


Abstract
Adjusting DNA structure via epigenetic modifications, and altering polyadenylation (pA) sites at which precursor mRNA is cleaved and polyadenylated, allows cells to quickly respond to environmental stress. Since polyadenylation occurs co-transcriptionally, and specific patterns of nucleosome positioning and chromatin modifications correlate with pA site usage, epigenetic factors potentially affect alternative polyadenylation (APA). We report that the histone H3K4 methyltransferase Set1, and the histone H3K36 methyltransferase Set2, control choice of pA site in Saccharomyces cerevisiae, a powerful model for studying evolutionarily conserved eukaryotic processes. Deletion of SET1 or SET2 causes an increase in serine-2 phosphorylation within the C-terminal domain of RNA polymerase II (RNAP II) and in the recruitment of the cleavage/polyadenylation complex, both of which could cause the observed switch in pA site usage. Chemical inhibition of TOR signaling, which causes nutritional stress, results in Set1- and Set2-dependent APA. In addition, Set1 and Set2 decrease efficiency of using single pA sites, and control nucleosome occupancy around pA sites. Overall, our study suggests that the methyltransferases Set1 and Set2 regulate APA induced by nutritional stress, affect the RNAP II C-terminal domain phosphorylation at Ser2, and control recruitment of the 3′ end processing machinery to the vicinity of pA sites.
INTRODUCTION
The basic structural unit of chromatin is the nucleosome, consisting of a histone octamer, around which 147 base pairs of DNA are coiled (1,2). Epigenetic modifications provide a higher level of chromatin structure by organizing it into either transcriptionally active euchromatin or inactive heterochromatin. The epigenetic machinery remodels nucleosomes and performs histone posttranslational modifications, which in turn control access of transcription regulatory proteins to DNA, and dictate the initiation and elongation rate of RNA Polymerase II (RNAP II) (3). Epigenetic factors include DNA methyltransferases, histone demethylases, methyltransferases (HMTs), deacetylases (HDACs), acetyltransferases (HATs), dephosphorylases, kinases, deubiquitinases, ubiquitinases and nucleosome remodelers which control nucleosome positioning (3,4). The cell easily modulates its gene expression by dynamic and reversible modifications of chromatin (5–7).
In addition to chromatin remodeling, the cell tunes its transcriptional regulation by switching polyadenylation (pA) sites (8–10). Most human genes have multiple pA sites located not only in their 3′ UTRs, which contain cis-regulatory elements, but also within introns and coding sequences. Choice of pA site determines the location at which the pre-mRNA is cleaved and polyadenylated leading to the production of different mRNA isoforms in a process called alternative polyadenylation (APA). Controlling 3′ UTR length may promote or repress mRNA degradation, nuclear export, and translation. Thus, APA leads to production of proteins with different expression levels, structure, function and subcellular localization so that the cell properly responds to different environmental stimuli (11–13). Recent advances have shown that APA and dysregulation of the epigenetic landscape are hallmarks of cancer (4,11,14–18), aging (19–24), DNA damage (8,25,26), differentiation (27–32), metabolism (33–35), heart failure (36,37), pulmonary fibrosis (38,39), neurodegenerative diseases (40,41) and immune responses (28,32,42–46). Due to the lack of knowledge about APA regulation, reversing such dysregulation remains a challenge.
Because polyadenylation occurs co-transcriptionally, epigenetic factors are likely to affect APA. Indeed, slowing the transcriptional elongation rate of RNAP II favors utilization of upstream pA sites (47). Furthermore, specific patterns of nucleosome positioning and chromatin modifications correlate with APA, and are more accurate in predicting functional pA sites than cis elements (48). More specifically, nucleosomes are depleted in the immediate vicinity of pA sites, and preferred pA sites show greater nucleosome occupancy upstream and downstream of the sites, along with RNAP II accumulation upstream of the pA site (48). Moreover, the presence of downstream nucleosomes correlates with the transcriptional termination sites of non-coding RNAs and cryptic transcripts (49). Histone H3K4 (48) and H3K36 (48,50) methylation is strongly associated with pA sites in humans. Methylation of both histone H3K4 and H3K36 has been shown to regulate alternative splicing (51), but it remains unknown whether they regulate APA.
To determine whether histone H3K4 and H3K36 HMTs control APA, we took advantage of Saccharomyces cerevisiae, a powerful model for studying evolutionarily conserved eukaryotic processes. In S. cerevisiae, Set1 co-transcriptionally methylates histone H3 on lysine 4 (52,53). Histone H3K4me3 is enriched at the 5′ end of genes, histone H3K4me2 in the middle, while histone H3K4me1 is prominent toward the 3′ end of a gene (54,55). Set1 is the HMT in the COMPASS complex, which also includes Bre2, Sdc1, Shg1, Spp1, Swd1, Swd2 and Swd3 (53,56,57). Set1-mediated methylation of histone H3K4 is regulated by the PAF complex (58). Although methylation of histone H3K4 is considered a mark of open chromatin (59–61), it can also repress transcription of rDNA (62), silent mating-type loci (63) and telomeres (63–65), as well as prevent cryptic transcription (63,66–68). Loss of Set1 in S. cerevisiae up-regulates the expression of stress responsive genes in a Rad53- and histone H3K4me1-dependent manner (69). Set1 recruits the early termination factor Nrd1, and cells not expressing Set1 and Nrd1 are severely defective for termination of snoRNAs and cryptic unstable transcripts (CUTs) (68). The histone H3K4 demethylase Jhd2 controls choice of pA site by recruiting the 3′ end processing machinery (70) However, the effects of Set1 on the choice of pA site have not been determined. Like nucleosomes, histone H3K4me1 is depleted around pA sites (48). Mammalian genes utilizing upstream pA sites have high levels of histone H3K4me3 at the upstream site, and histone H3K4me3 levels drop down downstream of the pA site (71).
Histone H3K36 methylation in S. cerevisiae is catalyzed solely by Set2 (72), and occurs co-transcriptionally (73–76). Like histone H3K4me1, histone H3K36me3 is enriched toward the 3′ end of genes (54,55,77,78). Histone H3K36 methylation in S. cerevisiae is regulated by several factors, such as Spt6 (79), the PAF complex (73,80), and the RNAP II C-terminal domain (CTD) Ser2 kinases Ctk1 (73,76,79,81), and Bur1 (80,82,83). Set2 binds RNAP II phosphorylated at Ser2 and Ser5 of its CTD (84). Like methylation of histone H3K4, methylation of histone H3K36 is considered a mark of open chromatin (85), but it can also repress transcription of certain genes (86), and prevent cryptic (87–89) and intragenic transcription (90) mainly via its interaction with the HDAC Rpd3S (88,89,91–93). The repression of intragenic transcription by the histone H3K36 HMT is also conserved in humans (94). Methylation of histone H3K36 and repression of cryptic transcription requires interaction of Set2 with residues of histones H4, H2A and H3 (95,96). Histone H3K36 demethylases increase RNAP II processivity (97), and cells not expressing Set2 or cells with a mutated histone H3K36 residue have elevated levels of RNAP II at the 3′ ends of genes. Preferred pA sites in human genes have high levels of histone H3K36me3 (71), and histone H3K36me3 levels are significantly higher at pA sites in genes with multiple pA sites compared to genes with a single pA site, suggesting a role for histone H3K36me3 in APA regulation (50). Interestingly, levels of histone H3K36me3 drop gradually downstream of pA sites, and the persistence of this mark may lead to increased pausing of RNAP II, which would give more time for processing at an upstream pA site (98,99).
Studies on mechanisms leading to APA have focused on regulation by changing levels of cleavage/polyadenylation proteins or factors that suppress or enhance recruitment of the cleavage/polyadenylation complex to specific pA sites (100–102). Chromatin structure has been recently shown to control alternative promoter choice and alternative splicing (103,104). Previous studies on APA have only demonstrated a correlation between chromatin modifications and pA site choice, but functional validation was not performed (48–50). To test the hypothesis that epigenetic factors affect alternative pA site usage, we looked at utilization of pA sites in S. cerevisiae cells not expressing the Set1 or Set2 HMTs. We demonstrate that deletion of SET1 or SET2 leads to changes in pA site choice, and negatively affects APA in response to nutritional stress. Furthermore, using a chromatin immunoprecipitation (ChIP) assay, we show that in set1Δ and set2Δ cells, the amount of 3′ end processing complex at pA sites preferred in the mutants is increased. In addition, set1Δ cells had increased RNAP II CTD Ser2 phosphorylation (Ser2-P) at pA sites, which enhances RNAP II’s association with polyadenylation factors (105). Deletion of SET1 or SET2 also increased the 3′ end processing efficiency at genes containing single pA sites.
MATERIALS AND METHODS
Yeast strains and culture
The yeast mutants were a generous gift from Dr. Hungjiun Liaw (106) and are listed in Table 1. Cells were grown in YPD medium (1% yeast extract, 2% peptone, and 2% glucose) containing 0.004% DMSO with agitation (220 rpm) at 30°C. To induce nutritional stress, cells were grown in the presence or absence of 10 nM rapamycin (AdipoGen, from a 0.25 mM stock dissolved in DMSO) for 2 h in the dark with agitation (220 rpm) at 30°C. For spot assays, yeast cells were cultured in 5 ml of YPD media for 16 h and then diluted to OD600 = 0.5, which is about 5 × 106 cells/ml. Ten-fold serial dilutions were spotted on YPD agar plates containing 2 nM rapamycin or DMSO as a solvent control.
Table 1.
Yeast strains used in this study
| Strains | Genotype and carried plasmids |
|---|---|
| W303α | MATα leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15 |
| set1Δ | MATα leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15 set1::KANMX6 |
| set2Δ | MATα leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15 set2::KANMX6 |
| WT H3 | MATα leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15 hht1hhf1::KAN, hht2hhf2::KAN, pRS415-HHF1-HHT1 |
| H3K4R | MATα leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15 hht1hhf1::KAN, hht2hhf2::KAN, pRS415-HHF1-H3K4R |
| H3K36R | MATα leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15 hht1hhf1::KAN, hht2hhf2::KAN, pRS415-HHF1-H3K36R |
Immunoblot analysis
Whole-cell lysates were prepared from exponentially growing cultures, using a modified version of the TCA method (107). Briefly, 10 ml of cells were collected by centrifugation and frozen at −80°C. The cell pellet was resuspended in 0.25 ml of cold 20% TCA, and transferred to a 1.5 ml microfuge tube. The cells were broken by vortexing at the highest speed for 3 min at 4°C with acid-washed glass beads. The cell lysate was transferred to a new microfuge tube, avoiding the glass beads. Two 0.5 ml volumes of cold 5% TCA were used to wash beads, and combined with the lysate. The lysate was mixed and the precipitated protein was collected by centrifugation at 14 000 rpm for 10 min at 4°C. The pellet was washed with cold 100% ethanol. Protein was re-suspended in 40 μl of 1M TrisCl (pH 8.0), and 80 μl of 2× SDS loading buffer (60 mM Tris (pH 6.8), 2% SDS, 10% glycerol, 0.2% bromopheonol blue, 100 mM DTT). Samples were heated for 5 min at 95°C before loading onto a 10% SDS-PAGE gel. Proteins were transferred to a polyvinylidene difluoride membrane by electroblotting. Antibodies used for immunoblotting are listed in Table 2.
Table 2.
Antibodies used in this study
| Specificity | Supplier | Catalog # |
|---|---|---|
| Anti-Histone H3 | Abcam | Ab1791 |
| Anti-Histone H4 | Abcam | Ab10156 |
| Anti-H3K4me1 | Abcam | Ab8895 |
| Anti-H3K36me3 | Abcam | Ab9050 |
| RNAP II CTD Ser2-P | ChromoTek | 3E10 |
| RNAP II pan-CTD (4H8) | Santa Cruz | sc-47701 |
| Rna15 | Dr H. Domdey | |
| Pta1 | Dr H. Domdey | |
| α-tubulin (YOL1/34) | Invitrogen | MA1-80189 |
qRT-PCR analysis
Total RNA from exponentially growing wild-type or mutant cells was isolated using the Hot Phenol Method (108) and Heavy Phase Lock Gel tubes (Quantabio), and treated with RQ1 DNase (Promega). DNA-free RNA was subjected to reverse transcription using SuperScript III (Invitrogen), and either anchored oligo(dT)20 primer for the 3′ end analysis, or with random hexamers for pA site read-through determination. The resulting cDNA samples were analyzed using the real-time PCR analysis performed in a 12 μl reaction with 417 nM of 10 μM forward and reverse primers, 5 μl of SYBR Green Supermix (BIO-RAD), 1 μl cDNA and 5 μl of distilled water. The primer sequences are listed in Table 3. The expression of the long mRNA isoform of a given gene was normalized to the expression of total mRNA of that gene, and normalized to wild-type.
Table 3.
Primers used in this study
| Primer name | Primer sequence 5′-3′ |
|---|---|
| Anchored oligo d(T) primer | TTTTTTTTTTTTTTTTTTTTVN |
| Random hexamer | NNNNNN |
| ISM1 Total Forward | AGCAAGCGATATCTCGCCAA |
| ISM1 Total Reverse | GTCCATGACAATCCCAGCCA |
| ISM1 pA4 Forward | CACCAAGCATCACCTCCCAT |
| ISM1 pA4 Reverse | ATCCTCTTCGGCTGAGTTGG |
| FAT1 Total/pA1 Forward | TCACGGTGGTTGCCTTGCGT |
| FAT1 Total/pA1 Reverse | TGGATGTGCGTGGCTCCTGT |
| FAT1 pA2 Forward | CAAAGGGTTTGGATGGAAATGACAC |
| FAT1 pA2 Reverse | TCCCAATCAGCAGCGGTCAAG |
| MDV1 Total/pA1 Forward | TCACACAGAGCTTCCTAACTTCCA |
| MDV1 Total/pA1 Reverse | ACCCCAGGCGGTATGAGAAATGA |
| MDV1 pA2 Forward | TGAGGGTCGTGAAAATGGGGAC |
| MDV1 pA2 Reverse | TCTTCAAATGGGTTGACTTGATTGC |
| RPB2 Total Forward | GCCTGTAGAGGGTAGATCGAG |
| RPB2 Total Reverse | TCAGCCCGCAAATACCACAA |
| RPB2 pA2 Forward | TCATTTGTGCTGATCTTGCCA |
| RPB2 pA2 Reverse | TGCTTGAAAGTTCTCTCTGCT |
| RRD2 Total Forward | GGGAAGAATCCCCAACAAGAGC |
| RRD2 Total Reverse | ACTGCTCATCTGTGAGAGAGGG |
| RRD2 pA2 Forward | TCTCCACCAAGAGGCCACATAC |
| RRD2 pA2 Reverse | AGTAGCCGCAATAGCGCTCG |
| RAD53 Total/pA1 Forward | ACCAAACCTCAAAAGGCCCCGA |
| RAD53 Total/pA1 Reverse | AGGGGCAGCATTTTCTATGGGT |
| RAD53 Long Isoform/Between pAs Forward | AACCCGTCTTATGCCTTCCGGG |
| RAD53 Long Isoform/Between pAs Reverse | GCCGCCTCCGCCCCTTAATC |
| PDC1 Forward | GCCAGTCTTCGATGCTCCAC |
| PDC1 Total Reverse | ATCGCTTATTGCTTAGCGTTGG |
| PDC1 pA Span Reverse | ACTGTCGGCAACTTCTTGTCTGG |
| RPP1B Total Forward | ACGCTAAGGCTTTGGAAGGTAAGGA |
| RPP1B Total Reverse | AACCGAAACCCATGTCGTCGTCAGA |
| RPP1B pA Span Forward | GACGACGACATGGGTTTCGGT |
| RPP1B pA Span Reverse | TCGTAGCCCTTTCGTATGGACA |
| RAD53 Promoter Forward | AGGTAAGAAAGCAGAAAAGGACGG |
| RAD53 Promoter Reverse | GCGTGGATTGCTGTGTGGGT |
| RAD53 CDS Forward | TCCTAACGGGCCACTTACCTTT |
| RAD53 CDS Reverse | GGGCCCTTCATGATATGAGCCTCT |
| RAD53 End of CDS Forward | GTCGGCTAAGAAGCCGCCAG |
| RAD53 End of CDS Reverse | CGGGGCCTTTTGAGGTTTGGTC |
| RAD53 pA2 Forward | AGAAGTTTGGGTAATTCGCTGCT |
| RAD53 pA2 Reverse | TCTTCCCTTACGTGGTAGGC |
| GRS2 Total Forward | ATAACGATGGCTTCCCCGCT |
| GRS2 Total Reverse | ACGTAAAGCCTGCGAGATCC |
| GRS2 pA2 Forward | ACAACCCTGATGAATCGGACTGGG |
| GRS2 pA2 Reverse | ACAGGCGACAGTCCAAATGTTGAT |
| RTG2 Total Forward | AGGGTGGTGTTCGAGAGGGTTC |
| RTG2 Total Reverse | AATGGAGCATAAGGACGGGACGC |
| RTG2 pA2 Forward | AGTGCTTCCGTTCGTTCCAGA |
| RTG2 pA2 Reverse | TGCACGCCAATTTTAACCCTCTCT |
| 18S Forward | GATGCCCTTAGACGTTCTGG |
| 18S Reverse | GGCCTCACTAAGCCATTCAA |
Chromatin immunoprecipitation
ChIPs were performed as described previously (109). Quantitative real-time PCR analysis was carried out using SYBR Green reagents (BioRad) with the primers listed in Table 3.
Statistical analysis
Statistical analysis was carried out using a Student's t-test. A two-tailed distribution was performed using a two sample equal variance test. *P < 0.05, **P < 0.01, ***P < 0.001. A P value < 0.05 was considered significant.
RESULTS
Set1 and Set2 influence usage of pA sites
While histone H3K4 and histone H3K36 methylation correlate with the usage of pA sites (48), it has not been determined whether they influence APA. To assess the role of the histone H3K4 HMT Set1 and the histone H3K36 HMT Set2 in the choice of pA site, we looked at polyadenylation of eight different yeast genes with two or more pA sites. These genes have pA sites located within their open reading frames (ISM1, FAT1, MDV1, RRD2, RTG2, GRS2), or in their 3′ UTRs (RAD53 and RPB2) (Figure 1A–F, Supplementary Figure S1A and B). Switch in pA site usage was measured using total RNA reversely transcribed with oligo d(T) primer to select for polyadenylated RNA. Primers specific for long and total mRNA isoforms were used for qRT-PCR. Long mRNA isoforms were normalized to total mRNA isoforms for a given gene. set1Δ and set2Δ cells show a strong decrease in use of the ISM1, FAT1, MDV1 and RPB2 downstream pA sites (Figure 1A–D), as measured by the 3′ end amplification assay. Set1 and Set2 had minimal effect on choice of pA site in GRS2 and RTG2 (Supplementary Figure S1A and B). The expression of total ISM1, FAT1 and MDV1 mRNA was measured with primer sets amplifying regions close to the upstream pA sites and normalized to 18S was increased in set1Δ and set2Δ cells (Supplementary Figure S2A–C), while that of RPB2 decreased. In contrast to the other sites that we examined, set1Δ and set2Δ cells show a strong decrease in use of the RRD2 and RAD53 upstream pA sites (Figure 1E and F). Total RAD53 mRNA normalized to 18S was decreased in set1Δ and set2Δ cells, while the total RRD2 mRNA normalized to 18S remained the same (Supplementary Figure S2D–F).
Figure 1.

Set1 and Set2 HMTs determine choice of pA site. (A–F) Wild-type and set1Δ, set2Δ, histone H3K4R and histone H3K36R mutants were cultured in YPD media and RNA harvested during exponential growth, followed by reverse transcription using anchored oligo d(T) primers. Total and long gene isoforms of the ISM1, FAT1, MDV1, RPB2, RRD2 and RAD53 genes were amplified for the 3′ end analysis via qRT-PCR using the primer pairs indicated above the bar graphs. The ratios of long to total mRNAs in the mutant strains were normalized relative to the ratio in the wild-type W303 strain. Stars depict pA sites. Three biological replicates were performed for each gene. Bars show average values ± SD. **P < 0.01, ***P < 0.001 (Student's t-test). pA site positions were determined by Graber et al. (8).
Histone H3K4R and H3K36R mutants, which cannot be methylated by Set1 or Set2, demonstrated similar switches in pA site usage, although to different degrees than set1Δ and set2Δ cells (Figure 1A–F). Together, these data demonstrate that the absence of Set1 or Set2, as well as mutations in the histone residues that are their targets, change pA site choice, and most often increase use of upstream sites.
A recent paper has reported that the deletion of SET1 or SET2 can lead to utilization of internal cryptic promoters (110). Depending on their position relative to our primer sets used to detect total and long mRNA isoforms, activation of these promoters could affect our analysis. Examination of the localization of the internal cryptic promoters reported by Wei et al. (110) revealed that there were internal promoters in the ISM1, MDV1, FAT1 and RAD53 genes that were significantly increased in set2Δ cells, but not in set1Δ cells (Supplementary Figure S3A–H). However, activation of the internal cryptic promoters in the ISM1, MDV1, and FAT1 genes would cause the ratio of long to total mRNA isoforms to increase, but we observe a decrease. In summary, other mechanisms, as described below, are likely to be responsible for the APA changes.
Loss of set1 or Set2 increases processing efficiency at single pA sites
Changes in pA site usage as indicated by the analysis described above can occur if the efficiency of cleavage/polyadenylation is altered. We tested whether Set1 and Set2 alter the efficiency of 3′ end processing in vivo by looking at the level of transcripts that contain sequence upstream and downstream of a pA site. These transcripts represent RNA that has not been processed, and can be detected by RT-qPCR with a primer pair that spans the pA site. We examined the RPP1B and PDC1 genes which have single pA sites. In addition, the genes downstream of RPP1B and PDC1 have the same transcriptional orientation, and are located 496 and 376 bp downstream from the coding sequence of RPP1B and PDC1, respectively. This gene organization minimizes the possibility of transcriptional interference influencing pA site usage. Absence of Set1 or Set2 decreased the amount of unprocessed RNA, which implies that these chromatin modifiers inhibit processing at the RPP1B and PDC1 pA sites (Figure 2A and B). Total RPP1B and PDC1 mRNA normalized to 18S was decreased in set1Δ and set2Δ cells, which minimizes the possibility that the decrease in transcripts that span the pA site is due to post-transcriptional stabilization of mature mRNA in these cells (Supplementary Figure S2D–F). Moreover, we were recently able to show a decrease in processing efficiency at single pA sites of RPP1B and PDC1 in the processing-defective ipa1–1 mutant (111), which further verifies that using these two pA sites we can measure both, an increase and a decrease, in the 3′ end processing. These data indicate that Set1 and Set2 negatively affect the efficiency of cleavage/polyadenylation at these single pA sites.
Figure 2.
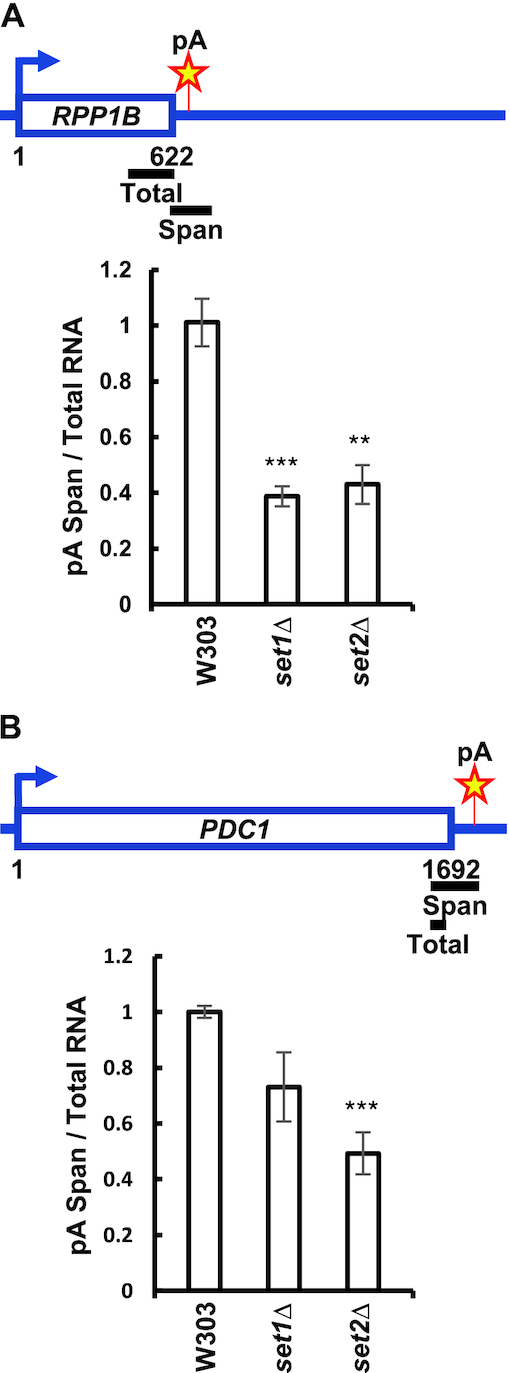
SET1 and SET2 deletion enhances utilization of single pA sites. (A, B) Schematic representation of primer pairs used for qRT-PCR analysis of transcripts reading through the PDC1 and RPP1B pA sites. Total RNA was reversely transcribed using random hexamers. qRT-PCR analysis of RNA was conducted using the primer pairs indicated above the bar graphs to determine the amount of total transcripts and that of transcripts spanning PDC1 and RPP1B pA sites, which represent unprocessed transcripts. The ratios of unprocessed to total transcripts in the mutant strains were normalized relative to the ratio in the wild-type W303 strain. Three biological replicates were performed for each gene. Bars show average values ± SD. **P < 0.01, ***P < 0.001 (Student's t-test).
Set1 and Set2 control nucleosome occupancy around pA sites
Nucleosome positioning and histone marks are both ways in which the cell alters its chromatin structure. However, only a few studies have addressed the interactions between these dynamic processes. Histone methylation patterns affect nucleosome occupancy (112). For example, cells not expressing Set1 have lower nucleosome occupancy at the PHO5 promoter (113,114). Likewise, Set2 has been shown to suppress histone exchange over transcribed regions and to suppress histone interactions with histone chaperones (115).
We examined histone occupancy in set1Δ and set2Δ cells using antibodies against histone H3 and H4. We focused our analysis on genes with single pA sites, or with well-spaced alternative pA sites to confidently measure changes associated with each pA site via ChIP assays. For this reason, we looked at the pA sites of FAT1, MDV1 and RAD53, which have a spacing of at least 500 bp between pA sites. Cells not expressing Set1 have a significant decrease in histone H3 and H4 occupancy around the FAT1, MDV1, PDC1, RPP1B and RAD53 pA sites compared to wild-type cells (Figure 3A–F). Cells not expressing Set2 have decreased histone H3 occupancy around the FAT1, MDV1 and RAD53 pA sites (Figure 3A, B and E), a moderate decrease at the RPP1B pA site, and no change at the PDC1 site (Figure 3C and D). The set2Δ cells also have a strong decrease in histone H4 in the vicinity of the FAT1, PDC1, RPP1B and RAD53 pA sites and moderate decrease at the MDV1 pA site (Figure 3A–D, F). Overall, the strongest decrease in nucleosome occupancy is seen in set1Δ cells (Figure 3A–F).
Figure 3.
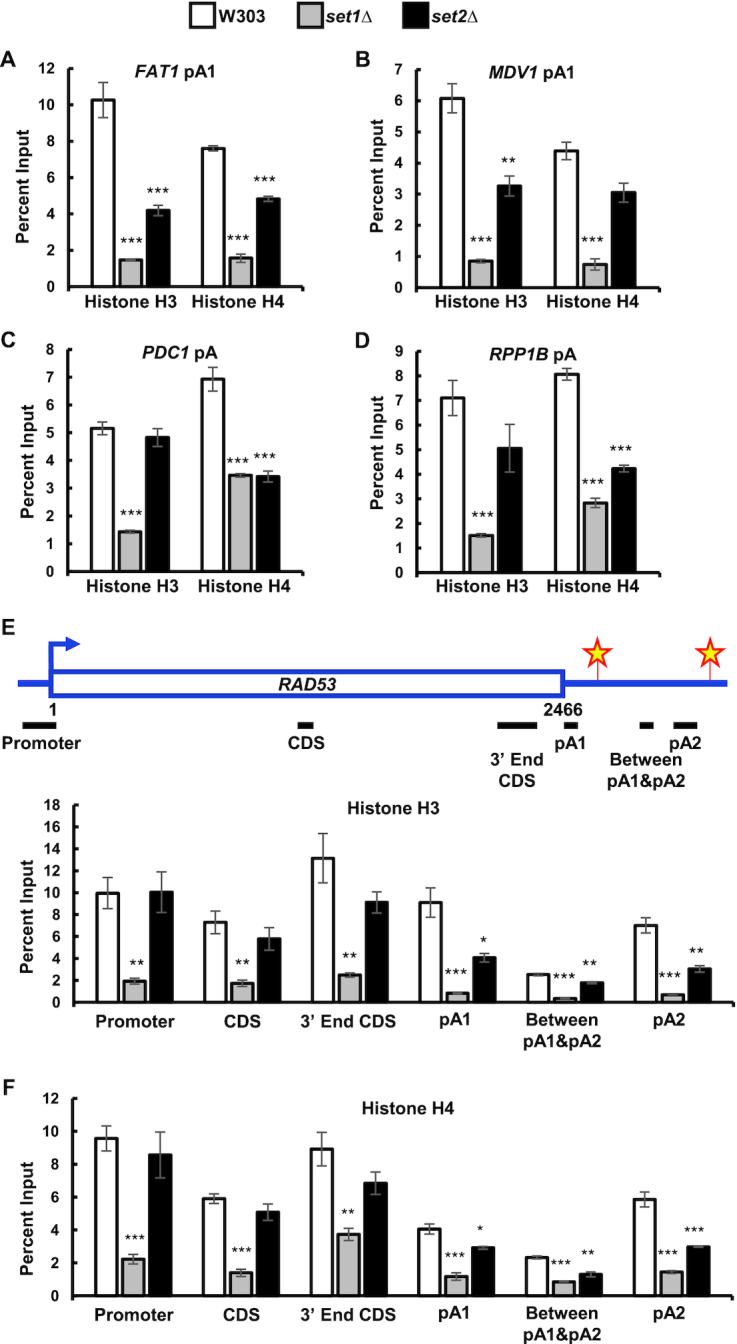
SET1 and SET2 deletion decreases nucleosome occupancy around pA sites. (A–D) ChIPs for histone H3 and histone H4 around FAT1, MDV1, PDC1 and RPP1B pA sites in wild-type (W303), set1Δ and set2Δ cells. (E) ChIPs for histone H3, and H4 (F) along the RAD53 gene in wild-type, set1Δ and set2Δ cells. Primer pairs indicated above the bar graphs were specific for the RAD53 promoter, coding sequence (CDS), 3′ end of coding sequence, pA1 site, region between pA1 and pA2 sites, as well as pA2 site. Two biological replicates were performed for each gene. Bars show average values ± SD. *P < 0.05, **P < 0.01, ***P < 0.001 (Student's t-test).
To see if the loss of histone H3 and H4 in set1Δ and set2Δ cells is specific to regions near pA sites, we looked at histone H3 and H4 occupancy along the RAD53 gene. In set1Δ cells, histone H3 and H4 levels significantly decreased across the whole gene (Figure 3E and F). SET2 deletion resulted in a significant loss of histone H3 and H4 occupancy only in the RAD53 3′ UTR (Figure 3E and F). The strong decrease in nucleosome occupancy seen in set1Δ cells was accompanied by a small decrease in total histone H3 and H4 levels as shown by western blot (Figure 4A). The set2Δ cells had unchanged total histone H3 levels, but the histone H4 levels were increased (Figure 4A). These data demonstrate that Set1 and Set2 regulate histone occupancy along genes, especially around pA sites.
Figure 4.

Set1 and Set2 modulate each other. (A) Protein levels of Set1, Set2, histone H3, histone H4, H3K4me1, H3K36me3, RNAP II, RNAP II Ser2-P, Rna15 and Pta1 in wild-type, set1Δ and set2Δ backgrounds. (B) Protein levels of histone H3K4me1 and H3K36me3 in wild-type histone H3, histone H3K4R and H3K36R backgrounds. For panel A and B, whole cell extracts from exponentially growing cells in YPD media in the presence of 10 nM rapamycin (for 2 hours) or DMSO were resolved by SDS-PAGE and analyzed by Western blot. α-tubulin was used as a protein loading control. (C–F) ChIP of histone H3K4me1 and H3K36me3 to FAT1, MDV1 and RAD53 pA sites in wild-type, set1Δ and set2Δ cells. (G–J) Same data as in (C–F) normalized to histone H3 ChIP from Figure 3. Two biological replicates were performed for each gene. Bars show average values ± SD. **P < 0.01, ***P < 0.001 (Student's t-test).
Set1 and Set2 modulate each other
To determine whether Set1 and Set2 influence each other, we looked at histone H3K4me1 in set2Δ in cells, and histone H3K36me3 in set1Δ cells. Interestingly, the absence of Set1 caused a strong decrease in total histone H3K36me3 levels and presence near pA sites, as measured by western blot and ChIP assay, respectively (Figure 4A, C–F). However, once normalized to total histone H3 levels, Δset1 cells had increased efficiency of histone H3K36 trimethylation (Figure 4G–J). The absence of Set2 caused a similar decrease in histone H3K4me1 near the FAT1 and MDV1 upstream pA sites, and the RAD53 downstream pA site, but not the RAD53 upstream pA site (Figure 4C–F). Once normalized to histone H3 ChIP signal, set2Δ cells had decreased monomethylation of histone H3K4 at the FAT1 and MDV1 upstream pA sites and decreased monomethylation of histone H3K4 at the downstream RAD53 pA site, while the H3K4me1 at the RAD53 upstream pA site remained unchanged (Figure 4G–J). These data suggest possible crosstalk between these different methylation machineries may in turn contribute to the effects of each on pA site choice.
Set1 and Set2 affect RNAP II Ser2-P and recruitment of cleavage/polyadenylation factors
RNAP II CTD Ser2, Tyr1 and Thr4 phosphorylation is enriched near the 3′ end of genes (116,117). RNAP II CTD Ser2-P is important for the recruitment of the cleavage/polyadenylation complex to the 3′ ends of genes (105). Previous reports showed that cells not expressing Set1 have increased RNAP II CTD Ser5 and Ser7 phosphorylation at the 5′-end of genes (118). We tested whether RNAP II CTD Ser2-P is affected by loss of the Set1 and Set2 HMTs. The amount of RNAP II and RNAP II Ser2-P in total protein extracts was not affected by Set1 depletion, but it was decreased by Set2 depletion (Figure 4A). The set1Δ cells had a strong increase in the RNAP II CTD Ser2-P occupancy at all the tested pA sites (Figure 5A–F). Deletion of SET2 increased RNAP II CTD Ser2-P only at the PDC1 and RPP1B pA sites, although to a lesser level than that seen in set1Δ cells (Figure 5C and D). These data show that the decrease in Set1, and to some degree in Set2, enhances the level of RNAP II CTD Ser2-P near pA sites.
Figure 5.

SET1 and SET2 deletion enhances the RNAP II CTD Ser2-P and the cleavage/polyadenylation complex levels at pA sites. (A–F) ChIP of the RNAP II CTD serine-2 phosphorylation, Rna15 and Pta1 to FAT1, MDV1, RAD53, PDC1 and RPP1B pA sites in wild-type, set1Δ and set2Δ cells. The values were normalized relative to RNAP II occupancy. Two biological replicates were performed for each gene. Bars show average values ± SD. *P < 0.05, **P < 0.01, ***P < 0.001 (Student's t-test).
The cleavage/polyadenylation complex in S. cerevisiae is composed of Cleavage Factor IA (CF IA), Cleavage Factor IB (CF IB), and holo-CPF, which contains core processing subunits and the Associated with Pta1 (APT) factor (119). To understand the mechanism by which Set1 and Set2 affect pA site choice, we tested whether the HMTs affect recruitment of cleavage/polyadenylation factors to the vicinity of pA sites via ChIP assay. We used the Rna15 subunit of CF IA and the Pta1 subunit of CPF as markers for the two factors, and examined the recruitment of these two proteins to the pA sites of FAT1, MDV1, RAD53, PDC1 and RPP1B. The expression of Rna15 and Pta1 in total protein extracts was not affected by Set1 or Set2 depletion (Figure 4A). However, set1Δ cells had increased recruitment of Rna15 to the FAT1 and MDV1 upstream pA sites, as well as the PDC1 and RPP1B pA sites (Figure 5A–D), and an increased recruitment of Pta1 to all of the pA sites (Figure 5A–F). The set2Δ cells had an increased presence of Rna15 only at the PDC1 and RPP1B pA sites but an increased recruitment of Pta1 to all of the pA sites (Figure 5A–F). These findings suggest that SET1 deletion enhances phosphorylation of the RNAP II CTD at Ser2, which in turn leads to increased recruitment of the 3′ end processing factors to the vicinity of pA sites, while SET2 depletion increases recruitment of the cleavage/polyadenylation complex mostly independent of RNAP II CTD Ser2-P.
The HMTs set1 and set2 control APA in response to rapamycin
To assess the role of Set1 and Set2 in the switch to alternative pA sites in response to environmental stress, we treated cells with the inhibitor of Target Of Rapamycin (TOR), rapamycin. TOR senses nutrients and regulates cell growth and aging. Mammalian TOR (mTOR) hyperactivation has been shown to lead to global mRNA 3′ UTR shortening in human cells, which upregulates translation of a subset of mRNAs (120). S. cerevisiae expresses two TOR proteins: Tor1 and Tor2, both of which are inhibited by rapamycin. Consistent with previous reports (56,121), Set1- and Set2-deficient cells, as well as histone H3K4R and H3K36R mutants, had increased sensitivity to rapamycin-induced nutrient stress (Figure 6A). Wild-type yeast grown for two hours in the presence of rapamycin switched to the upstream pA sites of ISM1, FAT1 and MDV1 (Figure 6B–D), and to the downstream pA site of RRD2 and RAD53 (Figure 6F and G). Rapamycin had minimal effect on RPB2 APA (Figure 6E).
Figure 6.

Histone H3K4 and H3K36 methylations are important for resistance to rapamycin. (A) set1Δ, set2Δ, H3K4R and H3K36R cells have increased sensitivity to rapamycin. Ten-fold serial dilutions of indicated strains were spotted on YPD agar containing 2 nM rapamycin or DMSO as a solvent control. (B–G) Set1 and Set2 mediate the alternative polyadenylation observed after rapamycin treatment. Wild-type and Set1, Set2, histone H3K4 and histone H3K36 mutants were exponentially grown in YPD media and shifted to media containing 10 nM rapamycin (for 2 h) or DMSO as a solvent control (DMSO data same as in Figure 1). Total RNA was reversely transcribed using anchored oligo d(T) primers. Total and long gene isoforms were amplified via qRT-PCR. The ratios of long to total mRNAs in the mutant strains and in the presence of rapamycin were normalized relative to the ratio in the wild-type W303 strain with DMSO. Three biological replicates were performed for each gene. Bars show average values ± SD. *P < 0.05, **P < 0.01, ***P < 0.001 (Student's t-test).
Cells not expressing Set1 or Set2 were not able to switch to alternative pA sites of ISM1, FAT1, MDV1 and RRD2 (Figure 6B–D, F). Cells expressing histone H3K4R were not able to switch to alternative pA sites of FAT1, MDV1, RPB2 and RRD2 (Figure 6C–F). Cells expressing histone H3K36R were not able to switch to alternative pA sites of ISM1, RPB2 and RRD2 (Figure 6B, E, F). The set1Δ and set2Δ cells, as well as mutants of histone H3K4 or H3K36 were still able to switch to the RAD53 downstream pA site following rapamycin treatment (Figure 6G). Thus, Set1 and Set2 are large contributors to the shift to alternative pA sites in the rapamycin-induced stress condition, at least in part via methylation of histone H3K4 and H3K36 residues.
Rapamycin alters chromatin structure around pA sites
Epigenetic modifications enable cells to quickly respond to environmental changes. For example, S. cerevisiae changes nucleosome occupancy and histone modifications in response to heat shock, osmostress, and different nitrogen conditions (5–7). We investigated whether nutritional stress caused by TOR inhibition affects histone occupancy and methylation within the vicinity of pA sites. We observed a significant decrease in histone H3 levels following rapamycin treatment at the FAT1 upstream pA site (Figure 7A), and the RAD53 pA sites (Figure 7C and D). The decrease in histone H3 levels was also detected in whole cell protein extracts via western blot (Figure 4A). Histone H3 levels at the MDV1 pA site were not as strongly affected by rapamycin treatment (Figure 7B). Histone H4 occupancy around pA sites varied according to the gene, with no change at the RAD53 upstream pA sites, an increase at the MDV1 upstream pA site, and a decrease at the RAD53 downstream pA site, and FAT1 pA site (Figure 7A–D). The total histone H4 protein levels were not affected by rapamycin treatment (Figure 4A).
Figure 7.

mTOR inhibition leads to epigenetic changes. (A–D) ChIPs for histone H3 and histone H4 around FAT1, MDV1, and RAD53 pA sites in wild-type S. cerevisiae exponentially growing in YPD media and shifted to media containing 10 nM rapamycin (for 2 h) or DMSO as a solvent control. (E–H) ChIPs for histone H3K4me1 and histone H3K36me3 near FAT1, MDV1 and RAD53 pA sites in wild-type cells from (A–D). (I–L) Same data as (E–H) normalized to total histone H3 levels from (A–D). Two biological replicates were performed for each gene. Bars show average values ± SD. *P < 0.05, **P < 0.01, ***P < 0.001 (Student's t-test).
Since histone H3K4me1 and H3K36me3 strongly correlate with pA site usage (48), and are found near the 3′ ends of genes (54,55), we examined these histone modifications around pA sites in cells undergoing nutritional stress. Rapamycin treatment decreased histone H3K4me1 around all pA sites (Figure 7E–H), despite Set1 expression being upregulated, and total histone H3K4me1 levels remaining unchanged (Figure 4A). Upon normalization to histone H3 levels, the monomethylation of histone H3K4 was unchanged around the FAT1 upstream pA site, and the RAD53 downstream pA site, while it increased around the RAD53 upstream pA site, and decreased around the MDV1 upstream pA site (Figure 7I–L). Histone H3K36me3 was decreased around RAD53 pA sites (Figure 7G and H), but not at the FAT1 and MDV1 pA sites (Figure 7E and F). Upon normalization to histone H3 levels, the trimethylation of histone H3K36 was unchanged around the MDV1 upstream pA site, but it was increased around the FAT1, and RAD53 upstream pA sites, and around the RAD53 downstream pA site (Figure 7I–L). Set2 expression, and total levels of histone H3K36me3 was downregulated following rapamycin treatment (Figure 4A). The decrease in histone H3K4me1 and H3K36me3 near pA sites can be explained by the overall decrease in nucleosome occupancy around pA sites. The decreased histone H3K36me3 total levels can be also attributed to the decrease in Set2 levels (Figure 4A). Overall, these findings show that rapamycin treatment changes nucleosome occupancy and epigenetic modifications around pA sites, as well as affects the expression of Set1 and Set2.
DISCUSSION
APA is dysregulated in many human diseases, but despite its relevance to health, the mechanisms regulating it remain a poorly understood aspect of biology. Most studies have looked at the impact of cleavage/polyadenylation factors and RNA-binding proteins on APA. The abundance of histone H3K4me1 and H3K36me3 modifications highly correlate with pA site positions, and they have been proposed to influence pA site selection (48,50). To establish whether these epigenetic processes are responsible for APA regulation, we examined mRNA 3′ end processing in yeast lacking Set1 or Set2 which methylate histone H3K4 and H3K36, respectively, as well as cells with mutations in the histone H3 residues that are targets of these two HMTs. Our findings support a model in which Set1 and Set2 affect RNAP II Ser2-P near pA sites, and hence the recruitment of cleavage/polyadenylation factors, and choice of pA site (Figure 8). H3K4R and H3K36R mutants show similar effects on pA site usage as the set1Δ and set2Δ mutants, respectively, confirming a role of these histone H3 modifications in APA.
Figure 8.

Model for Set1- and Set2-mediated choice of pA site. In wild-type cells, Set1 and Set2 mediate occupancy of histone H3K4me1 and H3K36me3 around pA sites. Methylation of histone H3K4 and H3K36 increases acetylation of nucleosomes, and assures an open chromatin structure, which allows RNAP II transcription to proceed to a downstream pA site (pA2), and production of long mRNA isoforms. Depletion of Set1 leads to loss of histone H3 and H4, reduction of histone H3K36me3, as well as increased phosphorylation of the RNAP II CTD Ser2 near pA1 site, resulting in recruitment of cleavage/polyadenylation factors. Loss of Set2 results in decreased nucleosome (Nuc) occupancy and histone H3K4me1 levels near the pA1 site which in turn enhances recruitment of the 3′ end processing complex.
In this study, we show that loss of Set1 or Set2 increases the 3′ end processing efficiency as measured by the decreased accumulation of unprocessed transcripts from genes with single pA sites. By decreasing the 3′ end processing efficiency, the presence of Set1 and Set2 may promote a switch to downstream pA sites, as fewer transcripts cleaved at the upstream pA site would increase the proportion of pre-mRNA processed at the downstream pA site. Indeed, this is the case, as cells lacking Set1 or Set2, or cells with H3K4 or H3H36 mutations, show a switch to upstream pA sites for the majority of genes that we have examined. Furthermore, discrepancies in the pA site utilization between set1Δ and H3K4 mutants, and between set2Δ and H3K36R mutants, are small, suggesting that Set1 and Set2 affect the choice of pA sites, at least in part, by methylation of their histone targets.
The changes in pA site choice in the absence of Set1 or Set2 could be caused in multiple ways. As summarized in Table 4, we find that loss of these epigenetic factors elicits several changes that could affect pA site usage. These include increased recruitment of the 3′ end processing machinery to transcribed genes, increased phosphorylation of the RNAP II CTD, and alterations in the occupancy of histone H3 and H4 around pA sites. While it is possible that Set1 and Set2 somehow affect the enzymatic activity of the cleavage/polyadenylation complex, such effects have not been reported.
Table 4.
Summary of key results
| FAT1 | MDV1 | RAD53 | PDC1 | RPP1B | ||
|---|---|---|---|---|---|---|
| set1Δ cells | Change in pA Site Usage | ↑↑↑ pA1 | ↑↑↑ pA1 | ↑↑↑ pA2 | No Change | ↓↓ Read-through |
| Histone H3 at pA1 or Single pA | ↓↓↓ | ↓↓↓ | ↓↓↓ | ↓↓↓ | ↓↓↓ | |
| Histone H3 at pA2 | ND | ND | ↓↓↓ | ND | ND | |
| Histone H4 at pA1 or Single pA | ↓↓↓ | ↓↓↓ | ↓↓↓ | ↓↓ | ↓↓ | |
| Histone H4 at pA2 | ND | ND | ↓↓↓ | ND | ND | |
| RNAP II Ser2-P at pA1 or Single pA | ↑↑↑ | ↑↑↑ | ↑↑↑ | ↑↑↑ | ↑↑↑ | |
| RNAP II Ser2-P at pA2 | ND | ND | ↑↑ | ND | ND | |
| Rna15 at pA1 or Single pA | ↑↑ | ↑↑ | No Change | ↑↑↑ | ↑ | |
| Pta1 at pA1 or Single pA | ↑ | ↑↑ | ↑ | ↑↑ | ↑↑↑ | |
| Pta1 at pA2 | ND | ND | ↑ | ND | ND | |
| set2Δ cells | Change in pA Site Usage | ↑↑↑ pA1 | ↑↑↑ pA1 | ↑ pA2 | ↓↓ Read-through | ↓↓ Read-through |
| Histone H3 at pA1 or Single pA | ↓↓ | ↓ | ↓↓ | No Change | No Change | |
| Histone H3 at pA2 | ND | ND | ↓↓ | ND | ND | |
| Histone H4 at pA1 or Single pA | ↓ | No Change | No Change | ↓↓ | ↓ | |
| Histone H4 at pA2 | ND | ND | ↓↓ | ND | ND | |
| RNAP II Ser2-P at pA1 or Single pA | No Change | No Change | No Change | ↑↑↑ | ↑↑↑ | |
| RNAP II Ser2-P at pA2 | ND | ND | No Change | ND | ND | |
| Rna15 at pA1 or Single pA | No Change | No Change | No Change | ↑↑↑ | ↑ | |
| Pta1 at pA1 or Single pA | ↑ | ↑ | ↑↑ | ↑↑ | ↑↑↑ | |
| Pta1 at pA2 | ND | ND | ↑↑ | ND | ND |
Not determined (ND).
Phosphorylation of RNAP II CTD Ser2 is required for the recruitment of the 3′ end processing factors (122). In this study, we show that loss of Set1 increases the level of RNAP II CTD Ser2-P at all analyzed pA sites, without affecting its total protein level. SET2 deletion upregulates RNAP II CTD Ser2-P only at single pA sites, and in contrast to Set1 loss, it decreases total RNAP II, and RNAP II Ser2-P levels. Consistent with the increased RNAP II CTD Ser2-P, set1Δ and set2Δ mutants have increased recruitment of Rna15 or Pta1 to pA sites, without changes in the overall expression of these subunits of the CF IA and CPF processing factors. However, the extent to which the recruitment of Rna15 and Pta1 to the tested pA sites changes is not always proportional to the increase in RNAP II CTD Ser2-P levels. This difference points to additional mechanisms that control 3′ end machinery recruitment, such as modifications of other RNAP II CTD residues (116,117,123) and interactions of the Rna15-containing CF IA factor with the Spt5 elongation factor and the RNAP II flap loop (124,125). For genes with multiple pA sites, the extent of RNAP II pausing downstream of the pA site and elongation rate of RNAP II between pA sites could also affect the time available for factors to be recruited to the upstream site. This timing could be modulated by changes in chromatin organization and modification.
Both Set1 and Set2 are positioned where they could affect the CTD phosphorylation status, which in turn, could affect recruitment of processing factors to the pA site region. For example, Set1 and Set2 physically interact with RNAP II, and both associate with the newly transcribed RNA (126–128). Set1 gets recruited to RNAP II phosphorylated at Ser5 of its CTD (58,129). However the highest level of Set1 binding to mRNA, as observed by UV crosslinking experiments, occurs right before the pA site, supporting the idea that Set1 will influence 3′ end processing (128). Set2 binds to RNAP II that is phosphorylated at Ser2 and Ser5 of its CTD (84). Set1 and Set2 may regulate RNAP II phosphorylation by controlling the expression, activity or recruitment of the RNAP II CTD kinases or phosphatases. Interestingly, deletion of SET1 in strains lacking the kinase Ctk1 (Ser2) (130), or RNAP II CTD phosphatases Glc7 (Tyr1) (131) and Rtr1 (Ser5) (118,132–135), and deletion of SET2 in strains lacking Ctk1 or the Ser2 phosphatase Fcp1 (135,136) alters the cell's fitness, suggesting that the two HMTs and RNAP II CTD modulators functionally interact.
Decreased RNAP II processivity has also been implicated as a mechanism for switching to upstream pA sites (47). It could not only prevent RNAP II from reaching downstream pA sites, but also allow more time for the 3′ end processing machinery to get recruited to, and to work at an upstream pA site. Furthermore, previous studies have indicated that a slow mutant of RNAP II results in increased CTD Ser2-P towards the 5′ of genes (137). Our observations are consistent with such a mechanism, as we find that loss of Set1 or Set2, two HMTs favoring open chromatin (59–61,85), increases RNAP II CTD Ser2-P around upstream, as well as single pA sites. Thus, Set1 and Set2 may also favor the use of downstream pA sites in most of the studied genes because they increase the processivity of RNAP II by altering chromatin structure and indirectly decreasing RNAP II CTD Ser2-P near upstream pA sites. The mechanism by which Set1 and Set2 induce a switch to some upstream pA sites is not clear, and may reflect the fact that these two HMTs promote closed chromatin structure in some genes (62–65,72,86,114,138).
Nucleosome positioning correlates with 3′ end formation (139), and the 3′ ends of genes, especially at pA sites, are depleted of nucleosomes (140). We found that the absence of Set1, and to a smaller degree Set2, decreases histone H3 and H4 occupancy around pA sites. These data are consistent with previous report that cells not expressing the histone H3K4 demethylase Jhd2 have higher histone H3 levels at the SRG1 3′ end (141). Likewise, deletion of the histone H3K4 demethylase KDM5B results in increased nucleosome occupancy at promoters in embryonic stem cells (142). Methylation of histone H3K4 and H3K36 is likely to affect nucleosome occupancy by affecting nucleosome turnover or remodeling, rather than deposition.
Histone H3K4 and H3K36 residues and HMTs that modify them may also affect pA site choice via interaction with other epigenetic factors. For example, histone H3K36me3 can be bound by NuA3 (143) and NuA4 HAT complexes. Likewise, H3K4me3 recruits HATs such as NuA3 (94), NuA4, SAGA and HBO1. Thus, another way in which a decrease in H3K4 and H3K36 methylation can lead to utilization of upstream pA sites is by decreasing the recruitment of HATs (143), which would then lead to condensed chromatin structure (3,4).
APA and epigenetic modifications allow cells to quickly adjust their RNA and protein composition (8,144,145). For example, nutritional stress results in switch to downstream pA sites in yeast (146), and increases utilization of the downstream CAT1 pA site, which in turn results in increased expression of the human amino acid transporter Cat1 (147). During cold shock, there is a global switch to upstream pA sites (148), while during heat shock there is a switch to the upstream HSP70.3 pA site, which results in increased translation of the heat shock protein HSP70.3 (149,150). Stress-induced APA occurs not only in animals and fungi; in plants, hypoxia induces a switch to upstream pA sites (151). The APA response also varies by species. For example, arsenic stress, which causes oxidative stress, leads to utilization of upstream pA sites in mouse (152), while in human cells, it leads to a switch to distal pA sites (9). Likewise, DNA damage in yeast results in a switch to downstream pA sites (8,25), while in human colon carcinoma RKO cells, it favors utilization of upstream pA sites (153).
Set1 and Set2 are important for the cell's proper response to cellular cues and environmental stress. Set2 regulates the proper response to carbon source (154), DNA damage (155–157), and longevity (23,158). Likewise, Set1 is important for the proper response to DNA damage (64,159–161). We have found that during the nutritional stress response induced by the TOR inhibitor rapamycin, several gene transcripts are alternatively polyadenylated. This rapamycin-induced APA correlates with a decrease in histone H3 levels, as well as a decrease in histone H3K4me1 and H3K36me3 around pA sites. Importantly, Set1 and Set2 were required for the rapamycin-induced switch to alternative pA sites. Thus, it is very likely that Set1 and Set2 mediate resistance to rapamycin in part by affecting the choice of pA sites.
Taken together, we present evidence that the epigenetic factors, Set1 and Set2, control choice of pA sites via modulation of RNAP II, and recruitment of the 3′ end processing machinery. As described above, changes in histone H3K4 and H3K36 methylation also affect APA during nutritional stress, and it will be interesting to see if they have similar roles in other cell responses. Set1 and Set2 increase nucleosome occupancy around pA sites, but it remains unknown whether this leads to changes in pA site selection. A better understanding of mechanisms regulating pA site choice, and APA’s role in human health, is required in order to manipulate it to affect disease outcomes.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Hungjiun Liaw (National Cheng Kung University, Tainan, Taiwan) for yeast strains. We also thank Susan Lee, Hui-Yun Liu, Srimoyee Mukherjee and Daniel Heller for critical discussion and suggestions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
American Cancer Society [132291-PF-18-054-01-RMC to K.K.M.]; National Science Foundation [MCB-1244043 to C.M.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Luger K., Mader A.W., Richmond R.K., Sargent D.F., Richmond T.J.. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997; 389:251–260. [DOI] [PubMed] [Google Scholar]
- 2. Richmond T.J., Davey C.A.. The structure of DNA in the nucleosome core. Nature. 2003; 423:145–150. [DOI] [PubMed] [Google Scholar]
- 3. Venters B.J., Pugh B.F.. How eukaryotic genes are transcribed. Crit. Rev. Biochem. Mol. Biol. 2009; 44:117–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Miremadi A., Oestergaard M.Z., Pharoah P.D., Caldas C.. Cancer genetics of epigenetic genes. Hum. Mol. Genet. 2007; 16:R28–R49. [DOI] [PubMed] [Google Scholar]
- 5. Shivaswamy S., Iyer V.R.. Stress-dependent dynamics of global chromatin remodeling in yeast: dual role for SWI/SNF in the heat shock stress response. Mol. Cell. Biol. 2008; 28:2221–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nadal-Ribelles M., Mas G., Millan-Zambrano G., Sole C., Ammerer G., Chavez S., Posas F., de Nadal E.. H3K4 monomethylation dictates nucleosome dynamics and chromatin remodeling at stress-responsive genes. Nucleic Acids Res. 2015; 43:4937–4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang P., Du G., Zou H., Xie G., Chen J., Shi Z., Zhou J.. Genome-wide mapping of nucleosome positions in Saccharomyces cerevisiae in response to different nitrogen conditions. Sci. Rep. 2016; 6:33970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Graber J.H., Nazeer F.I., Yeh P.C., Kuehner J.N., Borikar S., Hoskinson D., Moore C.L.. DNA damage induces targeted, genome-wide variation of poly(A) sites in budding yeast. Genome Res. 2013; 23:1690–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hollerer I., Curk T., Haase B., Benes V., Hauer C., Neu-Yilik G., Bhuvanagiri M., Hentze M.W., Kulozik A.E.. The differential expression of alternatively polyadenylated transcripts is a common stress-induced response mechanism that modulates mammalian mRNA expression in a quantitative and qualitative fashion. RNA. 2016; 22:1441–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gruber A.J., Zavolan M.. Alternative cleavage and polyadenylation in health and disease. Nat. Rev. Genet. 2019; 20:599–614. [DOI] [PubMed] [Google Scholar]
- 11. Mayr C., Bartel D.P.. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009; 138:673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tian B., Manley J.L.. Alternative polyadenylation of mRNA precursors. Nat Rev Mol Cell Biol. 2016; 18:18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Batra R., Manchanda M., Swanson M.S.. Global insights into alternative polyadenylation regulation. RNA Biol. 2015; 12:597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Akman B.H., Can T., Erson-Bensan A.E.. Estrogen-induced upregulation and 3′-UTR shortening of CDC6. Nucleic Acids Res. 2012; 40:10679–10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Matoulkova E., Sommerova L., Pastorek M., Vojtesek B., Hrstka R.. Regulation of AGR2 expression via 3′UTR shortening. Exp Cell Res. 2017; 356:40–47. [DOI] [PubMed] [Google Scholar]
- 16. Ni T.K., Kuperwasser C.. Premature polyadenylation of MAGI3 produces a dominantly-acting oncogene in human breast cancer. Elife. 2016; 5:e14730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miles W.O., Lembo A., Volorio A., Brachtel E., Tian B., Sgroi D., Provero P., Dyson N.. Alternative polyadenylation in triple-negative breast tumors allows NRAS and c-JUN to bypass PUMILIO posttranscriptional regulation. Cancer Res. 2016; 76:7231–7241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ye C., Zhou Q., Hong Y., Li Q.Q.. Role of alternative polyadenylation dynamics in acute myeloid leukaemia at single-cell resolution. RNA Biol. 2019; 16:785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mueller A.A., Cheung T.H., Rando T.A.. All's well that ends well: alternative polyadenylation and its implications for stem cell biology. Curr. Opin. Cell Biol. 2013; 25:222–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shen T., Li H., Song Y., Li L., Lin J., Wei G., Ni T.. Alternative polyadenylation dependent function of splicing factor SRSF3 contributes to cellular senescence. Aging (Albany NY). 2019; 11:1356–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen M., Lyu G., Han M., Nie H., Shen T., Chen W., Niu Y., Song Y., Li X., Li H. et al.. 3′ UTR lengthening as a novel mechanism in regulating cellular senescence. Genome Res. 2018; 28:285–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mangone M., Manoharan A.P., Thierry-Mieg D., Thierry-Mieg J., Han T., Mackowiak S.D., Mis E., Zegar C., Gutwein M.R., Khivansara V. et al.. The landscape of C. elegans 3′UTRs. Science. 2010; 329:432–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sen P., Dang W., Donahue G., Dai J., Dorsey J., Cao X., Liu W., Cao K., Perry R., Lee J.Y. et al.. H3K36 methylation promotes longevity by enhancing transcriptional fidelity. Genes Dev. 2015; 29:1362–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sen P., Shah P.P., Nativio R., Berger S.L.. Epigenetic mechanisms of longevity and aging. Cell. 2016; 166:822–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yu L., Volkert M.R.. UV damage regulates alternative polyadenylation of the RPB2 gene in yeast. Nucleic Acids Res. 2013; 41:3104–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Williamson A.K., Zhu Z., Yuan Z.M.. Epigenetic mechanisms behind cellular sensitivity to DNA damage. Cell Stress. 2018; 2:176–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ogorodnikov A., Levin M., Tattikota S., Tokalov S., Hoque M., Scherzinger D., Marini F., Poetsch A., Binder H., Macher-Goppinger S. et al.. Transcriptome 3′end organization by PCF11 links alternative polyadenylation to formation and neuronal differentiation of neuroblastoma. Nat. Commun. 2018; 9:5331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Qiu F., Fu Y., Lu C., Feng Y., Wang Q., Huo Z., Jia X., Chen C., Chen S., Xu A.. Small nuclear ribonucleoprotein polypeptide a-mediated alternative polyadenylation of STAT5B during Th1 cell differentiation. J. Immunol. 2017; 199:3106–3115. [DOI] [PubMed] [Google Scholar]
- 29. Spangenberg L., Correa A., Dallagiovanna B., Naya H.. Role of alternative polyadenylation during adipogenic differentiation: an in silico approach. PLoS One. 2013; 8:e75578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Grassi E., Santoro R., Umbach A., Grosso A., Oliviero S., Neri F., Conti L., Ala U., Provero P., DiCunto F. et al.. Choice of alternative polyadenylation sites, mediated by the RNA-Binding protein elavl3, plays a role in differentiation of inhibitory neuronal progenitors. Front. Cell Neurosci. 2018; 12:518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brumbaugh J., Di Stefano B., Wang X., Borkent M., Forouzmand E., Clowers K.J., Ji F., Schwarz B.A., Kalocsay M., Elledge S.J. et al.. Nudt21 controls cell fate by connecting alternative polyadenylation to chromatin signaling. Cell. 2018; 172:629–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shulman E.D., Elkon R.. Cell-type-specific analysis of alternative polyadenylation using single-cell transcriptomics data. Nucleic. Acids. Res. 2019; 47:10027–10039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brutman J.N., Zhou X., Zhang Y., Michal J., Stark B., Jiang Z., Davis J.F.. Mapping diet-induced alternative polyadenylation of hypothalamic transcripts in the obese rat. Physiol. Behav. 2018; 188:173–180. [DOI] [PubMed] [Google Scholar]
- 34. Etchegaray J.P., Mostoslavsky R.. Interplay between metabolism and epigenetics: a nuclear adaptation to environmental changes. Mol. Cell. 2016; 62:695–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sommerkamp P. Altamura S. Altamura S. Renders S., Narr A., Ladel L., Zeisberger P., Eiben P.L., Fawaz M., Rieger M.A., Cabezas-Wallscheid N. et al.. Alternative Polyadenylation Landscapes Mediate Hematopoietic Stem Cell Activation and Regulate Glutamine Metabolism. Cell Stem Cell. 2020; 10.1016/j.stem.2020.03.003. [DOI] [PubMed] [Google Scholar]
- 36. Creemers E.E., Bawazeer A., Ugalde A.P., van Deutekom H.W., van der Made I., de Groot N.E., Adriaens M.E., Cook S.A., Bezzina C.R., Hubner N. et al.. Genome-wide polyadenylation maps reveal dynamic mrna 3′-end formation in the failing human heart. Circ. Res. 2016; 118:433–438. [DOI] [PubMed] [Google Scholar]
- 37. Gilsbach R., Schwaderer M., Preissl S., Gruning B.A., Kranzhofer D., Schneider P., Nuhrenberg T.G., Mulero-Navarro S., Weichenhan D., Braun C. et al.. Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo. Nat. Commun. 2018; 9:391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Weng T., Ko J., Masamha C.P., Xia Z., Xiang Y., Chen N.Y., Molina J.G., Collum S., Mertens T.C., Luo F. et al.. Cleavage factor 25 deregulation contributes to pulmonary fibrosis through alternative polyadenylation. J. Clin. Invest. 2019; 129:1984–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang I.V., Schwartz D.A.. Epigenetics of idiopathic pulmonary fibrosis. Transl Res. 2015; 165:48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Patel R., Brophy C., Hickling M., Neve J., Furger A.. Alternative cleavage and polyadenylation of genes associated with protein turnover and mitochondrial function are deregulated in Parkinson's, Alzheimer's and ALS disease. BMC Med Genomics. 2019; 12:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Berson A., Nativio R., Berger S.L., Bonini N.M.. Epigenetic regulation in neurodegenerative diseases. Trends Neurosci. 2018; 41:587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Allis C.D., Jenuwein T.. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016; 17:487–500. [DOI] [PubMed] [Google Scholar]
- 43. Akman H.B., Erson-Bensan A.E.. Alternative polyadenylation and its impact on cellular processes. Microrna. 2014; 3:2–9. [DOI] [PubMed] [Google Scholar]
- 44. Singh I., Lee S.H., Sperling A.S., Samur M.K., Tai Y.T., Fulciniti M., Munshi N.C., Mayr C., Leslie C.S.. Widespread intronic polyadenylation diversifies immune cell transcriptomes. Nat. Commun. 2018; 9:1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jia X., Yuan S., Wang Y., Fu Y., Ge Y., Ge Y., Lan X., Feng Y., Qiu F., Li P. et al.. The role of alternative polyadenylation in the antiviral innate immune response. Nat. Commun. 2017; 8:14605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang Q., Cao X.. Epigenetic regulation of the innate immune response to infection. Nat. Rev. Immunol. 2019; 19:417–432. [DOI] [PubMed] [Google Scholar]
- 47. Cui Y., Denis C.L.. In vivo evidence that defects in the transcriptional elongation factors RPB2, TFIIS, and SPT5 enhance upstream poly(A) site utilization. Mol. Cell. Biol. 2003; 23:7887–7901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Khaladkar M., Smyda M., Hannenhalli S.. Epigenomic and RNA structural correlates of polyadenylation. RNA Biol. 2011; 8:529–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Roy K., Gabunilas J., Gillespie A., Ngo D., Chanfreau G.F.. Common genomic elements promote transcriptional and DNA replication roadblocks. Genome Res. 2016; 26:1363–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lee C.Y., Chen L.. Alternative polyadenylation sites reveal distinct chromatin accessibility and histone modification in human cell lines. Bioinformatics. 2013; 29:1713–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Luco R.F., Pan Q., Tominaga K., Blencowe B.J., Pereira-Smith O.M., Misteli T.. Regulation of alternative splicing by histone modifications. Science. 2010; 327:996–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Briggs S.D., Bryk M., Strahl B.D., Cheung W.L., Davie J.K., Dent S.Y., Winston F., Allis C.D.. Histone H3 lysine 4 methylation is mediated by Set1 and required for cell growth and rDNA silencing in Saccharomyces cerevisiae. Genes Dev. 2001; 15:3286–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Roguev A., Schaft D., Shevchenko A., Pijnappel W.W., Wilm M., Aasland R., Stewart A.F.. The Saccharomyces cerevisiae Set1 complex includes an Ash2 homologue and methylates histone 3 lysine 4. EMBO J. 2001; 20:7137–7148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li B., Carey M., Workman J.L.. The role of chromatin during transcription. Cell. 2007; 128:707–719. [DOI] [PubMed] [Google Scholar]
- 55. Woo H., Dam Ha S., Lee S.B., Buratowski S., Kim T.. Modulation of gene expression dynamics by co-transcriptional histone methylations. Exp. Mol. Med. 2017; 49:e326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nagy P.L., Griesenbeck J., Kornberg R.D., Cleary M.L.. A trithorax-group complex purified from Saccharomyces cerevisiae is required for methylation of histone H3. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Miller T., Krogan N.J., Dover J., Erdjument-Bromage H., Tempst P., Johnston M., Greenblatt J.F., Shilatifard A.. COMPASS: a complex of proteins associated with a trithorax-related SET domain protein. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:12902–12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Krogan N.J., Dover J., Wood A., Schneider J., Heidt J., Boateng M.A., Dean K., Ryan O.W., Golshani A., Johnston M. et al.. The Paf1 complex is required for histone H3 methylation by COMPASS and Dot1p: linking transcriptional elongation to histone methylation. Mol. Cell. 2003; 11:721–729. [DOI] [PubMed] [Google Scholar]
- 59. Santos-Rosa H., Schneider R., Bannister A.J., Sherriff J., Bernstein B.E., Emre N.C., Schreiber S.L., Mellor J., Kouzarides T.. Active genes are tri-methylated at K4 of histone H3. Nature. 2002; 419:407–411. [DOI] [PubMed] [Google Scholar]
- 60. Bernstein B.E., Humphrey E.L., Erlich R.L., Schneider R., Bouman P., Liu J.S., Kouzarides T., Schreiber S.L.. Methylation of histone H3 Lys 4 in coding regions of active genes. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:8695–8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Boa S., Coert C., Patterton H.G.. Saccharomyces cerevisiae Set1p is a methyltransferase specific for lysine 4 of histone H3 and is required for efficient gene expression. Yeast. 2003; 20:827–835. [DOI] [PubMed] [Google Scholar]
- 62. Bryk M., Briggs S.D., Strahl B.D., Curcio M.J., Allis C.D., Winston F.. Evidence that Set1, a factor required for methylation of histone H3, regulates rDNA silencing in S. cerevisiae by a Sir2-independent mechanism. Curr. Biol. 2002; 12:165–170. [DOI] [PubMed] [Google Scholar]
- 63. Nislow C., Ray E., Pillus L.. SET1, a yeast member of the trithorax family, functions in transcriptional silencing and diverse cellular processes. Mol. Biol. Cell. 1997; 8:2421–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Corda Y., Schramke V., Longhese M.P., Smokvina T., Paciotti V., Brevet V., Gilson E., Geli V.. Interaction between Set1p and checkpoint protein Mec3p in DNA repair and telomere functions. Nat. Genet. 1999; 21:204–208. [DOI] [PubMed] [Google Scholar]
- 65. Krogan N.J., Dover J., Khorrami S., Greenblatt J.F., Schneider J., Johnston M., Shilatifard A.. COMPASS, a histone H3 (Lysine 4) methyltransferase required for telomeric silencing of gene expression. J. Biol. Chem. 2002; 277:10753–10755. [DOI] [PubMed] [Google Scholar]
- 66. Kim T., Buratowski S.. Dimethylation of H3K4 by Set1 recruits the Set3 histone deacetylase complex to 5′ transcribed regions. Cell. 2009; 137:259–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pinskaya M., Gourvennec S., Morillon A.. H3 lysine 4 di- and tri-methylation deposited by cryptic transcription attenuates promoter activation. EMBO J. 2009; 28:1697–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Terzi N., Churchman L.S., Vasiljeva L., Weissman J., Buratowski S.. H3K4 trimethylation by Set1 promotes efficient termination by the Nrd1-Nab3-Sen1 pathway. Mol. Cell. Biol. 2011; 31:3569–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Seol J.H., Kim H.J., Yang Y.J., Kim S.T., Youn H.D., Han J.W., Lee H.W., Cho E.J.. Different roles of histone H3 lysine 4 methylation in chromatin maintenance. Biochem. Biophys. Res. Commun. 2006; 349:463–470. [DOI] [PubMed] [Google Scholar]
- 70. Blair L.P., Liu Z., Labitigan R.L., Wu L., Zheng D., Xia Z., Pearson E.L., Nazeer F.I., Cao J., Lang S.M. et al.. KDM5 lysine demethylases are involved in maintenance of 3′UTR length. Sci. Adv. 2016; 2:e1501662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ji Z., Luo W., Li W., Hoque M., Pan Z., Zhao Y., Tian B.. Transcriptional activity regulates alternative cleavage and polyadenylation. Mol. Syst. Biol. 2011; 7:534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Strahl B.D., Grant P.A., Briggs S.D., Sun Z.W., Bone J.R., Caldwell J.A., Mollah S., Cook R.G., Shabanowitz J., Hunt D.F. et al.. Set2 is a nucleosomal histone H3-selective methyltransferase that mediates transcriptional repression. Mol. Cell. Biol. 2002; 22:1298–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Krogan N.J., Kim M., Tong A., Golshani A., Cagney G., Canadien V., Richards D.P., Beattie B.K., Emili A., Boone C. et al.. Methylation of histone H3 by Set2 in Saccharomyces cerevisiae is linked to transcriptional elongation by RNA polymerase II. Mol. Cell. Biol. 2003; 23:4207–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Li B., Howe L., Anderson S., Yates J.R. 3rd, Workman J.L.. The Set2 histone methyltransferase functions through the phosphorylated carboxyl-terminal domain of RNA polymerase II. J. Biol. Chem. 2003; 278:8897–8903. [DOI] [PubMed] [Google Scholar]
- 75. Li J., Moazed D., Gygi S.P.. Association of the histone methyltransferase Set2 with RNA polymerase II plays a role in transcription elongation. J. Biol. Chem. 2002; 277:49383–49388. [DOI] [PubMed] [Google Scholar]
- 76. Xiao T., Hall H., Kizer K.O., Shibata Y., Hall M.C., Borchers C.H., Strahl B.D.. Phosphorylation of RNA polymerase II CTD regulates H3 methylation in yeast. Genes Dev. 2003; 17:654–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Barski A., Cuddapah S., Cui K., Roh T.Y., Schones D.E., Wang Z., Wei G., Chepelev I., Zhao K.. High-resolution profiling of histone methylations in the human genome. Cell. 2007; 129:823–837. [DOI] [PubMed] [Google Scholar]
- 78. Nojima T., Gomes T., Grosso A.R., Kimura H., Dye M.J., Dhir S., Carmo-Fonseca M., Proudfoot N.J.. Mammalian NET-Seq reveals genome-wide nascent transcription coupled to RNA processing. Cell. 2015; 161:526–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Youdell M.L., Kizer K.O., Kisseleva-Romanova E., Fuchs S.M., Duro E., Strahl B.D., Mellor J.. Roles for Ctk1 and Spt6 in regulating the different methylation states of histone H3 lysine 36. Mol. Cell. Biol. 2008; 28:4915–4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chu Y., Simic R., Warner M.H., Arndt K.M., Prelich G.. Regulation of histone modification and cryptic transcription by the Bur1 and Paf1 complexes. EMBO J. 2007; 26:4646–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Fuchs S.M., Kizer K.O., Braberg H., Krogan N.J., Strahl B.D.. RNA polymerase II carboxyl-terminal domain phosphorylation regulates protein stability of the Set2 methyltransferase and histone H3 di- and trimethylation at lysine 36. J. Biol. Chem. 2012; 287:3249–3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chu Y., Sutton A., Sternglanz R., Prelich G.. The BUR1 cyclin-dependent protein kinase is required for the normal pattern of histone methylation by SET2. Mol. Cell. Biol. 2006; 26:3029–3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Qiu H., Hu C., Hinnebusch A.G.. Phosphorylation of the Pol II CTD by KIN28 enhances BUR1/BUR2 recruitment and Ser2 CTD phosphorylation near promoters. Mol. Cell. 2009; 33:752–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kizer K.O., Phatnani H.P., Shibata Y., Hall H., Greenleaf A.L., Strahl B.D.. A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol. Cell. Biol. 2005; 25:3305–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Schaft D., Roguev A., Kotovic K.M., Shevchenko A., Sarov M., Shevchenko A., Neugebauer K.M., Stewart A.F.. The histone 3 lysine 36 methyltransferase, SET2, is involved in transcriptional elongation. Nucleic Acids Res. 2003; 31:2475–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Landry J., Sutton A., Hesman T., Min J., Xu R.M., Johnston M., Sternglanz R.. Set2-catalyzed methylation of histone H3 represses basal expression of GAL4 in Saccharomyces cerevisiae. Mol. Cell. Biol. 2003; 23:5972–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Li B., Jackson J., Simon M.D., Fleharty B., Gogol M., Seidel C., Workman J.L., Shilatifard A.. Histone H3 lysine 36 dimethylation (H3K36me2) is sufficient to recruit the Rpd3s histone deacetylase complex and to repress spurious transcription. J. Biol. Chem. 2009; 284:7970–7976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Joshi A.A., Struhl K.. Eaf3 chromodomain interaction with methylated H3-K36 links histone deacetylation to Pol II elongation. Mol. Cell. 2005; 20:971–978. [DOI] [PubMed] [Google Scholar]
- 89. Keogh M.C., Kurdistani S.K., Morris S.A., Ahn S.H., Podolny V., Collins S.R., Schuldiner M., Chin K., Punna T., Thompson N.J. et al.. Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell. 2005; 123:593–605. [DOI] [PubMed] [Google Scholar]
- 90. Venkatesh S., Li H., Gogol M.M., Workman J.L.. Selective suppression of antisense transcription by Set2-mediated H3K36 methylation. Nat. Commun. 2016; 7:13610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Carrozza M.J., Li B., Florens L., Suganuma T., Swanson S.K., Lee K.K., Shia W.J., Anderson S., Yates J., Washburn M.P. et al.. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell. 2005; 123:581–592. [DOI] [PubMed] [Google Scholar]
- 92. Li B., Gogol M., Carey M., Pattenden S.G., Seidel C., Workman J.L.. Infrequently transcribed long genes depend on the Set2/Rpd3S pathway for accurate transcription. Genes Dev. 2007; 21:1422–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Li B., Gogol M., Carey M., Lee D., Seidel C., Workman J.L.. Combined action of PHD and chromo domains directs the Rpd3S HDAC to transcribed chromatin. Science. 2007; 316:1050–1054. [DOI] [PubMed] [Google Scholar]
- 94. Carvalho S., Raposo A.C., Martins F.B., Grosso A.R., Sridhara S.C., Rino J., Carmo-Fonseca M., de Almeida S.F.. Histone methyltransferase SETD2 coordinates FACT recruitment with nucleosome dynamics during transcription. Nucleic Acids Res. 2013; 41:2881–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Du H.N., Fingerman I.M., Briggs S.D.. Histone H3 K36 methylation is mediated by a trans-histone methylation pathway involving an interaction between Set2 and histone H4. Genes Dev. 2008; 22:2786–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Du H.N., Briggs S.D.. A nucleosome surface formed by histone H4, H2A, and H3 residues is needed for proper histone H3 Lys36 methylation, histone acetylation, and repression of cryptic transcription. J. Biol. Chem. 2010; 285:11704–11713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Kim T., Buratowski S.. Two Saccharomyces cerevisiae JmjC domain proteins demethylate histone H3 Lys36 in transcribed regions to promote elongation. J. Biol. Chem. 2007; 282:20827–20835. [DOI] [PubMed] [Google Scholar]
- 98. Lian Z., Karpikov A., Lian J., Mahajan M.C., Hartman S., Gerstein M., Snyder M., Weissman S.M.. A genomic analysis of RNA polymerase II modification and chromatin architecture related to 3′ end RNA polyadenylation. Genome Res. 2008; 18:1224–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Khaladkar M., Smyda M., Hannenhalli S.. Epigenomic and RNA structural correlates of polyadenylation. RNA Biol. 2014; 8:529–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Bucheli M.E., He X., Kaplan C.D., Moore C.L., Buratowski S.. Polyadenylation site choice in yeast is affected by competition between Npl3 and polyadenylation factor CFI. RNA. 2007; 13:1756–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Gruber A.R., Martin G., Keller W., Zavolan M.. Means to an end: mechanisms of alternative polyadenylation of messenger RNA precursors. Wiley Interdiscip. Rev RNA. 2014; 5:183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Tian B., Manley J.L.. Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell Biol. 2017; 18:18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Luco R.F., Allo M., Schor I.E., Kornblihtt A.R., Misteli T.. Epigenetics in alternative pre-mRNA splicing. Cell. 2011; 144:16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Laverriere J.N., L’Hote D., Tabouy L., Schang A.L., Querat B., Cohen-Tannoudji J.. Epigenetic regulation of alternative promoters and enhancers in progenitor, immature, and mature gonadotrope cell lines. Mol. Cell. Endocrinol. 2016; 434:250–265. [DOI] [PubMed] [Google Scholar]
- 105. Ahn S.H., Kim M., Buratowski S.. Phosphorylation of serine 2 within the RNA polymerase II C-terminal domain couples transcription and 3′ end processing. Mol. Cell. 2004; 13:67–76. [DOI] [PubMed] [Google Scholar]
- 106. Su W.P., Hsu S.H., Chia L.C., Lin J.Y., Chang S.B., Jiang Z.D., Lin Y.J., Shih M.Y., Chen Y.C., Chang M.S. et al.. Combined interactions of plant homeodomain and chromodomain regulate NuA4 activity at DNA double-strand breaks. Genetics. 2016; 202:77–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Wright A.P., Bruns M., Hartley B.S.. Extraction and rapid inactivation of proteins from Saccharomyces cerevisiae by trichloroacetic acid precipitation. Yeast. 1989; 5:51–53. [DOI] [PubMed] [Google Scholar]
- 108. Ares M. Isolation of total RNA from yeast cell cultures. Cold Spring Harb. Protoc. 2012; 2012:1082–1086. [DOI] [PubMed] [Google Scholar]
- 109. Pearson E.L., Graber J.H., Lee S.D., Naggert K.S., Moore C.L.. Ipa1 is an RNA polymerase II elongation factor that facilitates termination by maintaining levels of the poly(A) site endonuclease Ysh1. Cell Rep. 2019; 26:1919–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Wei W., Hennig B.P., Wang J., Zhang Y., Piazza I., Pareja Sanchez Y., Chabbert C.D., Adjalley S.H., Steinmetz L.M., Pelechano V.. Chromatin-sensitive cryptic promoters putatively drive expression of alternative protein isoforms in yeast. Genome Res. 2019; 29:1974–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Lee S.D., Liu H.Y., Graber J.H., Heller-Trulli D., Kaczmarek Michaels K., Cerezo J.F., Moore C.L.. Regulation of the Ysh1 endonuclease of the mRNA cleavage/polyadenylation complex by ubiquitin-mediated degradation. RNA Biol. 2020; 17:689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Hainer S.J., Martens J.A.. Identification of histone mutants that are defective for transcription-coupled nucleosome occupancy. Mol. Cell. Biol. 2011; 31:3557–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Wang S.S., Zhou B.O., Zhou J.Q.. Histone H3 lysine 4 hypermethylation prevents aberrant nucleosome remodeling at the PHO5 promoter. Mol. Cell Biol. 2011; 31:3171–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Ramakrishnan S., Pokhrel S., Palani S., Pflueger C., Parnell T.J., Cairns B.R., Bhaskara S., Chandrasekharan M.B.. Counteracting H3K4 methylation modulators Set1 and Jhd2 co-regulate chromatin dynamics and gene transcription. Nat. Commun. 2016; 7:11949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Venkatesh S., Smolle M., Li H., Gogol M.M., Saint M., Kumar S., Natarajan K., Workman J.L.. Set2 methylation of histone H3 lysine 36 suppresses histone exchange on transcribed genes. Nature. 2012; 489:452–455. [DOI] [PubMed] [Google Scholar]
- 116. Kecman T., Heo D.H., Vasiljeva L.. Profiling RNA polymerase II phosphorylation genome-wide in fission yeast. Methods Enzymol. 2018; 612:489–504. [DOI] [PubMed] [Google Scholar]
- 117. Mayer A., Heidemann M., Lidschreiber M., Schreieck A., Sun M., Hintermair C., Kremmer E., Eick D., Cramer P.. CTD tyrosine phosphorylation impairs termination factor recruitment to RNA polymerase II. Science. 2012; 336:1723–1725. [DOI] [PubMed] [Google Scholar]
- 118. Soares L.M., Buratowski S.. Yeast Swd2 is essential because of antagonism between Set1 histone methyltransferase complex and APT (associated with Pta1) termination factor. J. Biol. Chem. 2012; 287:15219–15231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Kumar A., Clerici M., Muckenfuss L.M., Passmore L.A., Jinek M.. Mechanistic insights into mRNA 3′-end processing. Curr. Opin. Struct. Biol. 2019; 59:143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Chang J.W., Zhang W., Yeh H.S., de Jong E.P., Jun S., Kim K.H., Bae S.S., Beckman K., Hwang T.H., Kim K.S. et al.. mRNA 3′-UTR shortening is a molecular signature of mTORC1 activation. Nat. Commun. 2015; 6:7218. [DOI] [PubMed] [Google Scholar]
- 121. McDaniel S.L., Hepperla A.J., Huang J., Dronamraju R., Adams A.T., Kulkarni V.G., Davis I.J., Strahl B.D.. H3K36 methylation regulates nutrient stress response in saccharomyces cerevisiae by enforcing transcriptional fidelity. Cell Rep. 2017; 19:2371–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Hsin J.P., Manley J.L.. The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev. 2012; 26:2119–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Yurko N.M., Manley J.L.. The RNA polymerase II CTD “orphan” residues: Emerging insights into the functions of Tyr-1, Thr-4, and Ser-7. Transcription. 2018; 9:30–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Pearson E., Moore C.. The evolutionarily conserved Pol II flap loop contributes to proper transcription termination on short yeast genes. Cell Rep. 2014; 9:821–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Mayer A., Schreieck A., Lidschreiber M., Leike K., Martin D.E., Cramer P.. The spt5 C-terminal region recruits yeast 3′ RNA cleavage factor I. Mol. Cell. Biol. 2012; 32:1321–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Battaglia S., Lidschreiber M., Baejen C., Torkler P., Vos S.M., Cramer P.. RNA-dependent chromatin association of transcription elongation factors and Pol II CTD kinases. Elife. 2017; 6:e25637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Sayou C., Millan-Zambrano G., Santos-Rosa H., Petfalski E., Robson S., Houseley J., Kouzarides T., Tollervey D.. RNA binding by histone methyltransferases set1 and Set2. Mol. Cell. Biol. 2017; 37:e00165-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Luciano P., Jeon J., El-Kaoutari A., Challal D., Bonnet A., Barucco M., Candelli T., Jourquin F., Lesage P., Kim J. et al.. Binding to RNA regulates Set1 function. Cell Discov. 2017; 3:17040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Ng H.H., Robert F., Young R.A., Struhl K.. Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol. Cell. 2003; 11:709–719. [DOI] [PubMed] [Google Scholar]
- 130. Patturajan M., Conrad N.K., Bregman D.B., Corden J.L.. Yeast carboxyl-terminal domain kinase I positively and negatively regulates RNA polymerase II carboxyl-terminal domain phosphorylation. J. Biol. Chem. 1999; 274:27823–27828. [DOI] [PubMed] [Google Scholar]
- 131. Schreieck A., Easter A.D., Etzold S., Wiederhold K., Lidschreiber M., Cramer P., Passmore L.A.. RNA polymerase II termination involves C-terminal-domain tyrosine dephosphorylation by CPF subunit Glc7. Nat. Struct. Mol. Biol. 2014; 21:175–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Mosley A.L., Pattenden S.G., Carey M., Venkatesh S., Gilmore J.M., Florens L., Workman J.L., Washburn M.P.. Rtr1 is a CTD phosphatase that regulates RNA polymerase II during the transition from serine 5 to serine 2 phosphorylation. Mol. Cell. 2009; 34:168–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Bandyopadhyay S., Mehta M., Kuo D., Sung M.K., Chuang R., Jaehnig E.J., Bodenmiller B., Licon K., Copeland W., Shales M. et al.. Rewiring of genetic networks in response to DNA damage. Science. 2010; 330:1385–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Zhang K., Lin W., Latham J.A., Riefler G.M., Schumacher J.M., Chan C., Tatchell K., Hawke D.H., Kobayashi R., Dent S.Y.. The Set1 methyltransferase opposes Ipl1 aurora kinase functions in chromosome segregation. Cell. 2005; 122:723–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Costanzo M., VanderSluis B., Koch E.N., Baryshnikova A., Pons C., Tan G., Wang W., Usaj M., Hanchard J., Lee S.D. et al.. A global genetic interaction network maps a wiring diagram of cellular function. Science. 2016; 353:aaf1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Gopalakrishnan R., Marr S.K., Kingston R.E., Winston F.. A conserved genetic interaction between Spt6 and Set2 regulates H3K36 methylation. Nucleic. Acids. Res. 2019; 47:3888–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Fong N., Saldi T., Sheridan R.M., Cortazar M.A., Bentley D.L.. RNA pol II dynamics modulate co-transcriptional chromatin modification, CTD phosphorylation, and transcriptional direction. Mol. Cell. 2017; 66:546–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Lorenz D.R., Meyer L.F., Grady P.J., Meyer M.M., Cam H.P.. Heterochromatin assembly and transcriptome repression by Set1 in coordination with a class II histone deacetylase. Elife. 2014; 3:e04506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Fan X., Moqtaderi Z., Jin Y., Zhang Y., Liu X.S., Struhl K.. Nucleosome depletion at yeast terminators is not intrinsic and can occur by a transcriptional mechanism linked to 3′-end formation. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:17945–17950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Mavrich T.N., Ioshikhes I.P., Venters B.J., Jiang C., Tomsho L.P., Qi J., Schuster S.C., Albert I., Pugh B.F.. A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res. 2008; 18:1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Lee K.Y., Chen Z., Jiang R., Meneghini M.D.. H3K4 methylation dependent and independent chromatin regulation by JHD2 and SET1 in budding yeast. G3 (Bethesda). 2018; 8:1829–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Kurup J.T., Campeanu I.J., Kidder B.L.. Contribution of H3K4 demethylase KDM5B to nucleosome organization in embryonic stem cells revealed by micrococcal nuclease sequencing. Epigenet. Chromatin. 2019; 12:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Martin B.J., McBurney K.L., Maltby V.E., Jensen K.N., Brind’Amour J., Howe L.J.. Histone H3K4 and H3K36 Methylation Independently Recruit the NuA3 Histone Acetyltransferase in Saccharomyces cerevisiae. Genetics. 2017; 205:1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Fabrizio P., Garvis S., Palladino F.. Histone methylation and memory of environmental stress. Cells. 2019; 8:339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Xue Y., Acar M.. Mechanisms for the epigenetic inheritance of stress response in single cells. Curr. Genet. 2018; 64:1221–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Liu X., Hoque M., Larochelle M., Lemay J.F., Yurko N., Manley J.L., Bachand F., Tian B.. Comparative analysis of alternative polyadenylation in S. cerevisiae and S. pombe. Genome Res. 2017; 27:1685–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Aulak K.S., Mishra R., Zhou L., Hyatt S.L., de Jonge W., Lamers W., Snider M., Hatzoglou M.. Post-transcriptional regulation of the arginine transporter Cat-1 by amino acid availability. J. Biol. Chem. 1999; 274:30424–30432. [DOI] [PubMed] [Google Scholar]
- 148. Liu Y., Hu W., Murakawa Y., Yin J., Wang G., Landthaler M., Yan J.. Cold-induced RNA-binding proteins regulate circadian gene expression by controlling alternative polyadenylation. Sci. Rep. 2013; 3:2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Kraynik S.M., Gabanic A., Anthony S.R., Kelley M., Paulding W.R., Roessler A., McGuinness M., Tranter M.. The stress-induced heat shock protein 70.3 expression is regulated by a dual-component mechanism involving alternative polyadenylation and HuR. Biochim. Biophys. Acta. 2015; 1849:688–696. [DOI] [PubMed] [Google Scholar]
- 150. Tranter M., Helsley R.N., Paulding W.R., McGuinness M., Brokamp C., Haar L., Liu Y., Ren X., Jones W.K.. Coordinated post-transcriptional regulation of Hsp70.3 gene expression by microRNA and alternative polyadenylation. J. Biol. Chem. 2011; 286:29828–29837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. de Lorenzo L., Sorenson R., Bailey-Serres J., Hunt A.G.. Noncanonical alternative polyadenylation contributes to gene regulation in response to hypoxia. Plant Cell. 2017; 29:1262–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Zheng D., Wang R., Ding Q., Wang T., Xie B., Wei L., Zhong Z., Tian B.. Cellular stress alters 3′UTR landscape through alternative polyadenylation and isoform-specific degradation. Nat. Commun. 2018; 9:2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Devany E., Park J.Y., Murphy M.R., Zakusilo G., Baquero J., Zhang X., Hoque M., Tian B., Kleiman F.E.. Intronic cleavage and polyadenylation regulates gene expression during DNA damage response through U1 snRNA. Cell Discov. 2016; 2:16013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Kim J.H., Lee B.B., Oh Y.M., Zhu C., Steinmetz L.M., Lee Y., Kim W.K., Lee S.B., Buratowski S., Kim T.. Modulation of mRNA and lncRNA expression dynamics by the Set2-Rpd3S pathway. Nat. Commun. 2016; 7:13534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Carvalho S., Vitor A.C., Sridhara S.C., Martins F.B., Raposo A.C., Desterro J.M., Ferreira J., de Almeida S.F.. SETD2 is required for DNA double-strand break repair and activation of the p53-mediated checkpoint. Elife. 2014; 3:e02482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Jha D.K., Strahl B.D.. An RNA polymerase II-coupled function for histone H3K36 methylation in checkpoint activation and DSB repair. Nat. Commun. 2014; 5:3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Li F., Mao G., Tong D., Huang J., Gu L., Yang W., Li G.M.. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell. 2013; 153:590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Ryu H.Y., Rhie B.H., Ahn S.H.. Loss of the Set2 histone methyltransferase increases cellular lifespan in yeast cells. Biochem. Biophys. Res. Commun. 2014; 446:113–118. [DOI] [PubMed] [Google Scholar]
- 159. Schramke V., Neecke H., Brevet V., Corda Y., Lucchini G., Longhese M.P., Gilson E., Geli V.. The set1Delta mutation unveils a novel signaling pathway relayed by the Rad53-dependent hyperphosphorylation of replication protein A that leads to transcriptional activation of repair genes. Genes Dev. 2001; 15:1845–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Kanoh J., Francesconi S., Collura A., Schramke V., Ishikawa F., Baldacci G., Geli V.. The fission yeast spSet1p is a histone H3-K4 methyltransferase that functions in telomere maintenance and DNA repair in an ATM kinase Rad3-dependent pathway. J. Mol. Biol. 2003; 326:1081–1094. [DOI] [PubMed] [Google Scholar]
- 161. Faucher D., Wellinger R.J.. Methylated H3K4, a transcription-associated histone modification, is involved in the DNA damage response pathway. PLoS Genet. 2010; 6:e1001082. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.