

I. Introduction
The herpes simplex viruses (HSV) have co-evolved with their human hosts for millions of years: HSV-1 for an estimated 6 million years and HSV-2 for an estimated 1.6 million years. Humans and HSV have developed a successful equilibrium, where the viruses can establish a latent infection and persist in humans for the lifetime of the host organism. This homeostasis between host and virus involves host recognition of the virus infection and attempts to clear it, while the virus evolves to evade the host immune responses. Through this evolutionary chess game, the HSV viruses and the human host have reached an equilibrium. Disease may result if the viral load is too great or if the host has transient or genetic defects in the immune responses. The first line of protection against microbes is innate immunity, which encompasses all the mechanisms that provide barriers or restriction to infection and the signaling pathways that produce antiviral molecules. A number of innate immune mechanisms are invoked following HSV infection, and HSV in turn has evolved mechanisms to neutralize these host responses. In this article we will review the innate immune mechanisms that are known to impact HSV infection, the evasion strategies utilized by HSV to over come these responses, and the disease manifestations that result from immune deficiencies.
There are two species of HSV, HSV-1 and HSV-2. Viruses of both species cause ulcerative lesions at oral (usually HSV-1) and genital (usually HSV-2 but increasingly HSV-1) mucosae, and HSV innate immune evasion mechanisms likely contribute to viral spread and extent of disease at these mucosal sites.
HSV-1 undergoes a primary productive infection in the oral mucosa or oral cavity (gingivostomatitis) and then establishes a latent infection in sensory ganglia. HSV-1 reactivation from latency can lead to recurrent infections and lesions commonly known as cold sores or fever blisters. In addition to ulcers, oral herpes infections are associated with periodontal disease (Slots 2010), and oral HSV and bacterial infections may be associated with increased risk of cardiovascular disease (Vilkuna-Rautiainen et al. 2006). HSV-1 can also infect the cornea (herpes keratitis) as a result of primary infection or reactivation from latent infection. The host immune response to recurrent corneal infections can lead to progressive scarring of the cornea, clouding, and eventual blindness. HSV-1 can travel along neuronal pathways into the central nervous system during primary infection or reactivation, resulting in very serious encephalitis. Encephalitic disease, like ulcerative disease, is a composite of viral cytopathology and inflammatory effects. HSV-2 is acquired generally as a sexually transmitted disease, involving primary infection cells of the genital mucosa, spread into sensory neuron axons, and transport to sacral ganglia where it establishes a latent infection. Reactivation of latent virus causes the recurrent lesions associated with genital herpes. In addition to genital infection, HSV-2 can spread systemically to cause meningitis. More serious, however, is the intrauterine or peripartum transmission of HSV-2 from a productively infected mother to her child, which can result in encephalitis and/or disseminated herpes infection in the newborn. The mortality rate of infected newborns is high despite the availability of antivirals to limit infection, and survivors frequently experience life-long sequelae. Furthermore, genital herpes significantly raises the risk of human immunodeficiency virus (HIV) infection (summarized in Wald and Link 2002; Freeman et al. 2006). Therefore, herpes infections cause considerable morbidity and mortality, and new therapeutics and vaccines are needed to prevent and treat herpetic infections and the immunopathology caused by these infections.
Pattern recognition receptors, signaling, and effector pathways.
Antiviral responses in mammalian hosts are initiated by the interaction of germ-line encoded innate immune receptors that sense the presence of viral products within infected cells. These so-called pattern recognition receptors (PRRs) are known to recognize both pathogen-associated molecular patterns (PAMPs), including viral nucleic acids (vDNA, vRNA), viral proteins, and damage-associated molecular patterns (DAMPs) that are produced as a consequence of virus-induced tissue damage and cell death (Figure 1). Innate PRRs include members from several gene families including the Toll-like receptors (TLRs), cytosolic RNA sensors (RIG-I [retinoic acid-inducible gene I] and MDA5 [melanoma differentiation-associated protein 5]), and several classes of cytosolic DNA sensors, cGAS (cyclic GMP-AMP synthase), AIM2 (absent in melanoma 2), IFI16 (interferon gamma inducible protein16), and DHX (DEAH-box) proteins (reviewed in Thompson et al. 2011; Unterholzner 2013; Xiao and Fitzgerald 2013; Orzalli and Knipe 2014; Knipe 2015).
Figure 1.
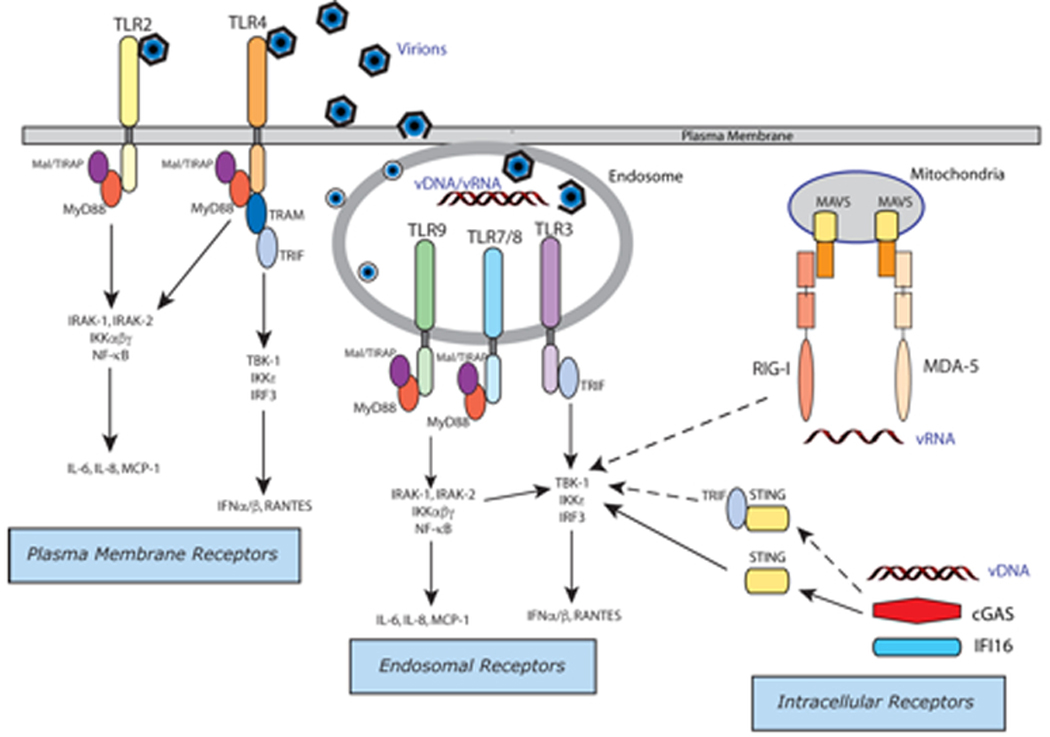
Antiviral Innate Immunity: Detection of viruses by pattern recognition receptors (Copyright, Evelyn Kurt-Jones and David Knipe)
Innate PRRs drive the production of type I interferon (IFN), cytokines, and chemokines. The type I IFNs (e.g., IFN-α and IFN-β) are essential antiviral proteins that drive the expression of IFN-stimulated genes (ISGs) which play crucial roles in controlling virus infections (Schoggins and Rice 2011). Viral nucleic acids are potent inducers of type I IFNs that are sensed via their interaction with TLR3/9, RIG-I/MDA-5, cGAS, or IFI16. TLR3 and its adapter TRIF (TIR [Toll/interleukin-1 receptor] domain-containing adaptor protein inducing IFN-β) are activated by double-stranded (ds)RNA in the endosome. RIG-I and MDA-5 receptors are cytosolic sensors for uncapped, 5’-tri-and di-phosphate single-stranded (ss)RNA and long ssRNA, respectively, and signal via the mitochondrial protein MAVS (mitochondrial antiviral-signaling protein). cGAS is a cytosolic DNA sensor that activates IFN production via second messenger signaling to the endoplasmic reticulum (ER)-associated protein STING (stimulator of IFN genes). The STING-TRIF interaction also contributes to IFN responses by an unknown mechanism (Wang et al. 2016). In addition to STING, IFI16 may function as a DNA sensor for IFN production and plays a role in viral DNA detection in both the cytosol and in the nucleus (Orzalli and Knipe 2014). All of these nucleic acid receptors with their associated adaptors trigger a canonical signaling pathway leading to type I IFN gene expression by activation/phosphorylation of TBK1 (TANK-binding kinase 1) leading to phosphorylation/activation of IRF3 (interferon regulatory factor 3) (and IRF7 in plasmacytoid dendritic cells [pDCs]). Phospho-IRF3 dimerizes and translocates to the nucleus where it binds to the IFN promoter and drives type I IFN gene expression.
Defects in type I IFN production and/or IFN-responsiveness result in unrestrained virus replication and are associated with severe herpes simplex encephalitis (HSE) in patients and in animal models. Binding of type I IFNs to the IFN-α/β receptor (IFNAR) induces the expression of hundreds of ISGs leading to dramatic cellular reprogramming for a coordinated antiviral state within mammalian hosts (Schoggins et al. 2014; Schneider et al. 2014). Cytokine and chemokine production downstream of PRRs plays an important role in viral disease, leading to the recruitment of immune cells (i.e., inflammatory response and leukocyte recruitment), the development of virus-specific adaptive immunity (i.e., maturation of antigen presenting cells), and the resolution of the response (i.e., anti-inflammatory cytokines and promotion of tissue repair). The inflammatory response is tightly regulated; excess inflammatory cytokine production during viral infection can damage host tissues and contribute to morbidity and mortality if unchecked.
Constitutive pathways that resist viral infection have sometimes been called intrinsic resistance or immunity mechanisms (Bieniasz 2004; Roizman et al. 2013). The classic example of such a restriction factor is rhesus TRIM5α (tripartite motif-containing protein 5α), which inhibits HIV replication (Stremlau et al. 2004). This is in contrast to innate immunity where a signaling pathway is induced to activate or affect antiviral mechanisms. However, these classes of pathways are often indistinguishable because IFN-α/β induce a number of the intrinsic resistance factors such as the TRIM proteins and IFI16.
II. Sensing and Signaling Pathways
A. Sensing of virion and infected cell proteins.
A number of receptors and innate sensors recognize HSV virions or infected cell proteins, and there are likely more that remain to be identified. One of the first opportunities for sensing HSV is the interaction of the virus with cell surface molecules. HSV-1 entry into infected cells is mediated by at least two different receptor proteins, herpes virus entry molecule (HVEM) and nectin-1, both of which bind the HSV glycoprotein D (gD). HVEM and nectin-1 vary in their tissue distribution and their relative importance depends on the route of infection (Taylor et al. 2007; Kopp et al. 2009; Karaba et al. 2012; Kopp et al. 2013; Kopp et al. 2014; Petermann et al. 2015). Nectin-1 is the major entry receptor for neuronal infection while HVEM plays an important role in corneal replication and neonatal neurologic disease; however, both receptors contribute to disease and nectin-1/HVEM double knockout mice are completely resistant to HSV-1 and HSV-2 disease (Kopp et al. 2013).
Toll-like receptor 2 (TLR2).
A number of mechanisms in HSV-infected cells can activate the nuclear factor κB (NF-κB) pathway. TLR2 is a major plasma membrane sensor of HSV interaction with the cell surface of antigen-presenting cells. HSV infection triggers TLR2-dependent activation of NF-κB and the production of inflammatory cytokines and chemokines in many cell types, including macrophages, monocytes, neutrophils, glial and neuronal cells, epithelial cells, and keratinocytes (Kurt-Jones et al. 2004; Aravalli et al. 2005; Wang et al. 2012; Gianni et al. 2013). Activation of TLR2 leads to MyD88-dependent induction of the NF-κB pathway and expression of pro-inflammatory cytokines and other cellular proteins (Kurt-Jones et al. 2004; Kurt-Jones et al. 2005). Soluble forms of two HSV glycoproteins, gH and gL, are sufficient to activate TLR2 signaling in cells (Gianni et al. 2013); thus, the same interactions of gH/gL on virions with TLR2 on infected cells is likely to be happening during viral infection of TLR2-positive cells. HSV strains and even different passages of the same strain show differences in ability to activate TLR2 (Sato et al. 2006; Kurt-Jones and Knipe, unpublished results). The basis for this variability is unknown, and it could be genetic or phenotypic. Furthermore, it could be due to differences in ability of HSV to activate TLR2 or the ability of viral functions to modulate TLR2 activation, as described below.
Additional TLR2 interactors play a role in the host response to HSV. The αvβ3-integrin has been reported to act in concert with TLR2 to elicit an innate response to HSV and lipopolysaccharide (Gianni et al. 2013). gH/gL bind to both TLR2 and αvβ3-integrin, thereby stabilizing the signaling complex and targeting it to lipid rafts for increased signaling (Gianni and Campadelli-Fiume 2014). The myeloid receptor CD200R1 plays a role in TLR2-driven innate immunity and in HSV-1 replication. CD200R1 expression licenses TLR2-dependent signaling in myeloid cells and enhances HSV-1-driven inflammatory cytokine responses (Soberman et al. 2012). In parallel with reduced cytokine induction, HSV-1 replication is reduced in CD200R1-deficient myeloid cells. CD200R1 also increases TLR2 signaling on mouse peritoneal macrophages infected with HSV-1 (Soberman et al. 2012).
TRIM5α.
Simian TRIM5α can restrict replication of HSV by limiting immediate early (IE) and early (E) gene expression (Reszka et al. 2010). Although human TRIM5α showed little effect on HSV replication, these observations provide part of the basis of the relatively inefficient replication of HSV in rhesus macaque cells.
Tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6).
HSV proteins can also bind to internal proteins and activate innate pathways. The HSV-1 UL37 tegument protein has a motif that binds to TRAF6 and activates it to autoubiquitinate (Yan et al., manuscript in preparation) and to activate NF-κB signaling at very early times post-infection (Liu et al. 2008).
Herpes virus entry molecule (HVEM).
During entry, HSV gD can bind to HVEM (herpesvirus entry molecule), a human cell surface receptor of the TNF-receptor superfamily, also known as TNF receptor superfamily member 14 (TNFRSF14). Binding of the natural ligand, TNFRSF or LIGHT (homologous to lymphotoxin, exhibits inducible expression and competes with HSV glycoprotein D for binding to herpesvirus entry mediator (HVEM), a receptor expressed on T lymphocytes), to HVEM leads to activation of the NF-κB pathway (Ware 2009). Soluble gD can activate NF-κB signaling (Medici et al. 2003), but it is not known if gD on the virion surface has the same activity.
B. Sensing of HSV DNA
Antiviral immunity and type I IFN production are triggered by innate sensing of HSV DNA. Defects in signaling pathways that are downstream from DNA sensors are associated with severe HSE in patients, suggesting that detection of herpes vDNA may be critical for host survival (reviewed in Sancho-Shimizu et al. 2011; Zhang and Casanova 2015). In this section, we will review the major DNA sensors that have been linked to HSV-1 disease and HSV-1 induced type I IFN production. As part of the incoming virion, HSV DNA is a “naked” linear dsDNA molecule that is protected from host sensors. However, when the virus introduces its DNA into the cell nucleus (Orzalli et al. 2012; Li et al. 2012), or under conditions where the viral capsid is disrupted before or during transit to the nucleus (Horan et al. 2013), it can be sensed by host DNA sensors.
Toll-like receptor 9 (TLR9).
Early studies demonstrated a role for TLR9 in IFN production by HSV-1 infected dendritic cells (Lund et al. 2003; Krug et al. 2004). TLR9 is an endosome-associated transmembrane protein receptor for unmethylated CpG-rich DNA motifs. In vitro, TLR9-deficient pDCs have a blunted type I IFN response to HSV-1 infection compared to wild type (wt) pDCs (Lund et al. 2003; Krug et al. 2004). TLR9-dependent IFN responses are cell type-specific, and TLR9 is not required to control HSV-1 infection in murine systems (Wang et al. 2012; Rasmussen et al. 2007; Rasmussen et al. 2009). In addition, TLR9-deficient mice do not exhibit obvious defects in their response to HSV-1, suggesting that TLR9 is redundant for controlling HSV-1 infection. Endosomal TLRs, including TLR9, must be transported to the endosome to function. Mutations in Unc93b disrupt TLR3, TLR7, TLR8 and TLR9 trafficking to endosomes (Brinkmann et al. 2007; Kim et al. 2008; Casrouge et al. 2006) and severely impair the IFN response to HSV-1 infection (Casrouge et al. 2006; Wang et al. 2012). In addition, patients with defects in Unc93b expression and Unc93b mutant mice are highly susceptible to HSE. Taken together, the data suggest that endosomal detection of HSV-1 nucleic acids by TLRs is critical for controlling HSV-1 infection and protection from HSE; however, the roles of individual TLRs and their mechanisms of activation in this process are unclear.
Cyclic guanosine monophosphate-adenosine monophosphate synthase (cGAS).
Recent studies have expanded the list of HSV DNA sensors to include intracellular DNA-binding sensors, including cGAS, IFI16, and AIM2. cGAS is a newly discovered DNA sensor that is critical for protection during HSV-1 infection. cGAS is an enzyme that catalyzes the synthesis of a mixed cyclic di-nucleotide second messenger molecule, cyclic G(2’−5’)pA(3’−5’)p (2’3’-cGAMP) (Sun et al. 2013; Ablasser et al. 2013; Diner et al. 2013). DNA binding to cGAS induces a conformational change in the cGAS enzyme, which triggers 2’3’-cGAMP synthesis. The 2’3’-cGAMP product binds to the ER-associated protein, STING. Activated STING in turn activates IRF3 and induces type I IFN production. STING is a cyclic di-nucleotide sensor and is the major adapter for cytosolic DNA sensing via cGAS/cGAMP second messenger (Li et al. 2013).
Despite the strong genetic evidence that cGAS is a critical component of the antiviral response to HSV infection, several questions remain in terms of the mechanistic role cGAS plays in this response. Is cGAS required for sensing of HSV DNA or for maintaining appropriate levels of basal sensing machinery? What is the ligand for cGAS in infected cells? HSV infection causes leakage of mitochondrial DNA (West et al. 2015), which could be the ligand for cGAS. If it is vDNA, where in an infected cell does cGAS sense that DNA? In contrast to the original studies that describe cGAS as a cytosolic protein (Li et al. 2013), we have observed cGAS both in the cytoplasm and in the nucleus of normal human fibroblasts and keratinocytes, two important cell types in HSV pathogenesis (Orzalli et al. 2015). However, further studies have been hampered by an inability to detect movement of cGAS to sites of incoming viral genomes (a phenotype observed in response to transfected DNA) or to detect 2’3’-cGAMP production after HSV-1 infection of these cell types (Orzalli et al. 2015).
PYHIN (PYRIN and HIN domain) family proteins.
IFI16.
IFI16 was first identified (Trapani et al. 1992) as a human homolog of the murine p202 and p204 proteins identified previously (Choubey et al. 1989). IFI16 was originally reported to be a cytosolic receptor for HSV DNA in THP-1 cells because it colocalized with a transfected HSV 60mer DNA (Unterholzner et al. 2010). However, immunofluorescence analysis has shown that IFI16 is primarily nuclear in many (e.g., human keratinocytes and fibroblasts) but not all cell types (e.g., THP-1 cells) (reviewed in (Veeranki and Choubey 2012)). Acetylation of IFI16 is at least part of the regulation of its intracellular location (Li et al. 2012). IFI16 is required for inflammasome activation in Kaposi’s sarcoma-associated herpes virus-infected endothelial cells (Kerur et al. 2011), and IFI16 was reported to translocate from the nucleus to the cytoplasm to organize the inflammasome (Kerur et al. 2011). IFI16 engagement by HSV DNA triggers type I IFN production via activation of IRF3 (Unterholzner et al. 2010; Orzalli et al. 2012; Li et al. 2012). In normal human fibroblasts, induction of IFN-β requires STING (Orzalli et al. 2012). Therefore, activation of IRF3 in the cytoplasm seemed to follow IFI16 sensing of HSV DNA in the nucleus and signaling through STING, TBK1, and IRF3 through an unknown signaling mechanism from nucleus to cytoplasm. In macrophages, proteasomal degradation of HSV capsids in the cytoplasm is believed to release HSV DNA for recognition by cytoplasmic IFI16 (Horan et al. 2013). IFI16 is thought to play a tissue-specific role in the anti-viral response to HSV-1 infection. In particular, IFI16 expression in epithelial cells is necessary for IRF3 activation and IFN-α production and is key to preventing systemic spread of HSV from primary infection of cornea or genital tract (Conrady et al. 2012).
In addition to its role as a cytosolic sensor, IFI16 also has broader anti-viral functions within the nucleus (Orzalli et al. 2013). IFI16 provides broad anti-viral protection by restricting the expression of foreign genes from either viral or transfected DNA. During HSV-1 infection, nuclear IFI16 binds to virion DNA and promotes its heterochromatinization resulting in epigenetic silencing of viral gene expression (Orzalli et al. 2013; Johnson et al. 2013). This effect was observed only with an ICP0- mutant HSV virus (see discussion of ICP0 below) in some cases (Orzalli et al. 2013; Cuchet-Lourenco et al. 2013), while others observed an inhibitory effect of IFI16 on wt virus (Conrady et al. 2012; Johnson et al. 2014). The restrictive effect of IFI16 was postulated to be due to IFI16 binding to the relatively unchromatinized HSV DNA (Orzalli et al. 2013; Li et al. 2012). Molecular biological studies have demonstrated that IFI16 binds to naked DNA and slides to form multimers and that the presence of nucleosomes prevents movement and coalescence of IFI16 (Stratmann et al. 2015), consistent with the earlier hypotheses. One report argued that IFI16 required BRCA1 (breast cancer 1) for binding to vDNA (Dutta et al. 2015).
Nuclear IFI16 could also play a direct regulatory role in innate immune gene expression following HSV-1 infection. Expression of type I IFN genes is enhanced in the presence of IFI16 (Thompson et al. 2014). IFI16 associates with the promoters of several innate immune genes, both basally and upon infection with virus, and IFI16-deficient cells are defective in their IFN responses to DNA, RNA, and viruses. Promoter occupancy and mRNA expression analysis of wt and IFI16-deficient cells suggest that IFI16 is a positive transcriptional regulator of several type I IFN antiviral genes including IFN-α (Thompson et al. 2014).
AIM2.
AIM2 was the first member of the PYHIN family identified as a sensor of viral DNA. In contrast to other cytosolic DNA sensors that drive IFN production, binding of DNA to AIM2 induces the assembly of a multi-protein complex, termed the inflammasome, which is responsible for processing pro-IL-1β and pro-IL-18 proteins into their mature, secreted forms via an ASC-and caspase-1 dependent mechanism (Hornung et al. 2009). IL-1β is a potent inflammatory cytokine and IL-18 is an important cytokine for the development of cell-mediated immunity, particularly type II IFN (IFN-γ) production and the maturation of T cells and NK cells. AIM2 is important for host responses to DNA viruses including murine cytomegalovirus (MCMV) and vaccinia virus, but the role of AIM2 in HSV infection is unclear (Rathinam et al. 2010). Despite the accumulation of HSV-1 DNA in the cytosol of infected macrophages (Horan et al. 2013), HSV-1 activation of inflammasomes is independent of AIM2. However, AIM2 is critical for IL-1β release in IFN-γ-primed human keratinocytes infected with HSV-1 (Strittmatter et al. 2016). Therefore, the role of AIM2 in HSV pathogenesis might be context-dependent.
Nuclear Domain 10 components.
The promyelocytic leukemia (PML) protein localizes with other cellular proteins, including Sp100, DAXX, and ATRX to nuclear structures called nuclear domain 10 (ND10) structures. These proteins have all been associated with a restriction of HSV gene expression (reviewed in Boutell and Everett 2013). PML levels and numbers of ND10 structures increase with IFN treatment, and the effects of PML are a 3–10 fold reduction in replication of an ICP0 mutant virus (Chee et al. 2003; Everett et al. 2006) but up to 1000-fold when induced by IFNs (Chee et al. 2003). Interestingly, some studies at low multiplicity of infection show that PML can enhance HSV replication (Xu et al. 2016; Merkl and Knipe, manuscript in preparation). Thus, PML is part of both a constitutive resistance mechanism and the antiviral mechanism of IFNs but may also support viral replication by other mechanisms.
Originally, viral genomes were believed to localize near ND10 structures for early transcription and vDNA replication (Ishov and Maul 1996); however, more recent results indicate that ND10 components localize near viral genomes (Everett and Murray 2005). In either case, the effect is to repress replication of ICP0-null viruses and inhibit viral gene expression (Glass and Everett 2013). The mechanism of inhibition of HSV gene expression has not been determined. Although ND10 components have been reported to surround viral genomes (Catez et al. 2012), there is no evidence of direct association of ND10 proteins with vDNA. This is an important area for future research.
C. Sensing of HSV RNA
RIG-I/Pol III and MDA5.
IFN induction by HSV infection in the Raw264.7 murine macrophage cell line requires RIG-I and RNA polymerase III and is thought to involve pol III transcribing the HSV genome followed by RIG-I recognition of the resulting ssRNA transcripts with 5’ triphosphates (Chiu et al. 2009). IFN induction in primary human macrophages infected with HSV also requires MDA-5, which recognizes long dsRNA, and the MDA-5 downstream adaptor MAVS (Melchjorsen et al. 2010).
Protein kinase RNA-activated (PKR).
PKR is an IFN-inducible dsRNA-binding protein that restricts virus replication. Upon recognition of dsRNA, PKR inhibits translation through phosphorylation of the eIF2alpha initiation factor. In addition, eIF2alpha phosphorylation promotes an antiviral autophagy response. The antiviral activities of PKR after HSV infection are observed in the absence of the viral ICP34.5 protein (mechanism reviewed in detail below). How PKR is activated during HSV infection remains unknown, although dsRNA molecules are observed in HSV-infected cells (Jacquemont and Roizman 1975) and may therefore be the ligand for HSV-1-induced PKR activation.
TLR3.
TLR3 may play an important role in the Unc93b mediated antiviral phenotype, as TLR3-deficient mice are more susceptible to HSV-2 infection, and astrocytes from these mice have reduced IFN responses following HSV infection in vitro (Reinert et al. 2012). However, the RNA ligand sensed by TLR3 in these cells following HSV infection is unknown.
D. Sensing of Viral Fusion
Innate immune responses are elicited by both PAMPs (i.e., viral nucleic acids) and by DAMPs, (e.g., mitoDNA, HMGB-1, uric acid) produced during virus infection (reviewed in Kumar et al. 2011; Labzin et al. 2016; Paludan et al. 2011). Innate antiviral immunity is also induced by perturbation of cellular membranes during virus-cell fusion. Type I IFN responses are induced by HSV-1 particles, independent of recognition of viral nucleic acids (Paladino et al. 2006; Collins et al. 2004; Holm et al. 2012; Hare et al. 2016). HSV-1-derived virus-like particles (VLPs) lacking viral genomes and capsid induce expression of IFN-β and the ISG CXCL10 via IRF3 activation (Holm et al. 2012). Fusion-defective VLPs (lacking gB) failed to induce ISG expression, suggesting that VLP-driven ISG expression is dependent on fusion with the cell membrane. Infection with enveloped viruses, including HSV-1, triggers intracellular Ca++ oscillations (Hare et al. 2016; Cheshenko et al. 2003) upon virus entry. Fusogenic liposomes trigger a similar Ca++ oscillation and induce a type I IFN response, suggesting that fusion during enveloped virus entry into cells and subsequent Ca++ signaling is a key trigger for anti-viral immunity. Disruption of Ca++ signaling abrogated IFN responses to both HSV-1 entry and to liposome fusion (Hare et al. 2016). Ca++ signaling is upstream of IRF3 activation and enhanced the detection of viral genomes by cytosolic nucleic acid sensors, suggesting that virus-cell membrane fusion is detected by the host and contributes to IFN anti-viral immune responses (Holm et al. 2012; Hare et al. 2016).
III. Cellular effector mechanisms.
A number of effector mechanisms are used by the host innate immune response to restrict HSV replication. The ISGs include PML, IFI16, and several other TRIM proteins that contribute to the coordinated anti-viral response induced by IFN. Cell death and autophagy pathways have also been implicated in the cellular response to HSV infection.
Cell death – Apoptosis and Necroptosis.
Apoptosis and necroptosis are forms of programmed cell death that serve to contain replication of HSV and other viruses by causing death of the infected cell (Guo et al. 2015). Apoptosis involves permeabilization of the outer membrane of mitochondria to release cytochrome c, which activates caspase-3 and −7 to activate cell death pathways. Necroptosis involves external signals such as tumor necrosis factor (TNF) inducing assembly of preexisting cytosolic components into a caspase-8-containing signaling complex that causes cell death (reviewed in Wallach et al. 2016). HSV induces both pathways, but this was revealed only when viral mutant strains that do not contain specific viral inhibitors were used for infection. The mechanisms of induction of apoptosis by HSV are not well defined, but it has been reported to involve the BH3 protein PUMA (p53 upregulated modulator of apoptosis. The early mechanism by which PUMA is activated by HSV infection remains to be defined (Papaianni et al. 2015). Apoptosis induction by HSV also differs depending on cell type (Tsalenchuck et al. 2016). Apoptosis is inhibited by induction of NF-κB by the virus. The induction of necroptosis by HSV infection is also not well defined but may involve the HSV-1 ICP6 protein binding to RIP1 (receptor-interacting protein kinase 1) and RIP3 in mouse cells to initiate necroptosis (Wang et al. 2014b). However, in human cells HSV-1 ICP6 blocks necroptosis by binding to RIP1 and RIP3 (Guo et al. 2015).
Autophagy.
Autophagy is a process in which proteins and organelles are degraded by engulfment by autophagosomes followed by fusion with lysosomes, which serves to limit viral replication in the cell. Autophagy is induced by PKR; thus, HSV infection may induce autophagy by inducing IFN and activating PKR. Autophagy is initiated by the dephosphorylation and activation of the ULK (unc-51-like) complexes, which phosphorylate Beclin-1. The autophagy-inducible Beclin-1 complex localizes to the site of phagosome initiation, where VPS34 in the Beclin-1 complex phosphorylates phosphatidylinositol, which leads to recruitment of the systems for autophagosome formation. Autophagy has been reported to have little effect on HSV replication in murine fibroblasts (Alexander et al. 2007). Nevertheless, autophagy plays a role in controlling the virus in vivo, particularly in neurons (Rosato and Leib 2015; Orvedahl et al. 2007; Yordy et al. 2012). Recently, the formation of non-canonical autophagic clusters was demonstrated in sensory neurons of mice infected with HSV (Katzenell and Leib 2016). This response was dependent on IRF3 and IFN signaling components and could be induced by treatment with recombinant IFN-β. Autophagic clusters were only observed in cells negative for HSV antigen, suggesting this cellular response may have a role in restricting viral gene expression. However, the role of these clusters in the antiviral response to HSV remains to be fully determined.
IV. Viral Evasion
There are a large number of potential effects of HSV gene products on innate immune mechanisms, and there are a number of reviews on this topic (Roizman et al. 2013; Rosato and Leib 2015; Su et al. 2016). We will review this area by focusing on the effects that are exerted by individual viral gene products.
Glycoprotein C (gC).
gC mediates the attachment of virions to cells by binding to glycosaminoglycans of heparan sulphate or to chondroitin sulfate. In addition, gC contains two domains involved in modulating complement activation: one binds C3, and the other is required for blocking C5 and properdin binding to C3, thereby blocking the classical and alternative complement pathways (Friedman et al. 1984; Friedman et al. 1986).
Glycoprotein E/glycoprotein I (gE/gI).
The glycoproteins gE and gI form a complex that promotes cell-to-cell spread of HSV. These glycoproteins also form an Fc receptor that binds the Fc region of IgG and blocks antibody neutralization of virions, antibody-dependent cytotoxicity, and phagocytosis (Dubin et al. 1991; Frank et al. 1989; Van Vliet et al. 1992).
US3.
US3 is a protein kinase that is present in the HSV virion and is expressed at late times after infection and phosphorylates a number of viral and cellular proteins to enhance its replication (Roizman et al. 2013). US3 phosphorylates KIF3 (Kinesin superfamily protein 3) to down-regulate CD1d expression, thereby inhibiting NKT (natural killer T) cell function (Xiong et al. 2010). US3 reduces type I IFN and ISG induction in HSV-1 infected human monocytes (Peri et al. 2008). In this study, Us3 blocked dimerization of IRF3, but the exact mechanism of inhibition has not been defined. In addition, US3 inhibits TLR2 signaling by reducing TRAF6 polyubiquitination through a mechanism dependent on the protein’s kinase activity (Sen et al. 2013). Furthermore, US3 hyperphosphorylates p65RelA to reduce NF-κB activation in response to TNFα or IL-1β stimulation (Wang et al. 2014a); although others have not observed a similar inhibitory activity in response to other NF-κB stimuli (Sen et al. 2013).
Vhs.
The virion host shut off (vhs) protein is a late viral gene product encoded by the UL41 gene that is incorporated into the virion tegument. Vhs contains RNAse activity that degrades both cellular and viral transcripts (Kwong and Frenkel 1989). Viruses that are genetically deficient in UL41 replicate more efficiently in the absence of the type I IFN response in vivo and in vitro cell culture models (Leib et al. 1999; Pasieka et al. 2008), indicating that vhs plays a role in overcoming the antiviral effector functions of IFN. In the absence of vhs, HSV-1 infected cells have augmented IFN-β production (Pasieka et al. 2008). In addition to blocking inducible antiviral responses, vhs promotes the loss of basal cellular proteins involved in antiviral immunity, such as TNF receptor 1 (Liang and Roizman 2006), viperin (Zenner et al. 2013; Shen et al. 2014), and IFI16 (Orzalli et al. 2016).
VP16.
Like vhs, VP16 is a late viral gene product and virion tegument protein that is introduced into the host cell during the initial stages of viral infection. VP16 may block IRF3 signaling by binding to IRF3 and blocking the recruitment of the CBP (CREB-binding protein) coactivator (Xing et al. 2013). In addition, Xing and colleagues propose that VP16 interacts with p65 to block NF-κB-dependent gene expression, but these experiments were mostly conducted in cells transfected with plasmids expressing VP16 rather than in infected cells. Thus, the biological significance remains to be demonstrated.
ICP0.
Infected cell protein 0 (ICP0) is an immediate-early viral regulatory protein that has an E3 ubiquitin ligase activity (Roizman et al. 2013). ICP0 is not essential for HSV replication, but ICP0 mutants replicate poorly in primary human cells and mouse models of virus infection. ICP0 counteracts host antiviral responses through multiple mechanisms. ICP0 promotes the degradation of a number of cellular proteins and thereby counters a number of host cell responses to HSV infection. ICP0 promotes the degradation of the nuclear domain 10 proteins PML and Sp100 that inhibit HSV gene expression (Chelbi-Alix and de The 1999; Everett et al. 1998). Disruption of ND10 bodies is sometimes assumed to promote epigenetic silencing, but there is no evidence for this and the mechanism of restriction by these proteins is completely unknown. ICP0 promotes the degradation of IFI16 in normal cells, which limits the innate signaling and epigenetic silencing functions of IFI16 (Orzalli et al. 2012; Orzalli et al. 2013; Orzalli et al. 2016). ICP0 blocks TLR2-induced NF-κB signaling by promoting the degradation of the Mal/TIRAP sorting adaptor and the MyD88 signaling adaptor proteins (van Lint et al. 2010). ICP0 promotes the degradation of DNA damage repair proteins, DNA protein kinase (Parkinson et al. 1999) and the histone E3 ubiquitin ligases RNF8 and RNF168, thereby inhibiting DNA damage responses to the incoming HSV genomes (Lilley et al. 2010).
In addition to promoting the degradation of cellular proteins, ICP0 can disrupt cellular innate responses independent of protein degradation. Nuclear ICP0 recruits activated IRF3 and CBP/p300 to nuclear foci, sequestering the complex away from cellular promoters (Melroe et al. 2004; Melroe et al. 2007; Orzalli et al. 2012; Orzalli and Knipe, unpublished results). In addition, ICP0 interacts with p65RelA and p50/NF-κB1 to inhibit TNF activation of NF-κB (Zhang et al. 2013). Furthermore, ICP0 may have additional cytoplasmic activities that inhibit antiviral signaling (Paladino et al. 2010; Taylor et al. 2014).
ICP27.
ICP27 is an immediate-early gene regulatory protein. ICP27 contributes to inhibition of host cell protein synthesis with vhs (Song et al. 2001) and to inhibition of host transcription (Rice and Knipe 1990), so it would be expected to play a role in inhibition of innate immune responses. In addition to its general role in inhibition of host transcription and translation, ICP27 specifically inhibits innate antiviral signaling pathways. ICP27 promotes the secretion of an unidentified heat stable, but protease sensitive, factor that blocks IFNAR signaling (Johnson et al. 2008; Johnson and Knipe 2010). Furthermore, ICP27 inhibits DNA-dependent IFN production by blocking the TBK-1-activated STING signalsome in infected macrophages (Christensen et al. 2016).
ICP34.5.
ICP34.5 is a leaky late gene that plays an important role in overcoming the host innate immune response through multiple mechanisms. ICP34.5-deficient virus replication is highly attenuated in vivo (Chou et al. 1990), but is enhanced in both IFN alpha/beta/gamma receptor and protein kinase R (PKR) deficient mice (Leib et al. 2000). PKR activation during viral infection results in phosphorylation of the eIF2α translation factor and subsequent inhibition of both cellular and viral protein synthesis (Roberts-Thomson et al. 1976). ICP34.5 alleviates translational arrest by promoting the dephosphorylation of eIF2α through an interaction with the PP1α host protein phosphatase (He et al. 1997). ICP34.5 also overcomes PKR-induced autophagy by binding the Beclin 1 protein and inhibiting its autophagy function (Orvedahl et al. 2007). ICP34.5 directly inhibits ISG expression by blocking the interaction of TBK1 with IRF3 through an interaction with TBK1 (Verpooten et al. 2009).
V. Mechanisms in Animal Models.
Animal models of HSV have greatly facilitated the investigation of the molecular and cellular events that are responsible for controlling HSV infection and preventing infection-induced tissue damage, morbidity, and mortality. HSV-1 infection has been studied primarily in mice, rabbits and guinea pigs (reviewed in Roizman et al. 2013). Numerous studies suggest that the innate immune response is critical for controlling HSV infection, but how individual innate receptors contribute to disease control is dependent on the route of infection. Different tissues and cells express distinct arrays of innate immune receptors and anti-viral effector molecules, and the variety of innate pathways engaged during HSV infection of different cell types affects how HSV is controlled and the infection resolved.
Cutaneous HSV-1 infection is established by inoculation of abraded skin. This model has been useful for studying antiviral therapies. It has also been used to study the development of adaptive immunity, particularly the priming and maturation of HSV-1-specific CD8+ cytolytic T cell (CTL) effectors. Carbone and colleagues have established a C57BL/6 mouse strain expressing a HSV gB-specific, CD8 T cell transgene that has enhanced our understanding of how the adaptive immune response to HSV-1 develops during primary and secondary cutaneous infection (van Lint et al. 2004; Stock et al. 2004; Fernandez et al. 2008). Cutaneous HSV-1 infection of mice has also been used to investigate the spread of replicating HSV-1 from local sites into peripheral nerves (Ma et al. 2014).
Genital herpes has been studied primarily in guinea pigs, where intravaginal inoculation with HSV-1 or HSV-2 leads to recurrent herpetic lesions (Hsiung et al. 1984). Studies in cotton rats and mice have also been used to evaluate antiviral therapies for genital infection. In recent years, intravaginal HSV infection in mice has been studied in C57BL/6 mice with defects in innate immune receptor expression and revealed that HSV-2 can be transported from the vaginal mucosa into neurons and ultimately into the spinal cord and brain stem of innate receptor-deficient mice (Sorensen et al. 2008; Reinert et al. 2012). HSV-1 can also travel from the genital tract to the dorsal root ganglia and spread to autonomic ganglia of the enteric nervous system. HSV-1 infection of enteric neurons of the colon leads to their destruction causing fecal retention and lethal toxic megacolon to develop in infected mice following genital inoculation (Khoury-Hanold et al. 2016).
Ocular infection and keratitis have been extensively studied by inoculation of virus on scarified corneas of mice and rabbits (Webre et al. 2012; Biswas and Rouse 2005; Stuart and Keadle 2012). Eye disease in mice and rabbits has many of the hallmarks of human eye disease; however, differences in the establishment of latency between rodents and humans have been noted. Despite their limitations, mouse models of ocular HSV infection have been very important for understanding how HSV spreads from epithelial cells into the trigeminal ganglion. Under some experimental conditions (e.g., high levels of virus replication, virulent virus strains, neutropenia), HSV is transported by neurons from the initial site of infection in the eye into the brain and the animals develop encephalitis (Kollias et al. 2015). Recently, a tree shrew model has been proposed to study HSV-1 latency in sensory neurons (Li et al. 2016).
Lethal HSE has been studied in mice by either intranasal (in) or intraperitoneal (ip) infection of very young mice (2–3 weeks of age). In older mice (>4 weeks of age), encephalitis develops only with very high doses of virus when the mice are infected by the ip route (reviewed in Kollias et al. 2015). Intracerebral inoculation with lower doses of HSV in older mice, albeit an unnatural route of infection, has proved useful for studies of the role of brain-intrinsic innate receptors in controlling virus replication and inflammatory responses as well as for evaluating HSV-targeted drugs (Kurt-Jones et al. 2004; Wang et al. 2012; Parker et al. 2015; Kollias et al. 2015). Intravenous (iv) inoculation with HSV does not cause encephalitis in wt mice, but replicating HSV can be detected in the brains of mice deficient in innate receptors that control type I IFN responses in brain (Parker et al. 2015; Li et al. 2013). Rabbit models of HSE have also been described with focal brain infection, similar to human disease. In the rabbit model, HSV infection is established in the olfactory bulb, either by direct inoculation or by infection of abraded epithelium innervated by the trigeminal ganglion (Kollias et al. 2015).
Animal models of TLR2 and Inflammation in HSE.
TLR2-deficient mice are protected from lethal HSE despite HSV-1 replication in cells and tissues, including brain (Kurt-Jones et al. 2004; Wang et al. 2012). While HSV-1 is present at high levels in their brains, TLR2-deficient mice do not develop brain inflammation. TLR2-deficient mice have a significant reduction in inflammatory cytokine production and a markedly reduced leukocyte infiltration into the infected brain compared to wt mice. On the other hand, type I IFN responses are normal in TLR2-deficient animals (Wang et al. 2012). Adaptive immune responses also develop normally in TLR2-deficient mice. Thus, TLR2 drives the inflammatory cytokine response to HSV-1 both in vivo and in vitro, and TLR2-dependent inflammation contributes to HSE mortality while TLR2-independent innate responses to HSV-1 are protective.
CD200R1 knockouts are hypo-responsive to HSV-1 in vivo. In vivo, CD200R1-deficient animals have a blunted inflammatory cytokine response to HSV-1 during brain infection and are protected from lethal HSE compared to wt mice. Mice lacking CD200R1 also exhibit a significant reduction in HSV-1 brain titers, suggesting that CD200R1, which is expressed in brain glial cells, is pro-viral and is necessary for sustained HSV-1 replication in the brain (Soberman et al. 2012). Both CD200R1 and TLR2 contribute to lethal HSE in mice and both receptors appear to have inter-related roles in brain inflammation and HSV-1 replication in myeloid cells; however, the mechanism(s) is unknown at present.
Animal models of type I IFN deficiency and HSE.
HSV infection studies performed in mice with targeted deletion of different immune genes have defined innate immune pathways that are responsible for both protective and detrimental anti-viral responses. Type I IFN has emerged as critical for the protection of animals from lethal HSV disease. Mice lacking the type I IFN-α/β receptor (IFNAR) are unable to respond to type I IFNs (IFN-α, IFN-β, IFN-ε) (de Weerd et al. 2007) and are highly susceptible to lethal HSE when infected either ip or ic, with significantly elevated viral burdens in the brain compared to wt controls (Wang et al. 2012). IFNAR KOs (but not wt mice) are also highly susceptible to corneal infection and rapidly succumb to HSV-1 with fulminant infection of liver and spleen and viremia (Pasieka et al. 2011). IFNAR expression in neurons themselves is critical to prevent HSE when HSV-1 infects corneal cells indicating that brain-intrinsic type I IFN responses are required for protection (Rosato and Leib 2015).
The cGAS-STING signaling pathway is essential for type I IFN responses to HSV-1 infection in vivo. Both STING and cGAS deficient mice are highly susceptible to HSV-1 and fail to mount a type I IFN response suggesting that cGAS activation of STING is the major driver of IFN response in animals (Ishikawa et al. 2009; Schoggins et al. 2015; Li et al. 2013; Parker et al. 2015; Royer and Carr 2016). cGAS knockouts rapidly succumb to HSV-1 with high brain viral titers following infection (Li et al. 2013). TRIF-deficient mice are also more susceptible to HSV-1 than wt controls, and recent studies have revealed an essential role for TRIF in STING activation during HSV-1 infection suggesting a novel pathway of STING activation leading to IFN production during HSV-1 infection (Menasria et al. 2013; Wang et al. 2016). TRIF is known to function as the adapter for TLR3-driven IFN responses, but the role of TLR3 in mouse models of HSV-1 induced HSE has not been defined. Knockdown of the murine homolog of IFI16, p204, increased viral titers shed in the tear film (Conrady et al. 2012), indicating a role for p204 in controlling local HSV replication. Inflammation also contributes to lethal HSV-1 disease independent of the IFN response, suggesting that HSV-1 induced NF-κB driven inflammation leads to enhanced pathology in animals with an intact IFN response (Kurt-Jones et al. 2004; Wang et al. 2012; Carty et al. 2014; Piret and Boivin 2015; Abe and Barber 2014).
VI. Manifestations in Humans
HSV infection causes a number of clinical manifestations in humans. Primary infection is usually mild and the reactivation of latent virus is associated with the development of cold sores that, although painful, are benign. On the other hand, reactivation of latent HSV infection in the eye can also cause blindness, while infection in the brain can cause encephalitis with significant neurologic sequelae and high mortality despite anti-viral therapy. HSE in adults is sporadic and the severity of disease in patients has been associated with both virus replication and with dysregulated innate immune activation, particularly IL-1Ra levels (Michael et al. 2016), suggesting that inflammatory response to HSV in the brain as well as the control of virus replication by innate antiviral immunity are linked to clinical outcomes.
Neonates are particularly susceptible to severe HSV disease upon primary infection. Infants infected with HSV can develop disseminated, multi-organ infections and encephalitis. Life-threatening HSV-1 infections are also found in young children with inborn defects in innate immune signaling linked to type I IFN production. Mutations in UNC93B1, TLR3, TRIF, TRAF3, TBK1, IRF3 and STAT1 are associated with the development of HSE during primary HSV-1 infection in childhood (Sancho-Shimizu et al. 2011; Herman et al. 2012; Guo et al. 2011; Ahmad et al. 2016). Although the mechanisms underlying the susceptibility of these patients to HSE are not known, these innate immune gene mutations reduce the magnitude of the IFN-β production and/or the expression of ISGs upon HSV infection of patient-derived cells in vitro (Lafaille et al. 2012; Guo et al. 2011; Herman et al. 2012). The genetic etiology of HSE suggests that the TLR3-TRIF/TRIF-STING-TBK1-IRF3 signaling axes for type I IFN responses are key to preventing HSE during primary HSV-1 infection in humans. Animal models support the hypothesis that type I IFN production and ISG responses reduce virus replication within the brain and, in the case of TLR3, may prevent spread of HSV to the brain by CNS-intrinsic immune sensor functions as well as by enhancing antigen presentation and the development of adaptive immunity in peripheral tissues (Wang et al. 2012; Menasria et al. 2013; Davey et al. 2010). A recent paper shows that these signaling pathways are more complex than previously thought. TRIF was shown to be required for STING signaling, and a specific TRIF mutation P625L associated with HSE could not support STING signaling (Wang et al. 2016). Therefore, these HSE patients could have defects in STING signaling.
Perspective.
This article describes many of the large number of innate immune mechanisms that the human host has evolved to recognize HSV infection and to control its replication. HSV has also evolved numerous functions that counter these innate immune mechanisms so that the virus can replicate and spread to a limited extent prior to establishing a latent infection. Some of the immune evasion mechanisms may also function during establishment and/or maintenance of latent infection. It is therefore easy to see how an evolutionary equilibrium has been achieved that allows HSV to persist in the human population. Genetic immunodeficiencies or immunosuppression may upset this equilibrium and limit control of HSV infection and replication, resulting in more disease. Further research into the mechanisms of innate immune responses to HSV and ways to augment or limit these responses will provide important therapeutics for treatment of viral and immune diseases caused by HSV infection.
Figure 2.

Major innate immune sensor pathways engaged by HSV-1
Acknowledgments
We thank Patrick T. Waters and Melanie Trombly for their assistance in preparation of the manuscript. Research in the laboratory of D.M.K. on this topic is supported by NIH grants AI106934, DE023909, and AI098681. MHO is supported by NIAID T32 training grant AI007512.
References
- Abe T, Barber GN (2014) Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-kappaB activation through TBK1. J Virol 88 (10):5328–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, Hopfner KP, Ludwig J, Hornung V (2013) cGAS produces a 2’−5’-linked cyclic dinucleotide second messenger that activates STING. Nature 498 (7454):380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad L, Zhang SY, Casanova JL, Sancho-Shimizu V (2016) Human TBK1: A Gatekeeper of Neuroinflammation. Trends Mol Med 22 (6):511–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander DE, Ward SL, Mizushima N, Levine B, Leib DA (2007) Analysis of the role of autophagy in replication of herpes simplex virus in cell culture. J Virol 81:12128–12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravalli RN, Hu S, Rowen TN, Palmquist JM, Lokensgard JR (2005) Cutting edge: TLR2-mediated proinflammatory cytokine and chemokine production by microglial cells in response to herpes simplex virus. J Immunol 175 (7):4189–4193. [DOI] [PubMed] [Google Scholar]
- Bieniasz PD (2004) Intrinsic immunity: A front-line defense against viral attack. Nat Immunol 5:1109–1115. [DOI] [PubMed] [Google Scholar]
- Biswas PS, Rouse BT (2005) Early events in HSV keratitis--setting the stage for a blinding disease. Microbes Infect 7 (4):799–810. [DOI] [PubMed] [Google Scholar]
- Boutell C, Everett RD (2013) Regulation of alphaherpesvirus infections by the ICP0 family of proteins. J Gen Virol 94:465–481. [DOI] [PubMed] [Google Scholar]
- Brinkmann MM, Spooner E, Hoebe K, Beutler B, Ploegh HL, Kim YM (2007) The interaction between the ER membrane protein UNC93B and TLR3, 7, and 9 is crucial for TLR signaling. J Cell Biol 177 (2):265–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carty M, Reinert L, Paludan SR, Bowie AG (2014) Innate antiviral signalling in the central nervous system. Trends Immunol 35 (2):79–87. [DOI] [PubMed] [Google Scholar]
- Casrouge A, Zhang SY, Eidenschenk C, Jouanguy E, Puel A, Yang K, Alcais A, Picard C, Mahfoufi N, Nicolas N, Lorenzo L, Plancoulaine S, Senechal B, Geissmann F, Tabeta K, Hoebe K, Du X, Miller RL, Heron B, Mignot C, Billette de Villemeur T, Lebon P, Dulac O, Rozenberg F, Beutler B, Tardieu M, Abel L, Casanova JL (2006) Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 314:308–312. [DOI] [PubMed] [Google Scholar]
- Catez F, Picard C, Held K, Gross S, Rousseau A, Theil D, Sawtell N, Labetoulle M, Lomonte P (2012) HSV-1 Genome Subnuclear Positioning and Associations with Host-Cell PML-NBs and Centromeres Regulate LAT Locus Transcription during Latency in Neurons. PLoS Pathog 8:e1002852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chee AV, Lopez P, Pandolfi PP, Roizman B (2003) Promyelocytic leukemia protein mediates interferon-based anti-herpes simplex virus 1 effects. J Virol 77:7101–7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelbi-Alix MK, de The H (1999) Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene 18:935–941. [DOI] [PubMed] [Google Scholar]
- Cheshenko N, Del Rosario B, Woda CV, Mercellimo D, Satlin LM, Herold BC (2003) Herpes simplex virus triggers activation of calcium-signaling pathways. J Cell Biol 163:283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu YH, Macmillan JB, Chen ZJ (2009) RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell Biochem Biophys 138:576–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou J, Kern ER, Whitley RJ, Roizman B (1990) Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 250 (4985):1262–1266. [DOI] [PubMed] [Google Scholar]
- Choubey D, Snoddy J, Chaturvedi V, Toniato E, Opdenakker G, Thakur A, Samanta H, Engel DA, Lengyel P (1989) Interferons as gene activators. Indications for repeated gene duplication during the evolution of a cluster of interferon-activatable genes on murine chromosome 1. J Biol Chem 264 (29):17182–17189. [PubMed] [Google Scholar]
- Christensen MH, Jensen SB, Miettinen JJ, Luecke S, Prabakaran T, Reinert LS, Mettenleiter T, Chen ZJ, Knipe DM, Sandri-Goldin RM, Enquist LW, Hartmann R, Mogensen TH, Rice SA, Nyman TA, Matikainen S, Paludan SR (2016) HSV-1 ICP27 targets the TBK1-activated STING signalsome to inhibit virus-induced type I IFN expression. EMBO J 35 (13):1385–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SE, Noyce RS, Mossman KL (2004) Innate cellular response to virus particle entry requires IRF3 but not virus replication. J Virol 78:1706–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrady CD, Zheng M, Fitzgerald KA, Liu C, Carr DJ (2012) Resistance to HSV-1 infection in the epithelium resides with the novel innate sensor, IFI-16. Mucosal Immunol 5:173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuchet-Lourenco D, Anderson G, Sloan E, Orr A, Everett RD (2013) The viral ubiquitin ligase ICP0 is neither sufficient nor necessary for degradation of the cellular DNA sensor IFI16 during herpes simplex virus 1 infection. J Virol 87 (24):13422–13432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey GM, Wojtasiak M, Proietto AI, Carbone FR, Heath WR, Bedoui S (2010) Cutting edge: priming of CD8 T cell immunity to herpes simplex virus type 1 requires cognate TLR3 expression in vivo. J Immunol 184 (5):2243–2246. [DOI] [PubMed] [Google Scholar]
- de Weerd N, Samarajiwa S, Hertzog P (2007) Type I interferon receptors: biochemistry and biological functions. J Biol Chem 282:20053–20057. [DOI] [PubMed] [Google Scholar]
- Diner EJ, Burdette DL, Wilson SC, Monroe KM, Kellenberger CA, Hyodo M, Hayakawa Y, Hammond MC, Vance RE (2013) The Innate Immune DNA Sensor cGAS Produces a Noncanonical Cyclic Dinucleotide that Activates Human STING. Cell reports 3 (5):1355–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubin G, Socolof E, Frank I, Friedman HM (1991) Herpes simplex virus type 1 Fc receptor protects infected cells from antibody-dependent cellular cytotoxicity. J Virol 65:7046–7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta D, Dutta S, Veettil MV, Roy A, Ansari MA, Iqbal J, Chikoti L, Kumar B, Johnson KE, Chandran B (2015) BRCA1 Regulates IFI16 Mediated Nuclear Innate Sensing of Herpes Viral DNA and Subsequent Induction of the Innate Inflammasome and Interferon-beta Responses . PLoS Pathog 11 (6):e1005030. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Everett RD, Freemont P, Saitoh H, Dasso M, Orr A, Kathoria M, Parkinson J (1998) The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110-and proteasome-dependent loss of several PML isoforms. J Virol 72:6581–6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD, Murray J (2005) ND10 components relocate to sites associated with herpes simplex virus type 1 nucleoprotein complexes during virus infection. J Virol 79 (8):5078–5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD, Rechter S, Papior P, Tavalai N, Stamminger T, Orr A (2006) PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J Virol 80:7995–8005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez MA, Evans IA, Hassan EH, Carbone FR, Jones CA (2008) Neonatal CD8+ T cells are slow to develop into lytic effectors after HSV infection in vivo. Eur J Immunol 38 (1):102–113. [DOI] [PubMed] [Google Scholar]
- Frank Y, Lim W, Kahn E (1989) Multiple ischemic infarcts in a child with acquired immunodeficiency syndrome, varicella zoster infection, and cerebral vasculitis. Pediatr Neurol 5:64–67. [DOI] [PubMed] [Google Scholar]
- Freeman EE, Weiss HA, Glynn JR, Cross PL, Whitworth JA, Hayes RJ (2006) Herpes simplex virus 2 infection increases HIV acquisition in men and women: Systematic review and meta-analysis of longitudinal studies. AIDS 20:73–83. [DOI] [PubMed] [Google Scholar]
- Friedman HM, Cohen GH, Eisenberg RJ, Seidel CA, Cines DB (1984) Glycoprotein C of herpes simplex virus 1 acts as a receptor for the C3b complement component on infected cells. Nature 309:633–635. [DOI] [PubMed] [Google Scholar]
- Friedman HM, Glorioso JC, Cohen GH, Hastings JC, Harris SL, Eisenberg RJ (1986) Binding of complement component C3b to glycoprotein gC of herpes simplex virus type 1: mapping of gC-binding sites and demonstration of conserved C3b binding in low-passage clinical isolates. J Virol 60:470–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianni T, Campadelli-Fiume G (2014) The epithelial alphavbeta3-integrin boosts the MYD88-dependent TLR2 signaling in response to viral and bacterial components. PLoS Pathog 10 (11):e1004477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianni T, Leoni V, Campadelli-Fiume G (2013) Type I interferon and NF-kappaB activation elicited by herpes simplex virus gH/gL via alphavbeta3 integrin in epithelial and neuronal cell lines. J Virol 87 (24):13911–13916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass M, Everett RD (2013) Components of promyelocytic leukemia nuclear bodies (ND10) act cooperatively to repress herpesvirus infection. J Virol 87:2174–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Omoto S, Harris PA, Finger JN, Bertin J, Gough PJ, Kaiser WJ, Mocarski ES (2015) Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe 17 (2):243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Audry M, Ciancanelli M, Alsina L, Azevedo J, Herman M, Anguiano E, Sancho-Shimizu V, Lorenzo L, Pauwels E, Philippe PB, Perez de Diego R, Cardon A, Vogt G, Picard C, Andrianirina ZZ, Rozenberg F, Lebon P, Plancoulaine S, Tardieu M, Valerie D, Jouanguy E, Chaussabel D, Geissmann F, Abel L, Casanova JL, Zhang SY (2011) Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity. J Exp Med 208:2083–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare DN, Collins SE, Mukherjee S, Loo YM, Gale M, Jr., Janssen LJ, Mossman KL (2016) Membrane Perturbation-Associated Ca2+ Signaling and Incoming Genome Sensing Are Required for the Host Response to Low-Level Enveloped Virus Particle Entry. J Virol 90 (6):3018–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Gross M, Roizman B (1997) The gamma (1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1 alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc Natl Acad Sci U S A 94:843–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman M, Ciancanelli M, Ou YH, Lorenzo L, Klaudel-Dreszler M, Pauwels E, Sancho-Shimizu V, Perez de Diego R, Abhyankar A, Israelsson E, Guo Y, Cardon A, Rozenberg F, Lebon P, Tardieu M, Heropolitanska-Pliszka E, Chaussabel D, White MA, Abel L, Zhang SY, Casanova JL (2012) Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood. J Exp Med 209 (9):1567–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm CK, Jensen SB, Jakobsen MR, Cheshenko N, Horan KA, Moeller HB, Gonzalez-Dosal R, Rasmussen SB, Christensen MH, Yarovinsky TO, Rixon FJ, Herold BC, Fitzgerald KA, Paludan SR (2012) Virus-cell fusion as a trigger of innate immunity dependent on the adaptor STING. Nat Immunol 13 (8):737–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horan KA, Hansen K, Jakobsen MR, Holm CK, Soby S, Unterholzner L, Thompson M, West JA, Iversen MB, Rasmussen SB, Ellermann-Eriksen S, Kurt-Jones E, Landolfo S, Damania B, Melchjorsen J, Bowie AG, Fitzgerald KA, Paludan SR (2013) Proteasomal degradation of herpes simplex virus capsids in macrophages releases DNA to the cytosol for recognition by DNA sensors. J Immunol 190 (5):2311–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA (2009) AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458:514–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiung GD, Mayo DR, Lucia HL, Landry ML (1984) Genital herpes: pathogenesis and chemotherapy in the guinea pig model. Reviews of Infectious Diseases 6 (1):33–50. [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Ma Z, Barber GN (2009) STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461:788–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishov AM, Maul GG (1996) The periphery of nuclear domain 10 (ND10) as site of DNA virus deposition. J Cell Biol 134 (4):815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont B, Roizman B (1975) Ribonucleic acid synthesis in cells infected with herpes simplex virus: characterization of viral high molecular weight nuclear RNA. J Gen Virol 29:155–165. [DOI] [PubMed] [Google Scholar]
- Johnson KE, Bottero V, Flaherty S, Dutta S, Singh VV, Chandran B (2014) IFI16 Restricts HSV-1 Replication by Accumulating on the HSV-1 Genome, Repressing HSV-1 Gene Expression, and Directly or Indirectly Modulating Histone Modifications. PLoS Pathog 10 (11):e1004503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KE, Chikoti L, Chandran B (2013) Herpes simplex virus 1 infection induces activation and subsequent inhibition of the IFI16 and NLRP3 inflammasomes. J Virol 87 (9):5005–5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KE, Knipe DM (2010) Herpes simplex virus-1 infection causes the secretion of a type I interferon-antagonizing protein and inhibits signaling at or before Jak-1 activation. Virology 396 (1):21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KE, Song B, Knipe DM (2008) Role for herpes simplex virus 1 ICP27 in the inhibition of type I interferon signaling. Virology 374 (2):487–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaba AH, Kopp SJ, Longnecker R (2012) Herpesvirus entry mediator is a serotype specific determinant of pathogenesis in ocular herpes. Proc Natl Acad Sci U S A 109 (50):20649–20654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzenell S, Leib DA (2016) Herpes Simplex Virus and Interferon Signaling Induce Novel Autophagic Clusters in Sensory Neurons. J Virol 90 (9):4706–4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P, Chandran B (2011) IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe 9 (5):363–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury-Hanold W, Yordy B, Kong P, Kong Y, Ge W, Szigeti-Buck K, Ralevski A, Horvath TL, Iwasaki A (2016) Viral Spread to Enteric Neurons Links Genital HSV-1 Infection to Toxic Megacolon and Lethality. Cell Host Microbe 19 (6):788–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YM, Brinkmann MM, Paquet ME, Ploegh HL (2008) UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature 452 (7184):234–238. [DOI] [PubMed] [Google Scholar]
- Knipe DM (2015) Nuclear sensing of viral DNA, epigenetic regulation of herpes simplex virus infection, and innate immunity. Virology 479–480:153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollias CM, Huneke RB, Wigdahl B, Jennings SR (2015) Animal models of herpes simplex virus immunity and pathogenesis. J Neurovirol 21 (1):8–23. [DOI] [PubMed] [Google Scholar]
- Kopp SJ, Banisadr G, Glajch K, Maurer UE, Grunewald K, Miller RJ, Osten P, Spear PG (2009) Infection of neurons and encephalitis after intracranial inoculation of herpes simplex virus requires the entry receptor nectin-1. Proc Natl Acad Sci U S A 106:17916–17920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp SJ, Karaba AH, Cohen LK, Banisadr G, Miller RJ, Muller WJ (2013) Pathogenesis of neonatal herpes simplex 2 disease in a mouse model is dependent on entry receptor expression and route of inoculation. J Virol 87 (1):474–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp SJ, Ranaivo HR, Wilcox DR, Karaba AH, Wainwright MS, Muller WJ (2014) Herpes simplex virus serotype and entry receptor availability alter CNS disease in a mouse model of neonatal HSV. Pediatr Res 76 (6):528–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug A, Luker GD, Barchet W, Leib DA, Akira S, Colonna M (2004) Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood 103:1433–1437. [DOI] [PubMed] [Google Scholar]
- Kumar H, Kawai T, Akira S (2011) Pathogen recognition by the innate immune system. Int Rev Immunol 30 (1):16–34. [DOI] [PubMed] [Google Scholar]
- Kurt-Jones, Knipe DM unpublished results.
- Kurt-Jones EA, Belko J, Yu C, Newburger PE, Wang J, Chan M, Knipe DM, Finberg RW (2005) The role of toll-like receptors in herpes simplex infection in neonates. J Infect Dis 191:746–748. [DOI] [PubMed] [Google Scholar]
- Kurt-Jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R, Arnold MM, Knipe DM, Finberg RW (2004) Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci U S A 101 (5):1315–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong AD, Frenkel N (1989) The herpes simplex virus virion host shutoff function. J Virol 63 (11):4834–4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labzin LI, Lauterbach MA, Latz E (2016) Interferons and inflammasomes: Cooperation and counterregulation in disease. J Allergy Clin Immunol 138 (1):37–46. [DOI] [PubMed] [Google Scholar]
- Lafaille FG, Pessach IM, Zhang SY, Ciancanelli MJ, Herman M, Abhyankar A, Ying SW, Keros S, Goldstein PA, Mostoslavsky G, Ordovas-Montanes J, Jouanguy E, Plancoulaine S, Tu E, Elkabetz Y, Al-Muhsen S, Tardieu M, Schlaeger TM, Daley GQ, Abel L, Casanova JL, Studer L, Notarangelo LD (2012) Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature 491:769–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leib DA, Harrison TE, Laslo KM, Machalek MA, Moorman NJ, Virgin HW (1999) Interferons regulate the phenotype of wild-type and mutant herpes simplex viruses in vivo. J Exp Med 189 (4):663–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leib DA, Machalek MA, Williams BR, Silverman RH, Virgin HW (2000) Specific phenotypic restoration of an attenuated virus by knockout of a host resistance gene. Proc Natl Acad Sci U S A 97 (11):6097–6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Li Z, Wang E, Yang R, Xiao Y, Han H, Lang F, Li X, Xia Y, Gao F, Li Q, Fraser NW, Zhou J (2016) Herpes Simplex Virus 1 Infection of Tree Shrews Differs from That of Mice in the Severity of Acute Infection and Viral Transcription in the Peripheral Nervous System. J Virol 90 (2):790–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Diner BA, Chen J, Cristea IM (2012) Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc Natl Acad Sci U S A 109 (26):10558–10563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ (2013) Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341 (6152):1390–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L, Roizman B (2006) Herpes simplex virus 1 precludes replenishment of the short-lived receptor of tumor necrosis factor alpha by virion host shutoff-dependent degradation of its mRNA. J Virol 80:7756–7759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley CE, Chaurushiya MS, Boutell C, Landry S, Suh J, Panier S, Everett RD, Stewart GS, Durocher D, Weitzman MD (2010) A viral E3 ligase targets RNF8 and RNF168 to control histone ubiquitination and DNA damage responses. EMBO J 29 (5):943–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Li K, Garofalo RP, Brasier AR (2008) Respiratory syncytial virus induces RelA release from cytoplasmic 100-kDa NF-kappa B2 complexes via a novel retinoic acid-inducible gene-I{middle dot}NF-kappa B-inducing kinase signaling pathway. J Biol Chem 283 (34):23169–23178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund J, Sato A, Akira S, Medzhitov R, Iwasaki A (2003) Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J Exp Med 198:513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma JZ, Russell TA, Spelman T, Carbone FR, Tscharke DC (2014) Lytic gene expression is frequent in HSV-1 latent infection and correlates with the engagement of a cell-intrinsic transcriptional response. PLoS Pathog 10 (7):e1004237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medici MA, Sciortino MT, Perri D, Amici C, Avitabile E, Ciotti M, Balestrieri E, De Smaele E, Franzoso G, Mastino A (2003) Protection by herpes simplex virus glycoprotein D against Fas-mediated apoptosis: role of nuclear factor kappaB. J Biol Chem 278:36059–36067. [DOI] [PubMed] [Google Scholar]
- Melchjorsen J, Rintahaka J, Soby S, Horan KA, Poltajainen A, Ostergaard L, Paludan SR, Matikainen S (2010) Early innate recognition of herpes simplex virus in human primary macrophages is mediated via the MDA5/MAVS-dependent and MDA5/MAVS/RNA polymerase III-independent pathways. J Virol 84 (21):11350–11358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melroe GT, DeLuca NA, Knipe DM (2004) Herpes simplex virus 1 has multiple mechanisms for blocking virus-induced interferon production. J Virol 78 (16):8411–8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melroe GT, Silva L, Schaffer PA, Knipe DM (2007) Recruitment of activated IRF-3 and CBP/p300 to herpes simplex virus ICP0 nuclear foci: Potential role in blocking IFN-beta induction. Virology 360:305–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menasria R, Boivin N, Lebel M, Piret J, Gosselin J, Boivin G (2013) Both TRIF and IPS-1 adaptor proteins contribute to the cerebral innate immune response against herpes simplex virus 1 infection. J Virol 87 (13):7301–7308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkl P, Knipe DM manuscript in preparation.
- Michael BD, Griffiths MJ, Granerod J, Brown D, Keir G, Wnek G, Cox DJ, Vidyasagar R, Borrow R, Parkes LM, Solomon T (2016) The Interleukin-1 Balance During Encephalitis Is Associated With Clinical Severity, Blood-Brain Barrier Permeability, Neuroimaging Changes, and Disease Outcome. J Infect Dis 213 (10):1651–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orvedahl A, Alexander D, Talloczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, Levine B (2007) HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1:23–35. [DOI] [PubMed] [Google Scholar]
- Orzalli MH, Broekema NM, Diner BA, Hancks DC, Elde NC, Cristea IM, Knipe DM (2015) cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc Natl Acad Sci U S A 112 (14):E1773–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orzalli MH, Broekema NM, Knipe DM (2016) Varying Roles of Herpes Simplex Virus 1 ICP0 and Vhs in Loss of Cellular IFI16 in Different Cell Types. J Virol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orzalli MH, Conwell SE, Berrios C, Decaprio JA, Knipe DM (2013) Nuclear interferon-inducible protein 16 promotes silencing of herpesviral and transfected DNA. Proc Natl Acad Sci U S A 110 (47):E4492–4501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orzalli MH, DeLuca NA, Knipe DM (2012) Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc Natl Acad Sci U S A 109 (44):E3008–3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orzalli MH, Knipe DM (2014) Cellular sensing of viral DNA and viral evasion mechanisms. Annu Rev Microbiol 68:477–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orzalli MH, Knipe DM unpublished results.
- Paladino P, Collins SE, Mossman KL (2010) Cellular localization of the herpes simplex virus ICP0 protein dictates its ability to block IRF3-mediated innate immune responses. PLoS ONE 5:e10428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paladino P, Cummings DT, Noyce RS, Mossman KL (2006) The IFN-independent response to virus particle entry provides a first line of antiviral defense that is independent of TLRs and retinoic acid-inducible gene I. J Immunol 177:8008–8016. [DOI] [PubMed] [Google Scholar]
- Paludan SR, Bowie AG, Horan KA, Fitzgerald KA (2011) Recognition of herpesviruses by the innate immune system. Nat Rev Immunol 11 (2):143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaianni E, El Maadidi S, Schejtman A, Neumann S, Maurer U, Marino-Merlo F, Mastino A, Borner C (2015) Phylogenetically Distant Viruses Use the Same BH3-Only Protein Puma to Trigger Bax/Bak-Dependent Apoptosis of Infected Mouse and Human Cells. PLoS One 10 (6):e0126645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker ZM, Murphy AA, Leib DA (2015) Role of the DNA Sensor STING in Protection from Lethal Infection following Corneal and Intracerebral Challenge with Herpes Simplex Virus 1. J Virol 89 (21):11080–11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson J, Lees-Miller SP, Everett RD (1999) Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J Virol 73:650–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasieka TJ, Collins L, O’Connor MA, Chen Y, Parker ZM, Berwin BL, Piwnica-Worms DR, Leib DA (2011) Bioluminescent Imaging Reveals Divergent Viral Pathogenesis in Two Strains of Stat1-Deficient Mice, and in alphassgamma Interferon Receptor-Deficient Mice. PLoS ONE 6 (9):e24018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasieka TJ, Lu B, Crosby SD, Wylie KM, Morrison LA, Alexander DE, Menachery VD, Leib DA (2008) Herpes simplex virus virion host shutoff attenuates establishment of the antiviral state. J Virol 82:5527–5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peri P, Mattila RK, Kantola H, Broberg E, Karttunen HS, Waris M, Vuorinen T, Hukkanen V (2008) Herpes simplex virus type 1 Us3 gene deletion influences toll-like receptor responses in cultured monocytic cells. Virol J 5:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann P, Rahn E, Thier K, Hsu MJ, Rixon FJ, Kopp SJ, Knebel-Morsdorf D (2015) Role of Nectin-1 and Herpesvirus Entry Mediator as Cellular Receptors for Herpes Simplex Virus 1 on Primary Murine Dermal Fibroblasts. J Virol 89 (18):9407–9416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piret J, Boivin G (2015) Innate immune response during herpes simplex virus encephalitis and development of immunomodulatory strategies. Rev Med Virol 25 (5):300–319. [DOI] [PubMed] [Google Scholar]
- Rasmussen SB, Jensen SB, Nielsen C, Quartin E, Kato H, Chen ZJ, Silverman RH, Akira S, Paludan SR (2009) Herpes simplex virus infection is sensed by both Toll-like receptors and retinoic acid-inducible gene-like receptors, which synergize to induce type I interferon production. J Gen Virol 90:74–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SB, Sorensen LN, Malmgaard L, Ank N, Baines JD, Chen ZJ, Paludan SR (2007) Type I interferon production during herpes simplex virus infection is controlled by cell-type-specific viral recognition through Toll-like receptor 9, the mitochondrial antiviral signaling protein pathway, and novel recognition systems. J Virol 81:13315–13324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, Hornung V, Vogel SN, Szomolanyi-Tsuda E, Fitzgerald KA (2010) The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol 11:395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinert LS, Harder L, Holm CK, Iversen MB, Horan KA, Dagnaes-Hansen F, Ulhoi BP, Holm TH, Mogensen TH, Owens T, Nyengaard JR, Thomsen AR, Paludan SR (2012) TLR3 deficiency renders astrocytes permissive to herpes simplex virus infection and facilitates establishment of CNS infection in mice. J Clin Invest 122 (4):1368–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reszka N, Zhou C, Song B, Sodroski JG, Knipe DM (2010) Simian TRIM5alpha proteins reduce replication of herpes simplex virus. Virology 398:243–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice SA, Knipe DM (1990) Genetic evidence for two distinct transactivation functions of the herpes simplex virus alpha protein ICP27. J Virol 64 (4):1704–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts-Thomson PJ, Esiri MM, Young AC, Maclennan IC (1976) Cerebrospinal fluid immunoglobulin quotients, kappa/lambda ratios, and viral antibody titres in neurological disease. J Clin Pathol 29 (12):1105–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roizman B, Knipe DM, Whitley RJ (2013) Herpes Simplex Viruses In: Knipe DM, Howley PM (eds) Fields Virology. 6th edn Lippincott Williams & Wilkins, Philadelphia, pp 1823–1897. [Google Scholar]
- Rosato PC, Leib DA (2015) Neuronal Interferon Signaling Is Required for Protection against Herpes Simplex Virus Replication and Pathogenesis. PLoS Pathog 11 (7):e1005028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer DJ, Carr DJ (2016) A STING-dependent innate-sensing pathway mediates resistance to corneal HSV-1 infection via upregulation of the antiviral effector tetherin. Mucosal Immunol 9 (4):1065–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho-Shimizu V, Perez de Diego R, Jouanguy E, Zhang SY, Casanova JL (2011) Inborn errors of anti-viral interferon immunity in humans. Curr Opin Virol 1 (6):487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato A, Linehan MM, Iwasaki A (2006) Dual recognition of herpes simplex viruses by TLR2 and TLR9 in dendritic cells. Proc Natl Acad Sci U S A 103:17343–17348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider WM, Chevillotte MD, Rice CM (2014) Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol 32:513–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, Mar KB, Richardson RB, Ratushny AV, Litvak V, Dabelic R, Manicassamy B, Aitchison JD, Aderem A, Elliott RM, Garcia-Sastre A, Racaniello V, Snijder EJ, Yokoyama WM, Diamond MS, Virgin HW, Rice CM (2014) Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505 (7485):691–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, Mar KB, Richardson RB, Ratushny AV, Litvak V, Dabelic R, Manicassamy B, Aitchison JD, Aderem A, Elliott RM, Garcia-Sastre A, Racaniello V, Snijder EJ, Yokoyama WM, Diamond MS, Virgin HW, Rice CM (2015) Corrigendum: Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 525 (7567):144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, Rice CM (2011) Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol 1 (6):519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen J, Liu X, Roller R, Knipe DM (2013) Herpes simplex virus US3 tegument protein inhibits Toll-like receptor 2 signaling at or before TRAF6 ubiquitination. Virology 439 (2):65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen G, Wang K, Wang S, Cai M, Li ML, Zheng C (2014) Herpes simplex virus 1 counteracts viperin via its virion host shutoff protein UL41. J Virol 88 (20):12163–12166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slots J (2010) Herpesviral-bacterial interactions in periodontal diseases. Periodontol 52:117–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soberman RJ, MacKay CR, Vaine CA, Ryan GB, Cerny AM, Thompson MR, Nikolic B, Primo V, Christmas P, Sheiffele P, Aronov L, Knipe DM, Kurt-Jones EA (2012) CD200R1 supports HSV-1 viral replication and licenses pro-inflammatory signaling functions of TLR2. PLoS One 7 (10):e47740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song B, Yeh KC, Liu JJ, Knipe DM (2001) Herpes simplex virus gene products required for viral inhibition of expression of G1-phase functions. Virology 290:320–328. [DOI] [PubMed] [Google Scholar]
- Sorensen LN, Reinert LS, Malmgaard L, Bartholdy C, Thomsen AR, Paludan SR (2008) TLR2 and TLR9 synergistically control herpes simplex virus infection in the brain. J Immunol 181:8604–8612. [DOI] [PubMed] [Google Scholar]
- Stock AT, Mueller SN, van Lint AL, Heath WR, Carbone FR (2004) Cutting edge: prolonged antigen presentation after herpes simplex virus-1 skin infection. J Immunol 173 (4):2241–2244. [DOI] [PubMed] [Google Scholar]
- Stratmann SA, Morrone SR, van Oijen AM, Sohn J (2015) The innate immune sensor IFI16 recognizes foreign DNA in the nucleus by scanning along the duplex. Elife 4:e11721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J (2004) The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427 (6977):848–853. [DOI] [PubMed] [Google Scholar]
- Strittmatter GE, Sand J, Sauter M, Seyffert M, Steigerwald R, Fraefel C, Smola S, French LE, Beer HD (2016) IFN-gamma Primes Keratinocytes for HSV-1-Induced Inflammasome Activation. J Invest Dermatol 136 (3):610–620. [DOI] [PubMed] [Google Scholar]
- Stuart PM, Keadle TL (2012) Recurrent herpetic stromal keratitis in mice: a model for studying human HSK. Clin Dev Immunol 2012:728480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su C, Zhan G, Zheng C (2016) Evasion of host antiviral innate immunity by HSV-1, an update. Virol J 13:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, Chen ZJ (2013) Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339 (6121):786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JM, Lin E, Susmarski N, Yoon M, Zago A, Ware CF, Pfeffer K, Miyoshi J, Takai Y, Spear PG (2007) Alternative entry receptors for herpes simplex virus and their roles in disease. Cell Host Microbe 2:19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor KE, Chew MV, Ashkar AA, Mossman KL (2014) Novel roles of cytoplasmic ICP0: proteasome-independent functions of the RING finger are required to block interferon-stimulated gene production but not to promote viral replication. J Virol 88 (14):8091–8101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson MR, Kaminski JJ, Kurt-Jones EA, Fitzgerald KA (2011) Pattern recognition receptors and the innate immune response to viral infection. Viruses 3 (6):920–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson MR, Sharma S, Atianand M, Jensen SB, Carpenter S, Knipe DM, Fitzgerald KA, Kurt-Jones EA (2014) Interferon gamma-inducible protein (IFI) 16 transcriptionally regulates type i interferons and other interferon-stimulated genes and controls the interferon response to both DNA and RNA viruses. J Biol Chem 289 (34):23568–23581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapani JA, Browne KA, Dawson MJ, Ramsay RG, Eddy RL, Show TB, White PC, Dupont B (1992) A novel gene constitutively expressed in human lymphoid cells is inducible with interferon-gamma in myeloid cells. Immunogenetics 36:369–376. [DOI] [PubMed] [Google Scholar]
- Tsalenchuck Y, Steiner I, Panet A (2016) Innate defense mechanisms against HSV-1 infection in the target tissues, skin and brain. J Neurovirol. [DOI] [PubMed] [Google Scholar]
- Unterholzner L (2013) The interferon response to intracellular DNA: Why so many receptors? Immunobiology 218 (11):1312–1321. [DOI] [PubMed] [Google Scholar]
- Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, Fitzgerald KA, Paludan SR, Bowie AG (2010) IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol 11 (11):997–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Lint AL, Ayers M, Brooks AG, Coles RM, Heath WR, Carbone FR (2004) Herpes simplex virus-specific CD8+ T cells can clear established lytic infections from skin and nerves and can partially limit the early spread of virus after cutaneous inoculation. J Immunol 172 (1):392–397. [DOI] [PubMed] [Google Scholar]
- van Lint AL, Murawski MR, Goodbody RE, Severa M, Fitzgerald KA, Finberg RW, Knipe DM, Kurt-Jones EA (2010) Herpes simplex virus immediate-early ICP0 protein inhibits Toll-like receptor 2-dependent inflammatory responses and NF-kappaB signaling. J Virol 84:10802–10811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Vliet KE, De Graaf-Miltenburg LA, Verhoef J, van Strijp JA (1992) Direct evidence for antibody bipolar bridging on herpes simplex virus-infected cells. Immunology 77:109–115. [PMC free article] [PubMed] [Google Scholar]
- Veeranki S, Choubey D (2012) Interferon-inducible p200-family protein IFI16, an innate immune sensor for cytosolic and nuclear double-stranded DNA: regulation of subcellular localization. Mol Immunol 49 (4):567–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verpooten D, Ma Y, Hou S, Yan Z, He B (2009) Control of TANK-binding kinase 1-mediated signaling by the gamma(1)34.5 protein of herpes simplex virus 1. J Biol Chem 284 (2):1097–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilkuna-Rautiainen T, Pussinen PJ, Roivainen M, Petays T, Jousilahti P, Hovi T, Vartiainen E, Asikainen S (2006) Serum antibody response to periodontal pathogens and herpes simplex virus in relation to classic risk factors of cardiovascular disease. Int J Epidemiol 35:1486–1494. [DOI] [PubMed] [Google Scholar]
- Wald A, Link K (2002) Risk of human immunodeficiency virus infection in herpes simplex virus type 2-seropositive persons: A meta-analysis. J Infect Dis 185:45–52. [DOI] [PubMed] [Google Scholar]
- Wallach D, Kang TB, Dillon CP, Green DR (2016) Programmed necrosis in inflammation: Toward identification of the effector molecules. Science 352 (6281):aaf2154. [DOI] [PubMed] [Google Scholar]
- Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, Wang X (2014a) Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 54 (1):133–146. [DOI] [PubMed] [Google Scholar]
- Wang JP, Bowen GN, Zhou S, Cerny A, Zacharia A, Knipe DM, Finberg RW, Kurt-Jones EA (2012) Role of specific innate immune responses in herpes simplex virus infection of the central nervous system. J Virol 86 (4):2273–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li Y, Liu S, Yu X, Li L, Shi C, He W, Li J, Xu L, Hu Z, Yu L, Yang Z, Chen Q, Ge L, Zhang Z, Zhou B, Jiang X, Chen S, He S (2014b) Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc Natl Acad Sci U S A 111 (43):15438–15443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Majumdar T, Kessler P, Ozhegov E, Zhang Y, Chattopadhyay S, Barik S, Sen GC (2016) STING Requires the Adaptor TRIF to Trigger Innate Immune Responses to Microbial Infection. Cell Host Microbe 20 (3):329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware CF (2009) Targeting the LIGHT-HVEM pathway. Adv Exp Med Biol 647:146–155. [DOI] [PubMed] [Google Scholar]
- Webre JM, Hill JM, Nolan NM, Clement C, McFerrin HE, Bhattacharjee PS, Hsia V, Neumann DM, Foster TP, Lukiw WJ, Thompson HW (2012) Rabbit and mouse models of HSV-1 latency, reactivation, and recurrent eye diseases. J Biomed Biotechnol 2012:612316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, Kaech SM, Smiley JR, Means RE, Iwasaki A, Shadel GS (2015) Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520 (7548):553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao TS, Fitzgerald KA (2013) The cGAS-STING pathway for DNA sensing. Mol Cell 51 (2):135–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing J, Ni L, Wang S, Wang K, Lin R, Zheng C (2013) Herpes simplex virus 1-encoded tegument protein VP16 abrogates the production of beta interferon (IFN) by inhibiting NF-kappaB activation and blocking IFN regulatory factor 3 to recruit its coactivator CBP. J Virol 87 (17):9788–9801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W, Candolfi M, Liu C, Muhammad AK, Yagiz K, Puntel M, Moore PF, Avalos J, Young JD, Khan D, Donelson R, Pluhar GE, Ohlfest JR, Wawrowsky K, Lowenstein PR, Castro MG (2010) Human Flt3L generates dendritic cells from canine peripheral blood precursors: implications for a dog glioma clinical trial. PLoS ONE 5 (6):e11074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Mallon S, Roizman B (2016) PML plays both inimical and beneficial roles in HSV-1 replication. Proc Natl Acad Sci U S A 113 (21):E3022–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Liu X, Knipe DM (manuscript in preparation) Herpesvirus UL37 tegument protein activates NF-κB signaling through TRAF6 ubiquitination. [Google Scholar]
- Yordy B, Iijima N, Huttner A, Leib D, Iwasaki A (2012) A neuron-specific role for autophagy in antiviral defense against herpes simplex virus. Cell Host Microbe 12 (3):334–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenner HL, Mauricio R, Banting G, Crump CM (2013) Herpes simplex virus 1 counteracts tetherin restriction via its virion host shutoff activity. J Virol 87 (24):13115–13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Wang K, Wang S, Zheng C (2013) Herpes simplex virus 1 E3 ubiquitin ligase ICP0 protein inhibits tumor necrosis factor alpha-induced NF-kappaB activation by interacting with p65/RelA and p50/NF-kappaB1. J Virol 87 (23):12935–12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SY, Casanova JL (2015) Inborn errors underlying herpes simplex encephalitis: From TLR3 to IRF3. J Exp Med 212 (9):1342–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]