

Abstract
Fibrosis is a common pathologic outcome of chronic disease resulting in the replacement of normal tissue parenchyma with a collagen-rich extracellular matrix produced by myofibroblasts. Although the progenitor cell types and cellular programs giving rise to myofibroblasts through mesenchymal transition can vary between tissues and diseases, their contribution to fibrosis initiation, maintenance, and progression is thought to be pervasive. Here we showed that the ability of transforming growth factor-β (TGFβ) to efficiently induce myofibroblast differentiation of cultured epithelial cells, endothelial cells, or quiescent fibroblasts is dependent upon the induced expression and activity of dimeric calpains, a family of non-lysosomal cysteine proteases that regulate a variety of cellular events through post-translational modification of diverse substrates. siRNA-based gene silencing demonstrated that TGFβ-induced mesenchymal transition of a murine breast epithelial cell line was dependent upon induction of expression of calpain 9 (CAPN9), an isoform previously thought to be restricted to the gastrointestinal tract. Mice lacking functional CAPN9 due to biallelic targeting of Capn9 were viable and fertile, but showed overt protection from bleomycin-induced lung fibrosis, carbon tetrachloride-induced liver fibrosis, and angiotensin II-induced cardiac fibrosis and dysfunction. A predicted loss-of-function allele of CAPN9 is common in Southeast Asia, with the frequency of homozygosity matching the prediction of Hardy-Weinberg equilibrium; together with the highly spatially-restricted pattern of CAPN9 expression under physiologic circumstances and the heartiness of the murine knockout, these data provide a strong signature for tolerance of therapeutic strategies for fibrosis aimed at CAPN9 antagonism.
One Sentence Summary:
Inhibition of calpain 9, a key mediator of myofibroblast differentiation and tissue fibrosis, protects against fibrosis in preclinical models.
Introduction
The replacement and distortion of tissue parenchyma with fibrillar collagens and other extracellular matrix (ECM) proteins – thereby compromising organ function – is a common feature of chronic disease and contributes to substantial number of deaths in the industrialized world (1–3). Although collagen deposition is an indispensable component of tissue homeostasis, chronic injury or dysregulation of wound healing can lead to pathologic scarring, a condition termed fibrosis (4). In some instances, provocations that induce tissue fibrosis have been identified, such as curtained genetic conditions (5–7), chemical exposures (8), and chronic inflammation secondary to autoimmune disorders (1). In other cases, such as the majority of idiopathic pulmonary fibrosis presentations, the specific driver of fibrosis is unknown.
Regardless of the initiating events, all fibrotic disorders show accumulation of activated fibroblasts that are invasive, synthetic, contractile, proliferative, and long-lived (9). The profibrotic cytokine transforming growth factor beta (TGFβ) can induce differentiation of a variety of progenitor lineages, including epithelial or endothelial cells, resident fibroblasts, or pericytes (10, 11), to so-called myofibroblasts in a process known as mesenchymal transition. The specific source of myofibroblasts in fibrotic diseases remains controversial and is likely varied, however the prevailing view is that mesenchymal transition plays a prominent role in most if not all fibrotic contexts (10). Typical alterations in cellular phenotype that accompany TGFβ-mediated epithelial- or endothelial-to-mesenchymal transition (EpMT or EnMT, respectively; EMT collectively) include down-regulation of markers of a mature polarized cell state (e.g., E-cadherin) and induction of mesenchymal markers – e.g., expression of a-smooth muscle actin (αSMA), vimentin, fibrillar collagens, and matrix metalloproteases (MMPs) 2 and 9 (12). Efforts to fate-map cells in fibrotic models of lung, liver, and heart fibrosis provide ample evidence for and against a role of EMT in the accumulation of myofibroblasts in vivo (13). Nevertheless, TGFβ-signaling induces canonical EMT transcription factors (12), and genetic deletion of EMT transcription factors in lung alveolar cells or in hepatocytes blunts experimentally-induced organ fibrosis (14, 15). Given the clear role of TGFβ in fibrosis, we reasoned that a distal molecular event that is critical for TGFβ-induced mesenchymal transition would be an attractive therapeutic target for multiple etiologies of fibrosis.
We were intrigued by the association in the literature between multiple EMT-related disease processes and the increased expression or activity of calpains – a family of calcium-dependent non-lysosomal cysteine proteases that cleave diverse substrates to regulate cell activities including differentiation, adhesion, invasion, migration, synthetic repertoire, and survival (16). For example, calpain activity has been mechanistically linked to the invasive behavior of epithelial tumors (17–20), normal wound healing (21), cardiac fibrosis after tissue injury (22–25), and lung fibrosis in response to bleomycin (26). These observations led to our hypothesis that specific calpain cleavage products are required for mesenchymal transition and that inhibition of calpain activity may have therapeutic value in fibrotic disorders.
Of the 15 calpain isoforms expressed by humans, CAPN1 and CAPN2 are the best characterized, and are termed the conventional classical calpains (27). Active CAPN1 and 2 enzymes consist of a heterodimer formed with small regulatory subunit CAPNS1. An alternative subunit, CAPNS2, is of unknown physiologic function. (28) The activity of these conventional dimeric calpains is tightly regulated by the endogenous calpain inhibitor calpastatin (CAST). CAST is thought to specifically inhibit all dimeric calpains. CAST binds near the active site cleft of dimeric calpains in the presence of calcium and prevents engagement of substrates but is protected from hydrolysis by not binding the active site itself (29–31). CAPN1, 2, S1, and CAST are ubiquitously expressed; however, other calpain isoforms are expressed primarily in specific tissues or organs. For example, large subunit CAPN9 is reportedly chiefly expressed in the gastrointestinal tract (32), whereas CAPNS2 reportedly shows predominant expression in the skin and esophagus (33).
In this study, we used complementary methods to implicate calpain 9 (Capn9) and calpain small subunit 2 (Capns2) in TGFβ-induced myofibroblast differentiation in vitro and in multiple experimentally induced models of fibrosis in vivo. Capn9 showed highly restricted physiologic expression but could be potently induced in naïve cell types by TGFβ, suggesting the potential of high tolerance for therapeutic strategies for fibrosis aimed at antagonism.
Results
Calpain inhibition prevents TGFβ-induced mesenchymal transition without affecting proximal signaling events
To explore the role of calpain proteases in mesenchymal transition, we initially turned to a robust cell culture-based TGFβ-induced EMT assay using the Namru Mouse Mammary Gland (NMuMG) epithelial cell line (34, 35). TGFβ-stimulated NMuMG cells demonstrated robust phosphorylation of SMAD2 (pSMAD2) accumulation at all assayed time points and expressed αSMA beginning at 48 h, indicating differentiation into myofibroblasts. Treatment with MDL-28170, a broad spectrum calpain and cathepsin inhibitor, resulted in a dose-dependent decrease in αSMA expression at 48 and 72 h post-TGFβ stimulation (fig. 1A). MDL-28170 did not affect TGFβ-dependent phosphorylation (fig. 1A) or nuclear accumulation (fig. S1A) of SMAD2, suggesting the relevance of events that occur distal to TGFβ receptor-mediated signal transduction.
Fig. 1. Broad spectrum calpain inhibition attenuates TGFβ-induced EMT in NMuMG cells.
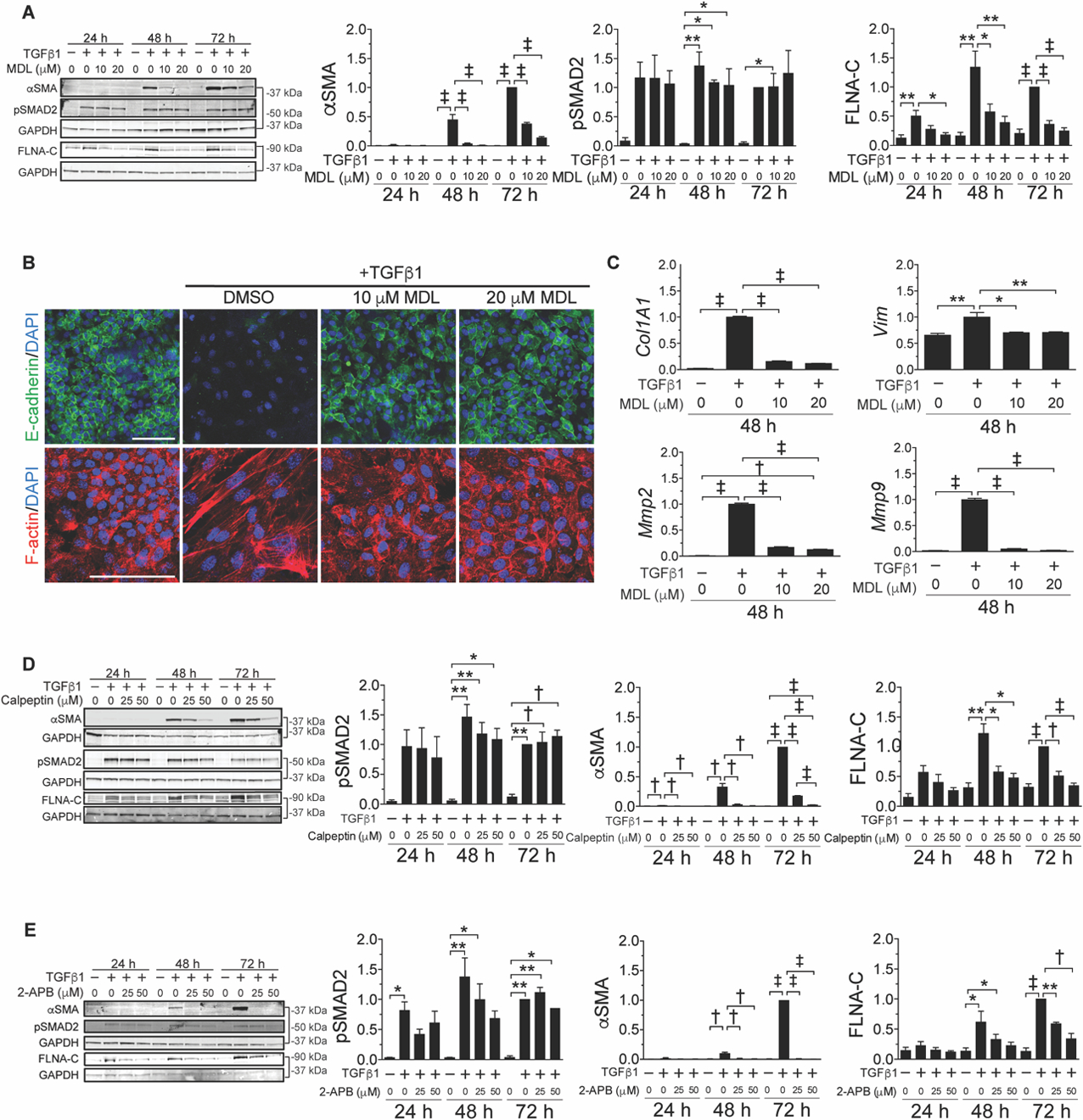
(A) Representative immunoblots (left, bracket indicates identical gel) and quantification (right, normalized to GAPDH) for the indicated proteins, time points after TGFβ stimulation, and concentrations of MDL-28170 (n =3 to 4). (B) Representative immunofluorescence images of TGFβ-induced EMT in NMuMG cells stained with E-cadherin (green), F-actin (red), and DAPI (blue). Scale bar: 100 µm. (C) Relative gene expression (normalized to Gapdh) in response to TGFβ with or without calpain inhibition (MDL) (n = 3). (D) Representative immunoblots (left) and quantification (right) for the indicated proteins, time points after TGFβ stimulation, and concentrations of calpeptin (n = 3). (E) Representative immunoblots (left) and quantification (right) for the indicated proteins, time points after TGFβ stimulation, and concentrations the calcium channel blocker 2-APB (n = 3). Data are expressed as mean ± s.e.m. *P < 0.05, **P < 0.01, †P < 0.005, ‡P < 0.001 by one-way ANOVA with Tukey’s post hoc test.
We observed that TGFβ induced calpain activity, as evidenced by production of a calpain-dependent filamin A C-terminal cleavage product (FLNA-C) (36). TGFβ stimulation induced FLNA-C accumulation in NMuMG cells that was attenuated by MDL-28170 in a dose-dependent manner (fig. 1A). We confirmed that accumulation of αSMA and FLNA-C related to canonical, SMAD-dependent TGFβ signaling in this system, because treatment with SB431542, a TGFβ receptor kinase inhibitor that precludes TGFβ receptor-dependent SMAD2/3 phosphorylation (fig. S1B), attenuated both.
Transition from an epithelial to a mesenchymal state was associated with downregulation of cell surface expression of E-cadherin, reorganization of F-actin from a cortical to a stress fiber distribution, and enhanced mRNA expression of type I collagen (Col1a1), vimentin (Vim), and MMP-2 and −9 (Mmp2,9) (fig. 1B,C). Treatment of NMuMG cells with MDL-28170 prior to administration of TGFβ attenuated each of these events.
A second broad spectrum calpain inhibitor, calpeptin (37), also inhibited TGFβ-induced EMT in NMuMG cells in association with decreased FLNA cleavage (fig. 1D). Similar results were obtained with 2-APB (38), a potent inhibitor of calcium channels and the calcium influx required for calpains to adopt a catalytically active conformation (fig. 1E) (39, 40). Calpeptin, MDL-28170, as well as other available broad-spectrum calpain inhibitors, also antagonize cathepsins, another class of calcium-dependent proteases. Evidence for the specific relevance of calpains included failure of CA-074-OMe, a cathepsin B and L inhibitor that does not cross react with calpains, to suppress TGFβ-induced EMT in NMuMG cells (fig. S1C) (41, 42).
As reported previously, mesenchymal differentiation of NMuMG cells is reversed when TGFβ is removed (34). Here, we found that myofibroblasts derived from NMuMG cells showed a decline in αSMA expression, reorganization of F-actin, and re-emergence of epithelial marker expression (i.e. E-cadherin) upon addition of MDL-28170 despite the continued presence of TGFβ (fig. S2).
Calpastatin-mediated inhibition of mesenchymal transition implicates dimeric calpain isoforms
We overexpressed CAST in NMuMG cells by transfecting with a CAST-IRES-GFP construct. The expression of CAST, as monitored by GFP abundance, was not influenced by the addition of TGFβ. Overexpression of CAST in TGFβ-stimulated NMuMG cells blocked the expression of αSMA and the cleavage of FLNA, but it did not alter SMAD2 phosphorylation (fig. 2A).
Fig. 2. Dimeric calpain isoforms CAPN9 and CAPNS2 mediate TGFβ-induced EMT in NMuMG cells.
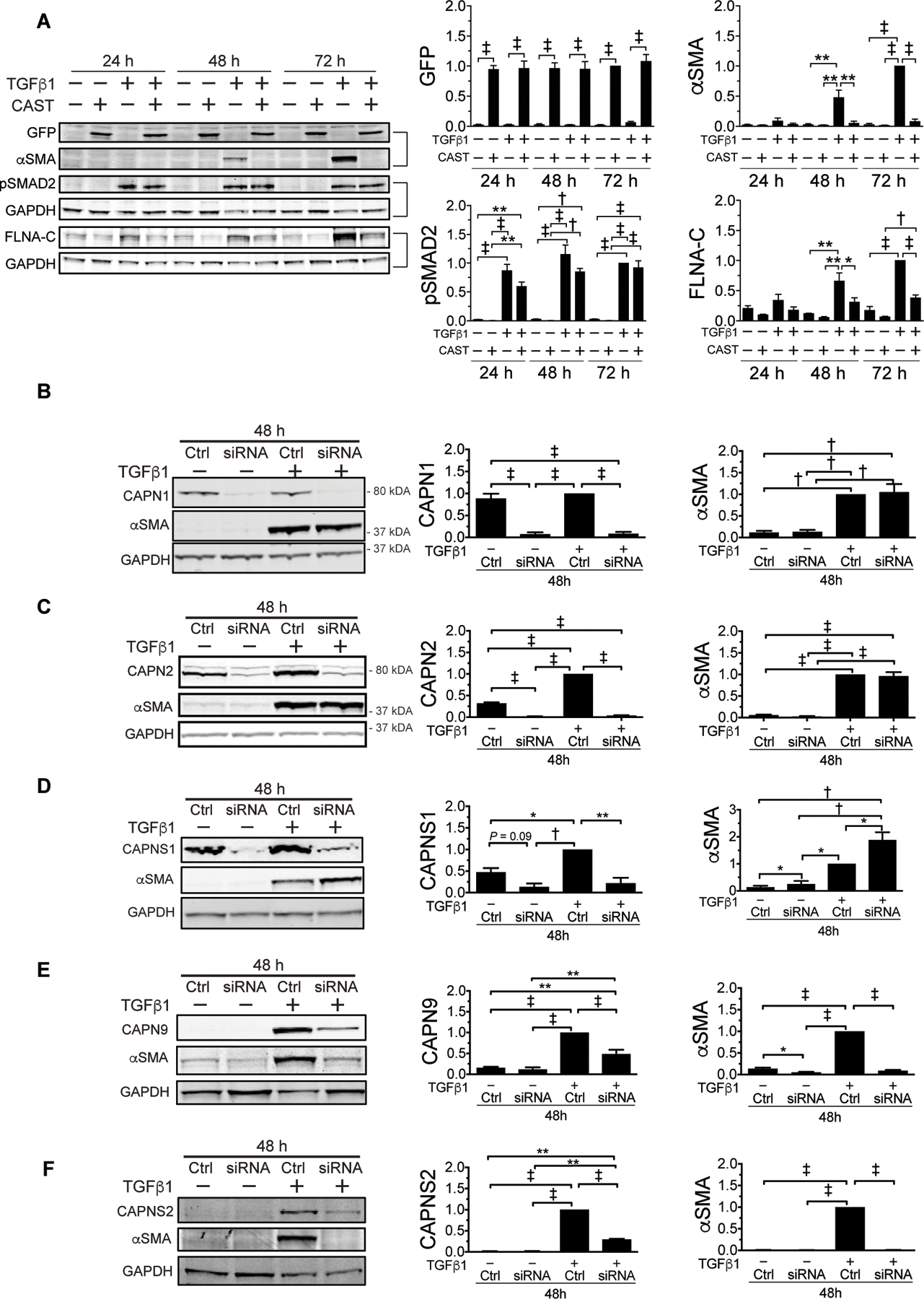
(A) Representative immunoblots (left, bracket indicates identical gel) and quantification (right, normalized to GAPDH) for indicated proteins and time points after TGFβ stimulation with or without CAST-IRES-GFP overexpression (n = 3). Representative αSMA immunoblots (left) for siRNA knockdown of dimeric calpain subunits. (B) CAPN1 (n = 4), (C) CAPN2 (n = 3), (D) CAPNS1 (n = 3), (E) CAPN9 (n = 5), and (F) CAPNS2 (n = 3) and quantification (right, normalized to GAPDH) with or without TGFβ stimulation. Data are expressed as mean ± s.e.m. *P < 0.05, **P < 0.01, †P < 0.005, ‡P < 0.001 by one-way ANOVA with Tukey’s post hoc test.
The suppressive activity of CAST suggested involvement of a dimeric calpain isoform in TGFβ-induced EMT. Given the potential for EMT and fibrosis to occur in many cell types and tissues, we initially suspected involvement of CAPN1, 2, or CAPNS1 given their ubiquitous expression. However, siRNA-mediated suppression of any of these calpain subunits in TGFβ-stimulated NMuMG cells failed to attenuate EMT, as indicated by the quantity of αSMA expression (fig. 2B–D). This observation was initially perplexing as the alternative dimeric calpain large subunit, CAPN9, is reportedly predominantly expressed in the gastrointestinal tract (43), whereas the remaining small subunit, CAPNS2, has been less well studied but reportedly also shows highly restricted expression (28). In keeping with these data, our survey showed predominant expression of Capn9 and Capns2 message in the gastrointestinal tract and skin, respectively (fig. S3A). Neither CAPN9 nor CAPNS2 protein is expressed in NMuMG cells at baseline, but both showed potent induction upon treatment with TGFβ (fig. 2E,F). Suppression of this effect using siRNA strongly attenuated EMT, as shown by reduced αSMA protein abundance (fig. 2E,F), but did not affect TGFβ-dependent induction of EMT transcription factors (fig. S3B). These findings suggest that calpain activity is required for a distal event in TGFβ-mediated myofibroblast differentiation and/or performance.
Calpain inhibition in multiple cells types diminishes TGFβ-induced mesenchymal transition
Considering the critical role played by calpains in TGFβ-induced EMT in the NMuMG epithelial cell line, we sought to explore the relevance of this mechanism in additional cell types. Consistent with our findings in NMuMG cells, pharmacologic inhibition of calpains with MDL-28170 in a second epithelial cell line, Madin-Darby Canine Kidney (MDCK) cells, attenuated mesenchymal transition in a dose dependent manner (fig. S4A). Treatment of normal human lung fibroblasts (NHLFs) with calpeptin had previously been shown to decrease the expression of TGFΒ1 and COL1A1 message in response to TGFβ (26). Here, calpain inhibition attenuated the expression of αSMA in NHLFs stimulated with TGFβ (fig. S4B).
Primary porcine aortic valve endothelial cells (PAVECs) undergo EnMT when stimulated with TGFβ, as evidenced by a mesenchymal transcriptional signature including expression of ACTA2 (encoding αSMA), VIM, MMP2, and MMP9 and downregulation of CDH1 (encoding E-cadherin) (fig. S3A). Each of these events abated upon either CAST overexpression or siRNA-mediated knockdown of CAPNS2 expression (fig. 3B). Inhibition of mesenchymal transition by PAVECs was associated with preservation of expression of E-cadherin and prevention of induction of vimentin (fig. 3C).
Fig. 3. Dimeric calpain inhibition suppresses TGFβ-induced mesenchymal transition in primary PAVECs.
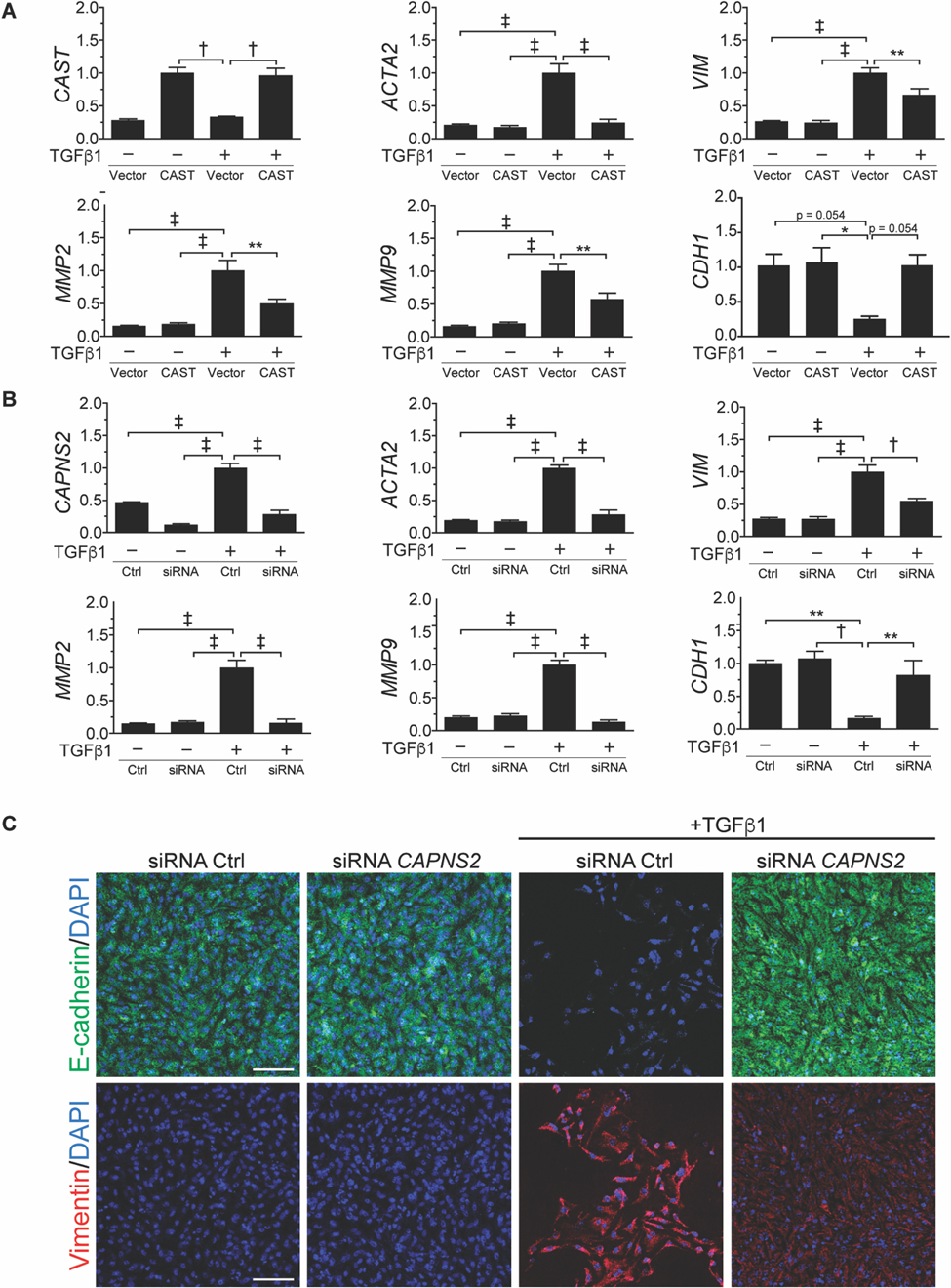
(A) Relative gene expression of PAVECs stimulated with TGFβ1 for 48 h with CAST overexpression normalized to GAPDH (n =3). (B) Relative gene expression (normalized to GAPDH) in response to TGFβ with or without siRNA knockdown of CAPNS2 (n = 3). (C) Representative immunofluorescence images of TGFβ-induced EnMT in PAVECs stained with E-cadherin (green), vimentin (red), and DAPI (blue). Scale bar: 100 µm. All quantitative data integrate each experiment performed in biologic quadruplicate. Data are expressed as mean ± s.e.m. *P < 0.05, **P < 0.01, †P < 0.005, ‡P < 0.001 by one-way ANOVA with Holm-Sidak post hoc test.
TGFβ induces CAPN9 expression by promoting translation with coordinated translation-dependent mRNA decay in NMuMG cells
We next sought to determine whether TGFβ-dependent induction of CAPN9 expression occurred at the level of transcription, translation, or both. We observed that upon TGFβ stimulation, Capn9 message diminished over time whereas CAPN9 protein accumulated (fig. S5A,B). Furthermore, cycloheximide-mediated translation inhibition attenuated TGFβ-dependent Capn9 message decay (fig. S5C); together, these data suggest translational regulation of Capn9 with coordinated translation-dependent mRNA decay. These findings parallel the regulation of expression of other EMT-inducing factors that have been demonstrated in NMuMG cells. For example, previous work showed that TGFβ induced the activation of AKT2, which in turn phosphorylated hnRNPE1, a factor that binds to hairpin structures in the 3’UTR of Ile1 and Dab2 (44). Upon phosphorylation, hnRNPE1 is displaced from these cis-regulatory elements allowing release of translation. To test the hypothesis that a similar mechanism is responsible for the regulation of CAPN9 expression in our system, we inhibited AKT2 phosphorylation with the competitive inhibitor CCT128930 in TGFβ-stimulated NMuMG cells. Contrary to our hypothesis, neither mRNA decay nor protein accumulation was prevented by AKT2 inhibition in the presence of TGFβ (fig. S5D,E). Curiously, there appears to be heterogeneity in the cellular mechanisms regulating Capn9 expression, as TGFβ induced Capn9 message in MDCK cells (fig. S5F). These phenomena are likely specific to TGFβ exposure, as Wnt activation via WNT3A exposure failed to promote EMT or CAPN9 protein expression in NMuMG cells (fig. S5G).
Capn9−/− mice are resistant to bleomycin-induced lung fibrosis
We next sought to define the therapeutic potential of calpain inhibition in animal models of fibrosis. We used mice lacking CAPN9 function that were generated by deleting Capn9 exon 3, which contains the catalytic cysteine in the peptidase domain (32). Deletion of the 119 nucleotides corresponding to exon 3 is predicted to create a frameshift and hence a premature termination codon (PTC) a short distance (6 codons) into exon 4 in mature mRNA. We confirmed these predictions using RT-PCR and sequencing of cDNA (fig. S6A). Contrary to prediction, the PTC did not initiate degradation of the mutant transcript via nonsense-mediated mRNA decay (fig. 6B). Loss of expression of CAPN9 protein was confirmed using one commercially available antibody (Abnova) but was not using a different reagent (Sigma), which detected a protein of the predicated mass for all dimeric calpains in both stomach and lung of wildtype and Capn9−/− animals, indicating lack of specificity for CAPN9. The Abnova antibody appears to be specific for CAPN9 but cross-reacts with a slightly smaller protein in the lung, highlighting the limitations of these reagents for certain applications (fig. S6C). Finally, we surveyed the expression of the dimeric calpain subunits Capn1/2/s1/s2 and the endogenous calpain inhibitor, Cast, in Capn9−/− animals. These data document that Capn9−/− animals can show a compensatory increase, but are not deficient in any of these calpain family members (fig. 6D).
Fig. 6. Capn9−/− mice are protected from Ang II-induced cardiac fibrosis.
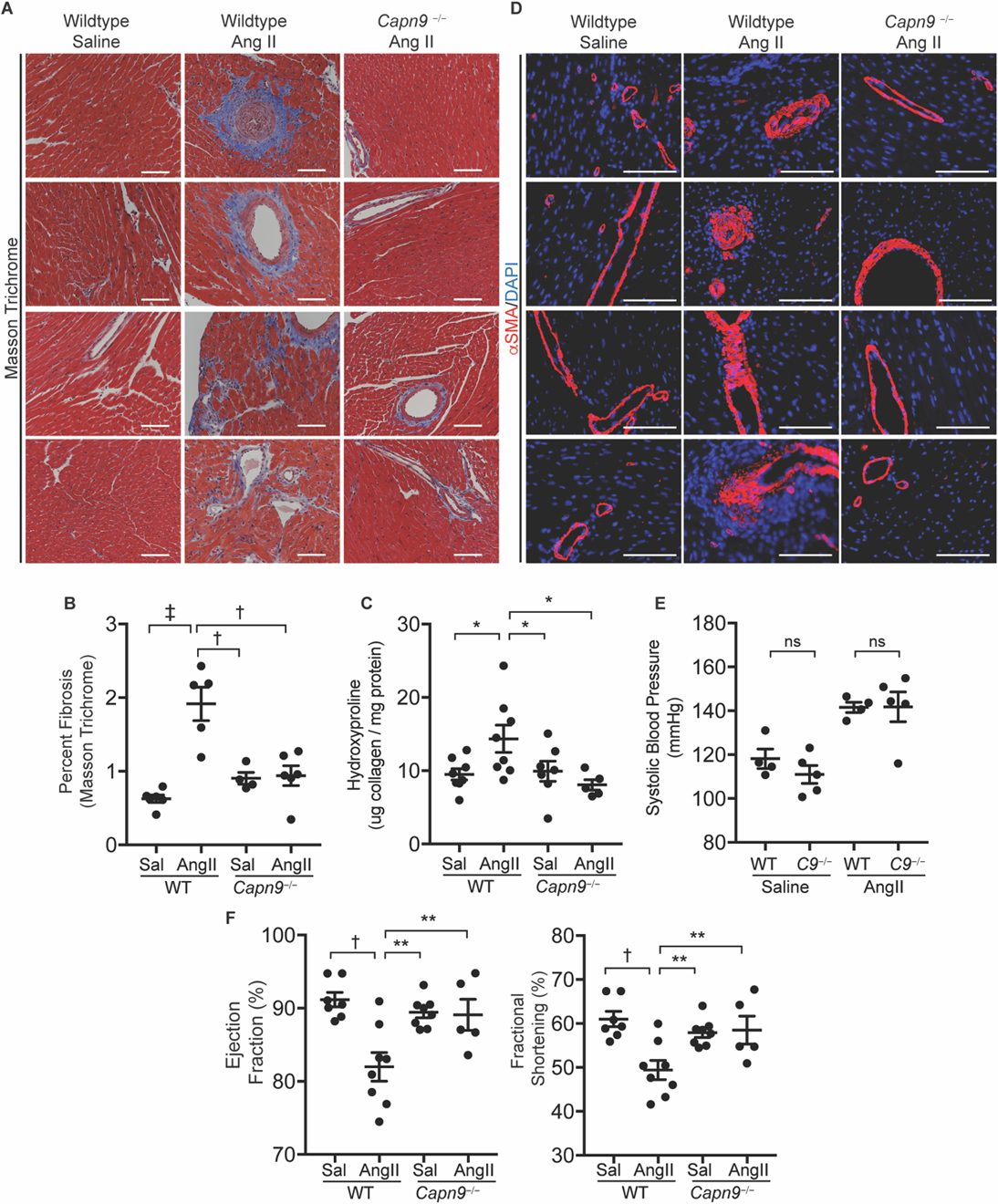
(A) Representative Masson trichrome stained slides of wildtype and Capn9-targeted mouse hearts from mice treated with saline or chronic Ang II infusion. Scale bar: 100 µm. (B) Quantification of blue stained collagen on trichrome slides by ImageJ (n = 4 to 6). (C) Collagen content in hearts from Capn9−/− or wildtype control mice treated with saline (Sal) or Ang II (n = 5 to 8). (D) Representative images of mouse hearts from groups as in (A), stained for SMA (red) or DAPI (blue). Scale bar: 100 µm. (E) Systolic blood pressure of wildtype and Capn9−/− (C9−/−) animals receiving Ang II infusion (n = 4 to 5). (F) Measures of left ventricular function including ejection fraction and fractional shortening in mice treated with Ang II (n = 5 to 8). Data are expressed as mean ± s.e.m. with statistical comparison between Ang II-treated wildtype mice and all other conditions. nsP > 0.05, *P < 0.05, **P < 0.01, †P < 0.005, ‡P < 0.001 by one-way ANOVA with Holm-Sidak post hoc test (B, D, F) and two-way ANOVA with Tukey’s post hoc test (E). Treatment effect was significant (P < 0.0001).
Experimentally-induced lung fibrosis is commonly achieved via exposure to the chemotherapeutic agent bleomycin. Systemic delivery of bleomycin by a subcutaneously implanted osmotic pump results in penetrant subpleural fibrosis (45). Compared to wildtype mice, Capn9−/− mice exposed to bleomycin were protected from fibrosis, as assessed histologically by Masson trichrome staining (fig. 4A), blinded histological observation (fig. 4B), and total lung collagen content (fig. 4C). Bleomycin lung injury in wildtype mice was associated with a dramatic mortality increase; however, Capn9−/− mice showed no mortality despite systemic delivery of bleomycin (fig. 4D) and exhibited preserved lung function tests, as monitored by respiratory resistance, total lung capacity (TLC), and lung compliance (fig. 4E–G). Fibrosis in wildtype animals exposed to bleomycin was accompanied by an increase in the number of αSMA-positive myofibroblasts in the lung interstitium, whereas αSMA immunoreactivity was restricted to airways and blood vessels in vehicle-treated wildtype or bleomycin-treated Capn9−/− animals (fig. 4H). Robust upregulation of Capn9 message, but not that for other dimeric calpains, accompanied myofibroblast accumulation in wildtype animals as detected by RT-PCR and in situ RNA hybridization (RNA-ISH) (fig. 4I). Cells that expressed Capn9 in bleomycin treated lungs also expressed Acta2, indicating upregulation of Capn9 expression in myofibroblasts (fig. 4J).
Fig. 4. Capn9−/− mice are protected from bleomycin-induced lung fibrosis.
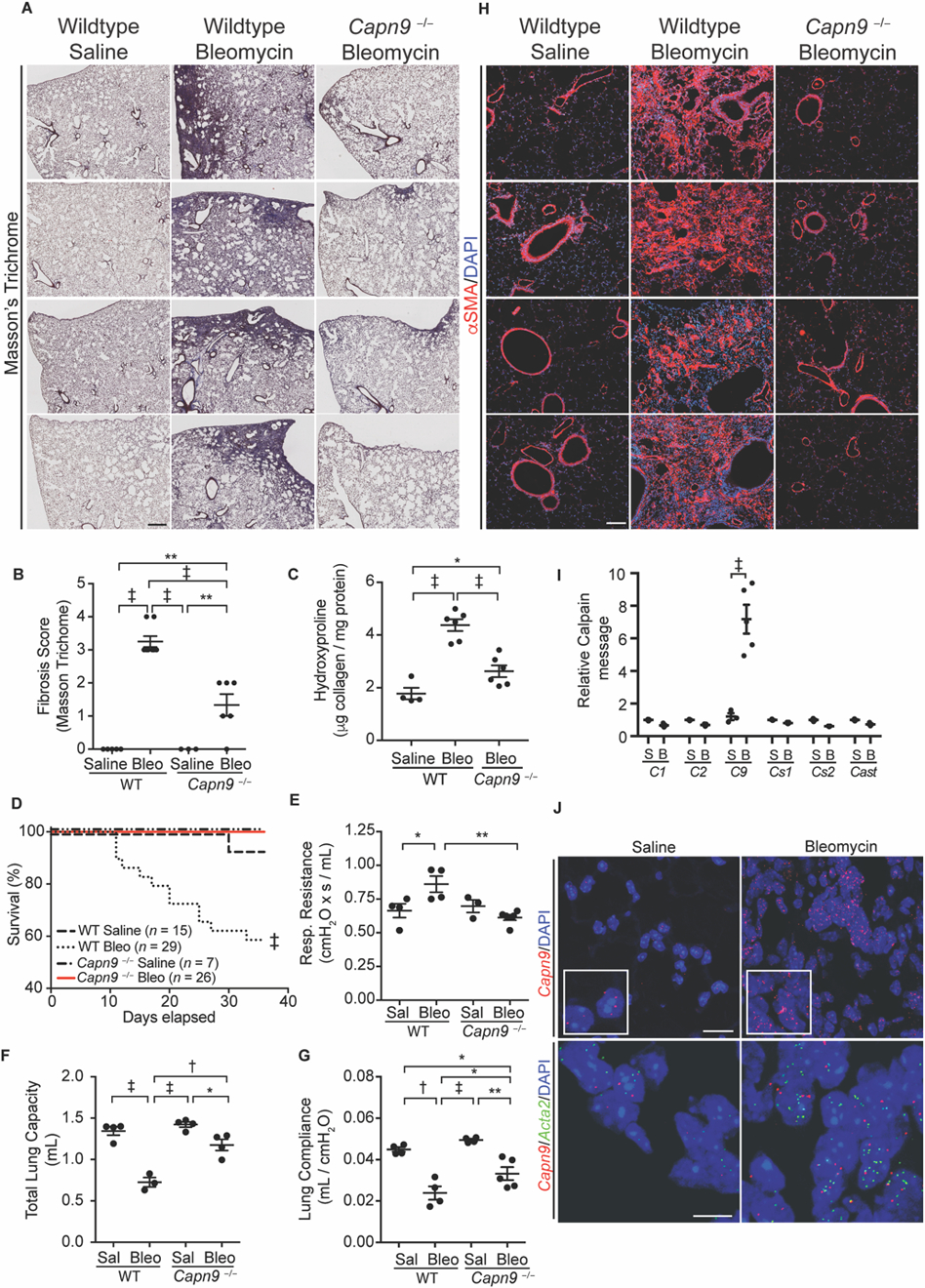
(A) Representative low power Masson trichrome stained lung from wildtype mice treated with saline or bleomycin and Capn9−/− mice treated with bleomycin. Scale bar: 500 µm. (B) Fibrosis scores of mouse lung stained with Masson trichrome (n = 3 to 8). (C) Collagen content of lungs from Capn9−/− or wildtype control mice treated with saline or bleomycin (Bleo) (n = 4 to 6). (D) Survival curves of Capn9−/− and wildtype control mice with saline or bleomycin treatment. Results are combined from three independent experiments. (E) Respiratory resistance (n = 3 to 6) (F) total lung capacity (n = 3 to 4) and (G) lung compliance (n = 3 to 6) of saline (Sal) and bleomycin treated wildtype and Capn9−/− mice. (H) Representative IF of mouse lung stained for αSMA (red) or DAPI (blue). Scale bar: 100 µm. (I) Calpain mRNA normalized to Gapdh in wildtype mouse lung with saline (S) or bleomycin (B) treatment (C1/2/9/s1/s1 are Capn1/2/9/s1/s2, respectively, n = 3 to 5), and (J) and representative Capn9 (top, scale bar: 20 µm, insets are 1.5x magnifications) and Acta2 and Capn9 (bottom, scale bar: 10 µm) RNA-ISH of mouse lungs. Dot plot data are expressed as mean ± s.e.m. *P < 0.05, **P < 0.01, †P < 0.005, ‡P < 0.001 by one-way ANOVA with Holm-Sidak post hoc test (B, C, E-G), log-rank test (D), and Student’s t-test (I).
To ensure that the protection from lung fibrosis afforded by CAPN9 deficiency is not due to relative immunosuppression of Capn9−/− animals, we administered intratracheal bleomycin to induce a robust inflammatory response to injury. One week after insult, both wildtype and Capn9−/− lungs showed intense inflammatory infiltrates (fig. S7A). Flow cytometric analyses demonstrated no differences in the proportions of CD45+ cell subsets in response to bleomycin injury in Capn9−/− compared to wildtype animals (fig. S7B).
Capn9−/− mice are protected from carbon tetrachloride (CCl4)-induced liver fibrosis
Chronic CCl4-induced liver injury drives periportal inflammation and fibrosis in mice (46). Wildtype mice treated with CCl4 for 4 weeks developed bridging fibrosis whereas Capn9−/− mice were protected from fibrosis, as shown by low-power bright-field picrosirius red (PSR) staining (fig. 5A), birefringence of mature collagen fibrils under polarized light (fig. 5B), blinded histologic scoring (fig. 5C), and total liver collagen content (fig. 5D). Robust upregulation of Capn9 message was detectable adjacent to fibrotic tracks in CCl4-treated wildtype animals by RNA-ISH (fig. 5E). These histologic changes were accompanied by markers of liver damage such as elevated serum concentration of the liver enzymes alanine transaminase (ALT) and aspartate transaminase (AST). Wildtype and Capn9−/− mice receiving injections of CCl4 had equal injury as indicated by liver function tests (fig. 5F); however, unlike wildtype controls, Capn9−/− animals developed minimal fibrosis.
Fig. 5. Capn9−/− mice are protected from CCl4-induced liver fibrosis.
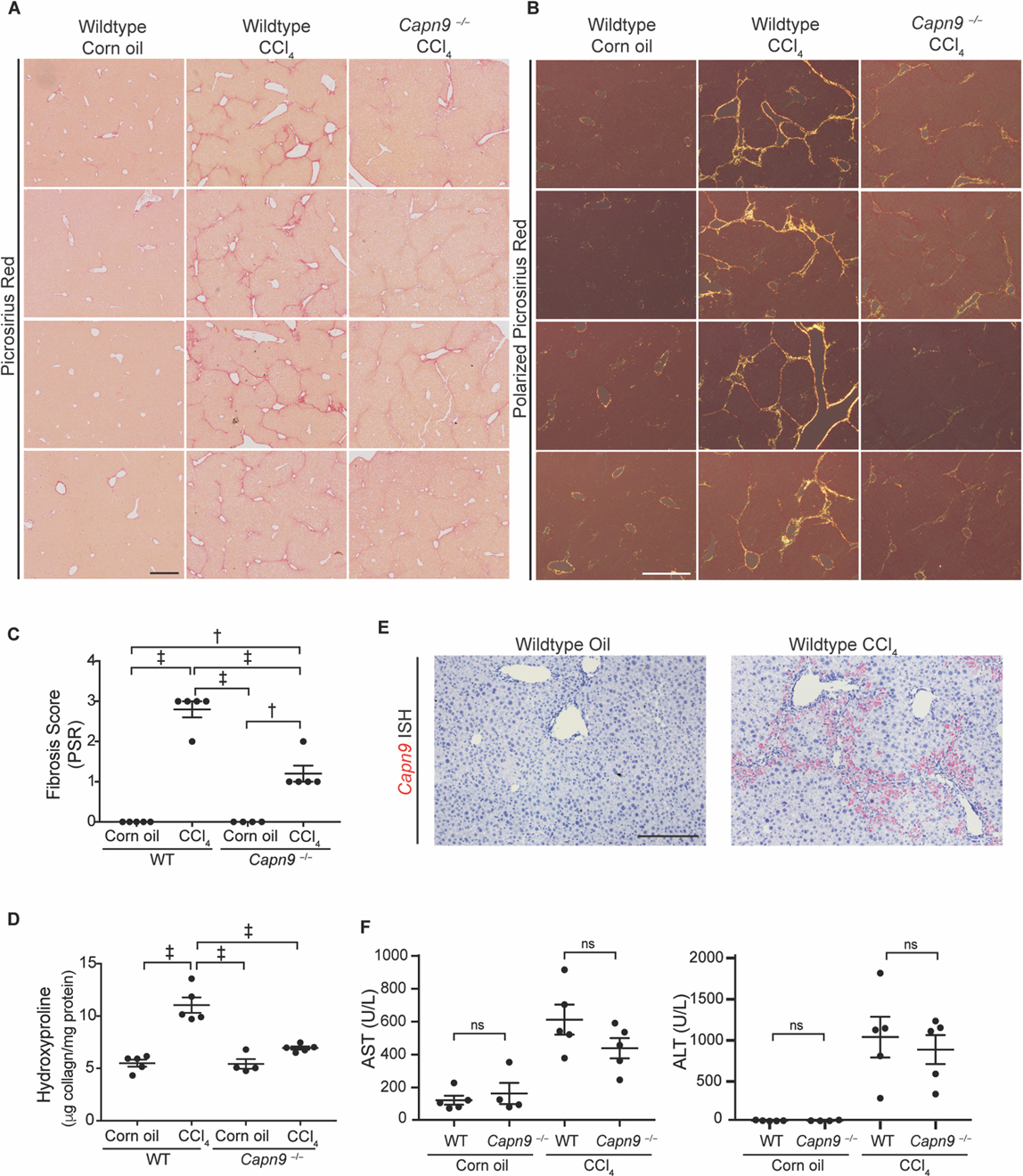
(A) Representative low power bright-field images of picrosirius red stained slides of mouse liver. Scale bar: 400 µm. (B) Representative picrosirius red stained livers under polarized light. Scale bar: 200 µm. (C) Fibrosis scores of mouse liver stained with picrosirius red (n = 4 to 5). (D) Collagen content in livers from Capn9−/− or wildtype control mice treated with corn oil or CCl4 (n = 4 to 5). (E) Representative RNA-ISH of Capn9 in wildtype control mouse liver from mice treated with corn oil or CCl4 (red, scale bar: 100 µm). (F) Serum liver function tests of mice with or without injury (n = 4 to 5). Data are expressed as mean ± s.e.m. nsP >0.05, *P < 0.05, **P < 0.01, †P < 0.005, ‡P < 0.001 by one-way ANOVA with Holm-Sidak post hoc test (C, D) and two-way ANOVA with Tukey’s post hoc test (F). Treatment effect was significant (P < 0.0001).
Capn9−/− mice are protected from angiotensin-2 (Ang II)-induced cardiac fibrosis
Ang II is capable of inducing a fibrotic synthetic repertoire in cardiac fibroblasts in vitro and in vivo (47–50). In wildtype animals, Ang II infusion results in cardiac arterial intimal proliferation and perivascular collagen accumulation with collagen fibers extending into the cardiac interstitium (51). Here we showed that Capn9−/− mice treated with Ang II were protected from perivascular and interstitial fibrosis, as shown by Masson trichrome staining (fig. 6A,B), total heart collagen content (fig. 6C), and αSMA immunoreactivity (fig. 6D). Despite systolic blood pressures equivalent to wildtype animals receiving Ang II (fig. 6E), Capn9 deficient animals showed preservation of Ang II-induced left ventricular cardiac function as monitored by echocardiography (fig. 6F).
Calpain inhibition reverses established lung fibrosis
To determine the therapeutic utility of calpain inhibition in clinically relevant disease models, we explored the ability of calpain inhibitors to suppress an established predisposition for lung fibrosis. Intratracheal delivery of bleomycin results in persistent lung fibrosis and physiologic defects for at least 6 months after the injury (52). Wild type mice receiving therapeutic dosing of MDL-28170 one week after intratracheal delivery of bleomycin showed full normalization of Col1a1 message expression, suggesting an overt arrest of ongoing fibrotic potential (fig. 7A,B).
Fig 7. Therapeutic dosing of MDL-28170 ameliorates early established lung fibrosis.
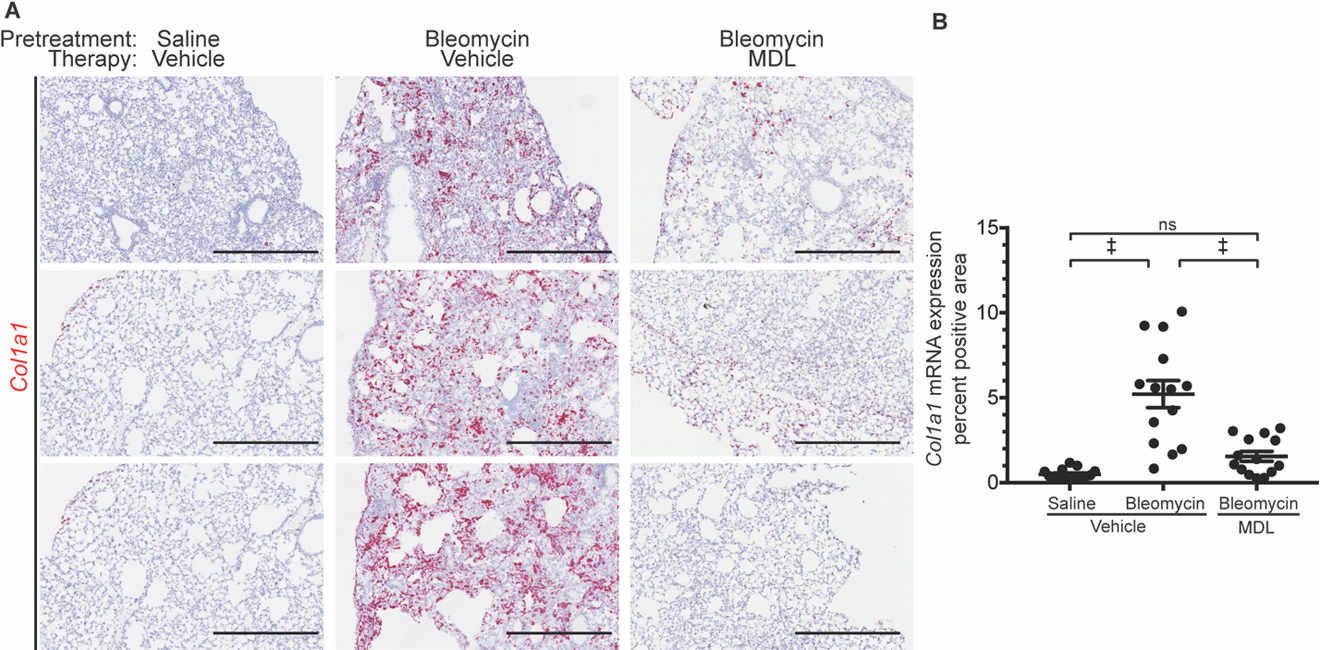
(A) Representative Col1a1 RNA-ISH slides of mouse lungs three weeks after saline or bleomycin treatment and therapeutic vehicle or MDL dosing (delivered one week after bleomycin). Scale bar: 500 µm. (B) Quantification of red Col1a1 RNA-ISH staining (n = 14 to 15). Data are expressed as mean ± s.e.m. nsP > 0.05, *P < 0.05, **P < 0.01, †P < 0.005, ‡P < 0.001 by one-way ANOVA with Holm-Sidak post hoc test.
Human genetics suggests tolerance for CAPN9 deficiency
In humans, loss-of-function CAPN9 variants do not associate with a signal for intolerance (probability for loss-of-function intolerance, pLI = 0.00) (53). A polymorphic variant (rs28359631) that substitutes the obligate A at the −2 position of the splice acceptor for exon 4 is predicted to cause exon skipping and a consequent frameshift, leading to nonsense-mediated mRNA decay. This allele is common in the Genome Aggregation Database (gnomAD) with a collective allele frequency of 1.0%. It is enriched in populations of East and South Asian ancestry (8.3% and 3.2% allele frequency, respectively) with homozygosity observed at rates compatible with Hardy-Weinberg equilibrium (0.8% and 0.1%, P = 0.47 and 0.43, respectively).
Discussion
Despite the substantial contribution of fibrosis to global disease burden, few therapies target the accumulation or function of the cell type primarily responsible for pathologic ECM production, the myofibroblast (9). The lack of effective therapies reflects the tremendous difficulty in targeting fibrosis, a pathology that develops from the co-optation of complex biological processes involved in tissue development and injury response, including EMT (54). Moreover, fibrosis is a common – and indolent – pathologic outcome of a group of highly heterogeneous disorders, compounding the challenge to achieve a comprehensive understanding of disease pathogenesis and vulnerabilities. Nevertheless, the recent observation that curative hepatitis treatment culminates in reversal of liver fibrosis establishes that a fibrotic extracellular matrix is a modifiable consequence of disease (55–57).
In this paper, we used in vitro assays of TGFβ-induced mesenchymal transition to investigate the role of calpains in the differentiation of myofibroblasts from various cell types. Our data show that TGFβ induces calpain activity whereas inhibition of calpains prevents the induction of a mesenchymal phenotype. In the setting of calpain inhibition, early TGFβ signaling events, such as pSMAD2 nuclear accumulation and the induction of EMT transcriptions factors like Snai1, are preserved. These findings suggest that the relevant calpain-dependent events mediating TGFβ-induced EMT are distal in the differentiation or maturation of myofibroblasts. We were initially surprised to observe that TGFβ can cause myofibroblast differentiation using calpain isoforms that are not normally expressed in the progenitor cell but are rapidly and potently induced by TGFβ. In retrospect, this appears to be an elegant strategy to tightly regulate a process that is necessary and productive in development and wound healing, but can prove deleterious if left unchecked. Restriction of this myofibroblast-promoting activity to calpains that are recruited after a fibrogenic stimulus, such as TGFβ, may prevent initiation of this pathway in response to physiological stimuli that normally result in activation of the constitutively expressed calpains. Indeed, prior work has demonstrated TGFβ-induced translational upregulation of other EMT-inducing factors in NMuMG cells including DAB2 and ILE1, albeit by a different mechanism than observed for CAPN9 (44). Our data do not preclude the involvement or sufficiency of other calpains or calpain-independent mechanisms in mesenchymal transition in other cell types and contexts; indeed, this appears to be required, given the observation of normal development and tissue homeostasis in Capn9 deficient animals (32). Moreover, a predicted loss-of-function mutation in humans is relatively common, found in homozygosity, and appears to be in Hardy-Weinberg equilibrium, suggesting tolerance for CAPN9 deficiency. Although congenital loss of a gene is an imperfect model of therapeutic antagonism, these observations suggest the potential for a broad therapeutic window.
Our data build upon previous studies that show protection from bleomycin-induced lung fibrosis (26, 58) and Ang II-induced cardiac fibrosis (22) upon broad spectrum calpain inhibition with calpeptin treatment or calpastatin overexpression, respectively. Both calpeptin and calpastatin are antagonists of the ubiquitous dimeric calpains, as well as CAPN9, and calpeptin inhibits other cysteine proteases including cathepsins (43, 59). Our demonstration of a specific role for Capn9 and Capns2 in fibrosis and in TGFβ-induced myofibroblast differentiation expands upon the known function of these dimeric calpain subunits. Indeed, existing literature describes CAPN9 function as largely restricted to the gastrointestinal tract, where co-expression of CAPN9 with CAPN8 in gastric pit cells forms a complex, termed G-calpain, that is involved in gastric mucosal injury response (32, 43). The tissue restricted alternative small subunit, CAPNS2, appears to be largely redundant with CAPNS1 in vitro. However, CAPNS2 binds to the large catalytic subunits with lower affinity, does not undergo activation-mediated autolysis, and conveys some substrate specificity differences in vivo (28, 60).
The development of clinically useful calpain small molecule inhibitors has been hampered by the challenge of developing agents with specificity for calpains over other cysteine proteases, such as some cathepsins. Despite these challenges, the existence of CAST, a highly specific inhibitor of dimeric calpains, indicates that the design of calpain specific inhibitors is possible. Crystal structures of CAPN2-CAST complexes reveal that although CAST is an unstructured protein in solution, in the presence of calcium it binds to calpain and wraps over the active site (31, 61). Calpain amino acid residues that interact with CAST near the active site are highly conserved in each of the CAST-inhibited isoforms, whereas CAPN3, which escapes CAST inhibition, is divergent, suggesting that calpain specificity may be achieved by inhibitors that display affinity for amino acids adjacent to the active site (62). Furthermore, crystal structures of the protease cores of CAPN1 and CAPN9 point to a number of differences that may be exploited for the development of CAPN9-specific antagonists (61).
Despite this progress, a number of limitations to this study should be considered. First, the observation that CAPN9-deficient NMuMG cells fail to support efficient TGFβ-induced EMT despite abundant expression of CAPN1, CAPN2, and CAPNS1 suggests the importance of a CAPN9 cleavage event that remains to be identified. Although many calpain substrates have been recognized, the identification of calpain isoform-specific substrates has proven challenging (59). Knowledge regarding the precise function of CAPN9 that influences fibrosis in model systems will inform the clinical utility of strategies aimed at its specific antagonism. As stated above, the lack of small molecule antagonists that display high bioavailability, potency, and specificity for CAPN9 – or indeed all dimeric calpains – limits the immediate clinical application of these findings. Finally, our observations are largely based on prophylactic modulation of chemically induced models of fibrosis in small animals, which may have a limited ability to predict efficacy in human presentations of disease. Such issues will be best addressed using pharmacologic manipulations in both preclinical models and human clinical trials.
Materials and Methods
Study design.
The purpose of this study was to identify and explore a pathway in fibrosis that may be amenable to pharmacologic antagonism. We used validated in vitro assays with complementary antagonism strategies to provide strong evidence for the role of dimeric calpains in TGFβ-induced mesenchymal transition. To implicate a specific calpain isoform, we used siRNA knockdown to find evidence that CAPN9 and CAPNS2 were required for the adoption of a mesenchymal phenotype in vitro. Because we found strong evidence for the importance of CAPN9 in in vitro assays in multiple cell types, we acquired animals lacking CAPN9 function and evaluated their response to experimentally induced lung, liver, and cardiac fibrosis.
Sample sizes were determined based on prior experience with similar studies and on pilot experiments. All mice used were males to eliminate the potential confounding influence of differences due to sex in response to fibrogenic agents. All mice were randomly assigned to treatment groups, and all analysis was performed blinded to genotype and treatment condition. Dosing and treatment duration for bleomycin, CCl4, and Ang II were determined based on prior literature. No surviving animals were excluded from analysis. No outliers were excluded. The number of biologic replicates is specified in the figure legends or represented by dot-plots.
Mice.
All mice were cared for in compliance with approved protocols from the Animal Care and Use Committee of the Johns Hopkins University School of Medicine. Capn9tm1Hiso mice were derived and backcrossed to C57Bl6/J previously (32). Murine Capn9 targeted sperm was acquired from the RIKEN BRC through the National Bio-Resource Project of the MEXT, Japan (RBRC04790), and targeted mice were generated by the Johns Hopkins Transgenics Core (Baltimore, MD) and maintained by sibling crosses. C57Bl6/J male control mice were acquired from the Jackson Laboratories (Bar Harbor, ME). Male mice were used for all experiments. All mice were kept in a specific pathogen free facility on a 12 h light-dark cycle and provided food and water ad libitum.
Cell culture.
All cells were cultured at 37°C and 5% CO2. NMuMG cells (ATCC, CRL-1636) were maintained in Dulbecco’s Modified Eagles Medium (DMEM) (Gibco, 11965–118), supplemented with 10% fetal bovine serum (FBS; Sigma, F4135), 2 mM L-glutamine (Thermo Fisher, 25030081) and 10 µg/mL insulin (Thermo Fisher, 12585014). Normal human lung fibroblasts (NHLF; Lonza, CC-2512) were cultured with Fibroblast Growth Media-2 and supplements (Lonza, C-3132). MDCK cells (ATCC, CCL-34) were cultured in Minimum Essential Media (MEM; Thermo Fisher, 11095098) supplemented with 10% FBS, 2 mM L-glutamine, and 1,000 U/mL penicillin-streptomycin. Porcine aortic valve endothelial cells (PAVECs) were isolated as previously described (63), grown in flasks coated with 50 µg/mL rat tail collagen I (BD Biosciences, 354236), and cultured in DMEM supplemented with 10% FBS, 1,000 U/mL penicillin-streptomycin, and 50 U/mL heparin (Sigma-Aldrich, H3393).
Small molecule inhibitors.
MDL-28170 (Enzo Lifesciences, BML-PI130), calpeptin (Tocris, 0448), 2-APB (Sigma, D9754), CA-074-OMe (Sigma, C5857), SB431542 (Sigma, S4317), CCT128930 (Selleckchem, S2635), cycloheximide (Cell Signaling Technologies (CST), 2112S) were prepared in stock solutions according to the manufacturer’s instructions.
In vitro EMT assays.
NMuMG cells and MDCK cells were cultured and grown to confluency as described above. To induce EMT, cells were starved in media containing 0.5% FBS and pretreated with pharmacological inhibitors 24 h prior to treatment with TGFβ1 at stated concentrations (R&D Systems, 240-B-010). Culture media was replaced every 24 h during all experiments. For some experiments, recombinant mouse Wnt3a (Abcam, ab81484) was used to stimulate serum starved NMuMG cells as above. For reversal, NMuMG cells were starved for 24 h, stimulated with TGFβ1 for 72 h, and then incubated with MDL-28170 at 10 or 20 µM in starvation media for an additional 72 h.
In vitro fibroblast-to-myofibroblast assay.
NHLFs were subcultured as described above, and then starved for 24 h in fibroblast growth media containing no supplements with MDL-28170 at indicated concentrations. Cells were stimulated with TGFβ1 (10 ng/mL) with or without the presence of calpain inhibitor for 24 h.
Transfection.
NMuMG cells were transfected with full-length mouse calpastatin (MGC 3710078) inserted into pCMV-IRES-AcGFP (Clontech, 632435) using Lipofectamine 2000 (Thermo Fisher, 11668019) according to the manufacturer’s instructions. Cells were transfected for 5 days, split, and selected for stably integrated clones using 400 µg/mL geneticin (Gibco, 10131027). A polyclonal cell line was confirmed to express AcGFP by live cell fluorescent microscopy. For siRNA knockdown, NMuMG cells were transfected with a SMARTpool mixture of 4 siRNAs (GE Healthcare), targeting Capn1 (M-062006–01-0010), Capn2 (M-043027–01-0010), Capns1 (M-048840–01-0010), Capns2 (M-014858–00-0010) or non-targeting control (D-001206–13-50). Two siRNAs targeting Capn9 were custom synthesized using previously validated sequences (59). Cells were transfected using Dharmafect 4 (GE Healthcare, T-2004–03) according to the manufacturer’s instructions at a final siRNA concentration of 100 nM. PAVECs were electroporated in solution using the Neon Transfection System (Invitrogen, MPK10025) as previously described (64). For CAPNS2 knockdown, siRNA targeting CAPNS2 (GE Healthcare, M-014858–00-0010) or non-targeting control (D-001206–13-50) were used. For CAST overexpression, PAVECs were electroporated in solution with plasmids encoding for either mouse calpastatin (pEF1α-Cast-IRES-IRFP) or empty vector.
Western blots.
Cultured cells were harvested using radio immunoprecipitation assay buffer (RIPA; Sigma, R0278), sonicated, and lysate protein concentration was quantified by colorimetric BCA Assay (Pierce, 23225). Equal quantity of protein was submitted to SDS-PAGE electrophoresis on 10% Bis-Tris gels (Bio-Rad, 3450113) in MOPS running buffer (Thermo Fisher, NP0001–02) and transferred onto a methanol-activated Immobilon-FL polyvinylidene fluoride membrane (Millipore, IPFL00010) in NuPage transfer buffer (Thermo Fisher, NP0006). Membranes were blocked (LI-COR Biosciences, 927–40000) and probed overnight at 4°C. Antibodies used were: mouse anti-αSMA (R&D clone 1A4, MAB 1420) at 1:1000, goat anti-GAPDH (Santa Cruz Biotechnology, sc-20457) at 1:5000, mouse anti-phosphoSMAD2 (Millipore clone A5S, 04–953) at 1:500, rabbit anti-filamin A-C-terminal fragment (Epitomics, 2242–1) at 1:15000, mouse anti-AcGFP (Clonetech clone JL-8, 632380) at 1:1000, rabbit anti-CAPN1 (CST, 2556) at 1:1000, rabbit anti-CAPN2 (CST, 2539) at 1:1000, mouse anti-CAPN9 (Abnova, H00010753-M02) at 1:1000, rabbit anti-CAPN9 (Sigma, HPA020398) at 1:1000, rabbit anti-CAPNS2 (Lifespan Biosciences, LS-C133503) at 1:1000, rabbit anti-phospho-β-catenin (Ser552) (CST, 9566). Species-appropriate secondary antibodies conjugated to IRdye-700 or IRdye-800 (LI-COR Biosciences) were used according to the manufacturer’s guidelines, and analyzed using the LI-COR Odyssey system. Quantification was performed on each blot by normalizing to GAPDH loading control.
Immunofluorescence.
NMuMG cells were plated and grown on sterilized glass coverslips and submitted to EMT assays as described above. Coverslips were fixed with 4% paraformaldehyde (PFA, EMS 15710) in PBS at RT and blocked in buffer (5% donkey serum, 0.1% Triton X-100, in PBS) at RT. Slides were incubated with mouse anti-E cadherin (BD Biosciences, 610405) 1:500 in blocking buffer overnight at 4°C, then probed with donkey anti-mouse Alexa 488 (Molecular Probes, A-21202) 1:500, rhodamine-phalloidin (Molecular Probes, R415) 1:500, and DAPI (Molecular Probes, D1306) 1:25000 for 1 h at RT in the dark. After washing with PBS, coverslips were mounted on slides with Prolong Gold Mountant (Molecular Probes, P10144). Images were obtained with a Zeiss LSM710. For pSMAD2, NMuMG cells were plated, incubated in starvation mediate with 20 µM MDL-28170 pretreatment for 24 h, stimulated with 10 ng/ml TGFβ1 for 1 h, and then washed and fixed in 4% PFA for 15 min at RT. Cells were permeabilized by incubating in 0.1% Triton X-100 for 5 min, blocked for 20 min, and incubated with a rabbit anti-phospho-Smad2 antibody (Millipore Sigma, 04–953, 1:1000) overnight in buffer (5% BSA, 0.1% Triton X-100 in PBS). The cells were then probed with donkey anti-rabbit IgG Alexa Fluor 594 (Thermo Fisher Scientific, A-21207) and mounted using VectaShield with DAPI (Vector Laboratories, H-1200). Images were obtained with a Zeiss LSM 780. PAVECs grown on sterilized glass coverslips were fixed in 4% PFA overnight at 4°C, permeabilized with 0.2% Triton X-100 for 10 min, incubated overnight at 4°C in 1% BSA followed by another 4°C overnight incubation with either rabbit anti-human E-cadherin 1:100 (Abcam, ab53033) or mouse anti-human vimentin 1:100 (Invitrogen, V9). Samples were exposed to species-appropriate secondary antibodies conjugated to Alexa Fluor 488 or 568 (Invitrogen) at 1:100 in 1% BSA for 2 h at RT then mounted with media containing DAPI (Vector Laboratories, H-1500). Formalin fixed paraffin embedded tissue was sectioned at 5 µm, deparaffinized in xylene, rehydrated, and blocked for 1 h at RT (PBS, 0.1% Tween 20, 5% normal goat serum). Blocked samples were incubated with mouse anti-αSMA-Cy3 (Sigma clone 1A4, C6198) 1:1000 overnight at 4°C. Slides were counter stained with DAPI and mounted. Images were acquired on a Zeiss LSM710 (lung) or a Leica DMi8 (heart). Post-acquisition processing was performed equally for all representative images in a figure panel.
RNA analysis.
RNA was harvested from cells using the RNeasy Kit (Qiagen, 74106) with DNase digestion (Qiagen, 79254). RNA was harvested from dissected mouse organs following homogenization in an automatic bead homogenizer (FastPrep24, MP Biomedicals) in Trizol (Thermo Fisher, 15596018) according to the manufacturer’s instructions. cDNA was generated using a high capacity RNA to cDNA kit (Applied Biosystems, 4387406). Quantitative PCR was done using TaqMan probes for mouse Col1A1 (Mm00801555_g1), Mmp2 (Mm00439498_m1), Mmp9 (Mm00442991_m1), Gapdh (Mm99999915_g1), Snai1 (Mm00441533_g1), Snai2 (Mm00441531_m1), Twist1 (Mm00442036_m1), Twist2 (Mm00492147_m1), Zeb1 (Mm00495564_m1), Zeb2 (Mm00497193_m1); porcine ACTA2 (Ss04245588_m1), CDH1 (Ss03377287_u1), VIM (Ss04330801_gH), MMP2 (Ss03394318_m1), MMP9 (Ss03392092_g1) and GAPDH (Ss03375629_u1). Canine qPCR was performed using Sybr Green (Thermo Fisher, 4309155) and canine CAPN9 primers were: sense (5’-GCAGAGACCTTCGCAACTAA-3’) and antisense (5’-GCTGCATTTCTGGTATCAATGG-3’); canine GAPDH primers were: sense (5’-AACATCATCCCTGCTTCCAC-3’) and antisense (5’-GACCACCTGGTCCTCAGTGT-3’). Samples were run on a QuantStudioTM 7 Flex Real-Time PCR system (Thermo Fisher). Ct values were corrected for loading and calculated using the 2−∆Ct method.
cDNA gel electrophoresis of Capn9 amplicons was performed by RNA isolation and total cDNA synthesis (as above). Amplification of mouse Capn9 cDNA was performed with Flash Phusion (Thermo Fisher, F548L) for 40 cycles with the following primers: exon 1 sense (5’-CTTTGTGTGGAAACGGCCAG-3’); exon 3 sense (5’-AGAAAGCACTGACCAGGGTG-3’); exon 5 sense (5’-TGGAAGACTTCACTGGGGGT-3’); exon 9 sense (5’-TGCAACCTCACACCTGATGC-3’); exon 5 antisense (5’-TCCATGGCTTCAATGGCACT-3’); exon 7 antisense (5’-CCCAAGGGTTACGGACTCTG-3’); exon 9 antisense (5’-AGCTTCCTTGGTGGATGGTC-3’); exon 11 antisense (5’-TCCCTACTCAGGTGTCCGTC-3’); exon 13 antisense (5’-TGTGGAGTCGGCTTTGGAAG-3’). Mouse Gadph cDNA amplicons were generated with primers (5’-CAGGAGAGTGTTTCCTCGTCC-3’) and (5’-TTCCCATTCTCGGCCTTGAC-3’). Band intensities were measured with ImageJ (NIH) and normalized to Gapdh loading control.
RNA in situ hybridization.
Formalin fixed, paraffin embedded histology sections were pretreated with the RNAScope Target Retrieval kit (ACD, 322000) then hybridized with the Mm-Capn9-O1 probe (ACD, 487221), Mm-Acta2 (ACD, 319531), or Mm-Col1a1 (ACD, 319371). Immunofluorescent detection was performed with the RNAScope Fluorescent Multiplex Detection kit (ACD, 320851); chromogenic detection was performed with the RNAScope 2.5 HD Red kit (ACD, 322350) according to the manufacturer’s instructions. Fluorescent maximum intensity projection images were captured on a Zeiss LSM780 using a 40x objective. Image analysis of Col1a1 stained lung sections was performed by OracleBio using the Indica Labs HALO platform. Following quality assessment, slides were scanned, and total lung tissue, excluding large bronchial and vascular structures, was selected for analysis. Data were output as Col1a1 message positivity (red staining) per total tissue area.
Histology.
Tissue was fixed with 10% formalin for 24 h prior to embedding in paraffin and sectioning for histological staining. Masson trichrome staining was conducted by AML Laboratories (St. Petersburg, FL) for the lung and liver histology and the Reference Histology Laboratory at Johns Hopkins University (Baltimore, MD) for the heart histology. Picrosirius red (PSR) staining was performed using standard techniques. Lung and liver histology images were captured by whole-slide scan performed by HistoTox Labs (Boulder, CO). Histological scoring of lung and liver histology was performed by a blinded histopathologist (LNH ToxPath Consulting, LLC).
Bleomycin delivery and induced lung fibrosis.
Bleomycin was delivered by osmotic pump as previously described (45). Briefly, osmotic minipumps (Alzet, 1007D) designed to deliver their contents at 0.5 µL/h for 7 days containing either 100 µL saline vehicle or pharmaceutical grade bleomycin (100 U/kg, Teva Generics, NDC# 00703–3155-01) were implanted. Pumps were removed on day 10 as recommended by the manufacturer and mice were sacrificed on day 35. Intratracheal bleomycin was delivered as previously described (65). Briefly, bleomycin suspended in PBS was delivered to anesthetized male mice six to seven weeks of age at a dose of 1.5 U/kg. One week following bleomycin delivery, animals were administered therapeutic treatment with 100 mg/kg of MDL-28170 twice daily by oral gavage (200 mg/kg total daily dose) or vehicle (0.5% methylcellulose) for 2 weeks.
Carbon tetrachloride (CCl4) induced hepatic fibrosis in mice.
Male C57BL/6 and Capn9−/−mice, aged 10–12 weeks, received intraperitoneal injections twice a week with 1 mL/kg of CCl4 diluted 1:7 in corn oil for 4 weeks. Mice were sacrificed 3 days after final injection and subject to whole body perfusion with PBS via cardiac puncture. Serum was isolated from whole blood using blood collection tubes (BD Biosciences, 365967) as per manufacturer’s protocol. Liver enzyme serum concentrations were determined by the Johns Hopkins Department of Comparative & Molecular Pathology Phenotyping core facility.
Ang II delivery and plethysmography.
Wildtype mice 6 weeks of age were anesthetized using isoflurane and an osmotic pump (Alzet, 2004) delivering Ang II (Sigma, A9525) at 1.2 µg/kg/min or saline was implanted beneath the mid-scapular loose skin. Pumps were left for 28 days and mice were sacrificed for tissue collection. Blood pressures were measured by tail cuff plethysmography in the week prior to sacrifice. Mice were habituated to the system, and, on following day, at least 3 blood pressure readings were obtained per mouse and averaged.
Hydroxyproline assay.
Total collagen content was determined in freshly harvested lung tissue using the hydroxyproline assay kit (Sigma, MAK008) and normalized to total protein content (Pierce, 23225). Liver and heart total collagen content was determined from formalin fixed paraffin embedded tissue samples (QuickZyme Biosciences, QZBtotcol1) and normalized to protein content (QuickZyme Biosciences, QZBtotprot1) according to the manufacturer’s protocols.
Pulmonary lung function testing.
Pulmonary function testing was performed on a randomly selected subset of animals using a flexiVent ventilator (SCIREQ) as previously described (66). Tidal volume was set to 0.2 mL of 100% oxygen at a rate of 150 Hz with a positive end expiratory pressure (PEEP) of 3 cmH2O.
Lung flow cytometry.
Wildtype and Capn9-targeted male animals received intratracheal bleomycin as described above. Seven days following bleomycin administration, animals were euthanized with ketamine (100 mg/kg) and xylazine (10 mg/kg) and perfused with 10 mL cold PBS. The right lung was tied off and removed for histology; the left lung was inflated with digestion solution containing 1.5 mg/mL collagenase A (Sigma-Aldrich, 10103586001), 0.4 mg/mL DNaseI (Sigma-Aldrich, 4716728001), 5% FBS, 10 mM HEPES in HBSS. Cell suspensions were prepared and single-cell analysis was performed as previously describe (67).
Left ventricle cardiac function testing.
Left ventricular heart function was determined as previously described (68). In brief, ventral hair was removed with Nair the day prior to echocardiograms. All echocardiograms were performed on unsedated mice using a Vevo 2100 (VisualSonics) equipped with a 40 MHz linear transducer. M-mode echocardiography was acquired from the parasternal short axis view of the left ventricle.
Statistical analysis.
Data are expressed as the mean ± s.e.m. Differences in measured variables were assessed by using a two-tailed Student’s t-tests for single comparisons or one-way or two-way ANOVA followed by indicated post hoc corrections for multiple-comparison testing. Given limited sample sizes, data were assumed to be normally distributed, but this was not statistically tested. No surviving animals were excluded from analysis. Hardy-Weinberg calculations were performed by comparing observed population frequencies with calculated expected population frequencies with a χ2 test with 1 degree of freedom. Results were considered statistically significant at P < 0.05. No statistical method was used to predetermine sample size; however sample sizes were based off of prior experience with similar studies. All measurements were performed blinded. Prism 7 (GraphPad Software) was used for all statistical analysis. Individual subject-level data are provided in data file S1.
Supplementary Material
Fig. S1. TGFβ stimulation induced pSMAD2 nuclear localization, Alk5 inhibition prevented EMT, and cathepsin B and L inhibition did not alter EMT
Fig. S2. Broad spectrum calpain inhibition induced mesenchymal-to-epithelial transition in NMuMG cells
Fig. S3. Expression of dimeric calpain message in mouse tissue and expression of EMT transcription factors in NMuMG cells
Fig. S4. Calpain inhibition suppressed mesenchymal transition in multiple cell types
Fig. S5. TGFβ induced expression of CAPN9 protein with coordinated translation-dependent mRNA decay in NMuMG cells
Fig. S6. Capn9−/− mice expressed stable Capn9 message that lack exon 3
Fig. S7. Bleomycin induced an equal inflammatory response in wildtype and Capn9−/− animals Data file S1. Individual subject-level data.
Acknowledgments:
We are thankful to C. Schaefer and other individuals at Blade Therapeutics for their contributions during the review process. We thank Dr. Hiroyuki Sorimachi and Dr. Shoji Hata (Tokyo Metropolitan Institute of Medical Science, Japan) for providing Capn9tm1His mice through RIKEN BRC.
Funding: This work was supported by the Howard Hughes Medical Institute, the Scleroderma Research Foundation, the National Institutes of Health (NIH, R01AR068379 and R01HL128745), the Charles T. Bauer Foundation, the Johns Hopkins University School of Medicine Medical Scientist Training Program (T32GM007309), the Predoctoral Training Program in Human Genetics (T32GM007814), and the Johns Hopkins University School of Medicine Cellular and Molecular Medicine Program. Confocal microscopes from the JHU Microscope Facility were funded by the NIH (S10RR024550 and S10OD016374).
Footnotes
Publisher's Disclaimer: This manuscript has been accepted for publication in Science Translational Medicine. This version has not undergone final editing. Please refer to the complete version of record at www.sciencetranslationalmedicine.org/. The manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior written permission of AAAS.
Competing interests: H.C.D. and D.H.K. are co-inventors on patent application PCT/US2015/048739 entitled “Targeting Capn9/Capns2 Activity as a Therapeutic Strategy for the Treatment of Myofibroblast Differentiation and Associated Pathologies”. H.C.D. is also a founder and scientific advisor to Blade Therapeutics Inc. and has equity in the company. D.H.K. is an employee of and owns share in Blade Therapeutics Inc.
Data and materials availability: All data associated with this study are present in the paper or supplementary materials. Capn9-target sperm was obtained from RIKEN BRC under a materials transfer agreement.
References and Notes:
- 1.Wynn TA, Ramalingam TR, Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med 18, 1028–1040 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wynn TA, Cellular and molecular mechanisms of fibrosis. J Pathol 214, 199–210 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mehal WZ, Iredale J, Friedman SL, Scraping fibrosis: expressway to the core of fibrosis. Nat Med 17, 552–553 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wynn TA, Vannella KM, Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 44, 450–462 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loeys BL, Gerber EE, Riegert-Johnson D, Iqbal S, Whiteman P, McConnell V, Chillakuri CR, Macaya D, Coucke PJ, De Paepe A, Judge DP, Wigley F, Davis EC, Mardon HJ, Handford P, Keene DR, Sakai LY, Dietz HC, Mutations in fibrillin-1 cause congenital scleroderma: stiff skin syndrome. Sci Transl Med 2, 23ra20 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nogee LM, Dunbar AE 3rd, Wert SE, Askin F, Hamvas A, Whitsett JA, A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med 344, 573–579 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Armanios M, Telomeres and age-related disease: how telomere biology informs clinical paradigms. J Clin Invest 123, 996–1002 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Swaminathan S, Horn TD, Pellowski D, Abul-Ezz S, Bornhorst JA, Viswamitra S, Shah SV, Nephrogenic systemic fibrosis, gadolinium, and iron mobilization. N Engl J Med 357, 720–722 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Friedman SL, Sheppard D, Duffield JS, Violette S, Therapy for fibrotic diseases: nearing the starting line. Sci Transl Med 5, 167sr161 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Nieto MA, Huang RY, Jackson RA, Thiery JP, Emt: 2016. Cell 166, 21–45 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Sun YB, Qu X, Caruana G, Li J, The origin of renal fibroblasts/myofibroblasts and the signals that trigger fibrosis. Differentiation 92, 102–107 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Gonzalez DM, Medici D, Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal 7, re8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim KK, Sheppard D, Chapman HA, TGF-beta1 Signaling and Tissue Fibrosis. Cold Spring Harb Perspect Biol 10, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rowe RG, Lin Y, Shimizu-Hirota R, Hanada S, Neilson EG, Greenson JK, Weiss SJ, Hepatocyte-derived Snail1 propagates liver fibrosis progression. Mol Cell Biol 31, 2392–2403 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Balli D, Ustiyan V, Zhang Y, Wang IC, Masino AJ, Ren X, Whitsett JA, Kalinichenko VV, Kalin TV, Foxm1 transcription factor is required for lung fibrosis and epithelial-to-mesenchymal transition. EMBO J 32, 231–244 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zatz M, Starling A, Calpains and disease. N Engl J Med 352, 2413–2423 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Bai DS, Dai Z, Zhou J, Liu YK, Qiu SJ, Tan CJ, Shi YH, Huang C, Wang Z, He YF, Fan J, Capn4 overexpression underlies tumor invasion and metastasis after liver transplantation for hepatocellular carcinoma. Hepatology 49, 460–470 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Braun C, Engel M, Seifert M, Theisinger B, Seitz G, Zang KD, Welter C, Expression of calpain I messenger RNA in human renal cell carcinoma: correlation with lymph node metastasis and histological type. Int J Cancer 84, 6–9 (1999). [DOI] [PubMed] [Google Scholar]
- 19.Chen B, Tang J, Guo YS, Li Y, Chen ZN, Jiang JL, Calpains are required for invasive and metastatic potentials of human HCC cells. Cell Biol Int 37, 643–652 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Zhang C, Bai DS, Huang XY, Shi GM, Ke AW, Yang LX, Yang XR, Zhou J, Fan J, Prognostic significance of Capn4 overexpression in intrahepatic cholangiocarcinoma. PLoS One 8, e54619 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nassar D, Letavernier E, Baud L, Aractingi S, Khosrotehrani K, Calpain activity is essential in skin wound healing and contributes to scar formation. PLoS One 7, e37084 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Letavernier E, Perez J, Bellocq A, Mesnard L, de Castro Keller A, Haymann JP, Baud L, Targeting the calpain/calpastatin system as a new strategy to prevent cardiovascular remodeling in angiotensin II-induced hypertension. Circ Res 102, 720–728 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Letavernier E, Zafrani L, Perez J, Letavernier B, Haymann JP, Baud L, The role of calpains in myocardial remodelling and heart failure. Cardiovasc Res 96, 38–45 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Li Y, Ma J, Zhu H, Singh M, Hill D, Greer PA, Arnold JM, Abel ED, Peng T, Targeted inhibition of calpain reduces myocardial hypertrophy and fibrosis in mouse models of type 1 diabetes. Diabetes 60, 2985–2994 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sandmann S, Yu M, Unger T, Transcriptional and translational regulation of calpain in the rat heart after myocardial infarction--effects of AT(1) and AT(2) receptor antagonists and ACE inhibitor. Br J Pharmacol 132, 767–777 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tabata C, Tabata R, Nakano T, The calpain inhibitor calpeptin prevents bleomycin-induced pulmonary fibrosis in mice. Clin Exp Immunol 162, 560–567 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ono Y, Sorimachi H, Calpains: an elaborate proteolytic system. Biochim Biophys Acta 1824, 224–236 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Schad E, Farkas A, Jekely G, Tompa P, Friedrich P, A novel human small subunit of calpains. Biochem J 362, 383–388 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wendt A, Thompson VF, Goll DE, Interaction of calpastatin with calpain: a review. Biol Chem 385, 465–472 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Hanna RA, Campbell RL, Davies PL, Calcium-bound structure of calpain and its mechanism of inhibition by calpastatin. Nature 456, 409–412 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Moldoveanu T, Gehring K, Green DR, Concerted multi-pronged attack by calpastatin to occlude the catalytic cleft of heterodimeric calpains. Nature 456, 404–408 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hata S, Abe M, Suzuki H, Kitamura F, Toyama-Sorimachi N, Abe K, Sakimura K, Sorimachi H, Calpain 8/nCL-2 and calpain 9/nCL-4 constitute an active protease complex, G-calpain, involved in gastric mucosal defense. PLoS Genet 6, e1001040 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Ponten F, Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Piek E, Moustakas A, Kurisaki A, Heldin CH, ten Dijke P, TGF-(beta) type I receptor/ALK-5 and Smad proteins mediate epithelial to mesenchymal transdifferentiation in NMuMG breast epithelial cells. J Cell Sci 112 (Pt 24), 4557–4568 (1999). [DOI] [PubMed] [Google Scholar]
- 35.Xie L, Law BK, Chytil AM, Brown KA, Aakre ME, Moses HL, Activation of the Erk pathway is required for TGF-beta1-induced EMT in vitro. Neoplasia 6, 603–610 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gorlin JB, Yamin R, Egan S, Stewart M, Stossel TP, Kwiatkowski DJ, Hartwig JH, Human endothelial actin-binding protein (ABP-280, nonmuscle filamin): a molecular leaf spring. J Cell Biol 111, 1089–1105 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsujinaka T, Kajiwara Y, Kambayashi J, Sakon M, Higuchi N, Tanaka T, Mori T, Synthesis of a new cell penetrating calpain inhibitor (calpeptin). Biochemical and Biophysical Research Communications 153, 1201–1208 (1988). [DOI] [PubMed] [Google Scholar]
- 38.Bootman MD, Collins TJ, Mackenzie L, Roderick HL, Berridge MJ, Peppiatt CM, 2-aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+ release. FASEB J 16, 1145–1150 (2002). [DOI] [PubMed] [Google Scholar]
- 39.Hosfield CM, Elce JS, Davies PL, Jia Z, Crystal structure of calpain reveals the structural basis for Ca(2+)-dependent protease activity and a novel mode of enzyme activation. EMBO J 18, 6880–6889 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Strobl S, Fernandez-Catalan C, Braun M, Huber R, Masumoto H, Nakagawa K, Irie A, Sorimachi H, Bourenkow G, Bartunik H, Suzuki K, Bode W, The crystal structure of calcium-free human m-calpain suggests an electrostatic switch mechanism for activation by calcium. Proc Natl Acad Sci U S A 97, 588–592 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Montaser M, Lalmanach G, Mach L, CA-074, but not its methyl ester CA-074Me, is a selective inhibitor of cathepsin B within living cells. Biol Chem 383, 1305–1308 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Steverding D, The Cathepsin B-Selective Inhibitors CA-074 and CA-074Me Inactivate Cathepsin L Under Reducing Conditions. The Open Enzyme Inhibition Journal 4, 11–16 (2011). [Google Scholar]
- 43.Hata S, Kitamura F, Yamaguchi M, Shitara H, Murakami M, Sorimachi H, A Gastrointestinal Calpain Complex, G-calpain, Is a Heterodimer of CAPN8 and CAPN9 Calpain Isoforms, Which Play Catalytic and Regulatory Roles, Respectively. J Biol Chem 291, 27313–27322 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chaudhury A, Hussey GS, Ray PS, Jin G, Fox PL, Howe PH, TGF-beta-mediated phosphorylation of hnRNP E1 induces EMT via transcript-selective translational induction of Dab2 and ILEI. Nat Cell Biol 12, 286–293 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee R, Reese C, Bonner M, Tourkina E, Hajdu Z, Riemer EC, Silver RM, Visconti RP, Hoffman S, Bleomycin delivery by osmotic minipump: similarity to human scleroderma interstitial lung disease. Am J Physiol Lung Cell Mol Physiol 306, L736–748 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Knabel MK, Ramachandran K, Karhadkar S, Hwang HW, Creamer TJ, Chivukula RR, Sheikh F, Clark KR, Torbenson M, Montgomery RA, Cameron AM, Mendell JT, Warren DS, Systemic Delivery of scAAV8-Encoded MiR-29a Ameliorates Hepatic Fibrosis in Carbon Tetrachloride-Treated Mice. PLoS One 10, e0124411 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Campbell SE, Katwa LC, Angiotensin II stimulated expression of transforming growth factor-beta1 in cardiac fibroblasts and myofibroblasts. J Mol Cell Cardiol 29, 1947–1958 (1997). [DOI] [PubMed] [Google Scholar]
- 48.Sadoshima J, Izumo S, Molecular characterization of angiotensin II--induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role of the AT1 receptor subtype. Circulation Research 73, 413–423 (1993). [DOI] [PubMed] [Google Scholar]
- 49.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R, Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med 13, 952–961 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Lijnen PJ, Petrov VV, Fagard RH, Angiotensin II-induced stimulation of collagen secretion and production in cardiac fibroblasts is mediated via angiotensin II subtype 1 receptors. J Renin Angiotensin Aldosterone Syst 2, 117–122 (2001). [DOI] [PubMed] [Google Scholar]
- 51.Nagpal V, Rai R, Place AT, Murphy SB, Verma SK, Ghosh AK, Vaughan DE, MiR-125b Is Critical for Fibroblast-to-Myofibroblast Transition and Cardiac Fibrosis. Circulation 133, 291–301 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Limjunyawong N, Mitzner W, Horton MR, A mouse model of chronic idiopathic pulmonary fibrosis. Physiol Rep 2, e00249 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG, Exome Aggregation C, Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kalluri R, Weinberg RA, The basics of epithelial-mesenchymal transition. J Clin Invest 119, 1420–1428 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bachofner JA, Valli PV, Kroger A, Bergamin I, Kunzler P, Baserga A, Braun D, Seifert B, Moncsek A, Fehr J, Semela D, Magenta L, Mullhaupt B, Terziroli Beretta-Piccoli B, Mertens JC, Direct antiviral agent treatment of chronic hepatitis C results in rapid regression of transient elastography and fibrosis markers fibrosis-4 score and aspartate aminotransferase-platelet ratio index. Liver Int 37, 369–376 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Marcellin P, Gane E, Buti M, Afdhal N, Sievert W, Jacobson IM, Washington MK, Germanidis G, Flaherty JF, Schall RA, Bornstein JD, Kitrinos KM, Subramanian GM, McHutchison JG, Heathcote EJ, Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: a 5-year open-label follow-up study. The Lancet 381, 468–475 (2013). [DOI] [PubMed] [Google Scholar]
- 57.Liaw YF, Reversal of cirrhosis: an achievable goal of hepatitis B antiviral therapy. J Hepatol 59, 880–881 (2013). [DOI] [PubMed] [Google Scholar]
- 58.Liu Y, Liu B, Zhang GQ, Zou JF, Zou ML, Cheng ZS, Calpain inhibition attenuates bleomycin-induced pulmonary fibrosis via switching the development of epithelial-mesenchymal transition. Naunyn Schmiedebergs Arch Pharmacol, (2018). [DOI] [PMC free article] [PubMed]
- 59.Chen CJ, Nguyen T, Shively JE, Role of calpain-9 and PKC-delta in the apoptotic mechanism of lumen formation in CEACAM1 transfected breast epithelial cells. Exp Cell Res 316, 638–648 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ma H, Nakajima E, Shih M, Azuma M, Shearer TR, Expression of calpain small subunit 2 in mammalian tissues. Curr Eye Res 29, 337–347 (2004). [DOI] [PubMed] [Google Scholar]
- 61.Davis TL, Walker JR, Finerty PJ Jr., Mackenzie F, Newman EM, Dhe-Paganon S, The crystal structures of human calpains 1 and 9 imply diverse mechanisms of action and auto-inhibition. J Mol Biol 366, 216–229 (2007). [DOI] [PubMed] [Google Scholar]
- 62.Ono Y, Saido TC, Sorimachi H, Calpain research for drug discovery: challenges and potential. Nat Rev Drug Discov 15, 854–876 (2016). [DOI] [PubMed] [Google Scholar]
- 63.Gould RA, Butcher JT, Isolation of valvular endothelial cells. J Vis Exp, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mahler GJ, Farrar EJ, Butcher JT, Inflammatory cytokines promote mesenchymal transformation in embryonic and adult valve endothelial cells. Arterioscler Thromb Vasc Biol 33, 121–130 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Walters DM, Kleeberger SR, Mouse models of bleomycin-induced pulmonary fibrosis. Curr Protoc Pharmacol Chapter 5, Unit 5 46 (2008). [DOI] [PubMed] [Google Scholar]
- 66.Collins SL, Chan-Li Y, Oh M, Vigeland CL, Limjunyawong N, Mitzner W, Powell JD, Horton MR, Vaccinia vaccine-based immunotherapy arrests and reverses established pulmonary fibrosis. JCI Insight 1, e83116 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu YR, O’Koren EG, Hotten DF, Kan MJ, Kopin D, Nelson ER, Que L, Gunn MD, A Protocol for the Comprehensive Flow Cytometric Analysis of Immune Cells in Normal and Inflamed Murine Non-Lymphoid Tissues. PLoS One 11, e0150606 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rouf R, MacFarlane EG, Takimoto E, Chaudhary R, Nagpal V, Rainer PP, Bindman JG, Gerber EE, Bedja D, Schiefer C, Miller KL, Zhu G, Myers L, Amat-Alarcon N, Lee DI, Koitabashi N, Judge DP, Kass DA, Dietz HC, Nonmyocyte ERK1/2 signaling contributes to load-induced cardiomyopathy in Marfan mice. JCI Insight 2, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. TGFβ stimulation induced pSMAD2 nuclear localization, Alk5 inhibition prevented EMT, and cathepsin B and L inhibition did not alter EMT
Fig. S2. Broad spectrum calpain inhibition induced mesenchymal-to-epithelial transition in NMuMG cells
Fig. S3. Expression of dimeric calpain message in mouse tissue and expression of EMT transcription factors in NMuMG cells
Fig. S4. Calpain inhibition suppressed mesenchymal transition in multiple cell types
Fig. S5. TGFβ induced expression of CAPN9 protein with coordinated translation-dependent mRNA decay in NMuMG cells
Fig. S6. Capn9−/− mice expressed stable Capn9 message that lack exon 3
Fig. S7. Bleomycin induced an equal inflammatory response in wildtype and Capn9−/− animals Data file S1. Individual subject-level data.