

Summary
The airway epithelium represents a physical barrier to the external environment acting as the first line of defence against potentially harmful environmental stimuli including microbes and allergens. However, lung epithelial cells are increasingly recognized as active effectors of microbial defence, contributing to both innate and adaptive immune function in the lower respiratory tract. These cells express an ample repertoire of pattern recognition receptors with specificity for conserved microbial and host motifs. Modern molecular techniques have uncovered the complexity of the lower respiratory tract microbiome. The interaction between the microbiota and the airway epithelium is key to understanding how stable immune homeostasis is maintained. Loss of epithelial integrity following exposure to infection can result in the onset of inflammation in susceptible individuals and may culminate in lung disease. Here we discuss the current knowledge regarding the molecular and cellular mechanisms by which the pulmonary epithelium interacts with the lung microbiome in shaping immunity in the lung. Specifically, we focus on the interactions between the lung microbiome and the cells of the conducting airways in modulating immune cell regulation, and how defects in barrier structure and function may culminate in lung disease. Understanding these interactions is fundamental in the search for more effective therapies for respiratory diseases.
Keywords: immunity, lungs, mucosal immunology, respiratory epithelium, respiratory microbiome
The respiratory epithelium provides a highly effective barrier acting as the first line of defence against potentially harmful environmental stimuli including microbes and allergens.
Abbreviations
- AEC1
alveolar epithelial type 1 cells
- AEC2
alveolar epithelial type 2 cells
- BAL
bronchoalveolar lavage
- CF
cystic fibrosis
- CLRs
C‐type lectin receptors
- COPD
chronic obstructive pulmonary disease
- DKK
Dikkopf
- EGFR
epidermal growth factor receptor
- GORD
gastro‐oesophageal reflux disease
- HHV
human herpes virus
- ICAM‐1
intracellular adhesion molecule 1
- IPF
idiopathic pulmonary fibrosis
- KRM
Kreme
- Man‐6‐P
mannose‐6‐phosphate
- NLRs
NOD‐like receptors
- NOD
nucleotide‐binding and oligomerization domain
- PAF
platelet‐activating factor
- PAMPs
pathogen‐associated molecular patterns
- PARs
protease‐activated receptors
- PRR
pattern recognition receptors
- RLRs
retinoic acid‐inducible gene‐I‐like receptors
- RSV
respiratory syncytial virus
- SCFA
short‐chain fatty acid
- TARC
thymus‐ and activation‐regulated chemokine
- TEER
transepithelial electrical resistance
- Th2
T helper 2
- TIR
toll/IL‐1 receptor
- TLRs
toll‐like receptors
- TRIF
TIR‐domain‐containing adapter‐inducing interferon‐β
- TSLP
thymic stromal lymphopoietin
The epithelial barrier
The human respiratory tract is the main portal of entry for the continuous immigration and elimination of a vast array of airborne microorganisms and particles, including viruses, bacteria and fungi. The lungs are continually exposed to a multitude of microorganisms, some of which are able to persist and colonize the respiratory tract. These microbes that reside in the lower airways constitute the lung microbiome, which is distinct in composition from the microbiota observed in the oral and nasal cavities. The surface of the lungs presents a continuous layer of epithelial cells that constitute a physical and biological barrier for inhaled substances and pathogens. The specialized respiratory epithelium is required for maintenance of immune homeostasis, and epithelial dysfunction is involved in the development of many inflammatory disorders of the airways and lungs. 1 The respiratory tract is a complex organ system, and its primary function is the exchange of oxygen and carbon dioxide. It is divided into the upper respiratory tract including the nasal cavity, pharynx, larynx, and the lower respiratory tract that comprises the conducting airways (trachea and bronchi), the small airways (bronchioles) and the respiratory zone (the alveoli). The adult human airways have a surface area of approximately 70 m2, which harbours a vast range of bacterial communities, with the highest bacterial load observed in the upper respiratory tract. 2 The respiratory tract spans different anatomical sites, and the types of epithelial cells that comprise these sites are varied both in composition and in structure, reflecting the distinct functions of the airway epithelial cells lining the conducting airways and the alveolar regions of the lungs (Fig. 1).
Figure 1.
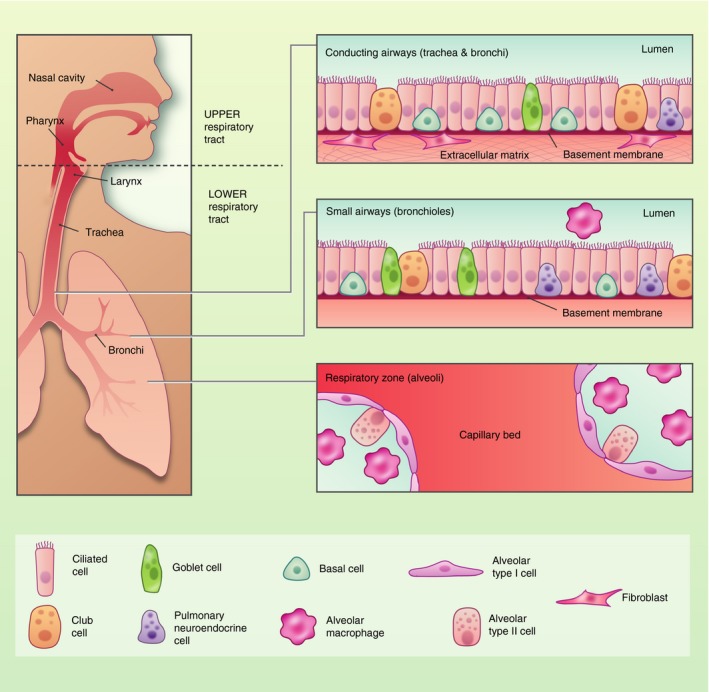
Composition of the respiratory epithelium in health. The human respiratory tract is divided into the upper respiratory tract including the nasal cavity, pharynx, larynx, and the lower respiratory tract that comprises the conducting airways (trachea and bronchi), the small airways (bronchioles) and the respiratory zone (the alveoli). The trachea and large airways are composed of ciliated cells, mucus‐secreting goblet cells, neuroendocrine cells and undifferentiated basal cells. Stromal cells including fibroblasts provide extracellular matrix that modulate airway epithelial cell turnover. The alveoli are comprised of alveolar macrophages and alveolar type I and type II cells.
In the lower respiratory tract, the respiratory epithelium provides a highly effective barrier acting as the first line of defence against potentially harmful environmental stimuli including microbes and allergens. 1 The pseudostratified epithelium of the conducting airways is primarily comprised of multi‐ciliated cells, mucus‐secreting goblet cells, neuroendocrine cells and basal cells, which secrete surfactant. 1 It constitutes the first site of interaction with inhaled environmental compounds, and is designed to facilitate effective mucociliary clearance of particles and microbes. 3 Multi‐ciliated cells have cilia on the apical surface that beat co‐ordinately to shuttle inhaled particulates and mucus out of the airways whilst goblet and secretory cells trap inhaled particulates and microorganisms. Basal cells are progenitor cells that act as resident stem cells for the trachea and proximal airways. These cells are capable of self‐renewal as well as repopulating the pseudostratified epithelium during homeostasis and after injury. 4 The tracheal region and large airways harbour stromal cells such as interstitial fibroblasts that aid in the regulation of the regenerative response of the airway epithelium after injury. 5 In contrast, the alveolar surfaces in the peripheral lung are lined by flat alveolar epithelial type 1 cells (AEC1) that form a continuous cell layer and are specialized in gas exchange, while cuboidal alveolar epithelial type 2 cells (AEC2) act as progenitor cells and secrete pulmonary surfactant, which reduces surface tension in order to prevent alveolar collapse during respiration. 6 , 7 A common feature shared by these cell types is the presence of intracellular tight junctions that are localized at the apical surface, and are pivotal for epithelial adhesion and barrier function. 8 These tight junctions ensure the cells adhere together to form a regulated impermeable barrier. This intracellular adhesion complex consists of interconnections of proteins and receptors including, zonular occluden (ZO)‐1, ‐2 and ‐3, occludins, claudins, and transmembrane junctional adhesion molecules. Tight junctions control paracellular permeability, and immediately below them are the adherens junctions, composed of β‐catenin and E‐cadherin, which mechanically connect the adjacent cells and initiate proliferation and differentiation. 8 , 9 The presence and function of these complexes can be influenced by cellular differentiation, exposure to a range of stimuli, such as allergens and pollutants, and may be altered in disease conditions, contributing to immunopathology. 10 , 11
The lung microbiota in health and disease
The long‐held, but incorrect, theory of sterility of the lung outside of clinical infection means historically little work has evaluated the role of the lung microbiota in chronic lung disease. Advances in high‐throughput molecular sequencing technologies have debunked this notion, demonstrating that the epithelial surfaces of the human respiratory tract are colonized by a complex and dynamic microbiota, termed the ‘lung microbiome’, which plays a role both in health and disease. 14 The most commonly used approach to the study of bacterial communities exploits high‐throughput sequencing of amplicons of the 16S rRNA gene, a highly conserved locus in the bacterial genome that contains specific variable regions that can be used for phylogenetic classification. 15 Sequencing studies have demonstrated stark differences in the lung microbiome between health and disease, and it is becoming increasingly apparent that the lung microbiome may not only contribute to explain the pathogenesis of these diseases, but also serve as a prognostic marker and therapeutic target. 16 , 17
In health, the lung microbiota has a low density but harbours a prominent diversity of interacting microbiota. The airway microbiota in healthy lungs is dominated by the Bacteroidetes, Firmicutes and Proteobacteria. Prominent genera in the lower airways include Prevotella, Veillonella and Streptococcus. 21 The composition of the lung microbiome is determined by three factors: microbial immigration; microbial elimination; and the relative reproduction rates of its members. The main routes of microbial immigration to the lungs are inhalation of air‐borne bacteria, direct dispersion along mucosal surfaces and microaspiration. 14 , 22 The similarities in community composition between the oral and lung microbiota suggest that microaspiration is likely to be the dominant route of immigration. 14 , 23 Microbial elimination is an active process that is achieved through mucociliary clearance, cough and host immune defences. 24 The environmental conditions necessary for microbial growth within the respiratory tract (e.g. pH, temperature, nutrient availability, oxygen tension and activation of host inflammatory cells) are heterogenous, and considerable regional variation can be observed in a single healthy lung. During lung disease, the balance between immigration and elimination is perturbed, leading to alterations in the lung microbiota, with bacteria exhibiting competitive advantages becoming predominant 14 (Table 1). This overgrowth of bacterial species leads to a decrease in overall richness, and is associated with the progression of chronic lung diseases such as asthma, chronic obstructive pulmonary disease (COPD), cystic fibrosis (CF) and idiopathic pulmonary fibrosis (IPF). 16 , 17 , 19 , 20 , 25 Furthermore, these studies have linked microbial dysbiosis with an increased morbidity and mortality in a multitude of chronic respiratory diseases.
Table 1.
Summary of studies illustrating changes in the respiratory microbiome that occur during chronic lung disease
| Disease | Taxonomic changes at the genus level 1 | Sample type | Cohort size | [Ref.] | |
|---|---|---|---|---|---|
| Asthma | Increased abundance of Haemophilus | Bronchoscopy | 11 | 16 | |
| Increased abundance of Haemophilus, Neisseria, Fusobacterium and Porphyromonas | Bronchoscopy | 42 | 29 | ||
| Increased abundance of Streptococcus | Nasopharyngeal | 234 | 30 | ||
| COPD | Increased abundance of Lactobacillus | Lung tissue | 24 | 17 | |
| Increased abundance of Fusobacteria, Leptotrichia and Fusobacterium | BAL | 32 | 31 | ||
| CF | Increased abundance of Burkholderia, Streptococcus and Staphylococcus | Sputum | 23 | 25 | |
| Increased abundance of Pseudomonas, Staphylococcus, Stenotrophomonas and Achromobacter | Sputum | 17 | 32 | ||
| Increased abundance of Staphylococcus, Streptococcus and Pseudomonas | BAL | 95 | 33 | ||
| IPF | Increased abundance of Haemophilus, Streptococcus, Neisseria and Veillonella | BAL | 65 | 19 | |
| Increased abundance of Staphylococcus and Streptococcus | BAL | 55 | 20 | ||
| Increased abundance of Streptococcus, Prevotella, Veillonella, Haemophilus and Pseudomonas | BAL | 35 | 26 | ||
BAL, bronchoalveolar lavage; CF, cystic fibrosis; COPD, chronic obstructive pulmonary disease; IPF, idiopathic pulmonary fibrosis.
Changes in the microbiota in cases compared with controls.
In addition to the bacterial component of the microbiome, there is evidence that other non‐bacterial organisms, including viruses and fungi, may play an important role in health and disease. Emerging evidence suggests that the lung virome and mycobiome have a significant impact on clinical outcome of chronic respiratory diseases such as asthma, COPD, CF and IPF. 27 , 34 , 35 It is also increasingly recognized that respiratory viruses are a major cause of acute exacerbations of chronic pulmonary diseases, and that the host response to viral infection is dysregulated in these diseases. For example, studies support a mechanistic role for viruses in the initiation and progression of IPF. The human herpes virus (HHV) family, which includes cytomegalovirus, Epstein−Barr virus, HHV‐7 and HHV‐8, has received considerable attention as either an aetiological or exacerbating agent of IPF. 36 In asthma, infection by respiratory viruses is a major trigger of wheezing in infants as well as exacerbations of asthma in older children. The infectious agents associated with these wheezing events include respiratory syncytial virus (RSV), human rhinovirus, parainfluenza, coronavirus and other viruses. 37 Despite the fact that very little is known about the lung mycobiome compared with the bacterial microbiome, in recent years specific fungi have been identified in respiratory diseases. 38 , 39 It has also been shown that commensal fungi can affect the immune system and also modulate bacterial communities, thus contributing to the restoration of the bacterial microbiota following antibiotic treatment. 40 , 41 Although studies into the composition and function of lung viral and fungal communities are gaining momentum, research in this field is still in its infancy, and future mechanistic work is needed to yield novel insights into their role in the pathogenesis of pulmonary diseases.
A homeostatic balance between the lung and its microbiota
Airway epithelial cells are in continuous contact with the environment, and this interaction is a crucial factor in maintaining stable homeostasis. 42 The three essential components that contribute to the barrier function of airway epithelium are: mucociliary escalators that trap and remove inhaled microbes and noxious stimuli from the airways; intracellular tight and adherens junctions that regulate epithelial paracellular permeability; and secreted antimicrobial peptides that kill inhaled pathogens. 1 The physiological features of the respiratory tract are pivotal for maintaining a balance between the lung and its microbiota. The pulmonary epithelium is composed of distinct cell types and is not continuous from the upper respiratory tract to the alveoli. 43 In the large airways, the submucosal gland harbours the mucus and serous cells that are responsible for the production of most airway mucus. 44 Towards the bronchioles, goblet and club cells produce mucus, whereas in the alveoli AEC1 and AEC2 secrete pulmonary surfactant. 45 , 46 Mucus is a gel composed mainly of water and glycosylated proteins, such as mucins. 48 Mucins are the large glycoprotein constituents of airway mucus. 49 MUC5AC from goblet cells and MUC5B from submucosal glands are the predominant gel‐forming mucins in the human airways. 50 , 51 Mucus is a major ecological niche for the microbiota; it promotes healthy interactions between microorganisms and epithelial cells through the formation of a thin mobile layer that is supported by the periciliary layer covering the cilia. 49 , 52 In health, the mucus layer provides an effective defence against epithelial injury; however, excessive mucus production is associated with increased susceptibility to viral infections, respiratory allergies, and contributes to obstruction in several respiratory diseases such as pneumonia, asthma, COPD and CF. 53 , 54 This obstruction hinders the ability of the cilia to transport mucus out of the lungs, and favours the selection of certain pathogenic bacteria that thrive in mucus and establish themselves in this obstructed niche. 55 Furthermore, microaspiration is commonly observed in chronic lung disease, and gastro‐oesophageal reflux disease (GORD) has emerged as a frequent co‐morbidity in chronic respiratory disorders. Although GORD is typically confined to the lower oesophagus, in some individuals it can be associated with pulmonary microaspiration of gastric contents. 56 The occurrence of aspiration can lead to increased levels of bile acid in the lungs, which can favour the establishment of microbial biofilms, composed of microbial cells that form structured communities embedded in an extracellular matrix. 57 Bacterial and fungal biofilms have emerged as a mechanism of virulence for opportunistic pathogens including Pseudomonas aeruginosa and staphylococcal species. 58 , 59 A hallmark of biofilms is their remarkable antimicrobial tolerance, which allows them to persist in the host and evade the host immune system. 59 Therefore, GORD‐derived bile and subsequent biofilm growth on host tissue may contribute to chronic respiratory infection. In addition, direct seeding of bacteria from the intestinal microbiota into the airways may play a role in influencing the interaction between the epithelium and the microbiome. 60
The intracellular tight and adherens junctions provide fundamental adhesive contacts between neighbouring epithelial cells. 61 Tight junctions are the main regulators of paracellular permeability, whereas the adherens junctions mechanically connect adjacent cells, and initiate the formation and maturation of cell−cell contacts. 8 , 9 During disease, barrier function is impaired leading to increased epithelial permeability and pathogen entry. 10 , 11 Upon infection, epithelial cells are able to respond by the release of humoral factors such as antimicrobial peptides that exert broad‐spectrum antimicrobial activity, and a number of these are upregulated in response to the microbial dysbiosis seen in disease. 62 Defensins are the most abundant antimicrobial peptides, and comprise α‐defensin in neutrophils and β‐defensin in epithelial cells. Human β‐defensin 1, 2, 3 and 4 are largely expressed on airway epithelial cells and function in airway mucosal defence by targeting a wide range of respiratory pathogens. Their expression in airway epithelia may be constitutive or inducible by bacterial products or proinflammatory cytokines. 63 Nutrient availability in each specific niche will also play a role in the maintenance and selection of the lung microbiota. Host compounds such as mucins, cytokines, defensins and lactoferrins are the nutrient source available for bacteria in the lungs. 1 , 64 Microbial growth rates can also be influenced by changes in oxygen tension. Within the lungs there is significant regional variation in oxygen tension that can alter population dynamics by affecting bacterial proliferation. 65 In chronic pulmonary diseases, an increased mucus production promotes bacterial growth and leads to the formation of zones with low oxygen concentration and high temperature favouring the selection and maintenance of particular bacteria. 14 Collectively, these differences in the cellular function and the physiological characteristics of the lungs (e.g. temperature, mucus, pulmonary surfactant and oxygen tension) have a major impact on the establishment and persistence of the bacterial communities.
The airway epithelium as a sensor of microbial presence
The airway epithelium not only represents a structural barrier but is also integral for innate host defence. Airway epithelial cells express several pattern recognition receptors (PRRs), and secrete antimicrobial molecules and mucins that aid in the defence against invading pathogens. 3 PRRs are germ‐line encoded receptors that recognize conserved pathogen‐associated molecular patterns (PAMPs) or cell damage‐associated molecular patterns. 66 , 67 , 68 These transmembrane receptors can be activated independently of the adaptive immune response and are classified into four families: the toll‐like receptors (TLRs), the cytoplasmic proteins retinoic acid‐inducible gene‐I‐like receptors (RLRs), the NOD‐like receptors (NLRs) and the C‐type lectin receptors (CLRs). 69
To date, 10 (TLR1−TLR10) have been identified in the human genome, and are characterized by N‐terminal leucine‐rich repeats, a transmembrane region and a cytoplasmic Toll/IL‐1 receptor (TIR) homology domain that mediates signalling via MyD88 and TIR‐domain‐containing adapter‐inducing interferon‐β (TRIF). 70 Toll‐like receptors play an important role in pathogen recognition and innate immunity. The main function of TLRs in mammals is to recognize distinct PAMPs, including components of microbial cell walls (TLR1, TLR2, TLR4 and TLR6), flagellin (TLR5), single‐stranded or double‐stranded viral RNA (TLR3, TLR7 and TLR8) or CpG‐containing DNA (TLR9). 71 , 72 , 73 , 74 The TLR3, 7, 8 and 9 are specifically localized in endosomes and detect pathogen nucleic acids following endocytosis. 75 , 76 Lastly, TLR10 is expressed in humans, but the specific ligand for it remains to be elucidated. 80 Signalling through TLRs is key in early inflammatory responses to bacteria through the activation and expression of host cytokines, chemokines and antimicrobial peptides. TLRs are localized on immune and inflammatory cells; however, given that airway epithelial cells come into contact with several potential pathogens, the expression of TLRs is also relevant to immunity in the airways. Indeed, human bronchial epithelial cells express most TLRs; TLR2, TLR3, TLR5 and TLR6 being the most highly expressed. 81
The PRR system can be also activated in response to allergens and viruses. 82 , 83 PRRs within the epithelium are able to recognize allergen‐associated danger signals via protease‐dependent and protease‐independent pathways. Protease‐dependent pathways specifically require direct cleavage of cell‐surface molecules or the activation of G‐protein‐coupled protease‐activated receptors (PARs). 85 , 86 , 87 , 88 RLRs are cytoplasmic receptors that recognize the presence of genomic RNA or double‐stranded RNA viruses or double‐stranded RNA intermediates from single‐stranded RNA viruses. Melanoma differentiation‐associated gene‐5 is expressed in human bronchial epithelial cells, and is involved in the detection of rhinovirus and other viruses. 89 NLRs are intracellular PRRs characterized by a central nucleotide‐binding and oligomerization domain (NOD) and C‐terminal leucine‐rich repeats. 90 More work is needed to define the role of NOD function in airway epithelial cells. Although the contribution of epithelial cells to antibacterial and antiviral immunity is well established, there is a paucity of information regarding the role of airway epithelial cells in immunity to fungi. CLRs belong to the dectin family and recognize fungal cell wall components. For example, Dectin‐1 is expressed in airway epithelial cells, and is responsible for their activation by mycobacterium, Aspergillus and allergens such as house dust mite. 91 , 92 Dectin receptors can be activated by β‐glucans via TLR4 signalling, TLR2‐dependant signalling, or induction of oxidative stress by intrinsic NADPH oxidase activity. 94 , 95 Because fungal spores are potent allergens and are associated with allergic and chronic diseases of the respiratory tract such as asthma, the importance of the lung mycobiome should not be overlooked. 97
Respiratory infections and the initiation of epithelial immune response
Infections play a crucial role in the induction and exacerbation of a multitude of chronic respiratory diseases such as asthma and COPD. 98 Such infections represent a major cause of morbidity and mortality globally, and may involve bacteria (e.g. Streptococcus pneumoniae, Haemophilus influenzae, Chlamydia pneumoniae) and viruses (e.g. RSV, influenza). 99 , 100 The initiation of infection typically occurs through the exposure of the respiratory tract to bacterial cells or viral particles (Fig. 2).
Figure 2.
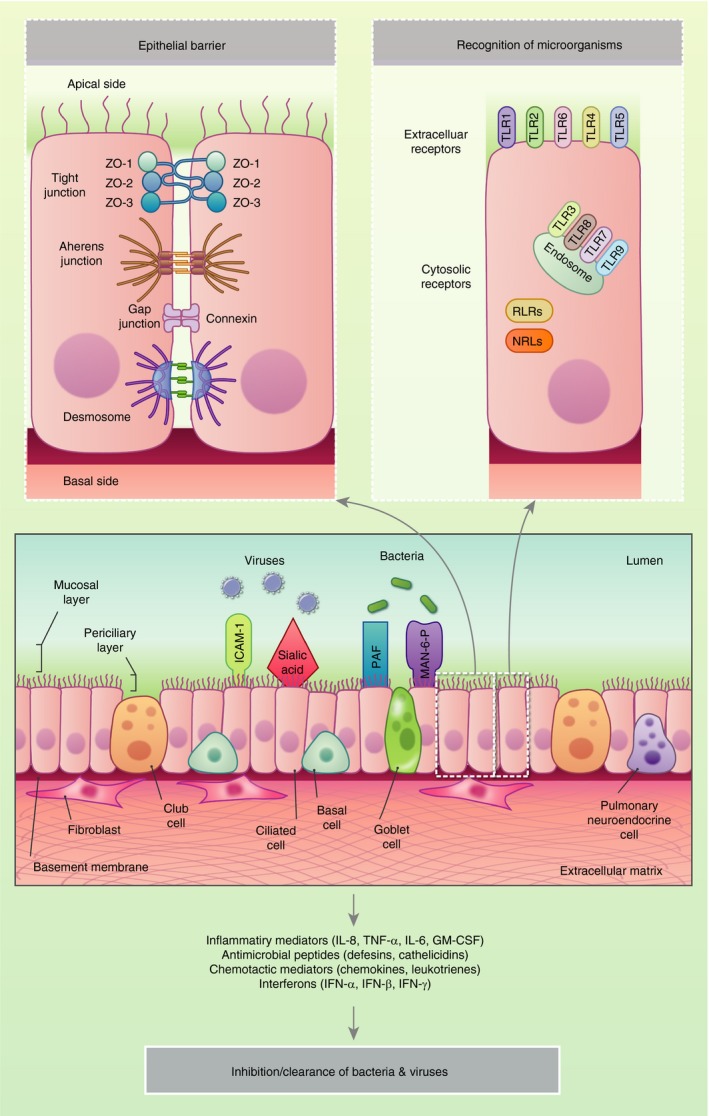
Response of the airway epithelium to infection. Bacteria and viruses bind to cellular receptors, including intercellular adhesion molecule 1 (ICAM‐1), sialic acid residues, platelet‐activating factor (PAF) and mannose‐6‐phosphate (Man‐6‐P). The pathogens are internalized in airway epithelial cells, which induces the production of innate immune defensins and the stimulation of intracellular and extracellular immune receptors such as the toll‐like receptors (TLR), retinoic acid‐inducible gene‐I‐like receptor (RLRs) and NOD‐like receptors (NLRs). In healthy individuals, pathogen clearance is achieved following the generation of pro‐inflammatory innate and adaptive immune responses.
During infection, viruses can bind to specific cellular receptors such as intracellular adhesion molecule 1 (ICAM‐1) and sialic acid residues, whereas bacteria bind to platelet‐activating factor (PAF) and mannose‐6 phosphate (Man‐6‐P) receptors. 101 The interaction between these infectious agents and the epithelial cells induces the stimulation of the PRRs, and the secretion of antimicrobial effector molecules, peptides, enzymes, reactive oxygen species together with a range of chemokines and growth factors. Collectively, these molecules facilitate the recruitment of and communication between immune cells, and contribute to the initiation of the innate immune responses that are pivotal for early control of an infection. In healthy individuals, this response promptly results in pathogen clearance. In contrast, in patients with chronic respiratory diseases these defences may be impaired leading to increased susceptibility to infection that drives inflammatory responses that in turn exacerbate the underlying disease. During chronic disease there is evidence that this barrier function is impaired, with disruption of tight junctions and increased epithelial permeability. This loss of barrier function leads to the entry of pathogens and other particles. Compromised barrier function is intimately linked to triggering of abnormal repair and remodelling. 102 Respiratory viruses, air pollutants and allergens all have the capacity to inflict such damage on the epithelium, either directly due to inherent proteolytic activity or indirectly by binding to PAR‐2 expressed on the apical surface of bronchial epithelial cells. For example, in vitro studies have shown that infection of the cells with rhinovirus results in the loss of ZO‐1 from the junctional complex resulting in decreased transepithelial electrical resistance (TEER) indicative of barrier ‘leakiness’. Both coxsackie and rhinovirus affect occludins 103 , 104 whereas RSV causes disassembly of junctional complexes. 105 In asthma, both decreases in ZO‐1 and TEER that correlate with asthma severity 106 or E‐cadherin and β‐catenin have been shown in vitro. 107 Furthermore, reduced expression of occludins and claudins has been shown in COPD. 108 In vivo, leakage of proteins such as albumin into the alveolar space has been reported following influenza infection. 109 Viruses, pollutants and allergens are all associated with junction dysfunction and exacerbated clinical symptoms across the spectrum of these diseases. 103 , 104 , 105
Defects in barrier structure and function have been observed in primary epithelial cell cultures from asthmatics even after being propagated for several weeks at air−liquid interface, suggesting that reduced barrier function is a stable property of these cells. 112 The type 2 cytokines IL‐4 and 13 induce barrier dysfunction by inhibiting expression of ZO‐1, occludins, E‐cadherin and β‐catenin. 113 It has been proposed that junctional disruption could be a T helper 2 (Th2) promoting signal in the airway to restore mucosal integrity. 114 This idea is supported by the fact that siRNA knockdown of E‐cadherin results in epidermal growth factor receptor (EGFR)‐dependent production of Th2‐promoting cytokines thymus‐ and activation‐regulated chemokine (TARC) and thymic stromal lymphopoietin (TSLP) by airway epithelial cells that explains the association between barrier dysfunction and Th2‐driven pulmonary disease. 115 If the wound healing response is not curtailed following restoration of mucosal integrity, detrimental airway remodelling can ensue. E‐cadherin, in addition to having a structural role at adherens junctions, is also the ligand for CD103, which is expressed on innate and adaptive immune cells and a subset of dendritic cells. Furthermore, it also binds to KLRG1, which is expressed by natural killer cells and regulatory T‐cells and group 2 innate lymphoid cells. E‐cadherin and occludin are known to be downregulated in asthmatics, 106 , 116 , 117 and the genes for E‐cadherin and protocadherin‐1 have been associated with airway hyperresponsiveness. 118 , 119 E‐cadherin anchors EGFR within adherens junctions, 120 preventing activation of the receptor. 121 , 122 EGFR can redistribute to the luminal surface of the cell as a result of reduced expression of E‐cadherin culminating in activation. 123 Excessive activation of EGFR can result in cell proliferation and goblet cell hyperplasia/metaplasia. 124 Increased expression of EGFR has been observed in asthma and correlated with impaired mucosal barrier function. 106
When dissociated from E‐cadherin, β‐catenin can translocate to the nucleus, activating Wnt signalling, which results in cell proliferation. 125 Dikkopf (DKK) proteins regulate Wnt signalling via interaction with Kreme (KRM) receptors, 126 and the Wnt/β‐catenin pathway has been implicated in IPF. 127 , 128 DKK1 expression is increased in lung and bronchoalveolar lavage (BAL) fluid of IPF patients. In addition to being located in hyperplastic alveolar cells, the strongest expression is observed in basal bronchial epithelial cells. KRM1 expression is also elevated in the lung tissue of IPF patients, particularly in areas of bronchiolization. 127 The phosphorylation signatures of β‐catenin in IPF are the same as in normal lung development, again suggesting that repair of lung injury involves reactivation of developmental programmes. 129 Wnt/β‐catenin signalling is also implicated in the pathogenesis of COPD, but in direct contrast to what is observed in IPF, signalling in the mesenchymal compartment is inactivated rather than activated. 130 Interestingly a shift from canonical Wnt signalling (β‐catenin‐dependent) to non‐canonical pathways is suggested to drive pathology in IPF. 131
Basal cells, which are candidate stem cells in the conducting airways, are found in highest concentration in the trachea, and numbers diminish further down the bronchial tree to the bronchioles. Upon damage these cells upregulate p63, a basal cell‐restricted transcription factor, which has been implicated in epithelial mesenchymal transition. 132 It has been shown that basal epithelial cells overlying fibroblastic foci in IPF acquire increased reactivity, determined by p63 expression, and gain a partial mesenchymal phenotype, 133 further evidence of the involvement of bronchial‐derived cells in the pathogenesis of IPF.
Collectively, it is likely that the dialogue between specific bacterial communities and host is a dynamic phenomenon. The installation and location of the bacterial communities within the lungs is affected by the cellular components within the lungs as well as the abiotic environment (temperature, pH, mucus and pulmonary surfactant). Specifically, loss of barrier function can influence the composition of the respiratory microbiota by allowing the entry of pathogens and other particles. However, it is evident that more information is needed to elucidate the nature of the dialogue occurring between the respiratory microbiota and epithelial cells in the lungs to further understand the function and mechanisms through which the microbiota shapes immunity in the lung.
The gut−lung axis
An increasing number of studies have provided evidence of the cross‐talk that occurs between the intestinal microbiota and the lungs, termed the ‘gut−lung axis’. Similar to the gut, the airway microbiota in healthy lungs is dominated by the short‐chain fatty acid (SCFA)‐producing bacteria: Bacteroidetes, Firmicutes and Proteobacteria. These are metabolically active bacteria that produce SCFAs via the bacterial fermentation of non‐digestible carbohydrates by the gut microbiota; acetate, propionate and butyrate being the most abundant in the human intestinal tract. 134 It is now well documented that the intestinal microbiota influences the microbial composition within the lung either by direct seeding of bacteria into the airways or by the distribution of SCFAs. These SCFAs not only act as a fuel for intestinal epithelial cells but also exert distinct physiological effects, acting as a link between the microbiota and the immune system, and playing a key role in maintaining mucosal immunity. Although there are limited studies investigating the impact of these SCFAs in the lungs, in the gut they have been shown to be integral for tight junction formation and to promote anti‐inflammatory mechanisms to sustain intestinal homeostasis. 135 Indeed, SCFAs interact with immune cells, and modulate their recruitment, differentiation, activation and survival at different tissues. 136 The proposed pathway of this gut−lung interaction involves the entry of the microbiota and its products from the airways into the intestinal mucosa. These are phagocytosed and transferred to the mesenteric lymph nodes by antigen‐presenting cells that stimulate the priming of B‐ and T‐cells. Once activated, these cells can then migrate back to the original site or to distal locations such as the lung epithelium where they can exert directly on their target. Another proposed pathway involves the direct migration of bacterial products from the intestinal mucosa to the lungs via the bloodstream where they act to stimulate the immune system. Similarly, the same route is proposed beginning in the lung and culminating in the intestinal mucosa, although the influence of the lung microbiota and its metabolites on the gut is yet to be elucidated. 137
The composition of a stable gut microbiota has been shown to have an influence on lung immunity. Mice devoid of their intestinal microbiota during early life are at an increased risk of developing allergic airway disease. 138 , 139 An increased risk of asthma has been associated with a predominance of Bacteroidetes fragilis and total anaerobes in early life, 140 decreases in the total abundances of Escherichia coli, Faecalibacterium, Lachnospira, Rothia and Veillonella bacterial genera. 141 In adults, taxa‐specific differences in the gut microbiota were observed, such as an increased relative abundance of Bifidobacterium adolescentis, which negatively correlated with the time since asthma diagnosis. 142 Furthermore, mice fed a peptide‐based enteral diet that increased the production of SCFAs attenuated elastase‐induced inflammation and emphysema in the lung. 143 The axis is by no means one‐directional, with respiratory infection driving perturbations in the gut microbiome, but further work is needed to validate this. 144 , 145 A study reported that the intranasal delivery of a non‐absorbable tracer into the nasal cavity of mice appeared in the gastrointestinal tract shortly after. 144 In addition, work by Sze et al. 146 reported that intratracheal administration of lipopolysaccharide not only disrupted the airway microbiota but also led to the translocation of airway bacteria belonging to the Clostridium and Lachnospiraceae genera into the bloodstream and affected the gut microbiota within 24 hr, resulting in an evident increase in total bacterial load. Collectively, these studies provide evidence of the bidirectional communication happening between these two sites. However, our understanding of this cross‐talk is still in its infancy, and there is a pressing need to uncover whether these changes observed in the microbiota are the cause or the effect of disease. Furthermore, there is growing evidence highlighting the use of oral antibiotics and their effect on lung immunity. Antibiotics could affect the lung microbiome either directly or indirectly through their effects on the gut microbiome and subsequently on lung immunity. To support this hypothesis, murine studies have shown that depletion of certain species within the gut microbiota due to antibiotic intake influences lung diseases and allergic inflammation. 147 For example, microbial dysbiosis due to the use of macrolides in early life correlated with an increased risk of asthma in Finnish pre‐school children. 148 It is clear that we need to appreciate the interactions between the gut and airway microbiota in order to understand the impact of antibiotics on lung health.
Conclusions and future directions
The epithelial surfaces of the lung, including the proximal and distal airways, are now considered to play a pivotal role in maintenance of immune homeostasis. The epithelium acts as the first line of defence against inhaled viral, bacterial and fungal pathogens. Studies have shown that the epithelial surfaces of the human respiratory tract are colonized by a complex and dynamic microbiota that plays a role both in health and disease. It is becoming increasingly recognized that the cellular and physiological characteristics of the lungs impact the establishment and persistence of these bacterial communities. Dysregulation of the epithelial immune response and barrier function along with microbial dysbiosis can contribute to chronic inflammatory lung diseases such as asthma, COPD, CF and IPF. However, the precise nature of the relationship between the respiratory microbiome and the epithelium in the lungs remains an active area of investigation. A better understanding of this interaction in maintaining airway homeostasis will provide new insights into the pathogenesis of several respiratory conditions as well as novel diagnostic and therapeutic opportunities. Emerging evidence, particularly involving SCFAs, has highlighted a link between the gut and the lung and the microbial communities within. These results highlight the need for future studies that will characterize the microbiota and metabolites comprising these two mucosal sites and determine their interaction with the host in order to develop more targeted treatments for lung diseases.
Disclosures
The authors have no competing interests or conflicts of interest to declare.
References
- 1. Hiemstra PS, McCray PB Jr, Bals R. The innate immune function of airway epithelial cells in inflammatory lung disease. Eur Respir J 2015; 45:1150–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weibel ER. Morphometry of the Human Lung. Berlin Heidelberg: Springer, 1963. [Google Scholar]
- 3. Leiva‐Juarez MM, Kolls JK, Evans SE. Lung epithelial cells: therapeutically inducible effectors of antimicrobial defense. Mucosal Immunol 2018; 11:21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rock JR, Randell SH, Hogan BL. Airway basal stem cells: a perspective on their roles in epithelial homeostasis and remodeling. Dis Model Mech 2010; 3:545–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sacco O, Silvestri M, Sabatini F, Sale R, Defilippi AC, Rossi GA. Epithelial cells and fibroblasts: structural repair and remodelling in the airways. Paediatr Respir Rev 2004;5(Suppl. A):S35–40. [DOI] [PubMed] [Google Scholar]
- 6. Whitsett JA, Weaver TE. Alveolar development and disease. Am J Respir Cell Mol Biol 2015; 53:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zepp JA, Morrisey EE. Cellular crosstalk in the development and regeneration of the respiratory system. Nat Rev Mol Cell Biol 2019; 20:551–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Niessen CM. Tight junctions/adherens junctions: basic structure and function. J Invest Dermatol 2007; 127:2525–32. [DOI] [PubMed] [Google Scholar]
- 9. Shen L, Weber CR, Raleigh DR, Yu D, Turner JR. Tight junction pore and leak pathways: a dynamic duo. Annu Rev Physiol 2011; 73:283–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Loxham M, Davies DE, Blume C. Epithelial function and dysfunction in asthma. Clin Exp Allergy 2014; 44:1299–313. [DOI] [PubMed] [Google Scholar]
- 11. Steed E, Balda MS, Matter K. Dynamics and functions of tight junctions. Trends Cell Biol 2010; 20:142–9. [DOI] [PubMed] [Google Scholar]
- 12. De Rose V, Molloy K, Gohy S, Pilette C, Greene CM. Airway epithelium dysfunction in cystic fibrosis and COPD. Mediators Inflamm 2018; 2018:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Aghapour M, Raee P, Moghaddam SJ, Hiemstra PS, Heijink IH. Airway epithelial barrier dysfunction in chronic obstructive pulmonary disease: role of cigarette smoke exposure. Am J Respir Cell Mol Biol 2018; 58:157–69. [DOI] [PubMed] [Google Scholar]
- 14. Dickson RP, Huffnagle GB. The lung microbiome: new principles for respiratory bacteriology in health and disease. PLoS Pathog 2015; 11:e1004923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jo JH, Kennedy EA, Kong HH. Research techniques made simple: bacterial 16S ribosomal RNA gene sequencing in cutaneous research. J Invest Dermatol 2016; 136:e23–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C et al Disordered microbial communities in asthmatic airways. PLoS ONE 2010; 5:e8578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sze MA, Dimitriu PA, Hayashi S, Elliott WM, McDonough JE, Gosselink JV et al The lung tissue microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2012; 185:1073–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang YJ, LiPuma JJ. The microbiome in cystic fibrosis. Clin Chest Med 2016; 37:59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Molyneaux PL, Cox MJ, Willis‐Owen SA, Mallia P, Russell KE, Russell AM et al The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2014; 190:906–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Han MK, Zhou Y, Murray S, Tayob N, Noth I, Lama VN et al Lung microbiome and disease progression in idiopathic pulmonary fibrosis: an analysis of the COMET study. Lancet Respir Med 2014; 2:548–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Salisbury ML, Han MK, Dickson RP, Molyneaux PL. Microbiome in interstitial lung disease: from pathogenesis to treatment target. Curr Opin Pulm Med 2017; 23:404–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gleeson K, Eggli DF, Maxwell SL. Quantitative aspiration during sleep in normal subjects. Chest 1997; 111:1266–72. [DOI] [PubMed] [Google Scholar]
- 23. Segal LN, Clemente JC, Tsay JC, Koralov SB, Keller BC, Wu BG et al Enrichment of the lung microbiome with oral taxa is associated with lung inflammation of a Th17 phenotype. Nat Microbiol 2016; 1:16031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mathieu E, Escribano‐Vazquez U, Descamps D, Cherbuy C, Langella P, Riffault S et al Paradigms of lung microbiota functions in health and disease, particularly, in asthma. Front Physiol 2018; 9:1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fodor AA, Klem ER, Gilpin DF, Elborn JS, Boucher RC, Tunney MM et al The adult cystic fibrosis airway microbiota is stable over time and infection type, and highly resilient to antibiotic treatment of exacerbations. PLoS ONE 2012; 7:e45001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Molyneaux PL, Cox MJ, Wells AU, Kim HC, Ji W, Cookson WO et al Changes in the respiratory microbiome during acute exacerbations of idiopathic pulmonary fibrosis. Respir Res 2017; 18:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Invernizzi R, Molyneaux PL. The contribution of infection and the respiratory microbiome in acute exacerbations of idiopathic pulmonary fibrosis. Eur Respir Rev 2019; 28:190 045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Invernizzi R, Barnett J, Rawal B, Nair A, Ghai P, Kingston S et al Bacterial burden in the lower airways predicts disease progression in idiopathic pulmonary fibrosis and is independent of radiological disease extent. Eur Respir J 2020; 55:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Durack J, Lynch SV, Nariya S, Bhakta NR, Beigelman A, Castro M et al Features of the bronchial bacterial microbiome associated with atopy, asthma, and responsiveness to inhaled corticosteroid treatment. J Allergy Clin Immunol 2017; 140:63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Teo SM, Mok D, Pham K, Kusel M, Serralha M, Troy N et al The infant nasopharyngeal microbiome impacts severity of lower respiratory infection and risk of asthma development. Cell Host Microbe 2015; 17:704–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pragman AA, Kim HB, Reilly CS, Wendt C, Isaacson RE. The lung microbiome in moderate and severe chronic obstructive pulmonary disease. PLoS ONE 2012; 7:e47305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Feigelman R, Kahlert CR, Baty F, Rassouli F, Kleiner RL, Kohler P et al Sputum DNA sequencing in cystic fibrosis: non‐invasive access to the lung microbiome and to pathogen details. Microbiome 2017; 5:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Frayman KB, Armstrong DS, Carzino R, Ferkol TW, Grimwood K, Storch GA et al The lower airway microbiota in early cystic fibrosis lung disease: a longitudinal analysis. Thorax 2017; 72:1104–12. [DOI] [PubMed] [Google Scholar]
- 34. Denning DW, Pashley C, Hartl D, Wardlaw A, Godet C, Del Giacco S et al Fungal allergy in asthma‐state of the art and research needs. Clin Transl Allergy 2014; 4:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Knutsen AP, Bush RK, Demain JG, Denning DW, Dixit A, Fairs A et al Fungi and allergic lower respiratory tract diseases. J Allergy Clin Immunol 2012; 129:280–91; quiz 292–283. [DOI] [PubMed] [Google Scholar]
- 36. Moore BB, Moore TA. Viruses in idiopathic pulmonary fibrosis. Etiology and exacerbation. Ann Am Thorac Soc 2015; 12(Suppl. 2):S186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Busse WW, Lemanske RF Jr, Gern JE. Role of viral respiratory infections in asthma and asthma exacerbations. Lancet 2010; 376:826–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Delhaes L, Monchy S, Frealle E, Hubans C, Salleron J, Leroy S et al The airway microbiota in cystic fibrosis: a complex fungal and bacterial community–implications for therapeutic management. PLoS ONE 2012; 7:e36313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Charlson ES, Diamond JM, Bittinger K, Fitzgerald AS, Yadav A, Haas AR et al Lung‐enriched organisms and aberrant bacterial and fungal respiratory microbiota after lung transplant. Am J Respir Crit Care Med 2012; 186:536–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Erb Downward JR, Falkowski NR, Mason KL, Muraglia R, Huffnagle GB. Modulation of post‐antibiotic bacterial community reassembly and host response by Candida albicans . Sci Rep 2013; 3:2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Marsland BJ, Gollwitzer ES. Host‐microorganism interactions in lung diseases. Nat Rev Immunol 2014; 14:827–35. [DOI] [PubMed] [Google Scholar]
- 42. Song X, Gao H, Lin Y, Yao Y, Zhu S, Wang J et al Alterations in the microbiota drive interleukin‐17C production from intestinal epithelial cells to promote tumorigenesis. Immunity 2014; 40:140–52. [DOI] [PubMed] [Google Scholar]
- 43. Evans SE, Xu Y, Tuvim MJ, Dickey BF. Inducible innate resistance of lung epithelium to infection. Annu Rev Physiol 2010; 72:413–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wine JJ, Joo NS. Submucosal glands and airway defense. Proc Am Thorac Soc 2004; 1:47–53. [DOI] [PubMed] [Google Scholar]
- 45. Rokicki W, Rokicki M, Wojtacha J, Dzeljijli A. The role and importance of club cells (Clara cells) in the pathogenesis of some respiratory diseases. Kardiochir Torakochirurgia Pol 2016; 13:26–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mason RJ, Williams MC. Type II alveolar cell. Defender of the alveolus. Am Rev Respir Dis 1977; 115:81–91. [DOI] [PubMed] [Google Scholar]
- 47. Fehrenbach H. Alveolar epithelial type II cell: defender of the alveolus revisited. Respir Res 2001; 2:33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Adler KB, Li Y. Airway epithelium and mucus: intracellular signaling pathways for gene expression and secretion. Am J Respir Cell Mol Biol 2001; 25:397–400. [DOI] [PubMed] [Google Scholar]
- 49. Fahy JV, Dickey BF. Airway mucus function and dysfunction. N Engl J Med 2010; 363:2233–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Proud D, Leigh R. Epithelial cells and airway diseases. Immunol Rev 2011; 242:186–204. [DOI] [PubMed] [Google Scholar]
- 51. Okuda K, Chen G, Subramani DB, Wolf M, Gilmore RC, Kato T et al Localization of secretory mucins MUC5AC and MUC5B in normal/healthy human airways. Am J Respir Crit Care Med 2019; 199:715–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Evans CM, Raclawska DS, Ttofali F, Liptzin DR, Fletcher AA, Harper DN et al The polymeric mucin Muc5ac is required for allergic airway hyperreactivity. Nat Commun 2015; 6:6281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Regnis JA, Robinson M, Bailey DL, Cook P, Hooper P, Chan HK et al Mucociliary clearance in patients with cystic fibrosis and in normal subjects. Am J Respir Crit Care Med 1994; 150:66–71. [DOI] [PubMed] [Google Scholar]
- 54. Smaldone GC, Foster WM, O'Riordan TG, Messina MS, Perry RJ, Langenback EG. Regional impairment of mucociliary clearance in chronic obstructive pulmonary disease. Chest 1993; 103:1390–6. [DOI] [PubMed] [Google Scholar]
- 55. Flynn JM, Niccum D, Dunitz JM, Hunter RC. Evidence and role for bacterial mucin degradation in cystic fibrosis airway disease. PLoS Pathog 2016; 12:e1005846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lee AL, Goldstein RS. Gastroesophageal reflux disease in COPD: links and risks. Int J Chron Obstruct Pulmon Dis 2015; 10:1935–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Flemming HC, Wingender J, Szewzyk U, Steinberg P, Rice SA, Kjelleberg S. Biofilms: an emergent form of bacterial life. Nat Rev Microbiol 2016; 14:563–75. [DOI] [PubMed] [Google Scholar]
- 58. Maurice NM, Bedi B, Sadikot RT. Pseudomonas aeruginosa biofilms: host response and clinical implications in lung infections. Am J Respir Cell Mol Biol 2018; 58:428–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Thurlow LR, Hanke ML, Fritz T, Angle A, Aldrich A, Williams SH et al Staphylococcus aureus biofilms prevent macrophage phagocytosis and attenuate inflammation in vivo. J Immunol 2011; 186:6585–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dickson RP, Erb‐Downward JR, Martinez FJ, Huffnagle GB. The microbiome and the respiratory tract. Annu Rev Physiol 2016; 78:481–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta 2008; 1778:660–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Molyneaux PL, Willis‐Owen SAG, Cox MJ, James P, Cowman S, Loebinger M et al Host‐microbial interactions in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2017; 195:1640–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schutte BC, McCray PB Jr. [beta]‐defensins in lung host defense. Annu Rev Physiol 2002; 64:709–48. [DOI] [PubMed] [Google Scholar]
- 64. Mallia P, Webber J, Gill SK, Trujillo‐Torralbo MB, Calderazzo MA, Finney L et al Role of airway glucose in bacterial infections in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol 2018; 142:815–23.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. O'Dwyer DN, Dickson RP, Moore BB. The lung microbiome, immunity, and the pathogenesis of chronic lung disease. J Immunol 2016; 196:4839–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mariathasan S, Monack DM. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol 2007; 7:31–40. [DOI] [PubMed] [Google Scholar]
- 67. Ye Z, Ting JP. NLR, the nucleotide‐binding domain leucine‐rich repeat containing gene family. Curr Opin Immunol 2008; 20:3–9. [DOI] [PubMed] [Google Scholar]
- 68. Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature 2007; 449:819–26. [DOI] [PubMed] [Google Scholar]
- 69. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell 2010; 140:805–20. [DOI] [PubMed] [Google Scholar]
- 70. Takeda K, Kaisho T, Akira S. Toll‐like receptors. Annu Rev Immunol 2003; 21:335–76. [DOI] [PubMed] [Google Scholar]
- 71. Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H et al A Toll‐like receptor recognizes bacterial DNA. Nature 2000; 408:740–45. [DOI] [PubMed] [Google Scholar]
- 72. Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K et al Small anti‐viral compounds activate immune cells via the TLR7 MyD88‐dependent signaling pathway. Nat Immunol 2002; 3:196–200. [DOI] [PubMed] [Google Scholar]
- 73. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double‐stranded RNA and activation of NF‐kappaB by Toll‐like receptor 3. Nature 2001; 413:732–8. [DOI] [PubMed] [Google Scholar]
- 74. Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR et al The innate immune response to bacterial flagellin is mediated by Toll‐like receptor 5. Nature 2001; 410:1099–103. [DOI] [PubMed] [Google Scholar]
- 75. de Bouteiller O, Merck E, Hasan UA, Hubac S, Benguigui B, Trinchieri G et al Recognition of double‐stranded RNA by human toll‐like receptor 3 and downstream receptor signaling requires multimerization and an acidic pH. J Biol Chem 2005; 280:38 133–45. [DOI] [PubMed] [Google Scholar]
- 76. Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF et al TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol 2004; 5:190–8. [DOI] [PubMed] [Google Scholar]
- 77. Lee J, Chuang TH, Redecke V, She L, Pitha PM, Carson DA et al Molecular basis for the immunostimulatory activity of guanine nucleoside analogs: activation of Toll‐like receptor 7. Proc Natl Acad Sci USA 2003; 100:6646–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Matsumoto M, Funami K, Tanabe M, Oshiumi H, Shingai M, Seto Y et al Subcellular localization of Toll‐like receptor 3 in human dendritic cells. J Immunol 2003; 171:3154–62. [DOI] [PubMed] [Google Scholar]
- 79. Nishiya T, DeFranco AL. Ligand‐regulated chimeric receptor approach reveals distinctive subcellular localization and signaling properties of the Toll‐like receptors. J Biol Chem 2004; 279:19 008–17. [DOI] [PubMed] [Google Scholar]
- 80. Oosting M, Cheng SC, Bolscher JM, Vestering‐Stenger R, Plantinga TS, Verschueren IC et al Human TLR10 is an anti‐inflammatory pattern‐recognition receptor. Proc Natl Acad Sci USA 2014; 111:E4478–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sha Q, Truong‐Tran AQ, Plitt JR, Beck LA, Schleimer RP. Activation of airway epithelial cells by toll‐like receptor agonists. Am J Respir Cell Mol Biol 2004; 31:358–64. [DOI] [PubMed] [Google Scholar]
- 82. Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN. House dust mite allergen induces asthma via Toll‐like receptor 4 triggering of airway structural cells. Nat Med 2009; 15:410–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hewson CA, Jardine A, Edwards MR, Laza‐Stanca V, Johnston SL. Toll‐like receptor 3 is induced by and mediates antiviral activity against rhinovirus infection of human bronchial epithelial cells. J Virol 2005; 79:12 273–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ritchie AI, Farne HA, Singanayagam A, Jackson DJ, Mallia P, Johnston SL. Pathogenesis of viral infection in exacerbations of airway disease. Ann Am Thorac Soc 2015; 12(Suppl. 2):S115–32. [DOI] [PubMed] [Google Scholar]
- 85. Ghaemmaghami AM, Gough L, Sewell HF, Shakib F. The proteolytic activity of the major dust mite allergen Der p 1 conditions dendritic cells to produce less interleukin‐12: allergen‐induced Th2 bias determined at the dendritic cell level. Clin Exp Allergy 2002; 32:1468–75. [DOI] [PubMed] [Google Scholar]
- 86. Adam E, Hansen KK, Astudillo Fernandez O, Coulon L, Bex F, Duhant X et al The house dust mite allergen Der p 1, unlike Der p 3, stimulates the expression of interleukin‐8 in human airway epithelial cells via a proteinase‐activated receptor‐2‐independent mechanism. J Biol Chem 2006; 281:6910–23. [DOI] [PubMed] [Google Scholar]
- 87. Kauffman HF, Tamm M, Timmerman JA, Borger P. House dust mite major allergens Der p 1 and Der p 5 activate human airway‐derived epithelial cells by protease‐dependent and protease‐independent mechanisms. Clin Mol Allergy 2006; 4:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Chiu LL, Perng DW, Yu CH, Su SN, Chow LP. Mold allergen, pen C 13, induces IL‐8 expression in human airway epithelial cells by activating protease‐activated receptor 1 and 2. J Immunol 2007; 178:5237–44. [DOI] [PubMed] [Google Scholar]
- 89. Wang Q, Nagarkar DR, Bowman ER, Schneider D, Gosangi B, Lei J et al Role of double‐stranded RNA pattern recognition receptors in rhinovirus‐induced airway epithelial cell responses. J Immunol 2009; 183:6989–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Lipinski S, Rosenstiel P. Debug your bugs ‐ how NLRs shape intestinal host‐microbe interactions. Front Immunol 2013; 4:479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lee HM, Yuk JM, Shin DM, Jo EK. Dectin‐1 is inducible and plays an essential role for mycobacteria‐induced innate immune responses in airway epithelial cells. J Clin Immunol 2009; 29:795–805. [DOI] [PubMed] [Google Scholar]
- 92. Sun WK, Lu X, Li X, Sun QY, Su X, Song Y et al Dectin‐1 is inducible and plays a crucial role in Aspergillus‐induced innate immune responses in human bronchial epithelial cells. Eur J Clin Microbiol Infect Dis 2012; 31:2755–64. [DOI] [PubMed] [Google Scholar]
- 93. Nathan AT, Peterson EA, Chakir J, Wills‐Karp M. Innate immune responses of airway epithelium to house dust mite are mediated through beta‐glucan‐dependent pathways. J Allergy Clin Immunol 2009; 123:612–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Trompette A, Divanovic S, Visintin A, Blanchard C, Hegde RS, Madan R et al Allergenicity resulting from functional mimicry of a Toll‐like receptor complex protein. Nature 2009; 457:585–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Page K, Lierl KM, Hughes VS, Zhou P, Ledford JR, Wills‐Karp M. TLR2‐mediated activation of neutrophils in response to German cockroach frass. J Immunol 2008; 180:6317–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Boldogh I, Bacsi A, Choudhury BK, Dharajiya N, Alam R, Hazra TK et al ROS generated by pollen NADPH oxidase provide a signal that augments antigen‐induced allergic airway inflammation. J Clin Invest 2005; 115:2169–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Salo PM, Arbes SJ Jr, Sever M, Jaramillo R, Cohn RD, London SJ et al Exposure to Alternaria alternata in US homes is associated with asthma symptoms. J Allergy Clin Immunol 2006; 118:892–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Hsu AC, See HV, Hansbro PM, Wark PA. Innate immunity to influenza in chronic airways diseases. Respirology 2012; 17:1166–75. [DOI] [PubMed] [Google Scholar]
- 99. Hansbro NG, Horvat JC, Wark PA, Hansbro PM. Understanding the mechanisms of viral induced asthma: new therapeutic directions. Pharmacol Ther 2008; 117:313–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Hansbro PM, Beagley KW, Horvat JC, Gibson PG. Role of atypical bacterial infection of the lung in predisposition/protection of asthma. Pharmacol Ther 2004; 101:193–210. [DOI] [PubMed] [Google Scholar]
- 101. Bosch AA, Biesbroek G, Trzcinski K, Sanders EA, Bogaert D. Viral and bacterial interactions in the upper respiratory tract. PLoS Pathog 2013; 9:e1003057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Ganesan S, Comstock AT, Sajjan US. Barrier function of airway tract epithelium. Tissue Barriers 2013; 1:e24997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Sajjan U, Wang Q, Zhao Y, Gruenert DC, Hershenson MB. Rhinovirus disrupts the barrier function of polarized airway epithelial cells. Am J Respir Crit Care Med 2008; 178:1271–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Comstock AT, Ganesan S, Chattoraj A, Faris AN, Margolis BL, Hershenson MB et al Rhinovirus‐induced barrier dysfunction in polarized airway epithelial cells is mediated by NADPH oxidase 1. J Virol 2011; 85:6795–08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Rezaee F, DeSando SA, Ivanov AI, Chapman TJ, Knowlden SA, Beck LA et al Sustained protein kinase D activation mediates respiratory syncytial virus‐induced airway barrier disruption. J Virol 2013; 87:11 088–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Xiao C, Puddicombe SM, Field S, Haywood J, Broughton‐Head V, Puxeddu I et al Defective epithelial barrier function in asthma. J Allergy Clin Immunol 2011; 128: 549–56.e12. [DOI] [PubMed] [Google Scholar]
- 107. Hackett TL, de Bruin HG, Shaheen F, van den Berge M, van Oosterhout AJ, Postma DS et al Caveolin‐1 controls airway epithelial barrier function. Implications for asthma. Am J Respir Cell Mol Biol 2013; 49:662–71. [DOI] [PubMed] [Google Scholar]
- 108. Shaykhiev R, Otaki F, Bonsu P, Dang DT, Teater M, Strulovici‐Barel Y et al Cigarette smoking reprograms apical junctional complex molecular architecture in the human airway epithelium in vivo. Cell Mol Life Sci 2011; 68:877–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Wonderlich ER, Swan ZD, Bissel SJ, Hartman AL, Carney JP, O'Malley KJ et al Widespread virus replication in alveoli drives acute respiratory distress syndrome in aerosolized H5N1 influenza infection of macaques. J Immunol 2017; 198:1616–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Coyne CB, Shen L, Turner JR, Bergelson JM. Coxsackievirus entry across epithelial tight junctions requires occludin and the small GTPases Rab34 and Rab5. Cell Host Microbe 2007; 2:181–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Molyneaux PL, Mallia P, Cox MJ, Footitt J, Willis‐Owen SA, Homola D et al Outgrowth of the bacterial airway microbiome after rhinovirus exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2013; 188:1224–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Grainge C, Dennison P, Lau L, Davies D, Howarth P. Asthmatic and normal respiratory epithelial cells respond differently to mechanical apical stress. Am J Respir Crit Care Med 2014; 190:477–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Saatian B, Rezaee F, Desando S, Emo J, Chapman T, Knowlden S et al Interleukin‐4 and interleukin‐13 cause barrier dysfunction in human airway epithelial cells. Tissue Barriers 2013; 1:e24333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Gause WC, Wynn TA, Allen JE. Type 2 immunity and wound healing: evolutionary refinement of adaptive immunity by helminths. Nat Rev Immunol 2013; 13:607–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Heijink IH, Kies PM, Kauffman HF, Postma DS, van Oosterhout AJ, Vellenga E. Down‐regulation of E‐cadherin in human bronchial epithelial cells leads to epidermal growth factor receptor‐dependent Th2 cell‐promoting activity. J Immunol 2007; 178:7678–85. [DOI] [PubMed] [Google Scholar]
- 116. de Boer WI, Sharma HS, Baelemans SM, Hoogsteden HC, Lambrecht BN, Braunstahl GJ. Altered expression of epithelial junctional proteins in atopic asthma: possible role in inflammation. Can J Physiol Pharmacol 2008; 86:105–12. [DOI] [PubMed] [Google Scholar]
- 117. Hackett TL, Singhera GK, Shaheen F, Hayden P, Jackson GR, Hegele RG et al Intrinsic phenotypic differences of asthmatic epithelium and its inflammatory responses to respiratory syncytial virus and air pollution. Am J Respir Cell Mol Biol 2011; 45:1090–100. [DOI] [PubMed] [Google Scholar]
- 118. Ierodiakonou D, Postma DS, Koppelman GH, Boezen HM, Gerritsen J, Ten Hacken N et al E‐cadherin gene polymorphisms in asthma patients using inhaled corticosteroids. Eur Respir J 2011; 38:1044–52. [DOI] [PubMed] [Google Scholar]
- 119. Koppelman GH, Meyers DA, Howard TD, Zheng SL, Hawkins GA, Ampleford EJ et al Identification of PCDH1 as a novel susceptibility gene for bronchial hyperresponsiveness. Am J Respir Crit Care Med 2009; 180:929–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Qian X, Karpova T, Sheppard AM, McNally J, Lowy DR. E‐cadherin‐mediated adhesion inhibits ligand‐dependent activation of diverse receptor tyrosine kinases. Embo J 2004; 23:1739–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Sorby M, Ostman A. Protein‐tyrosine phosphatase‐mediated decrease of epidermal growth factor and platelet‐derived growth factor receptor tyrosine phosphorylation in high cell density cultures. J Biol Chem 1996; 271:10 963–66. [DOI] [PubMed] [Google Scholar]
- 122. Symons JR, LeVea CM, Mooney RA. Expression of the leucocyte common antigen‐related (LAR) tyrosine phosphatase is regulated by cell density through functional E‐cadherin complexes. Biochem J 2002; 365:513–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Wendt MK, Smith JA, Schiemann WP. Transforming growth factor‐beta‐induced epithelial‐mesenchymal transition facilitates epidermal growth factor‐dependent breast cancer progression. Oncogene 2010; 29:6485–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Casalino‐Matsuda SM, Monzon ME, Forteza RM. Epidermal growth factor receptor activation by epidermal growth factor mediates oxidant‐induced goblet cell metaplasia in human airway epithelium. Am J Respir Cell Mol Biol 2006; 34:581–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Mazieres J, He B, You L, Xu Z, Jablons DM. Wnt signaling in lung cancer. Cancer Lett 2005; 222:1–10. [DOI] [PubMed] [Google Scholar]
- 126. Wang K, Zhang Y, Li X, Chen L, Wang H, Wu J et al Characterization of the Kremen‐binding site on Dkk1 and elucidation of the role of Kremen in Dkk‐mediated Wnt antagonism. J Biol Chem 2008; 283:23 371–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Pfaff EM, Becker S, Gunther A, Konigshoff M. Dickkopf proteins influence lung epithelial cell proliferation in idiopathic pulmonary fibrosis. Eur Respir J 2011; 37:79–87. [DOI] [PubMed] [Google Scholar]
- 128. Chilosi M, Poletti V, Zamo A, Lestani M, Montagna L, Piccoli P et al Aberrant Wnt/beta‐catenin pathway activation in idiopathic pulmonary fibrosis. Am J Pathol 2003; 162:1495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Sucre JMS, Deutsch GH, Jetter CS, Ambalavanan N, Benjamin JT, Gleaves LA et al A shared pattern of beta‐catenin activation in bronchopulmonary dysplasia and idiopathic pulmonary fibrosis. Am J Pathol 2018; 188:853–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Shi J, Li F, Luo M, Wei J, Liu X. Distinct roles of wnt/beta‐catenin signaling in the pathogenesis of chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Mediators Inflamm 2017; 2017:3 520 581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Baarsma HA, Skronska‐Wasek W, Mutze K, Ciolek F, Wagner DE, John‐Schuster G et al Noncanonical WNT‐5A signaling impairs endogenous lung repair in COPD. J Exp Med 2017; 214:143–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Oh JE, Kim RH, Shin KH, Park NH, Kang MK. DeltaNp63alpha protein triggers epithelial‐mesenchymal transition and confers stem cell properties in normal human keratinocytes. J Biol Chem 2011; 286:38 757–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Jonsdottir HR, Arason AJ, Palsson R, Franzdottir SR, Gudbjartsson T, Isaksson HJ et al Basal cells of the human airways acquire mesenchymal traits in idiopathic pulmonary fibrosis and in culture. Lab Invest 2015; 95:1418–28. [DOI] [PubMed] [Google Scholar]
- 134. Cummings JH, Pomare EW, Branch WJ, Naylor CP, Macfarlane GT. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 1987; 28:1221–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Peng L, Li ZR, Green RS, Holzman IR, Lin J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP‐activated protein kinase in Caco‐2 cell monolayers. J Nutr 2009; 139:1619–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Dang AT, Marsland BJ. Microbes, metabolites, and the gut‐lung axis. Mucosal Immunol 2019; 12:843–50. [DOI] [PubMed] [Google Scholar]
- 137. Samuelson DR, Welsh DA, Shellito JE. Regulation of lung immunity and host defense by the intestinal microbiota. Front Microbiol 2015; 6:1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Noverr MC, Falkowski NR, McDonald RA, McKenzie AN, Huffnagle GB. Development of allergic airway disease in mice following antibiotic therapy and fungal microbiota increase: role of host genetics, antigen, and interleukin‐13. Infect Immun 2005; 73:30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Fagundes CT, Amaral FA, Vieira AT, Soares AC, Pinho V, Nicoli JR et al Transient TLR activation restores inflammatory response and ability to control pulmonary bacterial infection in germfree mice. J Immunol 2012; 188:1411–20. [DOI] [PubMed] [Google Scholar]
- 140. Vael C, Nelen V, Verhulst SL, Goossens H, Desager KN. Early intestinal Bacteroides fragilis colonisation and development of asthma. BMC Pulm Med 2008; 8:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Arrieta MC, Stiemsma LT, Dimitriu PA, Thorson L, Russell S, Yurist‐Doutsch S et al infancy microbial and metabolic alterations affect risk of childhood asthma. Sci Transl Med 2015; 7:307ra152. [DOI] [PubMed] [Google Scholar]
- 142. Hevia A, Milani C, Lopez P, Donado CD, Cuervo A, Gonzalez S et al Allergic patients with long‐term asthma display low levels of bifidobacterium adolescentis. PLoS ONE 2016; 11:e0147809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Tomoda K, Kubo K, Dairiki K, Yamaji T, Yamamoto Y, Nishii Y et al Whey peptide‐based enteral diet attenuated elastase‐induced emphysema with increase in short chain fatty acids in mice. BMC Pulm Med 2015; 15:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Southam DS, Dolovich M, O'Byrne PM, Inman MD. Distribution of intranasal instillations in mice: effects of volume, time, body position, and anesthesia. Am J Physiol Lung Cell Mol Physiol 2002; 282:L833–39. [DOI] [PubMed] [Google Scholar]
- 145. Groves HT, Higham SL, Moffatt MF, Cox MJ, Tregoning JS. Respiratory viral infection alters the gut microbiota by inducing inappetence. mBio 2020; 11:2150–74511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Sze MA, Tsuruta M, Yang SW, Oh Y, Man SF, Hogg JC et al Changes in the bacterial microbiota in gut, blood, and lungs following acute LPS instillation into mice lungs. PLoS ONE 2014; 9:e111228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Metsala J, Lundqvist A, Virta LJ, Kaila M, Gissler M, Virtanen SM. Mother's and offspring's use of antibiotics and infant allergy to cow's milk. Epidemiology 2013; 24:303–9. [DOI] [PubMed] [Google Scholar]
- 148. Korpela K, Salonen A, Virta LJ, Kekkonen RA, Forslund K, Bork P et al Intestinal microbiome is related to lifetime antibiotic use in Finnish pre‐school children. Nat Commun 2016; 7:10 410. [DOI] [PMC free article] [PubMed] [Google Scholar]