

Abstract
Epithelial plasticity – reversible modulation of a cell’s epithelial and mesenchymal features – is associated with tumor metastasis and chemoresistance, leading causes of cancer mortality. While different master transcription factors and epigenetic modifiers have been implicated in this process in various contexts, the extent to which a unifying, generalized mechanism of transcriptional regulation underlies epithelial plasticity remains largely unknown. Here, through targeted CRISPR-Cas9 screening, we discovered two histone-modifying enzymes involved in the writing and erasing of H3K36me2 that act reciprocally to regulate epithelial-mesenchymal identity, tumor differentiation, and metastasis. Using a K-to-M histone mutant to directly inhibit H3K36me2, we found that global modulation of the mark is a conserved mechanism underlying the mesenchymal state in various contexts. Mechanistically, regulation of H3K36me2 reprograms enhancers associated with master regulators of epithelial-mesenchymal state. Our results thus outline a unifying epigenome-scale mechanism by which a specific histone modification regulates cellular plasticity and metastasis in cancer.
Keywords: Cellular plasticity, metastasis, epigenetics, histone modifications, EMT
INTRODUCTION
Cancer cells are capable of dramatic changes in phenotype and function, a phenomenon known as cellular plasticity (1). Epithelial-to-mesenchymal transition (EMT, i.e. epithelial plasticity), and its reverse process (MET), are among the most widely studied examples of cellular plasticity; these programs are utilized during embryonic development for tissue morphogenesis and are frequently reactivated during tumor progression (2). In particular, histological and transcriptomic signatures relevant to EMT are frequently associated with metastasis, therapeutic resistance, and other factors portending poor prognosis and patient mortality (3).
The cellular hallmarks of EMT, which include repression of epithelial proteins such as E-cadherin, Occludin, and Zo-1, and induction of mesenchymal proteins such as N-cadherin, Vimentin, and Fibronectin, are classically induced by a small cohort of master EMT-related transcription factors (“EMT-TFs”) including Snai1/2, Twist1/2, and Zeb1/2 (2). However, the relative importance of each of these transcription factors varies depending on experimental and clinical contexts, leading to seemingly conflicting reports on the roles they play in EMT and cancer progression (4–6). Such findings raise the possibility that a more generalized mechanism or common molecular feature might exist that could unify the activity of these EMT-TFs.
Epigenetic modifiers and complexes can enact broad, genome-wide changes in transcription; such global epigenetic alterations have been reported to underlie other cell fate changes, particularly during normal embryonic development (7,8). In tumor cells, several EMT-TFs have previously been reported to functionally cooperate with various epigenetic modifiers (9); however, such studies have focused on interactions at the promoters of pre-selected epithelial and mesenchymal genes (10–16). And while previous profiling efforts have identified genome-scale changes in DNA methylation (17) and certain histone marks (18) during experimentally-induced EMT, it remains uncertain whether these epigenetic changes are necessary and/or sufficient to induce cancer cell plasticity or whether they represent correlated (secondary) events. In the current study, we took an unbiased genetic approach to identify epigenome-wide mechanisms driving epithelial plasticity and metastatic progression, and directly interrogated the functional role of specific epigenetic modifications in cancer cell identity.
RESULTS
Modeling epithelial plasticity in vitro
To better understand epigenetic mechanisms underlying epithelial plasticity, we established an experimental system relying on stochastic changes in cell state rather than genetic or chemical induction. We began by utilizing a panel of single cell clones derived from the KPCY (LSL-KrasG12D; P53loxP/+; Pdx1-cre; LSL-RosaYFP/YFP) mouse model of pancreatic ductal adenocarcinoma (PDAC) (19). Clones retained both epithelial and mesenchymal subpopulations, as defined by the presence or absence of surface E-cadherin (Ecad) (20) and other classical epithelial or mesenchymal markers (Figures 1A–B and Supplementary Figure S1A). To confirm that epithelial plasticity is associated with global differences in chromatin landscape, we performed ATAC-seq on FACS-sorted Ecad+ versus Ecad− cells and identified chromatin regions exhibiting differential accessibility. Ecad− cells exhibited greater accessibility in genes associated with a mesenchymal phenotype and/or epithelial-mesenchymal transition (EMT) (Figure 1C and Supplementary Figure S1B). By contrast, Ecad+ cells exhibited greater accessibility in genes associated with an epithelial phenotype, including Ecad itself (Figure 1C). Thus, FACS sorting based on surface Ecad expression isolates cell populations with distinct chromatin profiles, reflecting transcriptional programs related to epithelial-mesenchymal state.
Figure 1: An epigenetic modifier-focused CRISPR-Cas9 screen identifies Nsd2 and Kdm2a as reciprocal regulators of epithelial-mesenchymal differentiation.
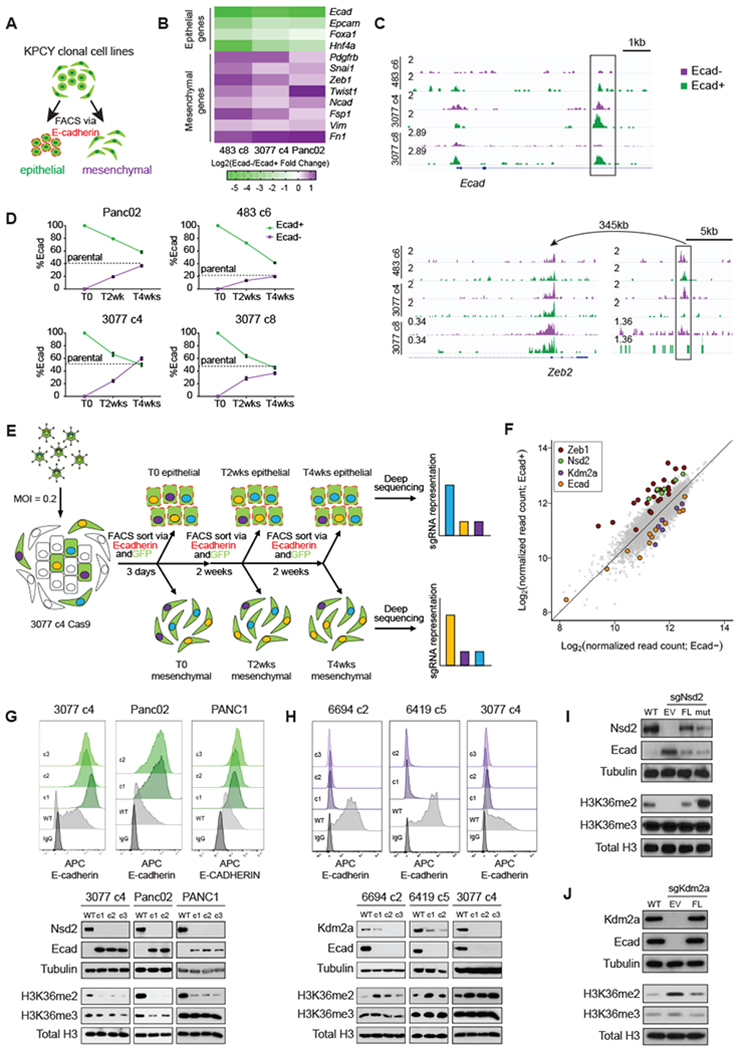
(A) Flow cytometry scheme for quantifying or sorting epithelial and mesenchymal subpopulations from clonal mouse pancreatic ductal adenocarcinoma (PDAC) cell lines. Epithelial subpopulations are identified by positive surface E-cadherin staining (Ecad+, outlined in red), while mesenchymal subpopulations are identified by the absence of surface E-cad (Ecad−).
(B) Heatmap summarizing triplicate qPCR experiments on sorted Ecad− and Ecad+ subpopulations for each of 3 cell lines. Increase and decrease in Ecad−/Ecad+ log2fold change is shown in purple and green, respectively.
(C) Representative ATAC-seq tracks of sorted Ecad− and Ecad+ subpopulations from 3 cell lines, shown in purple and green, respectively. Statistically significant (padj<0.1) enrichment peaks are boxed. Arrow indicates relationship between the Zeb2 promoter and a putative distal regulatory element.
(D) Summary of plasticity experiments performed on 4 cell lines. Sorted Ecad− and Ecad+ subpopulations were replated and passaged over the course of 4 weeks. Samples were reassessed for Ecad+% by flow cytometry every 2 weeks. Data are means ± SEM from biological replicates (n=3). Dotted lines represent the Ecad+% of the bulk, unsorted parental population of each cell line.
(E) Experimental outline for focused CRISPR screen. See materials and methods for a full description.
(F) sgRNA representation in Ecad− (x-axis) and Ecad+ (y-axis) populations as log2-transformed normalized read counts.
(G-H) Top: Representative Ecad flow histograms for clones expressing sgNsd2 (G)and sgKdm2a (H) compared to Cas9-only wildtype (WT) controls, as well as IgG-staining control for 3 cell lines. Bottom: Corresponding Western blots of cell lysates (top panels) and acid-extracted histones (bottom panels) with antibodies against the indicated proteins.
(I-J) Western blots of cell lysates (top panels) and acid-extracted histones (bottom panels) of Cas9-only wildtype (WT) controls and sgNsd2 (I) and sgKdm2a clones (J) expressing empty vector (EV), full-length (FL), or E1099K NSD2 (mut).
Next, we sought to ensure that the Ecad+ and Ecad− fractions do not represent fixed, non-plastic subpopulations. To this end, we sorted pure populations of Ecad+ or Ecad− cells. Over a 2-4-week period after initial sorting, all cultures exhibited phenotypic reversion (i.e. Ecad+ cells became Ecad−, and Ecad− cells became Ecad+) (Figure 1D). Remarkably, all cultures returned to an equilibrium that recapitulated the Ecad+/Ecad− ratio observed in the parental culture (Figure 1D, dotted line). Thus, these cell clones exhibit dynamic plasticity (i.e. both epithelial and mesenchymal transitions) under standard culture conditions.
CRISPR-Cas9 screen identifies epigenetic regulators important for EMT
To identify epigenetic modifiers important for plasticity, we carried out a focused CRISPR-Cas9 screen using an sgRNA library targeting ~600 epigenetic modifiers and controls in a clonal cell line (3077 c4) stably expressing Cas9 (Figure 1E). As expected, control sgRNAs targeting Zeb1 – which we had identified as the most potent EMT-TF in this cell line (Supplementary Figure S2A–D) – were significantly enriched in the Ecad+ population and depleted in the Ecad− fraction. Conversely, sgRNAs targeting Ecad exhibited the opposite pattern (Figure 1F). Furthermore, sgRNAs targeting ribosomal proteins were depleted in both fractions, while the representation of control non-targeting sgRNAs did not change (Supplementary Figures S3A–B).
Among the sgRNAs showing significant enrichment or depletion in the Ecad+ or Ecad− populations (Supplementary Table S1), nuclear receptor binding SET domain protein 2 (Nsd2) emerged as the top gene for which all targeting sgRNAs were significantly enriched in the Ecad+ subpopulation (Figure 1F and Supplementary Figure S3C), second only to Zeb1. Nsd2 is a histone methyltransferase that di-methylates histone H3 at lysine-36 (H3K36me2), a mark associated with actively transcribed genes (21), and acts as an oncogenic driver in multiple myeloma (22) and a regulator of invasion and metastasis in prostate cancer (23–25). Conversely, sgRNAs targeting lysine-specific demethylase 2A (Kdm2a) were significantly overrepresented in the Ecad− subpopulation (Figure 1F and Supplementary Figure S3D). Kdm2a is a demethylase that preferentially targets H3K36me2 (26–28). Both Nsd2 and Kdm2a have paralogs with similar catalytic activities, but none of these were enriched or depleted in our screen (Supplementary Figures S3C–F), possibly due to low levels of expression (Supplementary Figure S3G). These results suggest that Nsd2 and Kdm2a act non-redundantly on H3K36 as determinants of the mesenchymal or epithelial state, respectively.
To validate the role of Nsd2 and Kdm2a in epithelial plasticity, we performed gain- and loss-of-function studies in a panel of mouse and human PDAC cell lines. Nsd2 loss led to an increase in Ecad levels and decrease in H3K36me2 levels (Figure 1G and Supplementary Figure S4), while loss of Kdm2a resulted in reduced Ecad and increased H3K36me2 (Figure 1H). Both of these phenotypes could be rescued by re-expression of the appropriate gene (Figure 1I and J). Importantly, despite lower expression of the catalytically active mutant NSD2 (E1099K) (29), this mutant potently reversed the effects of Nsd2 loss, suggesting that Nsd2’s histone methyltransferase activity is critical for EMT. Notably, these manipulations had little effect on H3K36me3 levels, in line with previous observations (22,30), implying that gain or loss of the H3K36me2 mark is regulated independently of effects on higher order methylation. The opposing catalytic activities of Nsd2 and Kdm2a, together with their shared substrate specificity, thus implicate a mechanism wherein the writing and erasing of H3K36me2 regulates a cell’s position along the epithelial-mesenchymal spectrum.
Nsd2 and Kdm2a reciprocally regulate tumor differentiation and tumor initiation capacity
To understand the transcriptional changes regulated by Nsd2 and Kdm2a, we carried out RNA sequencing (RNA-seq) and gene set enrichment analyses (GSEA) in clonal wild type, sgNsd2, and sgKdm2a cells (Supplementary Tables S2 and S3). “EMT” was among the most significantly affected hallmark gene sets (Figure 2A), and many classic markers of EMT were downregulated with Nsd2 loss and upregulated with Kdm2a loss (Supplementary Figure S5A–D). Importantly, many other hallmark and curated gene sets enriched following Nsd2 loss were negatively enriched by Kdm2a loss, and vice versa (Supplementary Figure S6A–C), suggesting that these enzymes regulate a shared transcriptomic program in a reciprocal manner.
Figure 2: Nsd2 and Kdm2a have reciprocal effects on tumor differentiation and tumor initiation.
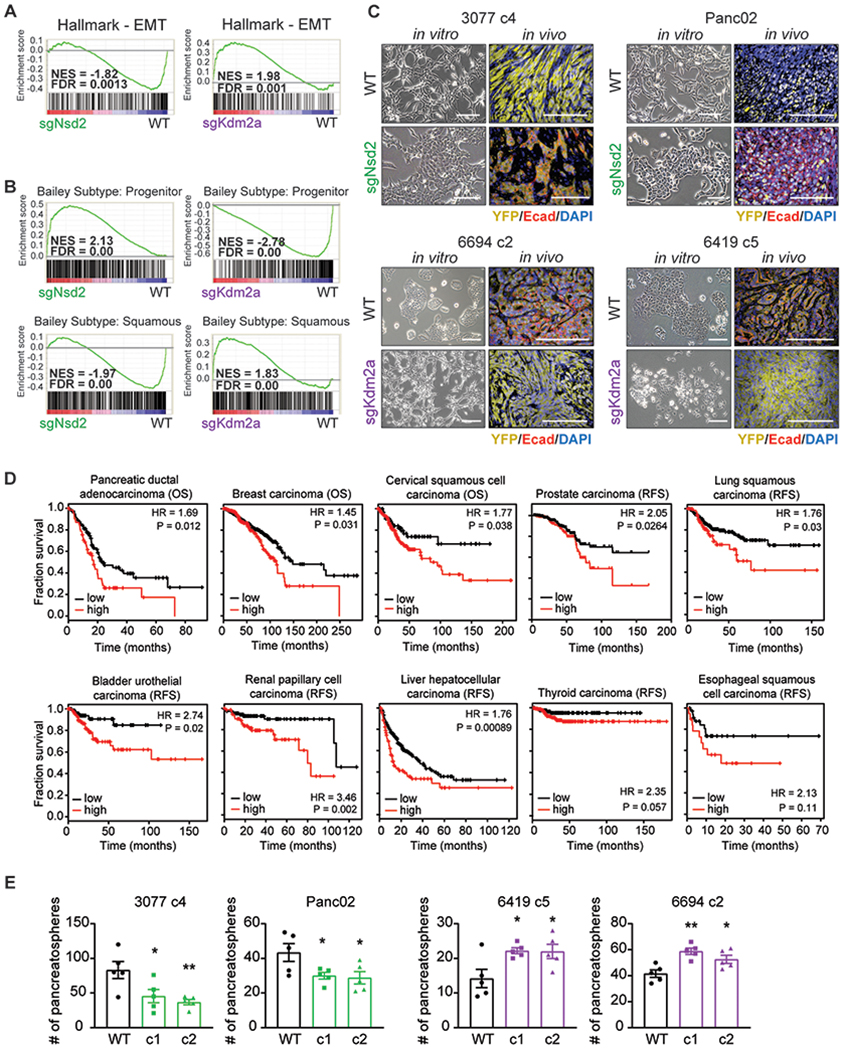
(A) Gene set enrichment analyses (GSEA) of WT versus sgNsd2 (left) or sgKdm2a (right) RNA-seq using the EMT Hallmark signature. Normalized enrichment score (NES) and false discovery rate (FDR) are shown alongside.
(B) GSEA plots evaluating the Bailey squamous and progenitor signatures upon loss of Nsd2 (left) or Kdm2a (right).
(C) Representative in vitro brightfield (BF, left) and in vivo immunofluorescent (IF, right) images of orthotopic tumors derived from cell lines with the indicated genotypes. Co-staining for Ecad (red), YFP (yellow), and DAPI (blue). Scale bars = 100μm.
(D) Kaplan-Meier plots of overall survival (OS) or relapse-free survival (RFS) of patients, stratified by high (red) or low (black) expression of a gene signature that represents the average expression level of NSD1, NSD2, and NSD3. Median expression of this gene signature was used as the cutoff. RNA-seq and clinical data was obtained from the KM plotter database and includes datasets deposited in TCGA and GEO (Nagy et al., 2018). P-value was calculated by log rank test.
(E) Number of pancreatospheres formed from 2,500 cells of the indicated cell lines and genotype (n=5 technical replicates, error bars indicate SEM). Statistical analysis by Student’s unpaired t-test with significance indicated (*, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001).
In the clinical setting, the squamous subtype of PDAC describes a subset of tumors that are poorly differentiated and of particularly poor prognosis (31–33). Using gene expression signatures that discriminate the squamous subtype from the well-to-moderately differentiated progenitor subtype in patient samples (34), we found that Nsd2 loss results in negative enrichment of the squamous signature and positive enrichment of the progenitor signature, with a converse pattern observed in the setting of Kdm2a loss (Figure 2B). These subtype changes are also reflected in the differentiation status of cultured cells and tumors arising from these cells (Figure 2C). These results suggest that Nsd2 and Kdm2a exert opposing effects on cellular transcriptomes, leading to changes in tumor subtype and differentiation.
Since Nsd2 appears to drive a PDAC subtype of particularly poor prognosis, we wished to evaluate its prognostic value across various cancer types. Nsd2 and its paralogs are all able to di-methylate H3K36 and are each catalytically dominant in different tissue types (35–37). We thus used a gene signature representing the average expression of NSD1, NSD2, and NSD3 to more broadly capture the consequences of increased H3K36me2 deposition on clinical outcomes. Upon applying this gene signature to transcriptomic databases across various carcinomas (38,39), we found that high expression of the signature predicted poor prognosis (Figure 2D). By contrast, the expression of individual members of the NSD family had substantially less predictive value (data not shown). These results suggest that elevated expression of writers of H3K36 di-methylation is associated with poor clinical outcome, underscoring the importance of considering shared catalytic activities or molecular functions in relation to tumor behavior.
Tumor initiating cells (TICs) – a subpopulation of tumor cells capable of seeding and propagating new tumors – are commonly enriched for cells that are transcriptionally characterized by EMT signatures (40). Furthermore, induction of an EMT program has been shown to directly regulate the TIC phenotype (10). Accordingly, we found that forcing cells to be epithelial by loss of Nsd2 decreased their ability to form 3D tumor spheres, while locking cells in the mesenchymal state by loss of Kdm2a increased their sphere-forming ability, suggesting that these enzymes reciprocally regulate tumor initiation capacity (Figure 2E). Collectively, these findings demonstrate that regulators of the histone mark H3K36me2 drive attributes of tumor progression that are associated with poor clinical outcome.
Nsd2 and Kdm2a-induced changes in epithelial plasticity have unique contributions to metastasis
Although epithelial-mesenchymal plasticity is thought to drive metastatic dissemination, recent studies have called this paradigm into question (5,6,41). To investigate the contributions of the epithelial and mesenchymal states to metastasis, we orthotopically transplanted tumor cells and found that Kdm2a-deficient tumors (locked in a mesenchymal state) released more circulating tumor cells (CTCs) into the bloodstream compared to Cas9 control tumors (Figure 3A), which is consistent with their increased migratory behavior in vitro (Supplementary Figures S7A–B). Conversely, Nsd2-deficient tumors (locked in an epithelial state) released few CTCs into the bloodstream (Figure 3A). These results support the notion that cells with a mesenchymal phenotype enter the bloodstream far more efficiently than cells with an epithelial phenotype.
Figure 3: Nsd2 and Kdm2a mediate reciprocal effects on metastatic progression.
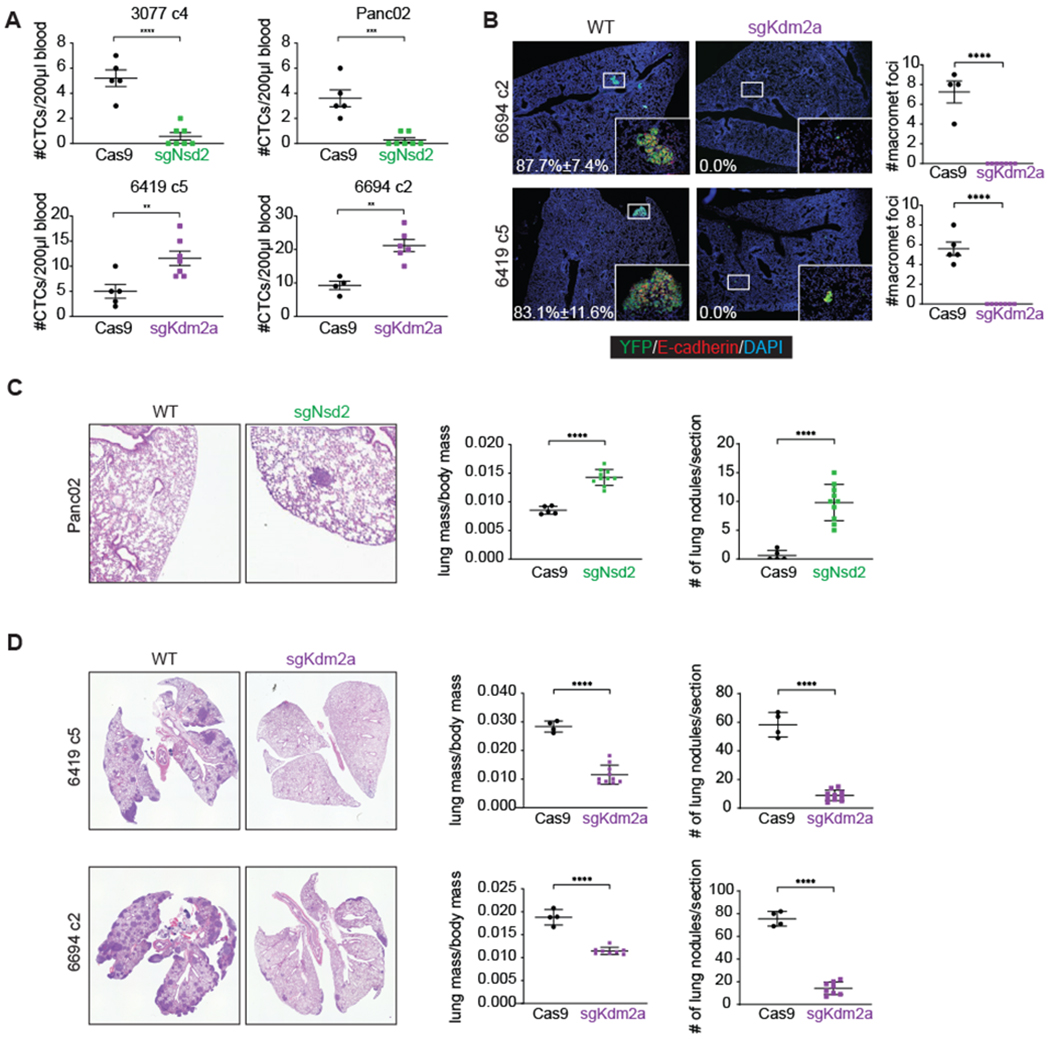
(A) Quantification of CTCs/200μl blood from mice harboring orthotopic tumors from cell lines with the indicated genotypes. For Cas9 controls, n=4-5 mice/cell line. For sgNsd or sgKdm2a, n=2 clones/genotype, n=3-5 mice/clone.
(B) Representative IF images (left) and quantification (right) of macrometastatic foci (≥ 100 cells) in lungs of mice bearing orthotopic tumors. Percentages of Ecad+YFP+ tumor cells out of total YFP+ tumor cells are indicated in white. Co-staining for Ecad (red), YFP (green), and DAPI (blue).
(C-D) Representative H&E images and quantifications of pulmonary metastatic burden 14 days after tail vein injection of sgNsd2 (C) and sgKdm2a (D) cells; pulmonary metastatic burden was determined by both lung/body mass ratio and number of lung nodules per section. For Cas9 controls, n=4-5 mice/cell line. For sgNsd or sgKdm2a, n=2 clones/genotype, n=4-5 mice/clone. Statistical analysis by Student’s unpaired t-test with significance indicated (*, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001; ns, not significant).
While Kdm2a-deficient tumor cells readily intravasated into circulation, the resulting metastatic lesions found in the lungs were significantly smaller than those arising in their wild type counterparts (Figure 3B). To further study the effect of epithelial-mesenchymal state on metastatic colonization, we injected Nsd2- and Kdm2a-deficient tumor cells directly into the tail veins of mice, thereby bypassing the first steps of the metastatic cascade. Nsd2-deficient cells were more efficient than control cells at forming large lung metastases (Figure 3C), while Kdm2a-deficient cells remained poor colonizers (Figure 3D). Importantly, these differences in metastatic competence were not due to differences in tumor growth or cell proliferation (Supplementary Figures S7C–D). Taken together, these results demonstrate that epithelial and mesenchymal phenotypes each have their own distinct advantages during metastatic spread, supporting the concept that EMT and MET contribute at different points in the metastatic cascade and that plasticity is more important for effective metastasis than either the epithelial or mesenchymal state alone (42–46).
Global increase in H3K36me2 is an epigenomic feature associated with the mesenchymal state
Nsd2 and Kdm2a act antagonistically to regulate H3K36me2, strongly indicating that this histone mark is a key determinant of epithelial identity. To test this hypothesis, we first performed histone mass spectrometry to look for global changes associated with epithelial plasticity. Remarkably, H3K36me2 was the only histone post-translational modification (PTM) to exhibit a significant difference (fold change>1.5, padj<0.05) between Ecad− and Ecad+ cells across multiple cell lines (Figure 4A and Supplementary Figures S8A–B). In this setting, the enrichment of H3K36me2 in mesenchymal cells was accompanied by higher expression of Nsd2 (Figure 4B). Differences in global levels of H3K36me2 were readily detected by western blots of sorted Ecad+ and Ecad− cells (Supplementary Figure S8C), and in cultured cells (Supplementary Figure S8D) and PDAC tumors (Supplementary Figure S8E) stained for H3K36me2. Thus, transition to a mesenchymal state is reproducibly associated with global accumulation of H3K36me2 in multiple in vitro and in vivo settings.
Figure 4: Upregulation of H3K36me2 is associated with the mesenchymal state.
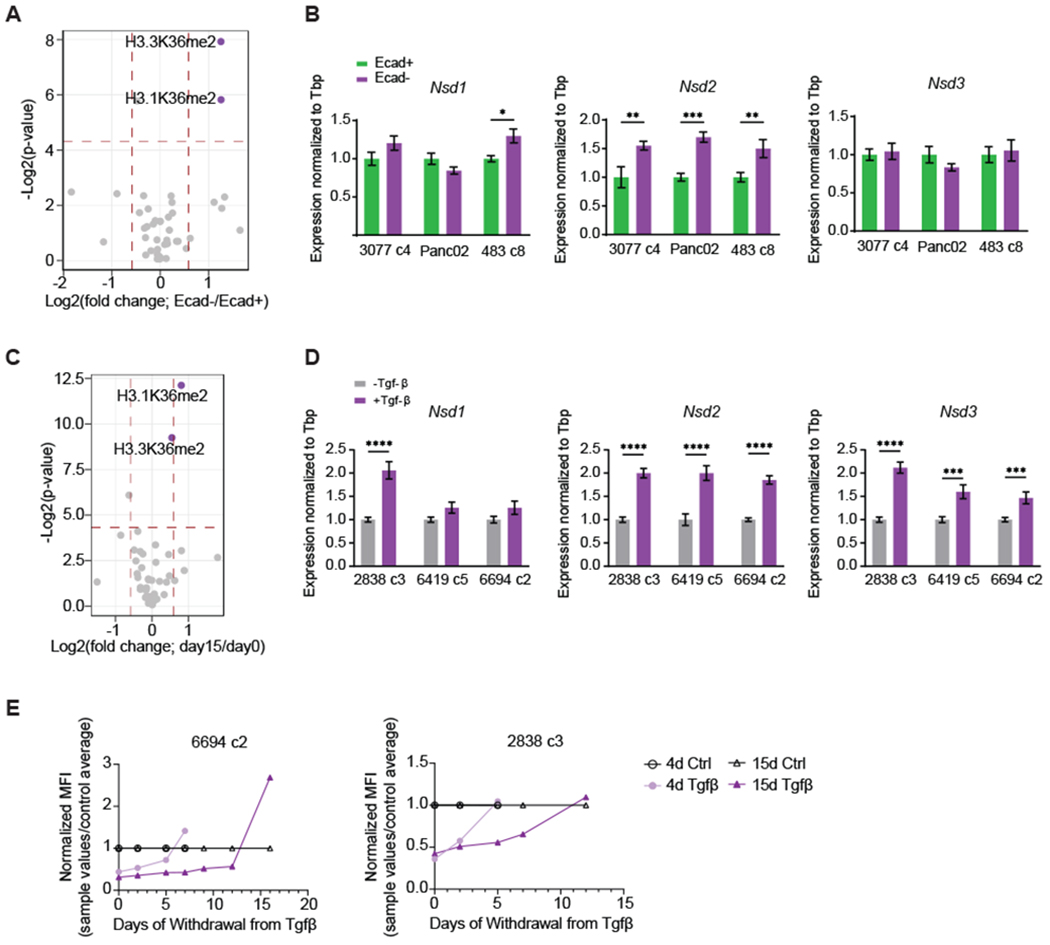
(A) Volcano plot presenting log2-transformed p-value and fold change of histone H3 and H4 single post-translational modification (PTM) relative abundance derived from mass spectrometry for Ecad− vs. Ecad+ sorted cells. Two variants for histone H3, the canonical H3.1 and non-canonical H3.3, which are both known to accumulate K36me2, are shown. Horizontal dotted line demarcates Student’s unpaired t-test p-value < 0.05, while vertical dotted lines demarcate ± 1.5-fold change (n=3 cell lines). H3K36me2 marks are labeled and represented in purple. When Holm Sidak correction for multiple testing is applied, Padj=0.05 (H3.1K36me2) and 0.028 (H3.3K36me2).
(B) Relative mRNA expression of Nsd1, Nsd2, and Nsd3 in sorted Ecad+ (green) and Ecad− (purple) cells. Results from 3 different cell lines are shown. Bars represent mean ± SEM.
(C) Volcano plot presenting log2-transformed p-value and fold change of histone H3 and H4 single post-translational modification (PTM) relative abundance derived from mass spectrometry for cells undergoing 15 days of Tgf-β treatment vs. citric acid control. Horizontal dotted line demarcates Student’s unpaired t-test p-value < 0.05, while vertical dotted lines demarcate ± 1.5-fold change (n=3 cell lines). When Holm Sidak correction for multiple testing is applied, Padj=0.00001 (H3.1K36me2 and H3.3K36me2).
(D) Relative mRNA expression of Nsd1, Nsd2, and Nsd3 in cells treated with Tgf-β for 15 days. Results from 3 different cell lines are shown. Bars represent mean ± SEM.
(E) Normalized mean fluorescence intensity (MFI) of Ecad in control cells and cells treated with Tgf-β for 4 or 15 days, followed by withdrawal of Tgf-β for the number of days indicated on the x-axis. MFI for the 4 and 15-day treated samples were normalized to their respective controls. Experiments were performed in 2 cell lines.
To determine whether differences in H3K36 methylation also occur in the setting of experimentally induced plasticity, we treated cells with Tgf-β, a potent EMT stimulus, and again found that H3K36me2 was the only histone PTM to exhibit a global increase across cell lines (Figure 4C). Upon Tgf-β stimulation, different cell lines upregulated the expression of different H3K36me2 methyltransferases, each of which can contribute to the observed global increase in H3K36me2 (Figure 4D). Interestingly, despite overt morphological changes within 4 days of Tgf-β treatment (Supplementary Figure S9A), a build-up of H3K36me2 was not apparent until 15 days of treatment (Supplementary Figures S9B–E), suggesting that global H3K36me2 accumulation may be a gradual phenomenon that helps to maintain the mesenchymal phenotype. To explore this possibility, we withdrew Tgf-β after 4 or 15 days of treatment and assessed how long it took cells to revert to an epithelial state. While the effects of short-term Tgf-β treatment were readily reversible upon Tgf-β withdrawal, cells exposed to Tgf-β for 15 days remained mesenchymal for much longer (Figure 4E). Eventually, when these cells finally reverted to their original epithelial state (Figure 4E and Supplementary Figure S10A), their H3K36me2 levels also began returning to baseline (Supplementary Figure S10B), demonstrating that epigenetic plasticity accompanies phenotypic plasticity. Collectively, these results suggest that Tgf-β-induced EMT involves an acute phase, likely mediated by the early activity of EMT-TFs, and a stable “memory” phase, reinforced by a broader regulation of H3K36me2 levels.
Upregulation of H3K36me2 is essential for EMT
Since Tgf-β is such a powerful EMT agent, we wondered whether Nsd2-deficient cells would fail to respond to this stimulus. Surprisingly, Nsd2-deficient cells treated with Tgf-β exhibited a typical EMT response, characterized by the repression of epithelial genes and induction of mesenchymal genes (Figure 5A). Strikingly, EMT was still accompanied by an increase in H3K36me2 levels despite total absence of Nsd2 (Figure 5B). This result suggests that in the presence of a strong EMT stimulus, Nsd2-deficient cells compensate by finding other ways to upregulate the mark, indicating that H3K36me2 accumulation itself is essential for the mesenchymal identity.
Figure 5: Upregulation of H3K36me2 is essential for EMT.
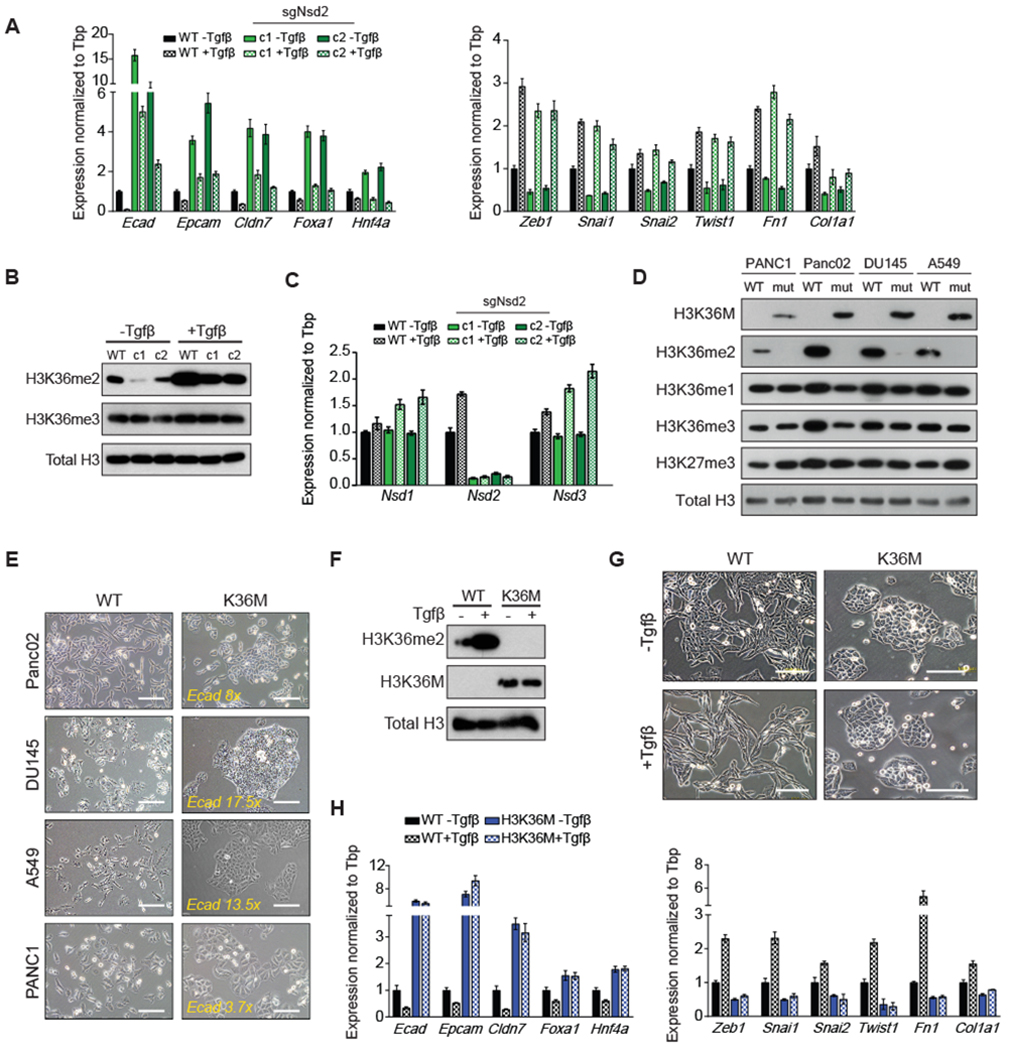
(A) Relative mRNA expression of epithelial genes (left) and mesenchymal genes (right) in WT and sgNsd2 clones, with or without 15 days of Tgf-β treatment, as determined by qPCR (n = 3, mean ± SEM).
(B) Western blots of acid-extracted histones from WT and sgNsd2 clones, with or without 15 days of Tgf-β treatment.
(C) Relative mRNA expression of Nsd1, Nsd2, and Nsd3 in WT and sgNsd2 clones, with or without 15 days of Tgf-β treatment, as determined by qPCR (n = 3, mean ± SEM).
(D-E) Western blots of acid-extracted histones (D) and BF images (E) from cell lines expressing WT or K36M H3F3A (scale bars = 100μm). Fold change increases in Ecad mRNA expression in K36M samples were determined by qPCR (see Supplementary Figure S11) and indicated in yellow.
(F-H) Western blots of acid-extracted histones (F), BF images (scale bars = 100μm) (G), and relative mRNA expression of epithelial genes (left) and mesenchymal genes (right) (H) from cells expressing WT or K36M H3F3A, with or without 15 days of Tgf-β treatment. mRNA expression was determined by qPCR (n = 3, mean ±SEM).
In the absence of Nsd2, other enzymes can still di-methylate H3K36. In fact, upon Tgf-β stimulation, Nsd2 paralogs were upregulated to a greater extent in the sgNsd2 clones compared to the wildtype (Figure 5C), potentially explaining how Nsd2-deficient cells treated with Tgf-β can still generate H3K36me2 and undergo EMT. To circumvent such compensatory mechanisms and more directly interrogate the biological role of this histone mark, we employed a mutant form of H3.3 with a Lys-to-Met substitution at position 36 (K36M), which reduces the overall abundance of H3K36me2 by dominantly inhibiting Nsd2 and other H3K36 methyltransferases (Figure 5D and refs. 36,47,48). In contrast to prior observations (35,36,47), we did not observe consistent decreases in H3K36me1 or H3K36me3.
Expression of H3.3K36M in cell lines from multiple tumor types, including PDAC (Panc02 and PANC1), lung carcinoma (A549), and prostate carcinoma (DU145), was sufficient to induce an epithelial phenotype (Figure 5E and Supplementary Figure S11). However, in contrast to Nsd2-deficient cells, K36M-expressing cells failed to accumulate H3K36me2 following Tgf-β treatment (Figure 5F) and were refractory to its EMT-inducing effects (Figures 5G–H). These results indicate that increased H3K36me2 is essential for stabilizing the mesenchymal state across various cancer types, and that failure to accumulate H3K36me2 impedes a cell’s ability to undergo EMT.
Genome-wide regulation of H3K36me2 reprograms enhancers associated with master EMT transcription factors
To better understand the epigenomic mechanisms by which H3K36me2 influences epithelial plasticity, we performed a series of ChIP-seq experiments. While loss of Nsd2 led to global reductions in H3K36me2, some genomic regions maintained this mark (Supplementary Figure S12A, boxed regions). We therefore performed separate analyses of genomic loci that exhibit dynamic changes in H3K36me2 (“H3K36me2-regulated”) versus loci that retain the mark (“H3K36me2-stable”) in Nsd2-deficient cells. H3K36me2-regulated domains were associated with loss of the activating marks H3K27ac and H3K4me1 and an increase in the repressive mark H3K27me3 (Figure 6A and Supplementary Figure S12B, top). By contrast, these changes were not observed in H3K36me2-stable regions (Figure 6B and Supplementary Figure S12B, bottom). This suggests that dynamically regulated H3K36me2 domains encompass a subset of H3K27ac-containing regulatory elements.
Figure 6: Changes in H3K36me2 levels reprogram enhancers associated with master EMT regulatory factors.
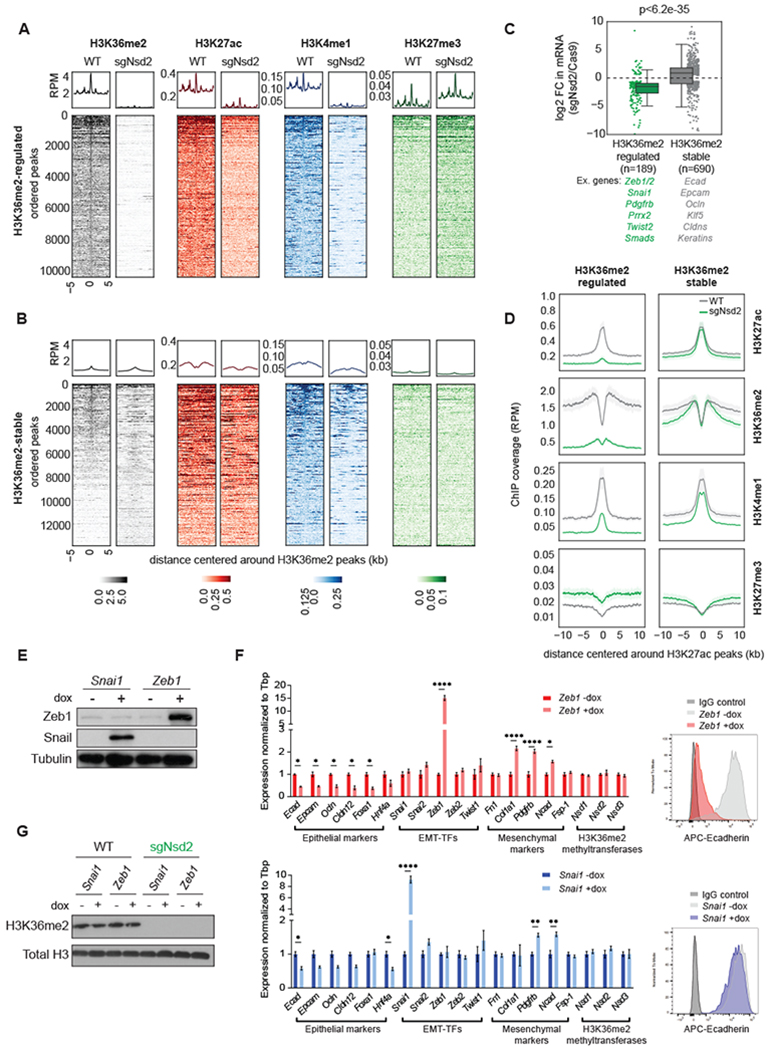
(A-B) Aggregate plots and heatmaps of ChIP signal of the indicated histone marks in WT and sgNsd2 samples, centered around H3K36me2 peaks that are lost with sgNsd2 (i.e. H3K36me2-regulated, n=10312 total peaks) (A) and H3K36me2 peaks that are not lost with sgNsd2 (i.e. H3K36me2-stable, n=13575 total peaks) (B).
(C) Boxplots of log2fold change of mRNA expression of genes associated with H3K36me2-regulated peaks (n=189 genes) and H3K36me2-stable peaks (n=690 genes). Examples are listed below each group.
(D) Aggregate plots comparing the average ChIP signal of the indicated histone marks in WT (grey) and sgNsd2 (green) samples centered around putative enhancers, defined here as H3K27ac peaks that are more than +/− 2.5kb outside of TSS.
(E) Western blots of cell lysates from sgNsd2 cells expressing dox-inducible Zeb1 or Snai1 expression vectors. Cells were treated with dox for 4 days or left untreated.
(F) Relative mRNA expression of epithelial markers, EMT-TFs, mesenchymal markers, and H3K36me2 methyltransferases (left) and flow plots of Ecad staining (right) in sgNsd2 cells expressing dox-inducible Zeb1 (top) or Snai1 (bottom) expression vectors. Cells were treated with dox for 4 days or left untreated. Bars represent mean ± SEM. Statistical analysis by Student’s unpaired t-test with significance indicated (*, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001).
(G) Western blots of acid-extracted histones from WT and sgNsd2 cells expressing dox-inducible Zeb1 or Snai1 expression vectors. Cells were treated with dox for 4 days or left untreated.
H3K27ac is a feature of both active promoters and enhancers. Since dynamic changes in H3K36me2 levels are most apparent in intergenic regions (refs. 22,36,47,49 and Supplementary Figure S12C), we hypothesized that H3K36me2 modulates epithelial plasticity by regulating enhancer activity. To test this, we examined the spatial distribution of H3K36me2, analyzing separately genomic regions near transcriptional start sites (putative promoters) versus more distal elements (putative enhancers). Consistent with our hypothesis, almost all H3K36me2-regulated domains were in distal elements, while a more substantial fraction of H3K36me2-stable domains was found in promoter regions (Supplementary Figure S12D). Similarly, H3K27ac peaks lost with Nsd2 depletion were more likely to be found in distal elements than those that were gained or unchanged (Supplementary Figure S12E). In addition, we compared H3K36me2 and H3K27ac coverage in distal elements and promoters in the presence or absence of Nsd2. Promoter-localized H3K36me2 domains and H3K27ac were maintained with loss of Nsd2 (Supplementary Figure 12F, right panels). By contrast, most H3K36me2 domains in distal elements disappeared following Nsd2 loss, a shift that was associated with large decreases in H3K27ac (Supplementary Figure 12F, left panels). These results suggest that H3K36me2-regulated domains are enriched for distal regulatory elements (i.e. enhancers), while H3K36me2-stable domains are enriched for promoters.
Finally, we examined the effects of H3K36me2 on gene expression by analyzing the transcriptional consequences of Nsd2 loss in H3K36me2-regulated and -stable regions. Genes associated with H3K36me2-regulated domains exhibited reduced expression following Nsd2 loss compared to genes associated with H3K36me2-stable domains (Figure 6C and Supplementary Figure S13A). Remarkably, although H3K36me2-regulated domains were widely dispersed across the genome (>10,000 peaks), we found them to be associated with relatively few genes (n=189), including many pro-metastatic and EMT master regulatory factors (Figure 6C and Supplementary Figures S13B–C). By contrast, the expression of genes associated with H3K36me2-stable domains was mostly unchanged or upregulated with Nsd2 loss and included markers of epithelial cell identity or differentiation (Figure 6C and Supplementary Figure S13B). Interestingly, H3K36me2-regulated and H3K36me2-stable genes are associated with distinct enhancer and promoter activities. Both enhancer (Figure 6D, left) and promoter (Supplementary Figure S13D, top) activities of H3K36me2-regulated genes are directly affected by H3K36me2 levels. And, as expected, the enhancer activities of H3K36me2-stable genes remained largely unchanged (Figure 6D, right). However, while H3K36me2 levels remained unchanged with Nsd2 loss at the promoters of H3K36me2-stable genes, including Ecad, H3K27ac coverage still increased at these promoters (Supplementary Figure S13D, bottom), suggesting that transcriptional activation at these sites is independent of H3K36me2 levels. These results indicate that H3K36me2 influences plasticity by altering enhancer activity, as well as corresponding promoter activity, to regulate the transcription of key regulatory factors (i.e. EMT-TFs).
Master EMT transcription factors act downstream of H3K36me2 activity
While previous studies have found that EMT-TFs like Snai1, Snai2, and Zeb1 physically and functionally interact with epigenetic modifiers to drive changes in gene expression (10–16), our analyses suggest that there is a level of epigenetic regulation upstream of EMT-TF activity. To test this, we overexpressed Snai1 or Zeb1 in cells lacking Nsd2 (Figure 6E). We found that either EMT-TF was sufficient to induce an EMT transcriptional program by downregulating epithelial markers and upregulating mesenchymal markers (Figure 6F). In agreement with our earlier finding that Zeb1 is the major driver of EMT in our PDAC cell lines, Snai1 was less effective than Zeb1 at rescuing the EMT phenotype. Interestingly, and in contrast to Nsd2 (Supplementary Figure S5), neither Zeb1 nor Snai1 appeared important for the expression of other EMT-TFs, suggesting that the level of H3K36 di-methylation, rather than transcription factor crosstalk, is responsible for broad EMT-TF upregulation during EMT. Finally, neither Zeb1 nor Snai1 overexpression induced the expression of Nsd1, Nsd2, or Nsd3 (Figure 6F) or resulted in increased H3K36me2 levels in Nsd2-deficient cells (Figure 6G). Collectively, these results indicate that these master regulatory factors act downstream of H3K36me2 activity to drive the EMT transcriptional program.
DISCUSSION
Epithelial plasticity is associated with the activity of multiple EMT-TFs, whose expression levels and functions vary across different developmental, homeostatic, and malignant settings. This context-specific variation has complicated efforts to establish unifying principles in their mode of action and downstream consequences. Here, we sought to understand how epigenetic regulators – and their molecular targets – influence epithelial differentiation state. Until recently, the biological functions of specific epigenetic modifications have been inferred from gain- and loss-of-function studies involving their putative modifying enzymes. However, many of these marks, particularly those found on histones, are regulated by multiple, redundant enzymatic writers and erasers. Furthermore, these enzymes have non-histone substrates and/or act as scaffolds to recruit other factors (50). Consequently, it has been challenging to directly assess the biological impact of these modifications (as opposed to the enzymes catalyzing their regulation). Recently, histone lysine-to-methionine (K-to-M) point mutations have emerged as a powerful tool to study a subset of histone modifications (36,47,51,52). Accordingly, we employed the K36M histone mutant to directly interrogate the function of H3K36me2 in cellular plasticity and its impact on other epigenomic features. Our study shows that gain of the histone mark H3K36me2 in carcinoma cells is a conserved epigenome-wide determinant of mesenchymal identity, thereby demonstrating that epigenomic reprogramming is not merely a consequence of EMT but instead is actively involved in enforcing and/or maintaining cellular states in cancer.
Our finding that the H3K36me2 mark is critical for reinforcing the mesenchymal state is consistent with work in human chondroblastoma, where mutant H3K36M acts as an oncogenic driver by dysregulating many of the pathways involved in normal mesenchymal differentiation (36,47). Similarly, loss of NSD2 is associated with Wolf-Hirschhorn syndrome (WHS), a condition characterized by a collection of midline phenotypes – craniofacial abnormalities, cardiac anomalies, and neurodevelopmental delays – indicative of defects in neural crest (53,54). EMT is essential for neural crest cells to leave the neural tube during development, and recent studies suggest that NSD2 plays a direct role in neural crest cell migration (55,56). Therefore, our finding that Nsd2 promotes a mesenchymal migratory phenotype in cancer may reflect an oncogenic adaptation of normal developmental processes involving this enzyme and its histone target, implicating the regulation of H3K36me2 in both physiologic and disease-related contexts.
We found that the enzymes involved in global writing and erasing of H3K36me2 play dramatic and distinct roles at different stages of metastatic progression. Specifically, Nsd2 loss inhibits EMT, thereby reducing invasion and bloodstream entry, while Kdm2a loss inhibits MET, thereby reducing metastatic colonization. Neither of these perturbations influence cell proliferation or tumor growth, suggesting that flexibility in cellular state, rather than primary tumor size, is a major determinant of metastatic progression. These results join a growing body of evidence implicating epigenetic deregulation in metastasis (57–59), where the phenotypic plasticity afforded by epigenetic reprogramming may allow cells to overcome the diverse selective pressures encountered at distinct stages of metastatic spread.
An inverse relationship between H3K36me2 and repressive polycomb activity has been proposed as a mechanism by which H3K36me2 regulates gene expression (30,36,47). While we also observed an inverse relationship between H3K36me2 and H3K27me3 (the target of polycomb), our study found that H3K27ac peaks residing within broad intergenic H3K36me2 domains are lost when H3K36me2 levels decrease, indicating that H3K36me2 also mediates its effects by modulating enhancer activity. We were surprised to find that despite the global distribution of H3K36me2, relatively few genes (<200) were transcriptionally impacted by dynamic changes in this histone mark. Remarkably, this list included virtually all key EMT-TFs, as well as several pro-metastatic factors, including members of the Zeb, Snail, Prrx, Sox, and Smad gene families. Our finding that Nsd2 regulates epithelial plasticity by altering enhancer activity expands on previous work implicating Nsd2 in the regulation of EMT and EMT TF expression in prostate cancer (23–25). By contrast, genes associated with the epithelial state, including various cell adhesion molecules (and Ecad itself), appeared to be regulated at their promoters independently of dynamic changes in H3K36me2. These results support a hierarchical model of epithelial plasticity involving two tiers of control: a first tier in which transcriptional regulation of epithelial and mesenchymal programs is achieved by the acute activity of EMT-TFs, and a higher order tier dependent on H3K36me2-mediated reprogramming of enhancers associated with these EMT-TFs, thereby achieving a more stable epigenetic memory of epithelial-mesenchymal state.
Finally, our results have translational implications for cancer therapy. In addition to its role in cancer metastasis, epithelial plasticity is a major mechanism of therapeutic resistance to chemotherapy and targeted agents (40). However, the signaling pathways that drive EMT and MET are highly heterogeneous and converge on diverse transcriptional regulators, making it challenging to develop strategies to inhibit these processes. Our finding that H3K36me2 regulation is a conserved and required feature of carcinoma-associated epithelial plasticity may represent a way around this barrier, as the enzymes mediating this histone PTM are all potentially targetable with small molecules. There is growing evidence that inhibitors of epigenetic modifiers can be both highly specific and potent (60), paving the way for strategies to target epithelial plasticity and other transcriptional processes. For example, other potential targets from our screen, such as KDM1A, which has been shown to interact with SNAIi1 to drive EMT (13), and DOT1L, which was recently demonstrated to regulate EMT in breast cancer (61), already have inhibitors that are currently being tested in clinical trials for various cancers. Nevertheless, in terms of targeting H3K36me2, the existence of at least five histone methyltransferases capable of catalyzing its formation – NSD1, NSD2, NSD3 , ASH1L, and SETMAR – suggests that redundant and/or context-dependent mechanisms may yet complicate such efforts, and further work will be needed to clarify their roles.
MATERIALS AND METHODS
Mice
KrasLSL-G12D; p53L/+; Pdx1-cre; Rosa26YFP/YFP (KPCY) mice have been described previously(19). Female 6- to 8-week-old C57BL/6J or NOD/SCID mice were purchased from The Jackson Laboratory and Charles River Laboratory, respectively, for tumor cell injection experiments. All vertebrate animals were maintained and experiments were conducted in compliance with the National Institutes of Health guidelines for animal research and approved by the University of Pennsylvania Institutional Animal Care and Use Committee.
Cell lines and culture conditions
Murine PDAC cell lines 3077c4, 483c6, and c8 were derived from primary KPCY tumors of mixed genetic background (20). 6419 c5 and 6694 c2 were derived from primary tumors derived from KPCY mice that have been backcrossed onto the C57BL/6J mouse strain(62). The murine Panc02 cell line was provided by Dr. Robert Vonderheide. The human PDAC PANC1 cell line was provided by Dr. Anil Rustgi. The human prostate cancer cell line DU145 was provided by Dr. Irfan Asangani, and the human lung adenocarcinoma cell line A549 was provided by Dr. Kathryn Wellen. All human lines were originally obtained from ATCC. All murine lines and the PANC1 cell line were single cell FACS to establish clonal cell lines.
3077 and 483 were cultured in PDEC media, as previously described (19). All other murine and human cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum, 10% glutamine, and 150 μg/mL gentamicin at 37°C, 5% CO2, 21% O2, and 100% humidity. Cell lines were maintained and passaged according to ATCC recommended procedures. Cells lines were regularly tested for mycoplasma using MycoAlert Mycoplasma Detection Kit (Lonza).
Plasmid construction and cloning
For studies interrogating individual genes, sgRNA-encoding oligonucleotides (Supplementary Table S4) were inserted into the pSpCas9(BB)-2A-Puro (PX459) V2.0 vector, a gift from Feng Zhang (Addgene plasmid #62988), using a BsmBI restriction site. shRNA oligonucleotides (Supplementary Table S5) were designed using the splashRNA algorithm (63), and cloned into the LT3REPIR vector (a gift from Scott Lowe, and modified from the LT3GEPIR vector - Addgene, plasmid #111177 - such that GFP is replaced by dsRed), as previously described (64). Briefly, XhoI and EcoRI restriction sites were PCR-cloned onto the shRNA oligonucleotides using the 5’ miR-XhoI primer – TACAATACTCGAGAAGGTATATTGCTGTTGACAGTGAGCG, the 3’ miR-EcoRI primer – TTAGATGAATTCTAGCCCCTTGAAGTCCGAGGCAGTAGGCA, and the NEBNext High-Fidelity 2x PCR Master Mix (New England Biolabs), according to manufacturer’s protocol. Amplified shRNA oligos and LT3REPIR vector were digested with XhoI and EcoRI and ligated together.
H3F3A K36M mutation was introduced to the pcDNA4/TO-Flag-H3.3 plasmid (a gift from Bing Zhu, Addgene plasmid #47980) by site-directed mutagenesis. The Q5 site-directed mutagenesis kit (New England Biolabs) was used according to the manufacturer’s protocol with the following mutagenic primers: forward - GGAGGGGTGATGAAACCTCATC and reverse – AGTAGAGGGCGCACTCTT. Following confirmation of the point mutation by Sanger sequencing, the epitope-tagged H3F3A K36M was cloned into the pCDH-EF1-FHC lentiviral vector (a gift from Richard Wood, Addgene plasmid #64874) for constitutive expression using NotI and BamHI restriction sites.
Flow cytometry and fluorescence-activated cell sorting (FACS)
Cultured tumor cells were dissociated into single cells with Hank’s based enzyme free cell dissociation solution (EMD Millipore) and washed in HBSS with 5% FBS and DNase I (Sigma). Cells were then stained with rat anti-Ecadherin (1:250, Takara, M108), followed by APC donkey anti-rat (1:100, Jackson Immunoresearch). For FACS, samples were filtered through a 70 μM strainer to form single cell suspensions. Flow cytometric analysis was performed on a LSR II flow cytometer (BD Biosciences) and analyzed using FlowJo software (Treestar). FACS was performed on a FACSAria II sorter (BD Biosciences).
Epigenetic modifier CRISPR sgRNA library construction
A list of sgRNA sequences directed against a comprehensive murine epigenetic modifier gene list was a gift from Scott Lowe (MSKCC), and a sgRNA list targeting the functional and catalytic domains of murine epigenetic modifiers was taken from a previous study (65). Additionally, 108 sgRNAs targeting EMT regulatory factors, 500 non-targeting sgRNAs, and 50 sgRNAs targeting essential genes were added. In total, the library contains 4803 sgRNAs. All sgRNAs were designed using design principles previously reported and were filtered to minimize predicted off-target effects (66). sgRNAs were synthesized in a pooled format on an array platform (CustomArray, Inc.) and then PCR cloned into the LRG2.1 vector, a gift from Christopher Vakoc (Addgene plasmid #108098) using previously described methods (67). To ensure proper representation of sgRNAs in the pooled lentiviral plasmids, the library was analyzed by deep-sequencing on a NextSeq500/550 (Illumina, 75 cycles High Output kit v2.0) and MAGeCK (68), which confirmed that 100% of the designed sgRNAs were cloned in the LRG2.1 vector with relatively even read count distribution.
Lentivirus production, transduction, and viral titering
Transfection of 293Ts for lentivirus production was performed using Opti-MEM I (GIBCO), DNA (expression plasmid, psPAX2, and pVSVg mixed in a 4:2:1 ratio), and polyethylenimine (PEI, Polysciences) at a 3:1 ratio with total DNA. Recipient cells were transduced with filtered, unconcentrated viral supernatant in the presence of 8 μg/mL polybrene. Appropriate antibiotic selection (2-8 μg/mL puromycin, 10 μg/mL blasticidin) was applied for at least 72 hours. Viral titering for sgRNA library was done by transduction at limiting dilution and determining the GFP+% 72 hours later using the GUAVA easyCyte HT BG flow cytometer (Millipore).
Gain of function retroviral transduction
Full length human NSD2 and E1099K NSD2 mutant pMSCV-puro expression plasmids were kindly provided by Irfan Asangani. Full length KDM2A (GFP-FBXL11) was a gift from Michele Pagano (Addgene plasmid # 126543). Full length murine Snai1 and Zeb1 were gifts from Thomas Brabletz and cloned into the pCW57.1 vector using AvrII/ BamHI and MluI/BamHI restriction sites, respectively. 293T cells were transfected using Opti-MEM I (GIBCO), DNA (expression plasmid, HIV gag-pol, and pVSVg mixed in a 4:2:2 ratio), and polyethylenimine (PEI, Polysciences) at a 3:1 ratio with total DNA. Virus collection and transduction were performed as previously detailed. Puromycin selection (5 μg/mL) was begun 48 hours post-transduction and continued for 1 week.
CRISPR-Cas9 screen
Cas9 was introduced to the 3077 c4 cell line by transduction of the lentiCas9-Blast construct (a gift from Feng Zheng, Addgene plasmid #52962), and selected with 10 μg/mL blasticidin. A clonal 3077 c4-Cas9 line was generated by single-cell FACS and was later transduced with the epigenetic modifier library at a MOI of 0.2 such that ~20% of cells were GFP positive. 210 million cells were transduced to yield 1000x coverage of the sgRNA library in both the Ecadherin+ and Ecadherin− subpopulations. T0 GFP+ Ecad+ and GFP+ Ecad− samples were collected by FACS 3 days post-transduction. GFP+ Ecad+ and GFP+ Ecad− samples were collected at 2 weeks (T2weeks) and 4 weeks (T4weeks) post-transduction. Enough cells were collected to maintain at least 500x coverage of the sgRNA library in each sample.
Genomic DNA was harvested using the QIAamp DNA mini kit (Qiagen), and libraries were constructed according to previously described protocols (69). Briefly, sgRNAs were amplified over 18 cycles with Herculase II fusion DNA polymerase (Agilent) per manufacturer specifications with PCR#1 forward and reverse primers (Supplementary Table S6). Each reaction included 3 μg of genomic DNA, and multiple PCR reactions were run in parallel such that all extracted genomic DNA was used to maintain library coverage. PCR reactions were then pooled for each sample, and 5 μl of each pooled PCR#1 sample was used as a template for PCR#2, which added Illumina P5/P7 adapters, barcodes, and staggers for nucleotide complexity. For PCR#2, template was amplified over 25 cycles with PCR#2 forward and reverse primers (Supplementary Table S6), and the resulting reactants were column purified with the QIAquick PCR purification kit (Qiagen) and gel extracted with the QIAquick gel extraction kit (Qiagen). The barcoded libraries were pooled at an equal molar ratio and sequenced on a NextSeq500/550 (Illumina, 75 cycles High Output kit v2.0) to generate 75 bp single-end reads.
MAGeCK was used for subsequent screen analysis (68). Briefly, the sequencing data were de-barcoded and the 20 bp sgRNA sequence was mapped to the reference sgRNA library without allowing for any mismatches. The read counts were calculated for each individual sgRNA and normalized to the non-targeting sgRNAs. Normalized read counts of sgRNAs in Ecad+ and Ecad− subpopulations were log2 transformed, and graphical representation was done using the R-package ggplot2. To exclude false positives due to off-target effects or inconsistencies in sample preparation, we only focused on hits for which >80% sgRNAs targeting a gene were enriched.
Pancreatosphere formation assay
Pancreatosphere assays were performed as described by Rovira et al., 2010 (ref. 70). Briefly, cell lines trypsinized and dissociated into single cells. 2500 cells were suspended in tumorsphere media: DMEM/F-12 with 1x B-27 supplement (Gibco), 3% FBS, 100 mM B-mercaptoethanol (Gibco), 1x non-essential amino acids (Sigma), 1x N2 supplement (Gibco), 20 ng/ml mouse EGF (Gibco), 20 ng/ml mouse FGF-basic (Gibco), and 10 ng/ml mouse LIF (Gibco). Cells in were plated in 6-well ultra-low attachment culture plates (Costar) and spheres were counted after 5 days. 5 independent wells were used per cell line.
Orthotopic implantation and circulating tumor cell (CTC) analysis
Mice were anaesthetized using continuous-flow isoflurane, and their abdomen was sterilized. An incision was then made over the left upper quadrant of the abdomen, and the pancreas was exteriorized onto a sterile field. 1.0x104 tumor cells in 50 μl sterile DMEM were injected into the tail of the pancreas via a 27 5/8” gauge needle. Successful injection was confirmed by the formation of a liquid bleb at the site of injection with minimal fluid leakage. The pancreas was then gently placed back into the peritoneal cavity, and the peritoneum and overlying skin were closed with 4-0 coated Vicryl violet FS-2 sutures (Ethicon). Tumors, lungs, and livers were harvested, weighed, and measured 4-6 weeks following implantation. 200 μl of blood was drawn from tumor-bearing animals via cardiac puncture with a 1 mL insulin syringe coated with 0.5M EDTA pH 8.0 (Gibco) to prevent coagulation and was immediately placed in a 150 mm gridded plate containing RBC lysis buffer (BD Biosciences). After 10 min. of lysis at room temperature, PBS was added to the plate and CTCs were directly visualized on a fluorescent microscope and counted.
Lung metastatic colonization assay
1.0x105 tumor cells in 100 μl sterile DMEM were injected into the tail veins of mice via a 27 5/8” gauge needle. After 2 weeks lungs were harvested and weighed.
Scratch assay
Cells were plated to 70% confluency in 6-well plates and grown in serum free media for 24-48 hours. A horizontal scratch was then made in each well with a 200 μL pipette tip. Cells were photographed at identical points at 0, 24, and 48 hours post-scratch. Cell migration was assessed by measuring the area of the scratch at each time point using ImageJ.
EdU analysis
2,000 cells were seeded into each well of a 96-well plate. EdU was spiked into the media at 10 μM, and 2 hours later cells were fixed and permeabilized with the fixation/permeabilization buffer set (eBioscience) for 30 min. at room temperature. Cells were then washed, and EdU was labeled with Alexa Fluor 647 azide (1:2000, ThermoFisher Scientific) by a click chemical reaction (100mM Tris-HCl pH 8.5, 4mM CuSO4, 100mM ascorbic acid) for 30 min. at room temperature, washed, and analyzed by flow cytometry.
Tgf-β treatment
100,000 cells were plated into 6-well plates and 24 hours later were treated with either citric acid (control) or 10 ng/mL Tgf-β (CST) over a given time course. Cells were continuously dosed every 3 days and were passaged in the given agents over longer time points.
RNA Isolation, real-time quantitative PCR (RT-qPCR), and RNA-seq
RNA was prepared from cultured tumor cells or sorted cells using RNeasy Mini Kit or RNeasy Micro Kit (Qiagen). For RT-qPCR, cDNA was generated using High-capacity cDNA Reverse Transcription Kit (Life Technologies). RT-qPCR analysis was performed with SsoAdvanced SYBR (Bio-Rad) using a CFX384 Real-Time System (Bio-Rad). Transcript quantities were determined using the difference of Ct method and values were normalized to the expression of Tbp. Primer sequences are listed in Supplementary Table S7.
Sequencing libraries were generated using the NEB Next Ultra RNA Library Prep kit and sequenced in technical duplicates on an Illumina HiSeq platform to generate 150bp paired-end reads (GSE137382) by Novogene (Beijing, China). Reads were aligned to the mm10 reference genome using STAR (v2.5.1) (71), and raw counts of gene transcripts were obtained using featureCounts (72), both with default settings. The raw count matrix was subsequently imported into R-studio and used as input for DESeq2 (73) for normalization and differential gene expression analysis. Salmon was used in parallel to normalize and quantify gene expression in transcripts per million (TPM) through quasi-alignment. To generate ranked gene lists for pre-ranked gene set enrichment analysis (GSEA) (http://www.broad.mit.edu/gsea/) genes were ranked by their DESeq2-derived Wald statistic. The gene signatures for Squamous and Progenitor subtypes were derived from(34). Briefly, the top 500 and bottom 500 protein-coding genes of each subtype were identified from RNA-seq data derived from patient samples(31), and used to define each subtype’s gene signature.
ATAC-seq and analysis
Library construction -
ATAC-seq samples were prepared by sorting 50,000 Ecad+ and 50,000 Ecad− subpopulations from three clonal cell lines. The libraries were prepared as previously described with minor modifications (74). Briefly, nuclei were isolated from sorted cells using a solution of 10 mM Tris-HC pH 7.4, 10 mM NaCl, 3 mM MgCl2, and 0.1% IGEPAL CA-630. Immediately following nuclei isolation, the transposition reaction was conducted using Tn5 transposase and TD buffer (Illumina) for 45 min. at 37oC. Transposed DNA fragments were purified using a Qiagen MinElute Kit, barcoded with primers based on Illumina TruSeq indices (Supplementary Table S8), and PCR amplified for 11 cycles using NEBNext High Fidelity 2x PCR master mix (New England Biolabs). Libraries were purified by extraction from a 6% TBE gel, followed by column purification with the Qiagen PCR Cleanup kit. Sequencing was performed on biological triplicates using a HiSeq2500 (Illumina) on rapid run mode to generate 50bp paired end reads (GSE137520).
Analysis –
After adapter trimming with cutadapt, reads were aligned to the mouse reference genome (GRCm38, May 23 2014), which was downloaded from the UCSC repository, by Bowtie2 (75). Picard was used to identify and discard reads that aligned to the mitochondrial genome, as well as reads mapping to multiple genomic loci were discarded from downstream analyses by Picard (Broad Institute). MACS2 with ‘-p 1e-7–nolambda–nomodel’ was applied on each ATAC-seq replicate separately to identify accessible chromatin. Peaks were subsequently merged using BEDTools (76) and ATAC-seq read counts were calculated in the merged peaks for every replicate. The resulting count table was used to identify differentially accessible loci with DESeq2 in R-studio, which were then associated with their putative target genes by GREAT (77). The GREAT algorithm associates genomic regions with genes by defining a ‘regulatory domain’ for each gene in the genome, then computes ontology term enrichments using a binomial test. Visualization of these loci was achieved by uploading the reads-per-million-aligned-read-normalized bigwig files to Integrative Genomics Viewer(78).
ChIP and ChIP sequencing
For ChIP, ~2x107 cells were crosslinked with 1% formaldehyde for 10 min. at room temperature, followed by quenching with 125 mM glycine for 5 min. at room temperature. They were then washed with PBS and lysed with nuclei isolation buffer. Samples were sonicated with a Diagenode Bioruptor at high intensity setting for 15 5 min. cycles of 30 sec on/off., to yield ~300bp fragments. After high-speed centrifugation, soluble chromatin from Drosophila S2 cells was spiked in at 2.5% of the mouse chromatin. The mixed soluble chromatin was diluted 10x and pre-cleared with magnetic protein A Dynabeads (Invitrogen). Immunoprecipitation with anti-H3K36me2 (CST, 2901S), anti-H3K27me3 (CST, 9733S), anti-H3K27ac (Abcam, ab4729), anti-H3K4me1 (Abcam, ab8895), or rabbit IgG Isotype (Abcam, ab171870) was performed overnight at 4°C with rotation, with 10% kept as input DNA. Immunocomplexes were recovered with magnetic Protein A dynabeads and washed sequentially using a low salt buffer, a high salt buffer, Lithium Chloride buffer, and TE buffer. Samples were eluted twice with sodium bicarbonate. Eluted ChIP DNA was treated with RNase A (0.2 mg/mL) overnight at 65°C, then incubated with Proteinase K (0.2 mg/mL) for 2 hours at 45°C. DNA samples were purified using QIAquick PCR Purification Kit (Qiagen). For ChIP sequencing, libraries were prepared according to the NEBNext Ultra II DNA Library Prep Kit (New England Biolabs) protocol, using barcoded primers from NEBNext Multiplex Oligos Index Primers Sets 1 and 2. Libraries were pooled and sequenced in technical duplicates on the NextSeq500/550 (Illumina, 75 cycles High Output kit v2.0) to generate 75 bp single-end reads (GSE137521).
ChIP-seq analysis
Sequencing reads were aligned to the mouse (GRCm38, May 23 2014) and the drosophila melanogaster reference genomes (dm6, August 2014) using STAR (v2.5.1) with default settings. Duplicate and low-quality reads (PHRED score <30) reads were removed using SAMtools (79). The normalization factor was determined from the drosophila reads as previously described(80).
The bam files were converted to ChIPRX-normalized bigwig tracks using deepTools (81). Single-end reads were extended up to the fragment length (200 bp) along the read direction. Enrichment peaks were called with MACS2 with default settings (H3K27ac and H3K4me1) and broad settings (H3K36me2 and H3K27me3). Blacklisted peaks (https://sites.google.com/site/anshulkundaje/projects/blacklists), were excluded in all subsequent analysis. To identify peaks differentially bound between WT and sgNsd2 samples we first identified the consensus peak set that includes the unique set of peaks enriched across both samples. Differentially bound peaks were then identified using DESeq2 with the ChIPRX normalized tag counts at the consensus peak regions as input and only peaks with adjusted p-value <0.001 were used for further analysis. Overlap analysis of peaks was performed using an in-house python script and peaks were annotated using HOMER (http://homer.ucsd.edu/homer/ngs/annotation.html).
Immunofluorescence and Hematoxylin & Eosin (H&E) staining
Tissues were fixed in Zinc-formalin and embedded in paraffin for histological analysis and immunofluorescence staining. Sections were deparaffinized, rehydrated, and prepared by antigen retrieval. For cell lines, cells were seeded into 8-well Nunc Lab-Tek II chamber slides (Thermo Scientific) and fixed in 4% paraformaldehyde for 15 mins. For immunofluorescence staining, sections or fixed cells were blocked in PBS with 0.3% Triton-X and 5% donkey serum for 1 hour, stained with primary and secondary antibodies, and mounted with Aqua Polymount (Polysciences, Inc). Primary antibodies used include goat anti-GFP (Abcam, ab6673), rat anti-E-cadherin (Takara Bio, M108), and rabbit anti-H3K36me2 (Abcam, ab9049). Slides were visualized using an Olympus IX71 inverted multicolor fluorescent microscope equipped with a DP71 camera. Images were quantified using ImageJ software.
For H&E staining, sections were deparaffinized, rehydrated, stained with hematoxylin, differentiated with acidic ethanol, stained for eosin, dehydrated, and mounted with Permount. Slides were visualized using the Keyence BZ-X710 all-in-one fluorescence microscope.
Histone extraction
Histones were acid extracted according to standard protocols. Briefly, nuclei were isolated from cells lysed with a hypotonic lysis buffer. Histones were extracted from the nuclei with 0.2N H2SO4, precipitated in 33% trichloroacetic acid (TCA), washed with acetone, and resuspended in diH2O.
Immunoblotting
For whole cell lysates, cells were washed with cold PBS and lysed in RIPA lysis buffer. Extracted histones or whole cell lysates were separated by SDS-PAGE, transferred to PVDF membrane, blocked in 5% non-fat milk in PBS plus 0.1% Tween-20, probed with primary antibodies, and detected with horseradish peroxidase-conjugated secondary antibodies (Jackson Immunoresearch). Primary antibodies used include: anti-H3 (CST, 9715S); anti-H3K36me2 (CST, 2901S); anti-H3K36me3 (Abcam, ab9050); anti-H3K36M (Millipore, ABE1447); anti-Nsd2 (Millipore, MABE191); anti-Kdm2a (Abcam, ab191387); anti-alpha tubulin (CST, 3873S); anti-E-cadherin (Takara, M108).
Histone derivatization, mass spectrometry, and PTM quantification
Derivatization -
Histones were derivatized according to standard protocol(82). Briefly, 5-10ug of acid-extracted histones were resuspended in 100mM ammonium bicarbonate (pH 8.0) and mixed with freshly prepared propionic anhydride with acetonitrile for 15 min. at 37 °C. Histones were then digested with trypsin (enzyme: sample ratio 1:20) overnight at 37 °C. The peptides were then desalted and stored dried. They were resuspended in 0.1% formic acid just before mass spectrometry.
Direct injection–MS -
Samples were placed in a TriVersa NanoMate (Advion) and acquired either manually or by using a sequence coordinated with MS acquisition by a contact closure. The NanoMate was set up with a spray voltage of 1.7 kV and a gas pressure of 0.5 psi. Samples were acquired in the Orbitrap Fusion Tribrid (Thermo Scientific). All scans were acquired in the orbitrap, at 240,000 resolution for the full MS and at 120,000 resolution for MS/MS. The AGC target for the tSIM-MSX scans was set to 10E6. The full description of the DI-MS acquisition method has been previously described(83).
Histone peptide quantification -
Raw files were searched with a modified version of the software EpiProfile 2.0 (84). Histone peptides are collected in MS scans, and isobaric peptides are collected in targeted pre-set MS/MS scans. The software reads the intensities from MS scans to calculate the percentage of all peptides with the same amino acid sequence. The unique fragment ions in the MS/MS scans are extracted to discriminate isobaric peptide intensities from the MS scans. The software EpiProfileLite is available on GitHub at https://github.com/zfyuan/EpiProfileLite, including the user guide.
Statistical Analysis
Comparisons between two groups were performed using Students’ unpaired t test. The Holm Sidak correction for multiple testing was applied where indicated. All statistical analyses were performed with Graphpad Prism 7 and 8 (GraphPad). Error bars show standard error of the mean (SEM), as indicated in the legends, and p<0.05 was considered statistically significant. * indicates p<0.05, ** p<0.01, *** p< 0.001, and **** p< 0.0001 unless otherwise indicated. ns denotes not significant.
For sequencing experiments, DESeq2 (73) used to generate adjusted p-values (FDR). And for TCGA survival analysis, KM plotter (40) was used to generate logrank P-values.
Data Resources
All sequencing data has been deposited in GEO under the series GSE137523, which includes the following subseries: GSE137382 (RNA-seq), GSE137520 (ATAC-seq), and GSE137521 (ChIP-seq).
Software
PRISM software was used for the statistical analysis and data visualization (http://www.graphpad.com). The R language and environment for statistical computing and graphics (https://www.r-project.org) was utilized in this study for the statistical and bioinformatics analysis of RNA-seq, ATAC-seq, and genome-seq data. The R packages used for the analysis described in the method section were obtained from the Bioconductor (https://www.bioconductor.org) and CRAN (https://cran.r-project.org/web/packages/).
Supplementary Material
STATEMENT OF SIGNIFICANCE.
Although epithelial plasticity contributes to cancer metastasis and chemoresistance, no strategies exist for pharmacologically inhibiting the process. Here, we show that global regulation of a specific histone mark, H3K36me2, is a universal epigenome-wide mechanism that underlies EMT and MET in carcinoma cells. These results offer a new strategy for targeting epithelial plasticity in cancer.
ACKNOWLEDGEMENTS
We thank John Tobias in the Penn Molecular Profiling Facility, Jonathan Schug in the Penn Next Generation Sequencing Core, Kathryn Sun in the Penn Pancreatic Cancer Research Center, Ken Zaret, Andres Blanco, and Liling Wan for helpful discussion of the study and manuscript, Grace Anderson and Donita Brady for technical guidance of the screen, and all members of the Stanger laboratory for helpful discussions.
This work was supported by NIH grants CA229803 and DK083355 (BZS), CA224970 (SY), CA196539 and AI118891 (BAG), CA249210 (IAA), a V Foundation Translational award (BZS), a Leukemia and Lymphoma Robert Arceci Award (BAG), and DOD W81XWH-17-1-0404 (IAA), the Abramson Family Cancer Research Institute, the Abramson Cancer Center, and the NIH/Penn Center for Molecular Studies in Digestive and Liver Diseases.
Footnotes
The authors declare no potential conflicts of interest.
REFERENCES
- 1.Yuan S, Norgard RJ, Stanger BZ. Cellular Plasticity in Cancer. Cancer Discov. United States; 2019;9:837–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nieto MA, Huang RY-J, Jackson RA, Thiery JP. EMT: 2016. Cell. United States; 2016;166:21–45. [DOI] [PubMed] [Google Scholar]
- 3.Lu W, Kang Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev Cell. United States; 2019;49:361–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krebs AM, Mitschke J, Lasierra Losada M, Schmalhofer O, Boerries M, Busch H, et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat Cell Biol. England; 2017;19:518–29. [DOI] [PubMed] [Google Scholar]
- 5.Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. England; 2015;527:525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong STC, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. England; 2015;527:472–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edwards JR, Yarychkivska O, Boulard M, Bestor TH. DNA methylation and DNA methyltransferases. Epigenetics Chromatin. England; 2017;10:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schuettengruber B, Bourbon H-M, Di Croce L, Cavalli G. Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell. United States; 2017;171:34–57. [DOI] [PubMed] [Google Scholar]
- 9.Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. United States; 2013;19:1438–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaffer CL, Marjanovic ND, Lee T, Bell G, Kleer CG, Reinhardt F, et al. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell. United States; 2013;154:61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong C, Wu Y, Yao J, Wang Y, Yu Y, Rychahou PG, et al. G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer. J Clin Invest. United States; 2012;122:1469–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong C, Wu Y, Wang Y, Wang C, Kang T, Rychahou PG, et al. Interaction with Suv39H1 is critical for Snail-mediated E-cadherin repression in breast cancer. Oncogene. England; 2013;32:1351–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin Y, Wu Y, Li J, Dong C, Ye X, Chi Y-I, et al. The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J. England; 2010;29:1803–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peinado H, Ballestar E, Esteller M, Cano A. Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol Cell Biol. United States; 2004;24:306–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herranz N, Pasini D, Diaz VM, Franci C, Gutierrez A, Dave N, et al. Polycomb complex 2 is required for E-cadherin repression by the Snail1 transcription factor. Mol Cell Biol. United States; 2008;28:4772–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang M-H, Hsu DS-S, Wang H-W, Wang H-J, Lan H-Y, Yang W-H, et al. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat Cell Biol. England; 2010;12:982–92. [DOI] [PubMed] [Google Scholar]
- 17.Cardenas H, Vieth E, Lee J, Segar M, Liu Y, Nephew KP, et al. TGF-beta induces global changes in DNA methylation during the epithelial-to-mesenchymal transition in ovarian cancer cells. Epigenetics. United States; 2014;9:1461–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McDonald OG, Wu H, Timp W, Doi A, Feinberg AP. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat Struct Mol Biol. United States; 2011;18:867–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, et al. EMT and dissemination precede pancreatic tumor formation. Cell. United States; 2012;148:349–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aiello NM, Maddipati R, Norgard RJ, Balli D, Li J, Yuan S, et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev Cell. United States; 2018;45:681–695.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wagner EJ, Carpenter PB. Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol. England; 2012;13:115–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuo AJ, Cheung P, Chen K, Zee BM, Kioi M, Lauring J, et al. NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol Cell. United States; 2011;44:609–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ezponda T, Popovic R, Shah MY, Martinez-Garcia E, Zheng Y, Min D-J, et al. The histone methyltransferase MMSET/WHSC1 activates TWIST1 to promote an epithelial-mesenchymal transition and invasive properties of prostate cancer. Oncogene. England; 2013;32:2882–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aytes A, Giacobbe A, Mitrofanova A, Ruggero K, Cyrta J, Arriaga J, et al. NSD2 is a conserved driver of metastatic prostate cancer progression. Nat Commun. England; 2018;9:5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li N, Xue W, Yuan H, Dong B, Ding Y, Liu Y, et al. AKT-mediated stabilization of histone methyltransferase WHSC1 promotes prostate cancer metastasis. J Clin Invest. United States; 2017;127:1284–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng Z, Cheung P, Kuo AJ, Yukl ET, Wilmot CM, Gozani O, et al. A molecular threading mechanism underlies Jumonji lysine demethylase KDM2A regulation of methylated H3K36. Genes Dev. United States; 2014;28:1758–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blackledge NP, Zhou JC, Tolstorukov MY, Farcas AM, Park PJ, Klose RJ. CpG islands recruit a histone H3 lysine 36 demethylase. Mol Cell. United States; 2010;38:179–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature. England; 2006;439:811–6. [DOI] [PubMed] [Google Scholar]
- 29.Swaroop A, Oyer JA, Will CM, Huang X, Yu W, Troche C, et al. An activating mutation of the NSD2 histone methyltransferase drives oncogenic reprogramming in acute lymphocytic leukemia. Oncogene. England; 2019;38:671–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhuang L, Jang Y, Park Y-K, Lee J-E, Jain S, Froimchuk E, et al. Depletion of Nsd2-mediated histone H3K36 methylation impairs adipose tissue development and function. Nat Commun. England; 2018;9:1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bailey P, Chang DK, Nones K, Johns AL, Patch A-M, Gingras M-C, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. England; 2016;531:47–52. [DOI] [PubMed] [Google Scholar]
- 32.Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. United States; 2011;17:500–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SGH, Hoadley KA, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet. United States; 2015;47:1168–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Somerville TDD, Xu Y, Miyabayashi K, Tiriac H, Cleary CR, Maia-Silva D, et al. TP63-Mediated Enhancer Reprogramming Drives the Squamous Subtype of Pancreatic Ductal Adenocarcinoma. Cell Rep. United States; 2018;25:1741–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Papillon-Cavanagh S, Lu C, Gayden T, Mikael LG, Bechet D, Karamboulas C, et al. Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas. Nat Genet. United States; 2017;49:180–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu C, Jain SU, Hoelper D, Bechet D, Molden RC, Ran L, et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science. United States; 2016;352:844–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Streubel G, Watson A, Jammula SG, Scelfo A, Fitzpatrick DJ, Oliviero G, et al. The H3K36me2 Methyltransferase Nsd1 Demarcates PRC2-Mediated H3K27me2 and H3K27me3 Domains in Embryonic Stem Cells. Mol Cell. United States; 2018;70:371–379. [DOI] [PubMed] [Google Scholar]
- 38.Nagy A, Lanczky A, Menyhart O, Gyorffy B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci Rep. England; 2018;8:9227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen X, Miao Z, Divate M, Zhao Z, Cheung E. KM-express: an integrated online patient survival and gene expression analysis tool for the identification and functional characterization of prognostic markers in breast and prostate cancers. Database (Oxford). England; 2018;2018: 10.1093/database/bay069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. England; 2017;14:611–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Somarelli JA, Schaeffer D, Marengo MS, Bepler T, Rouse D, Ware KE, et al. Distinct routes to metastasis: plasticity-dependent and plasticity-independent pathways. Oncogene. England; 2016;35:4302–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsai JH, Yang J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. United States; 2013;27:2192–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stankic M, Pavlovic S, Chin Y, Brogi E, Padua D, Norton L, et al. TGF-beta-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell Rep. United States; 2013;5:1228–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell. United States; 2012;22:725–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ocana OH, Corcoles R, Fabra A, Moreno-Bueno G, Acloque H, Vega S, et al. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell. United States; 2012;22:709–24. [DOI] [PubMed] [Google Scholar]
- 46.Chaffer CL, Brennan JP, Slavin JL, Blick T, Thompson EW, Williams ED. Mesenchymal-to-epithelial transition facilitates bladder cancer metastasis: role of fibroblast growth factor receptor-2. Cancer Res. United States; 2006;66:11271–8. [DOI] [PubMed] [Google Scholar]
- 47.Fang D, Gan H, Lee J-H, Han J, Wang Z, Riester SM, et al. The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas. Science. United States; 2016;352:1344–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang S, Zheng X, Lu C, Li G-M, Allis CD, Li H. Molecular basis for oncohistone H3 recognition by SETD2 methyltransferase. Genes Dev. United States; 2016;30:1611–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garcia-Carpizo V, Sarmentero J, Han B, Grana O, Ruiz-Llorente S, Pisano DG, et al. NSD2 contributes to oncogenic RAS-driven transcription in lung cancer cells through long-range epigenetic activation. Sci Rep. England; 2016;6:32952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hamamoto R, Saloura V, Nakamura Y. Critical roles of non-histone protein lysine methylation in human tumorigenesis. Nat Rev Cancer. England; 2015;15:110–24. [DOI] [PubMed] [Google Scholar]
- 51.Herz H-M, Morgan M, Gao X, Jackson J, Rickels R, Swanson SK, et al. Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science. United States; 2014;345:1065–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brumbaugh J, Kim IS, Ji F, Huebner AJ, Di Stefano B, Schwarz BA, et al. Inducible histone K-to-M mutations are dynamic tools to probe the physiological role of site-specific histone methylation in vitro and in vivo. Nat Cell Biol. England; 2019;21:1449–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nimura K, Ura K, Shiratori H, Ikawa M, Okabe M, Schwartz RJ, et al. A histone H3 lysine 36 trimethyltransferase links Nkx2–5 to Wolf-Hirschhorn syndrome. Nature. England; 2009;460:287–91. [DOI] [PubMed] [Google Scholar]
- 54.Bergemann AD, Cole F, Hirschhorn K. The etiology of Wolf-Hirschhorn syndrome. Trends Genet. England; 2005;21:188–95. [DOI] [PubMed] [Google Scholar]
- 55.Mills A, Bearce E, Cella R, Kim SW, Selig M, Lee S, et al. Wolf-Hirschhorn Syndrome-Associated Genes Are Enriched in Motile Neural Crest Cells and Affect Craniofacial Development in Xenopus laevis. Front Physiol. Switzerland; 2019;10:431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rutherford EL, Lowery LA. Exploring the developmental mechanisms underlying Wolf-Hirschhorn Syndrome: Evidence for defects in neural crest cell migration. Dev Biol. United States; 2016;420:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roe J-S, Hwang C-I, Somerville TDD, Milazzo JP, Lee EJ, Da Silva B, et al. Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell. United States; 2017;170:875–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McDonald OG, Li X, Saunders T, Tryggvadottir R, Mentch SJ, Warmoes MO, et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat Genet. United States; 2017;49:367–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chatterjee A, Rodger EJ, Eccles MR. Epigenetic drivers of tumourigenesis and cancer metastasis. Semin Cancer Biol. England; 2018;51:149–59. [DOI] [PubMed] [Google Scholar]
- 60.Mohammad HP, Barbash O, Creasy CL. Targeting epigenetic modifications in cancer therapy: erasing the roadmap to cancer. Nat Med. United States; 2019;25:403–18. [DOI] [PubMed] [Google Scholar]
- 61.Cho M-H, Park J-H, Choi H-J, Park M-K, Won H-Y, Park Y-J, et al. DOT1L cooperates with the c-Myc-p300 complex to epigenetically derepress CDH1 transcription factors in breast cancer progression. Nat Commun. England; 2015;6:7821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity. United States; 2018;49:178–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pelossof R, Fairchild L, Huang C-H, Widmer C, Sreedharan VT, Sinha N, et al. Prediction of potent shRNAs with a sequential classification algorithm. Nat Biotechnol. United States; 2017;35:350–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fellmann C, Zuber J, McJunkin K, Chang K, Malone CD, Dickins RA, et al. Functional identification of optimized RNAi triggers using a massively parallel sensor assay. Mol Cell. United States; 2011;41:733–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, Vakoc CR. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat Biotechnol. United States; 2015;33:661–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. United States; 2013;31:827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Anderson GR, Winter PS, Lin KH, Nussbaum DP, Cakir M, Stein EM, et al. A Landscape of Therapeutic Cooperativity in KRAS Mutant Cancers Reveals Principles for Controlling Tumor Evolution. Cell Rep. United States; 2017;20:999–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. England; 2014;15:554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. United States; 2014;343:84–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rovira M, Scott S-G, Liss AS, Jensen J, Thayer SP, Leach SD. Isolation and characterization of centroacinar/terminal ductal progenitor cells in adult mouse pancreas. Proc Natl Acad Sci. 2010;107:75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. England; 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. England; 2014;30:923–30. [DOI] [PubMed] [Google Scholar]
- 73.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. England; 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. United States; 2013;10:1213–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. United States; 2012;9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. England; 2010;26:841–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. United States; 2010;28:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat. Biotechnol. United States; 2011;29:24–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. England; 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Orlando DA, Chen MW, Brown VE, Solanki S, Choi YJ, Olson ER, et al. Quantitative ChIP-Seq normalization reveals global modulation of the epigenome. Cell Rep. United States; 2014;9:1163–70. [DOI] [PubMed] [Google Scholar]
- 81.Ramirez F, Dundar F, Diehl S, Gruning BA, Manke T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. England; 2014;42:187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sidoli S, Bhanu N V, Karch KR, Wang X, Garcia BA. Complete Workflow for Analysis of Histone Post-translational Modifications Using Bottom-up Mass Spectrometry: From Histone Extraction to Data Analysis. J Vis Exp. United States; 2016;111:e54112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sidoli S, Kori Y, Lopes M, Yuan Z-F, Kim HJ, Kulej K, et al. One minute analysis of 200 histone posttranslational modifications by direct injection mass spectrometry. Genome Res. United States; 2019;29:978–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yuan Z-F, Sidoli S, Marchione DM, Simithy J, Janssen KA, Szurgot MR, et al. EpiProfile 2.0: A Computational Platform for Processing Epi-Proteomics Mass Spectrometry Data. J Proteome Res. United States; 2018;17:2533–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.