

Summary
Protein kinase inhibitors are one of the most successful targeted therapies to date. Despite this progress, additional kinase inhibitors are needed to expand the target space as well as overcome drug resistance that has emerged in clinical setting. Here, we developed KiDNN (Kinase inhibitor prediction using Deep Neural Networks). KiDNN utilizes non-linear, multilayer feedforward network that mimics complex and dynamic kinase-driven signaling pathways. We used KiDNN to predict the effect of ∼200 kinase inhibitors on migration of breast and liver cancer cells. We show that the prediction accuracy of KiDNN outperformed other prediction tools based on linear models. We validated that an inhibitor of tyrosine kinase receptors, and an inhibitor of Src family kinases, decreased migration of triple-negative breast cancer cells, consistent with the role of these kinases in driving motility. Overall, we show that non-linear, DNN-based models provide a powerful approach to in silico screen hundreds of kinase inhibitors.
Subject Areas: Bioinformatics, Computational Bioinformatics, Neural Networks, Cancer
Graphical Abstract
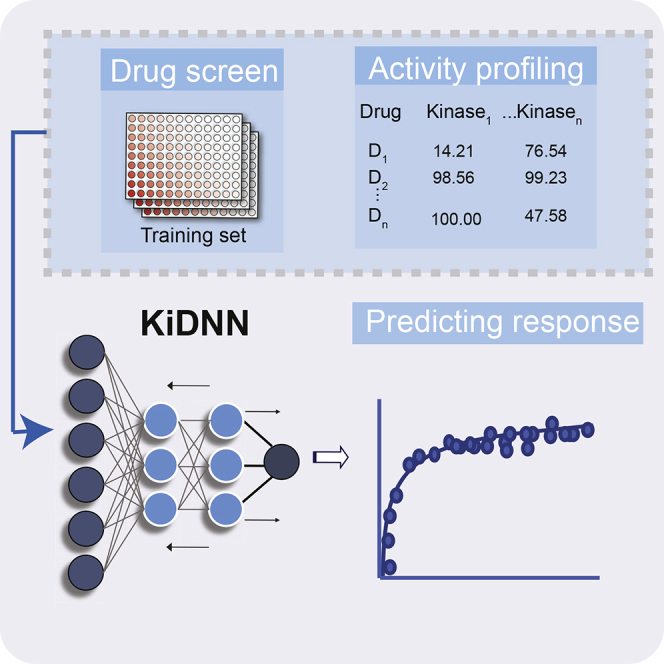
Highlights
-
•
Deep Neural Networks mimic non-linear, complex intracellular signaling pathways
-
•
Multi-phase grid search identified best networks with less computation time
-
•
Prediction accuracy of KiDNN outperformed linear models
-
•
KiDNN can accelerate drug discovery and development efforts
Bioinformatics; Computational Bioinformatics; Neural Networks; Cancer
Introduction
Kinase inhibitors represent a large class of US Food and Drug Administration-approved drugs and are currently in use for the treatment of various cancers. Advances in “omics” technologies have led to the development of high-throughput methods to efficiently and reliably profile the target selectivity of kinase inhibitors in vitro and in a cellular environment (Klaeger et al., 2017, Schmidlin et al., 2019, Zegzouti et al., 2016). Several groups have profiled hundreds of kinase inhibitors against sizable fractions of the ∼500 human protein kinases (Anastassiadis et al., 2011, Davis et al., 2011, Duong-Ly et al., 2016, Klaeger et al., 2017, Schmidlin et al., 2019). The resulting “kinase-inhibitor interaction maps” revealed that majority of kinase inhibitors bind multiple targets, despite the efforts to generate selective agents (Duong-Ly et al., 2016, Klaeger et al., 2017). In some cases, these unintended targets result in off-target toxicity, whereas in other instances, off-target binding is of clinical benefit, leading to the effect commonly referred to as polypharmacology. Although broadly acknowledged as a dominant mode of action, polypharmacology is currently poorly understood and difficult to predict.
We and others have developed linear regression-based approaches, such as Kinome Regularization (KiR), which exploit the polypharmacology of kinase inhibitors and rely on linear combinations of the contributions of kinases to cellular behavior to build models for predicting the effect of a particular inhibitor on specific cellular phenotypes (Gujral et al., 2014a, Gujral et al., 2014b). However, kinase-driven signaling pathways are highly complex and dynamic, and the outcome of a perturbation is difficult to predict from the linear combination of the individual parts. Here, we hypothesize that there is a complex, non-linear dependence between the activity profiles of inhibitors and their respective contribution to phenotypes, such as cell proliferation or migration. This suggests that a non-linear, multilayer feedforward network would exhibit improved performance over the linear approach. To test this hypothesis, we developed a deep neural network (DNN) based on non-linear principles called KiDNN for Kinase Inhibitor prediction using Deep Neural Networks.
KiDNN takes advantage of the DNN framework that mimics the human brain. DNNs incorporate processing nodes that are analogous to neurons (Zupan, 1994). In KiDNN, these nodes are connected by weighted links into a complex, multi-layered neural network. All nodes, except those comprising the input layer, receive weighted sums of the output from the nodes in the previous layer and transmit their output to nodes in successive layers until the final output layer is reached (Dongare et al., 2012). We applied KiDNN to predict the effect of ∼200 kinase inhibitors on migration of triple-negative breast cancer (TNBC) cell line (Hs578t) and liver cancer cells (FOCUS). We experimentally tested a subset of the inhibitors and determined that KiDNN predictions outperform predictions from the linear KiR model. As predicted by KiDNN, we showed that inhibition of the tyrosine kinase receptor including PDGFRb and VEGFR and nonreceptor tyrosine kinases Src decreases migration of Hs578t cells, consistent with the role of these kinases in driving motility of TNBC cells (Jechlinger et al., 2006, Simiczyjew et al., 2018, Van Swearingen et al., 2017). Overall, our results indicated that the application of models based on non-linear DNNs is superior to linear-based models for predicting the cellular response to kinase inhibitors with known activity profiles.
Results
Developing an Optimized KiDNN for Predicting Kinase Inhibitor Effect on Hs578t Cell Migration
Kinase inhibitors are widely used to identify cellular signaling pathways underlying complex phenotypes. Here, we developed a predictive DNN model and screened for kinase inhibitors that impaired migration of cancer cells as a disease-relevant phenotype (Figure 1). In our previous study, we demonstrated that a set of 32 kinase inhibitors as well as non-specific compounds provides >85% coverage of the 300 kinase targets (Gujral et al., 2014b). To develop and optimize KiDNN models, we used a previously generated experimental dataset for the quantitative effects of these 32 inhibitors on migration of Hs578t TNBC cells (Gujral et al., 2014b).
Figure 1.

The Development of KiDNN
A schematic illustrating the supervised learning approach to develop, evaluate, and predict kinase inhibitor-mediated changes in migration using KiDNN. The input variable is defined by a set of 32 kinase inhibitors and their target profiles against 300 kinases (32X300 matrix). The numbers in input variable indicate percent residual kinase activity. Output variable is defined by the response of 32 inhibitors on migration of cancer cells measured using wound healing assay in 96-well plates. The numbers in output variable indicate percent wound density. The development of KiDNN consisted of model development phase during which hyperparameters are optimized, model evaluation where LOOCV is employed to generate predictions, and model prediction where response to naive kinase inhibitors are predicted and experimentally validated.
To develop DNN models that effectively predict changes in cell migration in response to kinase inhibitors, we devised a five-phase strategy for optimizing the neural network hyperparameters for maximum predictive capability on Hs578t cell migration (Figure 2A). The first phase was deducing the range of overfitting, where we used the training and validation loss to identify the optimal range of epochs. In subsequent phases (phases 2–5), we performed optimization using Grid Search (Bergstra et al., 2011), in which numerous models were built with every combination of hyperparameter values, and the top models (and their respective combination of hyperparameter values) were identified. Various hyperparameters were optimized in each phase. Batch size and epochs were optimized in phase 2. In phase 3, activation function, weight initialization, and optimizers were tuned, whereas hidden layers, nodes per hidden layer, and dropout were tuned in phase 4 (Figure 2A). Table S1 lists the tested hyperparameter values, and the Transparent Methods outlines the definitions of the hyperparameters. When performing Grid Search, hyperparameters that were not undergoing optimization were kept constant according to a set of baseline hyperparameter values (see the Transparent Methods for details on how the baseline hyperparameter values are updated). In the last phase (phase 5), the top two hyperparameter values identified in phases 2, 3, and 4 were used to perform a final Grid Search that selects the single top combination of all eight hyperparameter values. This set of eight hyperparameter values was then used to build the final KiDNN model for predicting Hs578t cell migration (Figure 2A).
Figure 2.

Tuning Network Hyperparameters and Structure for Optimal KiDNN Performance
(A) A schematic showing the progressive, multi-phase approach of optimizing the network hyperparameters/structure for peak network performance.
(B) A plot showing the network validation MSE (k = 26) and training MSE as a function of the number of epochs. A polynomial fit (n = 5) of the validation error is also shown and the range of overfitting is indicated.
(C) A heatmap showing the respective LOOCV MSE of 42 networks built with selected combinations of batch sizes and epochs. Yellow regions indicate low relative errors.
(D) A 3D scatterplot illustrating respective LOOCV MSE of 300 networks built with selected combinations of activation functions, weight initializations, and optimizers. Darker spheres indicate low relative errors corresponding to specific combinations of hyperparameters.
(E) A 3D scatterplot illustrating respective LOOCV MSE of 120 networks built with selected combinations of hidden layers, nodes per hidden layer, and dropout rate. Darker blue spheres indicate low relative errors corresponding to specific combinations of hyperparameters. Complete list of hyperparameters tuned are listed in Table S1 and additional trials of the top five hyperparameters are evaluated in Tables S2–S4.
To evaluate and select the top hyperparameters in each phase, we applied leave-one-out-cross-validation (LOOCV) (Zhang, 1993). In LOOCV, each time the KiDNN model was trained on the activity profiles of 31 of the 32 inhibitors and their effects on cell migration (Gujral et al., 2014b) to predict the effect of the excluded inhibitor on cell migration. The process was repeated 33 times (including control), leaving out and predicting the effect on migration for each of the 32 inhibitors. The average mean squared error (MSE) of all 33 predictions was used to evaluate predictive accuracy of various networks and select the hyperparameters that produced the lowest LOOCV MSE.
In phase 1, we determined the range of model overfitting starting with an unoptimized baseline neural network. We trained the baseline neural network on the Hs578t migration responses to 26 randomly selected inhibitors (representing ∼80% of the data) to predict the effect on cell migration of the six excluded inhibitors (the remaining ∼20% of the data). The model was trained for 400 epochs, which is the total number of times KiDNN learned from the entire training dataset to optimize its weights. We plotted the training MSE for the 26 kinase inhibitors and the cross-validation MSE of predicted and observed migration for the six excluded kinase inhibitors as a function of the number of epochs (Figure 2B). A polynomial fit (n = 5) for the cross-validation error indicated that the network's predictive performance improved until 125 epochs, after which the network gradually started to overfit the data. Although too few epochs can cause underfitting of the data, suggesting the model stops learning too early, too many epochs can cause the model to overfit the training data such that the model cannot generalize to effectively predict the response to six left out kinase inhibitors on cell migration. Consequently, an optimal range of epochs where the MSE reached a global minimum was selected using the validation error: 50, 75, 100, 125, 150, 175, 200 (Figure 2B). Since batch size (optimized in phase 2) can affect the optimal number of epochs, a buffer of 75 epochs above and below the actual global minimum of 125 epochs ensured that the top combination of epochs and batch size was selected with variations in batch size in phase 2.
In phase 2, we evaluated the optimal combination of batch size and epochs. The batch size is the number of samples (individual observations in the dataset) input into KiDNN before updating the weights (Chollet, 2018). Using Grid Search, we constructed 42 networks representing the combinations of the seven epoch quantities and six batch sizes and evaluated the accuracy of the network by LOOCV MSE (Figure 2C). Our data showed that network error was minimized with larger batch sizes and lower quantities of epochs (Figure 2C, lower right quadrant). We re-evaluated the top five combinations of epochs and batch sizes an additional five times, which revealed that the top two combinations of batch size and epochs were 32-sample batch size and 75 epochs (average MSE 100.48) and 16-sample batch size and 50 epochs (average MSE 101.47) (Table S2).
In phase 3, we tuned the weight initialization, optimizer, and activation function hyperparameters. We built and evaluated 300 networks (5 activation functions x 10 weight initializations x 6 optimizers combinations) using Grid Search (Figure 2D, Table S1). Our data indicated that networks built with the TanH (hyperbolic tangent) and sigmoid activation functions were completely ineffective at predicting changes in cell migration (Figure 2D); thus, we excluded them from subsequent optimizations for activation function. We re-evaluated the top five combinations of weight initialization, optimizer, and activation function (Table S3). The top two combinations of hyperparameters were truncated normal initialization, Adagrad optimizer, and ELU activation function (average MSE 92.90) and truncated normal initialization, Adagrad optimizer, and ReLU activation function (average MSE 94.62).
In phase 4, we optimized the dropout rate, the number of hidden layers, and the number of nodes per hidden layer. With Grid Search, we built and evaluated 120 networks (5 dropout rates x 3 hidden layer quantities x 8 nodes per hidden layer). Networks built with lower dropout rates and more than one hidden layer had greater predictive power as indicated by their lower average MSE values (Figure 2E). These results confirm our initial hypothesis that the activity profiles of kinase inhibitors and their respective effect on Hs578t cell migration have a complex, non-linear dependence that Deep Neural Networks can better predict rather than shallow neural networks with one hidden layer or linear models. We re-evaluated the top five combinations of the hidden layer quantity, nodes per hidden layer, and dropout rate (Table S4). The top two combinations of the hyperparameters were two hidden layers, 300 nodes per hidden layer, and a dropout rate of 0 (average MSE 85.74) and two hidden layers, 200 nodes per hidden layer, and a dropout rate of 0 (average MSE 89.86).
In phase 5, we tested the top two sets of hyperparameters (shaded rows in Tables S2–S4), which yielded 8 (23) networks that combined the two sets of hyperparameters from each of the three phases. To identify the top performing model, we calculated the average LOOCV MSE and MAE (Mean Absolute Error) of five runs of each of the eight models (Table 1). The top two performing models both had similar MSE and MAE values (Table 1). Given the similarity in performance between the two top-performing models, we selected the model with the lowest average MSE (87.17) and simpler structure to build the final KiDNN model (KiDNN-Hs578t).
Table 1.
Evaluation of Networks from Combination of Top Hyperparameters Selected in Each Optimization Phase
| Epochs | Batch Size | Weight Initializer | Optimizer | Activation | Hidden Layers | Nodes per HL | Dropout Rate | Mean MSE | Mean MAE |
|---|---|---|---|---|---|---|---|---|---|
| 50 | 16 | Truncated Normal | Adagrad | ReLU | 2 | 300 | 0 | 108.742 | 6.6 |
| 50 | 16 | Truncated Normal | Adagrad | ReLU | 2 | 200 | 0 | 101.4 | 6.6 |
| 75 | 32 | Truncated Normal | Adagrad | ReLU | 2 | 300 | 0 | 116.341 | 6.2 |
| 75 | 32 | Truncated Normal | Adagrad | ReLU | 2 | 200 | 0 | 99.2 | 5.9 |
| 50 | 16 | Truncated Normal | Adagrad | ELU | 2 | 300 | 0 | 90.3 | 5.9 |
| 50 | 16 | Truncated Normal | Adagrad | ELU | 2 | 200 | 0 | 95.7 | 6.1 |
| 75 | 32 | Truncated Normal | Adagrad | ELU | 2 | 300 | 0 | 87.4 | 5.6 |
| 75 | 32 | Truncated Normal | Adagrad | ELU | 2 | 200 | 0 | 87.2 | 5.68 |
The network with the lowest average MSE used to build KiDNN is highlighted. ReLU, rectified linear unit; ELU, exponential linear unit; Adagrad, adaptive gradient.
The network architecture consisted of 300 input nodes (representing activity of each of the kinases from the in vitro kinase profiling work that tested 178 inhibitors against 300 kinases, Dongare et al., 2012), two hidden layers with 200 nodes per layer, and a single output node for predicted migration (Figure 3). The batch size of 32 equaled input sample size, commonly referred to as batch gradient descent, and the entire training dataset was input through KiDNN-Hs578t 75 times (epochs) in total. Because the final model included only 200 nodes in the hidden layer rather than the top-performing hyperparameter for the number of nodes in the hidden layer (300 nodes, Table S4), this last phase captured a further optimized combination of hyperparameters. Thus, our results support performing this last optimization phase rather than just building the final model from the top-performing hyperparameters (lowest MSE) identified in each phase.
Figure 3.

Optimized KiDNN Architecture
A schematic illustrating the final architecture of KiDNN after hyperparameter optimization using training dataset from Hs578t cell migration.
Evaluating the Predictive Capability of KiDNN Models for New Drugs
Having optimized the hyperparameters of KiDNN-Hs578t to yield the lowest validation MSE in predicting changes in migration of Hs578t cells in response to kinase inhibitors, we evaluated the predictive capability of KiDNN-Hs578t. First, we used LOOCV to predict the response to each of the 32 kinase inhibitors (entire training dataset) and compared those with experimentally observed migration (Figure 4A). The MSE of the predictions was 78.15, whereas the Pearson correlation was 0.88. On average, the model differed from experimentally observed changes in cell migration by 5.80% for any given prediction.
Figure 4.

Predicting Response to Naive Kinase Inhibitors
(A) A plot showing comparison of KiDNN-predicted and measured percent migration in response to 32 inhibitors in Hs578t cells using LOOCV. Migration in response to untreated/DMSO control was 70%. The MSE, MAE, and Pearson correlations are also indicated.
(B) KiDNN model was run for 10 iterations to generate predictions for all 32 inhibitors. Green circles denote the mean predicted migration, and the error bars show the standard deviation of predictions for each kinase inhibitor. The standard deviation is also listed.
(C) A plot showing KiDNN-predicted response to 178 small molecule kinase inhibitors (146 untested). The black circles denote the predicted percent migration, whereas blue circles denote the 32 experimentally validated kinase inhibitors. The kinase inhibitors are ranked by predicted migration.
(D) A heatmap showing correlation of kinase target profiles of the 10 most and 10 least effective inhibitors predicted by KiDNN.
(E) A table showing comparison of KiDNN-prediction and experimental changes in migration of Hs578t cells in response to eight kinase inhibitors. Each inhibitor was tested at multiple doses (10 nM–10 μM), and the effect of kinase inhibitor at 500 nM dose was calculated using the dose-response curves. Hs578t cells treated with DMSO control showed migration of 70%.
Although the accuracy of predictions is informative in evaluating a model, a low variance of predictions can ensure a high-precision network. A potential limitation of DNN models is that these models can produce extremely high variance among predictions, resulting in drastically varying output from one run to another. To test for variance in KiDNN-Hs578t, we determined LOOCV predictions of changes in cell migration in response to each of the 32 inhibitors in our training set during 10 different evaluations by KiDNN-Hs578t (Figure 4B). The mean standard deviation of all 33 (including control) predictions was 1.44, indicating an overall high prediction precision. With the low MSE and MAE (Figure 4A), this model has both high prediction precision and accuracy for the effect of kinase inhibitors on Hs578t cell migration.
The previous analysis determined the accuracy and precision using the observed Hs578t migration in response to inhibitors used in our training set, whereas we next used KiDNN-Hs578t to predict the effect of 178 compounds (for which in vitro activity profiles against 300 kinases have previously been determined [Anastassiadis et al., 2011]) on Hs578t cell migration. The experimentally measured effects of the 32 compounds in our training set closely aligned with the curve formed by the predicted migration effects of all 178 kinase inhibitors, indicating that the predicted effects are likely close to what one might expect to observe experimentally (Figure 4C, Table S5). Of the 178 kinase inhibitors predicted by the model, 175 had differences between the predicted and interpolated migrations (based on polynomial interpolation (n = 6) of the 32 kinase inhibitors' experimental migration) <10% and 137 had differences <5%.
To gain insight into the key signaling nodes targeted by the top KiDNN predicted inhibitors, we determined the correlation among the top 10 highly ranked and the least 10 effective inhibitors predicted by KiDNN to effect migration of Hs578t cells. Staurosporine, K252a, and CDK1/2 inhibitor II, the three most promiscuous inhibitors were omitted from this analysis. We found a strong positive correlation (r > 0.5 Pearson) among the top 10 highly ranked inhibitors and negative correlation (r < −0.5) in terms of their activity profiles across all 300 kinases (Figure 4C). Furthermore, unsupervised clustering of the kinases clearly showed that the kinases that are inhibited (<40% residual activity) by the top 10 most effective inhibitors are not inhibited (>90% residual activity) by any of the least 10 effective predicted inhibitors (Figure S1). These kinases include Src family kinases and several RTKs, including PDGFR, VEGFR, and FGFR1. Together, these data provide further support and biological rationale to KiDNN predicted most effective drugs that inhibit migration of Hs578t cells.
Experimental Validation of KiDNN-Hs578t Predictions in Hs578t Cells
A stringent test of any predictive model is its ability to forecast responses to a completely new dataset that was not used for training the model. To evaluate predictive accuracy of KiDNN, we experimentally tested the migration of Hs578t cells in response to eight kinase inhibitors that were not part of the 32 previously evaluated. The overall small differences between the observed and predicted migration of the Hs578t cells in response to the eight kinase inhibitors confirmed the high predictive performance of KiDNN (Figure 4D) with the predictions differing on average from the observed responses by 4.99% (Figure 4E). Response to five kinase inhibitors (Purvalanol A, Staurosporine N-benzoyl-, SU11652, PD98059, and Dovitinib) were predicted extremely well with a <5% difference (Figure 4D). The strong effect of Dovitinib, which inhibits Src family kinases (Bello and Gujral, 2018), and SU11652, which inhibits receptor tyrosine kinases, such as PDGFRb and VEGFR (Bello and Gujral, 2018), was evident in the wound migration assay (Figures 5A and 5B). With a 4.2% and 2.5% difference between the predicted effect and observed effect, respectively, these results indicate that KiDNN could accurately identify and predict migration in response to drugs that had a profound effect on the measured phenotype.
Figure 5.

Evaluating Predictive Capability of KiDNN
(A) A plot showing relative migration of Hs578t cells in response to DMSO control, SU11652 (1 μM), or Dovitinib (1 μM). Each point is a mean of three replicates, and the error bars denote SEM.
(B) Representative images of Hs578t cells at time 0 h (wounding) and 48 h post wounding. Wound area is highlighted in yellow, and the migrating cells in the wound area are shown in blue mask.
(C) A plot showing comparison of KiDNN-predicted and measured percent migration in response to 32 inhibitors in FOCUS cells using LOOCV. Migration in response to untreated/DMSO control was 70%.
(D) A plot showing KiDNN-predicted response to 178 small molecule kinase inhibitors (146 untested) in FOCUS cells. The black circles denote the predicted percent migration, whereas the blue circles and red circles denote the percent migration for 32 experimentally validated kinase inhibitors and 7 unseen inhibitors respectively. The kinase inhibitors are ranked by predicted migration.
In addition to identifying drugs that are effective, it is also desirable to identify kinase inhibitors with minimal effects or opposite effects from those sought for therapeutic intervention. Of the eight kinase inhibitors evaluated, four had a limited effect (70%) on Hs578t cell migration relative to control (Figure 4D). All four kinase inhibitors were predicted to have minimal effect by the model (>50% migration). Purvanol A, PD98059, and PDK1/Akt/Flt Dual Pathway Inhibitor were all predicted accurately (<10% error), whereas JAK1 inhibitor I was not predicted as accurately (>10% error). Together, these results validate that the KiDNN model can accurately predict changes in cell migration in response to both highly effective and ineffective kinase inhibitors, even with a minimal training dataset (∼18% of entire set of all 178 kinase inhibitors).
Comparing Predictions from KiDNN with Those from KiR
The initial application of KiDNN was to predict the effects of kinase inhibitors on TNBC Hs578t cell migration, whereas here we generated a KiDNN model for the effects of kinase inhibitors on liver cancer cells. Each KiDNN must be optimized and trained for the specific cell and phenotype under investigation. Previously, we applied the KiR approach to build a model based on a response to training set (32 inhibitors) and determined its ability to predict the effect of 178 kinase inhibitors on migration of liver cancer cells (FOCUS) (Gujral et al., 2014b). Here, we used the same dataset to build an optimized KiDNN model and compared the predictions of the KiDNN model with those of the KiR model. Using the same five-phase hyperparameter optimization strategy that we used to build the KiDNN model for predicting migration of Hs578t cells (Figure 2), we determined an optimal KiDNN architecture for the dataset collected on FOCUS cells.
The KiDNN-FOCUS architecture consisted of two hidden layers with 100 nodes per layer. Weights were updated in batch sizes of two (mini-batch gradient descent) with the entire dataset input through KiDNN-FOCUS a total of 120 times (epochs). The other network hyperparameters included the uniform weight initialization, SELU activation function, and the Adagrad optimizer. The only hyperparameters in common with KiDNN-Hs578t were the optimizer, the number of hidden layers, and the Dropout rate. The differences in the two KiDNN models are consistent with the cancer cells coming from different tissues and having different characteristics. As we did for KiDNN-Hs578t, we used LOOCV to evaluate the accuracy of the KiDNN-FOCUS predictions. Using the 32 inhibitors from the training set plus the control, the MSE of the LOOCV predictions was 114 and on average the model's predictions differed from experimentally observed migrations by 7.2% (Figure 5C).
We also used KiDNN-FOCUS to predict the effects of the seven kinase inhibitors that were not part of the original dataset. Using the analyses of these seven inhibitors, we compared the predictive capability of KiR (Gujral et al., 2014b) and KiDNN using the FOCUS models (Table 2). Our data showed that KiDNN predictions outperformed KiR predictions with KiDNN reducing the prediction error (MSE) by ∼40% compared with that of KiR. The average differences between KiR predictions and experimental observations (10.65%) was also greater than those between KiDNN predictions and experimental observations (7.58%).
Table 2.
Comparison of Predicted Migrations of the Seven Additional Inhibitors for Both KiDNN and KiR on FOCUS
| Kinase Inhibitor | Measured | Linear Regression Predictions | KiDNN Predictions | KiDNN LOOCV Predictions |
|---|---|---|---|---|
| Aminopurvalanol A | 66.8 | 60.3 | 64.2 | 66.3 |
| AMPK compound C | 64.8 | 54.5 | 57.1 | 56.6 |
| Cdk2 inhibitor IV, NU6140 | 70 | 52.7 | 66.3 | 63.8 |
| Dovitinib | 32.3 | 33.6 | 51.3 | 50.1 |
| GSK-3 inhibitor XIII | 59.5 | 53.7 | 59.9 | 62.0 |
| Staurosporine, N-benzoyl- | 37.9 | 30.9 | 55.3 | 48.0 |
| SU11652 | 47.0 | 20.7 | 49.4 | 50.9 |
| Mean squared error | 175.1 | 106.5 | 78.2 |
In the KiDNN Predictions column, KiDNN was trained with the 32 kinase inhibitors and applied to the 7 additional inhibitors, whereas in the KiDNN LOOCV Predictions column, each time 6 of the 7 additional inhibitors were added to the training set and the remaining inhibitor's migration was predicted for each of the 7 inhibitors. The MSE of the models' predictions are indicated.
Predictions of KiDNN and KiR models did not agree (>10% difference between the models) for four of the seven kinase inhibitors: Cdk2 Inhibitor IV (NU6140), Dovitinib, Staurosporine (N-benzoyl-), and SU11652. Of these, SU11652 and Cdk Inhibitor IV (NU6140) were better predicted by KiDNN; Dovitinib and Staurosporine (N-benzoyl-) were better predicted by KiR. In total, five inhibitors (Aminopurvanol A, AMPK Compound C, Cdk Inhibitor IV [NU6140], GSK-3 Inhibitor XIII, and SU11652) were better predicted by KiDNN with an average MSE of 17.09 and an average difference from observed migration of 3.36%. Dovitinib and Staurosporine (N-benzoyl-) were better predicted by KiR with an average MSE of 25.79 and an average difference from observed migration of 4.17%.
To further improve the KiDNN-FOCUS predictions, each time we included six of the seven inhibitors as part of the training set and predicted the excluded inhibitor's effect on migration of FOCUS cells (LOOCV). The MSE of KiDNN-FOCUS LOOCV prediction for the seven kinase inhibitors was 78.19, a ∼26% decrease in prediction error from KiDNN-FOCUS original predictions and a more than a 2-fold decrease in MSE compared with KiR. This decrease in prediction error demonstrates that addition of experimental data can drastically improve KiDNN performance enabling an iterative approach to build accurate KiDNN models. We used this improved KiDNN-FOCUS model to predict the effect on FOCUS cell migration of 178 inhibitors (139 untested) with previously known activity profiles (Anastassiadis et al., 2011) and reported effects on migration (Gujral et al., 2014b) (Figure 5D, see Table S6 for predicted and observed effects of each kinase inhibitor). Of the 178 kinase inhibitors' effect on FOCUS cell migration predicted by the model, 177 had differences between the predicted and interpolated migrations (based on polynomial interpolation (n = 7) of the 39 kinase inhibitors' experimental migration) <10% and 166 had differences <5%.
Discussion
Technological advancement in “omics”-based approaches for collecting large-scale datasets, particularly those involving drug-target profiling, gene expression, and protein abundance and modification, have unlocked the door to data-driven systems biology-based studies. Although studying a large number of interactions even in a network of closely related proteins is powerful in principle, it is often hard to find actionable or informative patterns in the data. Thus, system biologists rely on computational models to analyze and make scientific breakthroughs from large-scale datasets. In particular, machine learning and deep learning approaches are used to aid in model design and selection of compounds in pre-clinical drug discovery (Zhavoronkov et al., 2019). These computational approaches offer an effective strategy for prioritizing compounds that can lower the cost of pre-clinical trials by reducing the experimental search to smaller, model-predicted subsets (Zhang et al., 2019).
Here, we developed KiDNN, a multilayer DNN approach (Figure 1) and applied it to predict changes in migration of metastatic cancer cells in response to hundreds of kinase inhibitors. DNNs can capture non-linear relationships among features, such as the effects of multispecific inhibitors on kinase activities and the effects of kinase activities on complex cellular phenotypes like migration, and are better suited than linear regression-based approaches for investigating questions with a large number of samples and complex features. We applied KiDNN to predict kinase inhibitor effects on the migration of two cancer cell lines, one a TNBC cell line and the other a hepatocellular carcinoma cell line. For each cell line KiDNN is optimized and trained using existing data for the polypharmacology of a subset of kinase inhibitors and data on the effects of those inhibitors on migration of each cell line. We used LOOCV MSE to assess the predictive capability for each cell line-specific KiDNN and determined that KiDNN-Hs587t had an MSE of <80 with predictions that differed from observed effects by <6%, while KiDNN-FOCUS had an MSE of 114 with predictions that differed by 7%. These small differences indicated that the two KiDNN models had biologically acceptable performance. Furthermore, experimental validation showed that KiDNN predictions (MSE 78) were more accurate than KiR-based predictions (175) (Table 2), suggesting that using a multilayered DNN substantially improves overall accuracy and performance over linear models.
The molecular heterogeneity of cancer underscores a need for screening drug candidates in multiple patient-derived cell lines or animal models (Barretina et al., 2012). In oncology drug discovery and development, this is often a cost-prohibitive and laborious undertaking. To address this, we showed that DNN hyperparameters optimized on inhibitor responses in breast mesenchymal cells (Hs578t) coupled with a new training dataset can be used as a starting point to build a KiDNN model that accurately predicted inhibitor responses in liver mesenchymal (FOCUS) cancer cells. Such promising results suggested that the hyperparameters learned from one set of data can be used to generate KiDNN that predicts responses in multiple relevant cancer cell lines with minimal training data from each cell line.
Finally, we applied the KiDNN approach to predict the effect of ∼200 kinase inhibitors on migration of Hs578t and FOCUS mesenchymal cancer cells. The models predicted drugs that were effective at impairing migration and those that were ineffective. Such information is valuable in drug discovery. Noteworthy for the Hs578t cells, KiDNN-Hs578t accurately predicted response to Dovitinib (58.2% predicted versus 55.7% observed) and SU11652 (48.6% predicted versus 44.4%, observed) (Figures 5A and 5B). Dovitinib is a Src family kinase inhibitor (Anastassiadis et al., 2011), and SU11652 is an inhibitor of multiple receptor tyrosine kinases, including PDGFRb and VEGFR (Bello and Gujral, 2018). Both Src and PDGFRb are important for migration of TNBC cell lines (Ho-Yen et al., 2015, Jechlinger et al., 2006, Sausgruber et al., 2015, Simiczyjew et al., 2018, Van Swearingen et al., 2017), highlighting validating the relevance of the KiDNN predictions.
The high predictive accuracy for individual kinase inhibitors demonstrated that KiDNN is a powerful deep learning approach that overall performed significantly better than linear models for predicting the effects of a large panel of kinase inhibitors on a complex cellular phenotype. Furthermore, the models can be retrained on limited data from different cells. We predict that application of KiDNN modeling coupled with polypharmacology profiling will enable cheaper and more effective screening than exhaustive, unbiased testing of compound libraries. We anticipate future development of KiDNN models could integrate additional parameters, such as non-kinase targets of kinase inhibitors, chemical moieties, pharmacokinetic properties, and other medicinal chemistry properties, to enable de novo compound design, prioritization, and drug discovery. Furthermore, combining KiDNN predictions for different cellular outcomes, such as proliferation, apoptosis, and migration, could identify lead candidate drugs that produce desired outcomes without stimulating undesired ones.
Limitations of the Study
A particular limitation of KiDNN and Deep Neural Network models broadly is their lack of interpretability in predictions. Deep Neural Networks have traditionally been labeled black-box models as it is increasingly difficult to inspect how the neural network model reaches its predictions as its complexity increases. Although KiDNN prediction of cell migration of Hs578t and FOCUS cells closely followed experimental values, the precise biological basis behind the predictions in terms of specific kinases or their combination are difficult to extract. To address this limitation, future development of KiDNN could involve a feature importance algorithm to deduce the primary kinases the KiDNN model relies on to make predictions.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Taranjit S Gujral (tgujral@fredhutch.org).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Source code for KiDNN is available in Data S1.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported by the NIH/NCI (K22CA201229, P30CA015704), Fred Hutch Evergreen Fund, and the Sidney Kimmel Foundation (Kimmel Scholar Award). We thank Drs. Nancy Gough and Milka Kostic for helpful comments on the manuscript.
Author Contributions
T.S.G. and S.V conceived the project. S.V. developed the KiDNN algorithm. T.S.G performed experiments. S.V. and T.S.G. performed analysis. S.V. and T.S.G. wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: May 22, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101129.
Supplemental Information
References
- Anastassiadis T., Deacon S.W., Devarajan K., Ma H., Peterson J.R. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 2011;29:1039. doi: 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A.A., Kim S., Wilson C.J., Lehár J., Kryukov G.V., Sonkin D. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bello T., Gujral T.S. KInhibition: a kinase inhibitor selection portal. iScience. 2018;8:49–53. doi: 10.1016/j.isci.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstra, J.S., Bardenet, R., Bengio, Y., and Kégl, B. (2011). Algorithms for hyper-parameter optimization. Paper presented at: Advances in neural information processing systems.
- Chollet F. Astrophysics Source Code Library; 2018. Keras: the python Deep Learning Library. [Google Scholar]
- Davis M.I., Hunt J.P., Herrgard S., Ciceri P., Wodicka L.M., Pallares G., Hocker M., Treiber D.K., Zarrinkar P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011;29:1046. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- Dongare A., Kharde R., Kachare A.D. Introduction to artificial neural network. Int. J. Eng. Innov. Technol. 2012;2:189–194. [Google Scholar]
- Duong-Ly K.C., Devarajan K., Liang S., Horiuchi K.Y., Wang Y., Ma H., Peterson J.R. Kinase inhibitor profiling reveals unexpected opportunities to inhibit disease-associated mutant kinases. Cell Rep. 2016;14:772–781. doi: 10.1016/j.celrep.2015.12.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gujral T.S., Chan M., Peshkin L., Sorger P.K., Kirschner M.W., MacBeath G. A noncanonical Frizzled2 pathway regulates epithelial-mesenchymal transition and metastasis. Cell. 2014;159:844–856. doi: 10.1016/j.cell.2014.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gujral T.S., Peshkin L., Kirschner M.W. Exploiting polypharmacology for drug target deconvolution. Proc. Natl. Acad. Sci. U S A. 2014;111:5048–5053. doi: 10.1073/pnas.1403080111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho-Yen C.M., Jones J.L., Kermorgant S. The clinical and functional significance of c-Met in breast cancer: a review. Breast Cancer Res. 2015;17:52. doi: 10.1186/s13058-015-0547-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jechlinger M., Sommer A., Moriggl R., Seither P., Kraut N., Capodiecci P., Donovan M., Cordon-Cardo C., Beug H., Grünert S. Autocrine PDGFR signaling promotes mammary cancer metastasis. J. Clin. Invest. 2006;116:1561–1570. doi: 10.1172/JCI24652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaeger S., Heinzlmeir S., Wilhelm M., Polzer H., Vick B., Koenig P.-A., Reinecke M., Ruprecht B., Petzoldt S., Meng C. The target landscape of clinical kinase drugs. Science. 2017;358:eaan4368. doi: 10.1126/science.aan4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sausgruber N., Coissieux M., Britschgi A., Wyckoff J., Aceto N., Leroy C., Stadler M., Voshol H., Bonenfant D., Bentires-Alj M. Tyrosine phosphatase SHP2 increases cell motility in triple-negative breast cancer through the activation of SRC-family kinases. Oncogene. 2015;34:2272. doi: 10.1038/onc.2014.170. [DOI] [PubMed] [Google Scholar]
- Schmidlin T., Debets D.O., van Gelder C.A., Stecker K.E., Rontogianni S., van den Eshof B.L., Kemper K., Lips E.H., van den Biggelaar M., Peeper D.S. High-throughput assessment of kinome-wide activation states. Cell Syst. 2019;9:366–374.e5. doi: 10.1016/j.cels.2019.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simiczyjew A., Dratkiewicz E., Van Troys M., Ampe C., Styczeń I., Nowak D. Combination of EGFR inhibitor lapatinib and MET inhibitor foretinib inhibits migration of triple negative breast cancer cell lines. Cancers (Basel) 2018;10:335. doi: 10.3390/cancers10090335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Swearingen A.E., Sambade M.J., Siegel M.B., Sud S., McNeill R.S., Bevill S.M., Chen X., Bash R.E., Mounsey L., Golitz B.T. Combined kinase inhibitors of MEK1/2 and either PI3K or PDGFR are efficacious in intracranial triple-negative breast cancer. Neuro Oncol. 2017;19:1481–1493. doi: 10.1093/neuonc/nox052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zegzouti H., Hennek J., Goueli S.A. Using bioluminescent kinase profiling strips to identify kinase inhibitor selectivity and promiscuity. In: Zegzouti H., Goueli S.A., editors. Kinase Screening and Profiling. Springer; 2016. pp. 59–73. [DOI] [PubMed] [Google Scholar]
- Zhang P. Model selection via multifold cross validation. Ann. Stat. 1993;21:299–313. [Google Scholar]
- Zhang H., Ericksen S.S., Ching-Pei L., Ananiev G.E., Wlodarchak N., Mitchell J.C., Gitter A., Wright S.J., Hoffmann F.M., Wildman S.A. Predicting kinase inhibitors using bioactivity matrix derived informer sets. PLoS Comput. Biol. 2019;15:e1006813. doi: 10.1371/journal.pcbi.1006813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhavoronkov A., Ivanenkov Y.A., Aliper A., Veselov M.S., Aladinskiy V.A., Aladinskaya A.V., Terentiev V.A., Polykovskiy D.A., Kuznetsov M.D., Asadulaev A. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 2019;37:1038–1040. doi: 10.1038/s41587-019-0224-x. [DOI] [PubMed] [Google Scholar]
- Zupan J. Introduction to artificial neural network (ANN) methods: what they are and how to use them. Acta Chim. Slov. 1994;41:327. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source code for KiDNN is available in Data S1.