

Abstract
Bacterial biofilms are surface-attached communities of slow-growing and non-replicating persister cells that demonstrate high levels of antibiotic tolerance. Biofilms occur in nearly 80% of infections and present unique challenges to our current arsenal of antibiotic therapies, all of which were initially discovered for their abilities to target rapidly-dividing, free-floating planktonic bacteria. Bacterial biofilms are credited as the underlying cause of chronic and recurring bacterial infections. Innovative approaches are required to identify new small molecules that operate through bacterial growth-independent mechanisms to effectively eradicate biofilms. One source of inspiration comes from within the lungs of young Cystic Fibrosis (CF) patients, who often endure persistent Staphylococcus aureus infections. As these CF patients age, Pseudomonas aeruginosa co-infects the lungs and utilize phenazine antibiotics to eradicate the established S. aureus infection. Our group has taken a special interest in this microbial competition strategy and we are investigating the potential of phenazine antibiotic-inspired compounds and synthetic analogues thereof to eradicate persistent bacterial biofilms. To discover new biofilm-eradicating agents, we have established an interdisciplinary research program involving synthetic medicinal chemistry, microbiology and molecular biology. From these efforts, we have identified a series of halogenated phenazines (HP) that potently eradicate bacterial biofilms and future work aims to translate these preliminary findings into groundbreaking clinical advances for the treatment of persistent biofilm infections.
Keywords: bacterial biofilms, halogenated phenazines, drug discovery, biofilm-eradicating agents, chronic bacterial infection
Graphical Abstract
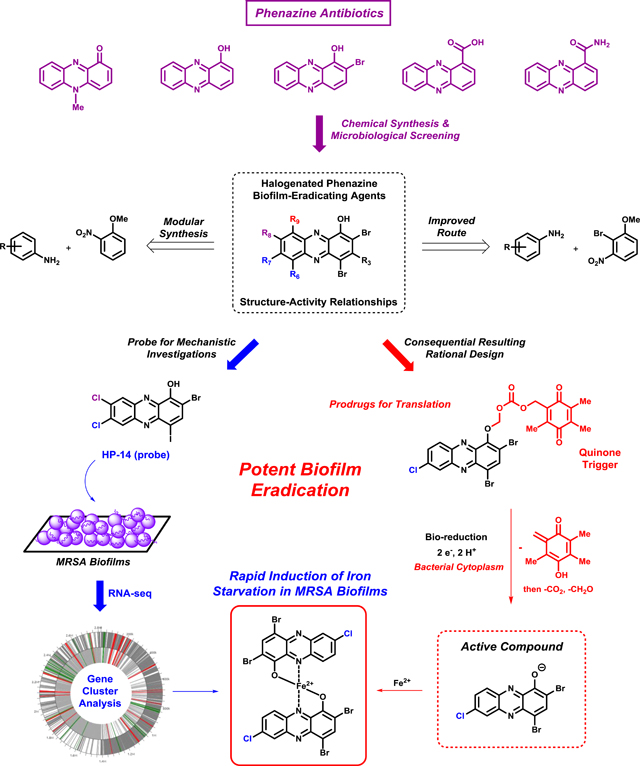
Inspired by phenazine antibiotic natural products, our group has discovered and developed a series of tunable halogenated phenazines that demonstrate potent biofilm eradication activities. Recently, one halogenated phenazine was used as a probe molecule in mechanistic studies in MRSA biofilms using RNA-seq technology, and ongoing efforts are directed at prodrug development for translational purposes.
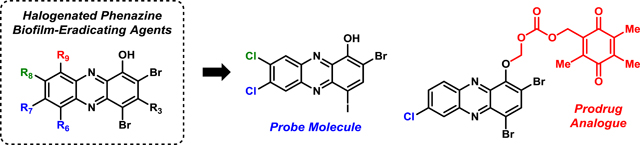
Frontispiece.
1. Introduction
We are facing significant clinical challenges regarding bacterial infections and our current antibiotic arsenal, which include: (1) acquired antibiotic resistance1–4, (2) innate antibiotic tolerance5–10, and (3) a lack of novel molecular scaffolds for antibacterial development.11–13 Each of these problems have been amplified as a result of pharmaceutical companies either downsizing or eliminating antibacterial programs due to failed discovery campaigns and limited investment returns.14,15 Despite a diminished antibacterial pipeline, groundbreaking innovations are needed to address the growing problems associated with bacterial infections.16,17
Bacteria acquire antibiotic resistance through one, or more, well-characterized mechanisms in response to selective pressures resulting from antibiotic treatments.1,3,18 Utilizing various tactics to neutralize the activities of antibacterial treatments, bacteria are known to avoid the effects of antibiotics through the following mechanisms: mutations (that lead to modifications to the antibiotic targets, decreased drug uptake, activation of efflux mechanisms, or global changes in important metabolic pathways of regulatory networks), horizontal gene transfer of mobile genetic elements bearing antibiotic resistant genes (i.e., plasmids), modification of antibiotic drugs (“deactivation” via chemical modification, or destruction), decreased antibiotic penetration and efflux, changes in target site (i.e., target protection) and resistance due to global cell adaption.18
Non-replicating bacteria (NRB) demonstrate antibiotic tolerance, which is a distinct biological phenomena from antibiotic resistance.19,20 A small percentage of free-floating planktonic cells enter into a quiescent, non-dividing state and are known as “persister” cells. These persister cells are able to survive antibiotic treatments after extensive exposure due to their reduced metabolisms and are able to restart growth following stress.5–7,21,22 As such, persister cells create significant clinical challenges in the treatment of infection. Surface-attached bacterial communities, known as “biofilms”, are encased within an extracellular polymeric substance23–26 (EPS, a matrix of biomolecules; Figure 1) and have enriched persister cell populations. Bacterial biofilms are credited as the underlying cause of chronic and recurring infections20–22,27 due to their ability to thrive in hostile environments by persisting through the onslaught of insults from host immune responses (to clear bacterial infection) and aggressive antibiotic treatments.28–34
Figure 1.

Free-floating planktonic bacteria and surface-attached biofilms enriched with persister, or non-dividing, bacterial communities. These bacterial phenotypes are associated with antibiotic resistance (planktonic) and antibiotic tolerance (biofilms with enriched persister cell populations).
It is important to note that each class of clinically used antibiotic was initially discovered as bacterial growth-inhibiting agents that target rapidly-dividing planktonic cells.35 As a result, conventional antibiotics are ineffective against bacterial biofilms enriched in non-replicating persister cells. This is cause for great concern as biofilms occur in ~80% of all bacterial infections and result in >500,000 deaths each year.35–37 These surface-attached communities of bacteria play a critical role in medicine and infectious disease, specific examples include: hospital-acquired infections (e.g., Staphylococcal infections)38, endocarditis39,40, immunocompromised patients41, periodontitis40, osteomyelitis39,40, skin/burn wounds42–44, implanted medical devices (i.e., heart valve45, prosthetic joint46,47), Cystic Fibrosis (chronic lung infections39,40), catheter-related infections40,48–51 (e.g., urinary catheters40, central venous catheters50, cerebrospinal fluid shunts51) and caries (tooth decay52).
With the many success stories of microbial warfare agents (antibiotics from fungus and bacteria) have had in bringing new antibiotics to the clinic53, our group reasoned that biofilm-eradicating natural products exist; however, they have not yet been harnessed for clinical applications.35 Innovative approaches to identify biofilm-eradicating agents are desperately needed to address the critical problems associated with chronic and persistent bacterial infections. This review provides some general background information regarding different types of anti-biofilm small molecules and corresponding activity profiles to orient the reader. However, we present a focused, in-depth review regarding the inspiration for investigating phenazine antibiotics54,55 and synthetic analogues regarding the potential to eradicate biofilms using medicinal chemistry strategies. The main contents this review covers include: inspiration and initial discovery of halogenated phenazines with potent antibacterial activities, chemical synthesis strategies regarding new halogenated phenazine analogues, antibacterial activity profiles, structure-activity relationships, prodrug efforts and RNA-seq investigations to determine the mechanism of action for HP-14 (halogenated phenazine analogue 14), a potent biofilm-eradicating agent. For those interested to learn more about bacterial biofilm inhibitors and dispersal agents, we recommend outstanding reviews by Melander37,56, Sintim57,58 and Wuest.59 In addition, for those interested in viewing comprehensive reports on biofilm-eradicating or persister cell killing agents, we recommend recently published reviews by our group35, Wood7, Hancock60 and Michiels.61
2. “Anti-biofilm Molecules”: Biofilm Inhibitors, Dispersal Agents and Biofilm-Eradicating Agents
It is important to understand the different types of “anti-biofilm” agents along with their activity profiles. Biofilm inhibitors, biofilm dispersal agents and biofilm-eradicating agents have different activities due to their modes of action. This section provides a brief overview to differentiate “anti-biofilm” activities. Select examples of different anti-biofilm compounds (biofilm inhibitors, dispersal agents and eradicating agents) will be presented for illustrative purposes and context.
Bassler and co-worker’s pioneering studies have demonstrated that bacteria utilize small organic signaling molecules, also referred to as autoinducers, in a complex communication system known as quorum sensing.62–67 Bacteria synthesize and secrete distinct signaling molecules into their surrounding environment, which facilitates communication (sensing) and enables the coordination of group behaviors, including biofilm formation.37,68–70 Quorum sensing systems utilize species-specific autoinducers; however, cross-talk between different kinds of bacteria also occurs.65,66,68–70 For excellent in-depth reviews on quorum sensing and small molecule modulators of bacterial communication, we recommend outstanding reviews by Bassler65,66,68,69, Blackwell71,72 and Spring.73,74
Quorum sensing pathways have been of interest regarding the identification of bacterial biofilm inhibitors (compounds that inhibit bacterial attachment to a surface and subsequent growth/development) and dispersal agents (compounds that promote the dispersion, or release, of planktonic cells from mature biofilms).73–79 Biofilm inhibitors and dispersal agents do not inhibit, or alter, bacterial growth and avoid placing a selective pressure on bacteria to develop resistance. Prominent classes of biofilm inhibitors and/or dispersal agents include: brominated furanones (derived from marine red algae; targeting LuxR signaling protein)73,74,76–78, homoserine lactones (quorum sensing mimics in Gram-negative bacteria)72–75 and 2-aminoimidazoles (derived from 2-aminoimidazole-pyrrole marine natural products37,56,80–83; target response regulator proteins that govern biofilm formation and antibiotic resistance84–86) (Figure 2). There are several methods to assess biofilm inhibitors and dispersal agents; however, crystal violet staining of bacterial biofilms (see Figure 4B) formed on the surfaces of 96-well plates has been widely used in combination with bacterial growth assessments (e.g., growth curve analysis; minimum inhibitory concentration assays for MIC value assessment) to ensure that biofilm inhibitors and dispersal agents do not inhibit planktonic growth.80,81
Figure 2.

Select classes of well-characterized quorum sensing modulators, biofilm inhibitors and biofilm dispersal agents, including: (A) brominated furanones, (B) homoserine lactones, and (C) 2-aminoimidazole analogues. These compounds operate through mechanisms that do not alter bacterial growth or kill bacteria.
Figure 4.

In vitro assays that enable the study of (A) antibacterial, (B) biofilm inhibition and dispersion, and (C) biofilm eradication activities of various small molecules. (D) Representative in vivo biofilm infection models that have been developed in animals. Bioluminescently engineered bacteria allow for real-time imaging and monitoring of biofilm during infections. Note: All microtiter assays (A-C) are shown with increasing concentrations of compound from left to right.
Unlike biofilm inhibitors and dispersal agents, biofilm-eradicating agents operate through mechanisms that kill bacterial biofilms and persister cells (Figure 3).35 With the clinical problems associated with bacterial biofilms, there is a significant interest for small molecules that can eradicate persistent biofilms7,35,60,61 and have the potential to be utilized as stand-alone clinical agents. Biofilm inhibitors and dispersal agents can be used as adjuvants and require combination therapy with antibiotics to treat biofilm infections.
Figure 3.

Select persister cells and bacterial biofilm-eradicating agents are presented and include: (A) quaternary ammonium cations, NH125 analogue and CD437 (membrane-lysing, or disrupting agents), (B) mitomycin C (DNA alkylating/crosslinking agent), and (C) ADEP-4, a ClpP-activating agent. These agents operate through mechanisms that kill and eradicate bacterial biofilms (and planktonic cells).
Membrane-lysing agents, including quaternary ammonium cations (QACs), have been shown to rapidly kill persister cells and eradicate bacterial biofilms (Figure 3A).87–92 Typically, potent QAC biofilm-eradicating agents demonstrate hemolysis, or the lysis of red blood cells, which is a concern for therapeutic development.87,91,92 However, there is hope for QAC compounds as therapeutic agents as these compounds operate through mechanisms in a similar manner to antimicrobial peptides (AMPs), which are naturally occurring membrane-active antimicrobial agents utilized by the mammalian immune response to eradicate microorganisms during infection.93 Despite the challenges of developing effective membrane-disrupting agents that selectively target bacterial membranes over mammalian membranes, progress has been made in this area.94,95 A new series of synthetic retinoid antibiotics, including CD437 (Figure 3A), was recently reported to effectively eradicate MRSA persister cells through selective bacterial membrane disruption in animal models.95
In addition to membrane disruptors, DNA and ClpP protease have proven to be promising biological targets for biofilm eradication (Figure 3B & C). Mitomycin C, a DNA crosslinking agent with a history of clinical use for cancer therapy96, has also been shown to eradicate persister cells and bacterial biofilms against a broad-spectrum of pathogens.9,97 ADEP4, a ClpP protease-activating agent, has also demonstrated promising persister cell killing activities and Staphylococcus aureus biofilm eradication in a mouse model in combination with rifampin.98
3. In vitro and In vivo Assessment of Biofilm-Eradicating Agents
Antibacterial agents are typically evaluated in vitro using minimum inhibitory concentration (MIC) assays (Figure 4A), which provide information regarding the growth-inhibitory activities of a compound towards rapidly-dividing planktonic cultures. MIC assays are operationally simple and can be carried out in 96-well plates to provide antibacterial potencies of multiple compounds following overnight incubation (16–24 hours). MIC assays do not provide information regarding biofilm eradication activities and alternative assays are required to establish bacterial biofilms on surfaces before compound treatment. Biofilm inhibition and dispersion assays typically involve crystal violet staining of bacterial biofilms following a predetermined incubation period and wash of microtiter wells (Figure 4B). Crystal violet staining is a common method used to quantify the amount of biofilm biomass formed, or remaining, in a microtiter well following compound treatment.80–82
Biofilm-eradicating agents are often evaluated in vitro using 96-well plate assays (e.g., standard assay plates87,99, or Calgary Biofilm Devices100–102) involving three distinct phases. The phases of this assay are separated by a removal of planktonic cells and media, followed by a gentle wash of surface-attached biofilms formed on surfaces. The three biofilm-eradication assay phases include: (1) biofilm establishment phase (inoculated media allow bacterial biofilms to establish on assay plate surfaces), (2) compound test phase (test compounds in media are added to established biofilms, allowing for biofilm eradication), and (3) biofilm recovery phase (fresh media only added to biofilms; viable biofilms will grow and disperse planktonic cells into media, which will lead to viable bacterial populations that give a final turbid readout in a microtiter well; eradicated biofilms will give a non-turbid readout in a microtiter well; Figure 4C).
We have found the Calgary Biofilm Device (CBD) to be a superior assay for obtaining minimum biofilm eradication concentration (MBEC) values for test compounds.100,103–105 CBD assays allow the simultaneous assessment of a test compound’s planktonic (minimum bactericidal concentration, MBC) and biofilm cell eradication from a single bacterial colony, or overnight culture of bacteria. CBD pegs can be removed from their specialized lid, sonicated and plated out to determine the colony forming units (CFU) of viable biofilm cells. From our investigations with methicillin-resistant S. aureus and S. epidermidis (MRSA, MRSE) biofilm-eradicating agents, we have determined that there is a ≥ 3-log reduction of biofilm cells (eradication of ≥ 99.9% biofilm cells) at the corresponding MBEC values of active biofilm-eradicating agents.100,104,105
There are several interesting in vivo biofilm infection models98,106–113, which can serve as important tools for determining the efficacy of new biofilm-eradicating agents during pre-clinical investigations (Figure 4D). Recently, Lewis and co-workers reported the in vivo activities of an ADEP4/rifampin combination in a deep-seated mouse thigh (biofilm) infection.98 In this chronic infection model, mice were made neutropenic by treatment with cyclophosphamide before a large dose of Staphylococcus aureus was delivered and allowed to develop biofilms for 24 hours, leading to a severe, deep-seated infection, which emulates difficult to treat immunocompromised patients. Histopathological assessment of the infected thigh revealed S. aureus biofilms adhered to muscle cells during these studies. Following the establishment of S. aureus biofilms in mice, an ADEP4/rifampin combination treatment led to complete sterilization of the biofilm infection (tissue) to the limit of detection whereas vancomycin was without effect following 24 and 48 hours of treatment. This deep-seated biofilm model was critical to establishing the clinical potential of activating the ClpP protease (ADEP4) for the treatment of chronic bacterial infections.
Bioluminescently engineered bacteria can be used for real-time imaging in mice, allowing for continuous monitoring of bacterial infections in live mouse models.112 A range of both Gram-positive and Gram-negative bacteria have been bioluminescently transformed and tested in vivo using standard animal models to establish this platform for basic research and drug development efforts. During initial efforts, acute bacterial infection models (soft tissue, sepsis, pneumonia) were the primary focus; however, investigators quickly realized that bioluminescently transformed bacterial could be highly useful for studying chronic, biofilm-associated infections.110–113 Kadurugamuwa and co-workers reported the use of bioluminescent Pseudomonas aeruginosa (Xen 5) and S. aureus (Xen 29) colonized Teflon catheters (103 to 105 colony forming units) that were implanted at subcutaneous sites in mice.111 In this model, growth of the biofilm was monitored by detecting photon emission over 20 days using an IVIS spectrum for bioluminescence imaging. This approach is particularly appealing as viable biofilms are monitored using a non-destructive and non-invasive method to allow detailed studies regarding chronic biofilm infections and efficacy studies regarding investigational therapeutics.
4. Phenazine Antibiotics: Inspiration for the Discovery of New Biofilm-Eradicating Agents
Young Cystic Fibrosis (CF) patients are often afflicted with persistent S. aureus lung infections.114–116 We believe these infections to be biofilm-associated as CF patients endure chronic lung infections that persist for years. As these CF patients age, Pseudomonas aeruginosa co-infects their lungs and eradicates the established S. aureus infection using a diverse array of redox-active phenazine antibiotic compounds that demonstrate antimicrobial activities.54,55 P. aeruginosa secretes high concentrations of the phenazine antibiotic pyocyanin114, a deep blue pigment, which is believed to be the primary eradicating agent of S. aureus in CF patient’s lungs. Interested by the competition strategy P. aeruginosa utilizes to eradicate S. aureus in the lung, our group was poised to explore phenazine antibiotics and synthetic analogues to determine if these compounds could target and eradicate biofilms.
Initially, we scanned the literature for routes capable of synthesizing electronically diverse phenazines with the aim of tuning redox properties of the phenazine heterocycle to act and kill bacterial biofilms. Interestingly, we found few modular synthetic routes to phenazine compounds; however, we were able to find reports that enabled the rapid synthesis of pyocyanin and other naturally occurring phenazine antibiotics, including 2-bromo-1-hydroxyphenazine.117 Our initial efforts enabled the rapid synthesis of 13 phenazines, including 5 naturally occurring and 8 synthetic phenazines for antibacterial assessment against S. aureus and S. epidermidis.118
4.1. Halogenated Phenazines: A New Series of Potent Antibacterial Agents that Eradicate Biofilms
Our initial collection of 13 phenazine compounds was evaluated for antibacterial activities against S. aureus and S. epidermidis. We were interested to find that pyocyanin demonstrated only moderate antibacterial activities (minimum inhibitory concentration, MIC = 50 μM), while structurally related analogue 1-hydroxyphenazine demonstrated no antibacterial activities against S. aureus or S. epidermidis (MIC > 100 μM).118 In addition, we were delighted to see that marine-derived 2-bromo-1-hydroxyphenazine (2-Br-1-OHPhz, MIC = 6.25 μM; Figure 5A) demonstrated an 8-fold increase in antibacterial potencies compared to pyocyanin. We also found synthetic analogue 2,4-dibromo-1-hydroxyphenazine (2,4-diBr-1-OHPhz, MIC = 1.56 μM against S. aureus) to demonstrate a 4-fold enhancement of antibacterial activities compared to the marine phenazine 2-Br-1-OHPhz.
Figure 5.

(A) Select analogues from initial work that led to the discovery of 2,4-diBr-1-OHPhz as an potent antibacterial agent. (B) Initial structure-activity relationship analysis of 2,4-diBr-1-OHPhz, which motivated the investigation of additional synthetic analogues of the HP scaffold.
The discovery of 2,4-diBr-1-OHPhz was followed up with analogue synthesis to probe the necessity of the 1-hydroxyl group.118 Substitutions with methoxy and amine groups led to completely inactive analogues (MIC > 100 μM, highest test concentration; Figure 5A) against Staphylococcus, which told us that the 1-hydroxyl group was critical for the antibacterial activity of 2,4-diBr-1-OHPhz. We also identified several ester analogues at the phenolic hydroxyl group of 2,4-diBr-1-OHPhz to be active in antibacterial assays, which we attributed to prodrug activation through ester cleavage (hydrolysis). These initial studies established 2,4-diBr-1-OHPhz as the minimal structural motif, or scaffold, required for potent antibacterial activity against S. aureus and S. epidermidis. We termed this focused series of compounds “halogenated phenazines” (HP). In addition, 2,4-diBr-1-OHPhz was found to have no cytotoxicity against HeLa cells at 100 μM, demonstrating efficient bacterial targeting by this HP.103
Following the identification of 2,4-diBr-1-OHPhz, we advanced this HP to biofilm eradication assays to determine if this small molecule was capable of killing surface-attached bacterial communities. Using Calgary Biofilm Device assays, we observed 2,4-diBr-1-OHPhz to eradicate methicillin-resistant S. aureus (MRSA; MBEC = 150 μM), methicillin-resistant S. epidermidis (MRSE; MBEC = 100 μM) and vancomycin-resistant Enterococcus faecium (VRE; MBEC = 9.38 μM) biofilms.100 In addition, 2,4-diBr-1-OHPhz demonstrates no hemolysis activity at 200 μM (highest test concentration), confirming that this HP was not eradicating biofilms through a membrane-lysis mechanism, similar to QAC compounds.100,103 Positive biofilm eradication without hemolytic, or membrane-lysing, activities provides some initial mechanistic insights and is ideal for the development of new therapeutic agents.100,103–105
Multiple comparator agents with known antibacterial modes of action have been investigated in biofilm eradication assays alongside HPs.100,103–105 QAC-10, a known biofilm-eradicating agent87 that operates through membrane lysis, reported an MBEC of 93.8 μM against MRSA-1707 (along with >99% hemolysis at 200 μM). In addition, front-running MRSA treatments vancomycin, daptomycin and linezolid were unable to eradicate MRSA-1707 biofilms (MBEC >2,000 μM) when tested alongside 2,4-diBr-1-OHPhz and QAC-10 (note: planktonic MRSA-1707 cells are susceptible to each of these conventional antibiotics).
4.2. Chemical Syntheses and Activity Profiles of Halogenated Phenazine Small Molecules
Once we found 2,4-diBr-1-OHPhz to eradicate biofilms, our team was motivated to explore the 6–9 positions of the HP scaffold to determine the biological impact various substituents at these positions have on antibacterial activities (Figure 5B).100,103–105 We were primarily interested to learn if these positions could improve biofilm eradication potencies of HP compounds. To investigate the 6–9 positions on the HP scaffold, we developed modular synthetic pathways using various (retrosynthetic analysis) disconnection strategies through the central ring of the phenazine heterocycle. This approach allowed us to generate a diversity of 1-methoxyphenazines differentially substituted at the 6–9 positions (Figure 6). Once in hand, 1-methoxyphenazines were subjected to a two-step sequence involving an initial boron tribromide (BBr3) demethylation step, then a final dibromination step using two equivalents of N-bromosuccinimide (NBS) to yield target HP analogues.100,103–105
Figure 6.

Overview of modular synthesis strategies that have been developed to synthesize diverse HP biofilm-eradicating agents.
Initial synthetic efforts involved a chloranil oxidation of 3-methoxycatechol to the corresponding quinone, which was immediately taken forward and condensed with a 4,5-disubstituted phenylenediamine in acetic acid to yield 7,8-disubstituted 1-methoxyphenazines (synthetic details not shown).103,104 The resulting 1-methoxyphenazine intermediates were then (1) demethylated using boron tribromide and (2) dibrominated using N-bromosuccinimide to generate 7,8-disubstutited halogenated phenazines for antibacterial studies. During these studies, our final bromination reaction proved troublesome and we modified the route to (1) selectively mono-halogenate the 4-position of the 1-methoxyphenazine scaffold, then (2) demethylate using boron tribromide to generate phenolic intermediates and (3) final mono-halogenation of the 2-position of the HP scaffold. This modified synthetic route enabled access to mixed halogenations at the 2- and 4-positions of HP. Although this route enabled valuable insights regarding 7,8-disubstituted HPs, significant limitations included: (1) relatively few phenylenediamines are readily available and (2) the 4,5-substitutents of the phenylenediamine starting materials had to be equivalent to avoid issues with regioisomeric products upon condensation to 1-methoxyphenazines.103,104
Despite these limitations, 7,8-dihalogenated HPs proved to be significantly more active in antibacterial and biofilm eradication assays compared to unsubstituted HP analogues at the 7,8-positions.103,104 In particular, HP-14 was found to demonstrate potent biofilm eradication activities against three MRSA isolates (MBEC = 6.25–9.38 μM), MRSE 35984 (MBEC = 2.35 μM) and VRE 700221 (MBEC = 0.20 μM; Figure 7 & 9). At the corresponding MBEC values, HP-14 was found to eradicate ≥99.9% MRSA and MRSE biofilm cells in addition to demonstrating slow (kinetic) killing of stationary MRSA-2 cultures at 12.5 μM (≥99.9% eradication of stationary cells after 24 hours; vancomycin proved to be inactive against stationary MRSA-2 cultures at 100 μM, 100 x MIC value).103 Despite the enhanced antibacterial and biofilm-killing activities of 7,8-disubstituted HPs, these compounds continued to show encouraging bacterial targeting as minimal HeLa cytotoxicity and hemolysis were observed (Figure 7).
Figure 7.

(A) Activity profiles of select halogenated phenazine biofilm-eradicating agents. (B) Identification of metal(II)-binding moiety involved in the mode of action for halogenated phenazine antibacterial agents.
Figure 9.

Select HP analogues with improved eradication activities against MRSA, MRSE and VRE biofilms.
During these investigations, we demonstrated that active HPs bind metal(II) cations (e.g., copper(II), iron(II) cations) using UV-vis spectroscopy.104 In addition, antibacterial activities of HPs were shown to be reduced or eliminated upon co-treatment with various metal(II) cations in MIC assays. HPs bind metal(II) cations through chelation that involves the 1-hydroxyl oxygen atom and adjacent nitrogen of the phenazine, which forms a stable 5-membered chelate (see Figure 7B). Several attempts have been made to investigate the involvement of reactive oxygen species (ROS) in the antibacterial activities displayed by HPs, similar to what is proposed for pyocyanin and other phenazine antibiotics; however, no experimental evidence to date has shown that HPs elicit antibacterial activities through redox activities.104
A second-generation synthesis utilized diverse aniline building blocks that were incorporated into the HP scaffold via a Wohl-Aue reaction to probe substituents in the 6–9 positions (Figure 6).105 The Wohl-Aue reaction is a base-promoted condensation reaction between an aniline and nitroarene to afford a phenazine.119 During these studies, 2-nitroanisole and 2-nitro-4-methylanisole were condensed with a diverse series of anilines using potassium hydroxide in refluxing toluene. These harsh conditions led to very low yields in the Wohl-Aue reaction (<10% average yield); however, we were able to synthesize enough material to advance through the (1) demethylation, (2) dibromination sequence to afford 20 new HPs for biological investigations. This approach enabled the synthesis of 6- and 8-mono-substituted HPs and 6,7- and 6,8-disubstituted HPs. From this library, we found several 6-substituted HPs (e.g., methyl, ethyl, chlorine, bromine) to be highly potent in antibacterial (MRSA-1707, MIC = 0.05–0.30 μM) and biofilm eradication (MRSA-1707, MBEC = 4.69–75 μM) assays. Additionally, 8-halogenated HPs (e.g., chlorine, bromine) demonstrated improved antibacterial activities compared to the unsubstituted parent HP; however, 8-methyl or 8-phenoxy HPs were found to be less active than the parent HP scaffold. Despite the identification of highly active HP analogues, the Wohl-Aue route requires significant improvements.
A third-generation, modular HP synthesis was developed utilizing a (1) palladium-catalyzed Buchwald-Hartwig cross coupling between anilines and 2-bromo-3-nitroanisole, followed by a (2) base-promoted reductive cyclization to afford 1-methoxyphenazine analogues (Figure 8).100 The initial Buchwald-Hartwig step utilized 6 mol% bis(dibenzylideneacetone)palladium(0) and enabled the coupling of 21 anilines with 2-bromo-3-nitroanisole to afford the desired nitroarene intermediates in 21–94% yield (70% average yield). The following base-promoted reductive cyclization step was performed using sodium borohydride and sodium ethoxide in ethanol to yield 1-methoxyphenazines in 15–97% (68% average yield). Treatment with BBr3 (89% average yield), then N-bromosuccinimide (NBS; 71% average yield) enabled the conversion of 1-methoxyphenazine intermediates to 18 target HPs for biological evaluation. This Buchwald-Hartwig/reductive cyclization pathway was dramatically more efficient and high yielding than our initial synthetic efforts. In addition, this chemistry enabled biological investigations of 7- and 9-substituted HPs, including several di- and tri-substituted HPs at positions 6–9 of the HP scaffold.
Figure 8.

An efficient Buchwald-Hartwig/reductive cyclization route to diverse HPs for biological investigations. Antibacterial activity for select HP analogues is against MRSA-1707.
From this series of HPs, we identified several highly potent biofilm-eradicating HPs that are substituted at the 7-position of the HP scaffold, including 7-phenoxy HP (MIC = 0.1 μM; MBEC = 2.35 μM against MRSA-1707) and 7-chloro HP (MIC = 0.1 μM, MBEC = 4.69 μM against MRSA-1707) (Figure 8).100 Interestingly, both 9-substituted HPs synthesized during these investigations were completely inactive against MRSA-1707 in MIC assays (>1,000-fold less potent than the 7-substituted HPs, Figure 8). We subjected 9-substituted HP analogues (9-methyl, 9-ethyl) to metal(II) binding experiments using UV-vis spectroscopy; however, neither HP was able to bind metal(II) cations, likely due to the methyl and ethyl substituents at the 9-position sterically disrupting metal-chelation. This important structure-activity relationship information provided further support that metal(II) binding was critical for the mode of action of HP biofilm-eradicating agents.
From our collective synthetic efforts aimed at probing the 6–9 positions of the HP scaffold, we have identified several new HPs with significantly enhanced biofilm-eradication activities against MRSA isolates, MRSE 35984 and VRE 700221 (Figure 9).100,103–105 When compared to the parent HP (2,4-diBr-1-OHPhz; MBEC = 150 μM), potent HP analogues mono-substituted at the 6- or 7-position of the HP scaffold have demonstrated 32- to 64-fold enhancements in biofilm-eradicating potencies against MRSA biofilms (MBEC = 2.35–4.69 μM). Against MRSE 35984 biofilms, 7,8-dichlorinated HP (bearing a 4-iodine; HP-14) and 6-ethyl HP reported a 43-fold enhancement of biofilm eradication activity (MBEC = 2.35 μM) compared to parent HP.103–105 VRE 700221 biofilms show enhanced overall sensitivities toward HPs (parent HP, MBEC = 9.38 μM); however, we have seen a 47-fold enhancement in biofilm eradication activities by HP-14 (MBEC = 0.20 μM) and a 24-fold enhancement with 7,8-dibromo HP analogue (MBEC = 0.39 μM).103,104
4.3. Structure Activity Relationships and Prodrug Design of Halogenated Phenazines
In addition to identifying more active biofilm-eradicating HPs through a diverse set of synthetic pathways, we established a detailed structure activity relationship for this exciting series of compounds (Figure 10A).100,103–105 Several mono-substituted analogues demonstrate significantly increased antibacterial and biofilm-killing properties through simple modifications at the 6- (R6)105 and/or 7-position (R7)100 of the HP scaffold. Alkyl groups (e.g., methyl, ethyl), chlorine or bromine at R6 significantly enhances antibacterial activities of the HP scaffold (≥24-fold enhancement of biofilm eradication activity against MRSA) compared to the parent HP. In a similar fashion, an alkyl group (e.g., methyl, ethyl, tert-butyl), chlorine or phenyl ether at R7 enhances antibacterial activities compared to the parent HP (≥32-fold enhancement of biofilm eradication activity against MRSA). In addition, 6,7-dimethylated and 6,7,8-trimethylated HPs demonstrate significantly enhanced antibacterial and biofilm eradication activities when compared to parent HP.
Figure 10.

Structure-activity relationships of halogenated phenazine biofilm-eradicating agents that informs the rational design of future analogues, including prodrug versions of potent HPs.
Mono-substitution at the 8-position (R8; Figure 10A) of the HP scaffold has led to analogues with both increased and decreased antibacterial activities.104,105 Mono-chlorinated or brominated HP at R8 results in HPs with increased antibacterial activities; however, methyl and phenyl ether analogues at R8 have resulted in HPs with decreased antibacterial activities compared to parent HP. Additional information is needed to make more general conclusions regarding 8-substituted analogues, but the increased CLogP values for the 8-halogenated HPs could enhance bacterial membrane penetration of these analogues to elicit their antibacterial activities. All substitutions of the HP at the 9-position (R9 = Me, Et, Br) has led to inactive analogues. As previously mentioned, the loss of antibacterial activity regarding 9-substituted HPs is likely due to steric interactions disrupting a critical metal(II)-chelation event required for antibacterial activity.100,105
With extensive SAR information in hand, we employed a series of diverse prodrugs designed to inactivate the critical metal-chelating moiety of the HP scaffold, to avoid off-target metal chelation and potential toxicity, until removal under particular conditions to liberate an active HP.100,105 Functionalization of the phenolic hydroxyl group of the HP scaffold leads to analogues that are unable to bind metal(II) cations; however, several analogues are active antibacterial agents due to the cleavage of prodrug moieties and liberation of active HP agent, which then elicits antibacterial activities. We have been able to develop various carbonate analogues containing PEG moieties aimed to enhance water solubility (Figure 10B). The carbonate prodrug analogues are designed to be cleaved by bacterial esterases and demonstrate significantly improved antibacterial potencies compared to the non-prodrug HP versions; however, biofilm-eradication activities of HP carbonate prodrugs are near equipotent as their corresponding HP. The carbonate HP prodrugs are highly potent antibacterial agents and we will continue to investigate these compounds; however, we feel that these carbonate prodrugs may have limitations due to non-specific esterase cleavage in a therapeutic context (i.e., cleavage from both mammalian and bacterial esterases) and alternative strategies are under investigation.
We designed an alternative prodrug strategy that relies on the reductive cytoplasm of bacterial cells for activation of a quinone moiety, which leads to unstable intermediates that will subsequently fragment and release an active HP molecule.100 This strategy is inspired by the natural product mitomycin C (Figure 3), which undergoes an initial bio-reduction of its quinone before DNA-crosslinking can occur.35,96 For our HP scaffold, we designed QuAOCOM (quinone alkyloxycarbonyloxymethyl; AOCOM prodrugs have been previously reported120,121) prodrugs that are appended to the phenolic oxygen to eliminate off-target metal-chelation, similar to carbonate HP prodrugs. We selected the electron-deficient quinone moiety since it is known to undergo a two-electron bio-reduction to an electron-rich hydroquinone in the reductive bacterial cytoplasm (Figure 11).9 Upon reduction of the quinone, the hydroquinone is primed to release an ortho quinone fragment, resulting in an unstable carbonic acid that spontaneously decarboxylates (- CO2) and finally deformylates (- CH2O) to yield the active HP molecule. Initial results for HP QuAOCOM analogues are promising as these prodrugs are active in antibacterial assays with minimal losses of antibacterial activities compared to the corresponding non-prodrug HP version. We attribute any slight loss in antibacterial activities to the time needed for processing the QuAOCOM prodrug moiety to liberate the active HP.
Figure 11.

Bio-reductive activation of HP QuAOCOM prodrugs with subsequent release of the active HP scaffold.
At this time, multiple prodrug strategies are being pursued to develop HPs for translational purposes. Additional studies are needed to determine the clinical applicability of various HP prodrugs; however, we are encouraged by initial results.100,105 Bringing an effective biofilm-eradicating agent to the clinic would be game changing in the way chronic and recurring bacterial infections are treated.
4.4. Identification of Halogenated Phenazines with Anti-Tuberculosis Activity
Mycobacterium tuberculosis (MtB) is a slow-growing bacterial pathogen122,123 and the leading killer of adults worldwide, causing >1.5 million deaths in 2017.124,125 MtB’s persistent nature, along with antibiotic resistance, has led to significant challenges regarding effective treatment options for individuals with TB infections. We have evaluated select HP analogues for antibacterial activity against MtB. We believed that HPs would have good antibacterial activities against MtB since bacterial biofilms are extremely slow or non-replicating bacterial communities.
From our series of HPs, several have demonstrated good antibacterial activities against MtB H37Ra.100,103–105 The parent HP demonstrated lower antibacterial activity (MIC = 25 μM; Figure 12) compared to various analogues with substituents at the 6–8 positions of the HP scaffold. Interestingly, several of the most potent HP analogues against MtB contain one, or more, chlorine atoms in the 6–8 positions (MIC = 3.13 μM; 1–2 μg/mL). In addition, we did note two MtB-active HPs do not have the 2-position halogenated, which is likely offset by the chlorine atoms in the 6- and 7-position(s). Multiple animal models can determine the efficacy of investigational drugs for MtB infections126, including lung infection models that deliver test compounds via aerosolized formulations.127,128 Collectively, HPs could serve as great starting points for new treatment options against persistent MtB infections.
Figure 12.

Halogenated phenazine analogues that have demonstrated antibacterial activities against the slow-growing, persistent human pathogen Mycobacterium tuberculosis (MtB).
4.5. Transcriptome Analysis of MRSA Biofilms Treated with HP-14
Transcriptome analysis of MRSA-1707 biofilms treated with HP-14 was performed using RNA-seq129 to define the mechanism of action for this class of biofilm-eradicating agents and establish cellular targets and pathways critical for biofilm survival.130 To do this, we treated established MRSA biofilms with potent biofilm-eradicating agent HP-14 at sub-MBEC concentrations (1/10 MBEC; 0.625 μM) for 20 hours. After that time, we isolated total RNA (from HP-14 treated and vehicle-treated MRSA biofilms) and performed RNA-sequencing to identify MRSA biofilm gene transcripts differentially expressed (up- and down-regulated) due to HP-14 treatment (Figure 13).
Figure 13.

(A) Work flow using HP-14 as a probe molecule in transcriptome analysis of MRSA biofilms. (B) Heatmap of MRSA biofilm transcripts that are up-regulated and down-regulated resulting from HP-14 treatment. (C) WoPPER analysis identifies six iron uptake gene clusters that HP-14 activates in MRSA biofilms.
From >2,700 MRSA-1707 biofilm gene transcripts analyzed using RNA-seq technology, 83 transcripts were down-regulated and 134 transcripts were up-regulated in response to HP-14.130 With >200 gene transcripts altered from HP-14 treatment, we utilized the WoPPER analysis tool131 to focus attention on gene cluster activation or inhibition. We found this approach to streamline our efforts as 37 gene clusters were either up- or down-regulated from HP-14 treatment, including six activated gene clusters involved in iron uptake (Figure 13). Real-time qPCR (RT-qPCR) experiments were carried out to validate initial WoPPER findings, confirming up-regulation of isd (iron-regulated surface determinant; heme iron acquisition), sbn (staphyloferrin B; siderophore), sfa (staphyloferrin A; siderophore), MW0695 (hypothetical protein, similar to ferrichrome ABC transporters), fhu (ferric hydroxamate uptake), spl (serine proteases), opp (oligopeptide transporters), and down-regulation of gap (glycolysis), arc (arginine deiminase), ure (urease), hem (heme biosynthesis), dnaC (DNA synthesis and repair).
A time-course assessment using RT-qPCR of MRSA biofilms treated with HP-14 (1/10 MBEC; 0.625 μM) revealed that four iron acquisition gene clusters (isd, sbn, sfa, MW0695) were activated in 1 hour.130 In addition, HP-14 treated MRSA biofilms displayed higher levels of activation regarding these iron acquisition gene clusters at 4 and 8 hours. Interestingly, metal-chelating agents TPEN and ETDA were unable to activate iron acquisition genes when tested alongside HP-14, aligning with our previous results demonstrating that these agents were unable to eradicate MRSA biofilms at high concentrations (EDTA & TPEN, MBEC >2,000 μM). We found the rapid activation of multiple iron uptake genes upon HP-14 treatment to be profound as biofilms are notoriously dormant. These findings demonstrate that although metabolically dormant, bacterial biofilms are highly sensitive and possess the ability to rapidly respond to threats to enable survival.
It is essential that bacteria acquire iron(III) from their environment132–135; however, iron(III) is unable to passively diffuse through bacterial membranes. Bacteria synthesize and secrete siderophore molecules with high binding affinities into their surrounding environment to sequester iron(III). Upon binding, the siderophore-iron(III) complex is recognized by protein transporters on the surface of bacterial cells, which deliver the siderophore-iron(III) complex into the bacterial cell. Once in the bacterial cell, iron(III) is reduced to iron(II) by the bacteria’s reductive cytoplasm. Upon reduction, the siderophore loses affinity and releases iron(II) for the bacterium to utilize in multiple functions, including: respiration, DNA biosynthesis and metalloprotein function.135
Based on our collective data, we believe HP-14 (CLogP = 6.25) rapidly diffuses through bacterial cells and directly binds iron(II), following its release from a siderophore or heme. The binding of iron(II) by HP-14 results in the rapid starvation of iron in MRSA biofilm cells, leading to the activation of iron uptake systems and eventual death (Figure 14).130 This mechanism is distinct from simple metal sequestration as EDTA and TPEN are unable to eradicate biofilms and activate iron uptake systems when tested alongside HP-14. We believe that HP-14 readily diffuses into biofilm cells, whereas EDTA and TPEN do not penetrate biofilms. These findings suggests that bacterial biofilms are highly sensitive and their viability relies on iron homeostasis. Future work aims to exploit this unique, iron starvation activity of HP small molecules for the eradication of persistent biofilms in the clinic.
Figure 14.

Model for the primary mechanism of action of biofilm eradication through rapid iron starvation by HP-14. WoPPER analysis identified additional gene clusters involved in the response of MRSA biofilms to HP-14; however, these are downstream to iron starvation.
5. Future Perspectives and Conclusions
Our work related to phenazine antibiotics has led to the identification of a series of halogenated phenazine small molecules that demonstrate potent biofilm-eradicating activities against multiple Gram-positive bacteria, including MRSA, MRSE and VRE. The modular chemical syntheses that have been employed during these studies have been critical in establishing activity profiles and structure activity relationships for halogenated phenazine antibacterial agents; however, additional synthesis efforts are ongoing and will be reported in due course. We have utilized the halogenated phenazine as inspiration to scaffold hop to an extensive parallel library of halogenated quinoline biofilm-eradicating agents136–140, which were not presented in this review, but enhance the translational potential of these compounds. HP-14 has been a valuable tool molecule for establishing the mode of action of HP biofilm-eradicating agents while defining critical iron acquisition targets and pathways involved in MRSA biofilm survival. Various prodrug strategies and animal studies are currently underway for lead HPs as we aim to establish the translational potential for these exciting compounds for biofilm infections and tuberculosis. Halogenated phenazines could have a significant impact on the treatment of chronic and recurring bacterial infections and we are motivated to optimize these compounds for the eradication of persistent, life-threatening infections to improve human health.
Acknowledgements
We thank the University of Florida for start-up funds to support initial work to establish our halogenated phenazine platform. We would also like to thank the National Institute of General Medical Sciences of the National Institutes of Health for providing financial support (R35GM128621 to RWH) related to halogenated phenazine biofilm-eradicating agents and probe molecules.
Biographical Sketch.

Robert Huigens received a B.A. in biology from UNC-Greensboro in 2003. He then completed a Ph.D. in chemistry with Prof. Christian Melander at N.C. State University in 2009. Dr. Huigens then moved to the University of Illinois of Urbana-Champaign as an American Cancer Society Postdoctoral Fellow under the guidance of Prof. Paul Hergenrother. In 2013, Prof. Huigens began his independent career as an assistant professor of medicinal chemistry at the University of Florida. The Huigens lab has drug discovery programs inspired by natural products that aim to: (1) identify bacterial biofilm-eradicating agents, and (2) rapidly generate complex and diverse compounds from available indole alkaloids.

Yasmeen Abouelhassan received a B.S. in Pharmaceutical Sciences from Cairo University, Egypt in 2012. She then went on to obtain a Ph.D. at the University of Florida in Medicinal Chemistry with Prof. Robert Huigens in 2018. Dr. Abouelhassan’s graduate research focused on the identification and microbiological investigations of novel biofilm-eradicating agents. Her work led to the discovery of a class of diverse and synthetically tunable halogenated phenazines, inspired by phenazine antibiotics, that demonstrate potent biofilm eradication activities. Dr. Abouelhassan’s work also utilized RNA-seq to determine HP-14 induced rapid iron starvation in methicillin-resistant S. aureus biofilms.

Hongfen Yang received a B.S. in chemistry from Hubei University, China in 2008. She then completed a M.S. in bioorganic chemistry under the guidance of Professor Wu at the University of Chinese Academy of Sciences in Beijing. Currently, Hongfen is a graduate student in the Medicinal Chemistry Department at the University of Florida under the supervision of Professor Huigens. Her doctoral work involves the design, synthesis and microbiological investigations of novel halogenated phenazine small molecules.
Footnotes
Conflicts of Interest
The authors declare no conflicts of interest.
References
- 1.Lewis K, Nat. Rev. Drug Discov. 2013, 12, 371–387. [DOI] [PubMed] [Google Scholar]
- 2.Ventola CL, P T. 2015, 40, 277–283. [PMC free article] [PubMed] [Google Scholar]
- 3.Rossiter SE, Fletcher MH, Wuest WM, Chem. Rev. 2017, 117, 12415–12474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Waglechner N, Wright GD, BMC Biol. 2017, 15, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lewis K, Nat. Rev. Microbiol. 2007, 5, 48–56. [DOI] [PubMed] [Google Scholar]
- 6.Lewis K, Annu. Rev. Microbiol. 2010, 64, 357–372. [DOI] [PubMed] [Google Scholar]
- 7.Wood TK, Biotechnol. Bioengineer. 2016, 113, 476–483. [DOI] [PubMed] [Google Scholar]
- 8.Conlon BP, Bioessays. 2014, 36, 991–996. [DOI] [PubMed] [Google Scholar]
- 9.Kwan BW, Chowdhury N, Wood TK, Environ. Microbiol. 2015, 17, 4406–4414. [DOI] [PubMed] [Google Scholar]
- 10.Brauner A, Fridman O, Gefen O, Balaban NQ, Nat. Rev. Microbiol. 2016, 14, 320–330. [DOI] [PubMed] [Google Scholar]
- 11.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL, Nat. Rev. Drug Discov. 2007, 6, 29–40. [DOI] [PubMed] [Google Scholar]
- 12.Payne DJ, Miller LF, Findlay D, Anderson J, Marks L, Phil. Trans. R. Soc. 2015, 370, 20140086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tommasi R, Brown DG, Walkup GK, Manchester JI, Miller AA, Nat. Rev. Drug Discov. 2015, 14, 529–542. [DOI] [PubMed] [Google Scholar]
- 14.Spellberg B, Crit. Care. 2014, 18, 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harbarth S, Theuretzbacher U, Hackett J, J. Antimicrob. Chemother. 2015, 70, 1604–1607. [DOI] [PubMed] [Google Scholar]
- 16.Kmietowicz Z, BMJ. 2017, 358, j4339. [Google Scholar]
- 17.Theuretzbacher U, Savic M, Outterson K, Nat. Rev. Drug Discov. 2017, 16, 744–745. [DOI] [PubMed] [Google Scholar]
- 18.Munita JM, Arias CA, Microbiol. Spectr. 2016, 4, VMBF-0016–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lewis K, Handb. Exp. Pharmacol. 2012, 211, 121–133. [DOI] [PubMed] [Google Scholar]
- 20.Olsen I, Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 877–886. [DOI] [PubMed] [Google Scholar]
- 21.Wood TK, Knabel SJ, Kwan BW, Appl. Environ. Microbiol. 2013, 79, 7116–7121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fisher RA, Gollan B, Helaine S, Nat. Rev. Microbiol. 2017, 15, 453–464. [DOI] [PubMed] [Google Scholar]
- 23.Fux CA, Costerton JW, Stweart PS, Stoodley P, Trends Microbiol. 2005, 13, 34–40. [DOI] [PubMed] [Google Scholar]
- 24.Flemming H-C, Neu TR, Wozniak DJ, J. Bacteriol. 2007, 189, 7945–7947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flemming H-C, Wingender J, Szewzyk U, Steinberg P, Rice SA, Kjelleberg S, Nat. Rev. Microbiol. 2016, 14, 563–575. [DOI] [PubMed] [Google Scholar]
- 26.Koo H, Allan RN, Howlin RP, Stoodley P, Hall-Stoodley L, Nat. Rev. Microbiol. 2017, 15, 740–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lewis K, Biochemistry (Moscow) 2005, 70, 267–274. [DOI] [PubMed] [Google Scholar]
- 28.Donlan RM, Clin. Inf. Dis. 2001, 33, 1387–1392. [DOI] [PubMed] [Google Scholar]
- 29.Donlan RM, Costerton JW, Clin. Microbiol. Rev. 2002, 15, 167–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hall-Stoodley L, Costerton JW, Stoodley P, Nat. Rev. Microbiol. 2004, 2, 95–108. [DOI] [PubMed] [Google Scholar]
- 31.Musk DJ, Hergenrother PJ, Curr. Med. Chem. 2006, 13, 2163–2177. [DOI] [PubMed] [Google Scholar]
- 32.Stewart PS, Davison WM, Steenbergen JN, Antimicrob. Agents Chemother. 2009, 53, 3505–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.López D, Vlamakis H, Kolter R, Cold Spring Harb. Perspect. Biol. 2010, 2, a000398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oubekka SD, Briandet R, Fontaine-Aupart M-P, Steenkeste K, Antimicrob. Agents Chemother. 2012, 56, 3349–3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garrison AT, Huigens RW III, Curr. Top. Med. Chem. 2017, 17, 1954–1964. [Google Scholar]
- 36.Wolcott R, Dowd S, Plast. Reconstr. Surg. 2011, 127, 28S–35S. [DOI] [PubMed] [Google Scholar]
- 37.Worthington RJ, Richards JJ, Melander C, Org. Biomol. Chem. 2012, 10, 7457–7474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Otto M, Curr. Top. Microbiol. Immunol. 2008, 322, 207–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paharik AE, Horswill AR, Microbiol. Spectr. 2016, 4, VMBF-0022–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gupta P, Sarkar S, Das B, Bhattacharjee S, Tribedi P, Arch. Microbiol. 2016, 198, 1–15. [DOI] [PubMed] [Google Scholar]
- 41.Weisser M, Schoenfelder SMK, Osasch C, Arber C, Gratwohl A, Frei R, Eckart M, Flückiger U, Ziebuhr W, J. Clin. Microbiol. 2010, 48, 2407–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Halstead FD, Rauf M, Moiemen NS, Bamford A, Wearn CM, Fraise AP, Lund PA, Oppenheim BA, Webber MA, PLoS One 2015, 10, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Phillips PL, Yang Q, Schultz GS, Int. Wound J. 2013, 10, 48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ganesh K, Sinha M, Mathew-Steiner SS, Das A, Roy S, Sen CK, Adv. Wound Care 2015, 4, 382–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim MK, Drescher K, Pak OS, Bassler BL, Stone HA, New J Phys. 2014, 16, 065024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peel TN, Buising KL, Choong PF, Curr. Opin. Infect. Dis. 2012, 25, 670–676. [DOI] [PubMed] [Google Scholar]
- 47.Fernández J, Greenwood-Quaintance KE, Patel R, Diagn. Microbiol. Infect. Dis. 2016, 85, 449–451. [DOI] [PubMed] [Google Scholar]
- 48.Heim CE, Hanke ML, Kielian T, Methods Mol. Biol. 2014, 1106, 183–191. [DOI] [PubMed] [Google Scholar]
- 49.Percival SL, Suleman L, Vuotto C, Donelli G, J. Med. Microbiol. 2015, 64, 323–334. [DOI] [PubMed] [Google Scholar]
- 50.Chauhan A, Ghigo JM, Beloin C, Nat. Protoc. 2016, 11, 525–541. [DOI] [PubMed] [Google Scholar]
- 51.Fux CA, Quigley M, Worel AM, Post C, Zimmerli S, Ehrlich G, Veeh RH, Clin. Microbiol. Infect. 2006, 12, 331–337. [DOI] [PubMed] [Google Scholar]
- 52.Marsh PD, Compend. Contin. Educ. Dent. 2009, 30, 76–78. [PubMed] [Google Scholar]
- 53.Brown ED, Wright GD, Nature 2016, 529, 336–343. [DOI] [PubMed] [Google Scholar]
- 54.Laursen JB, Nielsen J, Chem. Rev. 2004, 104, 1663–1685. [DOI] [PubMed] [Google Scholar]
- 55.Price-Whelan A, Dietrich LEP, Newman DK, Nat. Chem. Biol. 2006, 2, 71–78. [DOI] [PubMed] [Google Scholar]
- 56.Worthington RJ, Richards JJ, Melander C, Anti-Infective Agents 2014, 12, 120–138. [Google Scholar]
- 57.Rabin N, Zheng Y, Opoku-Temeng C, Du Y, Bonsu E, Sintim HO, Future Med. Chem. 2015, 7, 493–512. [DOI] [PubMed] [Google Scholar]
- 58.Rabin N, Zheng Y, Opoku-Temeng C, Du Y, Bonsu E, Sintim HO, Future Med. Chem. 2015, 7, 647–671. [DOI] [PubMed] [Google Scholar]
- 59.Fletcher MH, Jennings MC, Wuest WM, Tetrahedron 2014, 70, 6373–6383. [Google Scholar]
- 60.Wolfmeier H, Pletzer D, Mansour SC, Hancock REW, ACS Inf. Dis. 2018, 4, 93–106. [DOI] [PubMed] [Google Scholar]
- 61.Defraine V, Fauvart M, Michiels J, Drug Resist. Updat. 2018, 38, 12–26. [DOI] [PubMed] [Google Scholar]
- 62.Bassler BL, Greenberg EP, Stevens AM, J. Bacteriol. 1997, 179, 4043–4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen X, Schauder S, Potier N, Van Dorsselaer A, Pelczer I, Bassler BL, Hughson FM, Nature 2002, 415, 545–549. [DOI] [PubMed] [Google Scholar]
- 64.Zhu J, Miller MB, Vance RE, Dziejman M, Bassler BL, Mekalanos JJ, Proc. Natl. Acad. Sci. USA 2002, 99, 3129–3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bassler BL, Cell. 2002, 109, 421–424. [DOI] [PubMed] [Google Scholar]
- 66.Taga ME, Bassler BL, Proc. Natl. Acad. Sci. USA. 2003, 100, 14549–14554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mukherjee S, Moustafa DA, Stergioula V, Smith CD, Goldberg JB, Bassler BL, Proc. Natl. Acad. Sci. USA 2018,115, E9411–E9418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ng WL, Bassler BL, Annu. Rev. Genet. 2009, 43, 197–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Papenfort K, Bassler BL, Nat. Rev. Microbiol. 2016, 14, 576–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Eickhoff MJ, Bassler BL, Cell. 2018, 174, 1328–1328.e1. [DOI] [PubMed] [Google Scholar]
- 71.Praneenararat T, Palmer AG, Blackwell HE, Org. Biomol. Chem. 2012, 10, 8189–8199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Welsh MA, Blackwell HE, FEMS Microbiol. Rev. 2016, 40, 774–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Galloway WRJD, Hodgkinson JT, Bowden SD, Welch M, Spring DR, Chem. Rev. 2011, 111, 28–67. [DOI] [PubMed] [Google Scholar]
- 74.Galloway WRJD, Hodgkinson JT, Bowden SD, Welch M, Spring DR, Trends Microbiol. 2012, 20, 449–458. [DOI] [PubMed] [Google Scholar]
- 75.Geske GD, Wezeman RJ, Siegel AP, Blackwell HE, J. Am. Chem. Soc. 2005, 127, 12762–12763. [DOI] [PubMed] [Google Scholar]
- 76.Hentzer M, Givskov M, J. Clin. Invest. 2003, 112, 1300–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hentzer M, Riedel K, Rasmussen TB, Heydorn A, Andersen JB, Parsek MR, Rice SA, Eberl L, Molin S, Høiby N, Kjelleberg S, Givskov M, Microbiol. 2002, 148, 87–102. [DOI] [PubMed] [Google Scholar]
- 78.Wu H, Song Z, Hentzer M, Andersen JB, Molin S, Givskov M, Høiby N, J. Antimicrob. Chemother. 2004, 53, 1054–1061. [DOI] [PubMed] [Google Scholar]
- 79.Rémy B, Mion S, Plener L, Elias M, Chabriére E, Daudé D, Front. Pharmacol. 2018, 9, 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Huigens RW III, Richards JJ, Parise G, Ballard TE, Zeng W, Deora R, Melander C, J. Am. Chem. Soc. 2007, 129, 6966–6967. [DOI] [PubMed] [Google Scholar]
- 81.Huigens RW III, Ma L, Gambino C, Moeller PDR, Basso A, Cavanagh J, Wozniak DJ, Melander C, Mol. BioSys. 2008, 4, 614–621. [DOI] [PubMed] [Google Scholar]
- 82.Richards JJ, Ballard TE, Huigens RW III, Melander C, ChemBioChem 2008, 9, 1267–1279. [DOI] [PubMed] [Google Scholar]
- 83.Draughn GL, Allen CL, Routh PA, Stone MR, Kirker KR, Boegli L, Schuchman RM, Linder KE, Baynes RE, James G, Melander C, Pollard A, Cavanagh J, Drug Des. Devel. Ther. 2017, 11, 153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Thompson RJ, Bobay BG, Stowe SD, Olson AL, Peng L, Su Z, Actis LA, Melander C, Cavanagh J, Biochemistry 2012, 51, 9776–9778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Milton ME, Allen CL, Feldmann EA, Bobay BG, Jung DK, Stephens MD, Melander RJ, Theisen KE, Zeng D, Thompson RJ, Melander C, Cavanagh J, Mol. Microbiol. 2017, 106, 223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Milton ME, Minrovic BM, Harris DL, Kang B, Jung D, Lewis CP, Thompson RJ, Melander RJ, Zeng D, Melander C, Cavanagh J, Front. Mol. Biosci. 2018, 5, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jennings MC, Ator LE, Paniak TJ, Minibole KPC, Wuest WM, ChemBioChem 2014, 15, 2211–2215. [DOI] [PubMed] [Google Scholar]
- 88.Jennings MC, Minibole KPC, Wuest WM, ACS Infect. Dis. 2015, 1, 288–303. [DOI] [PubMed] [Google Scholar]
- 89.Al-Khalifa SE, Jennings MC, Wuest WM, Minbole KPC, ChemMedChem 2017, 12, 280–283. [DOI] [PubMed] [Google Scholar]
- 90.Kim W, Fricke N, Conery AL, Fuchs BB, Rajamuthiah R, Jayamani E, Vlahovska PM, Ausubel FM, Mylonakis E, Future Med. Chem. 2016, 8, 257–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Abouelhassan Y, Basak A, Yousaf H, Huigens RW III, ChemBioChem 2017, 18, 352–357. [DOI] [PubMed] [Google Scholar]
- 92.Basak A, Abouelhassan Y, Zuo R, Yousaf H, Ding Y, Huigens RW III, Org. Biomol. Chem. 2017, 15, 5503–5512. [DOI] [PubMed] [Google Scholar]
- 93.Brogden KA, Nat. Rev. Microbiol. 2005, 3, 238–250. [DOI] [PubMed] [Google Scholar]
- 94.Hurdle JG, O’Neill AJ, Chopra I, Lee RE RE, Nat. Rev. Microbiol. 2011, 9, 62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim W, Zhu W, Hendricks GL, Van Tyne D, Steele AD, Keohane CE, Fricke N, Conery AL, Shen S, Pan W, Lee K, Rajamuthiah R, Fuchs BB, Vlahovska PM, Wuest WM, Gilmore MS, Gao H, Ausubel FM, Mylonakis E, Nature 2018, 556, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tomasz M. Chem Biol. 1995, 2, 575–579. [DOI] [PubMed] [Google Scholar]
- 97.Cruz-Muñiz MY, López-Jacome LE, Hernández-Durán M, Franco-Cendejas R, Licona-Limón P, Ramos-Balderas JL, Martinéz-Vázquez M, Belmont-Díaz JA, Wood TK, García-Contreras R, Int. J. Antimicrob. Agents. 2017, 49, 88–92. [DOI] [PubMed] [Google Scholar]
- 98.Conlon BP, Nakayasu ES, Fleck LE, Isabella VM, Coleman K, Leonard SN, Smith RD, Adkins JN, Lewis K, Nature 2013, 503, 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Garrison AT, Bai F, Abouelhassan Y, Paciaroni NG, Jin S, Huigens RW III, RSC Adv. 2015, 5, 1120–1124. [Google Scholar]
- 100.Garrison AT, Abouelhassan Y, Kallifidas D, Tan H, Kim YS, Jin S, Luesch H, Huigens RW III, J. Med. Chem. 2018, 61, 3962–3983. [DOI] [PubMed] [Google Scholar]
- 101.Ceri H, Olson ME, Stremick C, Read RR, Morck D, Buret A, J. Clin. Microbiol. 1999, 37, 1771–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Harrison JJ, Stremick CA, Turner RJ, Allan ND, Olson ME, Ceri H, Nat. Protoc. 2010, 5, 1236–1254. [DOI] [PubMed] [Google Scholar]
- 103.Garrison AT, Abouelhassan Y, Kallifidas D, Bai F, Ukhanova M, Mai V, Jin S, Luesch H, Huigens RW III, Angew. Chem. Int. Ed. 2015, 54, 14819–14823. [DOI] [PubMed] [Google Scholar]
- 104.Garrison AT, Abouelhassan Y, Norwood IV VM, Kallifidas D, Bai F, Nguyen M, Rolfe M, Burch GM, Jin S, Luesch H, Huigens RW III, J. Med. Chem. 2016, 59, 3808–3825. [DOI] [PubMed] [Google Scholar]
- 105.Yang H, Abouelhassan Y, Burch GM, Kallifidas D, Huang G, Yousaf H, Jin S, Luesch H, Huigens RW III, Sci. Rep. 2017, 7, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Coenye T, Nelis HJ, J. Microbiol. Methods 2010, 83, 89–105. [DOI] [PubMed] [Google Scholar]
- 107.Lebeaux D, Chauhan A, Rendueles O, Beloin C, Pathogens 2013, 2, 288–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Balaban N, Cirioni O, Giacometti A, Ghiselli R, Braunstein JB, Silvestri C, Mocchegiani F, Saba V, Scalise G, Antimicrob. Agents Chemother. 2007, 51, 2226–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gurjala AN, Geringer MR, Seth AK, Hong SJ, Smeltzer MS, Galiano RD, Leung KP, Mustoe TA, Wound Repair Regen. 2011, 19, 400–410. [DOI] [PubMed] [Google Scholar]
- 110.Kadurugamuwa JL, Sin LV, Yu J, Francis KP, Kimura R, Purchio T, Contag PR, Antimicrob. Agents Chemother. 2003, 47, 3130–3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kadurugamuwa JL, Sin L, Albert E, Yu J, Francis K, DeBoer M, Rubin M, Bellinger-Kawahara C, Parr TR Jr, Contag PR, Infect. Immun. 2003, 71, 882–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kadurugamuwa JL, Francis KP, Methods Mol Biol. 2008, 431, 225–239. [DOI] [PubMed] [Google Scholar]
- 113.Agostinho Hunt AM, Gibson JA, Larrivee CL, O’Reilly S, Navitskaya S, Needle DB, Abramovitch RB, Busik JV, Waters CM, J. Wound Care 2017, 26, S24–S33. [DOI] [PubMed] [Google Scholar]
- 114.Machan ZA, Pitt TL, White W, Watson D, Taylor GW, Cole PJ, Wilson R, J. Med. Microbiol. 1991, 34, 213–217. [DOI] [PubMed] [Google Scholar]
- 115.Stone A, Saiman L, Curr. Opin. Pulm. Med. 2007, 13, 515–521. [DOI] [PubMed] [Google Scholar]
- 116.Junge S, Görlich D, den Reijer M, Wiedemann B, Tümmler B, Ellemunter H, Dübbers A, Küster P, Ballmann M, Koerner-Rettberg C, Große-Onnebrink J, Heuer E, Sextro W, Mainz JG, Hammermann J, Riethmüller J, Graepler-Mainka U, Staab D, Wollschläger B, Szczepanski R, Schuster A, Tegtmeyer FK, Sutharsan S, Wald A, Nofer JR, van Wamel W, Becker K, Peters G, Kahl BC, PLoS One 2016, 11, e0166220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Conda-Sheridan M, Marler L, Park EJ, Kondratyuk TP, Jermihov K, Mesecar AD, Pezzuto JM, Asolkar RN, Fenical W, Cushman M, J. Med. Chem. 2010, 53, 8688–8699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Borrero NV, Bai F, Perez C, Duong BQ, Rocca JR, Jin S, Huigens RW III, Org. Biomol. Chem. 2014, 12, 881–886. [DOI] [PubMed] [Google Scholar]
- 119.Pachter IJ, Kloetzel MC, J. Am. Chem. Soc. 1951, 73, 4958–4961. [Google Scholar]
- 120.Thomas JD, Sloan KB, Int. J. Pharm. 2009, 371, 25–32. [DOI] [PubMed] [Google Scholar]
- 121.Thomas JD, Majumdar S, Sloan KB, Molecules 2009, 14, 4231–4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Young DB, Perkins MD, Duncan K, Barry CE, J. Clin. Invest. 2008, 118, 1255–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Evangelopoulos D, McHugh TD, Chem. Biol. Drug Des. 2015, 86, 951–960. [DOI] [PubMed] [Google Scholar]
- 124.Keshavjee S, Amanullah F, Cattamanchi A, Chaisson R, Dobos KM, Fox GJ, Gendelman HE, Gordon R, Hesseling A, Hoi LV, Kampmann B, Kana B, Khuller G, Lewinsohn DM, Lewinsohn DA, Lin PL, Lu LL, Maartens G, Owen A, Protopopova M, Rengarajan J, Rubin E, Salgame P, Schurr E, Seddon JA, Swindells S, Tobin DM, Udwadia Z, Walzl G, Srinivasan S, Rustomjee R, Nahid P, Am. J. Respir. Crit. Care Med. 2018, in press. doi: 10.1164/rccm.201806-1053PP. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.World Health Organization. Tuberculosis fact sheet. 2018; http://www.who.int/mediacentre/factsheets/fs104/en/ Accessed November 18, 2018.
- 126.Flynn JL, Gideon HP, Mattila JT, Lin PL, Immunol. Rev. 2015, 264, 60–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gonzalez-Juarrero M, Woolhiser LK, Brooks E, DeGroote MA, Lenaerts AJ, Antimicrob. Agents Chemother. 2012, 56, 3957–3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Costa-Gouveia J, Pancani E, Jouny S, Machelart A, Delorme V, Salzano G, Iantomasi R, Piveteau C, Queval CJ, Song OR, Flipo M, Deprez B, Saint-André JP, Hureaux J, Majlessi L, Willand N, Baulard A, Brodin P, Gref R, Sci. Rep. 2017, 7, 5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang Z, Gerstein M, Snyder M, Nat. Rev. Genet. 2009, 10, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Abouelhassan Y, Zhang Y, Jin S, Huigens RW III, Angew. Chem. Int. Ed. 2018, 57, 15523–15528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Puccio S, Grillo G, Licciulli F, Severgnini M, Liuni S, Bicciato S, De Bellis G, Ferrari F, Peano C, Nucleic Acids Res. 2017, 45, W109–W115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hider RC, Kong X, Nat. Prod. Rep. 2010, 27, 637–657. [DOI] [PubMed] [Google Scholar]
- 133.Hammer ND, Skaar EP, Annu. Rev. Microbiol. 2011, 65, 129–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sheldon JR, Heinrichs DE, FEMS Microbiol. Rev. 2015, 39, 592–630. [DOI] [PubMed] [Google Scholar]
- 135.Bilitewski U, Blodgett JAV, Duhme-Klair A-K, Dallavalle S, Laschat S, Routledge A, Schobert R, Angew. Chem. Int. Ed. 2017, 56, 14360–14382. [DOI] [PubMed] [Google Scholar]
- 136.Abouelhassan Y, Garrison AT, Burch GM, Wong W, Norwood IV VM, Huigens RW III, Bioorg. Med. Chem. Lett. 2014, 24, 5076–5080. [DOI] [PubMed] [Google Scholar]
- 137.Basak A, Abouelhassan Y, Huigens RW III, Org. Biomol. Chem. 2015, 13, 10290–10294. [DOI] [PubMed] [Google Scholar]
- 138.Basak A, Abouelhassan Y, Norwood IV VM, Bai F, Nguyen M, Jin S, Huigens RW III, Chem. Eur. J. 2016, 22, 9181–9189. [DOI] [PubMed] [Google Scholar]
- 139.Garrison AT, Abouelhassan Y, Yang H, Yousaf HH, Nguyen T, Huigens RW III, Med. Chem. Commun. 2017, 8, 720–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Basak A, Abouelhassan Y, Kim KS, Norwood IV VM, Jin S, Huigens RW III, Eur. J. Med. Chem. 2018, 155, 705–713. [DOI] [PubMed] [Google Scholar]