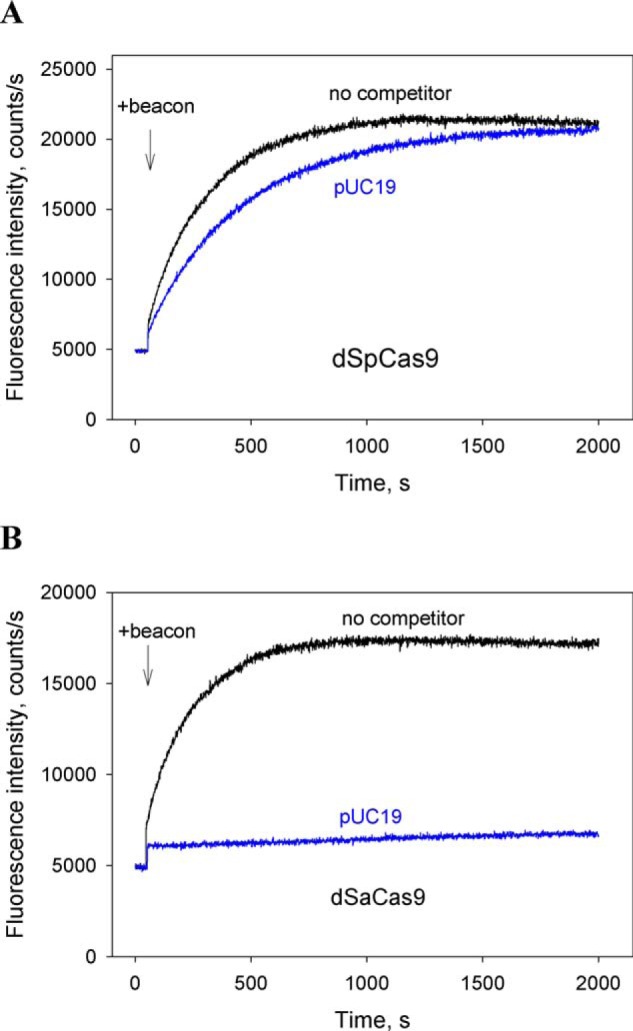
Competition assays with pUC19 DNA vector. Shown are the effects of pUC19 (3 nm) on the kinetics of dSpCas9–gRNA (A) and dSaCas9–gRNA (B) binding to beacons.
Interaction of FnCas9–gRNA with short DNA probes bearing its PAM sequence
Because FnCas9 recognizes the 5′-NGG sequence as an optimal PAM (43), we used the same competitor probes that were used with SpCas9 and SaCas9 (Fig. 5A) in competition beacon binding assays with FnCas9. We found that FnCas9–gRNA–bound probe 4.1 that bears no nucleotides upstream of PAM quite strongly (calculated Kd = 3.0 ± 0.6 nm; Fig. 7 and Fig. S9). In sharp contrast to results obtained with SpCas9 and SaCas9, extensions of the upstream edge of probe 4.1 by just 1 and 2 bp led to a considerable decrease in affinities (by 7- and 30-fold, respectively) (Fig. 7, probes 4.2 and 4.3). Probes 4.4–4.6, which bore longer segments upstream of PAM, and probe 4.8 containing truncated PAM, were very weak competitors (Fig. 7 and Fig. S7C). These data thus show a difference in the modes of FnCas9–gRNA and SpCas9–gRNA interactions with the segment of target proximal to PAM, implying differences in the process of DNA interrogation that requires destabilization of this segment of the target (24, 29).
Figure 7.
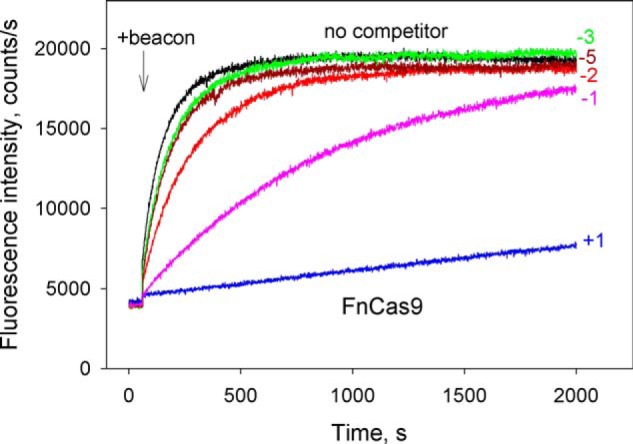
Competition assay with FnCas9 using DNA probes with upstream edges at different positions. Shown are the effects of competitor DNA probes 4.1–4.6 (shown in Fig. 5A) on the kinetics of FnCas9–gRNA binding to beacon. Positions of upstream edges of the probes are indicated in the panels. The concentration of all competitors was 200 nm.
Discussion
High editing efficiency is desired for many research and potential therapeutic applications of genome editing (44). We hypothesized that strong binding of a Cas9 effector to PAM during early steps of DNA interrogation might favor high efficiency of genome editing, for example, through acceleration of target location or facilitation of effector access to partially blocked targets. On the other hand, too high an affinity for PAM might delay the target search by increasing the effector's dwell time at multiple PAM sites located throughout the genome. To elucidate the extent of variation of Cas9 nucleases affinities for PAM sequences, we compared the ability of several Cas9 nucleases to bind to short DNA probes bearing functional PAM sequences. Overall, our results reveal large differences in the affinity for PAM among the Cas9 enzymes studied. In particular, we find that affinities of probes containing canonical SpCas9 and SaCas9 PAM sequences but noncomplementary to gRNA spacer are ∼20-fold lower for SpCas9 effector than for SaCas9. Given that SaCas9 was observed to form less stable R-loop complexes with target DNA than SpCas9 (45), the high affinity of SaCas9–gRNA for cognate PAM is somewhat surprising. These observations are, however, consistent with a previous study showing that R-loop stability is mainly determined by Cas9–gRNA interactions with protospacer, whereas PAM affects primarily the R-loop formation rate (46). A number of earlier studies showed that SaCas9 works with comparable efficiency to SpCas9 (7, 8). However, recently reported measurements of SpCas9 and SaCas9 genome editing activities found that SaCas9 was noticeably more efficient in inducing mutations than SpCas9 when targeting the same sites at similar conditions (47–49). We suggest that the higher efficiency of SaCas9 observed in these studies may be in part accounted by for its relatively stronger interaction with PAM.
Comparison of affinities of SpCas9–gRNA and its engineered variants for optimal and suboptimal PAM sequences also supports the idea that there is a relationship between the PAM affinity and the efficiency of genome editing. Our data indicate that Cas9–VQR–gRNA has much higher affinity for probes bearing TGAG PAM than for similar probes with bases other than G at the fourth PAM position, which is consistent with the preference for the NGAG PAM sequence observed in genome editing experiments (15). Further, we find that affinities of SpCas9–gRNA for suboptimal NAG and NGA PAMs are much lower than that for the canonical NGG PAM but higher than for nonfunctional PAM sequences. The finding that Cas9–NG–gRNA binds to short probes bearing the relaxed NGN PAM sequences stronger than xCas9–gRNA is also consistent with the correlation between PAM affinity and genome editing efficiency. Amino acid substitutions introduced into xCas9 and Cas9–NG are located in different protein domains (Table S1), implying that different structural features account for their relaxed PAM recognition (17, 18). The crystal structure of Cas9–NG–gRNA complex with target DNA suggests that the relatively high PAM affinity of Cas9–NG is due to non–base-specific interactions of the PAM duplex with substituted SpCas9 amino acid residues, which compensate the loss of the base-specific interaction with the third PAM nucleobase (18).
Our data show that relative affinities of SpCas9–gRNA and SaCas9–gRNA for probes bearing duplex segments noncomplementary to gRNA upstream of PAM gradually decrease with increasing length of the upstream segments in a quite similar way (Fig. 5, B and C). In contrast, in the case of FnCas9, we found that probe bearing no nucleotides upstream of PAM had high affinity for FnCas9–gRNA, but the affinities of probes bearing even short (2–3 bp) segments upstream of PAM dropped sharply (Fig. 7). These observations suggest a difference in the modes of interaction of these effectors with the adjacent to PAM DNA duplex during DNA interrogation. In the crystal structure of FnCas9–sgRNA-DNA R-loop complex, the PAM duplex is sandwiched between the WED and PI domains, which are connected by the WED–PI linker loop. As in SpCas9 and SaCas9, the backbone phosphate group between nucleotides at the +1 and −1 positions in the target DNA strand interacts with the phosphate lock loop, thereby stabilizing target DNA immediately upstream of PAM in an unwound conformation (43). The structure also shows that Arg1474 in the WED–PI linker forms a stacking interaction with the most upstream PAM base in the target DNA strand without hydrogen-bond formation (43). Such stacking interaction is not observed in structures of SpCas9 and SaCas9 ternary complexes. Consideration of the FnCas9–sgRNA–DNA R-loop complex structure suggests that a 1-bp duplex extension of the PAM segment in the upstream direction would likely result in a steric clash with Arg1474. We thus suggest that the observed obstruction of DNA binding to FnCas9–gRNA by an adjacent to PAM short duplex segment may be at least in part due to its clash with the WED–PI linker, although the involvement of other neighboring segments is also possible. This suggestion is in line with a recent study on specificity of FnCas9 binding to and cleavage of DNA, which proposes that FnCas9 may possess a distinct mode of DNA interrogation and rejecting off targets (10).
Experimental procedures
Purification of Cas9 proteins
The SpCas9 variants Cas9–VQR, xCas9 3.7, and SpCas9–NG used in this work were prepared essentially as described previously (15, 17, 18). Details of the purification procedure are described in supporting “Experimental procedures and Table S2.” dSaCas9 protein (saCas9 null mutant protein) was purchased from Applied Biological Materials, Inc. FnCas9 protein was purchased from Sigma–Aldrich. The commercial proteins showed a high batch-to-batch reproducibility in the beacon assay experiments. The concentrations of SpCas9 and its variants, SaCas9 and FnCas9, were determined spectrophotometrically at 280 nm using calculated extinction coefficients of 120,450, 116,900, and 199,750 m−1 cm−1, respectively.
Preparation of guide RNAs and DNA probes
Single guide RNAs (shown in Fig. S1) were in vitro transcribed from DNA templates (shown in Table S3) using a HiScribe T7 high-yield RNA synthesis kit (NEB) according to the manufacturer's protocol. The transcribed sgRNAs were purified by Bio-Gel P-30 spin-column (Bio-Rad). SpCas9 dual guide RNA (dgRNA) was formed by mixing equimolar amounts of SpCas9 trans-activating crRNA (Sigma–Aldrich) and crRNA (Integrated DNA Technologies) in a buffer containing 20 mm Tris, pH 7.5, 100 mm NaCl; heating for 30 s at 90 °C; and slowly cooling the reactions to 20 °C. In experiments with SpCa9 and its variants, we used dgRNA because it could be readily prepared in large quantities. Control beacon assay experiments with SpCas9 sgRNA and dgRNA yielded identical results. All gRNAs used in this work contained the same spacer segment (Fig. S1). Cas9 beacon constructs (Fig. S2) and competitor DNA probes (Fig. S3) were prepared from unmodified and chromophore-labeled DNA oligonucleotides synthesized by Integrated DNA Technologies by mixing equimolar amounts of synthetic complementary strands in a buffer containing 20 mm Tris (pH 7.5) and 100 mm NaCl heated for 2 min at 90 °C, with the reactions slowly cooled to 20 °C. The PAM-distal ends of the beacon target and nontarget strands were labeled with fluorescein and Iowa Black R-FQ, respectively.
Fluorometric measurements
Fluorescence measurements were carried out at 25 °C using a QuantaMaster QM4 spectrofluorometer (PTI) in binding buffer (20 mm Tris-HCl, pH 7.5, 100 mm NaCl, 5% glycerol, 0.1 mm DTT, and 1 mm MgCl2) containing 0.02% Tween 20. The MgCl2 concentration used was chosen because it is close to estimates of intracellular concentration of free Mg2+ (50). Final assay mixtures (800 μl) contained 5–10 nm Cas9 protein, gRNA in 2-fold excess over Cas9, 0.5–1 nm Cas beacon, and competitor DNA probes at various concentrations. The DNA probes were preincubated with Cas9–gRNA complexes for 15 min at 25 °C prior to the beacon addition. The fluorescein fluorescence intensities were recorded with an excitation wavelength of 498 nm and an emission wavelength of 520 nm. Time-dependent fluorescence changes were monitored using manual mixing; the mixing dead time was 15 s. Competition experiments were analyzed with Felix software (PTI), and the Kd values were calculated as described previously (29, 34). Details of the Kd calculation are also described in supporting “Experimental procedures.” The Kd values were determined as averages obtained from at least three independent experiments performed on different days, and the standard deviations were less than 30% of the corresponding mean values. Typical variations among replicate kinetic traces are shown in Fig. S5.
Electrophoretic mobility shift assay
DNA probe duplexes were formed by mixing 5′-32P-labeled target strand (110 nm) and unlabeled nontarget strand (125 nm) in nuclease-free duplex buffer (Integrated DNA Technologies), heating for 2 min at 95 °C, and slowly cooling to 20 °C. Reconstitution of the Cas9–gRNA complexes was carried out by mixing stock solutions of a dCas9 protein and dgRNA or sgRNA in 1:1.5 ratio and incubating at room temperature for 10 min. Target DNA binding assays were carried out in 10 μl of assay buffer (20 mm Tris-HCl, pH 8.0, 25 mm NaCl, 5 mm MgCl2, 0.25 mm tris(2-carboxyethyl)phosphine) containing 10 nm DNA probe and 8–400 nm dCas9–gRNA. Binding reactions were incubated for 10 min at 37 °C, and resolved by native 6% PAGE at 4 °C (0.5× TBE buffer with 2 mm MgCl2). DNA probes were visualized by phosphorimaging and quantified with ImageQuant (GE Healthcare).
Author contributions
V. M. and K. S. conceptualization; V. M. and K. K. investigation; V. M. and K. K. writing-original draft; K. S. supervision; K. S. writing-review and editing.
Supplementary Material
This work was supported by National Institutes of Health Grant GM104071 and Russian Science Foundation grant 19-14-00323 (to K. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supporting text, Tables S1–S3, and Figs. S1–S9.
- Cas
- CRISPR associated
- PAM
- protospacer adjacent motif
- gRNA
- guide RNA
- crRNA
- CRISPR RNA
- bp
- base pair(s)
- dgRNA
- dual guide RNA.
References
- 1. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J. A., and Charpentier E. (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 10.1126/science.1225829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mali P., Yang L., Esvelt K. M., Aach J., Guell M., DiCarlo J. E., Norville J. E., and Church G. M. (2013) RNA-guided human genome engineering via Cas9. Science 339, 823–826 10.1126/science.1232033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cong L., Ran F. A., Cox D., Lin S., Barretto R., Habib N., Hsu P. D., Wu X., Jiang W., Marraffini L. A., and Zhang F. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 10.1126/science.1231143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hwang W. Y., Fu Y., Reyon D., Maeder M. L., Tsai S. Q., Sander J. D., Peterson R. T., Yeh J. R., and Joung J. K. (2013) Efficient genome editing in zebrafish using a CRISPR–Cas system. Nat. Biotechnol. 31, 227–229 10.1038/nbt.2501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Doudna J. A., and Charpentier E. (2014) The new frontier of genome engineering with CRISPR–Cas9. Science 346, 1258096 10.1126/science.1258096 [DOI] [PubMed] [Google Scholar]
- 6. Hsu P. D., Lander E. S., and Zhang F. (2014) Development and applications of CRISPR–Cas9 for genome engineering. Cell 157, 1262–1278 10.1016/j.cell.2014.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ran F. A., Cong L., Yan W. X., Scott D. A., Gootenberg J. S., Kriz A. J., Zetsche B., Shalem O., Wu X., Makarova K. S., Koonin E. V., Sharp P. A., and Zhang F. (2015) In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191 10.1038/nature14299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Friedland A. E., Baral R., Singhal P., Loveluck K., Shen S., Sanchez M., Marco E., Gotta G. M., Maeder M. L., Kennedy E. M., Kornepati A. V., Sousa A., Collins M. A., Jayaram H., Cullen B. R., et al. (2015) Characterization of Staphylococcus aureus Cas9: a smaller Cas9 for all-in-one adeno-associated virus delivery and paired nickase applications. Genome Biol. 16, 257 10.1186/s13059-015-0817-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen F., Ding X., Feng Y., Seebeck T., Jiang Y., and Davis G. D. (2017) Targeted activation of diverse CRISPR–Cas systems for mammalian genome editing via proximal CRISPR targeting. Nat. Commun. 8, 14958 10.1038/ncomms14958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Acharya S., Mishra A., Paul D., Ansari A. H., Azhar M., Kumar M., Rauthan R., Sharma N., Aich M., Sinha D., Sharma S., Jain S., Ray A., Jain S., Ramalingam S., et al. (2019) Francisella novicida Cas9 interrogates genomic DNA with very high specificity and can be used for mammalian genome editing. Proc. Natl. Acad. Sci. U.S.A. 116, 20959–20968 10.1073/pnas.1818461116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen B., Gilbert L. A., Cimini B. A., Schnitzbauer J., Zhang W., Li G. W., Park J., Blackburn E. H., Weissman J. S., Qi L. S., and Huang B. (2013) Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 155, 1479–1491 10.1016/j.cell.2013.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gilbert L. A., Larson M. H., Morsut L., Liu Z., Brar G. A., Torres S. E., Stern-Ginossar N., Brandman O., Whitehead E. H., Doudna J. A., Lim W. A., Weissman J. S., and Qi L. S. (2013) CRISPR-mediated modularRNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451 10.1016/j.cell.2013.06.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gaudelli N. M., Komor A. C., Rees H. A., Packer M. S., Badran A. H., Bryson D. I., and Liu D. R. (2017) Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551, 464–471 10.1038/nature24644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jiang F., and Doudna J. A. (2017) CRISPR–Cas9 structures and mechanisms. Annu. Rev. Biophys. 46, 505–529 10.1146/annurev-biophys-062215-010822 [DOI] [PubMed] [Google Scholar]
- 15. Kleinstiver B. P., Prew M. S., Tsai S. Q., Topkar V. V., Nguyen N. T., Zheng Z., Gonzales A. P., Li Z., Peterson R. T., Yeh J. R., Aryee M. J., and Joung J. K. (2015) Engineered CRISPR–Cas9 nucleases with altered PAM specificities. Nature 523, 481–485 10.1038/nature14592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kleinstiver B. P., Prew M. S., Tsai S. Q., Nguyen N. T., Topkar V. V., Zheng Z., and Joung J. K. (2015) Broadening the targeting range of Staphylococcus aureus CRISPR–Cas9 by modifying PAM recognition. Nat. Biotechnol. 33, 1293–1298 10.1038/nbt.3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hu J. H., Miller S. M., Geurts M. H., Tang W., Chen L., Sun N., Zeina C. M., Gao X., Rees H. A., Lin Z., and Liu D. R. (2018) Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556, 57–63 10.1038/nature26155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nishimasu H., Shi X., Ishiguro S., Gao L., Hirano S., Okazaki S., Noda T., Abudayyeh O. O., Gootenberg J. S., Mori H., Oura S., Holmes B., Tanaka M., Seki M., Hirano H., et al. (2018) Engineered CRISPR–Cas9 nuclease with expanded targeting space. Science 361, 1259–1262 10.1126/science.aas9129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. He X., Wang Y., Yang F., Wang B., Xie H., Gu L., Zhao T., Liu X., Zhang D., Ren Q., Liu X., Liu Y., Gao C., and Gu F. (2019) Boosting activity of high-fidelity CRISPR/Cas9 variants using a tRNAGln-processing system in human cells. J. Biol. Chem. 294, 9308–9315 10.1074/jbc.RA119.007791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Anders C., Niewoehner O., Duerst A., and Jinek M. (2014) Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 513, 569–573 10.1038/nature13579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nishimasu H., Cong L., Yan W. X., Ran F. A., Zetsche B., Li Y., Kurabayashi A., Ishitani R., Zhang F., and Nureki O. (2015) Crystal structure of Staphylococcus aureus Cas9. Cell 162, 1113–1126 10.1016/j.cell.2015.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hsu P. D., Scott D. A., Weinstein J. A., Ran F. A., Konermann S., Agarwala V., Li Y., Fine E. J., Wu X., Shalem O., Cradick T. J., Marraffini L. A., Bao G., and Zhang F. (2013) DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31, 827–832 10.1038/nbt.2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang Y., Ge X., Yang F., Zhang L., Zheng J., Tan X., Jin Z. B., Qu J., and Gu F. (2014) Comparison of non-canonical PAMs for CRISPR/Cas9-mediated DNA cleavage in human cells. Sci. Rep. 4, 5405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sternberg S. H., Redding S., Jinek M., Greene E. C., and Doudna J. A. (2014) DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507, 62–67 10.1038/nature13011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Knight S. C., Xie L., Deng W., Guglielmi B., Witkowsky L. B., Bosanac L., Zhang E. T., El Beheiry M., Masson J. B., Dahan M., Liu Z., Doudna J. A., and Tjian R. (2015) Dynamics of CRISPR–Cas9 genome interrogation in living cells. Science 350, 823–826 10.1126/science.aac6572 [DOI] [PubMed] [Google Scholar]
- 26. Ma H., Tu L. C., Naseri A., Huisman M., Zhang S., Grunwald D., and Pederson T. (2016) CRISPR–Cas9 nuclear dynamics and target recognition in living cells. J. Cell Biol. 214, 529–537 10.1083/jcb.201604115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Singh D., Sternberg S. H., Fei J., Doudna J. A., and Ha T. (2016) Real-time observation of DNA recognition and rejection by the RNA-guided endonuclease Cas9. Nat. Commun. 7, 12778 10.1038/ncomms12778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jones D. L., Leroy P., Unoson C., Fange D., Ćurić V., Lawson M. J., and Elf J. (2017) Kinetics of dCas9 target search in Escherichia coli. Science 357, 1420–1424 10.1126/science.aah7084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mekler V., Minakhin L., and Severinov K. (2017) Mechanism of duplex DNA destabilization by RNA-guided Cas9 nuclease during target interrogation. Proc. Natl. Acad. Sci. U.S.A. 114, 5443–5448 10.1073/pnas.1619926114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pawluk A., Davidson A. R., and Maxwell K. L. (2018) Anti-CRISPR: discovery, mechanism and function. Nat. Rev. Microbiol. 16, 12–17 10.1038/nrmicro.2017.120 [DOI] [PubMed] [Google Scholar]
- 31. Maji B., Gangopadhyay S. A., Lee M., Shi M., Wu P., Heler R., Mok B., Lim D., Siriwardena S. U., Paul B., Dancík V., Vetere A., Mesleh M. F., Marraffini L. A., Liu D. R., et al. (2019) A high-throughput platform to identify small-molecule inhibitors of CRISPR–Cas9. Cell 177, 1067–1079.e19 10.1016/j.cell.2019.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. von Hippel P. H., and Berg O. G. (1986) On the specificity of DNA-protein interactions. Proc. Natl. Acad. Sci. U.S.A. 83, 1608–1612 10.1073/pnas.83.6.1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mekler V., Minakhin L., Semenova E., Kuznedelov K., and Severinov K. (2016) Kinetics of the CRISPR–Cas9 effector complex assembly and the role of 3′-terminal segment of guide RNA. Nucleic Acids Res. 44, 2837–2845 10.1093/nar/gkw138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mekler V., Kuznedelov K., Minakhin L., Murugan K., Sashital D. G., and Severinov K. (2019) CRISPR–Cas molecular beacons as tool for studies of assembly of CRISPR–Cas effector complexes and their interactions with DNA. Methods Enzymol. 616, 337–363 10.1016/bs.mie.2018.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kuznedelov K., Mekler V., Lemak S., Tokmina-Lukaszewska M., Datsenko K. A., Jain I., Savitskaya E., Mallon J., Shmakov S., Bothner B., Bailey S., Yakunin A. F., Severinov K., and Semenova E. (2016) Altered stoichiometry Escherichia coli Cascade complexes with shortened CRISPR RNA spacers are capable of interference and primed adaptation. Nucleic Acids Res., 44, 10849–10861 10.1093/nar/gkw914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hu X., Wang C., Fu Y., Liu Q., Jiao X., and Wang K. (2016) Expanding the range of CRISPR/Cas9 genome editing in rice. Mol. Plant 9, 943–945 10.1016/j.molp.2016.03.003 [DOI] [PubMed] [Google Scholar]
- 37. Anders C., Bargsten K., and Jinek M. (2016) Structural plasticity of PAM recognition by engineered variants of the RNA-guided endonuclease Cas9. Mol. Cell 61, 895–902 10.1016/j.molcel.2016.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhong Z., Sretenovic S., Ren Q., Yang L., Bao Y., Qi C., Yuan M., He Y., Liu S., Liu X., Wang J., Huang L., Wang Y., Baby D., Wang D., et al. (2019) Improving plant genome editing with high-fidelity xCas9 and noncanonical PAM-targeting Cas9–NG. Mol. Plant. 12, 1027–1036 10.1016/j.molp.2019.03.011 [DOI] [PubMed] [Google Scholar]
- 39. Zeng D., Li X., Huang J., Li Y., Cai S., Yu W., Li Y., Huang Y., Xie X., Gong Q., Tan J., Zheng Z., Guo M., Liu Y. G., and Zhu Q. (2019) Engineered Cas9 variant tools expand targeting scope of genome and base editing in rice. Plant Biotechnol. J. 10.1111/pbi.13293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Niu Q., Wu S., Yang X., Liu P., Xu Y., and Lang Z. (2019) Expanding the scope of CRISPR/Cas9-mediated genome editing in plants using an xCas9 and Cas9–NG hybrid. J. Integr. Plant. Biol. 62, 398–402 10.1111/jipb.12886 [DOI] [PubMed] [Google Scholar]
- 41. Kim H. K., Lee S., Kim Y., Park J., Min S., Choi J. W., Huang T. P., Yoon S., Liu D. R., and Kim H. H. (2020) High-throughput analysis of the activities of xCas9, SpCas9–NG and SpCas9 at matched and mismatched target sequences in human cells. Nat. Biomed. Eng. 4, 111–124 10.1038/s41551-019-0505-1 [DOI] [PubMed] [Google Scholar]
- 42. Jiang F., Taylor D. W., Chen J. S., Kornfeld J. E., Zhou K., Thompson A. J., Nogales E., and Doudna J. A. (2016) Structures of a CRISPR–Cas9 R-loop complex primed for DNA cleavage. Science 351, 867–871 10.1126/science.aad8282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hirano H., Gootenberg J. S., Horii T., Abudayyeh O. O., Kimura M., Hsu P. D., Nakane T., Ishitani R., Hatada I., Zhang F., Nishimasu H., and Nureki O. (2016) Structure and engineering of Francisella novicida Cas9. Cell 164, 950–961 10.1016/j.cell.2016.01.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang F. (2019) Development of CRISPR–Cas systems for genome editing and beyond. Q. Rev. Biophys. 52, e6 10.1017/S0033583519000052 [DOI] [Google Scholar]
- 45. Yourik P., Fuchs R. T., Mabuchi M., Curcuru J. L., and Robb G. B. (2019) Staphylococcus aureus Cas9 is a multiple-turnover enzyme. RNA 25, 35–44 10.1261/rna.067355.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Szczelkun M. D., Tikhomirova M. S., Sinkunas T., Gasiunas G., Karvelis T., Pschera P., Siksnys V., and Seidel R. (2014) Direct observation of R-loop formation by single RNA-guided Cas9 and Cascade effector co,mplexes. Proc. Natl. Acad. Sci. U.S.A. 111, 9798–9803 10.1073/pnas.1402597111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xie H., Tang L., He X., Liu X., Zhou C., Liu J., Ge X., Li J., Liu C., Zhao Qu J. J., Song Z., and Gu F. (2018) SaCas9 requires 5′-NNGRRT-3′ PAM for sufficient cleavage and possesses higher cleavage activity than SpCas9 or FnCpf1 in human cells. Biotechnol. J. 14, e1700561 10.1002/biot.201700561 [DOI] [PubMed] [Google Scholar]
- 48. Najm F. J., Strand C., Donovan K. F., Hegde M., Sanson K. R., Vaimberg E. W., Sullender M. E., Hartenian E., Kalani Z., Fusi N., Listgarten J., Younger S. T., Bernstein B. E., Root D. E., and Doench J. G. (2018) Orthologous CRISPR–Cas9 enzymes for combinatorial genetic screens. Nat. Biotechnol. 36, 179–189 10.1038/nbt.4048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Raitskin O., Schudoma C., West A., and Patron N. J. (2019) Comparison of efficiency and specificity of CRISPR-associated (Cas) nucleases in plants: an expanded toolkit for precision genome engineering. PLoS One 14, e0211598 10.1371/journal.pone.0211598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rink T. J., Tsien R. Y., and Pozzan T. (1982) Cytoplasmic pH and free Mg2+ in lymphocytes. J. Cell Biol. 95, 189–196 10.1083/jcb.95.1.189 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.