

Abstract
Gliomas, the most common malignant primary brain tumours, remain universally lethal. Yet, seminal discoveries in the past 5 years have clarified the anatomy, genetics and function of the immune system within the central nervous system (CNS) and altered the paradigm for successful immunotherapy. The impact of standard therapies on the response to immunotherapy is now better understood, as well. This new knowledge has implications for a broad range of tumours that develop within the CNS. Nevertheless, the requirements for successful therapy remain effective delivery and target specificity, while the dramatic heterogeneity of malignant gliomas at the genetic and immunological levels remains a profound challenge.
Editor’s Summary
This Review discusses how advances in our understanding of the immune system within the brain have implications for the successful implementation of immunotherapy to treat brain tumours, despite challenges such as effective delivery, target specificity and intratumour heterogeneity.
Introduction
Malignant primary brain tumours are a leading cause of cancer mortality in children and young adults, with few therapeutic options. In adults, glioblastomas, the most common primary brain tumours, remain uniformly lethal, with a median survival of <21 months, despite surgical resection, targeted radiation therapy, high-dose chemotherapy and novel approaches such as tumour-treating fields (TTFields)[1, 2, 3, 4, 5, 6, 7, 8]. Despite the early identification of genetic drivers of these tumours, they contain relatively few coding mutations, and their intratumour heterogeneity has been shown to be extraordinary[9, 10]. In addition, unique features of these tumours and of the host organ thwart immunotherapeutic approaches. The immune system in the brain follows different principles from the immune system elsewhere, whereby access to the tumours is limited by the blood–brain barrier (BBB), and the host is subjected to substantial endogenous and treatment-induced immune suppression. Thus, primary brain tumours are devastating diseases, and their unique features are a topic of considerable importance. In this Review, we discuss the immunological underpinnings of brain metastases only briefly (Box 1) but, rather, focus on the specifics of the immune privilege of the brain, the unique microenvironment of malignant glial tumours, and the limitations that the genetic underpinnings and standard therapies used place on the immunotherapeutic approaches and their success. We also summarize the existing immunotherapeutic approaches and biomarkers of response that are currently under evaluation.
Box 1. Brain metastasis and immune checkpoint blockade.
Subversion of endogenous and manipulated immune responses, long a hallmark of glioblastoma, is increasingly recognized among brain metastases, as well (reviewed in Farber et al.[230]). The brain has evolved a variety of mechanisms aimed at curbing inflammatory responses that might otherwise be harmful, limiting both the access and scope of T cell responses. Tumours of the intracranial compartment, in turn, are often able to usurp such mechanisms to restrict the antitumour immune response. Similar to glioblastoma, brain metastases frequently contain fewer T cell infiltrates than their peripherally located counterparts, and T cells that do successfully infiltrate are subject to further suppressive influences geared at promoting such dysfunction as tolerance and exhaustion[150, 231]. Tumour infiltrates within the brain are uniquely heavy in microglia and monocytes, which may function ultimately to dampen the cell-mediated immune response, a function employed by brain metastases[232, 233, 234].
While the systemic and local immunological consequences of CNS metastasis are not characterized as well as in primary brain tumours, current data suggest contributions by many of the same microenvironmental immunosuppressive factors, including regulatory T (Treg) cells[154, 235], and noteworthy expression of programmed cell death protein 1 ligand 1 (PDL1)[236]. Numerous studies have documented T cell ineffectiveness that correlates with the expression of immune checkpoints[150, 160]; however, unlike in the case of glioblastoma, the available evidence suggests that immune checkpoint blockade may proffer significant efficacy against intracranial metastases, particularly those from melanoma[151]. Nevertheless, future immunotherapeutic successes will depend on deepening our understanding of the interactions between metastatic tumours and immune populations within the brain specifically.
Brain tumour immunology is different
Immune privilege
The accessibility of the brain to the afferent and efferent arms of the immune system, and thus to immunotherapy, has been a matter of some debate for decades[11]. Conceptually, the term ‘immune privilege’ refers to the failure of a site to reject heterotopic tissue transplantation. In the case of the brain, these experiments are habitually attributed to Peter Medawar in the 1940s (however, Medawar himself references the earlier work of Shirai, Murphy and Tansley in his landmark 1948 paper)[12]. What is less well remembered of Medawar’s studies is that while homologous skin grafting into brains failed to elicit immunity, skin homografts did indeed succumb to rejection if their implantation followed preliminary grafting of foreign tissue elsewhere in the body. Thus, pre-existing immune states could extend to the brain to promote rejection, but such states could not be evoked by central nervous system (CNS) transplantation de novo. Medawar attributed this observed disparity to the perceived absence of a system for lymphatic drainage, and therefore for afferent immunity within the brain, a perception that has only recently been disproven[13, 14, 15, 16, 17, 18, 19].
In the many years since Medawar’s tissue-grafting experiments, newer techniques have increasingly revealed the CNS immune privilege paradigm to be overstated. Studies highlighting an existent afferent mechanism for CNS participation in regional lymphatics debunked the isolationist idea of the brain as immunologically silenced. For instance, routes of antigenic egress from the brain to the deep cervical lymph nodes (via the arachnoid sheath of the olfactory nerves, through the cribriform plate, and to the nasal mucosa) were uncovered beginning in the 1980s[13, 14, 15, 16]. Identification of the glial–lymphatic (glymphatic) pathway offered a mechanism for fluid and solute clearance from the brain, linking the parenchyma and interstitium to the cerebrospinal fluid (CSF) spaces: CSF flows into the brain along arterial perivascular spaces and then translocates into the interstitium via aquaporin 4 (AQP4) water channels, before exiting along venous perivascular spaces[20, 21]. Since 2015, the discovery of functional lymphatic vessels in the meninges has provided a direct drainage pathway for CSF containing solute and immune cells from the brain into the cervical lymph nodes[17, 18, 19].
Given these provisions for an operative afferent immune system, many have proposed that the brain is immunologically ‘distinct’ rather than ‘privileged’[22] (Fig. 1). Such notions are bolstered by newer paradigms of efferent CNS immunity, as well. Challenges for the efferent arm of CNS immunity have historically been ascribed to the BBB[23]. Formed by capillary tight junctions and the astrocytic glia limitans[24, 25], the BBB serves as a structural barrier to the passive transit of molecules between the brain and the systemic circulation, permitting immune cell passage only at the level of the post-capillary venules[26]. Ultimately, the CNS parenchyma was believed to encourage only limited immunity, as direct antigenic challenges instigated a modest immune infiltration when compared with systemic injections[27]. However, newer paradigms have documented T cell entry and immunosurveillance within the brain[26, 28, 29], disproving the notion that the BBB acts as a ‘hermetic seal’ to immune cell entry. Equally, damage incurred to the BBB in the context of gliomas and other tumours limits the restrictions normally proffered by the BBB[30, 31, 32].
Fig. 1. Immune privilege in the brain.
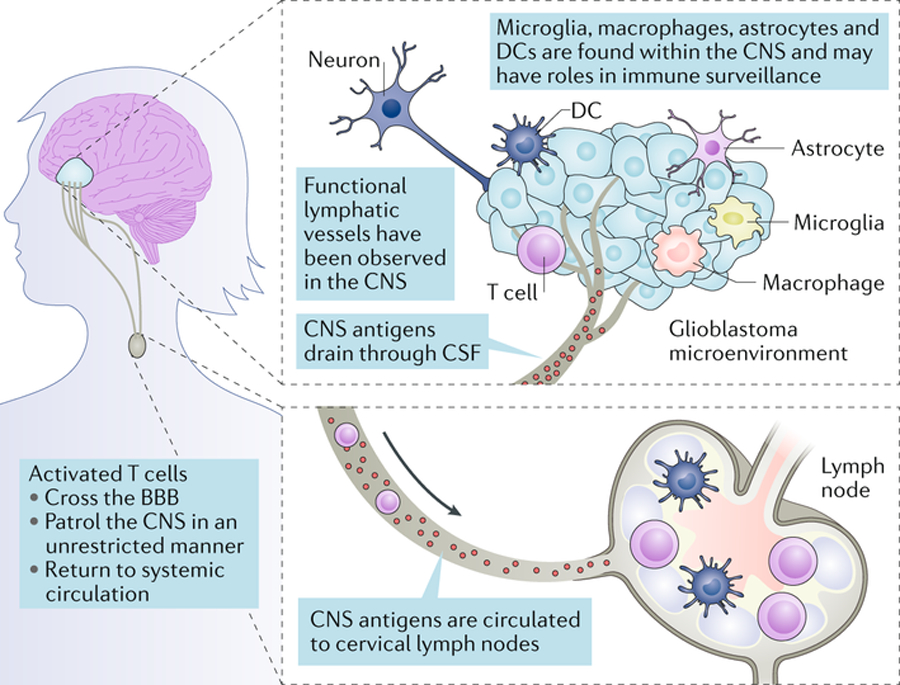
Historically, the central nervous system (CNS) was considered to be isolated immunologically. Features that contributed to this understanding were the presence of tight junctions in the blood–brain barrier, the absence of a classic lymphatic drainage system, and empirical data showing the ability of the CNS to target foreign tissues with minimal inflammatory responses. However, today the concept of immune privilege has been partially redefined. It is now clear that there are functional lymphatic vessels in the CNS, and that antigen-presenting cells (APCs) of varied types exist within the CNS, including microglia, macrophages, astrocytes and classic APCs such as dendritic cells (DCs). It is now known that the CNS is not isolated from activated T cells, which can patrol these compartments in an unrestricted manner, and that CNS antigens can be presented locally or in the draining cervical lymph nodes. While the immune system in the CNS remains different, it is not incapable. CSF, cerebrospinal fluid.
Another unique yet dynamic aspect of the brain immune environment is its population of resident myeloid cells[33]. At baseline, this population is composed almost entirely of microglia, the brain’s equivalent of tissue-resident macrophages. Microglia arise from yolk sac myeloid progenitors that seed neural tissues during development and are maintained through replication[34]. In the absence of inflammation, microglia maintain their yolk sac origin throughout life, with no contribution from bone marrow-derived cells. After an inflammatory stimulus, microglia undergo substantial phenotypic changes, while additional macrophage populations are recruited from circulating monocytes[35]. Brain environmental cues influence the phenotype and activity of both microglia and recruited macrophages; however, the functions of these resident and recruited myeloid populations appear to differ in ways we do not yet fully understand[36].
Despite the absence of the traditional manifestations and protections of immune privilege, brain tumours still possess the ability to prevent immunity and facilitate their own modes of immune evasion. For instance, a recent study has uncovered the capacity of intracranial tumours, specifically, to sequester T cells in the bone marrow in a sphingosine 1 phosphate receptor 1 (S1PR1)-dependent fashion, preventing their surveillance of the CNS and fostering antigenic ignorance[37]. Thus, while our historic concept of CNS immune privilege has perhaps become obsolete, the idiosyncrasies of the brain environment continue to offer unique and formidable challenges to immune-based platforms targeting those tumours harboured within[38].
Tumour microenvironment
Relative to other tumour types, CNS tumours display low numbers of tumour-infiltrating lymphocytes (TILs) and other immune effector cell types[39]. This ‘cold tumour’ phenotype is associated with poor responses to immune stimulatory therapies such as immune checkpoint blockade[40]. Even when T cell responses to CNS tumours are induced through means such as vaccination, antigen-specific TIL numbers can remain relatively low, and those cells that are present frequently display an exhausted phenotype[41]. The reduced quantity and limited activity of T cells in CNS tumours is largely owing to the unique immune environment of the brain[42]. Due to its solid enclosure and the potential for damage from increased intracranial pressure, unrestrained inflammation in the brain poses a threat not seen in peripheral organs. For this reason, the CNS may have evolved to be an environment in which both inflammatory and adaptive immune responses are tightly regulated. This regulation involves a variety of immunosuppressive mechanisms at both the molecular and cellular levels[43].
In response to inflammatory stimuli, including those derived from tumours, brain stromal cells produce remarkably high levels of the classic immunosuppressive cytokines transforming growth factor β (TGFβ) and IL-10, which counteract inflammatory cytokines to maintain homeostasis[44, 45]. Glioma cells produce large amounts of indolamine 2,3-dioxygenase (IDO), which both stimulates the accumulation of regulatory T (Treg) cells and suppresses T cell activity by depleting tryptophan from the microenvironment[46, 47]. Both microglia and tumour-infiltrating myeloid cells produce high levels of arginase, which inhibits T cell proliferation and function through the depletion of tissue arginine levels[48]. The brain appears to be particularly susceptible to amino acid deprivation, as CSF arginine and tryptophan levels depend on active transport across the BBB and are sustained at only ~10% of the concentrations found in plasma[49] (Fig. 2).
Fig. 2. Immunosuppressive mediators and therapeutic targets in the brain tumour microenvironment.
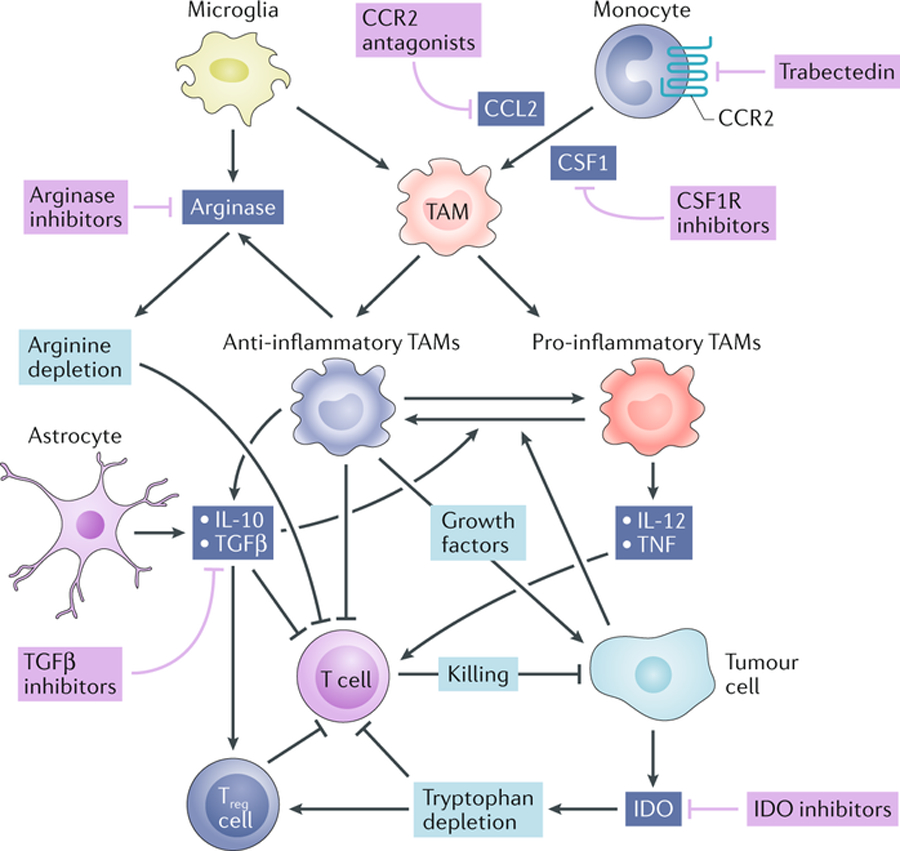
Tumour-associated macrophages (TAMs) play a central role in the brain tumour microenvironment. TAMs arise from circulating monocytes and, to a lesser extent, microglia. The recruitment of monocytes and their differentiation into TAMs is supported by the chemokine CC-chemokine ligand 2 (CCL2) and the cytokine colony-stimulating factor 1 (CSF1). TAMs can be activated towards either an inflammatory or an anti-inflammatory phenotype. Inflammatory TAMs inhibit tumour growth and support T cell-mediated tumour killing through the production of inflammatory cytokines such as IL-12 and tumour necrosis factor (TNF). Anti-inflammatory TAMs and astrocytes produce IL-10 and transforming growth factor β (TGFβ), both of which inhibit T cell effector functions and inflammatory TAM activities[45]. Anti-inflammatory TAMs and microglia produce arginase, which inhibits T cells through arginine depletion from the tumour microenvironment. Gliomas produce indolamine 2,3-dioxygenase (IDO), which acts to recruit regulatory T (Treg) cells and inhibit effector T cells through tryptophan depletion. Potential therapeutic strategies to target these immunosuppressive pathways are shown in purple.
The strategy of inhibiting specific immunosuppressive factors is now being tested in patients with primary brain tumours, typically in combination with other therapies. Targeting TGFβ with antisense oligonucleotides[50] or blocking antibodies[51], as well as the use of TGFβ receptor 1 (TGFβR1) kinase inhibitors[52], has failed to show survival benefits. Several ongoing studies are examining the use of IDO inhibitors in primary brain tumours (NCT02502708 (ref.[53]), NCT02052648 (ref.[54]), NCT02327078 (ref.[55])) (Table 1); however, enthusiasm for these agents has lessened since the phase III ECHO-301 trial (NCT02752074 (ref.[56])) showed no clinical benefit to adding IDO inhibition to immune checkpoint blockade in metastatic melanoma (all secondary sites)[57]. Several phase I and II clinical trials of arginase inhibitors for solid tumours are currently underway (NCT02903914 (ref.[58])), but none are specific for brain tumours. In general, the strategy of inhibiting specific immunosuppressive mediators in patients with brain tumours has not shown promise to date, despite its success in animal models, which may be related to the penetration of some of these agents into the brain parenchyma or the diversity of immunosuppressive mechanisms available to the tumour that can compensate for the loss of one.
Table 1.
Ongoing clinical trials in brain tumours
| Therapy | Cancer type | Phase | NCT identifier |
|---|---|---|---|
| Personalized neoepitope-based vaccine | Recurrent paediatric brain tumours | I | NCT03068832 (ref.[211]) |
| Personalized neoantigen DNA vaccine in combination with immune checkpoint blockade therapy | Newly diagnosed, unmethylated glioblastoma | I | NCT04015700 (ref.[212]) |
| IDO inhibitor (indoximod) in combination with the chemotherapy temozolomide | Progressive primary malignant paediatric brain tumours | I | NCT02502708 (ref.[53]) |
| Anti-LAG3 +/− anti-PD1 therapy nivolumab | Recurrent glioblastoma | I | NCT02658981 (ref.[168]) |
| Anti-TIM-3 monoclonal (TSR-022) +/− nivolumab | Advanced solid tumours | I | NCT02817633 (ref.[169]) |
| Personalized neoantigen-based vaccine + nivolumab, | Newly diagnosed, unmethylated glioblastoma | I | NCT03422094 (ref.[213]) |
| EGFRvIII-specific CAR T cells + anti-PD1 therapy pembrollizumab | Newly diagnosed, unmethylated glioblastoma | I | NCT03726515 (ref.[214]) |
| EGFR806-specific CAR T cells in combination with locoregional immunotherapy (EGFR 806 is a monoclonal antibody recognizing EGFRvIII and a subset of overexpressed wild type EGFRs) | EGFR-positive recurrent or refractory paediatric CNS tumours | I | NCT03638167 (ref.[215]) |
| HER2-specific CAR T cells in combination with locoregional immunotherapy | HER2-positive recurrent or refractory paediatric CNS tumours | I | NCT03500991 (ref.[216]) |
| HER2-specific CAR T cells | HER2-positive CNS tumours | I | NCT02442297 (ref.[217]) |
| IL-13Rα2-specific CAR T cells | Recurrent or refractory malignant glioma | I | NCT02208362 (ref.[218]) |
| CMV pp65 peptide DC vaccine | Recurrent medulloblastoma or malignant glioma | I | NCT03299309 (ref.[219]) |
| CMV pp65 peptide DC vaccine in combination with nivolumab | Recurrent brain tumours | I | NCT02529072 (ref.[220]) |
| IDH1 peptide vaccine | Recurrent grade II glioma | I | NCT02193347 (ref.[221]) |
| Personalized peptide vaccine in combination with TTFields | Glioblastoma | I | NCT03223103 (ref.[222]) |
| Tumour antigen-loaded DC vaccine | Brain tumours | I | NCT03914768 (ref.[223]) |
| IDO inhibitor in combination with temozolomide | Primary malignant brain tumours | I and II | NCT02052648 (ref.[54]) |
| IDO inhibitor (Epacadostat) in combination with nivolumab | Select advanced cancers including brain tumours | I and II | NCT02327078 (ref.[55]) |
| Arginase inhibitor (INCB001158) +/− immune checkpoint blockade therapy | Advanced or metastatic solid tumours | I and II | NCT02903914 (ref.[58]) |
| Autologous CMV-specific CTLs in combination with temozolomide | Glioblastoma | I and II | NCT02661282 (ref.[176]) |
| Nivolumab | IDH-mutant gliomas with and without hypermutator phenotype | II | NCT03718767 (ref.[97]) |
| WT1 peptide vaccine (DSP-7888) in combination with bevacizumab (Anti-VEGFA) | Recurrent or progressive glioblastoma following initial therapy | II | NCT03149003 (ref.[224]) |
| Tumour lysate-pulsed DC vaccine | Brain tumours | II | NCT01204684 (ref.[225]) |
| CMV RNA-loaded DC vaccine | Newly diagnosed WHO Grade IV unmethylated glioma | II | NCT03927222 (ref.[226]) |
| CMV RNA-loaded DC vaccine +/− anti-CD27 therapy varlilumab (to deplete Treg cells but also enhances activation via other mechanisms) | Glioblastoma | II | NCT03688178 (ref.[227]) |
| Temozolomide in combination with radiation +/− nivolumab | Newly diagnosed, unmethylated glioblastoma | III | NCT02667587 (ref.[148]) |
| Nivolumab or temozolomide in combination with radiation | Newly diagnosed, unmethylated glioblastoma | III | NCT02617589 (ref.[147]) |
CAR, chimeric antigen receptor; CMV, cytomegalovirus; CTLs, cytotoxic T lymphocytes; DC, dendritic cell; EGFRvIII, epidermal growth factor receptor variant III; HER2, human epidermal growth factor receptor 2; IDH, isocitrate dehydrogenase; IDO, indolamine 2,3 -dioxygenase; IL-13Rα2, interleukin 13 receptor α2; LAG3, lymphocyte activation gene 3; PD1, programmed cell death protein 1; TIM3, T cell immunoglobulin and mucin domain-containing protein 3; TTFields, tumour treating fields; WHO, World Health Organization; WT1, Wilms tumour 1.
An alternative to targeting specific immunosuppressive factors is to target the immunosuppressive cells within the tumour microenvironment that produce such factors. The major cellular component of this microenvironment is tumour-associated macrophages (TAMs), which can comprise up to 30% of the tumour mass[59]. In both mouse models and humans, the vast majority of brain tumour TAMs appear to arise from circulating monocytes, with a minor proportion (~15%) being microglia-derived[60, 61, 62]. TAMs are believed to promote tumour growth, and TAM numbers correlate with tumour grade[63]. TAM growth-promoting activity has been associated with the anti-inflammatory M2 macrophage phenotype[64]; however, this likely represents an oversimplification, as a much broader range of macrophage activation states occurs in vivo[65].
A variety of approaches have been explored as means to either reduce TAM numbers or reprogram them to be more inflammatory and immunogenic[66]. The cytokine macrophage colony-stimulating factor 1 (CSF1) promotes myeloid cell differentiation and survival[67]. In mice, CSF1 receptor (CSF1R) inhibition with a small molecule markedly increased survival in a genetically engineered platelet-derived growth factor β (PDGFβ)-driven model of glioma[68]; however, no objective response was observed in a phase II study (NCT01349036 (ref.[69])) of a CSF1R inhibitor in patients with recurrent glioblastoma[70]. The chemotherapy agent trabectedin has been shown to function, at least in part, by depleting the monocytes that serve as TAM precursors[71]. Other strategies, including the inhibition of CC-chemokine ligand 2 (CCL2)–CC-chemokine receptor 2 (CCR2)-mediated TAM accumulation with receptor antagonists[72], the reprogramming of TAMs with CD40 agonists or PI3Kγ inhibitors[73, 74], and enhancing TAM phagocytic activity with CD47 blockade[75], have the potential to be applied to patients with brain tumours, but each of these approaches presents considerable developmental hurdles[66]. For example, although CD47 antibodies increase the phagocytosis of tumour cells in mouse xenograft models, CD47 ligand affinity in humans differs substantially from that in mice, raising questions about its translational potential[76]. Thus, while targeting the brain tumour microenvironment represents an attractive therapeutic approach, its implementation may require the identification of more promising therapeutic targets.
Genomic factors
The most common malignant glioma, glioblastoma, presents as a genetically heterogeneous disease, despite a relatively low mutational burden[77, 78]. Clonal selection of driver mutations occurs as an early event, and it is the later acquisition of numerous passenger mutations that results in the intratumoural heterogeneity. Before treatment, glioblastoma has a neutral evolutionary pattern, whereby mutational variants expand at similar rates and accumulate[79]. Unfortunately, this situation is lost once patients commence therapy, when expansion of subclones is influenced by strong selection pressures and adaptation in response to treatment modalities occurs. In the setting of low-grade glioma, hypermutation induced by treatment with the alkylating chemotherapeutic agent temozolomide (TMZ) is observed in up to 60% of tumours at recurrence[80]. Hypermutations also occur in primary glioblastoma at recurrence, but at a substantially lower frequency of around 10% of patients after exposure to TMZ[81]. The culmination of these events is the rapid emergence of resistance to therapies, particularly those with a single target[82, 83].
In the context of immune-based therapies, these fundamental observations have a number of major implications. High tumour mutation burden (TMB) has emerged as a biomarker for immune checkpoint inhibitor (ICI) response, and TMB is believed to correlate with the level of pre-existing antitumour adaptive immunity[84, 85, 86, 87, 88]. This paradigm applies well to tumours for which the TMB is greater than 10 mutations/megabase, such as melanoma or some colorectal cancers; however, the median TMB in newly diagnosed glioma is around 1.5 mutations/megabase. Fewer than 2% of glioblastomas from patients in the newly diagnosed setting carry >10 mutations/megabase, and these usually occur in the context of germ-line mutations of mismatch repair genes, such as that observed in Lynch syndrome[89]. Thus, a relatively low TMB offers up few somatic mutations for T cell targets and could reduce the array of responding T cell clones. These observations may in part explain the poor performance of ICIs in the majority of patients with either newly diagnosed or recurrent glioblastoma.
Immunoediting is a process whereby the immune system eliminates tumour cells carrying immunogenic antigens, resulting in the selection of resistant subclones[90]. Evidence of this process has been shown in several cancer types, including glioma[91, 92, 93, 94, 95]. By comparing newly diagnosed specimens with those at recurrence from the same patients, the Glioma Longitudinal Analysis (GLASS) Consortium has found evidence in support of immunoediting in gliomas, suggesting that active immunosurveillance is ongoing in some adult patients[81]. In addition, supporting the possibility of immunoediting in some patients is a recent study that examined the whole exome and transcriptome after anti-programmed cell death protein 1 (PD1) therapy in recurrent malignant glioma samples. Those tumours defined as responsive were associated with a more diverse pattern of evolution, possibly due to elimination of neoantigens[96].
Several strategies seek to overcome these inherent mechanisms of resistance. For example, attempts to identify patients with hypermutated tumours at diagnosis are underway, in an attempt to enrich for patients who may respond to immune checkpoint inhibition (NCT03718767)[97]. However, the utility of such an approach remains unresolved, is perhaps over-simplistic, and may be adversely affected by numerous competing factors.
Glioblastoma often express a mutated form of the epidermal growth factor receptor (EGFR), which is constitutively active and enhances tumorigenicity by activating the RAS–SHC–GRB2 pathway.40 The most common mutant form of EGFR in glioblastoma is the EGFR class III variant (EGFRvIII), which has a truncated extracellular domain due to an 801-base-pair in-frame deletion of the wild-type receptor[98]. This deletion results in the fusion of the two ends of the peptide and the creation of an antigenic site that contains a novel glycine residue not included in the wild-type peptide[99]. Therefore, EGFRvIII serves as an ideal tumour-specific antigen for glioblastoma. EGFRvIII is expressed in approximately 30% of glioblastomas and is found to be expressed on 37–86% of tumour cells in a given tissue sample[100].
The loss of antigenic targets in response to therapy has severely limited immune-based therapies that target single antigens. Some of the best examples of this phenomenon are the targeting of EGFRvIII by vaccines or chimeric antigen receptor (CAR) T cell therapies, in response to which tumours re-emerge rapidly, having lost expression of the target, despite documented evidence of CAR T cell infiltration in several patients and preliminary evidence of reduced EGFRvIII transcript expression[101, 102, 103]. To overcome this induction of resistance to single-target immune-based therapies, emerging strategies include those that simultaneously target multiple antigens, such as the recently reported neoantigen vaccines[41, 104]; those that target ubiquitous viral antigens, such as those from cytomegalovirus (CMV); and those that aim to induce broad-based immune responses, such as innate immune targeting through oncolytic viruses[105, 106, 107, 108, 109].
Epigenetic impacts
There is emerging evidence that epigenetic events play a key role in modulating immune responses to tumours, and many brain tumours carry mutations of epigenetic drivers[110, 111, 112, 113, 114]. Key examples include isocitrate dehydrogenase (IDH) mutated gliomas; those carrying histone mutations such as in histones H3.1 and H3.3; a subset of primary glioblastomas, discovered more recently, that carry SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A-like protein 1 (SMARCAL1) mutations; and many paediatric tumours, such as atypical teratoid rhabdoid tumour (ATRT) driven by mutations in SMARCB1 (refs[110, 111, 112, 113, 114]). SWI/SNF complexes are important regulators of chromatin structure and transcription in several cancers, including brain tumours[115]. It is now clear in non-CNS tumours that mutations of components within these important protein complexes, such as AT-rich interactive domain 1A (ARID1A) or polybromo 1 (PBRM1; also known as BAF180), can drive both resistance and sensitivity to immune-based therapies through alterations in the transcription of downstream targets, such as chemokines that attract inflammatory cells[116, 117, 118, 119].
Similar epigenetic events also seem to be at play in IDH1 mutated glioma. Several groups have shown that IDH1 mutations can cause down-regulation of leukocyte chemotaxis, resulting in repression of the tumour-associated immune system[120]. This is related to reduced expression of cytotoxic T lymphocyte-associated genes and interferon-γ (IFNγ)-inducible chemokines, including CXC-chemokine ligand 10 (CXCL10), in IDH-mutated tumours compared with IDH-wild-type tumours[121]. In syngeneic mouse glioma models, the expression of mutant IDH1 also suppressed the accumulation of T cells in tumour sites via reduced secretion of CXCL10 (ref.[121]). In a separate study, IDH mutant glioma cells acquired resistance to natural killer (NK) cells through epigenetic silencing of the activating receptor NKG2D ligands UL16-binding protein 1 (ULBP1) and ULBP3. As well, hypomethylation mediated by the DNA methyltransferase inhibitor decitabine restores ULBP1 and ULBP3 expression in IDH-mutant glioma cells[122]. These data demonstrate a mechanism of immune evasion in IDH-mutated gliomas and suggest that specific inhibitors of mutant IDH may improve the efficacy of immunotherapy in patients with IDH-mutated gliomas[120, 121, 122] (Fig. 3).
Fig. 3. Epigenetic events play a key role in modulating immune responses to brain tumours.
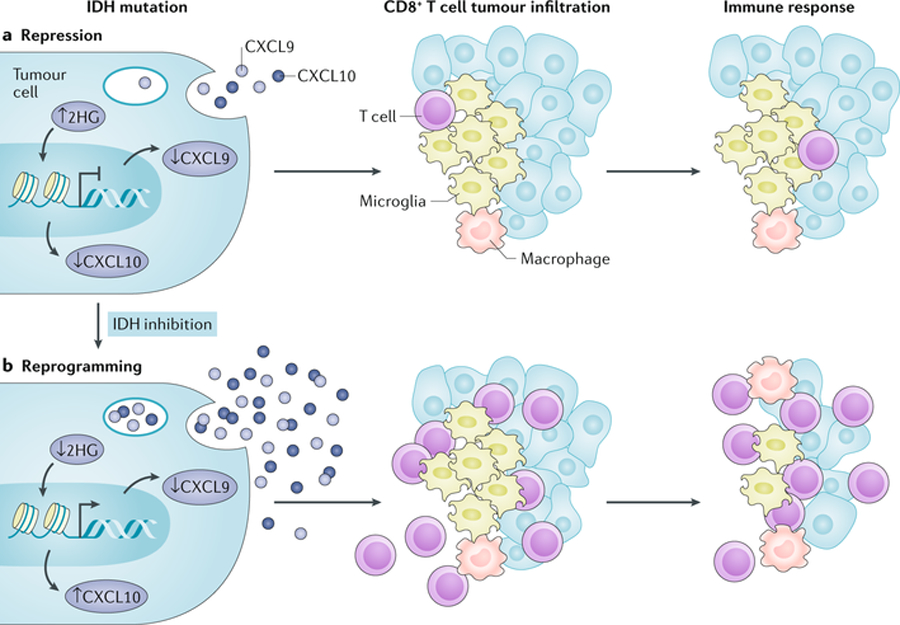
a | Isocitrate dehydrogenase 1 (IDH1) mutations in glioma can cause down-regulation of leukocyte chemotaxis, resulting in repression of the tumour-associated immune system. An IDH mutation results in the generation of the oncometabolite 2-hydroxyglutarate (2HG; via conversion from α-ketoglutarate), which in turn represses signal transducer and activator of transcription 1 (STAT1) expression, leading to reduced expression of interferon-γ (IFNγ)-inducible chemokines, including CXC chemokine ligand 9 (CXCL9) and CXCL10. As a consequence, IDH-mutated tumours suppress the infiltration and accumulation of T cells at tumour sites. b | In mice bearing IDH-mutated glioma, these effects can be reversed through pharmacological inhibition with IDH-C35, a specific inhibitor of mutant IDH. Adapted from ref.[228], Springer Nature Limited.
Future studies examining the role of epigenetic modulation of immune evasion in malignant gliomas and other brain tumours will be required. It is hypothesized that a number of epigenetic drugs, such as demethylating agents, bromodomain inhibitors and histone deacetylase inhibitors, may act to reverse the adverse immunological transcriptional profile seen in epigenetically driven tumours prior to the initiation of immune-based therapies. These are under investigation preclinically, but as yet they remain unpublished.
Impact of standard of care
Many patients benefit from multimodal therapy, including surgery, radiation therapy and chemotherapy such as TMZ. Although the limitations of immune-based therapy may be related to tumour-associated factors such as poor immunogenicity and tumour-induced immune tolerance, treatment-induced immune regulatory effects may also play major roles, both adverse and beneficial.
Many patients with brain tumours receive adjuvant TMZ. Although TMZ provides a survival benefit in patients with newly diagnosed glioblastoma[1], its major dose-limiting toxicity is myelosuppression[123]. Furthermore, patients treated with TMZ, especially in dose-intensified (DI) regimens, have an increased incidence of opportunistic infections and are immunosuppressed[124]. Indeed, treatment with the combination of radiation and TMZ is associated with changes in regulatory and effector mononuclear cells in the peripheral blood that tilt the balance towards an immune suppressive state. This shift could certainly affect the outcome of immune-based therapy following radiation and TMZ treatment and should be considered when designing any combination therapy regimens[125, 126]. However, preclinical and clinical data have also suggested that a state of lymphopenia upon recovery can induce reactive homeostatic proliferation and enhanced antitumour immune responses. As an example, in a clinical trial of glioblastoma comparing two different radiation and TMZ combination regimens, one using standard doses of TMZ at 5 days per month versus a more intensive regimen of 21 days of TMZ per month, whereas antigen-specific immune responses developed in all patients treated with either regimen, the more intensive regimen induced an increase in the proportion of immunosuppressive Treg cells, but the more intensive TMZ regimen also produced humoral and delayed-type hypersensitivity responses of greater magnitude[124, 127, 128]. Several published studies of vaccine approaches for brain tumours now support the hypothesis that lymphodepletion associated with the dose-dense 21-of-28-day TMZ regimen may enhance the immune response, and particularly the humoral immune response to tumour vaccines, which may ultimately translate into improved clinical benefit[100, 102]. Furthermore, data from extra-cranial solid tumours[129] such as melanoma have explored such strategies for adoptive immunotherapies. These studies suggest that well-timed lymphodepletion may be used to generate positive immunomodulatory effects, in part owing to the production of an immunostimulatory cytokine and chemokine environment[130, 131, 132, 133].
Current therapeutic approaches
Current approaches, unique observations
Immune checkpoint inhibitors
Immune checkpoint blockade targeting the PD1–PD1 ligand 1 (PDL1) axis and/or cytotoxic T lymphocyte-associated antigen 4 (CTLA4) has proffered dramatic successes against a variety of solid tumours[134, 135, 136, 137, 138] and has garnered US Food and Drug Administration (FDA) approval for a still increasing number of malignancies[139]; however, glioblastoma remains conspicuously absent from the list of approvals. Although numerous groups have reported preclinical successes for more than a decade[140, 141, 142, 143, 144, 145], a 2017 phase III trial (CheckMate-143) comparing the anti-PD1 therapy nivolumab with bevacizumab (anti-VEGFA) in the treatment of recurrent glioblastoma found no overall survival (OS) benefit to nivolumab[146]. More recently, the phase III CheckMate-498 study (NCT02617589 (ref.[147])) showed similarly disappointing results in newly diagnosed patients with O6-methylguanine-DNA-methytransferase (MGMT) promoter-unmethylated glioblastoma. In this study, nivolumab plus radiation was compared with the standard combination of radiation and TMZ. Failure to meet the primary end point of OS was announced by Bristol-Myers Squibb in May of 2019, although these results await publication. The outcome of CheckMate-548 (NCT02667587 (ref.[148])), which is evaluating nivolumab plus TMZ in patients with MGMT-methylated glioblastoma, is still pending. As a consequence, the focus has now shifted towards uncovering aetiological contributors to treatment failures, with the goal of removing barriers to successful immune checkpoint blockade[146, 149, 150].
One question that endures is whether the restrictive intracranial environs or simply glioblastoma itself presents the greatest challenge to therapeutic efficacy. A recent clue may be the modest but significant success demonstrated for immune checkpoint strategies targeting other intracranially situated tumours, such as melanoma brain metastases[151]. Glioblastoma, in turn, does involve its own set of obstacles. The hindrances to immune checkpoint blockade ascribable to glioblastoma include a low TMB and extensive intratumoural heterogeneity. Even individual cells within glioblastoma tumours prove varied in their expression of oncogenic transcriptional programs, reflecting a degree of intratumoural heterogeneity that proffers a unique challenge to immunological targeting[9]. Additional constraints include the restricted access of drugs and immune cells to the CNS, as we discussed above. (However, we should reiterate here that antigen-specific T cells suffer few constraints to their trafficking[26, 28, 29] and that the BBB is frequently disrupted by CNS tumours like glioblastoma[30, 31, 32].) Nevertheless, what remains less resolved is whether the CNS constraints for drug access or for immune cell access will prove to be more of a hindrance to effective immune checkpoint blockade. For instance, an unresolved question is whether PD1- and CTLA4-blocking antibodies must situate within tumours for their activity, versus simply acting on peripheral T cells prior to their CNS entry.
Perhaps the most salient restriction to effective immune checkpoint blockade in glioblastoma is rampant T cell dysfunction, with little baseline T cell activation to propagate during therapy[37, 150, 152, 153, 154, 155, 156]. A viable T cell compartment is a prerequisite for workable immune checkpoint blockade, which acts primarily to unbridle and perpetuate T cell activity. However, T cell dysfunction has long been identified as a hallmark of glioblastoma[38, 155, 156, 157, 158, 159], which imposes potent suppression of systemic immunity, despite its intracranial confines. As is seen in other solid tumours, resistance to immune checkpoint blockade at the T cell level is marked by the upregulation of multiple alternative immune checkpoints[160]. These can include T cell immunoglobulin and mucin domain-containing protein 3 (TIM3), lymphocyte activation gene 3 (LAG3), B and T lymphocyte attenuator (BTLA), 2B4 (also known as CD244), CD160, CD39 and T cell immunoreceptor with immunoglobulin and ITIM domains (TIGIT)[161, 162, 163]. Mounting expression of these less canonical immune checkpoints on T cells provides further mechanisms for T cell restraint, but it also serves to signal T cell transition into a dysfunctional, exhausted state[163, 164]. Although exhaustion may be reversible in early stages, it can rapidly progress beyond recovery[165, 166]. For example, Woroniecka et al. recently demonstrated comparatively severe T cell exhaustion amongst tumour-infiltrating lymphocytes (TILs) isolated from patients and mice with glioblastoma, and the corresponding inability of mice with gliomas to respond to PD1 blockade[150]. The increasing expression of alternative immune checkpoints may indicate a state of terminal exhaustion that cannot be reversed by traditional immune checkpoint blockade alone. However, preclinical work has highlighted a possible synergy between PD1, TIM3 and LAG3 blockade in mice with gliomas[167]. Clinical trials targeting TIM3 and LAG3, either alone or in combination with anti-PD1, are underway in glioblastoma (NCT02658981 (ref.[168]) and NCT02817633 (ref.[169])).
Nevertheless, a very recent clinical trial suggests that the situation may not be as dire as might perhaps be presumed. A simple modification to treatment regimens to include a single neoadjuvant administration of the anti-PD1 agent pembrolizumab extended median survival in patients with operable recurrent glioblastoma to 417 days, compared with 228.5 days in patients receiving more typical adjuvant dosing of pembrolizumab[170]. The modification appeared to capitalize on an improved capacity to generate antecedent T cell activation. Likewise, an additional group demonstrated that neoadjuvant nivolumab precipitated pro-inflammatory changes in the glioblastoma microenvironment, although the same clear survival benefits were not obtained[171].
It is clear that glioblastoma poses challenges to immune checkpoint blockade more paramount than those faced with somewhat more immunogenic tumours, such as melanoma. While these challenges have likely delayed the advent of truly viable immunotherapeutic approaches for glioblastoma, a newer focus on mechanistic considerations for treatment resistance and improving the functionality of the T cell compartment holds promise for providing important incremental advances to efficacy.
CARs and adoptive T cells
Adoptive T cell therapy holds considerable promise for the treatment of brain tumours. Earlier approaches involving nonspecific effector cells, such as NK cells or lymphokine-activated killer (LAK) cells, have now fallen out of favour, owing to their lack of efficacy[172]. The administration of autologous TILs has induced regressions in some tumour types[173] but is less feasible in glioblastoma, owing to the difficulty of isolating and expanding TILs from the CNS. A more feasible approach has been the administration of autologous cytomegalovirus (CMV)-specific T cells, whereby peripheral blood mononuclear cells can be isolated from the blood and expanded in vitro with synthetic CMV peptides to generate CMV-specific T cells, which can then be reinfused into the patient[174]. This strategy is based on the finding that the majority of glioblastomas, but not the surrounding normal brain tissues, express CMV antigens[175]. One study examining the administration of CMV-specific T cells for the treatment of glioblastoma is currently underway (NCT02661282 (ref.[176])).
The major advance in adoptive cell therapy in recent years has been the development of CAR T cells, which, in the case of CAR T cells targeting CD19, are approved for the treatment of B cell leukaemia and lymphoma[177]. For the treatment of glioblastoma, clinical trial results are available for CAR T cells targeting three antigens: EGFRvIII, human epidermal growth factor receptor 2 (HER2; also known as ERBB2) and IL-13 receptor α2 (IL-13Rα2)[103, 178, 179, 180] (Fig. 4). These trials have demonstrated that the use of CAR T cells for brain tumours is feasible, safe and potentially efficacious. As with other solid tumours, the use of CAR T cells for brain tumours still faces several substantial obstacles[181]. One major problem is heterogeneous expression of target antigens in tumour cells. Even in the case of uniformly expressed antigens, selective pressure can result in antigen loss and tumour recurrence. In the first clinical trial of EGFRvIII-directed CAR T cells for glioblastoma, a significant decrease in EGFRvIII expression, but not in wild-type EGFR, was seen in almost all patients in which tumour-infiltrating CAR T cells were observed[103]. In one patient with recurrent multifocal glioblastoma, intracranial administration of IL-13Rα2-targeted CAR T cells resulted in the regression of all intracranial and spinal lesions, but subsequent relapse was due to IL-13Rα2-negative tumours[180]. This suggests that successful CAR T cell therapy will require either targeting multiple antigens or the development of CAR T cell designs that induce significant epitope spreading. Either approach would lead to a broader immune response, which might also carry the risk of unintended reactivity against normal tissue. In mouse leukaemia and lymphoma models, CAR T cells engineered to express CD40L have been shown to circumvent tumour immune escape via antigen loss by stimulating CD40–CD40L-mediated cytotoxicity and the induction of robust endogenous immune responses[182]. In an intriguing study using intracranial mouse models of antigen-heterogeneous gliomas, Choi et al. examined tumour responses to EGFRvIII-directed CAR T cells that were engineered to also express bispecific T cell engagers (BiTEs) targeted to wild-type EGFR. These BiTE-expressing CAR T cells were able to recruit bystander T cells, eliminate heterogeneous tumours, and, despite the targeting of wild-type EGFR, displayed no toxicity towards EGFR-expressing normal tissues[183].
Fig. 4. Chimeric antigen receptor T cell immunotherapy in brain tumours.
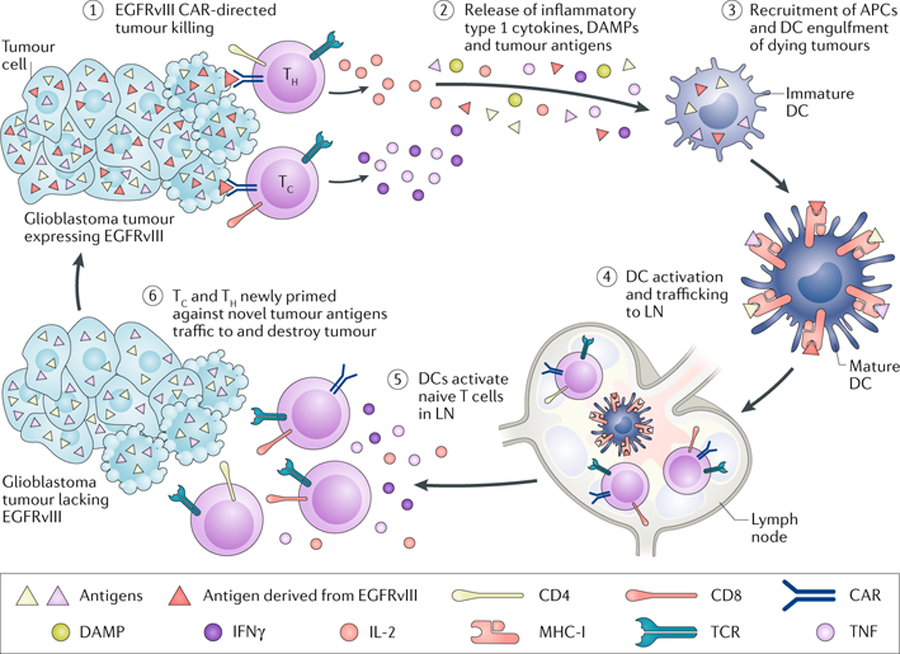
Chimeric antigen receptor (CAR) T cells have been used to target tumour-specific and tumour-associated antigens in malignant gliomas. There appears to be no impediment to these cells reaching and killing target antigen-expressing tumour cells in the central nervous system (CNS). The challenge remains for these highly specific and potent agents to target antigen-negative tumour cells directly within these highly heterogeneous tumours. APC, antigen-presenting cell; DAMP, damage-associated molecular pattern; DC, dendritic cell; EGFRvIII, epidermal growth factor receptor variant III; IFNγ, interferon-γ; LN, lymph node; MHC-I, major histocompatibility complex class I; Tc, cytotoxic T cell; TCR, T cell receptor; TH, T helper cell; TNF, tumour necrosis factor. Adapted from ref.[229] (Johnson, L. A., Sanchez-Perez, L., Suryadevara, C. M. & Sampson, J. H. Chimeric antigen receptor engineered T cells can eliminate brain tumors and initiate long-term protection against recurrence. Oncoimmunology 3, e944059 (2014)), with permission of the publisher (Taylor & Francis Ltd, http://www.tandfonline.com).
Another substantial issue, associated in particular with this type of therapy in brain tumours, is maximizing and maintaining the activity of the administered CAR T cells. CAR T cell administration appears to induce a compensatory immunosuppressive response in the brain, characterized by increased expression of immunosuppressive factors and an influx of Treg cells[103]. For this reason, successful therapies will likely require the development or engineering of CAR T cells resistant to such immunosuppression. Two recent studies of CD19 CAR T cells may be applicable to this problem. Fraietta et al. identified factors that were associated with clinical success in patients with chronic lymphocytic leukaemia (CLL)[184]. Clinical responses were not associated with any characteristics of the patients or tumours, but rather with the intrinsic properties of the administered cells. Markers of response included the expression of memory-related genes, IL-6–signal transducer and activator of transcription 3 (STAT3) signatures, robust ex vivo proliferation, and an increased frequency of CD27+ PD1– CD8+ CAR T cells expressing high levels of the IL-6 receptor (IL-6R). Poor responses were associated with the expression of genes involved in effector differentiation, glycolysis, exhaustion and apoptosis. This study strongly suggests that specific features of the CAR T cell product can predict clinical efficacy. This same group also described an extraordinary case in which complete remission of CLL was attributed to the massive in vivo expansion of a single CAR T cell clone[185]. Lentiviral integration of the CAR construct into the TET2 gene, encoding a methylcytosine dioxygenase, led to a null mutation of TET2 (ref.[185]), which normally regulates the expression of multiple genes[186]. TET2-mutated cells have a proliferative advantage and display increased inflammatory responses and memory T cell differentiation[187, 188]. These findings raise the possibility that inhibiting TET2 or similar enzymes in the administered CAR T cells could significantly increase their efficacy. Moreover, pharmacological inhibition of such enzymes would also have the potential to increase the antitumour activity of TAMs, as TET2-deficient macrophages have been shown to display increased inflammatory responses[189, 190].
While CAR T cell strategies remain a popular mainstay of engineered T cell therapies for brain tumours, alternative strategies exist and are being actively investigated. For instance, rather than equipping T cells with CAR constructs, cloned T cell receptors (TCRs) possessing a desired antigen specificity can likewise be transduced into T cells, which are then expanded and adoptively transferred. Such TCR-transduced T cells remain MHC-restricted and - dependent (unlike CARs), but the CAR requirement for surface antigen expression is obviated. Such strategies, then, are useful when targeting intracellular antigens. One recent example has been the engineering of a TCR targeting a mutated form of the H3 variant H3.3K27M, a common mutation in paediatric diffuse intrinsic pontine gliomas (DIPGs). T cells transduced with the relevant TCR demonstrated efficacy when transferred into mice bearing DIPG xenografts[191]. Interestingly, H3.3K27M DIPGs have been found to uniformly express the disialoganglioside GD2 at high levels and, in orthotopic H3K27M+-diffuse midline glioma xenograft models, GD2-targeted CAR T cells cleared the vast majority of tumours[192].
Vaccines
Therapeutic vaccination for brain tumours could be a promising potential therapeutic modality but is unproven as of yet. Although supportive data from phase III trials are still lacking, several vaccine strategies have shown good safety and immunogenicity in phase I and II clinical trials, with some vaccines having demonstrated improved survival relative to historical controls. Peptide vaccines targeting a single tumour antigen include those for EGFRvIII, IDH R132H, Wilms tumour 1 (WT1) and survivin. A vaccine against EGFRvIII in glioblastoma led to substantial increases in survival in uncontrolled phase II trials but to no such benefit in a randomized phase III trial[193, 194]. In a study of WT1 peptide vaccination, the development of anti-WT1 IgG responses was associated with increased survival in patients with glioblastoma in a nonrandomized trial[195]. A substantial problem with single-peptide vaccinations is the potential for tumour immune escape. In the EGFRvIII vaccine studies, the majority of patients who experienced recurrence had lost EGFRvIII expression[193]. For this reason, clinical trials have begun to investigate multi-peptide vaccines to target multiple glioma antigens, but none to date has given a clear indication as to efficacy[196, 197]. Two recent trials that have examined the use of personalized peptide vaccines in glioma, which are designed according to the mutations and gene expression patterns in an individual patient’s tumour, have provided several novel observations[41, 104]. First, administration of the corticosteroid dexamethasone, which is used to reduce cerebral oedema during vaccine priming, appeared to inhibit systemic antigen-specific T cell responses. Second, neoantigen vaccination could induce antigen-specific TILs, but these cells displayed an exhausted phenotype. Third, neoantigen-specific T cell responses occurred frequently, but primarily among CD4+ T cells, despite the selection of peptides based on MHC class I binding predictions[41, 104]. These findings suggest that corticosteroids should be avoided during the use of tumour vaccines, that vaccine efficacy may require additional measures to combat T cell exhaustion and that vaccine neoantigens should not be selected solely on the basis of MHC class I binding predictions.
Studies of dendritic cell (DC) vaccines for brain tumours have examined multiple types of loaded antigens. Antigen preparations loaded into DCs in clinical trials have included mixtures of autologous tumour peptides eluted from tumour-derived cell lines[198], autologous tumour lysates[199], peptides from known glioma antigens[200] and tumour-associated viral antigens such as CMV[127]. An initial trial of DCs loaded with six glioblastoma antigen peptides, termed ICT-107, suggested there was a survival benefit, but a randomized phase II trial showed no significant improvement in OS[201, 202]. One autologous tumour lysate-pulsed DC vaccine, DCVax-L, has advanced to a phase III trial, and preliminary results indicate that, on the basis of intent to treat, DCVax-L may improve OS[203]. However, concerns have been raised about this report, in that it presents only interim data, and the primary end point, the number of progression-free survival (PFS) events, was not reported.
Methods to improve the antigen-presenting cell (APC) function, cytokine production and migration to lymph nodes of DC vaccines have also been examined. Initial work focused on the optimization of conditions for in vitro DC generation in order to maximize DC immunogenicity[204]. The addition of agents such as type I interferon and the synthetic Toll-like receptor 3 (TLR3) ligand polyinosinic:polycytidylic acid (poly(I:C)) improves DC function in vitro[205]. In phase I and II trials in patients with gliomas, DC vaccines prepared in this manner increased post-vaccine cytokine production and were suggested to provide a survival benefit[200]. An alternative strategy has been to improve DC vaccine activity by additional treatments to patients. For example, inducing memory T cell activation with tetanus toxoid prior to vaccination with CMV antigen-loaded DCs increases migration of the administered DCs to lymph nodes and appears to improve patient survival[206]. Other strategies tested in humans to increase inflammation at the vaccine site have included the administration of granulocyte–macrophage colony-stimulating factor (GM-CSF)[127], poly(I:C)[200] and other TLR agonists[199]. In reality, the next major advance in DC vaccines is likely to come from their combination with other immunotherapies. As has been proposed by some[207], DC vaccination may be the best solution to the lack of efficacy seen with immune checkpoint blockade in tumours, such as glioblastoma, with low mutational burdens.
Biomarkers of response
In the context of immune-based therapies for all cancers, the prediction and monitoring of responses has been a major barrier to progress. Tumour biopsy-based techniques for measuring response, such as immunohistochemistry, flow cytometry-based techniques or sophisticated multi-omic approaches, remain unvalidated and are often limited in their use for brain tumours, owing to the clinical and ethical barriers related to longitudinal sample acquisition. Magnetic resonance imaging (MRI) has been the main modality used in the follow-up and monitoring of treatment response, but difficulties arise related to the differentiation between pseudoprogression, radiation-mediated necrosis and actual tumour progression[208, 209].
Three recent studies have focused efforts on in-depth analysis of glioblastoma tissue from patients treated with ICI therapy in a neoadjuvant setting[96, 170, 171]. These data revealed enhanced expression of chemokine transcripts, higher immune cell infiltration and augmented TCR clonal diversity among TILs, supporting a local immunomodulatory effect of treatment. Additionally, in a separate study, neoadjuvant PD1 blockade was associated with the upregulation of T cell- and IFNγ-related gene expression in immune cells and the downregulation of cell-cycle-related gene expression within the tumour[96, 170, 171]. Other observations have included focal induction of PDL1 in the tumour microenvironment, enhanced clonal expansion of T cells, decreased PD1 expression on peripheral blood T cells and a decreasing monocytic population[96, 170, 171]. These anti-PD1 therapies appear to lead to different responses in tumours with specific molecular alterations. Specifically, non-responders are associated with significant enrichment of PTEN mutations and immunosuppressive expression signatures, whereas responders are associated with an enrichment of MAPK pathway alterations (protein tyrosine phosphatase, non-receptor type 11 (PTPN11) and BRAF), suggesting a genotype–phenotype link[41, 170, 210]. Taken together, such data point to the need for continued development of predictive biomarkers for immune-based therapies in glioma.
Conclusions and perspective
Malignant brain tumours, particularly primary gliomas, remain universally lethal — immunotherapy has done little to improve this. What accounts for these failures? There are likely a complex set of interactions between the tumour, its host and the standard therapy used for these challenging lesions. These tumours tend to have few mutations that could be targeted immunotherapeutically, and rarely have even one mutation that is expressed homogeneously. Moreover, the genetic evolution of these tumours leads to tumour populations that are extremely heterogeneous, and therefore difficult to target with a therapy focused on a single antigen. Unfortunately, therapies targeted at tumour-associated antigens or multiple antigens risk unintended consequences to the brain and systemic tissues alike. The host also provides particular challenges. While the concept of CNS immune privilege has eroded over time, there are ways in which the brain treats a foreign body that are simply different from those in other organs in the host, and the opportunities we have to provide therapies that access the brain are limited. In addition, tumours in the brain seem able to promote bone marrow sequestration of the immune cells that could attack the tumour[37]. Finally, the standard therapies for this disease, principally radiation and chemotherapy, are both profoundly immunosuppressive and thwart many immunotherapeutic attempts. In the end, immunotherapy will only be successful if it is specific, can be successfully delivered to the brain and effectively deals with cellular heterogeneity.
Acknowledgements
This work was supported by grants from the National Institutes of Health to J.H.S.: R01-NS099463, P50-CA190991, P30-CA14236, P01-CA225622, R01-CA235612, U01-NS090284, R01-NS085412 and R01-CA175517. The funders had no role in the preparation of the manuscript or the decision to publish. The authors acknowledge T. Wright for her assistance with manuscript preparation.
Glossary
- Tumour-treating fields (TTFields)
External application of alternating electrical fields to disrupt the multiplication of tumour cells
- Glial–lymphatic (glymphatic) pathway
A recently characterized functional waste clearance pathway in the central nervous system that connects the brain interstitium to the cerebrospinal fluid spaces via aquaporin channels
- Glia limitans
A thin barrier of astrocyte foot processes associated with the parenchymal basal lamina surrounding the brain and spinal cord. The glia limitans plays a crucial role in regulating the movement of small molecules and cells into the brain parenchyma
- Microglia
The resident self-renewing population of macrophages in the central nervous system
- Lynch syndrome
Also known as hereditary non-polyposis colorectal cancer. A type of inherited cancer syndrome associated with a genetic predisposition to different cancer types, including glioblastoma
- Myelosuppression
A condition in which bone marrow activity is decreased, resulting in fewer red blood cells, white blood cells and platelets
- Lymphopenia
The condition of having an abnormally low level of lymphocytes in the blood
- Lymphokine-activated killer (LAK) cells
Lymphocytes cultured in the presence of IL-2 to stimulate their cytotoxic activity
- Pseudoprogression
A new or enlarging area of contrast agent enhancement occurring early after the end of radiotherapy or immunotherapy (for example, within 3–4 months), in the absence of true tumour growth, that subsides or stabilizes without a change in therapy
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Competing interests J.H.S. has an equity interest in Annias Immunotherapeutics, which has licensed intellectual property from Duke related to the use of the PEP-CMV vaccine in the treatment of glioblastoma. J.H.S. has an equity interest in Istari Oncology, which has licensed intellectual property from Duke related to the use of polio virus and D2C7 in the treatment of glioblastoma. J.H.S. has additional relationships with Celldex (intellectual property, royalties) and Medicenna Therapeutics (consulting). J.H.S. is an inventor on patents related to the PEP-CMV DC vaccine with tetanus, as well as to polio virus vaccine and D2C7 in the treatment of glioblastoma. D.M.A is an inventor on patents related to the use of polio virus. M.D.G is an inventor on patents related to cellular vaccines. Duke and certain Duke investigators could benefit financially if related therapies prove effective and are commercially successful. P.E.F. has no relevant disclosures.
References
- 1.Stupp R et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med 352, 987–996 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Stummer W et al. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: a randomised controlled multicentre phase III trial. Lancet Oncol 7, 392–401 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Walker MD et al. Randomized comparisons of radiotherapy and nitrosoureas for the treatment of malignant glioma after surgery. N. Engl. J. Med 303, 1323–1329 (1980). [DOI] [PubMed] [Google Scholar]
- 4.Hegi ME et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med 352, 997–1003 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Imperato JP, Paleologos NA & Vick NA Effects of treatment on long-term survivors with malignant astrocytomas. Ann. Neurol 28, 818–822 (1990). [DOI] [PubMed] [Google Scholar]
- 6.Gilbert MR et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med 370, 699–708 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stupp R et al. LTBK-01: prospective, multi-center phase III trial Tumor Treating Fields together temozolomide compared temozolomide alone patients newly diagnosed glioblastoma. Neuro Oncol 18 (Suppl. 6), i1 (2016).26705298 [Google Scholar]
- 8.Stupp R et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. JAMA 318, 2306–2316 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patel AP et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401 (2014).This study provides one of the most thorough documentations to date of glioblastoma heterogeneity at the single-cell level.
- 10.Vogelstein B et al. Cancer genome landscapes. Science 339, 1546–1558 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engelhardt B, Vajkoczy P & Weller RO The movers and shapers in immune privilege of the CNS. Nat. Immunol 18, 123–131 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Medawar PB Immunity to homologous grafted skin; the fate of skin homografts transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. Br. J. Exp. Pathol 29, 58–69 (1948).This landmark paper is often credited as the first to suggest the notion of CNS immune privilege.
- 13.Bradbury MW & Westrop RJ Factors influencing exit of substances from cerebrospinal fluid into deep cervical lymph of the rabbit. J. Physiol 339, 519–534 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cserr HF & Knopf PM Cervical lymphatics, the blood–brain barrier and the immunoreactivity of the brain: a new view. Immunol. Today 13, 507–512 (1992). [DOI] [PubMed] [Google Scholar]
- 15.Goldmann J et al. T cells traffic from brain to cervical lymph nodes via the cribroid plate and the nasal mucosa. J. Leukoc. Biol 80, 797–801 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Widner H et al. Scintigraphic method to quantify the passage from brain parenchyma to the deep cervical lymph nodes in rats. Eur. J. Nucl. Med 13, 456–461 (1987). [DOI] [PubMed] [Google Scholar]
- 17.Aspelund A et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med 212, 991–999 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Louveau A et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat. Neurosci 21, 1380–1391 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Louveau A et al. Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eide PK, Vatnehol SAS, Emblem KE & Ringstad G Magnetic resonance imaging provides evidence of glymphatic drainage from human brain to cervical lymph nodes. Sci. Rep 8, 7194 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iliff JJ et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci. Transl. Med 4, 147ra111 (2012). This research was the first to document the existence of the glymphatic pathway. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kipnis J Multifaceted interactions between adaptive immunity and the central nervous system. Science 353, 766–771 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bechmann I, Galea I & Perry VH What is the blood–brain barrier (not)? Trends Immunol 28, 5–11 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Huber JD, Egleton RD & Davis TP Molecular physiology and pathophysiology of tight junctions in the blood-brain barrier. Trends Neurosci 24, 719–725 (2001). [DOI] [PubMed] [Google Scholar]
- 25.Tietz S & Engelhardt B Brain barriers: crosstalk between complex tight junctions and adherens junctions. J. Cell Biol 209, 493–506 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Owens T, Bechmann I & Engelhardt B Perivascular spaces and the two steps to neuroinflammation. J. Neuropathol. Exp. Neurol 67, 1113–1121 (2008).Along with Aspelund et al. (2015), this is the first study to characterize CNS lymphatic vessels within the meninges.
- 27.Andersson PB, Perry VH & Gordon S The acute inflammatory response to lipopolysaccharide in CNS parenchyma differs from that in other body tissues. Neurosci 48, 169–186 (1992). [DOI] [PubMed] [Google Scholar]
- 28.Schlager C et al. Effector T-cell trafficking between the leptomeninges and the cerebrospinal fluid. Nature 530, 349–353 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Zamvil SS & Steinman L The T lymphocyte in experimental allergic encephalomyelitis. Annu. Rev. Immunol 8, 579–621 (1990). [DOI] [PubMed] [Google Scholar]
- 30.Long DM Capillary ultrastructure and the blood–brain barrier in human malignant brain tumors. J. Neurosurg 32, 127–144 (1970). [DOI] [PubMed] [Google Scholar]
- 31.Vajkoczy P & Menger MD Vascular microenvironment in gliomas. J. Neurooncol 50, 99–108 (2000). [DOI] [PubMed] [Google Scholar]
- 32.Watkins S et al. Disruption of astrocyte–vascular coupling and the blood-brain barrier by invading glioma cells. Nat. Commun 5, 4196 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Q & Barres BA Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol 18, 225–242 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Ginhoux F et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ajami B, Bennett JL, Krieger C, McNagny KM & Rossi FM Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat. Neurosci 14, 1142–1149 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Bennett FC et al. A combination of ontogeny and CNS environment establishes microglial identity. Neuron 98, 1170–1183 e1178 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chongsathidkiet P et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med 24, 1459–1468 (2018).This research uncovers the novel phenomenon of T cell sequestration within the bone marrow. The phenomenon is specific to tumours of the intracranial compartment and suggests a novel mechanism for CNS-imposed limitations to T cell access that tumours can usurp.
- 38.Fecci PE, Heimberger AB & Sampson JH Immunotherapy for primary brain tumors: no longer a matter of privilege. Clin. Cancer Res 20, 5620–5629 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gajewski TF, Schreiber H & Fu YX Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol 14, 1014–1022 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gajewski TF et al. Cancer immunotherapy targets based on understanding the T cell-inflamed versus non-T cell-inflamed tumor microenvironment. Adv. Exp. Med. Biol 1036, 19–31 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Keskin DB et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 565, 234–239 (2019).This study highlights some issues associated with using the neoantigen vaccine strategy for glioblastoma.
- 42.Quail DF & Joyce JA The microenvironmental landscape of brain tumors. Cancer Cell 31, 326–341 (2017).This excellent review article includes an in-depth discussion of the brain tumour microenvironment, relevant cell types, potential tumour-promoting mechanisms and possible therapeutics.
- 43.Perng P & Lim M Immunosuppressive mechanisms of malignant gliomas: parallels at non-CNS sites. Front. Oncol 5, 153 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vitkovic L, Maeda S & Sternberg E Anti-inflammatory cytokines: expression and action in the brain. Neuroimmunomodulation 9, 295–312 (2001). [DOI] [PubMed] [Google Scholar]
- 45.Gong D et al. TGFbeta signaling plays a critical role in promoting alternative macrophage activation. BMC Immunol 13, 31 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wainwright DA et al. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin. Cancer Res 18, 6110–6121 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Uyttenhove C et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med 9, 1269–1274 (2003). [DOI] [PubMed] [Google Scholar]
- 48.Zhang I et al. Characterization of arginase expression in glioma-associated microglia and macrophages. PLOS ONE 11, e0165118 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hawkins RA, O’Kane RL, Simpson IA & Vina JR Structure of the blood–brain barrier and its role in the transport of amino acids. J. Nutr 136, 218S–226S (2006). [DOI] [PubMed] [Google Scholar]
- 50.Bogdahn U et al. Targeted therapy for high-grade glioma with the TGF-beta2 inhibitor trabedersen: results of a randomized and controlled phase IIb study. Neuro Oncol 13, 132–142 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.den Hollander MW et al. TGF-beta antibody uptake in recurrent high-grade glioma imaged with 89Zr-fresolimumab PET. J. Nucl. Med 56, 1310–1314 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Brandes AA et al. A phase II randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro Oncol 18, 1146–1156 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.US National Library of Medicine. ClinicalTrials.gov, https://ClinicalTrials.gov/show/NCT02502708 (2015). [DOI] [PubMed]
- 54.US National Library of Medicine. ClinicalTrials.gov, https://ClinicalTrials.gov/show/NCT02052648 (2014). [DOI] [PubMed]
- 55.US National Library of Medicine. ClinicalTrials.gov, https://ClinicalTrials.gov/show/NCT02327078 (2014). [DOI] [PubMed]
- 56.US National Library of Medicine. ClinicalTrials.gov, https://ClinicalTrials.gov/show/NCT02752074 (2016). [DOI] [PubMed]
- 57.Long GV et al. Epacadostat (E) plus pembrolizumab (P) versus pembrolizumab alone in patients (pts) with unresectable or metastatic melanoma: results of the phase 3 ECHO-301/KEYNOTE-252 study. J. Clin. Oncol 36, 108–108 (2018). [Google Scholar]
- 58.US National Library of Medicine. ClinicalTrials.gov, https://ClinicalTrials.gov/show/NCT02903914 (2016). [DOI] [PubMed]
- 59.Graeber MB, Scheithauer BW & Kreutzberg GW Microglia in brain tumors. Glia 40, 252–259 (2002). [DOI] [PubMed] [Google Scholar]
- 60.Chen Z et al. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res 77, 2266–2278 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bowman RL et al. Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Rep 17, 2445–2459 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou W et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol 17, 170–182 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hambardzumyan D, Gutmann DH & Kettenmann H The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci 19, 20–27 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Komohara Y, Ohnishi K, Kuratsu J & Takeya M Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J. Pathol 216, 15–24 (2008). [DOI] [PubMed] [Google Scholar]
- 65.Murray PJ et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Majety M, Runza V, Lehmann C, Hoves S & Ries CH A drug development perspective on targeting tumor-associated myeloid cells. FEBS J 285, 763–776 (2018). [DOI] [PubMed] [Google Scholar]
- 67.Hume DA & MacDonald KP Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling. Blood 119, 1810–1820 (2012). [DOI] [PubMed] [Google Scholar]
- 68.Pyonteck SM et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med 19, 1264–1272 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.US National Library of Medicine. ClinicalTrials.gov, https://ClinicalTrials.gov/show/NCT01349036 (2011). [DOI] [PubMed]
- 70.Butowski N et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro Oncol 18, 557–564 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Germano G et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 23, 249–262 (2013). [DOI] [PubMed] [Google Scholar]
- 72.Vakilian A, Khorramdelazad H, Heidari P, Sheikh Rezaei Z & Hassanshahi G CCL2/CCR2 signaling pathway in glioblastoma multiforme. Neurochem. Int 103, 1–7 (2017). [DOI] [PubMed] [Google Scholar]
- 73.Vonderheide RH & Glennie MJ Agonistic CD40 antibodies and cancer therapy. Clin. Cancer Res 19, 1035–1043 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kaneda MM et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature 539, 437–442 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Weiskopf K Cancer immunotherapy targeting the CD47/SIRPalpha axis. Eur. J. Cancer 76, 100–109 (2017). [DOI] [PubMed] [Google Scholar]
- 76.Kwong LS, Brown MH, Barclay AN & Hatherley D Signal-regulatory protein alpha from the NOD mouse binds human CD47 with an exceptionally high affinity—implications for engraftment of human cells. Immunology 143, 61–67 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hodges TR et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro Oncol 19, 1047–1057 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bouffet E et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J. Clin. Oncol 34, 2206–2211 (2016). [DOI] [PubMed] [Google Scholar]
- 79.Muscat AM et al. The evolutionary pattern of mutations in glioblastoma reveals therapy-mediated selection. Oncotarget 9, 7844–7858 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Johnson BE et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 343, 189–193 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Barthel F et al. Longitudinal molecular trajectories of diffuse glioma in adults. Nature (in the press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kim H et al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res 25, 316–327 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang J et al. Clonal evolution of glioblastoma under therapy. Nat. Genet 48, 768–776 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schumacher TN & Schreiber RD Neoantigens in cancer immunotherapy. Science 348, 69–74 (2015). [DOI] [PubMed] [Google Scholar]
- 85.Van Allen EM et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350, 207–211 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Goodman AM et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol. Cancer Ther 16, 2598–2608 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hellmann MD et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N. Engl. J. Med 378, 2093–2104 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hellmann MD et al. Genomic features of response to combination immunotherapy in patients with advanced non-small-cell lung cancer. Cancer Cell 33, 843–852.e844 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Therkildsen C et al. Glioblastomas, astrocytomas and oligodendrogliomas linked to Lynch syndrome. Eur. J. Neurol 22, 717–724 (2015). [DOI] [PubMed] [Google Scholar]
- 90.Dunn GP, Old LJ & Schreiber RD The three Es of cancer immunoediting. Annu. Rev. Immunol 22, 329–360 (2004). [DOI] [PubMed] [Google Scholar]
- 91.Marty Pyke R et al. Evolutionary pressure against MHC class II binding cancer mutations. Cell 175, 416–428. e413 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.McGranahan N et al. Allele-Specific HLA loss and immune escape in lung cancer evolution. Cell 171, 1259–1271 e1211 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang Q et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 32, 42–56 e46 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rooney MS, Shukla SA, Wu CJ, Getz G & Hacohen N Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48–61 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dunn GP, Bruce AT, Ikeda H, Old LJ & Schreiber RD Cancer immunoediting: from immunosurveillance to tumor escape. Nat. Immunol 3, 991–998 (2002). [DOI] [PubMed] [Google Scholar]
- 96.Zhao J et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med 25, 462–469 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.US National Library of Medicine. ClinicalTrials.gov, https://ClinicalTrials.gov/show/NCT03718767 (2018). [DOI] [PubMed]
- 98.Bigner SH et al. Characterization of the epidermal growth factor receptor in human glioma cell lines and xenografts. Cancer Res 50, 8017–8022 (1990). [PubMed] [Google Scholar]
- 99.Humphrey PA et al. Anti-synthetic peptide antibody reacting at the fusion junction of deletion-mutant epidermal growth factor receptors in human glioblastoma. Proc. Natl Acad. Sci. USA 87, 4207–4211 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wikstrand CJ, McLendon RE, Friedman AH & Bigner DD Cell surface localization and density of the tumor-associated variant of the epidermal growth factor receptor, EGFRvIII. Cancer Res 57, 4130–4140 (1997). [PubMed] [Google Scholar]
- 101.Padfield E, Ellis HP & Kurian KM Current Therapeutic Advances Targeting EGFR and EGFRvIII in Glioblastoma. Front. Oncol 5, 5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Migliorini D et al. CAR T-Cell Therapies in glioblastoma: a first look. Clin. Cancer Res 24, 535–540 (2018). [DOI] [PubMed] [Google Scholar]
- 103.O’Rourke DM et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med 9, eaaa0984 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hilf N et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 565, 240–245 (2019). [DOI] [PubMed] [Google Scholar]
- 105.Cloughesy TF et al. Phase 1 trial of vocimagene amiretrorepvec and 5-fluorocytosine for recurrent high-grade glioma. Sci. Transl. Med 8, 341ra375 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Desjardins A et al. Recurrent glioblastoma treated with recombinant poliovirus. N. Engl. J. Med 379, 150–161 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fountzilas C, Patel S & Mahalingam D Review: oncolytic virotherapy, updates and future directions. Oncotarget 8, 102617–102639 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lang FF et al. Phase I study of DNX-2401 (Delta-24-RGD) oncolytic adenovirus: replication and immunotherapeutic effects in recurrent malignant glioma. J. Clin. Oncol 36, 1419–1427 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Markert JM et al. A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol. Ther 22, 1048–1055 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cai J et al. Loss of ATRX, associated with DNA methylation pattern of chromosome end, impacted biological behaviors of astrocytic tumors. Oncotarget 6, 18105–18115 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gusyatiner O & Hegi ME Glioma epigenetics: from subclassification to novel treatment options. Semin. Cancer Biol 51, 50–58 (2018). [DOI] [PubMed] [Google Scholar]
- 112.Hassan A, Mosley J, Singh S & Zinn PO A comprehensive review of genomics and noncoding RNA in gliomas. Top Magn. Reson. Imaging 26, 3–14 (2017). [DOI] [PubMed] [Google Scholar]
- 113.Johann PD et al. Atypical teratoid/rhabdoid tumors are comprised of three epigenetic subgroups with distinct enhancer landscapes. Cancer Cell 29, 379–393 (2016). [DOI] [PubMed] [Google Scholar]
- 114.Mao CX et al. The molecular classification of astrocytic tumors. Oncotarget 8, 96340–96350 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Loo Yau H, Ettayebi I & De Carvalho DD The cancer epigenome: exploiting its vulnerabilities for immunotherapy. Trends Cell Biol 29, 31–43 (2019). [DOI] [PubMed] [Google Scholar]
- 116.Pan D et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 359, 770–775 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Miao D et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 359, 801–806 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shu XS et al. The epigenetic modifier PBRM1 restricts the basal activity of the innate immune system by repressing retinoic acid-inducible gene-I-like receptor signalling and is a potential prognostic biomarker for colon cancer. J. Pathol 244, 36–48 (2018). [DOI] [PubMed] [Google Scholar]
- 119.Shen J et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat. Med 24, 556–562 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Amankulor NM et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev 31, 774–786 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kohanbash G et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J. Clin. Invest 127, 1425–1437 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhang X et al. IDH mutant gliomas escape natural killer cell immune surveillance by downregulation of NKG2D ligand expression. Neuro Oncol 18, 1402–1412 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Su YB et al. Selective CD4+ lymphopenia in melanoma patients treated with temozolomide: a toxicity with therapeutic implications. J. Clin. Oncol 22, 610–616 (2004). [DOI] [PubMed] [Google Scholar]
- 124.Sampson JH et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro Oncol 13, 324–333 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Fadul CE et al. Immune modulation effects of concomitant temozolomide and radiation therapy on peripheral blood mononuclear cells in patients with glioblastoma multiforme. Neuro Oncol 13, 393–400 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Grossman SA et al. Survival in patients with severe lymphopenia following treatment with radiation and chemotherapy for newly diagnosed solid tumors. J. Natl Compr. Canc. Netw 13, 1225–1231 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Batich KA et al. Long-term survival in glioblastoma with cytomegalovirus pp65-targeted vaccination. Clin. Cancer Res 23, 1898–1909 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Asavaroengchai W, Kotera Y & Mule JJ Tumor lysate-pulsed dendritic cells can elicit an effective antitumor immune response during early lymphoid recovery. Proc. Natl Acad. Sci. USA 99, 931–936 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nistico P et al. Chemotherapy enhances vaccine-induced antitumor immunity in melanoma patients. Int. J. Cancer 124, 130–139 (2009). [DOI] [PubMed] [Google Scholar]
- 130.Schaaf MB, Garg AD & Agostinis P Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis 9, 115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yang J, Yan J & Liu B Targeting VEGF/VEGFR to Modulate Antitumor Immunity. Front. Immunol 9, 978 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Manzoni M et al. Immunological effects of bevacizumab-based treatment in metastatic colorectal cancer. Oncology 79, 187–196 (2010). [DOI] [PubMed] [Google Scholar]
- 133.Martino EC et al. Immune-modulating effects of bevacizumab in metastatic non-small-cell lung cancer patients. Cell Death Discov 2, 16025 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Larkin J, Hodi FS & Wolchok JD Combined Nivolumab and Ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med 373, 1270–1271 (2015). [DOI] [PubMed] [Google Scholar]
- 135.Robert C et al. Pembrolizumab versus Ipilimumab in advanced melanoma. N. Engl. J. Med 372, 2521–2532 (2015). [DOI] [PubMed] [Google Scholar]
- 136.Motzer RJ et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med 373, 1803–1813 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Garon EB et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med 372, 2018–2028 (2015). [DOI] [PubMed] [Google Scholar]
- 138.Ferris RL et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N. Engl. J. Med 375, 1856–1867 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Immunotherapy Timeline of Progress, https://www.cancerresearch.org/immunotherapy/timeline-of-progress (2019).
- 140.Agarwalla P, Barnard Z, Fecci P, Dranoff G & Curry WT Jr Sequential immunotherapy by vaccination with GM-CSF-expressing glioma cells and CTLA-4 blockade effectively treats established murine intracranial tumors. J. Immunother 35, 385–389 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Fecci PE et al. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin. Cancer Res 13, 2158–2167 (2007).This study is the first to preclinically test immune checkpoint blockade in a brain tumour model.
- 142.Antonios JP et al. PD-1 blockade enhances the vaccination-induced immune response in glioma. JCI Insight 1, e87059 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Reardon DA et al. Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model. Cancer Immunol. Res 4, 124–135 (2016). [DOI] [PubMed] [Google Scholar]
- 144.Wainwright DA et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin. Cancer Res 20, 5290–5301 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zeng J et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int. J. Radiat. Oncol. Biol. Phys 86, 343–349 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Filley AC, Henriquez M & Dey M Recurrent glioma clinical trial, CheckMate-143: the game is not over yet. Oncotarget 8, 91779–91794 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.US National Library of Medicine. ClinicalTrials.gov, https://ClinicalTrials.gov/show/NCT02617589 (2015). [DOI] [PubMed]
- 148.US National Library of Medicine. ClinicalTrials.gov, https://ClinicalTrials.gov/show/NCT02667587 (2016). [DOI] [PubMed]
- 149.Wu A et al. Combination anti-CXCR4 and anti-PD-1 immunotherapy provides survival benefit in glioblastoma through immune cell modulation of tumor microenvironment. J. Neurooncol 143, 241–249 (2019). [DOI] [PubMed] [Google Scholar]
- 150.Woroniecka K et al. T-Cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin. Cancer Res 24, 4175–4186 (2018).This study is the first to reveal and characterize the severity of bona fide T cell exhaustion in patients with glioblastoma.
- 151.Tawbi HA et al. Combined nivolumab and ipilimumab in melanoma metastatic to the brain. N. Engl. J. Med 379, 722–730 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Brooks WH, Caldwell HD & Mortara RH Immune responses in patients with gliomas. Surg. Neurol 2, 419–423 (1974). [PubMed] [Google Scholar]
- 153.Brooks WH, Roszman TL & Rogers AS Impairment of rosette-forming T lymphocytes in patients with primary intracranial tumors. Cancer 37, 1869–1873 (1976). [DOI] [PubMed] [Google Scholar]
- 154.Fecci PE et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res 66, 3294–3302 (2006).This publication is the first to demonstrate the systemic immune impact of Treg cells in patients with glioblastoma.
- 155.Dunn GP, Fecci PE & Curry WT Cancer immunoediting in malignant glioma. Neurosurg 71, 201–222; discussion 222–223 (2012). [DOI] [PubMed] [Google Scholar]
- 156.Woroniecka KI, Rhodin KE, Chongsathidkiet P, Keith KA & Fecci PE T-cell dysfunction in glioblastoma: applying a new framework. Clin. Cancer Res 24, 3792–3802 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Dix AR, Brooks WH, Roszman TL & Morford LA Immune defects observed in patients with primary malignant brain tumors. J. Neuroimmunol 100, 216–232 (1999). [DOI] [PubMed] [Google Scholar]
- 158.Brooks WH, Netsky MG, Normansell DE & Horwitz DA Depressed cell-mediated immunity in patients with primary intracranial tumors. Characterization of a humoral immunosuppressive factor. J. Exp. Med 136, 1631–1647 (1972). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Roszman TL & Brooks WH Immunobiology of primary intracranial tumours. III. Demonstration of a qualitative lymphocyte abnormality in patients with primary brain tumours. Clin. Exp. Immunol 39, 395–402 (1980). [PMC free article] [PubMed] [Google Scholar]
- 160.Koyama S et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun 7, 10501 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Chauvin JM et al. TIGIT and PD-1 impair tumor antigen-specific CD8+ T cells in melanoma patients. J. Clin. Invest 125, 2046–2058 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Blackburn SD et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol 10, 29–37 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Woo SR et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res 72, 917–927 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kurtulus S et al. TIGIT predominantly regulates the immune response via regulatory T cells. J. Clin. Invest 125, 4053–4062 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 165.Angelosanto JM, Blackburn SD, Crawford A & Wherry EJ Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. J. Virol 86, 8161–8170 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wherry EJ & Kurachi M Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol 15, 486–499 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kim JE et al. Combination therapy with anti-PD-1, anti-TIM-3, and focal radiation results in regression of murine gliomas. Clin. Cancer Res 23, 124–136 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.US National Library of Medicine. ClinicalTrials.gov, https://ClinicalTrials.gov/show/NCT02658981 (2016). [DOI] [PubMed]
- 169.US National Library of Medicine. ClinicalTrials.gov, https://ClinicalTrials.gov/show/NCT02817633 (2016). [DOI] [PubMed]
- 170.Cloughesy TF et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med 25, 477–486 (2019).This report describes the first trial to show the efficacy of immune checkpoint blockade in patients with glioblastoma, which was accomplished via simple neoadjuvant administration prior to resection of recurrent tumours.
- 171.Schalper KA et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat. Med 25, 470–476 (2019). [DOI] [PubMed] [Google Scholar]
- 172.Law TM et al. Phase III randomized trial of interleukin-2 with or without lymphokine-activated killer cells in the treatment of patients with advanced renal cell carcinoma. Cancer 76, 824–832 (1995). [DOI] [PubMed] [Google Scholar]
- 173.Rosenberg SA & Dudley ME Cancer regression in patients with metastatic melanoma after the transfer of autologous antitumor lymphocytes. Proc. Natl Acad. Sci. USA 101, 14639–14645 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Schuessler A et al. Autologous T-cell therapy for cytomegalovirus as a consolidative treatment for recurrent glioblastoma. Cancer Res 74, 3466–3476 (2014). [DOI] [PubMed] [Google Scholar]
- 175.Cobbs CS et al. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res 62, 3347–3350 (2002). [PubMed] [Google Scholar]
- 176.US National Library of Medicine. ClinicalTrials.gov, https://ClinicalTrials.gov/show/NCT02661282 (2016). [DOI] [PubMed]
- 177.Maus MV, Grupp SA, Porter DL & June CH Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood 123, 2625–2635 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ahmed N et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol 3, 1094–1101 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Brown CE et al. Bioactivity and safety of IL13R alpha2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin. Cancer Res 21, 4062–4072 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Brown CE et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N. Engl. J. Med 375, 2561–2569 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Long KB et al. CAR T cell therapy of non-hematopoietic malignancies: detours on the road to clinical success. Front. Immunol 9, 2740 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kuhn NF et al. CD40 ligand-modified chimeric antigen receptor T cells enhance antitumor function by eliciting an endogenous antitumor response. Cancer Cell 35, 473–488 e476 (2019).This article describes a preclinical study in which the expression of CD40L on CAR T cells led to the killing of antigen-negative tumour cells and the elimination of tumour immune escape via antigen loss.
- 183.Choi BD et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol 37, 1049–1058 (2019). [DOI] [PubMed] [Google Scholar]
- 184.Fraietta JA et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med 24, 563–571 (2018).This study identifies predictors of clinical response to CAR T cell therapy as being exclusively intrinsic properties of the administered cells.
- 185.Fraietta JA et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 558, 307–312 (2018).This case report describes how fortuitous deletion of the TET2 gene in CARs led to complete remission of CLL.
- 186.Ichiyama K et al. The methylcytosine dioxygenase Tet2 promotes DNA demethylation and activation of cytokine gene expression in T cells. Immunity 42, 613–626 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Shlush LI Age-related clonal hematopoiesis. Blood 131, 496–504 (2018). [DOI] [PubMed] [Google Scholar]
- 188.Carty SA et al. The loss of TET2 promotes CD8+ T cell memory differentiation. J. Immunol 200, 82–91 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Cull AH, Snetsinger B, Buckstein R, Wells RA & Rauh MJ Tet2 restrains inflammatory gene expression in macrophages. Exp. Hematol 55, 56–70 e13 (2017). [DOI] [PubMed] [Google Scholar]
- 190.Zhang Q et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 525, 389–393 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Chheda ZS et al. Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J. Exp. Med 215, 141–157 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Mount CW et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M+ diffuse midline gliomas. Nat. Med 24, 572–579 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Sampson JH et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J. Clin. Oncol 28, 4722–4729 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Weller M et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol 18, 1373–1385 (2017). [DOI] [PubMed] [Google Scholar]
- 195.Oji Y et al. Association of WT1 IgG antibody against WT1 peptide with prolonged survival in glioblastoma multiforme patients vaccinated with WT1 peptide. Int. J. Cancer 139, 1391–1401 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Rampling R et al. A cancer research UK first time in human phase I Trial of IMA950 (novel multipeptide therapeutic vaccine) in patients with newly diagnosed glioblastoma. Clin. Cancer Res 22, 4776–4785 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Pollack IF et al. Antigen-specific immune responses and clinical outcome after vaccination with glioma-associated antigen peptides and polyinosinic–polycytidylic acid stabilized by lysine and carboxymethylcellulose in children with newly diagnosed malignant brainstem and nonbrainstem gliomas. J. Clin. Oncol 32, 2050–2058 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Liau LM et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin. Cancer Res 11, 5515–5525 (2005). [DOI] [PubMed] [Google Scholar]
- 199.Prins RM et al. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin. Cancer Res 17, 1603–1615 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Okada H et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with α-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J. Clin. Oncol 29, 330–336 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Phuphanich S et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol. Immunother 62, 125–135 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Wen PY et al. A randomized, double-blind, placebo-controlled phase 2 trial of dendritic cell (DC) vaccination with ICT-107 in newly diagnosed glioblastoma (GBM) patients. J. Clin. Oncol 32, 2005–2005 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Liau LM et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med 16, 142 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Van Brussel I, Berneman ZN & Cools N Optimizing dendritic cell-based immunotherapy: tackling the complexity of different arms of the immune system. Mediators Inflamm 2012, 690643 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Mailliard RB et al. Alpha-type-1 polarized dendritic cells: a novel immunization tool with optimized CTL-inducing activity. Cancer Res 64, 5934–5937 (2004). [DOI] [PubMed] [Google Scholar]
- 206.Mitchell DA et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 519, 366–369 (2015).This study demonstrates that the activation of memory T cells prior to DC vaccination leads to increased migration of DCs to lymph nodes and improved clinical responses.
- 207.Garg AD, Coulie PG, Van den Eynde BJ & Agostinis P Integrating next-generation dendritic cell vaccines into the current cancer immunotherapy landscape. Trends Immunol 38, 577–593 (2017). [DOI] [PubMed] [Google Scholar]
- 208.Nandu H, Wen PY & Huang RY Imaging in neuro-oncology. Ther. Adv. Neurol. Disord 11, 1756286418759865 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Aquino D, Gioppo A, Finocchiaro G, Bruzzone MG & Cuccarini V MRI in glioma immunotherapy: evidence, pitfalls, and perspectives. J. Immunol. Res 2017, 5813951 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Park J et al. Immune checkpoint inhibitor-induced reinvigoration of tumor-infiltrating CD8+ T cells is determined by their differentiation status in glioblastoma. Clin. Cancer Res 25, 2549–2559 (2019). [DOI] [PubMed] [Google Scholar]
- 211.US National Library of Medicine. ClinicalTrials.gov, https://ClinicalTrials.gov/show/NCT03068832 (2017). [DOI] [PubMed]
- 212.US National Library of Medicine. ClinicalTrials.gov, https://ClinicalTrials.gov/show/NCT04015700 (2019). [DOI] [PubMed]
- 213.US National Library of Medicine. ClinicalTrials.gov, https://ClinicalTrials.gov/show/NCT03422094 (2018). [DOI] [PubMed]
- 214.US National Library of Medicine. ClinicalTrials.gov, https://ClinicalTrials.gov/show/NCT03726515 (2018). [DOI] [PubMed]
- 215.US National Library of Medicine. ClinicalTrials.gov, https://ClinicalTrials.gov/show/NCT03638167 (2018). [DOI] [PubMed]
- 216.US National Library of Medicine. ClinicalTrials.gov, https://ClinicalTrials.gov/show/NCT03500991 (2018). [DOI] [PubMed]
- 217.US National Library of Medicine. ClinicalTrials.gov, https://ClinicalTrials.gov/show/NCT02442297 (2015). [DOI] [PubMed]
- 218.US National Library of Medicine. ClinicalTrials.gov, https://ClinicalTrials.gov/show/NCT02208362 (2014). [DOI] [PubMed]
- 219.US National Library of Medicine. ClinicalTrials.gov, https://ClinicalTrials.gov/show/NCT03299309 (2017). [DOI] [PubMed]
- 220.US National Library of Medicine. ClinicalTrials.gov, https://ClinicalTrials.gov/show/NCT02529072 (2015). [DOI] [PubMed]
- 221.US National Library of Medicine. ClinicalTrials.gov, https://ClinicalTrials.gov/show/NCT02193347 (2014). [DOI] [PubMed]
- 222.US National Library of Medicine. ClinicalTrials.gov, https://ClinicalTrials.gov/show/NCT03223103 (2017). [DOI] [PubMed]
- 223.US National Library of Medicine. ClinicalTrials.gov, https://ClinicalTrials.gov/show/NCT03914768 (2019). [DOI] [PubMed]
- 224.US National Library of Medicine. ClinicalTrials.gov, https://ClinicalTrials.gov/show/NCT03149003 (2017). [DOI] [PubMed]
- 225.US National Library of Medicine. ClinicalTrials.gov, https://ClinicalTrials.gov/show/NCT01204684 (2010). [DOI] [PubMed]
- 226.US National Library of Medicine. ClinicalTrials.gov, https://ClinicalTrials.gov/show/NCT03927222 (2019). [DOI] [PubMed]
- 227.US National Library of Medicine. ClinicalTrials.gov, https://ClinicalTrials.gov/show/NCT03688178 (2018). [DOI] [PubMed]
- 228.Peng D et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 527, 249–253 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Johnson LA, Sanchez-Perez L, Suryadevara CM & Sampson JH Chimeric antigen receptor engineered T cells can eliminate brain tumors and initiate long-term protection against recurrence. Oncoimmunology 3, e944059 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Farber SH et al. Embracing rejection: immunologic trends in brain metastasis. Oncoimmunology 5, e1172153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Fischer GM et al. Molecular profiling reveals unique immune and metabolic features of melanoma brain metastases. Cancer Discov 9, 628–645 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Badie B & Schartner JM Flow cytometric characterization of tumor-associated macrophages in experimental gliomas. Neurosurgery 46, 957–961; discussion 961–962 (2000). [DOI] [PubMed] [Google Scholar]
- 233.Bloch O et al. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin. Cancer Res 19, 3165–3175 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Parney IF, Waldron JS & Parsa AT Flow cytometry and in vitro analysis of human glioma-associated macrophages: laboratory investigation. J. Neurosurg 110, 572–582 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Sugihara AQ, Rolle CE & Lesniak MS Regulatory T cells actively infiltrate metastatic brain tumors. Int. J. Oncol 34, 1533–1540 (2009). [DOI] [PubMed] [Google Scholar]
- 236.Berghoff AS et al. Tumour-infiltrating lymphocytes and expression of programmed death ligand 1 (PD-L1) in melanoma brain metastases. Histopathology 66, 289–299 (2015). [DOI] [PubMed] [Google Scholar]