

Abstract
Thousands of apurinic/apyrimidinic (AP or abasic) sites form in each cell, each day. This simple DNA lesion can have profound consequences to cellular function, genome stability, and disease. As potent blocks to polymerases, they interfere with the reading and copying of the genome. Since they provide no coding information, they are potent sources of mutation. Due to their reactive chemistry, they are intermediates in the formation of lesions that are more challenging to repair including double-strand breaks, interstrand crosslinks, and DNA protein crosslinks. Given their prevalence and deleterious consequences, cells have multiple mechanisms of repairing and tolerating these lesions. While base excision repair of abasic sites in double-strand DNA has been studied for decades, new interest in abasic site processing has come from more recent insights into how they are processed in single-strand DNA. In this review, we discuss the source of abasic sites, their biological consequences, tolerance mechanisms, and how they are repaired in double and single-stranded DNA.
Keywords: Base Excision Repair, HMCES, DNA-protein crosslinks, genome stability, abasic site
DNA is under constant threat by endogenous and environmental DNA damaging agents. Apurinic/apyrimidinic (AP or abasic) are the most frequent DNA lesions. AP sites result from cleavage of the N-glycosylic bond between the nitrogenous base and the deoxyribose sugar, leaving an intact phosphodiester backbone. Base loss can happen because of spontaneous hydrolysis, base damage leading to destabilization of N-glycosyl bond, or the action of specialized DNA glycosylases. There are numerous evolutionarily conserved repair pathways for AP sites reflecting their physiological importance and mutagenic potential. AP site lesions that persist are roadblocks to transcription and DNA replication. Furthermore, these non-coding lesions are cytotoxic and a threat to genomic integrity since they can cause strand-breaks, interstrand DNA crosslinks, mutations, and DNA-protein crosslinks. Therefore, understanding mechanisms of formation and repair of AP sites are critically important. Here, we will review how AP sites are formed, how they are processed by different repair and tolerance mechanisms depending on context, and their consequences for genome stability.
1. AP Site Formation
AP sites are the most frequent lesion in cells with estimates of ~10,000–20,000 per human cell per day [1]. They form spontaneously, as a consequence of destabilization of the N-glycosyl bond by other types of DNA damage, and enzymatically by glycosylases (Fig. 1A).
Fig. 1.
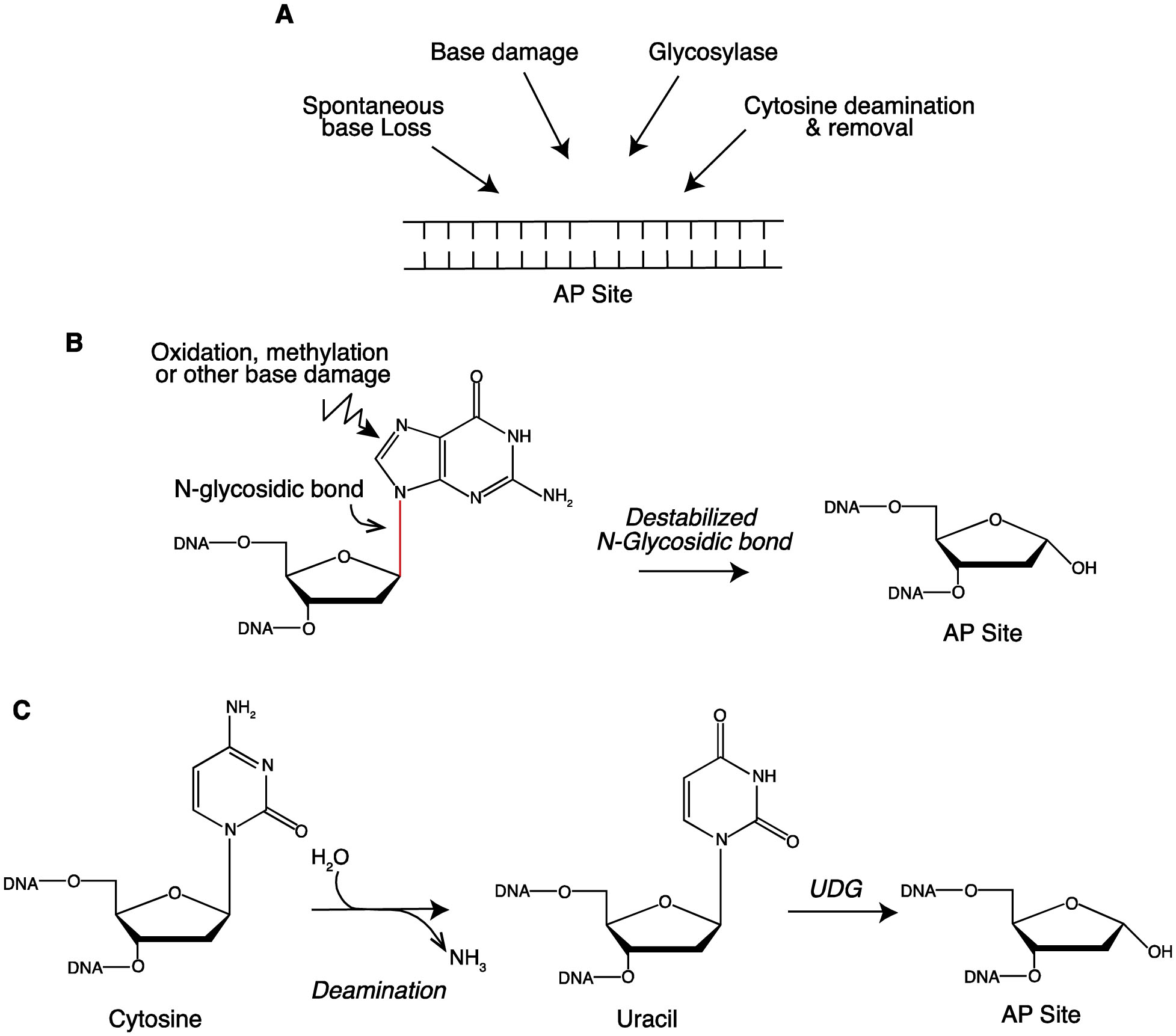
Mechanisms of AP site formation. A. Potential sources of AP site formation. B. Destabilization of N-glyosidic bond after base damage generates AP site. Spontaneous or catalyzed deamination of cytosine generates uracil. Subsequent action of UDG produces an AP site.
1.1. Spontaneous Base Loss
Although DNA is a relatively stable biomolecule, the N-glycosyl bond is prone to hydrolysis. Spontaneous depurination events occur at a rate of ~3 × 10−11 nucleotides per second in vitro under physiological conditions in duplex DNA [2], [3]. In E. coli, 0.5 depurinations occur per cell per generation, and in mammalian cells, approximately 10,000 purines are lost from the genome per day. Guanines are 1.5-times more likely to undergo depurination compared to adenines. Although the mechanism of base loss is similar for purines and pyrimidines, the rate of depyrimidination is 1/20th the rate for depurination since the N-glycosyl bond of pyrimidines are more stable [4]. Furthermore, abasic sites preferentially form at sites of DNA replication [5]. The stretches of single-stranded DNA (ssDNA) on the lagging strand are more vulnerable to chemical attack and spontaneous bass loss. In fact, the rate of depurination is accelerated more than four times in ssDNA versus double-stranded DNA (dsDNA) [2].
1.2. Base Damage and Destabilization of N-Glycosylic Bond
Several environmental and cancer therapeutic genotoxins including alkylating agents, oxidizing agents, ionizing radiation, and ultraviolet radiation can cause nucleobase loss and the formation of AP sites. Base alkylation often weakens the N-glycosyl bond by generating an unstable positive charge on the ring base. This protonation on the ring is stabilized via electron resonance leading to N-glycosyl bond cleavage [6]. Nitrogenous bases are predominately protonated on N3 and N7 of purines and O2 of pyrimidines [7],[8]. The most common alkylating agents used in anticancer therapies including methanesulfonate esters, nitrosoureas, and nitrogen mustards generate AP sites [9].
Oxidative damage is a significant burden to cells. There are more than 100 types of oxidized base modification present in mammalian DNA [10]. Reactive oxygen species such as superoxide, hydrogen peroxide, and hydroxyl radicals are by-products of cellular metabolism that can modify all four DNA bases [11]. Because of the low redox potential of guanine residues, 7,8-dihydro-8-oxoguanine (8-oxoG) is the most common oxidized lesion and is present in large quantities [12],[13]. In addition, the formamidopyrimidine lesions 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyG) and 4,6-diamino-5-hydroxy-5-formamidopyrimidine are other highly mutagenic oxidative lesions caused by fragmentation of the purine imidazole ring. Similar oxidative lesions are seen on adenine. Finally, free radicals tend to attack thymine and cytosines residues at the 5,6-double bond leading to various oxidized base products [11], [12]. These oxidized base derivates cause AP sites directly through destabilization of the N-glycosylic bond (Fig. 1B) or are recognized by DNA glycosylases leading to AP site formation.
Ionization radiation (IR) generates a broad spectrum of DNA damage products with double strand breaks the predominant lesion causing cellular lethality. However, IR-induced base damage can also generate AP sites. IR produces hydroxyl radicals that directly attack the N-glycosyl bond or react with the 5,6-double bond in nucleobases saturating the ring and decreasing N-glycosyl bond stability [14],[15]. Other ring-saturated derivates of IR, such as thymine glycol, are recognized by DNA glycosylases that then generate AP sites [16],[17].
Cyclobutane pyrimidine dimers (CPD) are the predominant lesion formed by ultraviolet (UV) radiation. However, other UV induced photoproducts can indirectly produce abasic site lesions [18]. The addition of water across the 5,6-double bond of cytosine generates cytosine hydrate. Cytosine hydrate undergoes deamination to uracil hydrate which is subsequently dehydrated to uracil [19]. This UV-induced product forms more readily in ssDNA than duplex DNA, and subsequent action of uracil DNA glycosylase generate AP sites [1]. Other notable UV induced photoproducts that can lead to abasic sites include thymine glycol, alkali labile purine lesions, and other pyrimidine hydrates [20],[13].
1.3. Action of Specialized DNA Glycosylases
DNA glycosylases produce abasic sites as an intermediate step during the repair of damaged DNA by the base excision repair pathway (discussed below). These specialized enzymes are generally well-conserved through all domains of life. DNA glycosylases catalyze the hydrolysis of the N-glycosidic bond between DNA base and sugar-phosphate, releasing the base. DNA glycosylases can be classified as monofunctional or bifunctional. Monofunctional glycosylases remove the base leaving only the intact AP site whereas bifunctional glycosylases have lyase activity that cleaves DNA 3’ of the AP site to generate a 3’ unsaturated aldehydic and a 5’-phosphorylated ends [13].
Some DNA glycosylases are specific for base-pair mismatches. Thymine DNA glycosylase (TDG) and methyl-CpG-binding domain protein 4 (MBD4) recognize G:T mismatch and G:U or G:T mismatch bases, respectively. Other DNA glycosylases are specialized for a variety of damaged bases, including 8-oxoguanine glycosylase (OGG1) that recognizes and removes 8-oxoG and FapyG lesions and N-methyl-purine DNA glycosylase (MPG) that repairs 3meA, 7meG, and 3meG lesions. Most single glycosylase knockouts are not lethal, suggesting overlapping substrate recognition amongst DNA glycosylases [13]. Most bifunctional glycosylases such as NEIL1, NEIL2, and NEIL3 (endonuclease VIII-like) and NTHL1 (endonuclease III-like) are specialized for oxidized pyrimidines and ring-opened purines [13].
Uracil DNA glycosylase is perhaps the most conserved among yeast, bacteria, and mammalian cells. All mammalian glycosylases that remove uracil are monofunctional. Mammalian uracil DNA glycosylase (UNG) travels with the replisome generating abasic sites. Uracils are incorporated into DNA via numerous mechanisms. DNA polymerases can incorporate dUMP instead of dTMP during replication[21]. While polymerases are highly selective, the genome size and much higher concentration of rNTPs than dNTPs in cells mean that more than 13,000 ribonucleotides are incorporated during each round of DNA replication in budding yeast and orders of magnitude larger numbers in the human genome [22]. Depletion of cellular dNTP pools, which can happen in response to drug treatments that interfere with ribonucleotide reductase and replication timing dysregulation caused by oncogenes, likely increases uracil incorporation [23].
Another prominent source of uracil incorporation in DNA is cytosine deamination (Fig. 1C). Spontaneous deamination of cytosine introduces ~100–500 uracils per day in human cells[24]. Cytosines are more prone to heat-induced degradation compared to other DNA bases. This deamination occurs spontaneously at neutral pH. The rate of cytosine deamination is higher in ssDNA than dsDNA [2],[25],[26], and approximately 1–2% of DNA in proliferating mammalian cells is in the single-stranded form at any given time, especially during the processes of replication and transcription [25]. Subsequent uracil removal by UNG generates significant numbers of AP sites. Furthermore, the additive action of enzymes, such as cytosine deaminases, can exacerbate the number of abasic sites in the genome. For example, the AID/APOBEC (activation-induced deaminase/apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like) family of DNA cytosine deaminases specifically act on ssDNA [27]. While APOBEC enzymes are usually retained in the cytoplasm and act to restrict invading nucleic acids from pathogens, cancer cells often exhibit elevated APOBEC expression and nuclear localization. Thus, cytosine deamination by APOBEC enzymes generates two mutational signatures in cancer. One of the signatures is the result of misincorporations across from abasic sites [28],[29]. This mutagenesis is biased towards the lagging strand since that is where the majority of ssDNA is present to be targeted by the APOBEC enzymes during replication [30],[31]. Thus, generation of uracils in DNA is a notable source of genome instability that drives tumorigenesis in part through production of abasic sites.
2. Consequences of AP sites for genome stability
AP sites have multiple deleterious consequences (Fig. 2). They interfere with the natural information content function of the DNA and are obstacles to RNA polymerases. They are reactive intermediates in the generation of interstrand and DNA protein crosslinks. When encountered during DNA replication, they can generate mutations. Finally, they are potent blocks to DNA polymerases, which can threaten the completion of DNA replication.
Fig. 2.
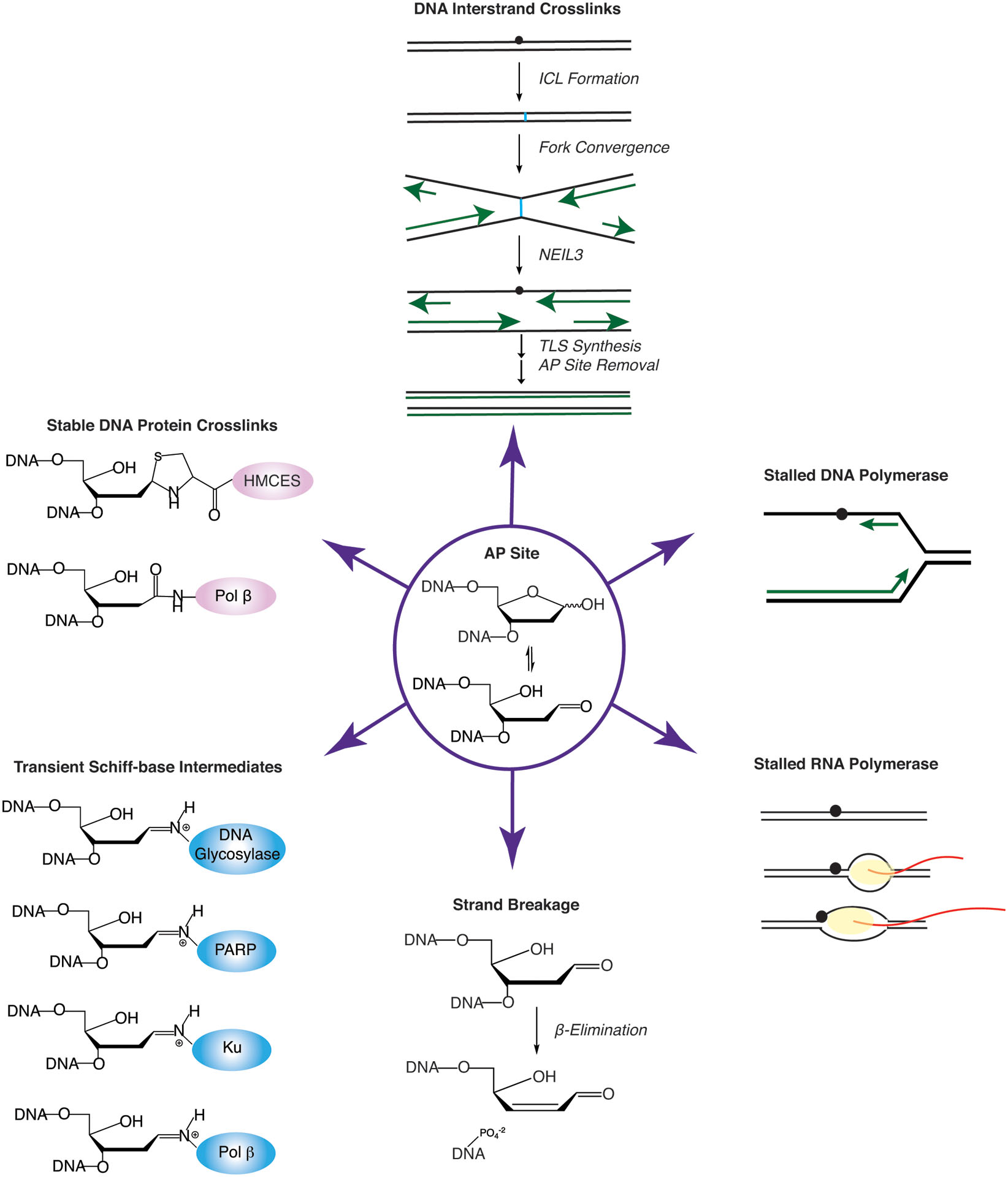
Consequences of unrepaired AP sites. AP sites (in center) react to form stable (pink) or transient (blue) DNA-protein crosslinks (DPCs) (left), generate intrastrand crosslinks (ICLs) (top), stall DNA polymerases and RNA polymerases (right), and can cause strand breakage via β-elimination (bottom).
2. 1. AP sites are intermediates in the formation of interstrand and DNA-protein crosslinks
Abasic sites exist as a sugar anomer in an equilibrating mixture of a closed-ring furanose (99%) and an open-ring aldehyde (1%) [32]. The open-ring aldehyde form is highly reactive [33]. AP sites can be converted into strand-breaks via a β-elimination reaction where the 3’ phosphodiester bond of the aldehyde form is hydrolyzed to generate a 3’-terminal unsaturated sugar and a terminal 5’-phosphate. The presence of nucleophilic molecules, including thiols, amines, polyamines, and basic proteins, further stimulates this reaction [34]. Furthermore, AP sites can generate DNA intra- and inter-strand crosslinks (ICLs) [35],[36]. The AP aldehyde can react with the exocyclic amino group on nucleobases, especially guanine [37],[35]. Interestingly, these abasic site ICLs are cleaved by the DNA glycosylase NEIL3. This NEIL3 mediated “unhooking” prevents DSB formation and curiously generates another AP site intermediate [38].
AP sites are also prone to forming crosslinks with multiple proteins. Several repair enzymes, including PARP1/2 [39],[40],[41], Pol β [42],[43], and KU [44],[45] can form DPCs with AP site lesions or BER intermediates (reviewed in [46],[47]) (Fig. 2). However, these DPCs are often thought to be suicidal, deleterious, or transient intermediates. PARP1/2, Pol β, and KU all form transient Schiff-base intermediates that can be trapped in the presence of NaBH4. Most Schiff-base intermediates are unstable and are quickly resolved by β-elimination to release the enzyme. Interestingly, Pol β also forms a dead-end suicide DPC with 2-deoxyribonolactone (dL), an oxidized AP site derivative [42]. This Pol β DPC occurs in the absence of NaBH4, forms in vivo, and is repaired in a proteasome-dependent mechanism[43]. Structures of other DNA enzymes that form DPCs such as topoisomerase and DNA methyltransferases are in the literature. However, these DPCs are often covalently trapped using mechanism-based compounds. Furthermore, at least in the case of topoisomerase cleavage complexes, their repair is mediated by a specific set of enzymes (ie-TDP1/TDP2) [48],[49].
2.2. AP sites are a strong block to replicative polymerases and generate mutations
AP sites preferentially form at sites of DNA replication due to the vulnerability of ssDNA to base damage[5]. Although AP sites can retain B-DNA structure [50], they are a potent block to replicative polymerases (Pol alpha, delta, and epsilon) [51],[52],[53]. AP sites are highly mutagenic, and their mutational capacity varies depending on the organism and system used[54]. In E. coli cells, replication past the AP site is characterized by the preferential incorporation of adenosine (A) opposite AP sites[55]. This “A-rule” predicts that depurinations should produce transversions, and depyrimindations should produce transitions. In transfection experiments in which single-stranded bacteriophage DNA was introduced into E. coli and recovered from cells, this was the exact mutational pattern observed [51],[56]. Furthermore, this rule is observed in systems using purified DNA polymerases and constructs containing abasic sites [53], [57], [58]. In Saccharomyces cerevisiae, using a dsDNA plasmid system, dAMP was preferentially inserted opposite AP sites[59]. However, other nucleotides have been reported to be preferentially inserted opposite abasic sites in other studies. Gibbs and colleagues observed a dCMP preference in a gapped duplex shuttle vector with a single abasic site located within a single-stranded region[60]. Similarly, Otsuka and colleagues using a short oligonucleotide transformation observed a preference for dCMP insertion opposite abasic sites[61]. In another study, using spontaneous mutagenesis of the SUP4-o gene in APN1 deficient yeast dGMP was preferential inserted[62]. Dissimilar results have also been obtained in eukaryotic cells suggesting that the A-rule may not always be operable[63],[64],[65]. In any case, the preference for nucleotide insertion across form AP sites is dependent on a multiplicity of factors including sequence context and cell type. Furthermore, peculiarities dependent on the assay such as aberrant DNA synthesis of transfected DNA vs. chromosomal replication may also play a role in observed differences in nucleotide preference.
3. Repair of AP sites in dsDNA
Cells must cope with the high levels of AP site lesions to avoid deleterious consequences. In dsDNA, AP sites can be repaired using the high-fidelity methods of Base Excision Repair (BER) and Nucleotide Excision Repair (NER) since an intact DNA template remains to provide coding information for repair synthesis. These repair systems are fast, accurate, and likely conduct the majority of AP site repair[13],[66],[67].
3.1. Repair of AP sites by Base Excision Repair
BER protects the genome from base damage by endogenous and exogenous sources. BER is a series of sequential reactions coordinated by multiple enzymes to remove small damaged or distorted DNA bases, AP sites, and AP site intermediates (reviewed here [13,68–71]). Briefly, the five major steps are: recognition and removal of incorrect or damaged DNA base, excision of AP site by AP endonuclease or AP lyase, cleaning up of DNA ends, repair synthesis by DNA polymerase, and sealing of the nick by DNA ligase. BER can be broken into two subcategories--short-patch repair for single-nucleotide lesions and long-patch repair for multiple-nucleotide lesions. In general, short-patch repair is more prevalent and utilizes APE1 and Pol β [72].
For short patch repair, the modified base is removed by a monofunctional DNA glycosylase. AP endonuclease incises the DNA 5’ to the AP lesion, leaving behind 3’ hydroxyl and 5’ terminal deoxyribose phosphate (5’ dRP) DNA ends. The resulting one-nucleotide gap in duplex DNA is then repaired by Pol β or Pol λ with the help of accessory factors poly(ADP-ribose) polymerase 1 (PARP) and X-ray repair cross-complementing 1 (XRCC1). Finally, DNA ligase I or 3 ligates the DNA ends to seal the nick. For long patch repair, Pol δ, ε, or λ perform strand displacement synthesis to remove 2–10 nucleotides. A 5’ flap is generated and then removed by flap endonuclease 1 (FEN1). This generates the DNA end required for ligation. Short and long-patch BER occur in parallel. Changes in cell type, cell cycle, the initiating glycosylase, or the ATP concentration all have been implicated in BER pathway choice [73],[74].
3.2. Nucleotide excision repair of AP sites
In addition to BER, nucleotide excision repair (NER) can also remove AP sites from DNA. Two types of NER are possible – global genome NER (GG-NER) or transcription-coupled NER (TC-NER) (reviewed here [66,67]). In GG-NER, large distortions in the DNA helix, such as pyrimidine dimers, promote the recruitment of NER machinery. NER enzymes generate single-strand nicks in the DNA 5’ and 3’ to damage to generate a ~25–30 nucleotide single-strand fragment[75]. The subsequent gap is repaired by the actions of DNA polymerase and DNA ligase. Generally, small base damage and AP sites are not thought to generate the needed helical distortion for recognition by GG-NER. However, in E. coli the central NER protein uvrABC can generate 3’ and 5’ nicks to an AP site in vitro [76],[77]. Similarly, eukaryotic NER proteins can recognize and process AP sites in vitro[78]. In yeast, NER inactivation increases sensitivity of AP endonuclease-deficient cells to MMS. Furthermore, the rate of AP site repair is decreased and the MMS-induced mutation frequency is increased in BER- and NER-deficient cells.[79] Other studies have also found an overlap or crosstalk between BER and NER pathways[80] suggesting that NER and BER have the ability to recognize and repair similar lesions.
TC-NER is a major repair mechanism for AP sites in cells. AP sites are a potent block to T7 RNAP and RNA polymerase II (RNAPII) during transcription when present on the actively transcribed strand[81]. The AP lesion structurally interferes with nucleotide incorporation, and subsequent nucleotide extension past the AP site is slow[82]. The stalling of transcription at AP sites is highly mutagenic[83], and AMP is preferentially inserted opposite AP sites by RNAPII[82]. Furthermore, highly transcribed sequences are more susceptible to AP site accumulation and are a significant source of transcription-associated mutagenesis[84], [85], [86], [87]. NER deficiencies cause increased transcriptional mutagenesis at the AP site [84], [88]. The paused RNAPII at lesions signals the recruitment of the NER machinery. Overall, the final steps of excision, repair, and ligation remain the same as GG-NER.
4. AP site repair in ssDNA context
AP sites in ssDNA present a different threat to genome stability that generally cannot be solved by excision repair. ssDNA AP sites can form wherever ssDNA exists such as at telomeres or in transcription bubbles. However, they are most prevalent during DNA replication due to either the unwinding of dsDNA containing an unrepaired AP site or the generation of a new AP site in the ssDNA on the lagging strand. Thus, most ssDNA abasic site repair or tolerance mechanisms operate in the context of replication. BER enzymes such as APE1 do have activity on AP sites in ssDNA, although AP-ssDNA processing by APE1 is ~20-fold less compared to AP-dsDNA[89]. Many DNA glycosylases also have activity against AP-ssDNA substrates [90],[91],[92]. There may be a function for BER repair in ssDNA at sites of transcription or replication [90]. However, since BER cleaves the DNA backbone, it generally would not be useful for AP sites in ssDNA since there would not be a template for repair. Translesion synthesis (TLS) polymerases provide one error-prone mechanism of AP site tolerance. Homologous recombination or fork reversal combined with template switching can provide an error-free mechanism. Finally, two additional initiating mechanisms for repair or tolerance of ssDNA-AP sites were recently described, which are mediated by Rad51 paralogs[93] or the evolutionarily conserved AP basic site shielding protein HMCES [94].
4. 1. TLS Bypass of AP Sites
TLS polymerases replicate past lesions that block replicative polymerases, thus allowing cells to tolerate DNA damage (Fig. 3A). These polymerases have a larger active site compared to replicative polymerase allowing accommodation of damaged or distorted bases. However, the low fidelity and lack of exonuclease proofreading activity of TLS polymerases combined with the lack of coding information on the template strand make bypass of DNA lesions error prone. Here we will focus on the abasic site specificity of TLS polymerases. The specificity of bypass for other lesions has been reviewed previously [95],[96].
Fig. 3.
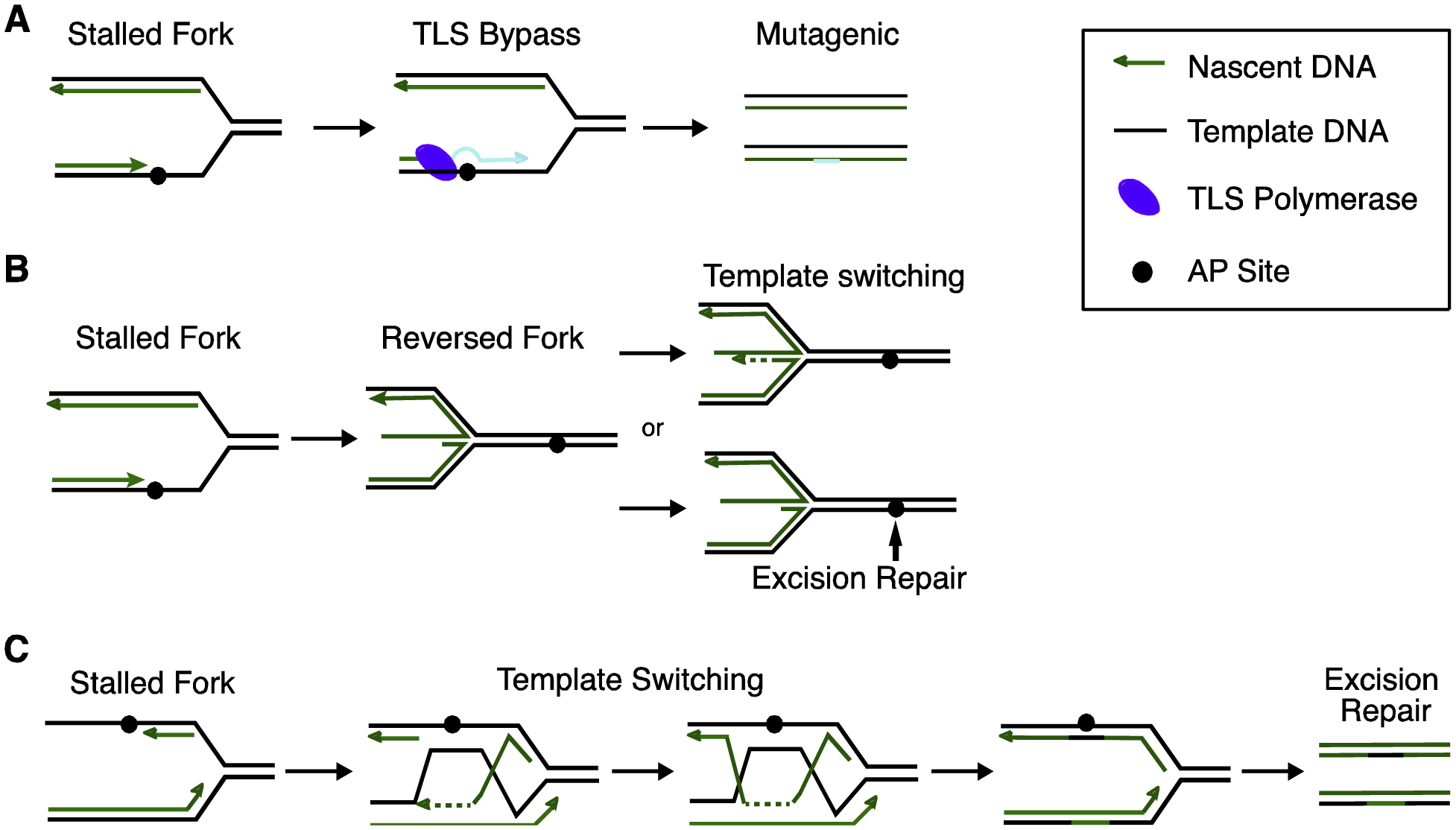
AP site repair in ssDNA. A. Translesion synthesis (TLS) polymerase bypass an AP lesion to promote error-prone damage tolerance. B. Fork reversal generates a four-way chicken foot structure that can facilitate template switching or excision repair. C. Template switching using a strand invasion step can place a ssDNA AP lesion in the context of dsDNA for error-free bypass and repair.
TLS polymerases bypass often follows a two-step process where one polymerase inserts a nucleotide across from the lesion and another polymerase extends past the lesion. Following bypass and extension by TLS, further extension is resumed by high fidelity polymerases. TLS polymerases involved in abasic site bypass include the Y family of polymerases including Pol eta (Rad30) and Rev1 in S. cerevisiae, Polη, Polι, Polκ, and Rev1 in humans, and DNA Pol IV (DinB) and Pol V (UmuC) in E. coli. The Y-family polymerases generally act as insertors across from the AP site lesions. B-family polymerases such as E. coli DNA pol II and eukaryotic Polζ usually perform extension synthesis from the inserted nucleobase opposite the abasic site. Other polymerases such as Polλ and Polμ are generally not thought to be involved in AP site bypass.
In vertebrates, much of the in vivo evidence for TLS bypass of AP sites comes from studies of immunoglobulin (Ig) somatic hypermutation (SHM). During B-cell development, B cells undergo a second round of antibody diversification involving the Ig locus. In this process, the rearranged IgG genes are hypermutated by activation-induced deaminase (AID), a member of the APOBEC (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like) family of proteins. AID intentionally generates deoxyuridines in ssDNA. A portion of these uracils are subsequently processed by uracil DNA glycosylase to generate abasic sites that are then bypassed by TLS polymerases. Data suggests a role for Polη Polι Rev1, and Polζ in SHM although the modest effects of genetic inactivation of Polη and Polι suggest there is significant redundancy or compensation.
Polη is adept at bypassing UV photoproducts. However, its overall contribution to AP site bypass is controversial. In yeast, pol eta (Rad30) deficiency is dispensable for AP site bypass [97]. In patients with a variant form of xeroderma pigmentosum (XPV), the frequency of SHM is normal [98]. Similarly, in Polη-deficient mice the frequency of mutations remains overall the same compared to wild-type[99]. However, the Ig mutational profile in both the XPV patients and Polη-deficient mice suggest that Polη contributes to mutations at A and T[98], [99]. In vitro, Polη can bypass AP sites but its efficiency may be low and dependent on sequence context and experimental conditions [100],[101],[102].
Polι may also have limited activity in AP site bypass. The overall frequency of SHM is decreased in BL2 Burkitt’s Lymphoma cells deficient for Polι [103]. However, mice with a natural nonsense mutation in the POLI gene have a normal frequency of Ig hypermutation [104]. Inactivation of Polι with simultaneous loss of Polη in mice does not alter the mutation pattern seen by Polη alone suggesting a marginal role for Polι in AP site bypass [105]. Polι can incorporate nucleotides opposite an abasic site lesion with preference for dGTP and dTTP over dATP and dCTP [106],[107] but it cannot do extension synthesis after nucleotide incorporation [108].
The REV1 Y family DNA polymerase, interacts with REV7 and inserts dCMP opposite AP sites due to its intrinsic dCMP transferase activity [109], [110]. Mice with a rev1 deficiency have a normal frequency of SHM but a decrease in C to G transversions suggesting a role for Rev1 in AP site bypass [111]. Similarly, disruption of the REV1 gene in DT40 cells causes defects in SHM [112]. Using a subtelomeric ssDNA reporter system, Chan and colleagues were able to assess the in vivo specificity of AP site bypass by different TLS polymerases in yeast [97]. By overexpressing APOBEC3G, they were able to selectively generate AP sites in ssDNA and show that Rev1 and Rev3 (the catalytic subunit of Polζ) were necessary for TLS bypass of AP sites [97]. Additionally, Rev1-deficient yeast exhibit a marked decrease in mutation frequency of AP-ssDNA [97]. Interestingly, catalytic dead Rev1 expressing cells have wild-type mutation frequencies suggesting it has a non-catalytic, scaffolding function for other TLS polymerases [97],[59],[113]. However, the composition of mutations in the Rev1-catalytic dead cells is markedly different [97]. Thus, Rev1 may be the predominant polymerase that inserts dCMP across from AP sites, which is consistent with other studies [60],[111],[112],[114],[115].
To date there does not seem to be a significant role for Polκ [116],[117] or Polθ in SHM, although Polθ can bypass AP sites in vitro [118],[119].The role of Polβ in SHM is controversial and its effects in B-cell development may be due to other DNA repair functions [120],[121],[122]. After insertion of a base across from the AP, site Polζ is involved in the majority of extension polymerization prior to switching to high fidelity polymerases [113]. Polζ is a complex of the catalytic subunit REV3 and an accessory subunit REV7. Error-prone synthesis by Polζ can continue for several hundred nucleotides[123]. In yeast, Rev3 is required for AP site induced mutagenesis [113],[97]. Studies based on transfection of plasmids with a single AP site show that Rev3 is necessary for the majority of mutagenic TLS bypass [114],[59]. Mice expressing rev3l anti-sense RNA have reduced mutation frequencies at all base pairs in the IgVH genes and impaired affinity maturation of memory B cells indicating a significant function for Polζ in SHM [124]. Thus, Polζis critical for TLS-mediated bypass of AP sites.
In E. coli, SOS induction increases replication-dependent AP site tolerance by increasing expression of TLS polymerases. The SOS-induced polymerases umuC and umuD (Pol V) and dinB (pol IV) can bypass AP sites. Pol V is thought to be the major lesion bypass DNA polymerase for AP sites in vivo and contributes to the A-rule by preferentially incorporating dAMP [125],[126],[127]. Deletion of umuDC operon causes a 3.5-fold decrease in spontaneous mutation frequency in BER deficient E. coli [128]. The UmuC-UmuD’ complex in conjunction with RecA and SSB (single-stranded DNA binding protein) act to rescue replicative polymerase stalled at AP sites [129],[130]. In the absence of UmuD’ and UmuC AP sites are skipped resulting in mostly single-nucleotide deletions [130]. Pol IV (dinB) is 5–10-fold more accurate than Pol V (umuCD), but it is inefficient at AP site bypass [125],[127], and DinB does not seem to play a major role in AP site bypass.
4.2. Recombination and Template-Switching repair of AP Sites
Homologous recombination (HR)-mediated repair and template switching have also been suggested as an error free mechanism of AP site repair. Certainly if AP sites are processed into DSBs or if AP sites are part of complex lesions such as those generated by radiation or free radicals, then homology-directed repair of the break including resection and strand invasion would be useful [131],[1]. HR-mediated repair may be particularly important during DNA replication. When a replication fork encounters an AP site, the helicase unwinds the DNA but the polymerase stalls leading to an accumulation of ssDNA. This increase of ssDNA could invoke HR and template switching pathways.
Using an engineered thymine DNA glycosylase to generate chromosomal AP sites, Otterle and colleagues demonstrated that AP site cytotoxic effects are enhanced in cells deficient in BER, TLS, and recombination repair [132]. Furthermore, chromosomal AP sites are cytotoxic and mutagenic in E. coli strains deficient in recombination or BER [133]. In S. cerevisiae, BER-deficient strains have an increased rate of HR [80]. When HR is diminished in the setting of BER deficiency, a stronger mutator phenotype results compared to loss of BER or HR alone. Likewise, when TLS and BER are inactivated, there is a synergistic increase in the recombination rate indicating that multiple, partly compensatory pathways can converge to accomplish AP site repair [80]. In mammalian cells, HR was shown to repair DNA gaps opposite an abasic site in a plasmid-based assay [134]
During replication, template switching can promote error-free repair of AP sites. In template switching the undamaged information on the sister chromatid is utilized to generate duplex DNA that can then be processed via one of the dsDNA AP site repair mechanisms. Template switching could occur via a fork reversal process in which a four-way junction is generated by annealing the two nascent DNA strands (Fig. 3B), or a strand invasion mechanism dependent on HR proteins like RAD51 or RAD52 [135] (Fig. 3C). In both cases, DNA synthesis followed by a resolution step allows bypass of the lesion without introduction of a mutation [136]. The AP site can then be repaired via BER at a later time. Alternatively, the lesion could be repaired while the fork is reversed to generate an intact template for continued DNA synthesis. In E. coli, repair of a gapped plasmid containing an abasic site was repaired 20% of the time by template switching [137]. Fork reversal is thought to be a frequent event in human cells [136], but whether it actually facilitates error-free AP site tolerance is unknown [138].
4.3. The Shu complex promotes damage tolerance of AP sites
The S. cerevisiae Shu complex is a heterotetrametric complex containing Shu2 (SWIM domain containing protein) and the RAD51 paralogs Csm2, Pys3, and Shu1. Based on subunit homologies to RAD51, it was originally characterized in HR repair of DSBs[139]. More recently, an activity in controlling ssDNA AP site processing was discovered (Fig. 4A). Shu mutant yeast are sensitive to MMS-induced replication blocking lesions and have an elevated mutation frequency consistent with an activity in promoting error-free lesion tolerance [140]. Indeed, Shu promotes error-free bypass of MMS-induced alkylation damage and abasic sites specifically when they are present on the lagging strand template during replication [140]. Csm2-Psy3 of the Shu complex binds double-flap DNA substrates with AP sites in vitro and accumulates on AP site associated chromatin in vivo. This binding of abasic sites blocks AP endonuclease activity thereby preventing DSB formation[93]. The Shu complex may both protect the ssDNA AP site from cleavage and promote RAD51-dependent template switching to prevent mutations[93]. A Shu2 protein (SWS1) is found in human cells [141]. SWS1 binds a RAD51 paralog called SWSAP1 [142]. Inactivation of these proteins increases cellular sensitivity to DNA damaging agents that stall forks and reduces fork restart [143]. However, it is not yet known if it has an ssDNA AP-site tolerance activity like the yeast Shu complex.
Fig. 4.
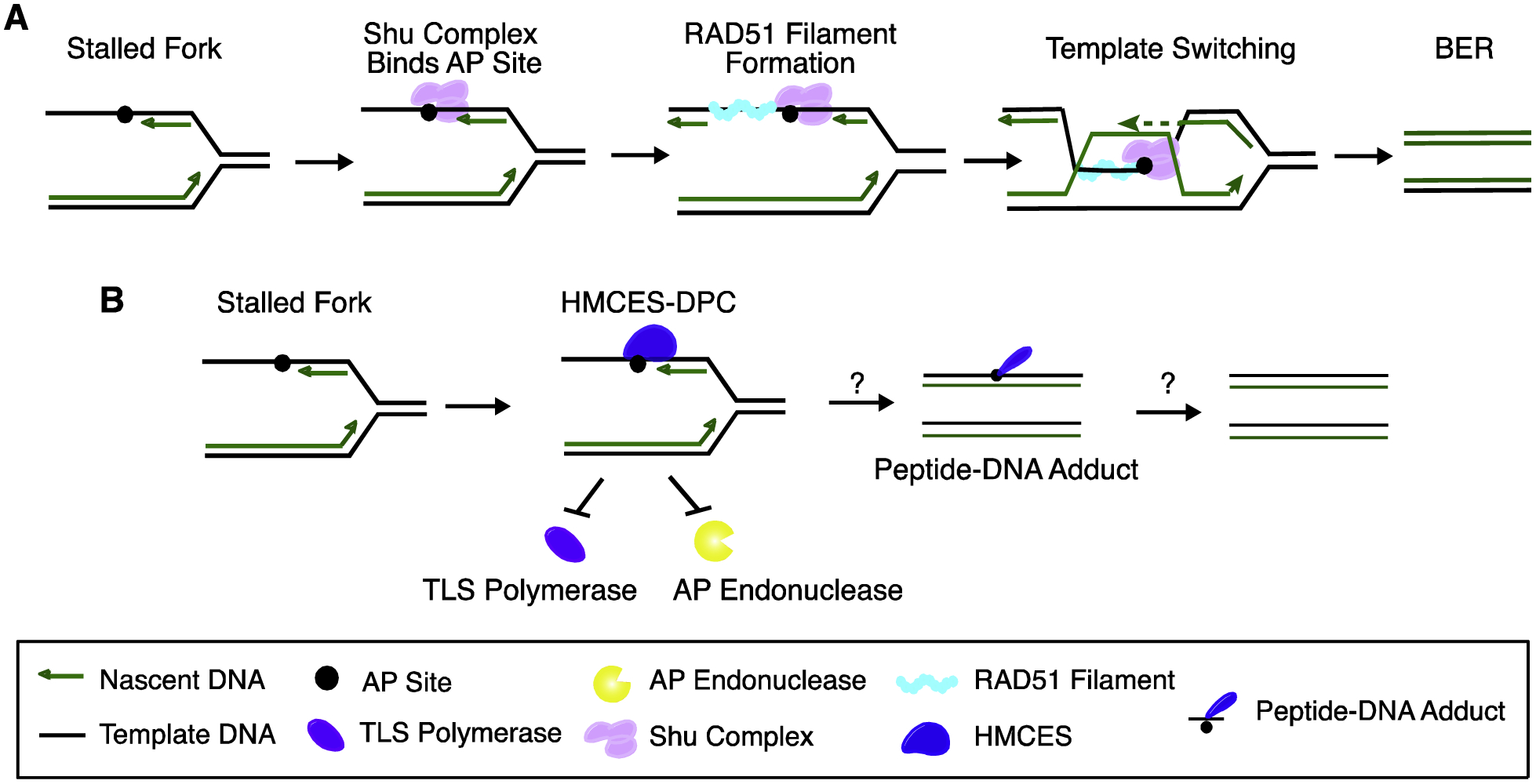
New mechanisms of AP site recognition and repair in ssDNA. A. Model of Shu-mediated error-free damage tolerance of AP site. Shu complex binding to an AP site promotes RAD51 filament formation and template switching using the sister chromatid. BER is used to repair AP site post-replicatively. B. HMCES initiates repair of AP sites by forming a DNA-protein crosslink to AP site in ssDNA especially in the context of DNA replication. The HMCES-AP site DPC prevents endonuclease cleavage and TLS bypass. The mechanisms by which the HMCES-DPC is ultimately repaired remain unknown.
4.4. HMCES initiates a new mechanism for ssDNA AP site recognition and repair
The evolutionary conserved protein HMCES (5-hydroxymethylcytosine embryonic stem cells specific) recently was discovered to initiate a pathway for recognition and repair of AP sites in ssDNA [94] (Fig. 4B). HMCES was found in a mass spectrometry search for candidate proteins that preferentially bind oxidized derivatives of methylcytosine including 5-hydroxymethylcytosine (5hmC) suggesting it might act in an epigenetic pathway of gene expression control [144]. It was also reported to act as a nuclease towards 5hmc-containing DNA [145]. However, other studies failed to replicate these findings [94]. Furthermore, HMCES-deficient or over-expressing human cells did not exhibit changes in levels of 5mC or 5hmC [94]. HMCES deficiency also does not cause a global change 5hmc in bone marrow cells [145] or adult mice [146], although some change was reported in embryonic stem cells [146]. Consistent with a lack of epigenetic function, very few changes in gene expression were found in HMCES-deficient cells [94]. Therefore, HMCES may be a misnomer. Instead, an increase in replication-dependent DNA damage in HMCES-deficient cells, and an increase in DNA damage-dependent transcripts suggests it acts in a replication-coupled DNA repair pathway [94].
HMCES contains an SOS response associated peptidase (SRAP) domain named because SRAP genes in bacteria are spatially linked to SOS response genes including mutagenic TLS polymerases [147]. SRAP domains are conserved through all domains of life and have a proposed thiol autopeptidase activity dependent on a conserved triad of residues (cysteine, glutamate, and histidine) [147]. This peptidase activity was suggested to be an autoproteolytic switch [145]. However, the only residue that would be removed by this proposed activity would be the N-terminal methionine since the cysteine is almost universally encoded by residue two, and amino-peptidases should typically remove the methionine anyway.
Supporting an activity in AP site recognition and processing, HMCES-deficient cells exhibit marked sensitivity to AP site inducing DNA damage agents, including IR, UV, MMS, and KBrO3 and have a defect in AP site resolution [94]. HMCES is recruited to chromatin in response to these agents selectively in S-phase cells, it is localized to replication forks, and directly interacts with PCNA [94]. Furthermore, HMCES-deficient cells are hypersensitive to the nuclear expression of APOBEC3A, further supporting that HMCES is responding to an AP lesion [148].
HMCES and its E.coli ortholog yedK recognize AP sites in ssDNA and form a stable thiazolidine linkage between the ring-opened AP site and the alpha-amino and sulfhydryl groups of the invariant second amino acid cysteine [149],[150],[151]. The HMCES DPC is stable, resistant to strand cleavage by AP endonuclease, and is resolved at least in part via a ubiquitin-dependent mechanism [149],[94]. The HMCES peptide-DNA adduct left after proteolysis is also resistant to AP endonuclease cleavage [149]. HMCES-deficient cells accumulate DNA double-strand breaks during DNA replication [148]. Crystal structures reveal that HMCES has a preference for AP sites at 3’ dsDNA-ssDNA junctions [149] (and reviewed in this issue [156]). The is the exact type of junction that would form when a replicative polymerase stalls at an AP site. Interestingly, HMCES-DPC formation reduces mutation frequencies, HMCES-deficient cells have elevated Polζ levels at replication forks, and TLS activity generates the slow replication elongation kinetics seen in the cells expressing nuclear APOBEC3A [94],[148].
Unifying these observations is the model that HMCES shields ssDNA AP sites from other pathways that would generate DSBs or mutations. In the context of replication, this would presumably let cells get on with replication and reduce genome instability. However, most DPC repair is thought to be an error-prone process as the final steps require TLS polymerization past a peptide-DNA adduct [152],[153]. How the HMCES-DPC could be resolved by an error-free manner is an unanswered question. Template switching or fork reversal could be involved similar to what is proposed for the Shu complex pathway. Alternatively, TLS activity could still work downstream of the HMCES-DPC, but the specific lesion being bypassed (a DPC vs. abasic site) may provide some advantage.
Finally, HMCES may have a DSB repair activity in addition to its activity in AP site processing. HMCES-deficient cells are hypersensitive to ionizing radiation (IR), but not sensitive to PARP inhibitors and they do not exhibit defects in HR or classical non-homologous end joining (NHEJ) repair of DSBs [94]. However, HMCES has been implicated in microhomology-mediated alternative end-joining (Alt-EJ) [146], and the top co-dependency with HMCES identified in the cancer dependency map project is the Alt-EJ factor Polθ [154]. The mechanism of HMCES function in this pathway is unclear and is reported to be independent of the catalytic cysteine residue [146]. This Alt-EJ activity is unlikely to explain the IR sensitivity of HMCES-deficient cells since cells expressing HMCES with a mutation in the cysteine remain IR sensitive [94]. Further studies will be needed to understand the relationship of this function of HMCES to its activity in AP site processing.
5. Conclusion
The prevalence, mutagenicity, and potential for cytotoxicity of AP sites explains why there are so many evolutionarily conserved mechanisms of AP site repair and tolerance. The context of when and where an AP site lesion occurs helps to determine which repair pathway is used, but additional regulation that remains poorly understood must also be important. Given the long history of AP site repair studies, the recent identification of new mechanisms mediated by proteins like HMCES and the Shu complex underscores how much we do not know. Further studies will undoubtedly generate new surprises. Furthermore, these studies have health relevance. In the context of cancer, exploiting AP site repair may be advantageous. For example, in human B-cell lymphoma there is a higher expression of AID and high AP site levels. In fact, treatment with an agent that binds covalently to AP sites is cytotoxic to these AP site high cancer cells but not to other cancer cells or normal cells [155]. Thus, a relatively simple DNA lesion has complex and important causes and consequences.
Acknowledgments
Research on abasic site repair in the Cortez lab is funded by R01ES030575. P.S.T is supported by F30CA228242.
Abbreviations:
- AP
Apurinic/apyrimidinic
- ssDNA
single-stranded
- DNA
dsDNA double-stranded DNA
- 8-oxoG
7,8-dihydro-8-oxoguanine
- FapyG
2,6-diamino-4-hydroxy-5-formamidopyrimidine
- IR
ionizing radiation
- CPD
Cyclobutane pyrimidine dimers
- UV
ultraviolet
- TDG
Thymine DNA glycosylase
- MBD4
methyl-CpG-binding domain protein 4
- OGG1
8-oxoguanine glycosylase
- MPG
N-methyl-purine DNA glycosylase
- NEIL1/2
Neil endonucleaseVIII-like
- NTHL1
E. coli nth endonuclease III-like
- UNG or UDG
uracil DNA glycosylase
- AID/APOBEC
activation-induced deaminase/apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like
- dL
2-deoxyribonolactone
- BER
Base Excision Repair
- NER
Nucleotide Excision Repair
- PARP
poly(ADP-ribose) polymerase
- XRCC1
X-ray repair cross-complementing 1
- FEN1
flap endonuclease 1
- GG-NER
global genome nucleotide excision repair
- TC-NER
transcription-coupled nucleotide excision repair
- RNAPII
RNA polymerase II
- TLS
translesion synthesis
- HMCES
5-hydroxymethylcytosine embryonic stem cells specific
- Ig
immunoglobulin
- SHM
somatic hypermutation
- SSB
single-stranded DNA binding protein
- HR
homologous recombination
- Shu2
SWIM domain containing protein 2
- 5hmC
5-hydroxy-methylcytosine
- SRAP
SOS response associated peptidase
- DPC
DNA-protein crosslink
- NHEJ
non-homologous end joining
- Alt-EJ
microhomology-mediated alternative end-joining
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
There are no conflicts of interest to declare.
Bibliography
- [1].Errol CF, Graham CW, Wolfram S, Richard DW, Roger AS, Tom E, DNA Repair and Mutagenesis, Second Edition, ASM Press, 2006. 10.1128/9781555816704. [DOI] [Google Scholar]
- [2].Lindahl T, Nyberg B, Rate of Depurination of Native Deoxyribonucleic Acid, Biochemistry. 11 (1972) 3610–3618. 10.1021/bi00769a018. [DOI] [PubMed] [Google Scholar]
- [3].Barnes DE, Lindahl T, REPAIR AND GENETIC CONSEQUENCES OF ENDOGENOUS DNA BASE DAMAGE IN MAMMALIAN CELLS, Annu. Rev. Genet 38 (2004) 445–76. 10.1146/annurev.genet.38.072902.092448. [DOI] [PubMed] [Google Scholar]
- [4].Lindahl T, Karlström O, Heat-Induced Depyrimidination of Deoxyribonucleic Acid in Neutral Solution, Biochemistry. 12 (1973) 5151–5154. 10.1021/bi00749a020. [DOI] [PubMed] [Google Scholar]
- [5].Chastain PD, Nakamura J, Rao S, Chu H, Ibrahim JG, Swenberg JA, Kaufman DG, Abasic sites preferentially form at regions undergoing DNA replication, FASEB J. 24 (2010) 3674–3680. 10.1096/fj.09-145276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Loeb LA, Preston BD, Mutagenesis by Apurinic/Apyrimidinic Sites, Annu. Rev. Genet 20 (1986) 201–230. 10.1146/annurev.ge.20.120186.001221. [DOI] [PubMed] [Google Scholar]
- [7].LAWLEY PD, BROOKES P, FURTHER STUDIES ON THE ALKYLATION OF NUCLEIC ACIDS AND THEIR CONSTITUENT NUCLEOTIDES, Biochem. J 89 (1963) 127–138. 10.1042/bj0890127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Singer B, All oxygens in nucleic acids react with carcinogenic ethylating agents, Nature. 264 (1976) 333–339. 10.1038/264333a0. [DOI] [PubMed] [Google Scholar]
- [9].McNeill DR, Lam W, DeWeese TL, Cheng YC, Wilson DM, Impairment of APE1 function enhances cellular sensitivity to clinically relevant alkylators and antimetabolites, Mol. Cancer Res 7 (2009) 897–906. 10.1158/1541-7786.MCR-08-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Croteau DL, Bohr VA, Repair of oxidative damage to nuclear and mitochondrial DNA in mammalian cells, J. Biol. Chem 272 (1997) 25409–25412. 10.1074/jbc.272.41.25409. [DOI] [PubMed] [Google Scholar]
- [11].Cadet J, Richard Wagner J, DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation, Cold Spring Harb. Perspect. Biol 5 (2013) a012559 10.1101/cshperspect.a012559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Whitaker AM, Schaich MA, Smith MS, Flynn TS, Freudenthal BD, Base excision repair of oxidative DNA damage: From mechanism to disease, Front. Biosci. - Landmark 22 (2017) 1493–1522. 10.2741/4555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Krokan HE, Bjørås M, Base excision repair, Cold Spring Harb. Perspect. Biol 5 (2013) 1–22. 10.1101/cshperspect.a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Téoule R, Radiation-induced DNA Damage and Its Repair, Int. J. Radiat. Biol. Relat. Stud. Phys 51 (1987) 573–589. 10.1080/09553008414552111. [DOI] [PubMed] [Google Scholar]
- [15].Dunlap B, Cerutti P, Apyrimidinic sites in gamma-irradiated DNA, FEBS Lett. 51 (1975) 188–190. 10.1016/0014-5793(75)80884-2. [DOI] [PubMed] [Google Scholar]
- [16].Melamede RJ, Hatahet Z, Kow YW, Ide H, Wallace SS, Isolation and Characterization of Endonuclease VIII from Escherichia coli, Biochemistry. 33 (1994) 1255–1264. 10.1021/bi00171a028. [DOI] [PubMed] [Google Scholar]
- [17].Higgins SA, Frenkel K, Cummings A, Teebor GW, Definitive characterization of human thymine glycol N-glycosylase activity, Biochemistry. 26 (1987) 1683–1688. 10.1021/bi00380a029. [DOI] [PubMed] [Google Scholar]
- [18].Lindahl T, DNA Glycosylases, Endonucleases for Apurinic/Apyrimidinic Sites, and Base Excision-Repair, Prog. Nucleic Acid Res. Mol. Biol (1979). 10.1016/S0079-6603(08)60800-4. [DOI] [PubMed] [Google Scholar]
- [19].Boorstein RJ, Hilbert TP, Teebor GW, Cunningham RP, Formation and Stability of Repairable Pyrimidine Photohydrates in DNA, Biochemistry. 29 (1990) 10455–10460. 10.1021/bi00498a004. [DOI] [PubMed] [Google Scholar]
- [20].Ganguly T, Duker NJ, Stability of DNA thymine hydrates, Nucleic Acids Res. 19 (1991) 3319–3323. 10.1093/nar/19.12.3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Joyce CM, Choosing the right sugar: How polymerases select a nucleotide substrate, Proc. Natl. Acad. Sci. U. S. A 94 (1997) 1619–1622. 10.1073/pnas.94.5.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Williams JS, Lujan SA, Kunkel TA, Processing ribonucleotides incorporated during eukaryotic DNA replication, Nat. Rev. Mol. Cell Biol 17 (2016) 350–363. 10.1038/nrm.2016.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tomilin NV, Aprelikova ON, Uracil-DNA Glycosylases and DNA Uracil Repair, Int. Rev. Cytol 114 (1989) 125–179. 10.1016/S0074-7696(08)60860-8. [DOI] [PubMed] [Google Scholar]
- [24].Lindahl T, Instability and decay of the primary structure of DNA, Nature. 362 (1993) 709–715. 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- [25].Lindahl T, DNA Glycosylases, Endonucleases for Apurinic/Apyrimidinic Sites, and Base Excision-Repair, Prog. Nucleic Acid Res. Mol. Biol 22 (1979) 135–192. 10.1016/S0079-6603(08)60800-4. [DOI] [PubMed] [Google Scholar]
- [26].Bhagwat AS, Hao W, Townes JP, Lee H, Tang H, Foster PL, Strand-biased cytosine deamination at the replication fork causes cytosine to thymine mutations in Escherichia coli, Proc. Natl. Acad. Sci. U. S. A 113 (2016) 2176–2181. 10.1073/pnas.1522325113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Conticello SG, Langlois MA, Yang Z, Neuberger MS, DNA Deamination in Immunity: AID in the Context of Its APOBEC Relatives, Adv. Immunol 94 (2007) 37–73. 10.1016/S0065-2776(06)94002-4. [DOI] [PubMed] [Google Scholar]
- [28].Rebhandl S, Huemer M, Greil R, Geisberger R, AID/APOBEC deaminases and cancer, Oncoscience. 2 (2015) 320–333. 10.18632/oncoscience.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Burns MB, Temiz NA, Harris RS, Evidence for APOBEC3B mutagenesis in multiple human cancers, Nat. Genet 45 (2013) 977–983. 10.1038/ng.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hoopes JI, Cortez LM, Mertz TM, Malc EP, Mieczkowski PA, Roberts SA, APOBEC3A and APOBEC3B Preferentially Deaminate the Lagging Strand Template during DNA Replication, Cell Rep. 14 (2016) 1273–1282. 10.1016/j.celrep.2016.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Seplyarskiy VB, Soldatov RA, Popadin KY, Antonarakis SE, Bazykin GA, Nikolaev SI, APOBEC-induced mutations in human cancers are strongly enriched on the lagging DNA strand during replication, Genome Res. 26 (2016) 174–182. 10.1101/gr.197046.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wilde JA, Bolton PH, Mazumder A, Manoharan M, Gerlt JA, Characterization of the Equilibrating Forms of the Aldehydic Abasic Site in Duplex DNA by 17O NMR, J. Am. Chem. Soc 111 (1989) 1894–1896. 10.1021/ja00187a062. [DOI] [Google Scholar]
- [33].Lhomme J, Constant JF, Demeunynck M, Abasic DNA structure, reactivity, and recognition, Biopolymers. 52 (1999) 65–83. . [DOI] [PubMed] [Google Scholar]
- [34].Talpaert-Borlè M, Formation, detection and repair of AP sites, Mutat. Res. - Fundam. Mol. Mech. Mutagen 181 (1987) 45–56. 10.1016/0027-5107(87)90286-7. [DOI] [PubMed] [Google Scholar]
- [35].Dutta S, Chowdhury G, Gates KS, Interstrand cross-links generated by abasic sites in duplex DNA, J. Am. Chem. Soc 129 (2007) 1852–1853. 10.1021/ja067294u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sczepanski JT, Jacobs AC, Greenberg MM, Self-promoted DNA interstrand cross-link formation by an abasie site, J. Am. Chem. Soc 130 (2008) 9646–9647. 10.1021/ja8030642. [DOI] [PubMed] [Google Scholar]
- [37].Price NE, Johnson KM, Wang J, Fekry MI, Wang Y, Gates KS, Interstrand DNA-DNA cross-link formation between adenine residues and abasic sites in duplex dna, J. Am. Chem. Soc 136 (2014) 3483–3490. 10.1021/ja410969x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Semlow DR, Zhang J, Budzowska M, Drohat AC, Walter JC, Replication-Dependent Unhooking of DNA Interstrand Cross-Links by the NEIL3 Glycosylase, Cell. 167 (2016) 498–511.e14. 10.1016/j.cell.2016.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Khodyreva SN, Prasad R, Ilina ES, Sukhanova MV, Kutuzov MM, Liu Y, Hou EW, Wilson SH, Lavrik OI, Apurinic/apyrimidinic (AP) site recognition by the 5′-dRP/AP lyase in poly(ADP-ribose) polymerase-1 (PARP-1), Proc. Natl. Acad. Sci. U. S. A 107 (2010) 22090–22095. 10.1073/pnas.1009182107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Prasad R, Horton JK, Chastain PD, Gassman NR, Freudenthal BD, Hou EW, Wilson SH, Suicidal cross-linking of PARP-1 to AP site intermediates in cells undergoing base excision repair., Nucleic Acids Res. 42 (2014) 6337–51. 10.1093/nar/gku288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kutuzov MM, Khodyreva SN, Ilina ES, Sukhanova MV, Amé JC, Lavrik OI, Interaction of PARP-2 with AP site containing DNA, Biochimie. 112 (2015) 10–19. 10.1016/j.biochi.2015.02.010. [DOI] [PubMed] [Google Scholar]
- [42].Demott MS, Beyret E, Wong D, Bales BC, Hwang JT, Greenberg MM, Demple B, Covalent trapping of human DNA polymerase 03B2 by the oxidative DNA lesion 2-deoxyribonolactone, J. Biol. Chem 277 (2002) 7637–7640. 10.1074/jbc.C100577200. [DOI] [PubMed] [Google Scholar]
- [43].Quiñones JL, Thapar U, Yu K, Fang Q, Sobol RW, Demple B, Enzyme mechanism-based, oxidative DNA-protein cross-links formed with DNA polymerase β in vivo, Proc. Natl. Acad. Sci. U. S. A 112 (2015) 8602–8607. 10.1073/pnas.1501101112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Roberts SA, Strande N, Burkhalter MD, Strom C, Havener JM, Hasty P, Ramsden DA, Ku is a 5′-dRP/AP lyase that excises nucleotide damage near broken ends, Nature. 464 (2010) 1214–1217. 10.1038/nature08926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Strande N, Roberts SA, Oh S, Hendrickson EA, Ramsden DA, Specificity of the dRP/AP lyase of ku promotes nonhomologous end joining (NHEJ) fidelity at damaged ends, J. Biol. Chem 287 (2012) 13686–13693. 10.1074/jbc.M111.329730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Quiñones JL, Thapar U, Wilson SH, Ramsden DA, Demple B, Oxidative DNA-protein crosslinks formed in mammalian cells by abasic site lyases involved in DNA repair, DNA Repair (Amst). 87 (2020) 102773 10.1016/j.dnarep.2019.102773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Quiñones JL, Demple B, When DNA repair goes wrong: BER-generated DNA-protein crosslinks to oxidative lesions, DNA Repair (Amst). 44 (2016) 103–109. 10.1016/j.dnarep.2016.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yang SW, Burgin AB, Huizenga BN, Robertson CA, Yao KC, Nash HA, A eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type I topoisomerases, Proc. Natl. Acad. Sci 93 (1996) 11534–11539. 10.1073/PNAS.93.21.11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ledesma FC, El Khamisy SF, Zuma MC, Osborn K, Caldecott KW, A human 5′-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage, Nature. 461 (2009) 674–678. 10.1038/nature08444. [DOI] [PubMed] [Google Scholar]
- [50].Cuniasse P, Sowers LC, Eritja R, Kaplan B, Goodman MF, Cognet JAH, Lebret M, Guschlbauer W, Fazakerley GV, An abasic site in DNA. Solution conformation determined by proton NMR and molecular mechanics calculations, Nucleic Acids Res. 15 (1987) 8003–8022. 10.1093/nar/15.19.8003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Schaaper RM, Kunkel TA, Loeb LA, Infidelity of DNA synthesis associated with bypass of apurinic sites., Proc. Natl. Acad. Sci. U. S. A 80 (1983) 487–491. 10.1073/pnas.80.2.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lockhart ML, Deutsch JF, Yamaura I, Cavalieri LF, Rosenberg BH, Termination of DNA synthesis in vitro at apurinic sites but not at ethyl adducts on the template, Chem. Biol. Interact (1982). 10.1016/0009-2797(82)90144-2. [DOI] [PubMed] [Google Scholar]
- [53].Sagher D, Strauss B, Insertion of Nucleotides Opposite Apurinic/Apyrimidinic Sites in Deoxyribonucleic Acid during in Vitro Synthesis: Uniqueness of Adenine Nucleotides, Biochemistry. 22 (1983) 4518–4526. 10.1021/bi00288a026. [DOI] [PubMed] [Google Scholar]
- [54].Loeb LA, Preston BD, Mutagenesis by Apurinic/Apyrimidinic Sites, Annu. Rev. Genet 20 (1986) 201–230. 10.1146/annurev.ge.20.120186.001221. [DOI] [PubMed] [Google Scholar]
- [55].Strauss BS, The “A” rule revisited: Polymerases as determinants of mutational specificity, DNA Repair (Amst). 1 (2002) 125–135. 10.1016/S1568-7864(01)00014-3. [DOI] [PubMed] [Google Scholar]
- [56].Kunkel TA, Schaaper RM, Loeb LA, Depurination-induced infidelity of DNA synthesis with purified DNA replication proteins in vitro, Biochemistry. 22 (1983) 2378–2384. 10.1021/bi00279a012. [DOI] [PubMed] [Google Scholar]
- [57].Randall SK, Eritja R, Kaplan BE, Petruska J, Goodman MF, Nucleotide insertion kinetics opposite abasic lesions in DNA., J. Biol. Chem 262 (1987) 6864–6870. [PubMed] [Google Scholar]
- [58].Boiteux S, Laval J, Coding Properties of Poly(deoxycytidylic acid) Templates Containing Uracil or Apyrimidinic Sites: In Vitro Modulation of Mutagenesis by Deoxyribonucleic Acid Repair Enzymes, Biochemistry. 21 (1982) 6746–6751. 10.1021/bi00269a020. [DOI] [PubMed] [Google Scholar]
- [59].Pagès V, Johnson RE, Prakash L, Prakash S, Mutational specificity and genetic control of replicative bypass of an abasic site in yeast, Proc. Natl. Acad. Sci. U. S. A 105 (2008) 1170–1175. 10.1073/pnas.0711227105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Gibbs PEM, Lawrence CW, Novel mutagenic properties of abasic sites in saccharomyces cerevisiae, J. Mol. Biol 251 (1995) 229–236. 10.1006/jmbi.1995.0430. [DOI] [PubMed] [Google Scholar]
- [61].Otsuka C, Sanadai S, Hata Y, Okuto H, Noskov VN, Loakes D, Negishi K, Difference between deoxyribose- and tetrahydrofuran-type abasic sites in the in vivo mutagenic responses in yeast, Nucleic Acids Res. 30 (2002) 5129–5135. 10.1093/nar/gkf666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kunz BA, Henson ES, Roche H, Ramotar D, Nunoshiba T, Demple B, Specificity of the mutator caused by deletion of the yeast structural gene (APN1) for the major apurinic endonuclease, Proc. Natl. Acad. Sci. U. S. A 91 (1994) 8165–8169. 10.1073/pnas.91.17.8165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Gentil A, Renault G, Madzak C, Margot A, Cabral-Neto JB, Vasseur JJ, Rayner B, Imbach JL, Sarasin A, Mutagenic properties of a unique abasic site in mammalian cells, Biochem. Biophys. Res. Commun (1990). 10.1016/S0006-291X(05)80092-0. [DOI] [PubMed] [Google Scholar]
- [64].Klinedinst DK, Drinkwater NR, Mutagenesis by apurinic sites in normal and ataxia telangiectasia human lymphoblastoid cells, Mol. Carcinog 6 (1992) 32–42. 10.1002/mc.2940060107. [DOI] [PubMed] [Google Scholar]
- [65].Cabral Neto JB, Caseira Cabral RE, Margot A, Le Page F, Sarasin A, Gentil A, Coding properties of a unique Apurinic/Apyrimidinic site replicated in mammalian cells, J. Mol. Biol 240 (1994) 416–420. 10.1006/jmbi.1994.1457. [DOI] [PubMed] [Google Scholar]
- [66].Shuck SC, Short EA, Turchi JJ, Eukaryotic nucleotide excision repair: from understanding mechanisms to influencing biology, Cell Res. 18 (2008) 64–72. 10.1038/cr.2008.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Spivak G, Nucleotide excision repair in humans, DNA Repair (Amst). 36 (2015) 13–18. 10.1016/j.dnarep.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].David SS, O’Shea VL, Kundu S, Base-excision repair of oxidative DNA damage, Nature. 447 (2007) 941–950. 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Wallace SS, Base excision repair: A critical player in many games, DNA Repair (Amst). 19 (2014) 14–26. 10.1016/j.dnarep.2014.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Beard WA, Horton JK, Prasad R, Wilson SH, Annual Review of Biochemistry Eukaryotic Base Excision Repair: New Approaches Shine Light on Mechanism, (2019). 10.1146/annurev-biochem-013118. [DOI] [PMC free article] [PubMed]
- [71].Kim Y-J, Wilson III DM, Overview of Base Excision Repair Biochemistry, Curr. Mol. Pharmacol 5 (2011) 3–13. 10.2174/1874467211205010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].V Sukhanova M, Khodyreva SN, Lebedeva NA, Prasad R, Wilson SH, Lavrik OI, Human base excision repair enzymes apurinic/apyrimidinic endonuclease1 (APE1), DNA polymerase beta and poly(ADP-ribose) polymerase 1: interplay between strand-displacement DNA synthesis and proofreading exonuclease activity., Nucleic Acids Res. 33 (2005) 1222–9. 10.1093/nar/gki266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Petermann E, Ziegler M, Oei SL, ATP-dependent selection between single nucleotide and long patch base excision repair., DNA Repair (Amst). 2 (2003) 1101–14. 10.1016/s1568-7864(03)00117-4. [DOI] [PubMed] [Google Scholar]
- [74].Fortini P, Dogliotti E, Base damage and single-strand break repair: Mechanisms and functional significance of short- and long-patch repair subpathways, DNA Repair (Amst). 6 (2007) 398–409. 10.1016/j.dnarep.2006.10.008. [DOI] [PubMed] [Google Scholar]
- [75].Sancar A, DNA Excision Repair, Annu. Rev. Biochem 65 (1996) 43–81. 10.1146/annurev.bi.65.070196.000355. [DOI] [PubMed] [Google Scholar]
- [76].Snowden A, Van Houten B, Kow YW, Van Houten B, Van Houten B, Damage Repertoire of the Escherichia coli UvrABC Nuclease Complex Includes Abasic Sites, Base-Damage Analogues, and Lesions Containing Adjacent 5′ or 3′ Nicks, Biochemistry. 29 (1990) 7251–7259. 10.1021/bi00483a013. [DOI] [PubMed] [Google Scholar]
- [77].Lin JJ, Sancar A, A New Mechanism for Repairing Oxidative Damage to DNA: (A)BC Excinuclease Removes AP Sites and Thymine Glycols from DNA, Biochemistry. 28 (1989) 7979–7984. 10.1021/bi00446a002. [DOI] [PubMed] [Google Scholar]
- [78].Reardon JT, Bessho T, Kung HC, Bolton PH, Sancar A, In vitro repair of oxidative DNA damage by human nucleotide excision repair system: Possible explanation for neurodegeneration in Xeroderma pigmentosum patients, Proc. Natl. Acad. Sci. U. S. A 94 (1997) 9463–9468. 10.1073/pnas.94.17.9463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Torres-Ramos CA, Johnson RE, Prakash L, Prakash S, Evidence for the Involvement of Nucleotide Excision Repair in the Removal of Abasic Sites in Yeast, Mol. Cell. Biol 20 (2000) 3522–3528. 10.1128/mcb.20.10.3522-3528.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Swanson RL, Morey NJ, Doetsch PW, Jinks-Robertson S, Overlapping Specificities of Base Excision Repair, Nucleotide Excision Repair, Recombination, and Translesion Synthesis Pathways for DNA Base Damage in Saccharomyces cerevisiae, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Tornaletti S, Maeda LS, Hanawalt PC, Transcription Arrest at an Abasic Site in the Transcribed Strand of Template DNA, Chem. Res. Toxicol 19 (2006) 1215–1220. 10.1021/tx060103g. [DOI] [PubMed] [Google Scholar]
- [82].Wang W, Walmacq C, Chong J, Kashlev M, Wang D, Structural basis of transcriptional stalling and bypass of abasic DNA lesion by RNA polymerase II, Proc. Natl. Acad. Sci. U. S. A 115 (2018) E2538–E2545. 10.1073/pnas.1722050115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Yu S-L, Lee S-K, Johnson RE, Prakash L, Prakash S, The Stalling of Transcription at Abasic Sites Is Highly Mutagenic, Mol. Cell. Biol 23 (2003) 382–388. 10.1128/mcb.23.1.382-388.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Kim N, Jinks-Robertson S, Abasic Sites in the Transcribed Strand of Yeast DNA Are Removed by Transcription-Coupled Nucleotide Excision Repair, Mol. Cell. Biol 30 (2010) 3206–3215. 10.1128/mcb.00308-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Kim N, Jinks-Robertson S, dUTP incorporation into genomic DNA is linked to transcription in yeast, Nature. 459 (2009) 1150–1153. 10.1038/nature08033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Sweder KS, Hanawalt PC, Preferential repair of cyclobutane pyrimidine dimers in the transcribed strand of a gene in yeast chromosomes and plasmids is dependent on transcription, Proc. Natl. Acad. Sci. U. S. A 89 (1992) 10696–10700. 10.1073/pnas.89.22.10696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Clauson CL, Oestreich KJ, Austin JW, Doetsch PW, Abasic sites and strand breaks in DNA cause transcriptional mutagenesis in Escherichia coli, Proc. Natl. Acad. Sci. U. S. A 107 (2010) 3657–3662. 10.1073/pnas.0913191107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Kitsera N, Rodriguez-Alvarez M, Emmert S, Carell T, Khobta A, Nucleotide excision repair of abasic DNA lesions, Nucleic Acids Res. 47 (2019) 8537–8547. 10.1093/nar/gkz558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Marenstein DR, Wilson DM, Teebor GW, Human AP endonuclease (APE1) demonstrates endonucleolytic activity against AP sites in single-stranded DNA, DNA Repair (Amst). 3 (2004) 527–533. 10.1016/j.dnarep.2004.01.010. [DOI] [PubMed] [Google Scholar]
- [90].Dou H, Mitra S, Hazra TK, Repair of Oxidized Bases in DNA Bubble Structures by Human DNA Glycosylases NEIL1 and NEIL2, J. Biol. Chem 278 (2003) 49679–49684. 10.1074/jbc.M308658200. [DOI] [PubMed] [Google Scholar]
- [91].Kavli B, Sundheim O, Akbari M, Otterlei M, Nilsen H, Skorpen F, Aas PA, Hagen L, Krokan HE, Slupphaug G, hUNG2 is the major repair enzyme for removal of uracil from U:A matches, U:G mismatches, and U in single-stranded DNA, with hSMUG1 as a broad specificity backup, J. Biol. Chem 277 (2002) 39926–39936. 10.1074/jbc.M207107200. [DOI] [PubMed] [Google Scholar]
- [92].Schormann N, Ricciardi R, Chattopadhyay D, Uracil-DNA glycosylases - Structural and functional perspectives on an essential family of DNA repair enzymes, Protein Sci. 23 (2014) 1667–1685. 10.1002/pro.2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Rosenbaum JC, Bonilla B, Hengel SR, Mertz TM, Herken BW, Kazemier HG, Pressimone CA, Ratterman TC, Macnary E, De Magis A, Kwon Y, Godin SK, Van Houten B, Normolle DP, Sung P, Das SR, Paeschke K, Roberts SA, Vandemark AP, Bernstein KA, The Rad51 paralogs facilitate a novel DNA strand specific damage tolerance pathway, Nat. Commun 10 (2019). 10.1038/s41467-019-11374-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Mohni KN, Wessel SR, Zhao R, Wojciechowski ACAC, Luzwick JWJW, Layden H, Eichman BFBF, Thompson PSPS, Mehta KPMKPM, Cortez D, HMCES Maintains Genome Integrity by Shielding Abasic Sites in Single-Strand DNA, Cell. 176 (2019) 144–153.e13. 10.1016/j.cell.2018.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Prakash S, Johnson RE, Prakash L, EUKARYOTIC TRANSLESION SYNTHESIS DNA POLYMERASES: Specificity of Structure and Function, Annu. Rev. Biochem 74 (2005) 317–353. 10.1146/annurev.biochem.74.082803.133250. [DOI] [PubMed] [Google Scholar]
- [96].Waters LS, Minesinger BK, Wiltrout ME, D’Souza S, Woodruff RV, Walker GC, Eukaryotic Translesion Polymerases and Their Roles and Regulation in DNA Damage Tolerance, Microbiol. Mol. Biol. Rev 73 (2009) 134–154. 10.1128/mmbr.00034-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Chan K, Resnick MA, Gordenin DA, The choice of nucleotide inserted opposite abasic sites formed within chromosomal DNA reveals the polymerase activities participating in translesion DNA synthesis, DNA Repair (Amst). 12 (2013) 878–889. 10.1016/j.dnarep.2013.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Zeng X, Winter DB, Kasmer C, Kraemer KH, Lehmann AR, Gearhart PJ, DNA polymerase η is an A-T mutator in somatic hypermutation of immunoglobulin variable genes, Nat. Immunol 2 (2001) 537–541. 10.1038/88740. [DOI] [PubMed] [Google Scholar]
- [99].Delbos F, De Smet A, Faili A, Aoufouchi S, Weill JC, Reynaud CA, Contribution of DNA polymerase η to immunoglobulin gene hypermutation in the mouse, J. Exp. Med 201 (2005) 1191–1196. 10.1084/jem.20050292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Masutani C, Kusumoto R, Iwai S, Hanaoka F, Mechanisms of accurate translesion synthesis by human DNA polymerase eta., EMBO J. 19 (2000) 3100–9. 10.1093/emboj/19.12.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Zhao B, Xie Z, Shen H, Wang Z, Role of DNA polymerase η in the bypass of abasic sites in yeast cells, Nucleic Acids Res. 32 (2004) 3984–3994. 10.1093/nar/gkh710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Haracska L, Washington MT, Prakash S, Prakash L, Inefficient Bypass of an Abasic Site by DNA Polymerase η, J. Biol. Chem 276 (2001) 6861–6866. 10.1074/jbc.M008021200. [DOI] [PubMed] [Google Scholar]
- [103].Faili A, Aoufouchi S, Flatter E, Guéranger Q, Reynaud CA, Weill JC, Induction of somatic hypermutation in immunoglobulin genes is dependent on DNA polymerase iota, Nature. 419 (2002) 944–947. 10.1038/nature01117. [DOI] [PubMed] [Google Scholar]
- [104].McDonald JP, Frank EG, Plosky BS, Rogozin IB, Masutani C, Hanaoka F, Woodgate R, Gearhart PJ, 129-Derived strains of mice are deficient in DNA polymerase ι and have normal immunoglobulin hypermutation, J. Exp. Med 198 (2003) 635–643. 10.1084/jem.20030767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Martomo SA, Yang WW, Vaisman A, Maas A, Yokoi M, Hoeijmakers JH, Hanaoka F, Woodgate R, Gearhart PJ, Normal hypermutation in antibody genes from congenic mice defective for DNA polymerase ι, DNA Repair (Amst). 5 (2006) 392–398. 10.1016/j.dnarep.2005.12.006. [DOI] [PubMed] [Google Scholar]
- [106].Nair DT, Johnson RE, Prakash L, Prakash S, Aggarwal AK, DNA Synthesis across an Abasic Lesion by Human DNA Polymerase ι, Structure. 17 (2009) 530–537. 10.1016/j.str.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Johnson RE, Washington MT, Haracska L, Prakash S, Prakash L, Eukaryotic polymerases [iota] and [zeta] act sequentially to bypass DNA lesions, Nature. 406 (2000) 1015–1019. [DOI] [PubMed] [Google Scholar]
- [108].Choi JY, Lim S, Kim EJ, Jo A, Guengerich FP, Translesion Synthesis across Abasic Lesions by Human B-Family and Y-Family DNA Polymerases α, δ, η, ι, κ, and REV1, J. Mol. Biol 404 (2010) 34–44. 10.1016/j.jmb.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Lin W, Xin H, Zhang Y, Wu X, Yuan F, Wang Z, The human REV1 gene codes for a DNA template-dependent dCMP transferase, Nucleic Acids Res. 27 (1999) 4468–4475. 10.1093/nar/27.22.4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Nelson JR, Lawrence CW, Hinkle DC, Deoxycytidyl transferase activity of yeast REV1 protein, Nature. 382 (1996) 729–731. 10.1038/382729a0. [DOI] [PubMed] [Google Scholar]
- [111].Jansen JG, Langerak P, Tsaalbi-Shtylik A, Van Den Berk P, Jacobs H, De Wind N, Strand-biased defect in C/G transversions in hypermutating immunoglobulin genes in Rev1-deficient mice, J. Exp. Med 203 (2006) 319–323. 10.1084/jem.20052227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Simpson LJ, Rev1 is essential for DNA damage tolerance and non-templated immunoglobulin gene mutation in a vertebrate cell line, EMBO J. 22 (2003) 1654–1664. 10.1093/emboj/cdg161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Haracska L, Unk I, Johnson RE, Johansson E, Burgers PMJ, Prakash S, Prakash L, Roles of yeast DNA polymerases δ and ζ of Rev 1 in the bypass of abasic sites, Genes Dev. 15 (2001) 945–954. 10.1101/gad.882301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Gibbs PEM, McDonald J, Woodgate R, Lawrence CW, The relative roles in vivo of Saccharomyces cerevisiae Pol η, Pol ζ, Rev1 protein and Pol32 in the bypass and mutation induction of an abasic site, T-T (6–4) photoadduct and T-T cis-syn cyclobutane dimer, Genetics. 169 (2005) 575–582. 10.1534/genetics.104.034611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Kim N, Mudrak SV, Jinks-Robertson S, The dCMP transferase activity of yeast Rev1 is biologically relevant during the bypass of endogenously generated AP sites, DNA Repair (Amst). 10 (2011) 1262–1271. 10.1016/j.dnarep.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Schenten D, Gerlach VL, Guo C, Velasco-Miguel S, Hladik CL, White CL, Friedberg EC, Rajewsky K, Esposito G, DNA polymerase K deficiency does not affect somatic hypermutation in mice, Eur. J. Immunol 32 (2002) 3152–3160. . [DOI] [PubMed] [Google Scholar]
- [117].Shimizu T, Shinkai Y, Ogi T, Ohmori H, Azuma T, The absence of DNA polymerase kappa does not affect somatic hypermutation of the mouse immunoglobulin heavy chain gene., Immunol. Lett 86 (2003) 265–70. 10.1016/s0165-2478(03)00046-4. [DOI] [PubMed] [Google Scholar]
- [118].Seki M, Masutani C, Yang LW, Schuffert A, Iwai S, Bahar I, Wood RD, High-efficiency bypass of DNA damage by human DNA polymerase Q, EMBO J. 23 (2004) 4484–4494. 10.1038/sj.emboj.7600424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Hogg M, Seki M, Wood RD, Doublié S, Wallace SS, Lesion bypass activity of DNA polymerase θ (POLQ) is an intrinsic property of the pol domain and depends on unique sequence inserts, J. Mol. Biol 405 (2011) 642–652. 10.1016/j.jmb.2010.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Poltoratsky V, Prasad R, Horton JK, Wilson SH, Down-regulation of DNA polymerase β accompanies somatic hypermutation in human BL2 cell lines, DNA Repair (Amst). 6 (2007) 244–253. 10.1016/j.dnarep.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Esposito G, Texido G, Betz UA, Gu H, Müller W, Klein U, Rajewsky K, Mice reconstituted with DNA polymerase beta-deficient fetal liver cells are able to mount a T cell-dependent immune response and mutate their Ig genes normally, Proc. Natl. Acad. Sci. U. S. A 97 (2000) 1166–1171. 10.1073/pnas.97.3.1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Wu X, Stavnezer J, DNA polymerase beta is able to repair breaks in switch regions and plays an inhibitory role during immunoglobulin class switch recombination, J. Exp. Med 204 (2007) 1677–1689. 10.1084/jem.20070756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Kochenova OV, Daee DL, Mertz TM, Shcherbakova PV, DNA Polymerase ζ-Dependent Lesion Bypass in Saccharomyces cerevisiae Is Accompanied by Error-Prone Copying of Long Stretches of Adjacent DNA, PLOS Genet. 11 (2015) e1005110 10.1371/journal.pgen.1005110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Diaz M, Verkoczy LK, Flajnik MF, Klinman NR, Decreased Frequency of Somatic Hypermutation and Impaired Affinity Maturation but Intact Germinal Center Formation in Mice Expressing Antisense RNA to DNA Polymerase ζ, J. Immunol 167 (2001) 327–335. 10.4049/jimmunol.167.1.327. [DOI] [PubMed] [Google Scholar]
- [125].Tang M, Pham P, Shen X, Taylor JS, O’Donnell M, Woodgate R, Goodman MF, Roles of E coli DNA polymerases IV and V in lesion-targeted and untargeted SOS mutagenesis, Nature. 404 (2000) 1014–1018. 10.1038/35010020. [DOI] [PubMed] [Google Scholar]
- [126].Reuven NB, Arad G, Maor-Shoshani A, Livneh Z, The mutagenesis protein UmuC is a DNA polymerase activated by UmuD’, RecA, and SSB and is specialized for translesion replication, J. Biol. Chem 274 (1999) 31763–31766. 10.1074/jbc.274.45.31763. [DOI] [PubMed] [Google Scholar]
- [127].Maor-Shoshani A, Hayashi K, Ohmori H, Livneh Z, Analysis of translesion replication across an abasic site by DNA polymerase IV of Escherichia coli, DNA Repair (Amst). 2 (2003) 1227–1238. 10.1016/S1568-7864(03)00142-3. [DOI] [PubMed] [Google Scholar]
- [128].Janion C, Sikora A, Nowosielska A, Grzesiuk E, coli E BW535, a triple mutant for the DNA repair genes xth, nth, and nfo, chronically induces the SOS response, Environ. Mol. Mutagen 41 (2003) 237–242. 10.1002/em.10154. [DOI] [PubMed] [Google Scholar]
- [129].Rajagopalan M, Lu C, Woodgate R, O’Donnell M, Goodman MF, Echols H, Activity of the purified mutagenesis proteins UmuC, UmuD’, and RecA in replicative bypass of an abasic DNA lesion by DNA polymerase III, Proc. Natl. Acad. Sci. U. S. A 89 (1992) 10777–10781. 10.1073/pnas.89.22.10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Reuven NB, Tomer G, Livneh Z, The mutagenesis proteins UmuD′ and UmuC prevent lethal frameshifts while increasing base substitution mutations, Mol. Cell 2 (1998) 191–199. 10.1016/S1097-2765(00)80129-X. [DOI] [PubMed] [Google Scholar]
- [131].Ma W, Westmoreland JW, Gordenin DA, Resnick MA, Alkylation Base Damage Is Converted into Repairable Double-Strand Breaks and Complex Intermediates in G2 Cells Lacking AP Endonuclease, PLoS Genet. 7 (2011) e1002059 10.1371/journal.pgen.1002059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Otterlei M, Warbrick E, Nagelhus TA, Haug T, Slupphaug G, Akbari M, Aas A, Steinsbekk K, Bakke O, Krokan HE, Post-replicative base excision repair in replication foci, EMBO J. 18 (1999) 3834–3844. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1171460/pdf/003834.pdf (accessed March 9, 2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Otterlei M, Kavli B, Standal R, Skjelbred C, Bharati S, Krokan HE, Repair of chromosomal abasic sites in vivo involves at least three different repair pathways, EMBO J. 19 (2000) 5542–5551. 10.1093/emboj/19.20.5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Adar S, Izhar L, Hendel A, Geacintov N, Livneh Z, Repair of gaps opposite lesions by homologous recombination in mammalian cells, Nucleic Acids Res. 37 (2009) 5737–5748. 10.1093/nar/gkp632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Cortez D, Replication-Coupled DNA Repair, Mol. Cell 74 (2019) 866–876. 10.1016/j.molcel.2019.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Bhat KP, Cortez D, RPA and RAD51: Fork reversal, fork protection, and genome stability, Nat. Struct. Mol. Biol 25 (2018) 446–453. 10.1038/s41594-018-0075-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Izhar L, Goldsmith M, Dahan R, Geacintov N, Lloyd RG, Livneh Z, Analysis of strand transfer and template switching mechanisms of DNA gap repair by homologous recombination in E. coli: predominance of strand transfer, (2008). 10.1016/j.jmb.2008.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Neelsen KJ, Lopes M, Replication fork reversal in eukaryotes: from dead end to dynamic response, Nat. Rev. Mol. Cell Biol 16 (2015) 207–220. 10.1038/nrm3935. [DOI] [PubMed] [Google Scholar]
- [139].Martino J, Bernstein KA, The Shu complex is a conserved regulator of homologous recombination, FEMS Yeast Res. 16 (2016) fow073 10.1093/femsyr/fow073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Godin SK, Zhang Z, Herken BW, Westmoreland JW, Lee AG, Mihalevic MJ, Yu Z, Sobol RW, Resnick MA, Bernstein KA, The Shu complex promotes error-free tolerance of alkylation-induced base excision repair products, Nucleic Acids Res. 44 (2016) 8199–8215. 10.1093/nar/gkw535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Martín V, Chahwan C, Gao H, Blais V, Wohlschlegel J, Yates JR 3rd, McGowan CH, Russell P, Sws1 is a conserved regulator of homologous recombination in eukaryotic cells, EMBO J. 25 (2006) 2564–2574. 10.1038/sj.emboj.7601141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Liu T, Wan L, Wu Y, Chen J, Huang J, hSWS1·SWSAP1 is an evolutionarily conserved complex required for efficient homologous recombination repair., J. Biol. Chem 286 (2011) 41758–66. 10.1074/jbc.M111.271080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Martino J, Brunette GJ, Barroso-González J, Moiseeva TN, Smith CM, Bakkenist CJ, O’Sullivan RJ, Bernstein KA, The human Shu complex functions with PDS5B and SPIDR to promote homologous recombination, Nucleic Acids Res. 47 (2019) 10151–10165. 10.1093/nar/gkz738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Spruijt CG, Gnerlich F, Smits AH, Pfaffeneder T, Jansen PWTC, Bauer C, Münzel M, Wagner M, Müller M, Khan F, Eberl HC, Mensinga A, Brinkman AB, Lephikov K, Müller U, Walter J, Boelens R, Van Ingen H, Leonhardt H, Carell T, Vermeulen M, Dynamic readers for 5-(Hydroxy)methylcytosine and its oxidized derivatives, Cell. 152 (2013) 1146–1159. 10.1016/j.cell.2013.02.004. [DOI] [PubMed] [Google Scholar]
- [145].Kweon SM, Zhu B, Chen Y, Aravind L, Xu SY, Feldman DE, Erasure of Tet-Oxidized 5-Methylcytosine by a SRAP Nuclease, Cell Rep. (2017). 10.1016/j.celrep.2017.09.055. [DOI] [PubMed] [Google Scholar]
- [146].Shukla V, Halabelian L, Balagere S, Samaniego-Castruita D, Feldman DE, Arrowsmith CH, Rao A, Aravind L, HMCES Functions in the Alternative End-Joining Pathway of the DNA DSB Repair during Class Switch Recombination in B Cells, Mol. Cell 77 (2020) 384–394.e4. 10.1016/j.molcel.2019.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Aravind L, Anand S, Iyer LM, Novel autoproteolytic and DNA-damage sensing components in the bacterial SOS response and oxidized methylcytosine-induced eukaryotic DNA demethylation systems, Biol. Direct 8 (2013). 10.1186/1745-6150-8-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Mehta KPM, Lovejoy CA, Zhao R, Heintzman DR, Cortez D, HMCES maintains replication fork progression and prevents double-strand breaks in response to APOBEC deamination and abasic site formation, Under Revi (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Thompson PS, Amidon KM, Mohni KN, Cortez D, Eichman BF, Protection of abasic sites during DNA replication by a stable thiazolidine protein-DNA cross-link, Nat. Struct. Mol. Biol 26 (2019) 613–618. 10.1038/s41594-019-0255-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Halabelian L, Ravichandran M, Li Y, Zeng H, Rao A, Aravind L, Arrowsmith CH, Structural basis of HMCES interactions with abasic DNA and multivalent substrate recognition., Nat. Struct. Mol. Biol 26 (2019) 607–612. 10.1038/s41594-019-0246-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Wang N, Bao H, Chen L, Liu Y, Li Y, Wu B, Huang H, Molecular basis of abasic site sensing in single-stranded DNA by the SRAP domain of E. coli yedK., Nucleic Acids Res. (2019). 10.1093/nar/gkz744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Stingele J, Bellelli R, Boulton SJ, Mechanisms of DNA-protein crosslink repair, Nat. Rev. Mol. Cell Biol 18 (2017) 563–573. 10.1038/nrm.2017.56. [DOI] [PubMed] [Google Scholar]
- [153].Vaz B, Popovic M, Ramadan K, DNA–Protein Crosslink Proteolysis Repair, Trends Biochem. Sci 42 (2017) 483–495. 10.1016/J.TIBS.2017.03.005. [DOI] [PubMed] [Google Scholar]
- [154].Ghandi M, Huang FW, Jané-Valbuena J, V Kryukov G, Lo CC, McDonald ER, Barretina J, Gelfand ET, Bielski CM, Li H, Hu K, Andreev-Drakhlin AY, Kim J, Hess JM, Haas BJ, Aguet F, Weir BA, V Rothberg M, Paolella BR, Lawrence MS, Akbani R, Lu Y, Tiv HL, Gokhale PC, de Weck A, Mansour AA, Oh C, Shih J, Hadi K, Rosen Y, Bistline J, Venkatesan K, Reddy A, Sonkin D, Liu M, Lehar J, Korn JM, Porter DA, Jones MD, Golji J, Caponigro G, Taylor JE, Dunning CM, Creech AL, Warren AC, McFarland JM, Zamanighomi M, Kauffmann A, Stransky N, Imielinski M, Maruvka YE, Cherniack AD, Tsherniak A, Vazquez F, Jaffe JD, Lane AA, Weinstock DM, Johannessen CM, Morrissey MP, Stegmeier F, Schlegel R, Hahn WC, Getz G, Mills GB, Boehm JS, Golub TR, Garraway LA, Sellers WR, Next-generation characterization of the Cancer Cell Line Encyclopedia, Nature. 569 (2019) 503–508. 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Wei S, Perera MLW, Sakhtemani R, Bhagwat AS, A novel class of chemicals that react with abasic sites in DNA and specifically kill B cell cancers, PLoS One. 12 (2017). 10.1371/journal.pone.0185010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Amidon KM and Eichman BF, The structural biology of native protein-DNA crosslinks to abasic sites, DNA Repair X (2020) X-X [DOI] [PMC free article] [PubMed] [Google Scholar]