

Abstract
As a key element in the construction of complex organic scaffolds, the formation of C–C bonds remains a challenge in the field of synthetic organic chemistry. Recent advancements in single-electron chemistry have enabled new methods for the formation of various C–C bonds. Disclosed herein is the development of a novel single-electron reduction of acyl azoliums for the formation of ketones from carboxylic acids. Facile construction of the acyl azolium in situ followed by a radical-radical coupling was made possible using merged NHC-photoredox catalysis. The utility of this protocol in synthesis was showcased in the late-stage functionalization of a variety of pharmaceutical compounds. Preliminary investigations using chiral NHCs demonstrate that enantioselectivity can be achieved, showcasing the advantages of this protocol over alternative methodologies.
Keywords: N-heterocyclic carbine, acyl azolium, photochemistry, ketone, radical coupling
Graphical Abstract
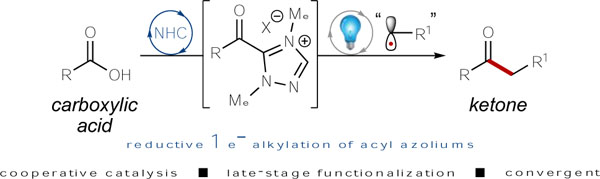
The conversion of carboxylic acids to ketones using combined photoredox/NHC catalysis has been developed. In situ activation of a carboxylic acid followed by generation of an acyl azolium allows for productive radical-radical coupling to afford ketones in good-to-excellent yields. This single-electron, reductive alkylation was then applied in the late-stage functionalization of various pharmaceutical compounds.
N-heterocyclic carbenes (NHCs) have emerged as unique Lewis basic catalysts that harness umpolung (polarity reversal) reactivity to mediate a wide range of organic transformations.[1] The majority of NHC-catalyzed processes are initiated by carbene addition into a carbonyl. Subsequent proton transfer affords the Breslow intermediate,[2] a species that is nucleophilic at a typically electrophilic carbonyl carbon. While the utility of two-electron NHC reactivity has continued to expand since the field’s inception, the scope of NHC-derived operators is limited by their inability to engage sp3 electrophiles, thus highlighting the potential opportunity for single-electron NHC operators.
Early work from our group showcased a mild oxidation of allylic alcohols[2b] and aldehydes[3] to esters using an NHC and MnO2 (Scheme 1A). Oxidation of the Breslow intermediate with stoichiometric MnO2 was employed to access an acyl azolium intermediate, and subsequent displacement by an alcohol afforded the desired C–O bond. Shortly thereafter, Studer and coworkers developed an NHC-catalyzed oxidation of aldehydes to esters mediated by 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO; Scheme 1A).[4] To broaden the scope of substrates, Studer followed up in 2010 with a report featuring 3,3’,5,5’-tetra-tert-butyldiphenoquinone as the stoichiometric oxidant in place of TEMPO (Scheme 1A).[5] While these NHC-catalyzed radical functionalizations have set the precedent for a variety of other transformations,[6] the use of toxic and wasteful stoichiometric oxidants has limited their overall utility in synthesis.[7]
Scheme 1.

Oxidations of the Breslow intermediate and expansion to one-electron reduction of acyl azoliums
To circumvent the need for stoichiometric oxidants, Boydston developed a direct NHC-catalyzed anodic oxidation of aldehydes for the formation of esters in 2012 (Scheme 1B).[8] Similarly, Studer employed air as the terminal oxidant in a cooperative NHC- and metal redox esterification of aldehydes (Scheme 1B).[9] While significant improvements in NHC-catalyzed processes have been made over the past decade, the majority of these strategies are confined to the formation of C–X bonds (X = O, N, etc.). In 2019, however, Ohmiya reported an NHC-catalyzed decarboxylative alkylation of aldehydes using N-(acyloxy)-phthalimide derivatives to afford ketones with quaternary-alpha centers (Scheme 1C).[10] Single-electron transfer (SET) of the redox-active ester was induced by the Breslow intermediate [E1/2 ≈ −0.95 V vs. saturated calomel electrode (SCE)],[11] and subsequent radical-radical coupling afforded the desired ketone. Following this initial report, similar modes of reactivity have been developed for the synthesis of ketones.[12]
In line with our experience in oxidations of the Breslow intermediate and our growing interest in photoredox catalysis,[13] we envisioned an opportunity for the development of novel reactivity at the interface of NHC catalysis and photochemistry. Our work in the field of cooperative NHC catalysis in addition to the contributions by other groups[14] has resulted in a variety of new transformations featuring the combination of NHCs with Lewis acids,[15] Brønsted acids,[16] and transition metals.[17] Similar tenets of cooperative catalysis have recently been exploited in photoredox chemistry, where the combination of organocatalysts,[18] Lewis acids,[19] Brønsted acids,[20] and transition metals[21] has enabled the expedient construction of synthetically tractable molecules. A limited number of reports explore the combination of NHC catalysis with photoredox catalysis.[22] As such, we aimed to further bridge the existing gap between the fields of NHC catalysis and photocatalysis by leveraging their unique redox properties. Herein we report the facile synthesis of ketones from readily available carboxylic acids via a combined photoredox-NHC catalyzed process.
We hypothesized that in situ activation of a carboxylic acid followed by NHC addition would afford an acyl azolium, which have been used extensively in NHC-redox acylations for the preparation of esters, amides, and carboxylic acids.[1a, 23] Single-electron reduction of the resulting species would provide an azolium radical ion that, when coupled with an alkyl radical, would furnish synthetically valuable ketones (Scheme 1D). Traditional methods for accessing acyl radicals, which are functionally equivalent to achiral azolium radicals, involve the use of aldehydes,[24] α-keto acids,[25] and others,[26] many of which suffer from significant drawbacks (i.e. toxicity, instability, superstoichiometric additive requirements, etc.). Additionally, carboxylic acids have recently gained attention for their ability to generate acyl radicals via a decarboxylation-carbonylation strategy[27] or pre-functionalization with an activating agent (e.g. dimethyldicarbonate,[28] PR3,[29] etc.[30]). However, carboxylic acid-derived acyl radicals have primarily been employed in Giese-type additions to activated alkenes.[26] Limited reports describe the coupling of an acyl radical with an alkyl radical,[31] thus presenting an opportunity to explore and develop new reactivity. As a complimentary approach to recent advancements in acyl radical chemistry, the work described herein showcases the coupling of an alkyl radical derived from easily prepared Hantzsch esters with an “acyl radical surrogate” accessed from readily available carboxylic acids.
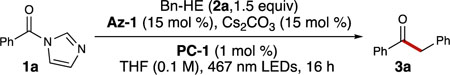
Our initial search for the desired reactivity was guided by semi-high-throughput experimentation (HTE), which allowed for numerous reaction components to be screened in parallel.[32] Semi-HTE enabled facile and rapid identification of the most optimal alkyl radical precursor for this transformation. While limited reactivity was achieved with alkyl silanes,[33] silicates,[34] and potassium trifluoroborate salts (R–BF3K),[35] significant conversion was observed using Hantzsch esters[36] as the alkyl radical source. Hantzsch esters have recently gained attention for their use as mild alkyl radical sources that are easily prepared and generate inert byproducts.[37] Of the azolium radical precursors that were screened, including perfluorophenyl esters and acyl chlorides, acyl imidazoles performed the best (only trace products were observed with acid halides). Reaction optimization thus ensued using phenyl acyl imidazole (1a) and benzyl Hantzsch ester (Bn-HE, 2a; E1/2 = +1.00 V vs SCE)[38] as radical coupling partners, PC-1 as the photocatalyst, dimethytriazolium (Az-1) as the NHC precursor, and cesium carbonate as the base in THF (Table 1).
Table 1.
Optimization of reaction conditionsa
 | ||
|---|---|---|
| entry | deviation from standard | GC yield (%)a,b |
| 1 | none | 63 |
| 2 | PC-2 instead of PC-1 | 11 |
| 3 | PC-3 instead of PC-1 | 0 |
| 4 | Az-2 instead of Az-1 | 14 |
| 5 | Az-3 instead of Az-1 | 0 |
| 6 | Az-4 instead of Az-1 | 0 |
| 7 | Az-5 instead of Az-1 | 11 |
| 8 | CsOAc instead of Cs2CO3 | 38 |
| 9 | K2CO3 instead of Cs2CO3 | 8 |
| 10 | Li2CO3 instead of Cs2CO3 | 0 |
| 11 | CH3CN instead of THF | 72 |
| 12 | CH2CI2 instead of THF | 41 |
| 13 | DMF instead of THF | 65 |
| 14 | no light | 0 |
| 15 | no photocatalyst | 0 |
| 16 | no Az | 0 |
| 17 | no base | 0 |
 | ||
Gas chromatography (GC) yield is based on a calibration curve using 1,3,5-trimethoxybenzene as the internal standard.
Reaction conditions: 1a (0.10 mmol), Bn–HE 2a (0.15 mmol), Az (0.015 mmol), base (0.015 mmol), PC (1 μmol), solvent (0.1 M; THF, tetrahydrofuran; DMF, dimethyformamide) for 16 h.
A brief survey of photocatalysts, azolium catalysts, bases, and solvents allowed for identification of optimal reaction conditions. Due to its broad potential range (E1/2 IrIII*/IrII - E1/2 IrIII/IrII = +1.21 - –1.37 V vs SCE),[39] iridium catalyst PC-1 was found to be the best photocatalyst. The use of strongly reducing catalysts (PC-2: E1/2 IrIII*/IrII - E1/2 IrIII/IrII = +0.31 - –2.10 V vs SCE)[40] or strongly oxidizing catalysts (PC-3: E1/2 PC*/PC– - E1/2 PC/PC– = +2.17 - –0.50 V vs SCE)[41] resulted in lower yields, presumably because the photocatalysts were unable to perform both redox events efficiently (Table 1, entries 1–3). It should be noted that 2,4,5,6-tetra(9H-carbazol-9-yl)isophthalonitrile (4CzIPN), an easily-accessible organophotocatalyst, afforded 3a with a slightly diminished yield compared to PC-1, thus offering a cost-effective alternative (see Supporting Information). Examination of different NHC precursors revealed Az-1 to be the only azolium catalyst to efficiently afford the desired ketone via the phenyl acyl azolium intermediate (E1/2 = –1.29 V vs SCE; see Supporting Information), with all other NHC precursors having at least a three-fold decrease in reactivity (Table 1, entries 4–7). Moreover, only cesium bases allowed for significant conversion to product, with cesium carbonate being the most suitable base; all other bases screened showed less than a 10% GC yield (Table 1, entries 8–10). Finally, a brief solvent screen revealed acetonitrile to be the best solvent, providing a ten percent increase in yield compared to THF (Table 1, entries 11–13).
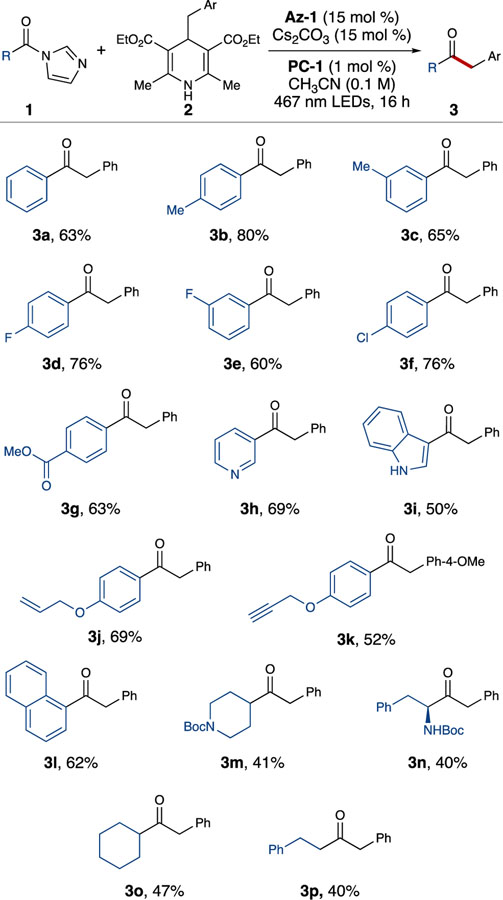
With these optimized reaction conditions, the scope of acyl imidazoles amenable to alkylation was surveyed (Table 2). A variety of electron-withdrawing and electron-donating substituents were tolerated on aryl acyl imidazoles. Methyl-substituted aryl ketones (3b-c) were isolated in good-to-high yields, and halogenated aryl acyl imidazoles afforded the desired ketones (3d-f) in good yields. Of note, alkylation of ester-substituted acyl imidazole to afford 3g occurred in good yield, showing tolerance to functional groups that traditional methods for ketone formation (e.g. Grignard reaction) may affect.
Table 2.
Acyl imidazole-derived substrate scopea,b
 |
See supporting information for reaction details.
Conversion of heteroaromatic substrates, such as 4-pyridinyl acyl imidazole and indole acyl imidazole, to the respective ketones (3h-i) was accomplished in good yields. Products containing pi systems (3j-k) as well as sterically-encumbering substituents (3l) were also synthesized using this method. Notably, the reaction with aliphatic substrates was achieved in modest yields (3m-p). Potentially due to the instability of aliphatic azolium radicals, methods featuring similar modes of NHC-mediated reactivity that have been developed to date have been unsuccessful when applied to aliphatic substrates.[10, 12a]
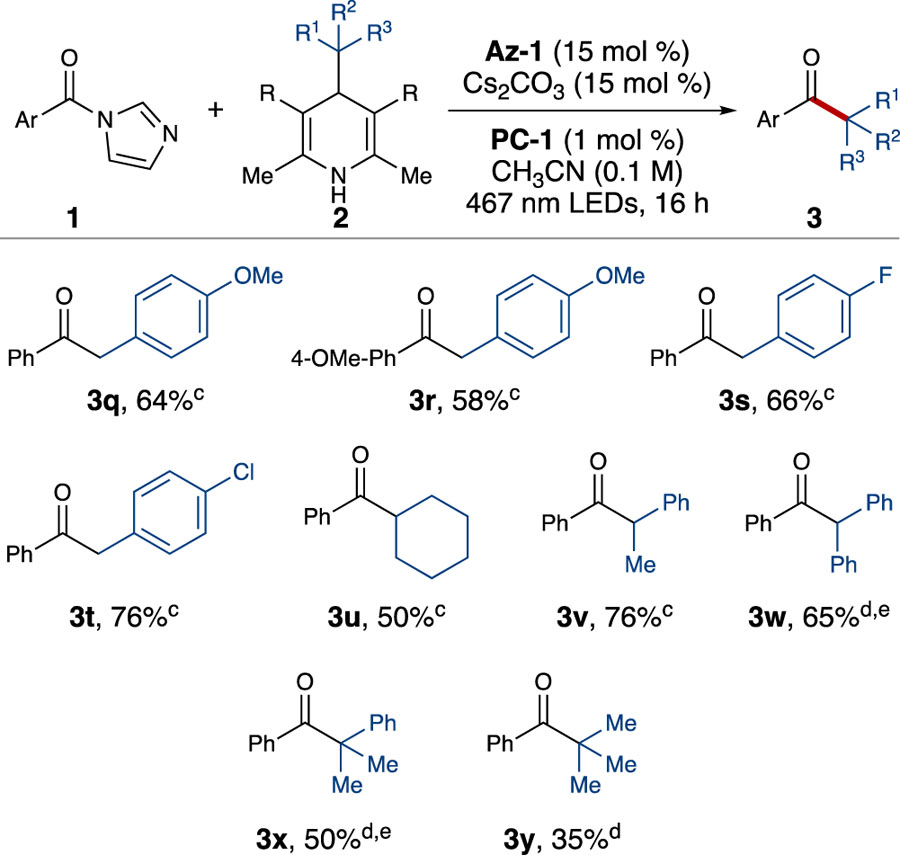
A variety of Hantzsch esters were also successfully employed for the conversion of phenyl acyl imidazole (1a) to an array of ketones (Table 3). Substituted benzyl Hantzsch esters bearing electron-withdrawing and electron-donating groups, including halogenated and methoxy substituents, were tolerant of the reaction conditions (3q-3t). Moreover, cyclohexyl Hantzsch ester productively served as an alkyl radical precursor, suggesting that the use of non-benzylic alkyl radicals for the formation of aliphatic ketones is possible (3u). Various Meyer nitrile[42] derivatives were also examined as alkyl radical precursors and allowed for the synthesis of ketones containing ⍺-tertiary (3v-3w) and ⍺-quaternary centers (3x-3y).
Table 3.
 |
See supporting information for reaction details.
Isolated yield.
R = CO2Et.
R = CN.
4-Cz-IPN instead of PC-1.
The successful formation of product 3v presented an opportunity to explore controlling enantioselectivity using chiral NHCs. When Az-6 was employed instead of Az-1, modest enantioselectivity was observed (Scheme 2). To the best of our knowledge, this preliminary result is a novel enantioselective acyl-like radical-radical coupling for the formation of ketones bearing an ⍺-stereogenic center, thus differentiating this work from other acyl radical processes.[24c, 26, 31, 43] Efforts to increase the selectivity and scope of this process are currently underway.
Scheme 2.

Enantioselective reaction with a chiral NHC.
To demonstrate the ease and practicality of this method, the standard reaction to make deoxybenzoin (3a) was performed starting from benzoic acid (Table 4). In situ generation of phenyl acyl imidazole using carbonyldiimidazole (CDI) was confirmed by gas chromatography-mass spectrometry (GC-MS) and subsequent subjection to the reaction conditions furnished 3a in 65% yield (compared to 63% when starting from phenyl acyl imidazole). To further evaluate the utility of this transformation, the reaction conditions were then employed for the late-stage functionalization (LSF) of various pharmaceutical compounds. As a critical component of many medicinal chemistry campaigns or total syntheses, LSF allows for the incorporation of important functional groups in the final steps of a synthesis, thus creating the need for efficient methodologies.[44] When the in situ reaction conditions were applied to the LSF of telmisartan, a carboxylic acid-containing drug used for the treatment of hypertension,[45] the desired ketone product (4a) was isolated in 91% yield. This direct, one-step alkylation was applied to various other pharmaceutical compounds to afford ketone products (4b-c) in moderate-to-good yields (Table 4).
Table 4.
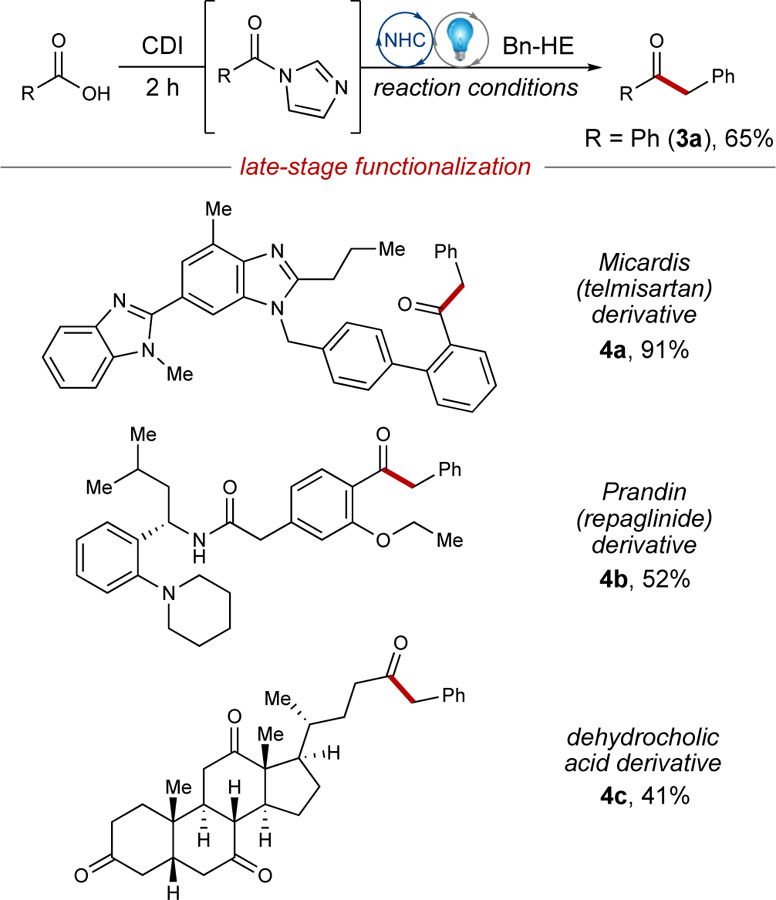 |
Reaction conditions: carboxylic acid component (0.10 mmol), Bn-HE 2a (0.15 mmol), Az-1 (0.015 mmol), Cs2CO3 (0.015 mmol), PC-1 (1 μmol), CH3CN (0.1 M) for 36 h.
Isolated yield.
To probe the mechanism of this reaction, control reactions and radical-trapping experiments were performed. No product was observed in the absence of base, azolium catalyst, photocatalyst, or light, suggesting that an NHC-mediated and photoredox-catalyzed process occurs (Table 1, entries 14–17). Additionally, the reaction did not proceed under standard conditions using TEMPO as a radical trap. Only the TEMPO-benzyl mass adduct was observed by ultra-performance liquid chromatography-mass spectrometry (UPLC-MS), thus confirming a radical mechanism and suggesting that the Hantzsch ester is oxidized prior to reduction of the acyl azolium (Scheme 3A). Moreover, the potentials of PC-1 (E1/2 IrIII*/IrIV - E1/2 IrIV/IrIII = –0.89 – 1.69 V vs SCE) do not support reduction of the acyl azolium prior to oxidation of the Hantzsch ester, as the reduction potential of the acyl azolium is outside the range of the photocatalyst. It is also reasonable to assert that Hanztsch ester oxidation occurs first due to the limited amount of acyl azolium present at a given time relative to superstoichiometric Hantzsch ester.
Scheme 3.

Mechanistic studies and proposed mechanism
Our proposed reaction pathway involves initial oxidation of the Hantzsch ester to the radical cation by the photoexcited photocatalyst (IrIII*). Fragmentation of the Hantzsch ester affords the benzyl radical, and single-electron reduction of the acyl triazolium provides the azolium radical while regenerating the ground-state photocatalyst (IrIII). Loss of the NHC and radical-radical combination affords the desired ketone (Scheme 3B). The recent mechanistic studies of Breslow intermediates and acyl azoliums reported by Bertrand and Martin suggest that definitive evidence of radical intermediates in thermal NHC-catalyzed processes does not exist.[46] In contrast, this NHC-mediated reaction is conducted under photochemical conditions. Moreover, the observed enantioselectivity (vide supra) provides additional evidence that an NHC-bound radical species is most likely involved in this process.
In summary, we have developed a reductive single-electron alkylation of acyl azoliums to form ketones from carboxylic acids. Activation of readily available carboxylic acids with CDI followed by addition of the NHC catalyst produces the acyl azolium intermediate in situ. This combined NHC and photoredox catalysis enabled a one-electron reduction of the acyl azolium, and subsequent radical-radical combination allowed for the facile construction of a C–C bond to furnish a ketone. The utility of this method in synthesis was showcased in the direct, one-step late-stage functionalization of pharmaceutical compounds. Importantly, preliminary results using a chiral NHC demonstrated that enantioselectivity is possible using this process, thus highlighting the potential advantage of using acyl azolium radicals in acyl radical transformations.
Supplementary Material
Acknowledgements
We thank Northwestern University and the National Institute of General Medical Sciences (GM073072 and GM131431) for support of this work. The authors thank Purav Vagadia of the Center for Molecular Innovation and Drug Discovery at Northwestern University for assistance with purification. We also thank Joshua Zhu and Ada Kwong (NU) for assistance with cyclic voltammetry experiments and HRMS, respectively.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].a) Hopkinson MN, Richter C, Schedler M, Glorius F, Nature 2014, 510, 485–496; [DOI] [PubMed] [Google Scholar]; b) Flanigan DM, Romanov-Michailidis F, White NA, Rovis T, Chem. Rev 2015, 115, 9307–9387; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhang C, Hooper JF, Lupton DW, ACS Catal 2017, 7, 2583–2596. [Google Scholar]
- [2].a) Breslow R, J. Am. Chem. Soc 1958, 80, 3719–3726; [Google Scholar]; b) Maki BE, Chan A, Phillips EM, Scheidt KA, Org. Lett 2007, 9, 371–374; [DOI] [PubMed] [Google Scholar]; c) Ukai T, Tanaka R, Dokawa T, J. Pharm. Soc. Jpn 1943, 63, 296–300. [Google Scholar]
- [3].Maki BE, Scheidt KA, Org. Lett 2008, 10, 4331–4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Guin J, De Sarkar S, Grimme S, Studer A, Angew. Chem. Int. Ed 2008, 47, 8727–8730. [DOI] [PubMed] [Google Scholar]
- [5].De Sarkar S, Grimme S, Studer A, J. Am. Chem. Soc 2010, 132, 1190–1191. [DOI] [PubMed] [Google Scholar]
- [6].a) Izquierdo J, Hutson GE, Cohen DT, Scheidt KA, Angew. Chem. Int. Ed 2012, 51, 11686–11698; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) De Sarkar S, Biswas A, Samanta RC, Studer A, Chemistry 2013, 19, 4664–4678. [DOI] [PubMed] [Google Scholar]
- [7].Delidovich I, Palkovits R, Green Chem 2016, 18, 590–593. [Google Scholar]
- [8].Finney EE, Ogawa KA, Boydston AJ, J. Am. Chem. Soc 2012, 134, 12374–12377. [DOI] [PubMed] [Google Scholar]
- [9].Zhao J, Mück-Lichtenfeld C, Studer A, Adv. Synth. Catal 2013, 355, 1098–1106. [Google Scholar]
- [10].Ishii T, Kakeno Y, Nagao K, Ohmiya H, J. Am. Chem. Soc 2019, 141, 3854–3858. [DOI] [PubMed] [Google Scholar]
- [11].a) Nakanishi I, Itoh S, Suenobu T, Inoue H, Fukuzumi S, Chem. Lett 1997, 26, 707; [Google Scholar]; b) Nakanishi I, Itoh S, Chem. Commun 1997, 1927–1928;; c) Nakanishi I, Itoh S, Suenobu T, Fukuzumi S, Angew. Chem. Int. Ed 1998, 37, 992–994; [DOI] [PubMed] [Google Scholar]; d) Nakanishi I, Itoh S, Fukuzumi S, Chem. Eur. J 1999, 5, 2810–2818. [Google Scholar]
- [12].a) Ishii T, Ota K, Nagao K, Ohmiya H, J. Am. Chem. Soc 2019, 141, 14073–14077; [DOI] [PubMed] [Google Scholar]; b) Li J-L, Liu Y-Q, Zou W-L, Zeng R, Zhang X, Liu Y, Han B, He Y, Leng H-J, Li Q-Z, Angew. Chem. Int. Ed 2019, 58, 2–10. [Google Scholar]
- [13].a) McDonald BR, Scheidt KA, Org. Lett 2018, 20, 6877–6881; [DOI] [PubMed] [Google Scholar]; b) Betori RC, McDonald BR, Scheidt KA, Chem. Sci 2019, 10, 3353–3359; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Betori RC, Scheidt KA, ACS Catal 2019, 9, 10350–10357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang MH, Scheidt KA, Angew. Chem. Int. Ed 2016, 55, 14912–14922. [DOI] [PubMed] [Google Scholar]
- [15].a) Jia Q, Li Y, Lin Y, Ren Q, Catalysts 2019, 9, 863; [Google Scholar]; b) Cardinal-David B, Raup DEA, Scheidt KA, J. Am. Chem. Soc 2010, 132, 5345–5347; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Raup DEA, Cardinal-David B, Holte D, Scheidt KA, Nat. Chem 2010, 2, 766–771; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Cohen DT, Scheidt KA, Chem. Sci 2012, 3, 53–57; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Mo J, Chen X, Chi YR, J. Am. Chem. Soc 2012, 134, 8810–8813; [DOI] [PubMed] [Google Scholar]; f) Jin Z, Xu J, Yang S, Song B-A, Chi YR, Angew. Chem. Int. Ed 2013, 52, 12354–12358. [DOI] [PubMed] [Google Scholar]
- [16].Tong Y-F, Mao J-H, Wu S, Zhao Y, Cheng Y, J. Org. Chem 2014, 79, 2075–2081. [DOI] [PubMed] [Google Scholar]
- [17].a) Bai Y, Xiang S, Leow ML, Liu XW, Chem. Commun 2014, 50, 6168–6170; [DOI] [PubMed] [Google Scholar]; b) Liu K, Hovey MT, Scheidt KA, Chem. Sci 2014, 5, 4026–4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Terrett JA, Clift MD, Macmillan DWC, J. Am. Chem. Soc 2014, 136, 6858–6861; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pirnot MT, Rankic DA, Martin DBC, Macmillan DWC, Science 2013, 339, 1593–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yoon TP, Acc. Chem. Res 2016, 49, 2307–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].a) Rono LJ, Yayla HG, Wang DY, Armstrong MF, Knowles RR, J. Am. Chem. Soc 2013, 135, 17735–17738; [DOI] [PubMed] [Google Scholar]; b) Cheng WM, Shang R, Fu Y, ACS Catal 2017, 7, 907–911. [Google Scholar]
- [21].a) Huo H, Shen X, Wang C, Zhang L, Rose P, Chen LA, Harms K, Marsch M, Hilt G, Meggers E, Nature 2014, 515, 100–103; [DOI] [PubMed] [Google Scholar]; b) Lee KN, Ngai M-Y, Chem. Commun 2017, 53, 13093–13112; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gutierrez-Bonet A, Tellis JC, Matsui JK, Vara BA, Molander GA, ACS Catal 2016, 6, 8004–8008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].a) >DiRocco DA, Rovis T, J. Am. Chem. Soc 2012, 134, 8094–8097; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dai L, Ye S, Org. Lett 2020; [DOI] [PubMed]; c) Mavroskoufis A, Rajes K, Golz P, Agrawal A, Ruß V, Götze JP, Hopkinson MN, Angew. Chem. Int. Ed 2020, 59, 3190–3194. [DOI] [PMC free article] [PubMed]
- [23].Vora HU, Wheeler P, Rovis T, Adv. Synth. Catal 2012, 354, 1617–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].a) Iqbal N, Choi S, You Y, Cho EJ, Tetrahedron Lett 2013, 54, 6222–6225; [Google Scholar]; b) Mukherjee S, Garza-Sanchez RA, Tlahuext-Aca A, Glorius F, Angew. Chem. Int. Ed 2017, 56, 14723–14726; [DOI] [PubMed] [Google Scholar]; c) Zhang X, Macmillan DWC, J. Am. Chem. Soc 2017, 139, 11353–11356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].a) Liu J, Liu Q, Yi H, Qin C, Bai R, Qi X, Lan Y, Lei A, Angew. Chem. Int. Ed 2014, 53, 502–506; [DOI] [PubMed] [Google Scholar]; b) Chu L, Lipshultz JM, MacMillan DW, Angew. Chem. Int. Ed 2015, 54, 7929–7933; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhang M, Xi J, Ruzi R, Li N, Wu Z, Li W, Zhu C, J. Org. Chem 2017, 82, 9305–9311. [DOI] [PubMed] [Google Scholar]
- [26].Ngai M-Y, Banerjee A, Lei Z, Synthesis 2019, 51, 303–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhou Q-Q, Guo W, Ding W, Wu X, Chen X, Lu L-Q, Xiao W-J, Angew. Chem. Int. Ed 2015, 54, 11196–11199. [DOI] [PubMed] [Google Scholar]
- [28].a) Bergonzini G, Cassani C, Wallentin C-J, Angew. Chem. Int. Ed 2015, 54, 14066–14069; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pettersson F, Bergonzini G, Cassani C, Wallentin C-J, Chem. Eur. J 2017, 23, 7444–7447; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhang M, Ruzi R, Xi J, Li N, Wu Z, Li W, Yu S, Zhu C, Org. Lett 2017, 19, 3430–3433; [DOI] [PubMed] [Google Scholar]; d) Zhang M, Li N, Tao X, Ruzi R, Yu S, Zhu C, Chem. Commun 2017, 53, 10228–10231; [DOI] [PubMed] [Google Scholar]; e) Ruzi R, Zhang M, Ablajan K, Zhu C, J. Org. Chem 2017, 82, 12834–12839. [DOI] [PubMed] [Google Scholar]
- [29].a) Zhang M, Xie J, Zhu C, Nat. Commun 2018, 9, 3517; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Stache EE, Ertel AB, Rovis T, Doyle AG, ACS Catal 2018, 8, 11134–11139; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Martinez Alvarado JI, Ertel AB, Stegner A, Stache EE, Doyle AG, Org. Lett 2019, 21, 9940–9944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wang J, Cary BP, Beyer PD, Gellman SH, Weix DJ, Angew. Chem. Int. Ed 2019, 58, 12081–12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ni S, Padial NM, Kingston C, Vantourout JC, Schmitt DC, Edwards JT, Kruszyk MM, Merchant RR, Mykhailiuk PK, Sanchez BB, Yang S, Perry MA, Gallego GM, Mousseau JJ, Collins MR, Cherney RJ, Lebed PS, Chen JS, Qin T, Baran PS, J. Am. Chem. Soc 2019, 141, 6726–6739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].a) Krska SW, DiRocco DA, Dreher SD, Shevlin M, Acc. Chem. Res 2017, 50, 2976–2985; [DOI] [PubMed] [Google Scholar]; b) Isbrandt ES, Sullivan RJ, Newman SG, Angew. Chem. Int. Ed 2019, 58, 7180–7191; [DOI] [PubMed] [Google Scholar]; c) Mennen SM, Alhambra C, Allen CL, Barberis M, Berritt S, Brandt TA, Campbell AD, Castañón J, Cherney AH, Christensen M, Damon DB, Eugenio de Diego J, García-Cerrada S, García-Losada P, Haro R, Janey J, Leitch DC, Li L, Liu F, Lobben PC, MacMillan DWC, Magano J, McInturff E, Monfette S, Post RJ, Schultz D, Sitter BJ, Stevens JM, Strambeanu II, Twilton J, Wang K, Zajac MA, Org. Process Res. Dev 2019, 23, 1213–1242. [Google Scholar]
- [33].a) Chatgilialoglu C, Lalevee J, Molecules 2012, 17, 527–555; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chatgilialoglu C, Acc. Chem. Res 1992, 25, 188–194. [Google Scholar]
- [34].Corce V, Chamoreau LM, Derat E, Goddard JP, Ollivier C, Fensterbank L, Angew. Chem. Int. Ed 2015, 54, 11414–11418. [DOI] [PubMed] [Google Scholar]
- [35].Tellis JC, Primer DN, Molander GA, Science 2014, 345, 433–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wang P-Z, Chen J-R, Xiao W-J, Org. Biomol. Chem 2019. [DOI] [PubMed]
- [37].a) Chen W, Liu Z, Tian J, Li J, Ma J, Cheng X, Li G, J. Am. Chem. Soc 2016, 138, 12312–12315; [DOI] [PubMed] [Google Scholar]; b) Huang W, Cheng X, Synlett 2017, 28, 148–158; [Google Scholar]; c) de Assis FF, Huang X, Akiyama M, Pilli RA, Meggers E, J. Org. Chem 2018, 83, 10922–10932. [DOI] [PubMed] [Google Scholar]
- [38].Buzzetti L, Prieto A, Roy SR, Melchiorre P, Angew. Chem. Int. Ed 2017, 56, 15039–15043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lowry MS, Goldsmith JI, Slinker JD, Rohl R, Pascal RA, Malliaras GG, Bernhard S, Chem. Mater 2005, 17, 5712–5719. [Google Scholar]
- [40].Tamayo AB, Alleyne BD, Djurovich PI, Lamansky S, Tsyba I, Ho NN, Bau R, Thompson ME, J. Am. Chem. Soc 2003, 125, 7377–7387. [DOI] [PubMed] [Google Scholar]
- [41].a) Margrey KA, Nicewicz DA, Acc. Chem. Res 2016, 49, 1997–2006; [DOI] [PubMed] [Google Scholar]; b) Wilger DJ, Grandjean J-MM, Lammert TR, Nicewicz DA, Nat. Chem 2014, 6, 720–726. [DOI] [PubMed] [Google Scholar]
- [42].Meyer E. v., J. Prakt. Chem 1908, 78, 497–534. [Google Scholar]
- [43].a) Wang C, Qin J, Shen X, Riedel R, Harms K, Meggers E, Angew. Chem 2016, 128, 695–698; [DOI] [PubMed] [Google Scholar]; b) Liu Y, Liu X, Li J, Zhao X, Qiao B, Jiang Z, Chem. Sci 2018, 9, 8094–8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Moir M, Danon JJ, Reekie TA, Kassiou M, Expert Opin. Drug Discov 2019, 14, 1137–1149. [DOI] [PubMed] [Google Scholar]
- [45].Battershill AJ, Scott LJ, Drugs 2006, 66, 51–83. [DOI] [PubMed] [Google Scholar]
- [46].Regnier V, Romero EA, Molton F, Jazzar R, Bertrand G, Martin D, J. Am. Chem. Soc 2019, 141, 1109–1117. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.