

Abstract
Background and Aim:
Our previous study of H. pylori-induced apoptosis showed the involvement of Bcl-2 family proteins and cytochrome c release from mitochondria. Here, we examine the release of other factors from mitochondria, such as apoptosis inducing factor (AIF), and upstream events involving caspase-8 and Bid.
Methods:
Human gastric adenocarcinoma (AGS) cells were incubated with a cagA-positive H. pylori strain for 0, 3, 6 and 24 h and either total protein or cytoplasmic, nuclear and mitochondrial membrane fractions were collected.
Results:
Proteins were immunoblotted for AIF, Bid, poly adenosine ribose polymerase (PARP), caspase-8 and β-catenin. H. pylori activated caspase-8, caused PARP cleavage, and attenuated mitochondrial membrane potential. A time-dependent decrease in β-catenin protein expression was detected in cytoplasmic and nuclear extracts, coupled with a decrease in β-actin. An increase in the cytoplasmic pool of AIF was seen as early as 3 h after H. pylori exposure, and a concomitant increase was seen in nuclear AIF levels up to 6 h. A band corresponding to full-length Bid was seen in both the cytoplasmic and nuclear fractions of controls, but not after H. pylori exposure. Active AIF staining was markedly increased in gastric mucosa from infected persons, compared to uninfected controls.
Conclusion:
H. pylori might trigger apoptosis in AGS cells via interaction with death receptors in the plasma membrane, leading to the cleavage of procaspase-8, release of cytochrome c and AIF from mitochondria, and activation of subsequent downstream apoptotic events, as reported previously for chlorophyllin (1). This is consistent with AIF activation that was found in the gastric mucosa of humans infected with H. pylori. Hence, the balance between apoptosis and proliferation in these cells may be altered in response to injury caused by H. pylori infection, leading to an increased risk of cancer.
Keywords: AIF, H. pylori, gastric cells, apoptosis, mitochondria
INTRODUCTION
Each year, approximately 24,000 new cases of gastric cancer are diagnosed in the United States, and there are about 700,000 new cases worldwide. There will be an estimated 12,400 deaths from this type of cancer in 2008 in the United States. Several studies have provided evidence that Helicobacter pylori is a major risk factor for gastric carcinogenesis. H. pylori has been shown to induce apoptosis in gastric epithelial cells in vitro and in vivo (1–6).
Many H. pylori strains contain a group of genes known as the cag pathogenicity island (PAI) (7). Early reports indicated that persons infected with strains possessing the PAI (cag-positive strains) display less apoptosis and more mucosal inflammation and cell injury than persons infected with cag-negative strains (8, 9). Other studies, however, showed more apoptosis (as well as more mucosal inflammation and cell injury) in persons infected with cag-positive strains (1, 3, 10–14). This discrepancy may be explained by differences between the patient populations studied, including the ethnicities of the study population. In vitro studies have demonstrated that H. pylori can induce apoptosis in different gastric epithelial cell lines (1, 3, 12–14). In vitro induction of apoptosis has also been reported to be dependent on the presence of the PAI (15). Differences in the amount of apoptosis induced by H. pylori may be dependent on several factors, including cell type, H. pylori strain, and cell culture conditions. H. pylori–induced apoptosis is associated with an increase in Bak expression both in gastric biopsies from patients colonized by H. pylori and in vitro (3). Konturek et al. reported induction of apoptosis with evidence of bax upregulation and bcl2 down-regulation in duodenal ulcer patients with H. pylori infection (13). The induction of apoptosis by H. pylori may be important in the development of peptic ulcer disease and in gastric carcinogenesis. However, the molecular processes controlling and executing H. pylori-induced apoptosis are still poorly understood.
Apoptosis is defined as an active physiological process of cellular self-destruction, with specific morphological and biochemical changes (16). Caspases are proenzymes of the aspartate-specific cysteine protease family that play an important role in apoptosis (14, 17–19). Execution of apoptosis in eukaryotic cells is usually dependent on the activation of caspases, which fall into three subfamilies: the CPP32 subfamily (caspase-3, −6, −8, −9, and −10), the ICH-1/Nedd2 (caspase-2) subfamily, and the ICE3 subfamily (caspase-1,−4, and −5). Although members of ICE-like and CPP32-like subfamilies have different subunit specificity, all caspases are cleaved at specific Asp residues. On the basis of that observation, a model of hierarchical activation of caspases has been proposed (20). They are all expressed as proenzymes (30–50 kDa) that contain three domains: an NH2-terminal domain, a large subunit (∼20 kDa), and a small subunit (∼10 kDa). In the above-proposed model, caspase-8 has been termed an initiator protease, which activates executioner proteases such as caspase-3 or caspase-6. Once an executioner caspase is activated by upstream signals, it can activate other downstream factors, including PARP (poly adenosine-diphosphate–ribose polymerase) and DFF45 (DNA fragmentation factor 45). Cleavage of PARP, catalyzed by caspase-3 and other death substrates, is required for apoptosis to proceed in various cell lines (21–23). The hierarchical activation of these caspases ultimately results in the cleavage of key nuclear, cytoplasmic, and membrane-associated proteins, as well as the activation of DNases in many cases, leading to the morphological and biochemical changes characteristic of apoptotic cell death (24, 25).
The apoptotic process often involves the mitochondria. When the mitochondrial outer membrane is permeabilized and/or ruptured (22, 23, 26), pro-apoptotic proteins normally confined to the intermembrane space are released into the cytosol (27). The release of protein from mitochondria triggers catabolic reactions that result in cell death. For example, the release of cytochrome c from the mitochondrion to the cytosol activates the caspase-3/9 complex (28). Activation of downstream caspases is preceded by a loss of mitochondrial membrane potential and release of cytochrome c (29). In addition, cytochrome c and other proteins are released from the mitochondria into the cytosol (3, 30–32). Upon entering the cytosol, cytochrome c promotes the assembly of a multiprotein complex that induces proteolytic processing and activation of cell death by caspase.
We previously investigated the mechanisms of apoptosis and showed that gastric epithelial cells exposed to H. pylori underwent growth arrest and apoptosis after 24 h, with evidence for the formation of a sub-G1 peak in the attached cell population and nuclear condensation (1, 33). In the present study, we continued investigating the mechanisms by which H. pylori induces gastric cell injury in human gastric cancer cells, identifying a pathway of cell death involving the release of apoptosis inducing factor (AIF) from mitochondria. Human gastric cancer cells were treated with H. pylori. There was a time-dependent attenuation of mitochondrial membrane potential (Δψm) with activation of the caspase-8 pathway. In addition, AIF was released from mitochondria into the cytosol and leaking in to the nucleus, leading to cleavage of nuclear lamins. Upstream mediators of this apoptosis pathway were identified as caspase-8/caspase-6 and tBid acting in conjunction with other pro-apoptotic members of the Bcl-2 family, such as Bak or Bax (3, 33, 34). These findings suggested that H. pylori triggers apoptosis via interaction with so-called death receptors in the plasma membrane of cancer cells, leading to cleavage of procaspase-8, release of AIF from mitochondria, and activation of subsequent downstream events leading to the destruction of nuclear lamins.
MATERIALS AND METHODS
Gastric cell culture
A gastric adenocarcinoma cell line (AGS) purchased from the American Type Culture Collection (Manassas, VA) was used for these experiments. AGS cells were cultured in DMEM medium supplemented with 10% fetal bovine serum without antibiotics in 5% CO2 at 37°C in a humidified water-jacketed CO2 incubator.
Transient transfection of AGS with Bcl-2 (pcDNA3-Bcl2) was performed using Fugene 6 (Roche) as per the manufacturer’s protocol. Cells were incubated overnight, washed, and infected with H. pylori as described below.
H. pylori culture
A cag-positive H. pylori strain (J117), a gift from Dr. Timothy Cover (35), was cultured on trypticase soy agar with 5% sheep’s blood (Curtin Matheson, Jessup, MD) with Skirrow’s selective antibiotic supplement (Prolab Inc., Scarborough, Canada) at 37°C in a CO2/O2 water-jacketed incubator (Forma Scientific, Marietta, OH) under microaerophilic conditions (10% CO2, 7.5% O2, 82.5% N2). Cultures were maintained for 3 days prior to passage and bacteria were used between passage 5 and 15 for these experiments to ensure consistency in their adherence to AGS cells (36). The adherence was visualized using microscopy.
Subcellular fractionation and immunoblot analysis
Cytoplasmic, nuclear and mitochondrial membrane fractions of AGS cells were prepared according to our previous study (33, 37). Briefly, AGS cells were overlaid with culture medium containing H. pylori (strains J117, cagA+) for 24 hours. PBS-washed cells were resuspended in extraction buffer [10 mM Tris-HCl (pH 7.6), 10 mM KCl, and 5 mM MgCl2] with protease inhibitors (0.2 mM PMSF, 10 μg/ml pepstatin A, and 10 μg/ml leupeptin) and incubated on ice for 10 min. Cells were lysed on ice with digitonin (Sigma-Aldrich), nuclei were removed by centrifugation, and nuclear proteins were extracted with radioimmunoprecipitation (RIA) lysis buffer [10 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% NP40, 0.1% SDS, 0.5% sodium deoxycholate, and 1 mM DTT] supplemented with protease inhibitor cocktail (each tablet was dissolved and used according to the manufacturer recommendations (Roche)). For whole-cell extractions, samples were washed with 1 ml of PBS, and ice-cold RIA lysis buffer (2 ml) was added to the cell monolayer followed by gentle scraping on ice. Cell lysates were transferred to a microcentrifuge tube containing the mixture of protease inhibitor cocktail according to the manufacturer recommendations (Roche). Samples were left on ice for 30 min and centrifuged at 15,000 RPM for 15 min. Supernatants were taken for protein quantification, determined as described by Peterson (27). Protein (100 μg) was mixed with 60 μl of gel sample buffer [0.125 M Tris-HCl (pH 6.8; Sigma), 20% (v/v) glycerol, 2% (w/v) SDS, 10% (v/v) β-mercaptoethanol, and 0.25% (w/v) bromophenol blue], and the tubes were placed in a hot plate at 95°C for 5 min. For SDS-PAGE, the stacking gel was 4.5%, and resolving gels were made up at 10%, 12.5%, or 15% as required. Gels were run in a buffer solution containing 192 mM glycine (Sigma), 25 mM Tris (Sigma), and 1% SDS and then electroblotted onto Immobilon polyvinylidine difluoride membrane (Millipore, Bedford, MA) in a transfer buffer of 192 mM glycine (Sigma), 10 mM Tris (Sigma), and 20% (v/v) methanol (Fisher).
Cells were lysed in chilled RIA lysis buffer (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP40, 0.1% SDS, 0.5% sodium deoxycholate, and 1 mM DTT, Roche). Protein was measured in whole-cell lysates or in cytoplasmic, nuclear and mitochondrial membrane fractions, then lysates or fractions were boiled for 5 minutes in equal volumes of 2x Laemmli buffer and 100 μg protein was electrophoresed on 10% SDS-PAGE. Proteins were electrotransferred onto polyvinylidine (Immobilon-P, Millipore, Bedford, MA) membranes blocked with 10% non-fat dry milk in TBS at 37ºC, and incubated for 4 h at room temp or overnight at 4°C in TBS containing one of the following antibodies: (a) β-actin mouse monoclonal (Sigma) 1:4,000 dilution; (b) PARP rabbit polyclonal (Santa Cruz Biotech., Santa Cruz, CA) 1:250 dilution; (c) AIF goat polyclonal clone D-20 (sc-9416; Santa Cruz Biotechnology), 1:400 dilution; (d) Bid rabbit polyclonal (#2002; Cell Signalling), 1:500 dilution; (e) PARP mouse monoclonal (sc-8007; Santa Cruz Biotechnology), 1:500 dilution; (f) caspase 6 rabbit polyclonal (#9762; Cell Signaling), 1:500 dilution; (g) caspase 8 mouse monoclonal (#9746; Cell Signalling), 1:500 dilution; (h) lamin B goat polyclonal (Santa Cruz), 1:100 dilution in TBS (Tris buffered saline) containing 1% bovine serum album); or (i) β-catenin mouse monoclonal (Transduction Lab), 1:400 dilution. After washing in TBS-T (TBS containing 0.5% Tween-20), the membranes were incubated with horseradish peroxidase (HRP)-conjugated appropriate antibodies (sheep anti-mouse or sheep anti-rabbit, or donkey anti-goat antibodies 1:3,000) for 2 h. The antigen-antibody complexes were detected by chemiluminescence (ECL-Plus New England Nuclear). Equal protein loading was confirmed by Ponceau-S staining.
Mitochondrial membrane potential assay
To measure the mitochondrial membrane potential (Δψm), we resuspended AGS cells (1 × 106 cells/ml) in 10 μg/ml solution of the cationic, lipophilic dye, tetramethyl-rhodamine ethyl ester (TMRE) (Molecular Probes), and DMSO-working concentrations were made in water at 1 mg/ml. For estimation of Δψm, cells were incubated with 100 nM TMRE for 15 minutes at room temperature in PBS. Cells were maintained in a stable concentration of the dye throughout the time of measurement. After incubation, cells were immediately analyzed by flow cytometry. Dead cells were excluded by forward and side scatter gating. Data were accumulated by analyzing an average population of 20,000 cells. TMRE fluorescence was detectable in the PI channel (red fluorescence, emission at 590 nm).
Caspase assay
Gastric cells were overlaid with culture medium with or without H. pylori and incubated for up to 48 h. At various time points during this 48-h period, soluble gastric cell protein was extracted and treated or not treated with 1 mM caspase-8 inhibitor. Caspase-8 activities were assayed by release of IETD containing synthetic peptides, according to the manufacturer’s protocol (Clontech) using a Cytofluor instrument (Applied Biosystems, Foster City, CA), per the manufacturer’s instructions. Typically, 1 × 106 cells (exposed or unexposed to H. pylori) were lysed with 50 μl of lysis buffer, IETD (1 mM) used as caspase-8 substrate, and the samples were read at 400 nm excitation/505 nm emission using the Cytofluor. Caspase-8 inhibitor IETD–fmk standard reactions was used (Clontech).
Trypan blue staining
For the exclusion dye experiment, H. pylori-infected and-uninfected AGS cells were exposed to trypan blue (10% v/v) for 5 min. The number of viable cells was determined by light microscopy (40–100× magnification) by counting those cells that excluded the dye. Cells were counted in a randomized manner using a hemocytometer.
Human gastric tissue
Archival formalin-fixed, paraffin-embedded gastric biopsies obtained from patients undergoing upper gastrointestinal endoscopy at Howard University Hospital and Tehran University hospital after IRB approval was obtained for the human subjects. They were sectioned onto glass slides for immunohistochemical staining for these experiments. Silver or Giemsa staining was used to confirm the previous diagnosis of gastric tissue infection with H. pylori. Gastric tissue from persons not infected with H. pylori was used as a control.
Silver staining
Silver staining was performed on tissue sections to detect H. pylori. Sections were de-waxed in xylene, rehydrated through graded alcohols, and placed in PBS. Sections were impregnated with 1% silver nitrate in PBS at 55 °C for 1 h. Slides were then placed in 0.15% hydroquinone 5% gelatin at 55 °C for approximately 3 min to develop. Afterward, slides were rinsed for 5 min in warm tap water, lightly counterstained with Harris hematoxylin, dehydrated through graded alcohols, cleared in xylene, and mounted with a cover glass using permount (Biomeda, Foster City, CA).
Immunohistochemistry and fluorescent microscopy
AIF was detected in tissue sections using goat polyclonal IgG antibodies raised against epitope near the N-terminus of AIF of human origin (sc-9417; Santa Cruz, 1:100) using LSAB+ kit (DAKO, Carpenteria, CA). Slides containing the gastric tissue (infected and uninfected with H. pylori) were washed with PBS and fixed with 3.7% formaldehyde, 0.05% gluteraldehyde, and 0.5% Triton X100. Tissues were blocked for 10 min in 10% donkey serum in PBS and then incubated with AIF antibody in 5% donkey serum in PBS for 1 h. Tissues were washed twice with PBS and incubated for 30 min with the FITC-conjugated donkey anti goat antibody, and washed again. The cells were then stained for 5 min with DAPI (Invitrogen) and cover slips were mounted with Prolong Gold anti-fade coverslip media (Invitrogen). Slides were analyzed with Olympus IX-70 fluorescent microscope. Sections or cultured cells were examined with a fluorescence microscope (Olympus AX70, Olympus America) equipped with appropriate filters to observe the DAPI and FITC signals. Colocalization of apoptotic cells with nucleus was identified by merged images.
For bright-field microscopy with 3,3′-diaminobenzidine tetrahydrochloride (DAB), separate series of sections were used, quenched with 0.5% H2O2 for 30 min, rinsed in PBS containing 0.3% Triton-X, and then transferred for 30 min to a PBS–Triton-X solution containing 5% donkey serum albumin to block nonspecific binding sites. After a second 30-min wash, the sections were incubated overnight at room temperature with goat anti-AIF polyclonal antibodies for 1 h. The cells/sections were then rinsed, incubated with biotinylated secondary antibody for 2 h at room temperature, washed and exposed for 30 min to ABC Elite reagent (ABC kit; Vector Laboratories, Burlingame, CA) and washed with PBS Triton-X. A solution of 0.01% H2O2, 0.003% NICl2, and 0.02% DAB in PBS was used as the substrate for the peroxidase.
AIF expression were determined by immunohistochemical staining using goat polyclonal IgG antibodies raised against an epitope near the N-terminus of human AIF (sc-9417; Santa Cruz, 1:100) using an LSAB+ kit (DAKO, Carpenteria, CA). Antigen retrieval was carried out by heating sections in proteinase K for 15 min in a microwave oven. Endogenous peroxidase activity was quenched by incubation in 3% H2O2 in methanol for 5 min. Sections were then incubated for 1 hr at room temperature with antibody provided in the kit. Immunohistochemical results were evaluated for intensity of staining of cytoplasmic components, and intensity of overall staining for each section. Antibody was left out as an appropriate negative control. The intensity of staining was graded as 0 (negative), 1 (weak), 2 (moderate), 3 (strong), 4 (very strong), and we estimated the percentage of cells that were stained in each section.
Statistics
For analysis of paired observations before and after H. pylori exposure, we used a paired t-test for normally distributed data. Each experiment was repeated three times.
RESULTS
Mitochondrial depolarization is caused by H. pylori infection and is blocked by Bcl-2
Previously, we performed TUNEL assays using flow cytometry and documented apoptosis induction in gastric cells exposed to H. pylori (Fig.1A). AGS cells were exposed to H. pylori for 24 hours and analyzed by trypan blue exclusion dye to count the dead cells. Exposure of gastric cells to H. pylori for 24 hours significantly increased the number of dead cells from 2% to 63% (P < 0.05) by trypan blue assay (figure 1A). Incubation of cells with H. pylori provokes profound damage to mitochondria. To further assess the death pathway targeting mitochondria, we measured mitochondrial membrane depolarization as a marker of mitochondrial dysfunction, using the fluorescent dye TMRE (33). Flow cytometry studies revealed a decrease in Δψm after H. pylori treatment, using TMRE as a marker of mitochondrial membrane integrity (figure 1B). Mitochondria begin to depolarize within 3 hours of incubation with H. pylori, as measured by loss of fluorescence compared to untreated control. Loss of the mitochondrial membrane potential (Δψm) inhibited by forced Bcl-2 expression in gastric cells treated with H. pylori. These findings predicted that apoptosis-inducing factors might be released from mitochondria, such as AIF, and that Bcl-2-family proteins might be involved upstream.
Fig. 1.
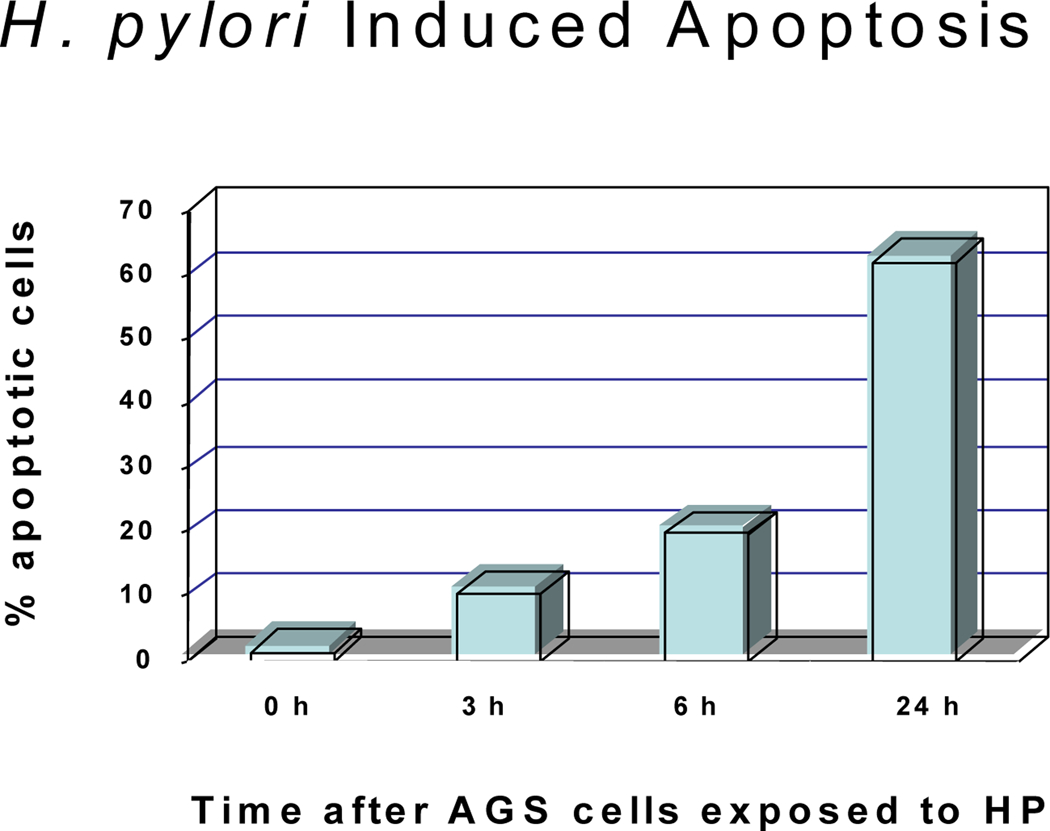

H. pylori-induced cell death and loss of mitochondrial membrane potential (Δψm). (A) Cell death detected in AGS cells exposed to H. pylori for 3, 6, and 24 hours. The cells were exposed to trypan blue (10% v/v) for 5 min. The number of viable cells was determined by light microscopy (40–100X magnification) by counting those cells that excluded the dye. Cells were counted in a randomized manner using a hemocytometer. (B). H. pylori, and mitochondrial membrane potential loss was inhibited by forced Bcl-2 expression. AGS cells (1 × 106 cells/ml) were resuspended in 10 μg/ml TMRE. After incubation cells were immediately analyzed by flow cytometry. Dead cells were excluded by forward and side scatter gating. Data were accumulated by analyzing an average population of 20,000 cells. TMRE fluorescence was detectable in the PI channel (red fluorescence, emission at 590 nm). In parallel experiments AGS cells transfected with Bcl-2 (to inhibit apoptosis) were stained as explained above. Loss of Δψm was quantified by flow cytometry analysis of untransfected cells versus Bcl-2 transfected gastric cells after H. pylori treatment for indicated time periods. Bcl-2 transfected cells exhibit a significantly reduced Δψm at 3 hr after H. pylori treatment (p<0.05). In contrast, Bcl-2 transfected cells exhibit a much slower decline in membrane potential. Each sample was performed in duplicates, and the figure is representative of three assays.
H. pylori induces the activation of caspase-8 with cleavage of nuclear lamin
Because the mechanism of apoptosis induced by H. pylori in AGS cells apparently involved the cytochrome c→caspase-9→caspase-3→PARP pathway (33, 34), additional caspases were examined, including those implicated in membrane “death receptor” pathways. H. pylori induced cell death (Fig. 1) and produced the active, cleaved form of caspase-8 (Fig. 2A), at a bacteria:cell concentration of 100:1. Lamin B, a major target of caspase-8, was cleaved by H. pylori, and cleaved lamin products of 45 and 40 kDa were detected in AGS cells treated with H. pylori for 24 hours. Accordingly, these cleaved lamin products were also found in cells treated with staurosporine (as a positive control), and full-length lamin B was seen in nuclear fractions of untreated AGS cells (as a negative control; Fig. 2B). There was extensive accumulation of lamin B cleavage products (Fig. 2B), consistent with our previous results obtained in AGS cells exposed to H. pylori that resulted in DNA laddering (38) under the same conditions used here. In addition, PARP cleavage was observed in cells treated with H. pylori (Fig. 2C). These results clearly establish that H. pylori-induced apoptosis in AGS cells involves the mitochondria.
Fig. 2.
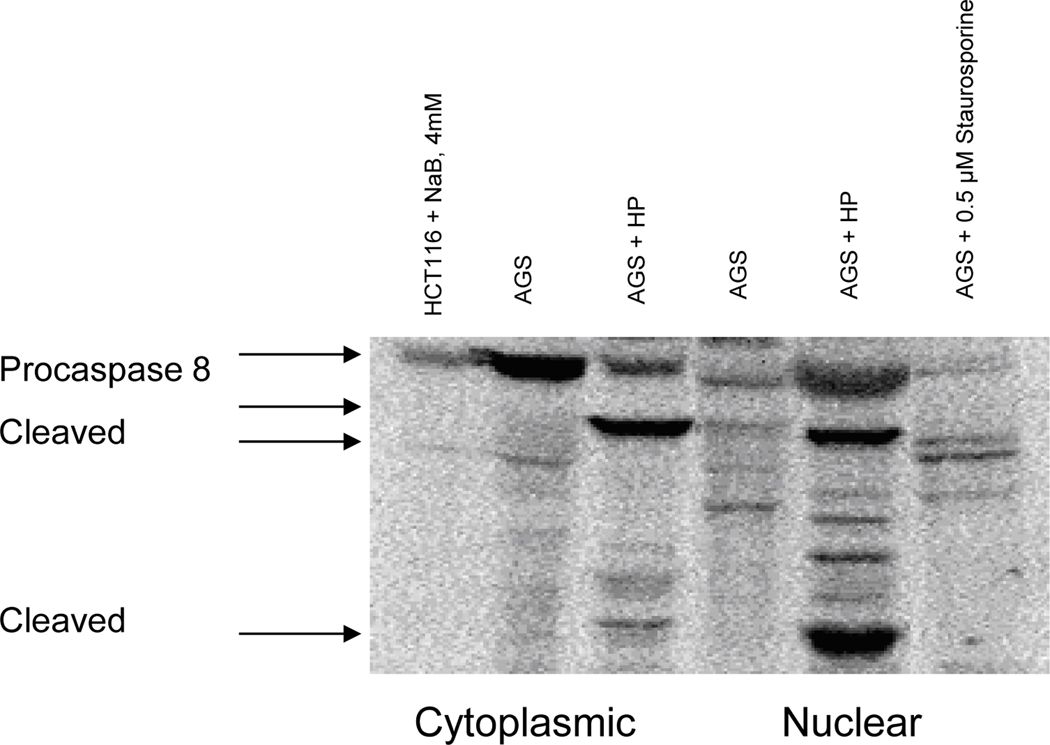
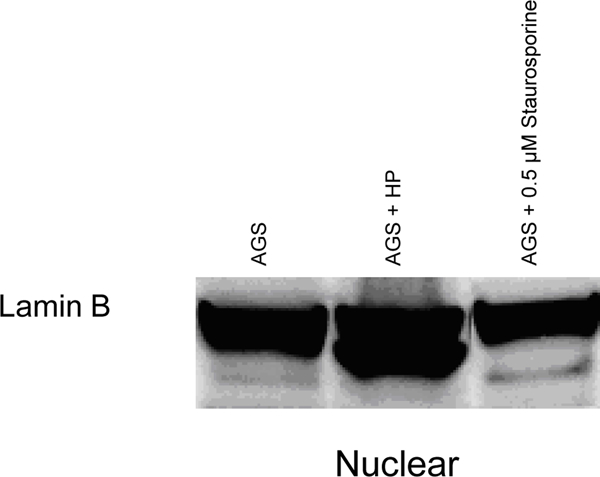
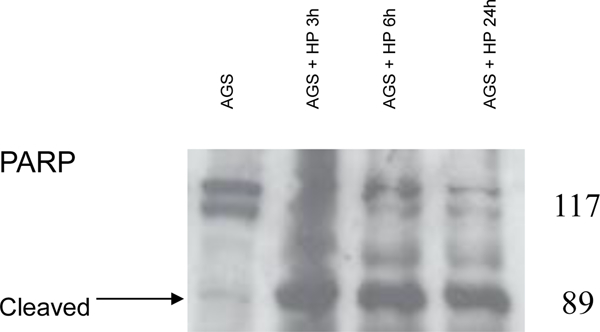
H. pylori-induced apoptosis in gastric cells involves the activation of procaspase-8 with cleavage of nuclear lamins and PARP. AGS cells were overlaid with culture medium containing H. pylori (at 100 bacteria per gastric cell, strains J117, cagA+) for 24 hours. (A) Cytoplasmic and nuclear proteins were run on SDS-PAGE and probed for anti-human caspase-8. Presence of activated caspase-8 protein observed in AGS cells treated with H. pylori. HCT116 and AGS cells treated with NaB or Staurosporine were used as positive controls. (B) Immunoblot showing the presence of cleaved nuclear lamin B fragments in cells treated with H. pylori but not in untreated AGS or in cells treated with staurosporine. (C) Cleaved PARP protein fragments observed in total protein lysates of H. pylori-treated cells but not in untreated controls. Results in A-C are representative of data obtained from two or more separate experiments.
H. pylori causes attenuation of the mitochondrial membrane potential (Δψm) and release and Translocation of AIF
To clarify the mechanism(s) of apoptosis by H. pylori, we focused initial experiments on the cytochrome c/caspase-8 pathway and downstream targets that might account for this phenomenon. Membrane potential was lost for up to 3 hours in a time-dependent manner in AGS cells overlaid with culture medium containing H. pylori (strains J117, cagA+) (data not shown). In a parallel study, the translocation of AIF from mitochondria to the cytoplasm and nucleus was observed (Fig 3) up to 24 hours. Release of AIF into the cytoplasmic fraction peaked within 3 hours, and translocation of AIF to the nucleus peaked at 6 hours. These increases occurred despite an apparent loss of β-actin (Fig 3). We previously examined likely candidates that might be released from mitochondria. Cytochrome c was released from mitochondria and was detected in the cytosol in AGS cells treated with H pylori (33, 39). Interestingly, AIF, which is normally localized within the mitochondria, was elevated in both the cytosol and nucleus in AGS cells treated with H. pylori (Fig. 3). The release of AIF from mitochondria was consistent with our previously detectable cytochrome c in the cytosolic fraction (37). This result was expected and suggested a detailed study of caspases normally regulated by cytochrome c/Apaf-1 as well as those activated via alternative mechanisms.
Fig. 3.
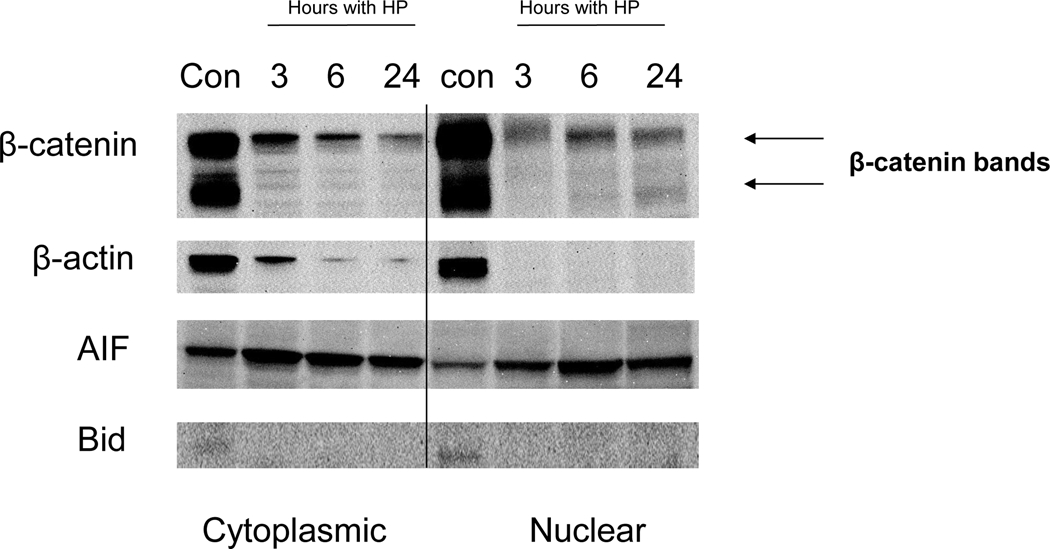
H. pylori-induced release of AIF. Western blots of AIF expression in cytosolic and nuclear fractions at 3, 6, 24 h after AGS treatment with H. pylori. Translocation of AIF from mitochondria to the cytoplasm and nucleus periphery was observed. Release of AIF into the cytoplasmic fraction peaked within 3 h, and translocation of AIF to the nucleus peaked at 6 h. The increase in cytoplasmic AIF occurred despite an apparent loss of β-actin. A band corresponding to full-length Bid was seen both in the cytoplasmic and nuclear fractions of controls, but not in cells treated with H. pylori. A time-dependent decrease in β-catenin protein expression was detected in cytoplasmic and nuclear extracts, as seen for β-actin, suggesting concentration-dependent protein cleavage with induction of apoptosis. H. pylori-induced apoptosis in AGS gastric cells involves cleavage of Bid. Immunoblot showing the presence of Bid in cells untreated cells. Cleaved Bid appeared only in the AGS cells treated with H. pylori. Cytoplasmic and nuclear proteins were run on SDS-PAGE and probed with anti-human Bid.
H. pylori cleaves β-catenin in a time-dependent manner and induces cleavage of Bid
AGS cells were overlaid with culture medium containing H. pylori (strains J117, cagA+) for 3, 6, and 24 hours. A time-dependent decrease in β-catenin protein expression was detected in cytoplasmic and nuclear extracts, as seen for β-actin, suggesting concentration-dependent protein cleavage with induction of apoptosis (Fig. 3). AGS cells were overlaid with culture medium containing H. pylori (strains J117, cagA+) for 3, 6, and 24 hours. A band corresponding to full-length Bid was seen both in the cytoplasmic and nuclear fractions of controls (Fig. 3), but not in cells treated with H. pylori.
H. pylori causes release and translocation of AIF
Immunohistochemical and immnuoflorescent staining of AIF in gastric tissue infected with H. pylori revealed the translocation and accumulation of AIF (Fig. 4 and 5). Immunohistochemistry showed the staining of AIF in the apoptotic cells. Bright field and fluorescent images of gastric tissue infected with H. pylori (fig. 4B, C, D) is shown in comparison to control tissue (fig. 4A). We also merged the DAPI with the FITC staining, showing the accumulation of AIF (fig. 5). DAPI penetration is evidenced by nuclei staining (fig. 5A). Analysis of 44 gastric adenocarinoma patients in tissue microarray showed a strong or very strong staining for AIF antibody in 28 (63%) of cases (Fig. 6C). In parallel experiment silver staining of the H. pylori positive tissue indicated the presence of the bacteria (6A) and in uninfected patients lack of brown color indicates absence of cytoplamic staining for AIF (Fig. 6B). Negative staining was detected in 9 (20%) of these cases. The remaining were stained weakly (11%) or moderately (7%).
Fig. 4.
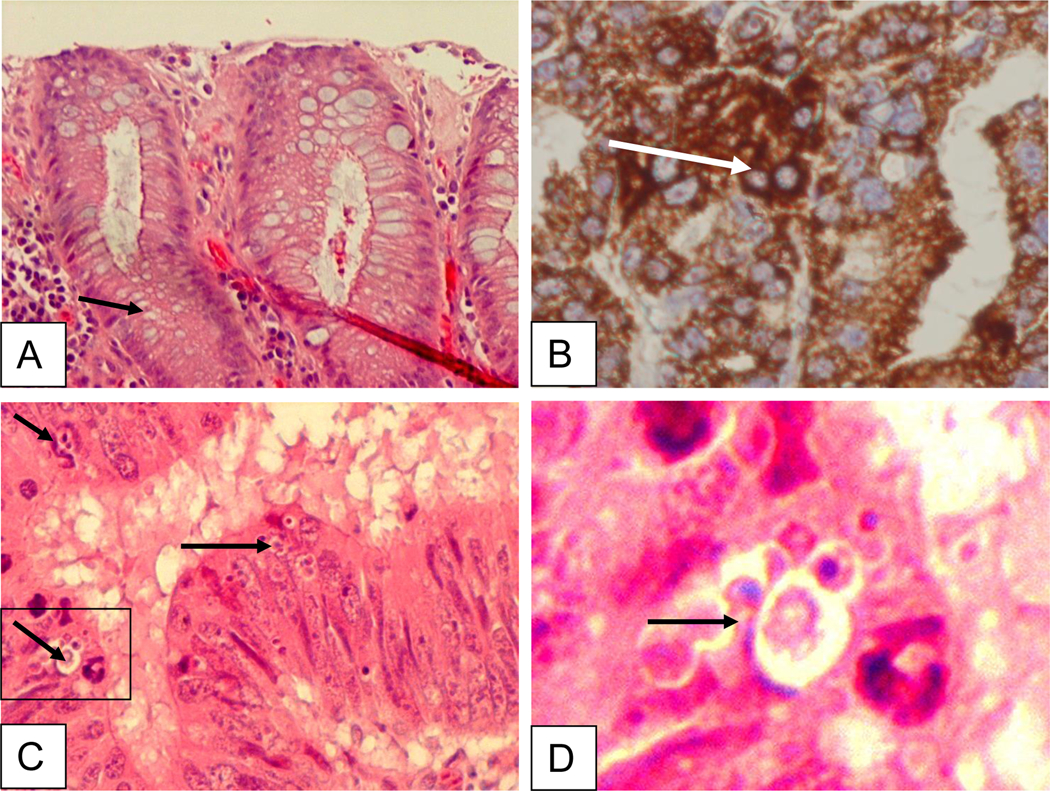
Immunohistochemical staining of gastric tissue infected with H. pylori. Bright field images of gastric tissue infected with H. pylori (B-D) vs. control (A). The tissue was stained with H & E stain, arrows show healthy (A), AIF cytoplasmic staining (B) or apoptotic cells (C, D). Cells were immunostained with primary AIF-specific antibody, (AIF goat polyclonal, clone D-20; sc-9416; Santa Cruz Biotechnology) and donkey anti-goat horseradish peroxidase conjugated secondary antibody for DAB staining. Adequate antibody penetration and cell architecture preservation is evidenced by cytoplasmic staining.
Fig. 5.
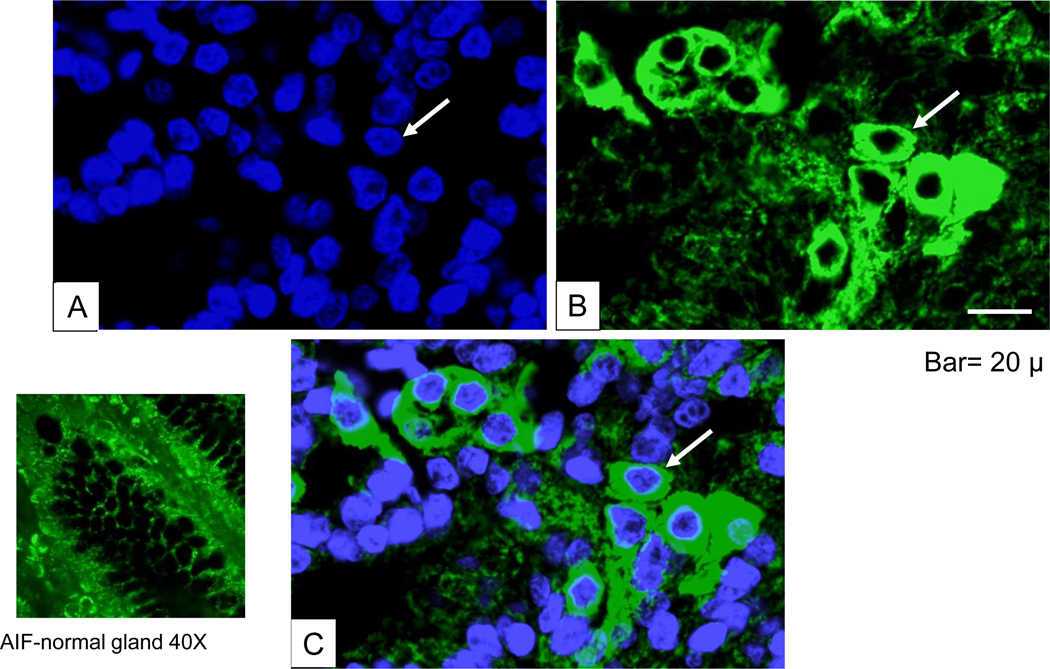
Immunofluorescent analysis of AIF. (A) DAPI-staining to visualize nucleus. (B) Immunohistochemical staining with rabbit polyclonal AIF primary antibody (sc-9417) followed by FITC-conjugated donkey anti-goat secondary antibody. (C) Merged image for viewing colocalization of AIF with DAPI-stained cytoplasmic and nuclear periphery staining. Bar =50 μm.
Fig. 6.
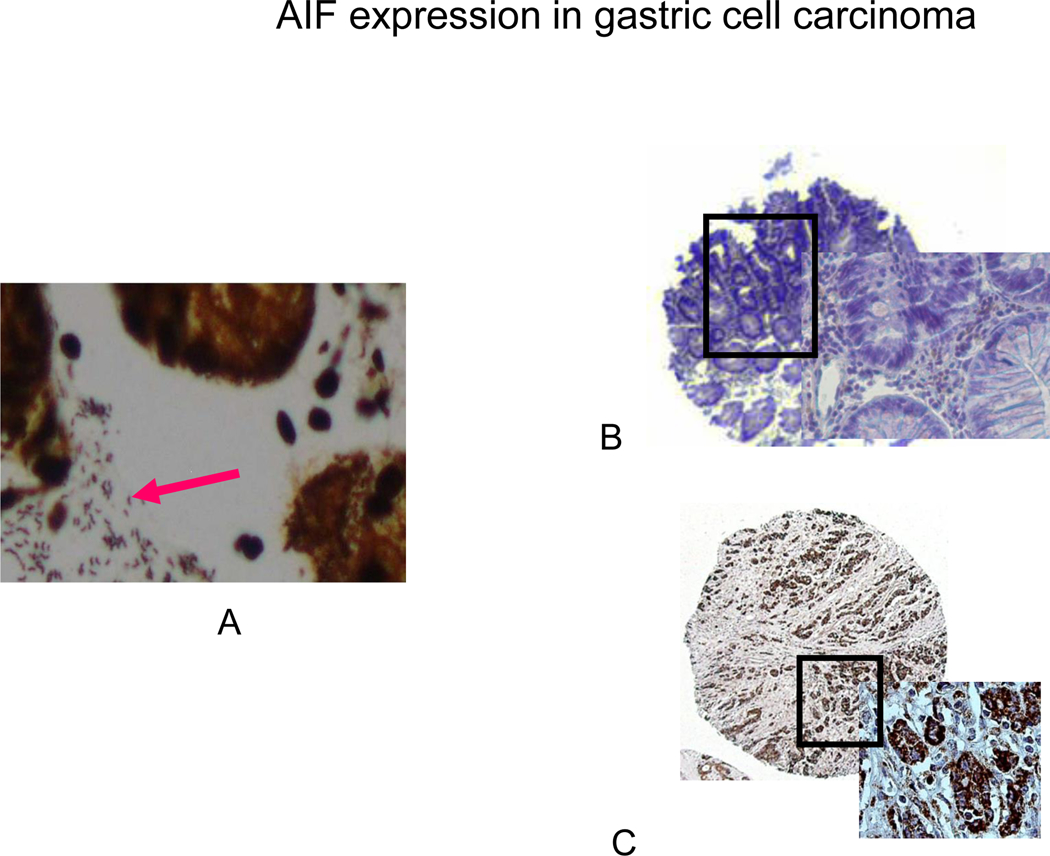
Silver (A) and Immunohistochemical staining of H. pylori and AIF in human tissue microarray (B, C). (A) Positive silver staining for H. pylori is evident in all of the glands in biopsy specimens from patients infected with H. pylori. (B) In uninfected patients, lack of brown color indicates absence of cytoplamic staining for AIF. (C) In patients infected with H. pylori, 28/44 (63%) of the cases showed strong cytoplasmic staining for AIF in the malignant epithelial cells.
CONCLUSIONS
In AGS cells, H. pylori is known to be involved in the ability to induce programmed cell death (33). Here we have shown that H. pylori might trigger apoptosis in AGS cells via interaction with death receptors in the plasma membrane, leading to the cleavage of procaspase-8, lamin B, and Bid, the release of cytochrome c and AIF from mitochondria, and activation of subsequent downstream apoptotic events, as reported previously for chlorophyllin (37).
Apoptosis is defined as an active physiological process of cellular self-destruction, with specific morphology and biochemical changes (16). Apoptosis and proliferation are tightly regulated processes within cells. Among apoptosis-related genes, p53, Bax, and Fas are of particular importance. The involvement of the Fas pathway has been shown along with involvement of Bcl-2 proteins in H. pylori-induced apoptosis(13, 40–42). Cleavage of caspase-3 and −8, and release of cytochrome c, have been shown to occur in H. pylori-induced apoptosis (36, 43, 44). Cytochrome c release from the mitochondria may contribute to the activation of caspases. Upon entering the cytosol, cytochrome c promotes the assembly of a multi-protein complex that induces proteolytic processing and activation of cell death by caspases (32, 45). Once activated by cytochrome c, caspase-3 triggers activation of various important cellular substrates leading to apoptosis (18, 46, 47).
The release of cytochrome c in response to H. pylori is an early event in gastric epithelial cells. H. pylori can cause stress signaling to transduce the mitochondria and cause the release of cytochrome c (48, 49). Activation of Bax and its translocation to the mitochondria followed by membrane depolarization, mitochondrial fragmentation and cytochrome c release are associated with H. pylori-induced apoptosis, which is consistent with various studies demonstrating decreased levels of Bcl-2 and increased levels of Bak in AGS cells exposed to H. pylori (3). When cytochrome c is released, it binds together with dATP and apoptosis activating factor-1 to procaspase-9, resulting in the activation of downstream caspases (50).
While qualitative differences occur between cag-positive and cag-negative H. pylori strains, most studies show no significant difference in the ability of these strains to induce apoptosis or to stimulate pro-apoptotic factors. Therefore, our experiments used only a cag-positive strain. In prior experiments, we have shown that H. pylori-induced apoptosis is associated with accumulation of p53 protein and a decrease in Bcl-2 (1). In addition, we demonstrated (36) that H. pylori-induced apoptosis activated caspase-8 and caspase-3, which in turn degrade PARP and DFF-45, consistent with the data reported here.
Several lines of in vitro evidence suggest that the induction of caspase-8, −9, and −3 may play a role in the etiology of H. pylori-associated ulcer disease and may be involved in early processes of gastric carcinoma (48, 51). After H. pylori infection, cytochrome c release from the mitochondria activates caspase-8, −9, and −3, as confirmed by cleavage of PARP (48, 51). Different H. pylori cytotoxic factors, including vacA, LPS and/or the PAI, may be important in Bax translocation and the induction of cytochrome c from mitochondria, which induces oligomerization of Apaf-1, allowing activation of procaspase-9 to promote apoptosis (50).
Here, we demonstrated not only the forced expression of Bcl-2 blocks cell death but also indicates the significant mitochondrial involvement in H. pylori-induced apoptosis. We previously showed that during apoptosis induced by H. pylori infection, the mitochondrial morphology in respective cell types switches from a reticulo-tubular to a punctiform mitochondrial phenotype, compatible with organelle fragmentation (33).
In the present study, H. pylori caused a reduction in the attached cell yield (Fig. 1) and marked changes in several of the molecular markers examined, including activation of caspase-8 (Fig.2A), cleavage of lamin B (Fig. 2B), increased cytosolic and nuclear AIF levels (Fig. 3), and inhibition of B-catenin and cleavage of BID (Fig. 3). Thus, many of the molecular and biochemical changes were induced in AGS cells treated with H. pylori. Importantly, several of the key molecular markers of apoptosis altered by H. pylori in vivo or in vitro, consistent with corresponding changes in cell growth characteristics and apoptosis (33, 36). The present study has shown that in AGS cells treated with H. pylori, there was a marked increase in the proportion of floating cells, and that these cells exhibited morphological hallmarks of apoptosis, including nuclear condensation and formation of a sub-G1 peak during FACS analysis (1, 33, 36, 37). H. pylori produced changes in the mitochondrial membrane potential (Δψm) and release of AIF in treated AGS cells, and cytochrome c was detected in the cytosolic fraction (Fig. 3) and upstream players such as caspase-8, caspase-3, and PARP were affected (Fig. 2). Bacteria other than H. pylori may have the ability to activate host apoptotic processes in response to infection and inflammation. Bax translocation has also been demonstrated in Chlamydia psittaci-infected cells where it was found to be independent of caspase activation (52). It is not clear whether the activation of the caspase pathway is generally beneficial to the host or the pathogen. Some pathogens including Chlamydia trachomattis and Rickettsia rickettsii do not induce apoptosis, which may allow these organisms to grow and persist intercellularly (52, 53). Streptococcus pyogenes causes mitochondrial dysfunction that leads to the release of cytochrome c from the mitochondria via Bax translocation (54). Basu et al. showed that tumor necrosis factor α-induced cell death in HeLa cells was augmented by rottlerin through a cytochrome c-independent pathway, and this involved release of mitochondrial AIF into the cytosol (55). In addition, rottlerin enhanced the activation of caspase-8 and cleavage of Bid (55), as found in the present studies with H. pylori (Figs. 2 and 3). Thus, it is possible that the major route for H. pylori-induced apoptosis involves caspase-8-mediated destruction of nuclear lamins via caspase-6, with concomitant cleavage of Bid to form tBid, the release of AIF from mitochondria, and additional elimination of nuclear lamins (34, 56). tBid can interact with anti-apoptotic Bcl-2 proteins and displace Bax, initiating pore formation in mitochondria (57), which is in line with our previous study showing that Bax was markedly induced by H. pylori treatment translocated to mitochondria (33). BAX translocation might explain, in part, the apparent release of cytochrome c from mitochondria in response to H. pylori treatment. However, tBid has also been reported to induce the oligomerization of pro-apoptotic Bak, as well as form homo-oligomers, resulting in cytochrome c release from mitochondria (58). The fact that tBid and BAX increased in response to H. pylori treatment is supporting evidence for AIF and cytochrome c release from mitochondria. The involvement of specific Bcl-2 family members in the apoptotic mechanism and the nature of the (Δψm) transition remain to be clarified, and we are investigating the time course for tBid formation and AIF appearance in the cytosol and nucleus. When we analyzed AIF translocation in primary gastric cancer patients infected with H. pylori, 63% of the cases showed strong cytoplasmic staining for AIF in the malignant epithelial cells. This further confirmed the correlation of the AIF staining in the tumors with cell death.
Another important observation from the present study was the finding that H. pylori decreased the expression of B-catenin and induced lamin cleavage, which are markers of cell cytoskeleton. In tissues undergoing continuous regeneration, such as the stomach, homeostatic mechanisms seek to balance the birth of cells in the lower half of the crypt with programmed cell death in the upper half of the crypt. Deregulation of this homeostatic balance, via a relative increase in the rate of cell proliferation or a decrease in the rate of apoptosis, provides a mechanism for tumorigenesis, and many chemopreventive agents in the stomach act by enhancing apoptosis (59, 60). However, at later stages, it is the differentiation status of the cancer that frequently dictates the response to chemopreventive and chemotherapeutic agents (61–63). The present study indicates, for the first time, that human gastric carcinoma cells exposed to H. pylori undergo cell death through AIF release from mitochondria. Additional work is now in progress to define the induction of death receptors and cell signaling changes by H. pylori and determine early changes that occur before the induction of apoptosis in mitochondria. However, the results presented here for B-catenin and lamin suggest that the activation of caspase-8 also leads to the formation of tBid, which, in conjunction with changes in Bak and other Bcl-2 family members, facilitates the release of AIF from mitochondria via a (Δψm) transition that may cause release of cytochrome c. Subsequently, AIF enhances the cleavage of nuclear lamins and activates the process of nuclear condensation, although the specific timing and relative contributions of AIF versus the caspase-8/caspase-6 pathway remain to be determined. The proposed model for H. pylori-induced cell death (Fig. 7) might provide a molecular rationale for future management of H. pylori infection.
Fig. 7.
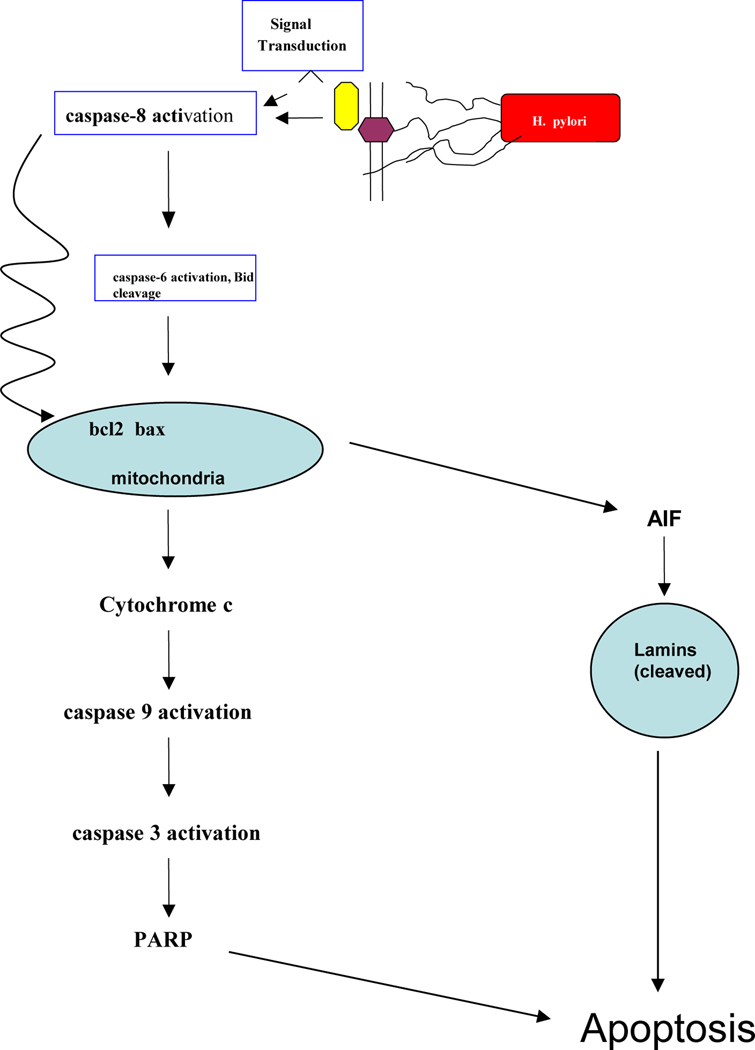
Proposed model for the mechanism of apoptosis induction by H. pylori in AGS gastric epithelial cells. Activation of death receptors by H. pylori results in the initial cleavage of procaspase-8 to generate caspase-8. Caspase-8 represents a branch point, activating caspase-6 in one direction and cleaving Bid in another direction. Increased levels proapoptotic Bcl-2 family members (e.g., Bak) facilitate a change in mitochondrial membrane potential (Δψm), leading to the release of AIF. As a consequence, nuclear lamins are cleaved, leading to nuclear condensation and apoptosis.
Acknowledgments:
We thank Mr. Daremipouran for preparing and culturing the AGS cells. The first two authors contributed equally. This work was supported by Public Health Service grants from the National Institutes of Health, DK53713 and DK56664.
Abbreviations:
- AGS
human gastric adenocarcinoma
- PARP
poly adenosine ribose polymerase
- TMRE
Tetramethylrhodamine
References:
- 1.Ahmed A, Smoot D, Littelton G, et al. Helicobacter pylori inhibits gastric cell cycle. Microbes and Infection 2000;2:1–11. [DOI] [PubMed] [Google Scholar]
- 2.Bechi P, Balzi M, Becciolini A, et al. Helicobacter pylori and cell proliferation of the gastric mucosa: possible implications for gastric carcinogenesis. Am. J. Gastroenterology 1996;91:271–276. [PubMed] [Google Scholar]
- 3.Chen G, Sordillo EM, Ramey WG, et al. Apoptosis in gastric epithelial cells is induced by Helicobacter pylori and accompanied by increased expression of BAK. Biochem Biophys Res Commun 1997;239:626–32. [DOI] [PubMed] [Google Scholar]
- 4.Chow K, Bank S, Ahn J, et al. Helicobacter pylori infection does not increase gastric antrum mucosal cell proliferation. Am. J. Gastroenterology 1995;90:64–66. [PubMed] [Google Scholar]
- 5.Fan X, Crowe SE, Behar S, et al. The effect of class II major histocompatibility complex expression on adherence of Helicobacter pylori and induction of apoptosis in gastric epithelial cells: a mechanism for T helper cell type 1-mediated damage. J Exp Med 1998;187:1659–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moss S, Calam J, Agarwal B, et al. Helicobacter pylori infection induces gastric epithelial apoptosis in vivo. Gastro 1995;108:A171. [Google Scholar]
- 7.Covacci A, Telford JL, Del Giudice G, et al. Helicobacter pylori virulence and genetic geography. Science 1999;284:1328–33. [DOI] [PubMed] [Google Scholar]
- 8.Graham DY, Yamaoka Y. Disease-specific Helicobacter pylori virulence factors: the unfulfilled promise. Helicobacter 2000;5:S3–9; discussion S27–31. [DOI] [PubMed] [Google Scholar]
- 9.Jang TJ, Kim JR. Proliferation and apoptosis in gastric antral epithelial cells of patients infected with Helicobacter pylori [see comments]. J Gastroenterol 2000;35:265–71. [DOI] [PubMed] [Google Scholar]
- 10.Moss SF, Sordillo EM, Abdalla AM, et al. Increased gastric epithelial cell apoptosis associated with colonization with cagA + Helicobacter pylori strains. Cancer Res 2001;61:1406–11. [PubMed] [Google Scholar]
- 11.Rokkas T, Ladas S, Liatsos C, et al. Relationship of Helicobacter pylori CagA status to gastric cell proliferation and apoptosis. Dig Dis Sci 1999;44:487–93. [DOI] [PubMed] [Google Scholar]
- 12.Ashktorab h, Rees B, Allen C, et al. Regulation of Apoptosis by Differential Expression of waf1, p53 and bcl2 in Gastric Epithelial Cells Exposed to Helicobacter Pylori. Gastroenterology 1999;112:(4)A59. [Google Scholar]
- 13.Konturek PC, Pierzchalski P, Konturek SJ, et al. Helicobacter pylori induces apoptosis in gastric mucosa through an upregulation of Bax expression in humans. Scand J Gastroenterol 1999;34:375–83. [DOI] [PubMed] [Google Scholar]
- 14.Shirin H, Sordillo EM, Oh SH, et al. Helicobacter pylori inhibits the G1 to S transition in AGS gastric epithelial cells. Cancer Res 1999;59:2277–81. [PubMed] [Google Scholar]
- 15.Le’Negrate G, Ricci V, Hofman V, et al. Epithelial intestinal cell apoptosis induced by Helicobacter pylori depends on expression of the cag pathogenicity island phenotype. Infect Immun 2001;69:5001–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steller H. Mechanisms and genes of cellular suicide. Science 1995;267:1445–9. [DOI] [PubMed] [Google Scholar]
- 17.Cohen GM. Caspases: the executioners of apoptosis. Biochem J 1997;326:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salvesen GS, Dixit VM. Caspases: intracellular signaling by proteolysis. Cell 1997;91:443–6. [DOI] [PubMed] [Google Scholar]
- 19.Wolter KG, Hsu YT, Smith CL, et al. Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol 1997;139:1281–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Srinivasula SM, Fernandes-Alnemri T, Zangrilli J, et al. The Ced-3/interleukin 1beta converting enzyme-like homolog Mch6 and the lamin-cleaving enzyme Mch2alpha are substrates for the apoptotic mediator CPP32. J Biol Chem 1996;271:27099–106. [DOI] [PubMed] [Google Scholar]
- 21.Simbulan-Rosenthal CM, Rosenthal DS, Iyer S, et al. Transient poly(ADP-ribosyl)ation of nuclear proteins and role of poly(ADP-ribose) polymerase in the early stages of apoptosis. J Biol Chem 1998;273:13703–12. [DOI] [PubMed] [Google Scholar]
- 22.Green DR, Amarante-Mendes GP. The point of no return: mitochondria, caspases, and the commitment to cell death. Results Probl Cell Differ 1998;24:45–61. [DOI] [PubMed] [Google Scholar]
- 23.Green DR, Reed JC. Mitochondria and apoptosis. Science 1998;281:1309–12. [DOI] [PubMed] [Google Scholar]
- 24.Enari M, Sakahira H, Yokoyama H, et al. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 1998;391:43–50. [DOI] [PubMed] [Google Scholar]
- 25.Sakahira H, Enari M, Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature 1998;391:96–9. [DOI] [PubMed] [Google Scholar]
- 26.Green DR. Apoptotic pathways: paper wraps stone blunts scissors. Cell 2000;102:1–4. [DOI] [PubMed] [Google Scholar]
- 27.Patterson SD, Spahr CS, Daugas E, et al. Mass spectrometric identification of proteins released from mitochondria undergoing permeability transition. Cell Death Differ 2000;7:137–44. [DOI] [PubMed] [Google Scholar]
- 28.Yang J, Liu X, Bhalla K, et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked [see comments]. Science 1997;275:1129–32. [DOI] [PubMed] [Google Scholar]
- 29.Nagahara Y, Ikekita M, Shinomiya T. Immunosuppressant FTY720 induces apoptosis by direct induction of permeability transition and release of cytochrome c from mitochondria [In Process Citation]. J Immunol 2000;165:3250–9. [DOI] [PubMed] [Google Scholar]
- 30.Momand J, Zambetti GP. Mdm-2: “big brother” of p53. J Cell Biochem 1997;64:343–52. [PubMed] [Google Scholar]
- 31.Lee BM, Jang JJ, Kim HS. Benzo[a]pyrene diol-epoxide-I-DNA and oxidative DNA adducts associated with gastric adenocarcinoma. Cancer Lett 1998;125:61–8. [DOI] [PubMed] [Google Scholar]
- 32.Zou H, Henzel WJ, Liu X, et al. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3 [see comments]. Cell 1997;90:405–13. [DOI] [PubMed] [Google Scholar]
- 33.Ashktorab H, Frank S, Khaled AR, et al. Bax translocation and mitochondrial fragmentation induced by Helicobacter pylori. Gut 2004;53:805–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Potthoff A, Ledig S, Martin J, et al. Significance of the caspase family in Helicobacter pylori induced gastric epithelial apoptosis. Helicobacter 2002;7:367–77. [DOI] [PubMed] [Google Scholar]
- 35.Peek RM Jr., Blaser MJ, Mays DJ, et al. Helicobacter pylori strain-specific genotypes and modulation of the gastric epithelial cell cycle. Cancer Res 1999;59:6124–31. [PubMed] [Google Scholar]
- 36.Ashktorab H, Neapolitano M, Bomma C, et al. In vivo and in vitro activation of caspase-8 and −3 associated with Helicobacter pylori infection. Microbes Infect 2002;4:713–22. [DOI] [PubMed] [Google Scholar]
- 37.Diaz GD, Li Q, Dashwood RH. Caspase-8 and apoptosis-inducing factor mediate a cytochrome c-independent pathway of apoptosis in human colon cancer cells induced by the dietary phytochemical chlorophyllin. Cancer Res 2003;63:1254–61. [PubMed] [Google Scholar]
- 38.Ashktorab H, Ahmed A, Littleton G, et al. p53 and p14 increase sensitivity of gastric cells to H. pylori-induced apoptosis. Dig Dis Sci 2003;48:1284–91. [DOI] [PubMed] [Google Scholar]
- 39.Basak C, Pathak SK, Bhattacharyya A, et al. The secreted peptidyl prolyl cis,trans-isomerase HP0175 of Helicobacter pylori induces apoptosis of gastric epithelial cells in a TLR4-and apoptosis signal-regulating kinase 1-dependent manner. J Immunol 2005;174:5672–80. [DOI] [PubMed] [Google Scholar]
- 40.Houghton J, Korah RM, Condon MR, et al. Apoptosis in Helicobacter pylori-associated gastric and duodenal ulcer disease is mediated via the Fas antigen pathway. Dig Dis Sci 1999;44:465–78. [DOI] [PubMed] [Google Scholar]
- 41.Houghton J, Macera-Bloch LS, Harrison L, et al. Tumor necrosis factor alpha and interleukin 1beta up-regulate gastric mucosal Fas antigen expression in Helicobacter pylori infection. Infect Immun 2000;68:1189–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rudi J, Kuck D, Strand S, et al. Involvement of the CD95 (APO-1/Fas) receptor and ligand system in Helicobacter pylori-induced gastric epithelial apoptosis. J Clin Invest 1998;102:1506–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Galmiche A, Rassow J, Doye A, et al. The N-terminal 34 kDa fragment of Helicobacter pylori vacuolating cytotoxin targets mitochondria and induces cytochrome c release. Embo J 2000;19:6361–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maeda S, Yoshida H, Mitsuno Y, et al. Analysis of apoptotic and antiapoptotic signalling pathways induced by Helicobacter pylori. Gut 2002;50:771–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu X, Zou H, Slaughter C, et al. DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell 1997;89:175–84. [DOI] [PubMed] [Google Scholar]
- 46.Smaili SS, Hsu YT, Sanders KM, et al. Bax translocation to mitochondria subsequent to a rapid loss of mitochondrial membrane potential. Cell Death Differ 2001;8:909–20. [DOI] [PubMed] [Google Scholar]
- 47.Desagher S, Martinou JC. Mitochondria as the central control point of apoptosis. Trends Cell Biol 2000;10:369–77. [DOI] [PubMed] [Google Scholar]
- 48.Obst B, Wagner S, Sewing KF, et al. Helicobacter pylori causes DNA damage in gastric epithelial cells. Carcinogenesis 2000;21:1111–5. [PubMed] [Google Scholar]
- 49.Smoot DT, Elliott TB, Verspaget HW, et al. Influence of helicobacter pylori on reactive oxygen-induced gastric epithelial cell injury. Carcinogenesis 2000;21:2091–5. [DOI] [PubMed] [Google Scholar]
- 50.Ruiz-Vela A, Gonzalez de Buitrago G, Martinez AC. Nuclear Apaf-1 and cytochrome c redistribution following stress-induced apoptosis. FEBS Lett 2002;517:133–8. [DOI] [PubMed] [Google Scholar]
- 51.Shibayama K, Doi Y, Shibata N, et al. Apoptotic signaling pathway activated by Helicobacter pylori infection and increase of apoptosis-inducing activity under serum-starved conditions. Infect Immun 2001;69:3181–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fan T, Lu H, Hu H, et al. Inhibition of apoptosis in chlamydia-infected cells: blockade of mitochondrial cytochrome c release and caspase activation. J Exp Med 1998;187:487–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Clifton DR, Goss RA, Sahni SK, et al. NF-kappa B-dependent inhibition of apoptosis is essential for host cellsurvival during Rickettsia rickettsii infection. Proc Natl Acad Sci U S A 1998;95:4646–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakagawa I, Nakata M, Kawabata S, et al. Cytochrome c-mediated caspase-9 activation triggers apoptosis in Streptococcus pyogenes-infected epithelial cells. Cell Microbiol 2001;3:395–405. [DOI] [PubMed] [Google Scholar]
- 55.Basu A, Johnson DE, Woolard MD. Potentiation of tumor necrosis factor-alpha-induced cell death by rottlerin through a cytochrome-C-independent pathway. Exp Cell Res 2002;278:209–14. [DOI] [PubMed] [Google Scholar]
- 56.Daugas E, Susin SA, Zamzami N, et al. Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. Faseb J 2000;14:729–39. [PubMed] [Google Scholar]
- 57.Wei MC, Lindsten T, Mootha VK, et al. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev 2000;14:2060–71. [PMC free article] [PubMed] [Google Scholar]
- 58.Grinberg M, Sarig R, Zaltsman Y, et al. tBID Homooligomerizes in the mitochondrial membrane to induce apoptosis. J Biol Chem 2002;277:12237–45. [DOI] [PubMed] [Google Scholar]
- 59.Dashwood RH, Xu M, Orner GA, et al. Colonic cell proliferation, apoptosis and aberrant crypt foci development in rats given 2-amino-3-methylimidaz. Eur J Cancer Prev 2001;10:139–45. [DOI] [PubMed] [Google Scholar]
- 60.Hague A, Manning AM, Hanlon KA, et al. Sodium butyrate induces apoptosis in human colonic tumour cell lines in a p53-independent pathway: implications for the possible role of dietary fibre in the prevention of large-bowel cancer. Int J Cancer 1993;55:498–505. [DOI] [PubMed] [Google Scholar]
- 61.Lu J, Imamura K, Nomura S, et al. Chemopreventive effect of peroxisome proliferator-activated receptor gamma on gastric carcinogenesis in mice. Cancer Res 2005;65:4769–74. [DOI] [PubMed] [Google Scholar]
- 62.Leung WK, Bai AH, Chan VY, et al. Effect of peroxisome proliferator activated receptor gamma ligands on growth and gene expression profiles of gastric cancer cells. Gut 2004;53:331–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lipkin M. Gastrointestinal cancer: pathogenesis, risk factors and the development of intermediate biomarkers for chemoprevention studies. J Cell Biochem Suppl 1992;16G:1–13. [DOI] [PubMed] [Google Scholar]