

Abstract
Long non-coding RNAs (lncRNAs) have recently been found to be important in gene regulation. lncRNA H19 has been reported to play an oncogenic role in many human cancers. Its specific regulatory role is still elusive. In this study, we developed a novel analytic approach by integrating the synergistic regulation among lncRNAs (e.g., H19), transcription factors (TFs), target genes, and microRNAs (miRNAs) and then applied it to the pan-cancer expression datasets from The Cancer Genome Atlas (TCGA). Using linear regression models, we identified 88 H19-TF-gene co-regulatory triplets, in which 93% of the TF-gene pairs were related to cancer, indicating that our approach was effective to identify disease-related lncRNA-TF-gene co-regulation mechanisms. lncRNAs can function as miRNA sponges. Our further experiments found that H19 might regulate SP1-TGFBR2 through let-7b and miR-200b, ETS1-TGFBR2 through miR-29a and miR-200b, and STAT3-KLF11 through miR-17 in breast cancer cell lines. Our work suggests that miRNA-mediated lncRNA-TF-gene co-regulation is complicated yet important in cancer.
Keywords: H19, pan-cancer, miRNA, lncRNA-TF-gene, lncRNA, co-regulation, breast cancer, regulation triplet
Graphical Abstract
Li et al. developed a novel analytic approach by integrating the synergistic regulation among lncRNAs, miRNAs, TFs, and genes, and then applied it to pan-cancer expression data from TCGA. Using linear regression models, they identified 88 H19-TF-gene co-regulatory triplets. Their study shows that lncRNAs can interfere with miRNA-mediated TF-gene interactions.
Introduction
Long non-coding RNA (lncRNA) refers to a class of transcripts that are longer than 200 nt (bp) and are not translated to protein.1 lncRNA has been recently found to have many biological functions such as transcriptional regulation, epigenetic modification, and cell fate determination.2 It has been involved in many diseases, including cancer.3 For example, oncogenic lncRNAs may downregulate cancer cell antigen presentation and intrinsic tumor suppression,4 and they can serve as potential biomarkers for cancer diagnosis and therapeutic strategy development.5,6 Among thousands of lncRNA molecules discovered so far, H19 is one that is highly expressed.7, 8, 9 Accumulating data have suggested that lncRNA H19 plays a critical role in tumor initiation, progression, and recurrence in various human cancers.10 H19 has been reported to control cell cycle progression through regulating RB-E2F signaling in colorectal cancer.11 It plays an essential role in the exosome-mediated phenotype of endothelial liver cancer cells.12 H19 competitively binds a microRNA, miR-17-5p, to regulate YES1 gene expression in thyroid cancer.13 Moreover, H19-derived miR-675 contributes to bladder cancer cell proliferation through regulating p53 activity.14
lncRNA can act as a competing endogenous RNA (ceRNA) to interact with other protein-coding RNA transcripts, both transcription factor (TF) and non-TF genes.15,16 Hereafter, we refer to genes as protein-coding genes to separate them from non-coding genes. By sharing the common miRNA-binding sites with mRNAs or directing miRNA degradation, lncRNA competes with the miRNA target genes (TFs or non-TF genes) through interacting with miRNA; consequently, the expression of miRNA-targeted genes will be upregulated. This type of lncRNA-miRNA-gene competing co-regulation (triplets) has been discovered in humans and several other species.17 lncRNA may also interfere with the classic TF-gene regulation by acting as a ceRNA.
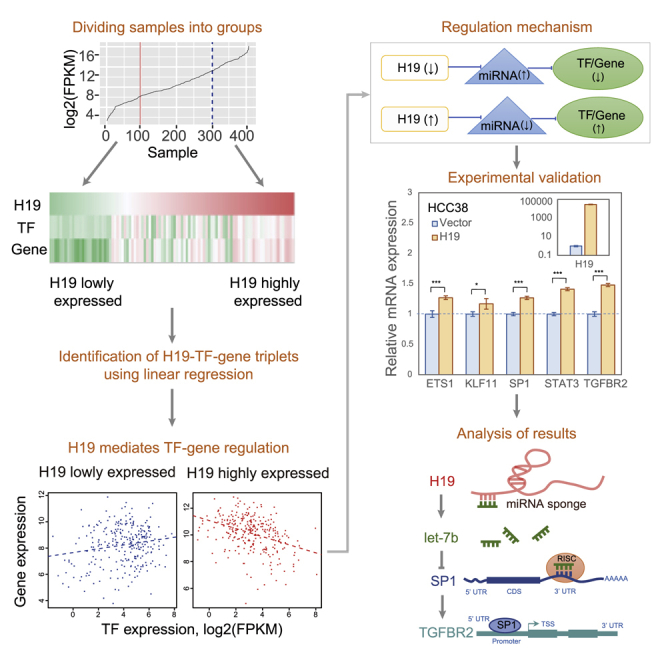
Although lncRNA is important in cancer, how it plays its regulatory roles in the complex and dynamic cellular systems remains largely unknown, especially at the pan-cancer level. In this work, we developed an analytical strategy to explore the synergistic regulation among lncRNAs, miRNAs, TFs, and genes. We applied this approach to The Cancer Genome Atlas (TCGA) pan-cancer datasets. We specifically examined H19, one of the highly expressed lncRNAs in cancer (Figure S1). Among the 24 cancer types we examined, we found that H19 was highly expressed in 21 cancer types, although not in the other three types (lower grade glioma [LGG], prostate adenocarcinoma [PRAD], and thyroid carcinoma [THCA]). As mentioned above, lncRNA might act as miRNA sponges to play roles in TF-gene regulation. Thus, we hypothesized that H19 could mediate TF-gene regulation through controlling miRNAs. After investigating the regulatory relationship among H19, miRNAs, TFs, and genes, we pinpointed three co-regulation triplets (H19-SP1-TGFBR2, H19-ETS1-TGFBR2, and H19-STAT3-KLF11) to validate our hypothesis in breast cancer cell lines. Our experimental results revealed that H19 might mediate (1) SP1-TGFBR2 (TF-gene) interaction through let-7b and miR-200b, (2) ETS1-TGFBR2 (TF-gene) interaction through miR-29a and miR-200b, and (3) STAT3-KLF11 (TF-gene) interaction through miR-17. Such regulatory triplets can be used to predict the potential function of lncRNAs and miRNAs in cancer. Our study showed that lncRNA could interfere with miRNA-mediated TF-gene interactions. This critical regulation, if universal in various cancer types, will be important for understanding the molecular mechanisms of cancer initiation and progression, and the molecules included in the interaction can serve as biomarkers for cancer diagnosis, drug development, and therapeutic strategy development.
Results
Classifying Samples by H19 Expression Level
lncRNA H19 is highly expressed in many cancer types and is actively involved in all stages of tumorigenesis.10 To better understand the role of H19 in cancer, we analyzed the expression level of H19 in TCGA pan-cancer datasets and grouped the samples into “low,” “middle,” and “high” by H19 expression level. For instance, in breast invasive carcinoma (BRCA) samples, we reordered all of the samples by H19 expression and named the top 25% as the high group and the bottom 25% as the low group of H19 expression (Figure 1A). Taking the same criterion, we evaluated the H19 expression level in the remaining 23 cancer types and found that 25% is an appropriate percentage to set as the threshold (Table S1; Figure 2). In most of these 24 cancer types, the H19 expression profile within the middle 50% of samples showed a steady line with a mild slope. This indicated that these samples had similar and stable levels of H19 expression. However, outside the middle section, the profile lines showed significant change at the inflection points. As shown in Figure 2, samples falling into the zone from zero to the end point of rapid increases (the red vertical solid line) are considered as the H19 low group, whereas samples with the H19 level in the top section, from the start of dramatic increases (the blue vertical dashed line) to the end, are designated as the H19 high group. Interestingly, in a previous study, Li et al.18 also used the 25th percentile as the threshold for dividing the top and bottom samples in their study. After having these cutoff thresholds, we further identified the H19 class labels of samples for BRCA TF expression data.
Figure 1.

Analytical Pipeline for Identification of lncRNA-TF-Gene Triplets and H19 Is Used as an Example
(A) Analysis of lncRNA, TF and gene expression, followed by ordering the samples according to H19 expression. Expression data for lncRNAs, TFs, and genes in 24 cancer types were extracted from TCGA. TF-gene interaction pairs were retrieved from two databases, TRANSFAC and TRRUST. On each plot, samples were ordered by H19 expression from low to high. Samples that were within 25% of the lowly (highly) expressed samples were considered as a lowly (highly) expressed group. (B) Linear regression analysis of H19 expression with the TF-gene pairs, leading to H19-TF-gene triplets. The effect of copy number variation (CNV) on gene expression was filtered out. (C) Test of the hypothesis that H19 may act as a miRNA sponge in lncRNA-TF-gene triplet regulation. In the low expression H19 case, loss of H19 weakens the inhibition of miRNAs, leading to the downregulation of miRNA target TFs and/or genes; in the high expression H19 case, enhanced H19 inhibits miRNA expression, leading to upregulation of miRNA target TFs and/or genes. ↑, Upregulated; ↓, downregulated.
Figure 2.

All Tested Cancer Types, with Samples Classified into Low, Moderate, and High Expression Groups by H19 Level
Twenty-four cancer types are represented. Classifications of samples are shown as low (left of the solid line), moderate (middle), and high (right of the dotted line).
Identification of H19-TF-Gene Triplets
We first focused on explaining the results from TCGA BRCA samples; the results of other cancer types are provided in Table S1. Based on the FPKM (fragments per kilobase of exon per million mapped reads) score, we filtered out 694 TFs and 11,867 non-TF genes from TCGA BRCA dataset. Using two databases, TRANSFAC (release 2016.4)19 and TRRUST (version 2.0),20 we obtained a total of 13,263 TF-target gene pairs (interactions), of which 8,181 TF-target gene pairs (interactions) were found to be expressed (FPKM score of at least 50% of samples was greater than 1) in these samples, corresponding to 625 unique TFs and 2,198 unique non-TF genes.
To evaluate the impact of H19 on TF-gene regulation, we used linear regression to obtain the expression profile of TFs and genes after excluding the effect of copy number variations (CNVs) on their expression (Figure 1B; Equation 1). Next, these new expression profiles were fed to the second linear regression model (Figure 1B; Equation 2) to assess the effect of H19 on TF or gene expression. In BRCA, 679 TF-gene regulation interactions were found to be affected by H19 (required both false discovery rate [FDR]EXPTF,ξ:GroupH19 <0.05 and pEXPTF,ξ <0.05) (Table 1). Of note, to reduce the false-positive rate (FDR), we required that these significant H19-TF-gene triplets should present in at least two TCGA cancer types. By following the analysis procedure above, we obtained a total of 88 triplets (Table S2). Figure 3 demonstrates the four most significant triplets (−log10(pEXPTF,ξ) > 9) whose TF-gene regulation was affected by the change of H19 expression. The remaining triplets are presented in Figure S2. In Figure 3, all TF-gene regulations affected by the change of H19 expression were statistically significant (FDR < 0.05). For instance, in the presence of high H19 expression, the correlation between MYBL2 and COL1A1 was significantly changed (positive correlation with p = 3.31 × 10−3 to negative correlation with p = 2.74 × 10−10).
Table 1.
TF-gene Regulation Interactions Significantly Affected by the Expression Alteration of H19
| Cancer Type | Number |
||||
|---|---|---|---|---|---|
| TFs | Genes | TF-Gene Pairs | TF-Gene Pairs (FDR < 0.05)a | Tripletsb | |
| BLCA | 607 | 2,103 | 7,723 | 1 | 0 |
| BRCA | 625 | 2,198 | 8,181 | 874 | 679 |
| CESC | 634 | 2,161 | 8,057 | 0 | 0 |
| COAD | 631 | 2,187 | 8,123 | 148 | 123 |
| ESCA | 655 | 2,205 | 8,277 | 3 | 1 |
| GBM | 636 | 2,156 | 7,755 | 2 | 0 |
| HNSC | 621 | 2,140 | 7,866 | 78 | 49 |
| KIRC | 628 | 2,172 | 7,889 | 246 | 160 |
| KIRP | 591 | 2,030 | 7,130 | 12 | 5 |
| LGG | 589 | 1,996 | 6,837 | 179 | 112 |
| LIHC | 546 | 1,964 | 6,988 | 2 | 1 |
| LUAD | 621 | 2,233 | 8,223 | 610 | 462 |
| LUSC | 653 | 2,281 | 8,444 | 0 | 0 |
| OV | 622 | 2,159 | 8,030 | 0 | 0 |
| PAAD | 651 | 2,369 | 8,758 | 1,165 | 945 |
| PCPG | 562 | 1,896 | 6,438 | 1 | 0 |
| PRAD | 624 | 2,170 | 7,889 | 1 | 0 |
| SARC | 602 | 2,053 | 7,353 | 24 | 13 |
| SKCM | 585 | 1,982 | 7,055 | 0 | 0 |
| STAD | 648 | 2,252 | 8,460 | 207 | 159 |
| TGCT | 620 | 2,202 | 8,042 | 574 | 466 |
| THCA | 587 | 2,020 | 7,284 | 1,095 | 916 |
| THYM | 602 | 2,055 | 7,270 | 0 | 0 |
| UCEC | 625 | 2152 | 8,092 | 0 | 0 |
Number of TF-gene pairs with regression FDR <0.05.
Number of triplets after CNV filtration.
Figure 3.

TF-Gene Regulation as Affected by H19 Expression Level
Linear regression was used to evaluate the association between TF expression and its target gene expression.
We found that 173 of 186 (93%) TF-gene pairs had direct or indirect evidence to support their relationship to cancer (Table S3). According to this high rate, we thought that the remaining 13 TF-gene pairs might play roles in cancer as well and warrant further studies. These 13 TF-gene pairs are as follows: LUAD, CTCF-IPO13; KIRC, SP3-EDF1; PAAD, USF1-FMR1; STAD, EZH2-DACT3; TGCT, FOXO1-HYOU1; THCA, CTCF-IPO13, NFKB1-CHUK, NFYB-EDF1, RELA-BGN, RUNX1-SYMPK, SP1-ME1, SP1-SIGIRR, and SP3-EDF1. Interestingly, most of them (8 out of 13) were found in THCA, suggesting that this cancer type might have additional regulatory roles. These results indicate that our approach is effective in identifying TFs or genes that are related to cancer.
H19 Regulates TF-Gene Function through the Related miRNAs
Given that one of the important lncRNA functions is modulating miRNAs, we speculated that H19 might mediate TF-gene interactions through the regulation of miRNAs. Through a comprehensive literature search (Table S4), we identified 29 miRNAs (let-7a, let-7b, let-7g, let-7i, miR-106a, miR-130b-3p, miR-138-5p, miR-139, miR-141, miR-152-3p, miR-152-5p, miR-17-5p, miR-181d-3p, miR-181d-5p, miR-18a, miR-194-5p, miR-196a, miR-19a, miR-19b-1, miR-200b, miR-200c, miR-20a, miR-22, miR-29a, miR-29b, miR-342-3p, miR-630, miR-874, and miR-92a-1) that were targeted by H19 (Table S4). Next, miRNA target genes of these 29 miRNAs were collected from the miRNA target prediction databases using the SpidermiR R tool.21 Among these target genes, 85 overlaid with the H19-TF-gene triplets that we identified in the earlier regression analysis. We selected eight triplets (H19-ETS1-TGFBR2, H19-FLI1-TGFBR2, H19-FOXO1-TXNIP, H19-KLF6-TXNIP, H19-NFYB-SP3, H19-PPARA-KLF11, H19-SP1-TGFBR2, and H19-STAT3-KLF11) and investigated the impact of these 29 miRNAs on them (Table S5). After having confirmed the target TF and non-TF genes of the H19-mediated miRNAs, we mainly focused on two cases (cases 1 and 2) in BRCA (Figure 1C).
Case 1
First, we chose 56 matched (common) H19 low expression group samples along with the corresponding 56 matched normal samples from TCGA BRCA miRNA/TF/non-TF gene expression database. Next, we applied the limma-voom R statistical tool22 and obtained the lists of upregulated (UPR) and downregulated (DWR) miRNAs/TFs/non-TF genes from this pool. As the result (i.e., for the H19 low expression group samples), we identified five cases as UPRmiRH19low (let-7b, p = 1.51 × 10−6; miR-29a, p = 6.77 × 10−6; miR-200b, p = 2.67 × 10−3; miR-17, p = 1.42 × 10−2; and miR-29b, p = 4.49 × 10−2), one case as DWRmiRH19low (miR-130b, p = 4.60 × 10−4), five cases as DWRTFH19low (ETS1, p = 8.40 × 10−4; PPARA, p = 8.16 × 10−4; STAT3, p = 3.29 × 10−8; NFYB, p = 4.58 × 10−8; and SP1, p = 4.63 × 10−3), and two cases as DWRGeneH19low (SP3, p = 2.84 × 10−14; and TGFBR2, p = 2.99 × 10−3). Neither TFs nor genes were found as UPRTFH19low or UPRGeneH19low, respectively (Table 2). The specific definitions of the terms such as UPRmiRH19low and DWRmiRH19low are provided in Materials and Methods.
Table 2.
Genes, TFs, and miRNAs Were Upregulated or Downregulated.
| Group | Molecule | Log2FC | p | Regulation | |
|---|---|---|---|---|---|
| H19 lowly expressed | miRNA | let-7b | 0.92885 | 1.51 × 10−6 | upregulated |
| miR-130b | −3.6149 | 4.60 × 10−4 | downregulated | ||
| miR-17 | 0.90485 | 1.42 × 10−2 | upregulated | ||
| miR-200b | 2.54065 | 2.67 × 10−3 | upregulated | ||
| miR-29a | 1.903 | 6.78 × 10−6 | upregulated | ||
| miR-29b-2 | 2.4692 | 4.49 × 10−2 | upregulated | ||
| TF | ETS1 | −1.66925 | 8.41 × 10−4 | downregulated | |
| NFYB | −2.8769 | 4.58 × 10−8 | downregulated | ||
| PPARA | −2.2694 | 8.16 × 10−4 | downregulated | ||
| SP1 | −0.89175 | 4.64 × 10−3 | downregulated | ||
| SP3 | −1.1854 | 2.85 × 10−14 | downregulated | ||
| STAT3 | −1.9026 | 3.29 × 10−8 | downregulated | ||
| gene | TGFBR2 | −2.41715 | 3.00 × 10−3 | downregulated | |
| H19 highly expressed | miRNA | miR-141 | 1.12925 | 5.86 × 10−5 | upregulated |
| miR-200b | −2.5865 | 4.46 × 10−3 | downregulated | ||
| miR-29b-2 | 1.13575 | 1.42 × 10−2 | upregulated | ||
| TF | NFYB | −1.8413 | 1.25 × 10−2 | downregulated | |
| SP1 | −0.9865 | 2.07 × 10−5 | downregulated | ||
| SP3 | 0.0436 | 6.47 × 10−8 | upregulated | ||
| STAT3 | −1.3972 | 2.86 × 10−3 | downregulated | ||
| gene | KFLI1 | −0.992 | 5.65 × 10−4 | downregulated | |
FC, fold change.
Case 2
Similarly, we selected 40 matched cancer and normal samples in the H19 high expression group from TCGA BRCA miRNA/TF/non-TF gene expression database. The limma-voom tool was utilized to identify the upregulated and downregulated miRNAs/TFs/non-TF genes in this set. For this H19 high expression group samples, we identified two cases as UPRmiRH19high (miR-141, p = 5.86 × 10−5; and miR-29b, p = 1.42 × 10−2), one case as DWRmiRH19high (miR-200b, p = 4.45 × 10−3), and four cases as DWRTFH19high (SP1, p = 2.06 × 10−5; FL11, p = 5.65 × 10−4; STAT3, p = 2.86 × 10−3; and NFYB, p = 1.24 × 10−2). Neither TFs nor genes were found as UPRTFH19high, UPRGeneH19high, or DWRGeneH19high (Table 2).
Statistically Evaluated Interesting Gene Regulation Cascades of Triplets
We proposed two scenarios to explain the regulation between every pair of two participating biomolecules within a triplet under different conditions, as follows: (1) H19 (↓)-UPRmiRH19low (↑)-DWRTFH19low/DWRGeneH19low (↓): the downregulated expression of H19 causes the upregulation of its target miRNAs, which results in the downregulated expression of their target TFs or genes. (2) H19 (↑)-DWRmiRH19low (↓)-UPRTFH19low/UPRGeneH19low (↑): the upregulated expression of H19 causes the downregulation of its target miRNAs, which in turn causes upregulated expression of their target TFs or genes. Accordingly, we found five triplets that met the regulation scenarios above: (1) H19 (↓)-let-7b (↑)-SP1 (↓)-TGFBR2 (↓); (2) H19 (↓)-miR-29a (↑)-ETS1 (↓)-TGFBR2 (↓); (3) H19 (↓)-miR-200b (↑)-ETS1 (↓)-TGFBR2 (↓); (4) H19 (↓)-miR-200b (↑)-SP1 (↓)-TGFBR2 (↓); and (5) H19 (↓)-miR-17 (↑)-STAT3 (↓)-KLF11 (↓). If we could prove that the status changes met our prediction, it would be helpful to improve the network validation and understanding the overall effect on expression patterns of the miRNAs/TFs/genes due to different expression levels of H19.
Experimental Validation
In order to examine whether H19 serves as a mRNA sponge to regulate the expression of TFs and their downstream targets through antagonizing let-7b, miR-17, miR-200b, and miR-29a,13,23, 24, 25, 26, 27, 28 breast cancer cell lines BT-549, HCC38, MCF7, and MDA-MB-231 were used with ectopically expressing inducible H19.29 Compared to the control cells, the overexpression of H19 could upregulate the distinct levels of mRNA expression of let-7b/miR-200b-regulated SP1, miR-29a/miR-200b-regulated ETS1, and miR-17-regualted STAT3 in the breast cancer cell lines (Figures 4A–4D). Consequently, the expression of the SP1/ETS1 transcriptional target TGFBR2 and STAT3 target KLF11 was also increased to different extents upon H19 induction. However, there was no obvious induction of TGFBR2 detected in BT549 or MD-MB231 cells, which could be due to the low expression of SP1 and ETS in such cells or dysregulation of other transcriptional co-regulators (Figures 4A and 4C). Taken together, these lines of experimental evidence support our notion of miRNA-mediated H19-TF-gene triplet regulation, indicating that H19 upregulates transcription factors and their downstream effects by sequestering miRNAs in cancer.
Figure 4.

H19 Functions as a miRNA Sponge to Relieve miRNA-Mediated Suppression of Transcription Factors and Their Targets
Transcription factor targets included H19-SP1-TGFBR2, H19-ETS1-TGFBR2, and H19-STAT3-KLF11. (A–D) H19 led to an upregulated expression of let-7b/miR-17/miR-29a/miR-200b-controlled SP1, ETS1 and STAT3 (three TF genes), and their transcriptional targets TGFBR2 and KLF11 in four breast cancer cell lines (A, BT-549; B, HCC38; C, MCF7; and D, MDA-MB-231). All samples were analyzed in triplicate and normalized to GAPDH expression. The top-right panel shows relative H19 expression. Quantitative real-time PCR data are presented as mean ± SE (standard error). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. (E) The H19-let7b-SP1-TGFBR2 interaction and its involvement with breast cancer. H19 acts as a miRNA sponge for let-7b that targets TF gene SP1. Let-7b inhibits the expression of SP1, whose overexpression inhibits breast cancer cell migratory and invasive abilities. SP1 regulates TGFBR2, whose abnormal expression or mutation is related to breast cancer. CDS, coding DNA sequence; RISC, RNA-induced silencing complex; TSS, transcription start site; UTR, untranslated region.
Discussion
Identification of cancer-associated lncRNAs and uncovering their molecular mechanisms are currently challenging but important tasks. Traditionally, studies of gene expression deregulation and alterations in genomic sequences in tumor cells have led to the identification of cancer-associated lncRNAs.5,30 Subsequent in vitro and in vivo studies have directly associated some of the identified lncRNAs with specific cancer phenotypes. In this study, we identified 88 H19-TF-gene triplets based on TCGA pan-cancer data using linear regression models. Most of these TF-gene pairs (93%) had direct or indirect evidence to support their relationship to cancer (Table S3). The remaining TF-gene pairs could serve as potential candidates and warrant further investigation. Our results demonstrate that this analytical, co-regulation-based approach is promising to identify TFs or genes related to cancer. To investigate the potential regulatory mechanism, we hypothesized that H19 acts as a miRNA sponge to diminish certain miRNAs, which in turn preserves the corresponding TF-gene function. Our quantitative real-time PCR experimental results suggest that H19 mediates SP1-TGFBR2 (TF-gene) regulation through let-7b and miR-200b (miRNA), ETS1-TGFBR2 regulation through miR-29a and miR-200b, and STAT3-KLF11 regulation through miR-17 in BT-549, HCC38, MCF7, and MDA-MB-231 breast cancer cell lines. Our approach can help identify lncRNAs, miRNAs, TFs, and genes that are potentially cancer associated and uncover their complex regulatory mechanisms. This approach effectively extends the previous miRNA-TF-gene co-regulation approach that has been well studied in various cancer types or other disease.31, 32, 33
TRANSFAC and TRRUST are representative databases for annotations of TF-target gene pairs. The annotations are based on stringent criteria, including both experimental evidence and statistical tests. After filtration, we obtained 13,263 TF-target gene pairs from these two databases. Although the number of TF-gene pairs is smaller than that from other databases, such as ENCODE,34 we decided to use them for reducing false-positive results. For the large-scale data analysis, false-positive data would have more potential problems than false-negative data, while our goal is to find those enriched signals (e.g., regulatory networks) that have reliable evidence.
The samples studied were classified into three groups by H19 expression, that is, low, middle, and high. In such a way, we could observe the TF-gene status change along with the changes of H19 expression. For example, we used linear regression to describe the association between ETS1 (TF) expression and TGFBR2 expression. The p values of linear regression were 1.69 × 10−31 in the H19 low expression group and 2.09 × 10−52 in the H19 high expression group. Additionally, this classification method significantly improved ETS1-TGFBR2 regulation (p < 10−9). This suggests that such an integrative grouping approach is effective in investigating the relationship between variation of lncRNA expression and TF-gene regulation.
Figure 2 shows a smooth slope of the curves for the patients with intermediate levels of H19 expression. This is because all of the samples were ordered by their H19 expression, and most of these samples had H19 expression without large fluctuation. For the H19 highly or lowly expressed groups, the curves displayed sharp slopes. These sharp slopes suggested that H19 was expressed with strong variation in these two groups, which is biologically useful to investigate the effect of H19 expression changes on TF-gene regulation.
Linear regression is a widely used approach to predict the output values of a biological process under specific conditions.35,36 Some studies use linear regression to predict the expression of genes.37 However, to our knowledge, there are few studies that use linear regression to investigate the effect of lncRNAs on TF-gene regulation. In this study, we used a linear regression model that was fit on non-TF gene expression changes in tumor samples as the response variable using a linear combination of the input variables, including TF expression, sample groups distinguished by H19 expression level, and interaction between TF and group (Equation 2). Within a H19-TF-gene triplet, this requires the TF to regulate the target gene, and H19 to affect TF expression and consequently regulate the target gene. Therefore, we required that such triplets should satisfy both FDREXPTF,ξ:GroupH19 <0.05 and pEXPTF,ξ <0.05.
CNVs contribute largely to gene expression.38 Structural variants, including CNVs, in cancer genomes can lead to significantly reduced or increased gene expression in cancer cells.39 Therefore, we removed the effect of CNVs on gene expression and reconsidered the mediation of H19 on the TF-gene regulation relationship.
The H19-let7b-SP1-TGFBR2 interaction we identified by our bioinformatics approach was supported by actual biological experiments (Figure 4E). The lncRNA H19 can act as a sponge to bind let-7b to mediate breast cancer cell plasticity.28 The let7b-SP1 interaction was verified from chimeric reads.40 DNA precipitation, electrophoretic mobility shift assays, and promoter analysis confirmed that the TGFBR2 promoter was bound by SP1.41
We observed the expression variation of SP1 and TFFBR2 in the presence of a high level of H19 in breast cancer cell lines. The H19-let7b-SP1-TGFBR2 interaction can be used to elucidate such a correlation. The abnormal expression of H19 and let-7b might lead to abnormal expression of SP1 and TGFBR2 and, consequently, lead to the development of breast cancer. The molecules included in the interaction can provide candidate diagnosis biomarkers and targets for therapy.
The synergistic regulatory relationship between lncRNAs, miRNAs, and protein-coding RNAs implemented in our approach can better infer the potential function of non-coding RNAs. The H19-let7b-SP1-TGFBR2 interaction can be used to interpret the potential functions of lncRNAs and miRNAs (Figure 4E). In mice, an overexpression of SP1 has been reported to suppress migratory and invasive abilities of breast cancer cells.42 TGFBR2 is upregulated in basal-like breast cancer cell lines.43 The minor allele homozygote (GG) of rs1078985, an intronic single nucleotide polymorphism (SNP) in TGFBR2, had a 24% lower risk of having breast cancer compared with major allele carriers (AG or AA).44 H19 and let-7b interact with SP1 and TGFBR2. Therefore, H19 and let-7b might be involved in breast cancer in terms of cell migratory and invasive abilities, and they could serve as potential biomarkers. This regulation can also be verified by the following evidence in literature: (1) H19 enhances breast cancer cell proliferation through positive control by E2F1;45 (2) overexpression of an ectopic H19 gene promotes the tumorigenic properties of breast cancer cells;46 and (3) let-7b expression in breast cancer patients was inversely associated with tumor lymph node metastasis and patient overall survival.47
The study has several limitations, and there is scope for future work to be carried out. For instance, the current work only focused on one lncRNA, H19. In fact, our approach can be applied to any other lncRNAs. We focused on H19 in this study because it is one of the most promising lncRNAs and is highly expressed in most of the cancer types we examined. Moreover, this study cannot exclude false-positive results: some H19-TF-gene triplets are possibly not the real or impactful regulatory correlations, and some TFs or genes may not directly cause or be related to the specific cancer under investigation. To address this problem, we can use a lower FDR threshold to minimize a false-positive rate or validate them using various biological experiments. Finally, cancer is highly heterogeneous, and its development is dynamic in the cellular system. Our approach, like many others, cannot consider real-time or dynamic regulation in cancer and matched normal cells. However, our approach of uncovering lncRNA molecular functions contributes to identify and functionally annotate these cancer related genes, making these genes the attractive targets. These regulatory units can also better explain cancer biology. Finally, in this study, we first examined lncRNA (H19) expression in pan-cancer and then explored how it potentially regulated genes, including TF genes. Alternatively, we may analyze H19-miRNA pairs first and examine which miRNAs might be altered by H19 in the datasets. We will explore this analytical approach in the future.
Materials and Methods
Data Collection
TCGA pan-cancer data consisting of 24 cancer types such as BLCA, BRCA, CESC, COAD, ESCA, GBM, HNSC, KIRC, KIRP, LGG, LIHC, LUAD, LUSC, OV, PAAD, PCPG, PRAD, SARC, SKCM, STAD, TGCT, THCA, THYM, and UCEC (full names are summarized in Table S1), whose numbers of samples were at least 90, were used for this study (Figure 1A). Tissue-specific data contained RSEM48 gene FPKM data that included the lncRNA expression profile, TF expression profile, and gene (non-TF) expression profile. The data were collected through the UCSC (University of California, Santa Cruz) Xena database (https://xenabrowser.net/). The BRCA RSEM gene FPKM expression data contained a total of 60,499 genes and a total of 1,212 samples. For the remaining tissue-specific data, such information was provided in the UCSC Xena database (Table S1). An expression dataset of miRNAs was also collected from the UCSC Xena database. It consisted of 744 miRNAs and 10,818 samples. The curated clinical (phenotype) data that provided the list of the primary tumor samples were collected from the UCSC Xena database. In addition, the interactions between TFs and genes were collected from TRANSFAC (release 2016.4)19 and TRRUST (version 2.0).20 Furthermore, the interactions between miRNAs and (validated or predicted) target genes were obtained from the SpidermiR R tool21 by using six target prediction databases. Among these databases, the miRTar49 and miRWalk50 databases provided only validated target genes, whereas the DIANA,51 miRanda,52 PicTar,53 and TargetScan54 databases supplied predicted target genes. H19-targeted miRNAs were identified from the published literature. The detailed descriptions regarding the association of H19 and its targeted miRNAs are demonstrated in Table S4.
Data Preprocessing
First, the biomolecules (lncRNAs, TFs, and genes) whose FPKM score of at least 50% of samples was greater than 1 were selected, whereas the remaining biomolecules were excluded from further analyses (Figure 1A). After this step, we partitioned the whole gene expression data into several subparts according to the category of the genes such as filtered TF expression data, filtered non-TF gene expression data, and filtered lncRNA (H19) expression data. The interactions between TFs and target genes were determined from two well-known databases, TRANSFAC (release 2016.4)19 and TRRUST (version 2.0).20 We first obtained 800 TFs and 3,470 genes from the TRANSFAC and TRRUST databases and then applied these TFs and genes to filter results from these databases and obtained 13,263 TF-target gene pairs. Using the resultant interactions, we further filtered the TF expression data in a way such that the participating (interacting) TFs belonging to the TF-gene interactions (obtained by TRANSFAC and TRRUST), which were listed in the TF expression data, were only considered in the resultant filtered TF expression data. Similarly, we again filtered the non-TF gene expression data in a way such that the target genes belonging to the TF-gene interactions that were mentioned in the non-TF gene expression data were only selected in the resultant filtered non-TF gene expression data. Next, to determine the type of expression of existing samples, we ordered the expression data of H19 underlying all of these samples based on the expression values from low to high. Here, a certain percentage of the lowly expressed samples was considered as the first group (lowly expressed group), whereas the same percentage of the highly expressed samples was treated as the second group (highly expressed group). The remaining samples were used as the third group (“middle” group). According to the resultant class labels of H19 samples, the samples of the other molecules such as gene and TF had been classified.
Linear Regression and Copy Number Variation Factor
CNVs contribute largely to gene expression.38 We removed the effect of CNVs on gene expression and then reconsidered the mediation of H19 on the TF-gene regulation relationship (Figure 1B). First, we obtained the residuals of expression of the TF and genes through the linear regression (“Stats” R tool)55 using Equation 1, and then used the residuals of expression of the TF and genes to evaluate the mediation of H19 on the TF-gene regulation relationship through Equation 2 as follows:
| (1) |
where EXPTF/Gene symbolizes the expression data of TF or gene, and CNVTF/Gene denotes the CNV data of the TF or gene; and
| (2) |
where EXPGene,ξ refers to the expression data of the gene after removing the effect of CNVs, EXPTF,ξ denotes the expression data of TF after removing the effect of CNVs, and GroupH19 symbolizes the class labels of samples (low, middle, and high), whereas EXPTF,ξ:GroupH19 refers to the interaction effect between the TF and the group. Finally, we determined the triplets using the criteria (1) the adjusted p value should be less than 0.05 (FDREXPTF,ξ:GroupH19 < 0.05), and (2) the p value corresponding to coefficient of EXPTF,ξ should be less than 0.05 (pEXPTF,ξ < 0.05).
Regulatory Mechanism
After obtaining the triplets (H19-TF-gene) through linear regression, we conducted an extensive literature search to determine the interactions between H19 and miRNAs. We identified target miRNAs that interacted with H19. Next, the target genes (including TFs as well as non-TF genes) of the employing miRNAs had been identified using the SpidermiR R tool.21 After finding the target genes of the H19-mediated miRNAs, we had mainly focused on two cases (Figure 1C), that is, cases 1 and 2.
Case 1
First we chose the matched (common) H19 lowly expressed group samples along with the matched normal samples from TCGA BRCA miRNA/TF/non-TF gene expression data. Then, we applied the limma-voom R statistical tool22 to determine which miRNAs/TFs/non-TF genes were upregulated (denoted as UPRmiRH19low, UPRTFH19low, and UPRGeneH19low, respectively), downregulated (denoted as DWRmiRH19low, DWRTFH19low, and DWRGeneH19low, respectively), or not differentially expressed.
Case 2
Similarly, we selected the matched (common) H19 highly expressed group samples along with the matched normal samples from TCGA BRCA miRNA or gene expression data. Then, we used the limma-voom R statistical tool22 to determine which miRNAs or genes were upregulated (UPRMiRH19high, UPRTFH19high, and UPRGeneH19lhigh, respectively), downregulated (DWRMiRH19high, DWRTFH19high, and DWRGeneH19high, respectively), or not differentially expressed.
Cell Lines
All cell lines were obtained from the American Type Culture Collection (Manassas, VA, USA), independently validated by STR DNA fingerprinting at the University of Texas MD Anderson Cancer Center (Houston, TX, USA) and determined to be negative for mycoplasma contamination. HEK293T (RRID:CVCL_0063) cells and human breast cancer cell lines BT-549 (RRID:CVCL_1092), HCC38 (RRID:CVCL_1267), MCF7 (RRID:CVCL_0031), and MDA-MB-231 (RRID:CVCL_0062) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS) supplemented with 100 IU/mL penicillin and 100 μg/mL streptomycin. Cells were maintained at 37°C in a humidified 5% CO2 incubator.
Generation of H19 Lentivirus and Infection
2 × 106 HEK293T cells were seeded in 10-cm tissue culture plates and maintained in DMEM complete medium for 20 h. Media were discarded and replaced with 3 mL of Opti-MEM. 6 μg of pCMV-dR8.2-dvpr, 2 μg of pCMV-VSV-dvpr, 8 μg of TetO-FUW H1929 or rtTA, and 32 μg of polyethyleneimine (PEI) were added into 400 μL of Opti-MEM. The transfection mixture was incubated at room temperature for 15 min and then added into HEK293T cells. After a 3-h incubation, 3 mL of DMEM complete medium was added. After overnight incubation, the medium was replaced with 6 mL of DMEM complete medium. The viruses were collected at 48–72 h after transfection and concentrated by Amicon Ultra-15 (Millipore). For viral infection, BT-549, HCC38, MCF7, and MDA-MB-231 cells were seeded, respectively, in six-well plates and infected with either control or H19 lentivirus. One μg/mL doxycycline was used to turn on the expression of H19. After 48–72 h of induction, cells were collected to examine H19, ETS1, KLF11, SP1, STAT3, and TGFBR2 gene expression for quantitative real-time PCR.
Quantitative Real-Time PCR
Total RNA was extracted using TRIzol reagent (Thermo Fisher Scientific, CA, USA) following the manufacturer’s instructions. The RNA samples were qualified using a NanoDrop spectrophotometer (Thermo Fisher Scientific, CA, USA). 1 μg of mRNA was used for reverse transcription using the iScript cDNA synthesis kit (Bio-Rad, CA, USA). A 20-μL quantitative real-time PCR reaction solution was composed of 1 μL of cDNA, 1 μL each of 10 μM forward and reverse qPCR primers, 10 μL of SYBR Green PCR master mix (Bio-Rad, CA, USA), and 7 μL of RT-PCR-grade water. qPCR reactions were performed on a CFX96 machine (Bio-Rad). All reactions were run in triplicate. The relative ETS1, KLF11, SP1, STAT3, and TGFBR2 mRNA expression levels were normalized to their corresponding GAPDH mRNA expression. Primers used for quantitative real-time PCR detection are listed in Table S6.
Statistical Analysis
The limma-voom R statistical tool22 using an empirical Bayes statistical test was applied to identify the differentially expressed miRNAs, TFs, and non-TFs in the (common) H19 lowly or highly expressed group samples versus the matched normal samples using TCGA breast cancer gene miRNA and mRNA expression datasets. For quantitative real-time PCR, all grouped data are presented as mean ± SE (standard error). A Student’s t test was used to assess statistical significance between the two groups.
Author Contributions
Z.Z. and P.J. conceived the study. A.L. and S.M. collected the data and conducted the bioinformatics analysis. H.L. and D.-F.L. conducted laboratory experiments. A.L., S.M., H.L., D.-F.L., and Z.Z. verified the results and wrote the manuscript. All authors revised and approved the final manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We thank the laboratory members of Bioinformatics and Systems Medicine Laboratory (SBML) for useful discussion, and Dr. Guangchun Han for initial analysis of lncRNA in TCGA datasets. A.L. was partially supported by the Natural Science Basic Research Plan in Shaanxi Province (2017JM6024), the Natural Science Foundation of Shaanxi Provincial Department of Education (17JK0572), and by the Teaching Research Foundation of Xi’an University of Technology (XJY1866). D.-F.L. is a CPRIT Scholar in Cancer Research and was supported by Cancer Prevention & Research Institute of Texas (CPRIT) grant RR160019. We thank the technical support from CPRIT cores (RP180734 and RP170668). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.05.028.
Contributor Information
Dung-Fang Lee, Email: dung-fang.lee@uth.tmc.edu.
Zhongming Zhao, Email: zhongming.zhao@uth.tmc.edu.
Supplemental Information
References
- 1.Iyer M.K., Niknafs Y.S., Malik R., Singhal U., Sahu A., Hosono Y., Barrette T.R., Prensner J.R., Evans J.R., Zhao S. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015;47:199–208. doi: 10.1038/ng.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li M., Izpisua Belmonte J.C. Roles for noncoding RNAs in cell-fate determination and regeneration. Nat. Struct. Mol. Biol. 2015;22:2–4. doi: 10.1038/nsmb.2946. [DOI] [PubMed] [Google Scholar]
- 3.Mitra R., Chen X., Greenawalt E.J., Maulik U., Jiang W., Zhao Z., Eischen C.M. Decoding critical long non-coding RNA in ovarian cancer epithelial-to-mesenchymal transition. Nat. Commun. 2017;8:1604. doi: 10.1038/s41467-017-01781-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hu Q., Ye Y., Chan L.-C., Li Y., Liang K., Lin A., Egranov S.D., Zhang Y., Xia W., Gong J. Oncogenic lncRNA downregulates cancer cell antigen presentation and intrinsic tumor suppression. Nat. Immunol. 2019;20:835–851. doi: 10.1038/s41590-019-0400-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu S., Mitra R., Zhao M.-M., Fan W., Eischen C.M., Yin F., Zhao Z. The potential roles of long noncoding RNAs (lncRNA) in glioblastoma development. Mol. Cancer Ther. 2016;15:2977–2986. doi: 10.1158/1535-7163.MCT-16-0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cui W., Qian Y., Zhou X., Lin Y., Jiang J., Chen J., Zhao Z., Shen B. Discovery and characterization of long intergenic non-coding RNAs (lincRNA) module biomarkers in prostate cancer: an integrative analysis of RNA-seq data. BMC Genomics. 2015;16(Suppl 7):S3. doi: 10.1186/1471-2164-16-S7-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matouk I.J., Raveh E., Abu-lail R., Mezan S., Gilon M., Gershtain E., Birman T., Gallula J., Schneider T., Barkali M. Oncofetal H19 RNA promotes tumor metastasis. Biochim. Biophys. Acta. 2014;1843:1414–1426. doi: 10.1016/j.bbamcr.2014.03.023. [DOI] [PubMed] [Google Scholar]
- 8.Matouk I.O.P., Ayesh S., Sidi A., Czerniak A., Groot N.D., Hochberg A. The oncofetal H19 RNA in human cancer, from the bench to the patient. Cancer Ther. 2005;3:249–266. [Google Scholar]
- 9.Matouk I.J., DeGroot N., Mezan S., Ayesh S., Abu-lail R., Hochberg A., Galun E. The H19 non-coding RNA is essential for human tumor growth. PLoS ONE. 2007;2:e845. doi: 10.1371/journal.pone.0000845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raveh E., Matouk I.J., Gilon M., Hochberg A. The H19 long non-coding RNA in cancer initiation, progression and metastasis—a proposed unifying theory. Mol. Cancer. 2015;14:184. doi: 10.1186/s12943-015-0458-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ling H., Ohtsuka M., Ivan C., Pichler M., Chen M., Slaby O., Goel A., Radovich M., Calin G. Oncogenic function and molecular mechanism of H19 noncoding RNA in colorectal cancer. Cancer Res. 2017;77(Suppl):2548. doi: 10.1158/1538-7445.AM2017-2548. [DOI] [Google Scholar]
- 12.Conigliaro A., Costa V., Lo Dico A., Saieva L., Buccheri S., Dieli F., Manno M., Raccosta S., Mancone C., Tripodi M. CD90+ liver cancer cells modulate endothelial cell phenotype through the release of exosomes containing H19 lncRNA. Mol. Cancer. 2015;14:155. doi: 10.1186/s12943-015-0426-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu L., Yang J., Zhu X., Li D., Lv Z., Zhang X. Long noncoding RNA H19 competitively binds miR-17-5p to regulate YES1 expression in thyroid cancer. FEBS J. 2016;283:2326–2339. doi: 10.1111/febs.13741. [DOI] [PubMed] [Google Scholar]
- 14.Liu C., Chen Z., Fang J., Xu A., Zhang W., Wang Z. H19-derived miR-675 contributes to bladder cancer cell proliferation by regulating p53 activation. Tumour Biol. 2016;37:263–270. doi: 10.1007/s13277-015-3779-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang P., Ning S., Zhang Y., Li R., Ye J., Zhao Z., Zhi H., Wang T., Guo Z., Li X. Identification of lncRNA-associated competing triplets reveals global patterns and prognostic markers for cancer. Nucleic Acids Res. 2015;43:3478–3489. doi: 10.1093/nar/gkv233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tay Y., Rinn J., Pandolfi P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505:344–352. doi: 10.1038/nature12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salmena L., Poliseno L., Tay Y., Kats L., Pandolfi P.P. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353–358. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y., Li L., Wang Z., Pan T., Sahni N., Jin X., Wang G., Li J., Zheng X., Zhang Y. LncMAP: pan-cancer atlas of long noncoding RNA-mediated transcriptional network perturbations. Nucleic Acids Res. 2018;46:1113–1123. doi: 10.1093/nar/gkx1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matys V., Kel-Margoulis O.V., Fricke E., Liebich I., Land S., Barre-Dirrie A., Reuter I., Chekmenev D., Krull M., Hornischer K. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34:D108–D110. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han H., Cho J.-W., Lee S., Yun A., Kim H., Bae D., Yang S., Kim C.Y., Lee M., Kim E. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018;46(D1):D380–D386. doi: 10.1093/nar/gkx1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cava C., Colaprico A., Bertoli G., Graudenzi A., Silva T.C., Olsen C., Noushmehr H., Bontempi G., Mauri G., Castiglioni I. SpidermiR: an R/bioconductor package for integrative analysis with miRNA data. Int. J. Mol. Sci. 2017;18:E274. doi: 10.3390/ijms18020274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Law C.W., Chen Y., Shi W., Smyth G.K. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kallen A.N., Zhou X.-B., Xu J., Qiao C., Ma J., Yan L., Lu L., Liu C., Yi J.S., Zhang H. The imprinted H19 lncRNA antagonizes let-7 microRNAs. Mol. Cell. 2013;52:101–112. doi: 10.1016/j.molcel.2013.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang Z., Lei W., Hu H.B., Zhang H., Zhu Y. H19 promotes non-small-cell lung cancer (NSCLC) development through STAT3 signaling via sponging miR-17. J. Cell. Physiol. 2018;233:6768–6776. doi: 10.1002/jcp.26530. [DOI] [PubMed] [Google Scholar]
- 25.Jia P., Cai H., Liu X., Chen J., Ma J., Wang P., Liu Y., Zheng J., Xue Y. Long non-coding RNA H19 regulates glioma angiogenesis and the biological behavior of glioma-associated endothelial cells by inhibiting microRNA-29a. Cancer Lett. 2016;381:359–369. doi: 10.1016/j.canlet.2016.08.009. [DOI] [PubMed] [Google Scholar]
- 26.He H., Wang N., Yi X., Tang C., Wang D. Long non-coding RNA H19 regulates E2F1 expression by competitively sponging endogenous miR-29a-3p in clear cell renal cell carcinoma. Cell Biosci. 2017;7:65. doi: 10.1186/s13578-017-0193-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou J., Zhou Y., Wang C.X. nlcRNA-MIAT regulates fibrosis in hypertrophic cardiomyopathy (HCM) by mediating the expression of miR-29a-3p. J. Cell. Biochem. 2018;120:7265–7275. doi: 10.1002/jcb.28001. [DOI] [PubMed] [Google Scholar]
- 28.Zhou W., Ye X.-l., Xu J., Cao M.-G., Fang Z.-Y., Li L.-Y., Guan G.-H., Liu Q., Qian Y.-H., Xie D. The lncRNA H19 mediates breast cancer cell plasticity during EMT and MET plasticity by differentially sponging miR-200b/c and let-7b. Sci. Signal. 2017;10 doi: 10.1126/scisignal.aak9557. eaak9557. [DOI] [PubMed] [Google Scholar]
- 29.Lee D.-F., Su J., Kim H.S., Chang B., Papatsenko D., Zhao R., Yuan Y., Gingold J., Xia W., Darr H. Modeling familial cancer with induced pluripotent stem cells. Cell. 2015;161:240–254. doi: 10.1016/j.cell.2015.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huarte M. The emerging role of lncRNAs in cancer. Nat. Med. 2015;21:1253–1261. doi: 10.1038/nm.3981. [DOI] [PubMed] [Google Scholar]
- 31.Sun J., Gong X., Purow B., Zhao Z. Uncovering microRNA and transcription factor mediated regulatory networks in glioblastoma. PLoS Comput. Biol. 2012;8:e1002488. doi: 10.1371/journal.pcbi.1002488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li A., Jia P., Mallik S., Fei R., Yoshioka H., Suzuki A., Iwata J., Zhao Z. Critical microRNAs and regulatory motifs in cleft palate identified by a conserved miRNA-TF-gene network approach in humans and mice. Brief. Bioinform. 2019:bbz082. doi: 10.1093/bib/bbz082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mitra R., Edmonds M.D., Sun J., Zhao M., Yu H., Eischen C.M., Zhao Z. Reproducible combinatorial regulatory networks elucidate novel oncogenic microRNAs in non-small cell lung cancer. RNA. 2014;20:1356–1368. doi: 10.1261/rna.042754.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu Q.H., Stephen S., Taylor J., Helliwell C.A., Wang M.B. Long noncoding RNAs responsive to Fusarium oxysporum infection in Arabidopsis thaliana. New Phytol. 2014;201:574–584. doi: 10.1111/nph.12537. [DOI] [PubMed] [Google Scholar]
- 36.GTEx Consortium Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chiang C., Scott A.J., Davis J.R., Tsang E.K., Li X., Kim Y., Hadzic T., Damani F.N., Ganel L., Montgomery S.B., GTEx Consortium The impact of structural variation on human gene expression. Nat. Genet. 2017;49:692–699. doi: 10.1038/ng.3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stranger B.E., Forrest M.S., Dunning M., Ingle C.E., Beazley C., Thorne N., Redon R., Bird C.P., de Grassi A., Lee C. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848–853. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jia P., Zhao Z. Impacts of somatic mutations on gene expression: an association perspective. Brief. Bioinform. 2017;18:413–425. doi: 10.1093/bib/bbw037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Helwak A., Kudla G., Dudnakova T., Tollervey D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell. 2013;153:654–665. doi: 10.1016/j.cell.2013.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Song K., Wang H., Krebs T.L., Kim S.-J., Danielpour D. Androgenic control of transforming growth factor-β signaling in prostate epithelial cells through transcriptional suppression of transforming growth factor-β receptor II. Cancer Res. 2008;68:8173–8182. doi: 10.1158/0008-5472.CAN-08-2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li L., Gao P., Li Y., Shen Y., Xie J., Sun D., Xue A., Zhao Z., Xu Z., Zhang M. JMJD2A-dependent silencing of Sp1 in advanced breast cancer promotes metastasis by downregulation of DIRAS3. Breast Cancer Res. Treat. 2014;147:487–500. doi: 10.1007/s10549-014-3083-7. [DOI] [PubMed] [Google Scholar]
- 43.Breunig C., Erdem N., Bott A., Greiwe J.F., Reinz E., Bernhardt S., Giacomelli C., Wachter A., Kanthelhardt E.J., Beißbarth T. TGFβ1 regulates HGF-induced cell migration and hepatocyte growth factor receptor MET expression via C-ets-1 and miR-128-3p in basal-like breast cancer. Mol. Oncol. 2018;12:1447–1463. doi: 10.1002/1878-0261.12355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma X., Beeghly-Fadiel A., Lu W., Shi J., Xiang Y.-B., Cai Q., Shen H., Shen C.Y., Ren Z., Matsuo K. Pathway analyses identify TGFBR2 as potential breast cancer susceptibility gene: results from a consortium study among Asians. Cancer Epidemiol. Biomarkers Prev. 2012;21:1176–1184. doi: 10.1158/1055-9965.EPI-12-0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berteaux N., Lottin S., Monté D., Pinte S., Quatannens B., Coll J., Hondermarck H., Curgy J.J., Dugimont T., Adriaenssens E. H19 mRNA-like noncoding RNA promotes breast cancer cell proliferation through positive control by E2F1. J. Biol. Chem. 2005;280:29625–29636. doi: 10.1074/jbc.M504033200. [DOI] [PubMed] [Google Scholar]
- 46.Lottin S., Adriaenssens E., Dupressoir T., Berteaux N., Montpellier C., Coll J., Dugimont T., Curgy J.J. Overexpression of an ectopic H19 gene enhances the tumorigenic properties of breast cancer cells. Carcinogenesis. 2002;23:1885–1895. doi: 10.1093/carcin/23.11.1885. [DOI] [PubMed] [Google Scholar]
- 47.Ma L., Li G.Z., Wu Z.S., Meng G. Prognostic significance of let-7b expression in breast cancer and correlation to its target gene of BSG expression. Med. Oncol. 2014;31:773. doi: 10.1007/s12032-013-0773-7. [DOI] [PubMed] [Google Scholar]
- 48.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hsu J.B.-K., Chiu C.-M., Hsu S.-D., Huang W.-Y., Chien C.-H., Lee T.-Y., Huang H.D. miRTar: an integrated system for identifying miRNA-target interactions in human. BMC Bioinformatics. 2011;12:300. doi: 10.1186/1471-2105-12-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dweep H., Sticht C., Pandey P., Gretz N. miRWalk—database: prediction of possible miRNA binding sites by “walking” the genes of three genomes. J. Biomed. Inform. 2011;44:839–847. doi: 10.1016/j.jbi.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 51.Maragkakis M., Vergoulis T., Alexiou P., Reczko M., Plomaritou K., Gousis M., Kourtis K., Koziris N., Dalamagas T., Hatzigeorgiou A.G. DIANA-microT Web server upgrade supports Fly and Worm miRNA target prediction and bibliographic miRNA to disease association. Nucleic Acids Res. 2011;39:W145-8. doi: 10.1093/nar/gkr294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Enright A.J., John B., Gaul U., Tuschl T., Sander C., Marks D.S. MicroRNA targets in Drosophila. Genome Biol. 2003;5:R1. doi: 10.1186/gb-2003-5-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krek A., Grün D., Poy M.N., Wolf R., Rosenberg L., Epstein E.J., MacMenamin P., da Piedade I., Gunsalus K.C., Stoffel M., Rajewsky N. Combinatorial microRNA target predictions. Nat. Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 54.Bartel D.P. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pinheiro J., Bates D., DebRoy S., Sarkar D. Linear and nonlinear mixed effects models. R package version. 2007;3:1–89. https://cran.r-project.org/web/packages/nlme/index.html [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.