

SUMMARY
Structural variants (SVs) underlie important crop improvement and domestication traits. However, resolving the extent, diversity, and quantitative impact of SVs has been challenging. We used long-read nanopore sequencing to capture 238,490 SVs in 100 diverse tomato lines. This panSV-genome, along with 14 new reference assemblies, revealed large-scale intermixing of diverse genotypes, as well as thousands of SVs intersecting genes and cis-regulatory regions. Hundreds of SV-gene pairs exhibit subtle and significant expression changes, which could broadly influence quantitative trait variation. By combining quantitative genetics with genome editing, we show how multiple SVs that changed gene dosage and expression levels modified fruit flavor, size, and production. In the last example, higher-order epistasis among four SVs affecting three related transcription factors allowed introduction of an important harvesting trait in modern tomato. Our findings highlight the underexplored role of SVs in genotype-tophenotype relationships and their widespread importance and utility in crop improvement.
Keywords: structural variation, long-read sequencing, tomato, introgression, QTL, domestication, copy number variation, cis-regulatory, dosage, cryptic variation, epistasis, crop, breeding
Graphical Abstract
INTRODUCTION
Phenotypic variation in crop plants is shaped by genetic variation from their wild ancestors, as well as the selection and maintenance of collections of mutations that impact agricultural adaptations and human preferences (Meyer and Purugganan, 2013; Olsen and Wendel, 2013). The majority of this variation is quantitative, and now more than ever a major goal of genetics is to identify and understand how specific genes and variants contribute to quantitative trait variation. In particular, this knowledge is necessary for designing and engineering favored alleles in crop improvement, enabled by genome editing (Chen et al., 2019; Rodríguez-Leal et al., 2017; Wallace et al., 2018). While high-throughput short-read sequencing accelerated the discovery of natural genetic variants among diverse germplasm of major crops, it has also introduced an unavoidable bias: characterized variants are disproportionately skewed towards single nucleotide polymorphisms (SNPs) and small indels (De Coster and Van Broeckhoven, 2019). However, decades of research have shown that structural variations (SVs: large deletions, insertions, duplications, and chromosomal rearrangements) are important in plant evolution and agriculture, affecting traits such as shoot architecture, flowering time, fruit size, and stress resistance (Lye and Purugganan, 2019). Compared to SNPs, SVs can cause large-scale perturbations of cis-regulatory regions and are therefore more likely to quantitatively change gene expression and phenotypes. SVs can also modify expression levels by directly altering gene copy number. However, despite their importance, identifying SVs with short-read sequencing is notoriously difficult and unreliable, leaving the vast majority of SVs poorly resolved and their molecular and phenotypic impacts largely hidden (Ho et al., 2020; Sedlazeck et al., 2018a).
High-throughput Oxford Nanopore Technology (ONT) long-read sequencing now enables a broad survey of population-scale SV landscapes. Such resources that capture the diversity of SVs, in combination with expression profiling and genome editing, immediately allow for the direct interrogation of the molecular and phenotypic consequences of SVs. Here, we present the most comprehensive panSV-genome for a major crop and study its significance in evolution, domestication, quantitative genetics, and breeding. We used ONT long-read sequencing to identify SVs from a collection of 100 diverse wild and domesticated tomato accessions. Tomato, in addition to its agricultural and economic importance, has extensive genetic resources, well-described phenotypic diversity, and efficient genome editing, making it an ideal system to investigate the broad significance of SVs in both fundamental plant biology and agriculture. Our long-read data provided continuous long-range information that allowed for the sequence resolved inference of more than 200,000 SVs, the majority being transposons and related repeat sequences. Patterns of SV distribution revealed extensive admixture and population-scale introgressions. RNA sequencing showed that gene expression is widely impacted by SVs affecting both coding and cis-regulatory regions. Establishing high-quality de novo genome assemblies for 14 selected genotypes allowed us to resolve hidden genomic complexity involving SVs. To demonstrate the value of this panSV-genome, we directly linked these complex alleles with multiple domestication and improvement traits affecting fruit flavor, size and productivity. For two of these traits, modest changes in expression originated from gene copy number variation, and we used CRISPR-Cas9 genome editing to demonstrate causal quantitative relationships between gene dosage and phenotype. Our work uncovers the prevalence and importance of SVs in plant genomes and demonstrates the underexplored roles of SVs in trait variation.
RESULTS
Long-read Sequencing of 100 Tomato Accessions Establishes a PanSV-Genome
To deeply survey the landscape of natural structural variation in tomato, we collected long-read sequencing data from a representative population-scale tomato panel (Figure 1A and Table S1A). To this end, we first used available short-read sequencing data to call SVs from over 800 tomato accessions and then applied the SVCollector algorithm to optimally select 51 diverse modern and early domesticated samples that maximize SV diversity (Sedlazeck et al., 2018b). We then separately selected an additional 49 wild species and modern accessions that are used by tomato research and breeding communities (Table S1A). Our final set of 100 accessions captures phylogenetic diversity spanning the closest wild relatives of domesticated tomato [S. pimpinellifolium (SP), S. cheesmaniae (CHE), S. galapagense (GAL)], early domesticated forms [S. lyc. var. cerasiforme (SLC)], and ‘vintage’ cultivars and modern varieties [S. lycopersicum, (SLL)] (Figures 1A and S1A–B) (Table S1B).
Figure 1. The tomato panSV-genome.
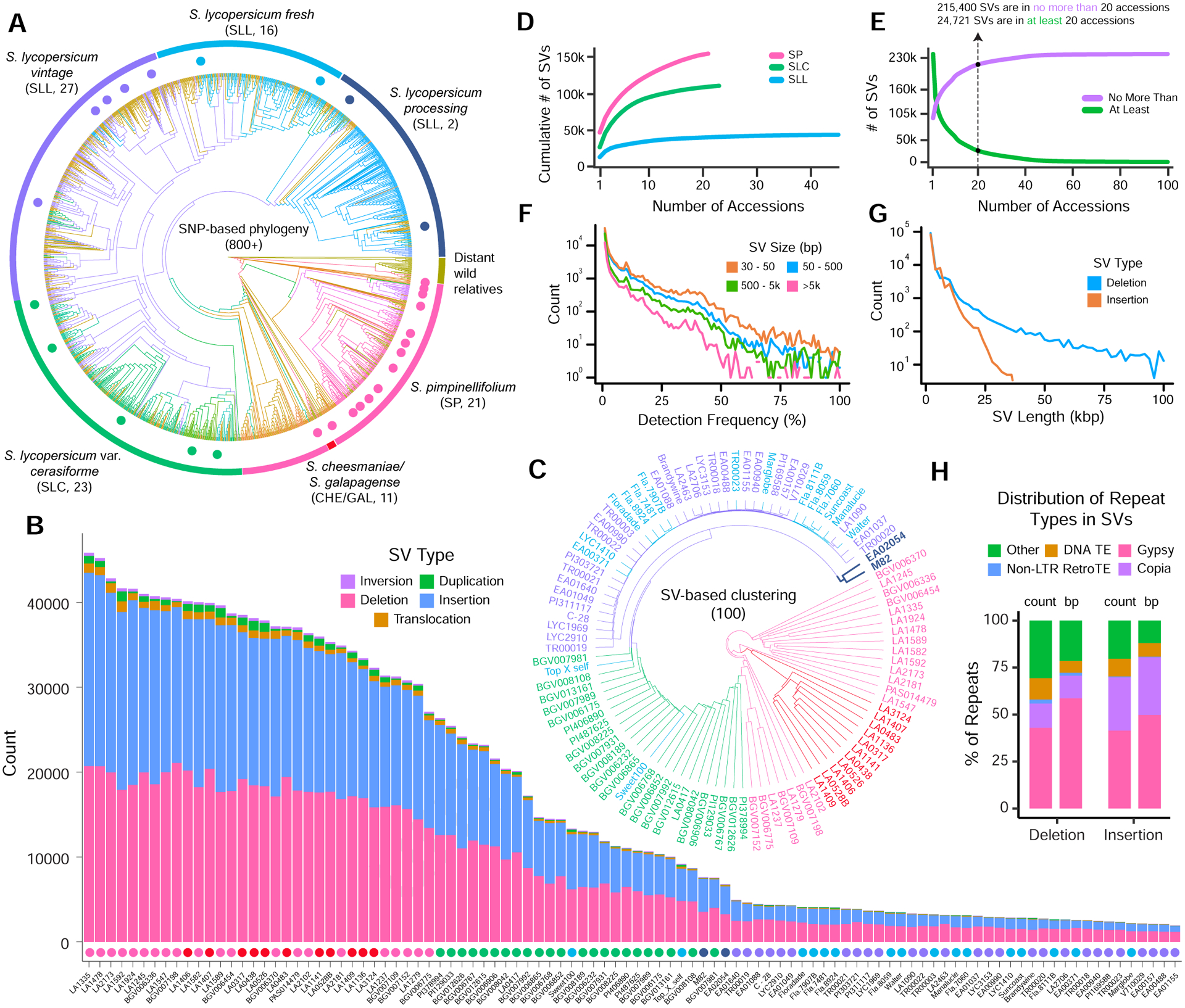
(A) SNP-based phylogenetic tree based on short-read sequencing of more than 800 tomato accessions. Major taxonomic groups are marked by colored lines along the circumference. Colored dots indicate a subset of the 100 accessions selected for long-read sequencing.
(B) Stacked bar graph showing SV number and type from the 100 accessions. Colored dots indicate the taxonomic group of each accession, corresponding to colors in (A).
(C) Hierarchical clustering dendrogram of the SV presence/absence matrix across the 100 accessions, with colors corresponding to (A). Bold branches and names highlight an outgroup of two SLL processing tomato accessions.
(D) SVCollector curves of SVs in the three major taxonomic groups. The “greedy” algorithm determines the order of accessions and depicts the cumulative number of SVs as a function of the number of accessions included.
(E) Graph showing the number of SVs (y-axis) in “no more than” or “at least” the number of accessions indicated on the x-axis.
(F) Histograms of detection frequencies for different SV sizes.
(G) Histogram of SV sizes for insertions and deletions.
(H) Annotation of the panSV-genome. The proportion of repeat types for all insertions and deletions annotations is shown in stacked bar graphs. “Count” shows the proportion of individual repeat annotations, and “bp” shows the proportion of cumulative repeat (not indel) sequence length. “Other” refers to other repeat types. Only indels at least 100 bp in size were considered. See also Figure S1.
For each of the 100 accessions, we used Oxford Nanopore long-read sequencing to generate a minimum of 40X genome coverage, achieving a total of 7.77 Tb of long-read data with an average read length N50 of 19.6 kbp (Table S1C). Reads were aligned to the recently released SL4.0 reference genome (Heinz 1706, SLL) with NGMLR, and SVs were called with Sniffles (Figure S1C and S1D)(Hosmani et al., 2019; Sedlazeck et al., 2018a). We then filtered, sequence resolved, and merged all 100 sets of SV calls, revealing 238,490 total SVs (defined in this study as >30bp) that comprise the most comprehensive sequence-resolved panSV-genome in plants (see STAR Methods). Importantly, we confirmed that the majority of these variants would not have been revealed using solely short-read sequencing data (Figure S1E).
Individual accessions had between 1,928 and 45,840 SVs, with the wild SP, GAL and CHE accessions harboring the most structural variation relative to the Heinz reference genome (Figure 1B). Insertions and deletions were the most common SV type, though we also found dozens to hundreds of inversions, duplications, and translocations in all samples. SVs are with respect to the reference genome and do not necessarily reflect underlying evolutionary context. Clustering of the SV presence/absence matrix revealed a structure that mirrored the larger SNP-based tomato phylogeny, with accessions clustering within their known taxonomic groups (Figure 1C). Interestingly, the SLL “cherry” variety Sweet100 grouped with the SLCs, and the only two processing cultivars, M82 and EA02054, form a distinct group from the SLLs, suggesting admixture. Comparative analysis of the long-read SVs showed that SP and SLC have more SV diversity compared to SLL, consistent with the loss of genetic variation during the domestication and improvement of tomato (Figure 1D and S1F) (Aflitos et al., 2014; Lin et al., 2014). This analysis also indicated that even sequencing 100 accessions, many SVs remain to be discovered (Figure 1E). Consistently, the majority of SVs are singletons, or are otherwise rare, although tens of thousands of SVs are common (>5% detection frequency) (Figure 1F). We evaluated SV length distribution, which showed that most SVs were relatively small: 30.5%: 30–50 bp; 30.5%: 50–200 bp; 39%: >200 bp (Figure 1G). We note that our method has limited ability to detect larger insertions, since, unlike deletion calling, such detection is bounded by read length (see STAR Methods). SVs are typically composed of, or generated by, transposons and related repeats (Audano et al., 2019; Fuentes et al., 2019), and annotation of our panSV-genome showed 84% of deletions and 76% of insertions larger than 100 bp match at least one repeat. Retrotransposon sequences, especially from Gypsy and Copia elements, are the most prevalent among the annotated SVs (Figure 1H).
Fourteen New High-Quality Tomato Reference Genomes
To supplement the panSV-genome with additional genomic resources, we selected 14 diverse accessions for genome assembly and annotation (Table S2D). Combining long and short-read sequencing data, de novo assemblies using the MaSuRCA hybrid assembler yielded an average contig N50 of 1.9 Mbp (Figures S2A and S2B and Table S2A) (see STAR Methods) (Zimin et al., 2017). Reference-guided scaffolding with RaGOO produced chromosome-scale pseudomolecules that contained, on average, a single copy of 96% of complete benchmarking universal single-copy orthologues (BUSCO) genes (Figures S2C–S2P) (Table S2B) (Alonge et al., 2019; Simão et al., 2015). Repeats were annotated using REPET, and genes annotations were “lifted-over” from reference annotations using geneLift (see STAR Methods) (Flutre et al., 2011). We used these new reference genomes (referred to as “MAS2.0”) to validate SVs in the same 14 accessions, of which 90% were also found in the assemblies (see STAR Methods). Owing to the diversity of these assemblies, which represent multiple SP, SLC and SLL accessions, we anchored 22% of recently discovered “pan-genome” genes that are missing from the ITAG reference annotation (Figures S2Q and S2R) (Table S2C) (Gao et al., 2019). These MAS2.0 genomes were critical to link complex SV loci with functional consequences shown below.
SV Distribution Reveals Extensive Admixture and Introgression
The chromosomal distributions of SVs from our panSV-genome revealed several hypervariable genomic regions relative to the Heinz reference shared among subsets of SLL accessions (designated SV “hotspots”) (Figure 2A). Since SP accessions have more structural variants than those of SLL, SV hotspots in SLL could reflect admixture and introgression between wild and domesticated accessions, which was previously partially explored using SNPs (Aflitos et al., 2014, 2015; Sato et al., 2012). Introgression is a common practice in tomato breeding, through which disease resistance genes and other desirable traits from wild donors are introduced into SLL breeding germplasm (Aflitos et al., 2014). We found that SV hotspots in SLL correlated with genomic regions that show high similarity with SP and/or SLC based on the Jaccard similarity of SV content between accessions (Figure S3A–L) (Tables S3A–L). For example, multiple SV hotspots exist on chromosome 4, including a 2 Mbp region common to all SLL accessions that corresponds to a known unique introgression in the Heinz reference genome (Figure 2A) (Sato et al., 2012). Most SP accessions show a decrease in SV frequency in this region, indicating these accessions are closely related to the introgression donor. We also found a large introgression block shared by five SLLs that occupies two-thirds of the chromosome (Figure 2B). Notably, two of these accessions are M82 and EA02054, which also carry large introgression blocks that span nearly all of chromosomes 5 and 11 (Figure 2A, S3E and S3K), explaining their distinct grouping in SLL and their relatively large number of SVs compared to Heinz 1706, which is also a processing type (Figure 1B and 1C).
Figure 2. SV distribution reveals large-scale admixture and introgression between wild and domesticated genotypes.
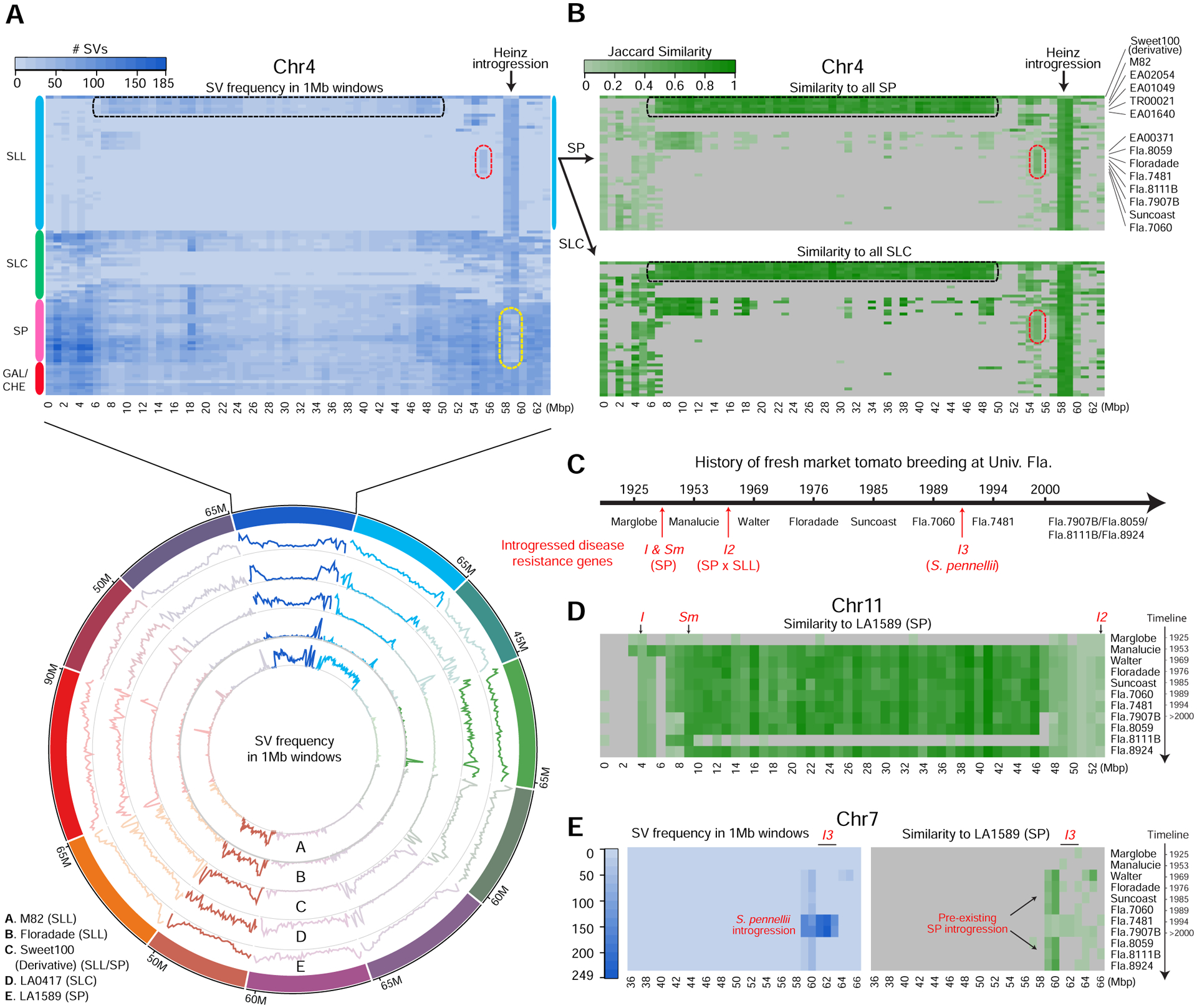
(A) Heatmap (top) showing SV frequency in 1 Mbp windows (columns) of chromosome 4 relative to the reference genome. Accessions (rows) are grouped by taxonomic group (colored bars). Dotted colored lines mark three notable regions: black, a large SV hotspot for 5 SLLs; red, a small hotspot shared by most UFL SLL lines; yellow, a SP group with reduced SV frequency, reflecting a small SP introgression in the reference genome. Circos plot (bottom) depicting genome-wide SV frequency for five notable accessions. Rings depict line plots showing the SV number in successive 1Mbp windows (y-axes are not shared between rings). Chromosomes 4, 5, 7 and 11 are highlighted to show regions of high SV frequency.
(B) Heatmaps showing admixture and introgressions on chromosome 4 measured by Jaccard similarity between accessions of SLL and SP (top) and SLC (bottom) in the same row-order as (A, top). For each 1 Mbp window, the SVs for a given SLL accession are compared to the SVs for all SP (top) or SLC (bottom) accessions, and the maximum Jaccard similarity is reported. Windows with fewer than 5 SVs in the SLL set are excluded and colored grey. Black and red dotted regions correlate with marked SV hotspots in (A, top).
(C) Timeline of UFL fresh market variety release over the last century. Approximate periods of introgression of key disease resistance genes are shown in red, along with major donor genotypes for Fusarium wilt (I, I2, I3) and grey leaf-spot (Sm).
(D) Jaccard similarity for chromosome 11 between the UFL lines (ordered chronologically) and LA1589, the closest SP to this introgression. Locations of I, Sm and I2 are shown in red.
(E) The UFL varieties on chromosome 7 showing a small SP introgression in all but two accessions; Fla.7481 and Fla.7907B carry a unique SV hotspot (left) due to introgression of the I3 resistance gene (red) from S. pennellii.
See also Figure S3.
Expecting that our panSV-genome would illuminate how breeding and introgression have shaped SV content, we examined 11 SLLs included in our 100 genomes from the University of Florida (UFL) tomato breeding program, which has a well-documented history of disease resistance gene introgression (Scott, 1999a). The devastating fungal disease Fusarium wilt first emerged in the 1930s, and the resistance genes I and I2 (from SP donors) and I3 (from S. pennellii) against three races of this disease were successively introduced into UFL breeding material between the 1930s and 1980s (Figure 2C) (Bohn and Tucker, 1939; Scott and Jones, 1989; Strobel et al., 1969). Furthermore, the Sm resistance gene against Grey leaf spot was introduced in the 1950s (Walter and Kelbert, 1953). Molecular mapping and gene cloning have shown that I and Sm are located on the opposite arms from I2 on chromosome 11. The variants from our panSV-genome demonstrated overlapping introgressions from multiple donors, including those contributing resistance to other diseases (Foolad and Panthee, 2012), accounting for the large introgression block in the UFL accessions (Figure 2D). Interestingly, the modern breeding line Fla.8111B carries the I, I2 and Sm resistance genes, but lacks a large portion of this introgression, suggesting this region was later purged during selection.
The I3 introgression on chromosome 7 was introduced in the 1980s (Figure 2C). The modern breeding lines Fla.7481 and Fla.7907B that carry I3 resistance show a 5 Mbp SV hotspot with low similarity to SP and SLC at the I3 locus, consistent with the donor being the distant green-fruited wild species S. pennelli (Figure 2E). Interestingly, UFL lines lacking I3 resistance have a 2 Mbp introgression from SP or SLC that first appeared in the 1960s and overlaps the I3 introgression. The I3 introgression is negatively implicated with several horticultural characteristics, including reduced fruit size and increased sensitivity to bacterial spot (Hutton et al., 2014; Li et al., 2018; Scott, 1999b). The earlier introduced SP introgression may have provided tolerance to bacterial spot or benefitted other traits, as is likely for many other putative SP or SLC introgressions revealed by our panSV-genome (Figure S3A–S3L) (Tables S3A–L). The large number of SVs from wild species introduced in breeding could have broad functional consequences.
SVs Associated with Genes Have Widespread Impacts on Expression
SVs may influence the expression of nearby genes, by altering the sequence or copy number of a gene or by changing the composition or position of cis-regulatory sequences (Chiang et al., 2017; Yang et al., 2019). We explored this relationship with the comprehensive catalog of SVs across our tomato panSV-genome. Candidate SVs that could potentially impact gene expression were abundant in our collection. Nearly 50% (112,114) of SVs overlap genes and/or flanking regulatory sequences (+/− 5 kbp of coding sequence), and among 34,075 annotated genes, 95% have at least one SV within 5 kbp of coding sequences across the 100 genomes, with the majority found in cis-regulatory regions (Figures 3A and 3B). To explore the impact of SVs on gene expression, we performed 3’ RNA-sequencing (RNA-seq) on three tissues (cotyledons, roots, apical meristems) for 23 accessions that capture 44,358 gene-associated SVs. We evaluated a total of 21,156 SV-gene pairs, and found hundreds of significant expression changes (Figure 3C) (Tables S4A and S4B) (see STAR Methods). Nearly half of the SVs affecting coding sequences (Deletions of CDS start, Deletions of exons, and Duplications) are significantly associated with differences in expression, with many substantially reducing or eliminating expression (Table S4). In regulatory regions, 1,534 SV-gene pairs (7.3%) showed significant differential expression across all tissues, and overall these differences were subtler compared to SVs in coding regions (mean log2 fold change 1.36 and 2.47, respectively) (Figure S4A).
Figure 3. Gene associated SVs impact expression.
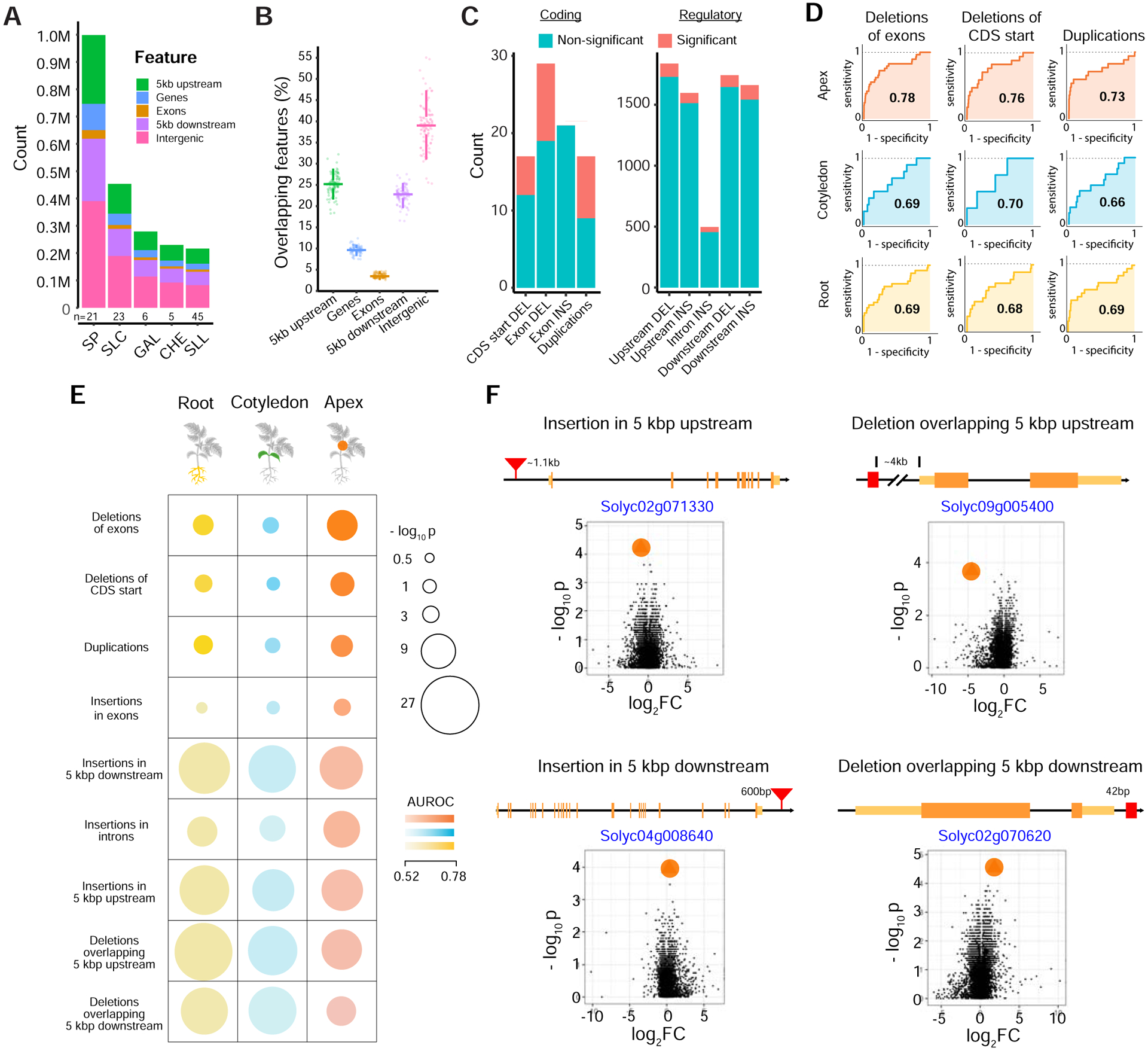
(A) Stacked bar chart showing total counts of SVs overlapping different genomic features in major taxonomic groups. N represents the number of accessions in each taxonomic group.
(B) Percentage of SVs overlapping different genomic features in 100 accessions. Each point is one sample. Fewer SVs are found within genes compared to surrounding regulatory regions.
(C) Stacked bar charts showing numbers of differentially expressed genes affected by insertion, deletion, and duplication SVs overlapping coding sequences (left) and regulatory regions (right)*. Differential expression was tested on common SVs in the 23 accessions used for RNA-sequencing (frequency between 0.2 and 0.8) (see STAR Methods).
(D) ROC curves for the top three SV annotation types, with high AUROC (Area Under the Receiver Operating Characteristics) scores across the three tissues demonstrating the ability to identify genes containing SVs using changes in expression across the accession split. The AUROC is specified within the ROC curve in each case. The steep rise of the curves in the top panel correspond to a near-perfect identification of a large fraction of the genes containing SVs based on differential expression. CDS, coding sequence.
(E) Differential expression significantly predicts genes with SVs. Overall performance of using “SV splits” and differential expression to predict associated gene(s) (see STAR Methods). Analyses are broken down into 9 categories across three tissues. Each category is defined based on SV type and relative position to genes. Circle sizes and colors represent the significance of performance (−log10 p-value) the magnitude of AUROC, respectively. SV categories are ranked in decreasing order of average AUC (Area Under the Curve) across the three tissues. Note that the significance of performance for each SV type is enhanced by the number of annotated SV-gene pairs (for example, p < 1×10−4 for ≈ 16 duplications, while p < 1×10−4 for ≈ 468 insertions in introns).
(F) Volcano plots for four regulatory SV-gene pair examples with the highest AUROC score highlight the extent of differential expression of SV-containing genes (marked in orange circles), compared to all expressed genes (black dots). Additional examples are presented in Figure S4F. p-values and expression fold changes are computed across two groups of accessions (with and without the indicated SV). Data shown for apex tissue. Exons (orange), UTRs (yellow), and SVs (red) are not drawn to scale. Distances between genes and SVs are shown.
* Significance is defined as an adjusted p-value less than 0.05. See also Figure S4.
Knowing that a substantial fraction of population-scale expression variation is explained by cis-eQTL (Aguet et al., 2017; Kawakatsu et al., 2016), we next formulated a classification task that uses changes in gene expression to predict the presence of a nearby SV (see STAR Methods) (Figure S4B). This classifier complements standard fold-change measurements among known SV-gene pairs and its performance can quantify the extent to which global expression changes are associated with SVs. Notably, this test is robust to population structure because global changes in expression or confounding variants can only serve to weaken any oneto-one relationship between gene expression variation and the existence of a nearby variant.
Broadly, differential expression significantly predicts genes with associated SVs (Figures 3D–3E and S4C–S4E) (Tables S4C–L). As expected, this classifier performs best on the coding sequence SVs (e.g. Deletions of exons, apex tissue expression, AUROC > 0.78, FDR < 0.05), as reflected by the sharp initial rise in ROC curves (Figure 3D). The strength of this signature indicates that indirect effects (e.g. trans regulation) do not dominate the observed relationship, and also demonstrates the high accuracy of our variant calls. Importantly, we also observe subtle but significant effects of regulatory SVs on gene expression (e.g. deletions overlapping 3’ flanking sequence, apex tissue expression, AUROC > 0.53, FDR < 0.05). The AUROC captures the individual cis-regulatory effect size, which is small on a per variant basis. However, in aggregate, these variants have a large impact on expression variation (Figure 3E), suggesting they globally shape expression profiles. Overall, our results show that SVs can impact gene expression in both substantial and subtle ways, and that many such variants in our panSV-genome may be functionally relevant (Figure 3F and S4F).
New Reference Genomes Resolve Multiple Haplotypes for the Smoky Volatile Locus
Our panSV-genome, new MAS2.0 assemblies, and expression dataset could help to reveal genes and variants underlying quantitative trait variation that has been masked by hidden genomic complexity. Many fruit aroma volatile QTLs that contribute to flavor have been identified through GWAS, but only a few have been functionally characterized (Tieman et al., 2017; Zhu et al., 2018). One such QTL involves the metabolically linked volatiles guaiacol and methylsalicyate, whose “smoky” or “medicinal” flavors negatively influence consumer appeal. A previous GWAS study identified a candidate gene E8 (Solyc09g089580), encoding a putative negative regulator of ethylene biosynthesis involved in fruit ripening (Tieman et al., 2017). While transcriptional knockdown of E8 resulted in accumulation of guaiacol and methylsalicylate, other volatiles were also modified. Furthermore, no causal mutations were identified, likely due to two large gaps flanking E8 in the reference genome at the time (SL3.0).
A separate study found that mutations in the NON-SMOKY GLYCOSYLTRANSFERASE1 (NSGT1) and NSGT2 paralogous genes, which are physically close on chromosome 9, cause an accumulation of guaiacol (Figure 4A) (Tikunov et al., 2013). Whereas NSGT2 shows little expression and is believed to be non-functional, upregulation of NSGT1 during ripening converts guaiacol to non-cleavable triglycosides, preventing guaiacol volatilization (Tikunov et al., 2013). To investigate if NSGT genes could be linked to the smoky QTL, we inspected the previous reference genome SL3.0 and found a partial sequence of NSGT1 near the gap at the chromosome 9 GWAS locus and another NSGT1 fragment at a second GWAS peak on an unanchored contig (Figure 4B) (Tieman et al., 2017). Consistently, a recent short-read k-mer-based analysis also linked the two smoky GWAS peaks and suggested hidden structural complexity (Voichek and Weigel, 2019). However, all these studies failed to resolve this locus. Importantly, our new MAS2.0 assemblies not only filled the gaps flanking E8 with these two NSGT paralogs but also further revealed coding sequence variants and SVs that are resolved into five haplotypes (Figure 4B and 4C) (see STAR Methods).
Figure 4. New reference genomes anchor candidate genes and resolve multiple SV and coding sequence haplotypes for the “smoky” volatile GWAS locus.
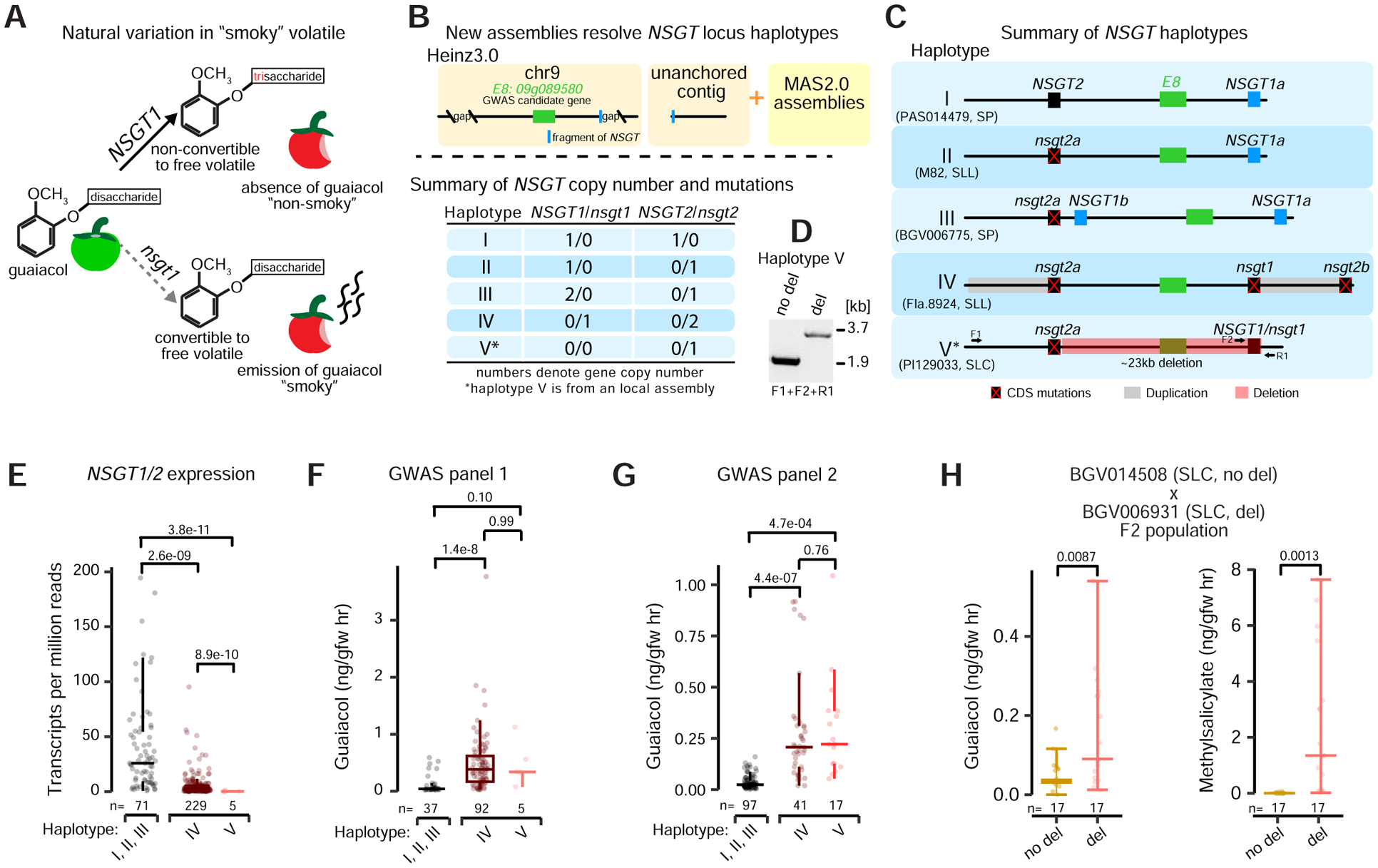
(A) Schematic showing a key step of the metabolic pathway underlying the “smoky” aroma trait. During fruit ripening, activation of glycosyltransferase NSGT1 prevents release of smoky-related volatiles by converting them into non-cleavable triglycosides (top). nsgt1 mutations result in the release of the smoky volatile guaiacol.
(B) Genomic resources used to resolve the GWAS locus for guaiacol (top) and summary of haplotypes (bottom). The published locus mapped to a region of chromosome 9 with one candidate gene and multiple gaps, and also to an unanchored contig with a fragment of an NSGT gene (top). MAS2.0 assemblies revealed multiple haplotypes that include copy number variation for the NSGT1 and NSGT2 paralogs and loss-of-function mutations (Bottom). A local assembly revealed haplotype V (asterisk) (see STAR Methods).
(C) Schematics depicting the five resolved haplotypes. The assemblies and major taxonomic groups from which the haplotypes were identified are shown below. Red “X”s mark coding sequence (CDS) mutations. Grey bars mark duplication in haplotype IV. Red rectangle marks a large deletion in haplotype V.
(D) PCR confirmation of the deletion in haplotype V. Primers (F1, F2, R1) are shown in (C).
(E) Quantification of NSGT1/2 expression by RNA-sequencing. Haplotypes are grouped according to functional NSGT1 (I, II, III), nsgt1 CDS mutation (IV) and nsgt1 deletion (V) (see STAR Methods). Expression data are from pericarp tissue of ripe fruit (Zhu et al., 2018).
(F-G) Guaiacol content of fruits from a previous GWAS study (F) (Tieman et al., 2017) and a new GWAS analysis using a collection of 155 SP and SLC accessions (G). Mutations in NSGT1 are associated with guaiacol accumulation. Accessions are grouped as in (E).
(H) Quantification of guaiacol and methylsalicylate content in an SLC x SLC F2 population segregating for the haplotype V 23 kbp deletion.
In (E-H), n represents sample size in each group. All p-values are based on two-tailed, two-sample t-tests.
Haplotype I is likely ancestral with the two paralogous NSGT1 and NSGT2 genes flanking E8. While an NSGT2 coding sequence mutation is found in all other haplotypes, haplotypes II and III have intact NSGT1, with the latter carrying two copies of NSGT1 (Figure 4C). Finally, copy number and functional variation are extended in haplotypes IV and V; haplotype IV has a 7 kbp duplication including mutant nsgt2 that disrupted NSGT1, rendering it non-functional, and haplotype V has a large 23 kbp deletion that removes both NSGT1 and E8, leaving only a single mutated copy of nsgt2 (Figure 4D).
These haplotypes, along with the previous characterization of NSGT1 (Tikunov et al., 2013), suggest that multiple mutant alleles of nsgt1 are responsible for natural variation in guaiacol (and methylsalicylate) accumulation and the smoky flavor. Using gene expression and metabolite data from fruits of more than 300 accessions (Tieman et al., 2017; Zhu et al., 2018), we tested associations between functional (I, II, III), coding sequence non-functional (IV) and deletion non-functional (V) NSGT1 haplotypes and guaiacol accumulation (see STAR Methods). Accessions carrying the mutant haplotypes IV and V, which emerged early in domestication in the SLCs (Table S5A), exhibited lower combined NSGT1/2 expression levels compared to accessions with functional haplotypes, with no NSGT1/2 expression detected in the five accessions carrying the haplotype V deletion (Figure 4E) (see STAR Methods). Consistently, both mutant haplotypes accumulated more guaiacol, though the effect from the rare haplotype V showed weak statistical significance (Figure 4F). We validated these findings using a new GWAS panel of 155 accessions comprised primarily of SP and SLC genotypes (Razifard et al., 2020). Again, both nsgt1 coding and deletion mutation haplotypes accumulate significantly more guaiacol than functional haplotypes (Figure 4G). Finally, we generated an F2 population between two SLCs segregating for haplotype V and functional NSGT1, which confirmed the deletion, lacking both NSGT1 and E8, is associated with accumulation of both guaiacol and methylsalicylate (Figure 4H). Together, our results anchored two NSGT genes to the smoky GWAS QTL and show that multiple nsgt1 mutations largely explain natural variations of the smoky flavor. This example demonstrates how our high quality long-read genome assemblies can resolve complex haplotypes and reveal causative variants for poorly understood QTLs.
The Fruit Weight QTL fw3.2 Resulted from a Tandem Duplication of a Cytochrome P450 Gene
A substantial increase in fruit weight was a major feature of tomato domestication(van der Knaap et al., 2014). The genes underlying five major fruit weight QTL have been identified, with the responsible mutations being either SVs or SNPs (Chakrabarti et al., 2013; Frary et al., 2000; Mu et al., 2017; Muños et al., 2011; Xu et al., 2015). Among these is fw3.2, which is strongly associated with a SNP in the promoter of the cytochrome P450 gene SlKLUH, a known regulator of organ size in multiple species (Anastasiou et al., 2007; Chakrabarti et al., 2013; Miyoshi et al., 2004). The promoter SNP was proposed to account for higher (2–3 fold) SlKLUH expression (Figure 5A), and transcriptional knockdown of this gene results in smaller fruits, but a causative role for the SNP was unclear.
Figure 5. The fruit weight QTL fw3.2 resulted from a tandem duplication that increased expression of a cytochrome P450 gene.
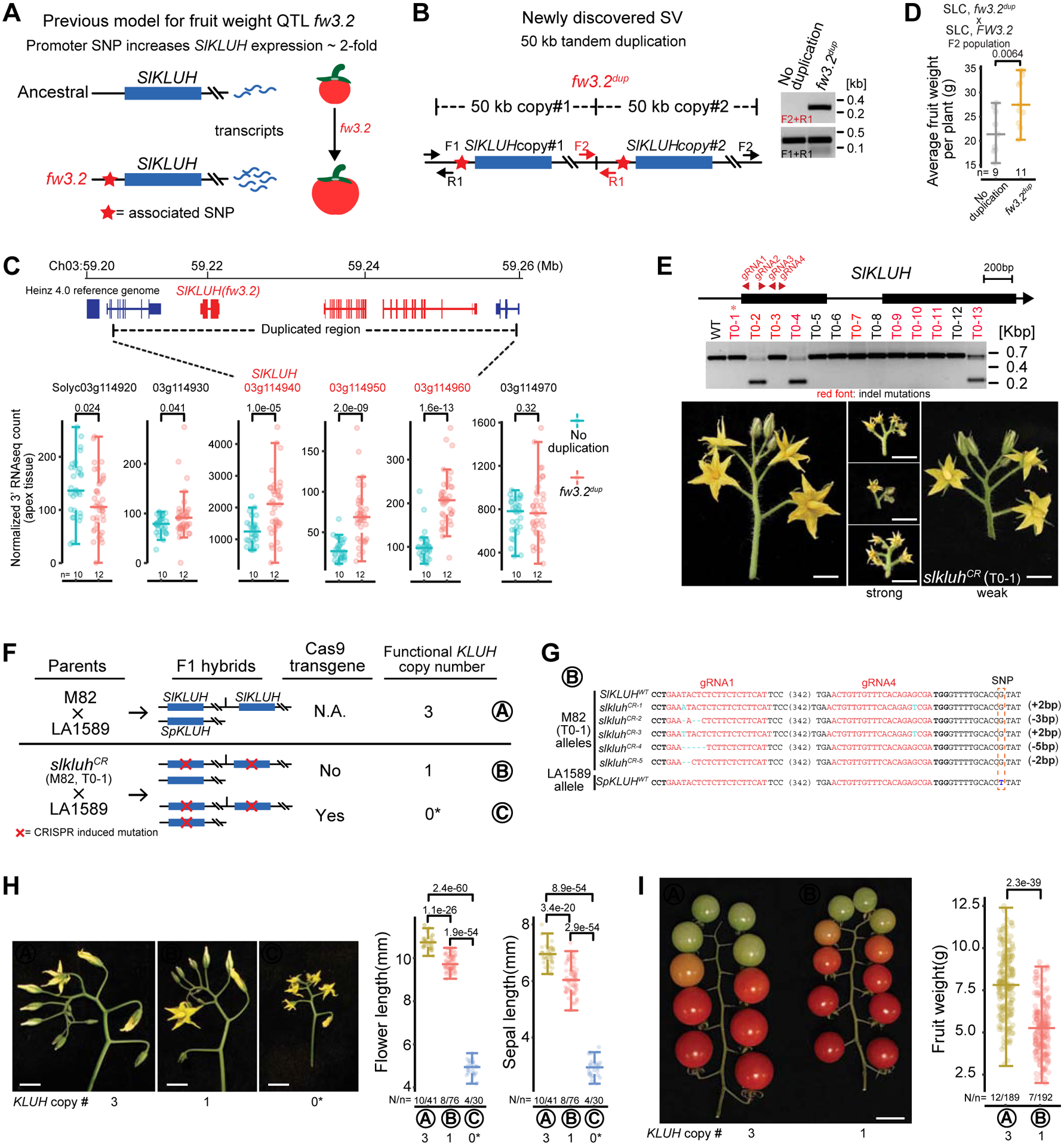
(A) Published mechanism for fw3.2 positing that a SNP in the promoter of the cytochrome P450 gene SlKLUH increased expression ~2-fold, resulting in larger fruits.
(B) SV analyses revealed a 50 kb tandem duplication at the fw3.2 locus that included SlKLUH (left). PCR validation of the duplication (right). Primers (F1, F2, R1) are labeled on the left. “No duplication” refers to the accession without this duplication and “fw3.2dup” refers to the accession that carries the duplicated copy of fw3.2 as shown by the PCR product across the duplication junction (F2 + R1).
(C) Expressions of genes within the fw3.2 duplication are ~2-fold higher. Gene coordinates and the duplication region (top), and RNA-seq box plots of duplicated and flanking genes (bottom) are shown. Each point is one biological replicate from one accession (see STAR Methods). n, number of accessions.
(D) An SLC x SLC F2 population segregating for the fw3.2 duplication, but fixed for the promoter SNP (see STAR Methods). Increased fruit weight is associated with the duplication.
(E) CRISPR-Cas9 mutagenesis of SlKLUH in the M82 background. SlKLUH gene model with gRNA targets (top), PCR genotyping (middle) and representative inflorescences (bottom) of slkluhCR T0 plants. The three slkluhCR T0 plants shown have mutations in all four copies of SlKLUH and exhibit similar tiny inflorescences, suggesting a null phenotype. Strong phenotypes were also observed for other T0 plants with sequenced indels (red font) except T0–1, which showed a weaker phenotype and was fertile, allowing a genetic test of dosage.
(F) Altering tomato KLUH gene dosage shows that copy number variation explains fw3.2. Schematic showing the M82/M82CR slkluh T0–1 (SL) x LA1589 (SP) crossing scheme used to test the phenotypic effects of altering tomato KLUH functional copy number in an F1 hybrid isogenic background. Genotypic groups A and B are isogenic for M82 x LA1589 genome-wide heterozygosity and differ only in having 3 or 1 functional copies of tomato KLUH, respectively. Genotypic group C effectively has 0 functional copies due to inheritance of the single insertion Cas9 transgene that targets the single SpKLUH allele in trans.
(G) Mutated slkluh alleles and the SpKLUH allele in genotypic group B. Red font, guide RNA targets. Cyan font, mutations. An LA1589 SNP (blue font) permits distinction of KLUH allele parent-of-origin. All SpKLUH sequences in genotypic group B are wild type.
(H) Decreasing tomato KLUH functional copy number reduces flower organ size. Representative inflorescences (left) and quantifications of flower and sepal length (right) from all three genotypic groups.
(I) Decreasing tomato KLUH functional copy number reduces fruit weight. Representative fruits (left) and fruit weight quantification (right) from genotypic groups A and B. Reducing tomato KLUH copy number from three to one reduces fruit size by 30%. Genotypic group C plants with mutated SpKLUH alleles fail to produce fruits.
Scale bar is 1 cm in (E and H) and is 2 cm in (I). In (H and I), N indicates plant number; n indicates flower/fruit number. All p-values are based on two-tailed, two-sample t-tests. See also Figure S5.
Our panSV-genome revealed a ~50 kbp tandem duplication at the fw3.2 locus containing three genes including two identical copies of SlKLUH (designated fw3.2dup) (Figure 5B). Although SNPs in promoters can affect expression by modifying cis-regulatory elements, we explored whether fw3.2dup is the causative variant, with the hypothesis that an increase in gene copy number explains the higher expression. In support of this, our expression analyses showed that all three intact genes within the duplication are expressed approximately two-fold higher in accessions carrying fw3.2dup (Figure 5C and S5A). To disentangle the effects of these variants on fruit weight, we generated F2 populations segregating for fw3.2dup, but fixed for the promoter SNP and other known fruit weight QTLs. Higher fruit weight co-segregated with the duplication allele (Figure 5D and S5B). In contrast, there was no association between the promoter SNP and fruit weight in F2 populations segregating only for the SNP (Figure S5C and S5D).
Our results suggested that the duplication carrying SlKLUH could explain fw3.2 due to an increase in gene copy number, and therefore dosage. We tested this by CRISPR-Cas9 targeting SlKLUH in the processing cultivar M82 (carrying fw3.2dup and therefore four functional copies of SlKLUH) with multiple gRNAs. PCR genotyping and sequencing of independent T0 plants showed large deletions and small indels in the target sites. The majority of these plants, including three confirmed to lack wild type (WT) alleles, were much smaller than control plants, had tiny inflorescences, and flowers that were infertile (Figure 5E and S5E).
Fortuitously, one fertile plant (slkluhCR T0–1) showed a weaker phenotype from having both WT and mutant alleles, allowing us to directly test how changes in SlKLUH dosage affect fruit weight. To work in an isogenic background with uniform “cherry” type fruits that allows for a robust assessment of fruit size, we crossed the slkluhCR T0–1 with the SP accession LA1589. As LA1589 has only two copies of SpKLUH (Figure 5F), the M82 x LA1589 F1 isogenic hybrids have three gene copies of KLUH (2 copies SlKLUH and 1 copy SpKLUH). These control F1 hybrids (group A) were compared with F1 progeny resulting from the cross between slkluhCR T0–1 and LA1589 (see STAR Methods). Several F1 hybrid plants that inherited the Cas9 transgene produced small organs and were infertile (group C), which we confirmed was due to inheritance of mutated and further trans-targeting of all KLUH copies (Figure S5F and S5G). Among F1 plants lacking the Cas9 transgene, a subset inherited two mutated alleles of SlKLUH and a single functional allele of SpKLUH (group B) (Figure 5F, 5G and S5H). Notably, these group B plants produced 15% smaller flowers and 30% smaller fruits compared to group A plants (1 vs 3 functional alleles of KLUH) (Figure 5H and 5I). Thus, our panSV-genome and functional genetic dissection using CRISPR-Cas9 genome editing show that the duplication including KLUH, and the corresponding increase in gene dosage and expression, underlies fw3.2.
Genetic Interactions Involving Four SVs Allowed Jointless Breeding.
We revealed thousands of genes with expression variation that could be caused by SVs. These variants might have little or no phenotypic consequences; however, many may be “cryptic”, having little or no effect on their own but causing phenotypic changes in the context of other variants (Paaby and Rockman, 2014; Sackton and Hartl, 2016). The “jointless” fruit pedicel is an important tomato harvesting trait that originated by different mutations from wild and domesticated accessions (Soyk et al., 2017). The jointless trait allows complete separation of fruits from other floral parts, and is caused by a transposon insertion that eliminates functional transcripts of the MADS-box transcription factor gene JOINTLESS2 (J2). A cryptic insertion in the related ENHANCER OF J2 (EJ2) gene reduces functional transcripts and causes excessive inflorescence branching with reduced fruit production following introduction of the jointless trait (Figure 6A). Breeders overcame this negative interaction and restored normal inflorescences by exploiting two natural “suppressor of branching” (sb) QTLs that we designated sb1 and sb3 (Soyk et al., 2019). We recently showed that sb3 is an 83 kbp duplication that includes ej2w, which causes a dose-dependent increase of weak allele expression that compensates for the reduced functional transcripts (Figure 6A).
Figure 6. Four SVs in three MADS-box genes were required to breed for the jointless trait.
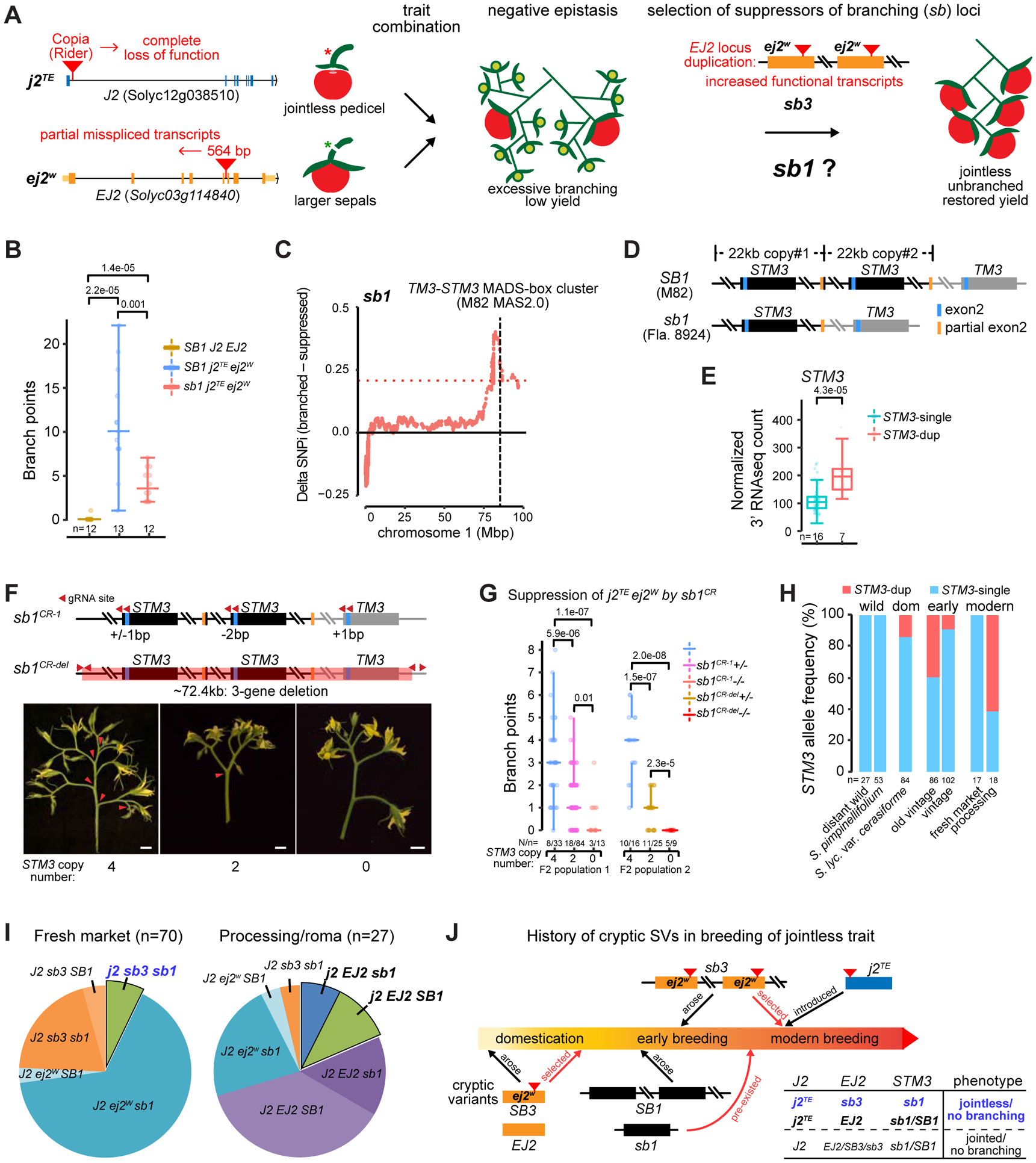
(A) Genetic suppressors were selected to overcome a negative epistatic interaction on yield caused by mutations in two MADS-box genes. The SV mutation j2TE causes a desirable jointless pedicel that facilitates harvesting. Introducing j2TE in backgrounds carrying the cryptic SV mutation ej2w results in excessive inflorescence branching and low fertility. The sb1 and sb3 QTLs were selected to suppress j2TE ej2w negative epistasis. sb3 is an 83 kb duplication harboring ej2w. sb1 is cloned in this study.
(B) Quantification of sb1 partial suppression of branching in the j2TE ej2w background. The SB1 j2TE ej2W and sb1 j2TE ej2W genotypes were derived from F3 families. Each data point is one inflorescence from F4 plants (n).
(C) Delta SNP index (deltaSNPi, QTL-seq) plot shows the sb1 locus contains the TM3-STM3 MADS-box gene cluster (see STAR Methods).
(D) Schematic of the TM3-STM3 locus in the SLL genotypes M82 and Fla.8924, with M82 having a ~22 kb tandem duplication (designated SB1) containing STM3.
(E) RNA-seq showing increased expression of STM3 from the SB1 duplication compared to sb1.
(F) CRISPR-Cas9 mutagenesis of the TM3-STM3 cluster (sb1CR) suppresses branching in the j2TE ej2w background. Schematics at top depict two CRISPR lines with indel mutations in the STM3 and TM3 genes (sb1CR−1) and a large deletion spanning all three genes (sb1CR-del) (top). Representative inflorescences from the indicated genotypes (bottom). Arrowheads mark branch points.
(G) Quantification and comparison of suppression of inflorescence branching by homozygous and heterozygous sb1CR−1 and sb1CR-del mutations in the background of j2TE ej2w. Genotypes were derived from F2 populations (see STAR Methods). N, plant number. n, inflorescence number.
(H) STM3 duplication allele frequency in wild tomato species (distant relatives, SP), early domesticates and cultivars (SLC, SLL vintage) and modern cultivars (SLL fresh market and processing).
(I) Distribution of J2 EJ2 SB1 genotypes in fresh market and processing/roma tomato types. All j2 fresh market genotypes carry sb1 and sb3, whereas processing/roma genotypes have SB1 or sb1, because EJ2 is functional.
(J) Schematic showing the history of breeding for the jointless trait, including when SVs in EJ2 and STM3 arose. The pre-existing sb1 cryptic variant (single copy STM3) mitigated the severity of branching caused by introduction of j2TE in varieties carrying the cryptic variant ej2w. Selection of the sb3 cryptic variant (two copies of ej2w) resulted in the complete suppression of branching and restoration of normal yield. Gradient colored bar represents timeline. The table summarizes genotypic combinations. Blue and black bold fonts indicate solutions for jointless breeding in fresh market and processing/roma types, respectively (I and J).
In (B, E, H and I), n represents sample size. P-values in (B and G) are based on two-tailed, two-sample t-tests. See also Figure S6.
The cryptic sb1 locus is a partial suppressor of branching, and our previous QTL mapping positioned sb1 to a 6 Mbp interval on chromosome 1 (Figure 6B and 6C). We searched for candidate genes and focused on two neighboring MADS-box paralogs, TM3 (Solyc01g093965) and SISTER OF TM3 (STM3, Solyc01g092950) (Figure S6A). Notably, STM3 showed approximately two-fold higher expression in the branched parental line (M82 j2TE ej2W) compared to the suppressed parent (Fla.8924 j2TE ej2W) (Figure S6B). There were no obvious coding or regulatory mutations in this gene; however, the Heinz 4.0 reference genome has gaps in that area. Our MAS2.0 assemblies filled the gaps and revealed copy number variation for STM3, with an extra copy of the gene in the branched parent due to a near perfect 22 kbp tandem duplication (Figure 6D and S6C). Consistently, genotypes with four copies of STM3 showed two-fold higher expression compared to two copy genotypes (Figure 6E).
To test if lower dosage and expression from a single STM3 gene is responsible for the sb1 QTL, we used CRISPR-Cas9 to generate mutant alleles disrupting the complex STM3-TM3 locus. A CRISPR construct with two gRNAs gave small indel mutations in all copies of the identical TM3/STM3 exon 2 (sb1CR−1), while a second construct with four gRNAs deleted the entire locus (sb1CR-del) (Figure 6F and S6D). Both sb1CR−1 and sb1CR-del plants were slightly late flowering, but their inflorescences were normal (Figure S6E). We then introduced each allele into the highly branched M82 j2TE ej2w double mutants and identified j2TE ej2w sb1CR−1 and j2TE ej2w sb1CR-del triple mutants from segregating F2 populations. Importantly, all of these plants (0 functional copies of STM3) showed practically complete suppression of branching compared to j2TE ej2w double mutants (4 functional copies of STM3) (Figure 6F, 6G and S6F). Moreover, j2TE ej2w plants that were heterozygous for the CRISPR alleles (2 functional copies of STM3) showed partial suppression of inflorescence branching, mimicking the effect of sb1 (e.g. Fla.8924, 2 functional copies of STM3) (Figure 6F, 6G and S6F). Thus, a single-copy STM3, and the corresponding lower gene expression, explains sb1.
Short-read based genotyping of more than 500 accessions spanning tomato taxonomic groups showed that the duplication of STM3 arose early in domestication, but the ancestral single gene has remained common in tomato germplasm (Figure 6H and S6G) (Table S5B). In fact, the majority of vintage and modern fresh-market accessions have single-copy STM3, indicating that a lower dosage and expression level provided partial suppression of branching upon the introduction of j2TE into lines carrying ej2w. The duplication of ej2w, and the resulting increased expression of this weak allele, arose later and was likely selected to achieve complete suppression of branching. In support, all jointless fresh market accessions carry both sb1 (single-copy STM3) and sb3 (duplicated ej2w) (Figure 6I). In contrast, breeding for jointless in processing tomato accessions was achieved by selecting against ej2w (Figure 6I). Consistent with this, sb1 and SB1 (duplicated STM3) are present at equal frequencies in processing tomato accessions, maintaining cryptic variation in the context of inflorescence development (Figure 6I and 6J). Our analysis reveals STM3 as a new regulator of tomato inflorescence development, and the dissection of sb1 shows that the path of jointless breeding depended on four SVs affecting the expression levels of three MADS-box genes, and further illustrate how functional consequences of structural variation can remain hidden.
DISCUSSION
Raising the Curtain on Structural Variation
Advancements in genome sequencing technologies continue to revolutionize biology by providing an increasingly comprehensive view of the genetic changes underlying phenotypic diversity. The recent development of high-throughput Oxford Nanopore long-read sequencing has provided the opportunity to rapidly reveal the breadth and depth of previously hidden SVs in complex genomes and across populations (Beyter et al., 2019). Taking advantage of the expansive genetic diversity of wild and domesticated tomatoes, we sequenced a collection of 100 accessions and resolved hundreds of thousands of SVs. These SVs were shaped predominately by transposons, are abundant across all chromosomes, frequently reside within or in close proximity to genes, are often associated with expression, and likely contribute to phenotypic variation. Integrating our panSV-genome, de novo assemblies, and expression data with genome-editing enabled us to resolve and functionally link SVs to three major domestication and breeding traits. The smoky and sb1 loci in particular demonstrate how these resources were essential to resolve complex haplotypes underlying QTLs where previous assemblies were thwarted by repeats, especially highly similar long and local duplications. Moreover, our analyses of the smoky and fw3.2 loci show that presumed causative variation may be incomplete or incorrect. More broadly, most QTLs discovered by GWAS in model and crop plants reside in regions with multiple candidate genes and variants. In addition to improving GWAS statistical power, long-read based discovery of abundant, sometimes complex, SVs, may immediately pinpoint high confidence candidate genes and variants for functional analyses. Similar progress in understanding functional impacts of SVs will likely emerge from generating population-scale panSV-genomes in other species (Danilevicz et al., 2020; Song et al., 2020; Sun et al., 2018; Yang et al., 2019; Zhou et al., 2019).
Duplications, Gene Copy Number Variation, and Dose-dependent Phenotypes
Our pan-SV genome revealed that fw3.2 and sb1 were both associated with previously hidden duplications. In both plants and animals, duplications that alter copy number and expression of dosage-sensitive genes were found to modify phenotypic diversity, including traits important in domestication and breeding (Lye and Purugganan, 2019). Large tandem recent duplications are one of the most challenging SVs to resolve, and even when a strong candidate gene is present, as with SlKLUH in the fw3.2 duplication, directly testing how modified gene dosage and expression impacts quantitative variation is challenging. Enabled by CRISPR-Cas9 genome editing, we generated plants with different gene copy numbers, and therefore dosages, for SlKLUH and STM3 in the fw3.2 and SB1 duplications, respectively. Establishing a dosage series of isogenic genotypes not only confirmed the causality of the duplications and the specific genes, but also directly demonstrated their quantitative impact. In particular, heterozygotes of sb1CR alleles (2 copies of STM3 on 1 chromosome) suppressed inflorescence branching of j2TE ej2W plants to a similar degree as the natural dosage effect from single-copy STM3 (1 copy of STM3 on each chromosome). Similarly, reducing functional KLUH copy number from three to one recapitulated the natural quantitative effect on fruit size of having four or two copies. Manipulating gene copy number by genome editing now provides a way to systematically interrogate and explore dosage to phenotype relationships (Veitia et al., 2013), which will be important for guiding the design and engineering of specific dosages for crop improvement.
Cis-Regulatory SVs and Quantitative Variation
Our panSV-genome showed that the majority of gene-associated SVs are in cis-regulatory regions, and many are associated with subtle changes in expression. Expanding long-read sequencing and expression analyses to a wider population will reveal even more such SVs. This raises the question to what extent cis-regulatory SVs affect phenotypes. For genes that are dosage-sensitive, such as those encoding components of molecular complexes or involved in signaling networks, a subtle change in expression could alter phenotype (Veitia et al., 2013). However, the magnitude of phenotypic effect may depend on a threshold change in expression and could be weak, making detection challenging in population genetics studies where other mutations and alleles influence trait variation. Genome editing could be used to study the effects of gene-associated SVs, by recreating specific mutations or mimicking the expression effects of natural cis-regulatory SVs in isogenic backgrounds. Our previous work characterizing collections of CRISPR-Cas9 engineered promoter alleles in multiple developmental genes showed that deletion and inversion SVs can affect expression and phenotypic outputs in various, often unpredictable, ways (Rodríguez-Leal et al., 2017). As SVs could be cryptic, a more powerful and informative approach would therefore be to combine natural cis-regulatory SVs with engineered SVs in the same promoter or with engineered mutations in related, potentially redundant genes. Resolving the functional impacts of SVs, particularly those whose effects are subtle or cryptic, will advance our understanding of genotype-to-phenotype relationships and facilitate the exploitation of natural and engineered SVs in crop improvement.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Zachary B. Lippman (lippman@cshl.edu).
Materials Availability
This study did not generate new unique reagents. Plasmids and transgenic plants generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
All sequencing data generated in this study have been deposited at the Sequence Read Archive (http://ncbi.nlm.nih.gov/sra) under BioProject PRJNA557253. Github repositories for software presented in this work are listed as follows: https://github.com/malonge/DupCheck, https://github.com/mkirsche/Jasmine, https://github.com/srividya22/geneLift, https://github.com/malonge/CallIntrogressions. All genome assemblies/annotations and SV VCF files are available at the Solanaceae Genomics Network (https://solgenomics.net/projects/tomato100).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Plant material and growth conditions
A hundred tomato accessions were collected from TGRC (Tomato Genetics Resource Center), USDA (United State Department of Agriculture), University of Florida, EU-SOL (The European Union-Solanaceae project), INRA (The National Institute for Agricultural Research), IVF-CAAS (The Institute of Vegetables and Flowers, Chinese Academy of Agricultural Science) and our own stocks. The landrace collection (S. lycopersicum var. cerasiforme) was from the seed stocks of E. van der Knaap. Seeds of S. pimpinellifolium (LA1589), S. lycopersicum cv. M82 (LA3475), and j2TE ej2w mutant are from Lippman lab. All accessions used in this study are listed in Table S1B.
Seeds were either germinated on moistened filter paper at 28 °C in the dark or directly sown in soil in 96-cell plastic flats. Plants were grown under long-day conditions (16-h light/8-h dark) in a greenhouse under natural light supplemented with artificial light from high-pressure sodium bulbs (~250 μmol m−2 s−1). Daytime and nighttime temperatures were 26–28 °C and 18–20 °C, respectively, with a relative humidity of 40–60%.
Quantification of fruit guaiacol and methylsalicylate contents in this study were conducted from plants grown in North Florida Research and Education Center-Suwannee Valley near Live Oak, Florida. Analyses of fruit weight in F2 segregation populations were conducted on plants grown at the University of Georgia (Athens, GA). Analyses of floral organ size, fruit weight of F1 hybrid plants and inflorescence branching in F4 generation were conducted on plants grown in the fields at Cold Spring Harbor Laboratory (CSHL), Cold Spring Harbor, NY. Seeds were germinated in 96-cell flats and grown for 32 d in the greenhouse before being transplanted to the field. Plants were grown under drip irrigation and standard fertilizer regimes. Analyses of inflorescence branching in two sbCR j2TE ej2W F2 populations were conducted on plants grown in the greenhouses at CSHL and Weizmann Institute of Science, Israel.
METHOD DETAILS
Short-Read Structural Variant Calling and Sample Selection
Publicly available short-read data came from a total of four sources (Aflitos et al., 2014; Lin et al., 2014; Tieman et al., 2017; Zhu et al., 2018). Phylogenetic trees derived from some of these data have been adapted from their original publication and are shown in Figure 1A, S1A and S1B (Razifard et al., 2020; Soyk et al., 2019). Phylogenetic classifications (branch coloring) were manually curated according to these previous phylogenetic studies and based on knowledge of tomato types and breeding classes. First, the raw reads were trimmed with Trimmomatic (v0.32, LEADING:30 TRAILING:30 MINLEN:75 TOPHRED33) (Bolger et al., 2014b). Reads we aligned to the SL4.0 reference genome with bwa mem (v0.7.10-r789, -M) (Hosmani et al., 2019; Li and Durbin, 2009) Alignments were then compressed, sorted and indexed with samtools view, sort, and index respectively (v0.1.19–44428cd) (Li et al., 2009). Next, PCR duplicates were marked with Picard (v1.126) (https://broadinstitute.github.io/picard/). We removed any samples that had less than 5X alignment coverage or any samples that had a duplication rate >= 20%. If a given accession had more than one associated BAM file, they were merged with samtools.
An ensemble approach was used to call SVs from these short-read alignments. We and others have found that a consensus among multiple short-read SV callers can achieve higher precision without substantially decreasing sensitivity (Zarate et al., 2018). We used 3 independent tools to call SVs: Delly (v0.7.3, -q 20), Lumpy (v0.2.13, -mw 4 -tt 0.0) and Manta (v1.0.3, -j 15 -m local -g 30) (Chen et al., 2016; Layer et al., 2014; Rausch et al., 2012). For each accession, SV call sets from Delly, Lumpy and Manta were then merged with SURVIVOR (v1.0.7, minimum distance of 1kbp, types must match, and a minimum length of 10bp) (Jeffares et al., 2017). Only SVs called by at least 2 of the 3 tools were retained. In total, we produced short-read SV calls for 847 accessions.
We then used SVCollector to select our first set of accessions for long-read sequencing (Sedlazeck et al., 2018b). For SVCollector, we further filtered short-read SV calls to only include SVs that intersect genes (+/− 5 kbp of flanking sequence). These filtered SVs were then used as input into SVCollector (greedy), and the top-ranked SLL (29) and SLC (22) accessions for which we had available seeds were selected (Table S1A). Aside from these 51 accessions selected with SVCollector, we selected an additional 49 accessions for long-read sequencing. These included SLL, SP, GAL and CHE accessions which were not included in the short-read SV analysis. A list of all accessions and their associated SVCollector ranks (where applicable) is available in Table S1A.
Tissue collection and high molecular weight DNA extraction
For extraction of high molecular weight DNA, young leaves were collected from 21-day-old light-grown seedlings. Prior to tissue collection, seedlings were etiolated in complete darkness for 48 h. Flash-frozen plant tissue was ground using a mortar and pestle and extracted in four volumes of ice-cold extraction buffer 1 (0.4 M sucrose, 10 mM Tris-HCl pH 8, 10 mM MgCl2, and 5 mM 2-mercaptoethanol). Extracts were briefly vortexed, incubated on ice for 15 min, and filtered twice through a single layer of Miracloth (Millipore Sigma). Filtrates were centrifuged at 4000 rpm for 20 min at 4 °C, and pellets were gently resuspended in 1 ml of extraction buffer 2 (0.25 M sucrose, 10 mM Tris-HCl pH 8, 10 mM MgCl2, 1% Triton X-100, and 5 mM 2-mercaptoetanol). Crude nuclear pellets were collected by centrifugation at 12,000g for 10 min at 4 °C and washed by resuspension in 1 ml of extraction buffer 2 followed by centrifugation at 12,000g for 10 min at 4 °C. Nuclear pellets were re-suspended in 500 μl of extraction buffer 3 (1.7 M sucrose, 10 mM Tris-HCl pH 8, 0.15% Triton X-100, 2 mM MgCl2, and 5 mM 2-mercaptoethanol), layered over 500 μl extraction buffer 3, and centrifuged for 30 min at 16,000g at 4 °C. The nuclei were resuspended in 2.5 ml of nuclei lysis buffer (0.2 M Tris pH 7.5, 2 M NaCl, 50 mM EDTA, and 55 mM CTAB) and 1 ml of 5% Sarkosyl solution and incubated at 60 °C for 30 min. To extract DNA, nuclear extracts were gently mixed with 8.5 ml of chloroform/isoamyl alcohol solution (24:1) and slowly rotated for 15 min. After centrifugation at 4000 rpm for 20 min, ~ 3 ml of aqueous phase was transferred to new tubes and mixed with 300 μl of 3 M NaOAC and 6.6 ml of ice-cold ethanol. Precipitated DNA strands were transferred to new 1.5 ml tubes and washed twice with ice-cold 80% ethanol. Dried DNA strands were dissolved in 100 μl of elution buffer (10 mM Tris-HCl, pH 8.5) overnight at 4 °C. Quality, quantity, and molecular size of DNA samples were assessed using Nanodrop (Thermofisher), Qbit (Thermofisher), and pulsed-field gel electrophoresis (CHEF Mapper XA System, Biorad) according to the manufacturer’s instructions.
Short-read DNA sequencing
Aside from the publicly available data used for short-read-based SV calling, we produced additional short-read data in-house for use in genome assembly for all but 2 (M82 and Fla.8924) MAS2.0 accessions. Short-read sequencing was performed according to Soyk et al. Nature Plants, 2019 (Soyk et al., 2019). In brief, libraries were prepared with the Illumina TruSeq DNA PCR-free prep kit from 2 μg genomic DNA sheared to 550 bp insert size. DNA libraries were sequenced on an Illumina NextSeq500 platform at the Cold Spring Harbor Laboratory Genome Center.
Long-read DNA Sequencing
Libraries for Oxford Nanopore genome sequencing were constructed using high-quality HMW DNA. DNA was sheared to ~20 kb using Covaris g-tubes or ~75 kb using Megarupter (Diagenode) and purified with a 1× AMPure XP bead cleanup. Next, DNA size selection was performed using the Short Read Eliminator kit (Circulomics). Library preparation was performed with 1.5 μg of size-selected HMW DNA, using the Ligation Sequencing Kit SQK-LSK109 (Oxford Nanopore Technologies) following manufacturer’s guidelines. Libraries were loaded on MinION or PromethION flow cells and sequenced according to standard protocols. Runs were basecalled with either Albacore v2.3 or with Guppy v2.1 through 3.2. Basecalling was performed using the PromethION r9.4.1 model, with recommended settings for the SQK-LSK109 kit and the FLO-PRO001 or FLO-PRO002 flowcells. At least 40G of data with mean read quality above or equal to Q7 were produced for each sample. Statistics describing the long-reads for all 100 accessions can be found in Table S1C.
Long-read Structural Variant Calling, Filtering, and Merging
A diagram describing the SV calling pipeline is depicted in Figure S1C. For each of our 100 accessions selected for long-read sequencing, we aligned a maximum of 60X coverage to the SL4.0 reference genome. The SL4.0 reference genome is a recently published preprint that improves to the previous (SL3.0) tomato reference genome (Hosmani et al., 2019). This PacBio long-reads assembled genome is the most complete and accurate representation of the Heinz 1706 reference genome to date. ITAG4.0, the reference gene models used in this study, are the accompanying reference gene annotation set. To call SVs relative to this reference, we aligned reads with NGMLR (v0.2.7, -x ont --bam-fix) and called SVs with Sniffles (v1.0.11)(--cluster --min_homo_af 0.7 -n 1000) (Sedlazeck et al., 2018a). As is convention, SV labels (insertions, deletions, duplications, inversions and translocations) are defined with respect to this single reference genome and do not necessarily define the underlying mutations causing the genetic variation. We further note that long insertions are somewhat underrepresented since Sniffles’ power to call insertions is bounded by read-length. For read sets exceeding 60X coverage, the longest set of reads achieving 60X was used. We then filtered SVs to remove potentially spurious calls. First, we identified regions of the reference genome prone to producing false SV calls and removed any SVs intersecting these regions (a total of 2,961,888 bp of the SL4.0 reference genome). To define these regions, we simulated ONT reads using SURVIVOR from the SL4.0 reference genome and called SVs with Sniffles. We performed this simulation a total of 9 times and merged the 9 VCF files with SURVIVOR (minimum distance of 1kbp, types must match, and a minimum length of 50bp). We then masked any region of the reference implicated in any SV from this simulation, including 2.5 kbp of flanking sequence. Next, we removed any SVs mapping to the ambiguous reference “chromosome 0” (SL4.0ch00). We also removed SVs larger than 100 kbp or SVs with a “0/0” genotype.
Using this same process described above, we also aligned Heinz 1706 PacBio reads to the SL4.0 reference genome to assess the propensity of the reference genome to produce false positives (Hosmani et al., 2019). We called only 75 from these alignments, suggesting that spurious false positives due to reference bias in our panSV-genome are rare.
For some accessions, duplications were filtered by observing short-read coverage across putative duplications. To do this, we wrote a custom tool similar to CNVnator’s genotyping functionality (Abyzov et al., 2011). First, for each accession, we calculated short-read coverage in non-overlapping 200bp windows of the reference genome using bedtools (Quinlan and Hall, 2010). The same reads and alignments as described in “Short-Read Structural Variant Calling and Sample Selection” were used here. Coverage was then corrected for GC bias using a custom version of the algorithm outlined in Yoon et al., 2019 (Yoon et al., 2009). The global mean coverage was calculated by first removing outliers (using the 1.5 x IQR rule) then fitting a Gaussian distribution to the coverages using SciPy (stats.norm.fit) (Virtanen et al., 2020). Finally, in order to verify a duplication, we required that the coverage roughly spanning the duplication boundaries must be greater than 1.75X the global mean coverage. Only duplications at least 1 kbp in size were considered. In order to calculate the coverage of the duplicated region, adjacent 200 bp windows were merged together via averaging to obtain 1 window close to the true duplication size. The coverage for this window, aligned to the original duplication coordinates (rounded to the nearest 200bp interval) was then compared to the global mean coverage. The above duplication filtering was only performed on samples for which we had short-read data available. The source code for duplication filtering can be found on GitHub (https://github.com/malonge/DupCheck).
By default, Sniffles provides supporting reads for each insertion call but reports the insertion sequence from a single noisy read. To associate each insertion with an accurate sequence, we used Iris (v1.0.1)(https://github.com/mkirsche/Iris). Iris extracts the reads supporting the insertion sequencing using samtools, computes their consensus using Racon (Vaser et al., 2017), and then replaces the original insertion sequence with the polished consensus. Finally, we used Jasmine to merge SVs across all accessions (v1.0.1, min_support=1 max_dist=500 k_jaccard=8 min_seq_id=0.25 spec_len=30)(see “Merging SVs with Jasmine” below). We used the default distance metric for merging, which is Euclidean distance. Briefly, 2-dimensional coordinates for each SV are given by (SV start position, SV length). SVs may be candidates for merging if their Euclidean distance between these 2D points is <= 500. The primary SV set was merged across all 100 accessions, though we also produced group-specific merged call sets for SLL, SLC, and SP using the same parameters.
Merging SVs with Jasmine
We developed a new SV merging tool called Jasmine, which is available open-source on GitHub (https://github.com/mkirsche/Jasmine). Jasmine constructs a graph G in which nodes represent SVs from individual samples. Edges connect pairs of SVs that may be merged based on criteria such as the distance between their breakpoints, and in the case of insertions, their sequence similarity. Next, the variants are partitioned based on reference sequence, SV type, and strand. In order to compute the best possible set of SV merges for a given group, Jasmine computes a forest on the graph which has a few key properties: 1) The edges in the forest are a subset of the edges in G, 2) No tree in the forest contains multiple nodes representing SVs from the same sample, 3) There are no unused edges in G which can be added to the forest while maintaining the previous properties, and 4) The sum of the breakpoint distances of edges in the forest is minimized. To do this, Jasmine uses a variant of Kruskal’s algorithm for computing minimum spanning trees. By considering the edges in non-decreasing order of edge weight, Jasmine greedily adds edges to the forest if they will not violate any of the required properties. To avoid storing this potentially very large network in memory, the network is computed dynamically by finding low-weight edges for each node with a KD-tree. Initially, a small constant number of edges incident to each node is stored, and as these are processed in increasing order of edge weight, new edges to process are added to the set by finding the next nearest neighbors for each node. As a result of this optimization, Jasmine is efficient in terms of both memory and runtime and can merge the entire set of over 1.7 million tomato SV calls in less than ten minutes on a single thread of a laptop.
We tested the efficacy of Jasmine on a simulated dataset. In this experiment, we use our merged tomato panSV-genome as our “ground truth”. This provides us with a realistic distribution of allele frequencies, SV types, and SV genomic positions. From this merged SV set, we then derived 100 individual SV sets, essentially reversing the merging process. When assigning variants to their original individual set, we added noise to the SV genomic position. The noise was modeled with a uniform distribution centered at 50 bp for both the start positions and lengths. In addition, the sequences of insertions were changed to model 10% sequencing error. Then, we reran Jasmine (using the same parameters as those used for our panSV-genome) on these noisy individual call sets and compared the results to the original merging. 98.98% of the 19.4 million variant pairs which were merged initially were also merged in the simulated results, while only 0.93% of the merged pairs from the simulation were unmerged in the original dataset. We also found that of the 238k variant calls which originally consisted of merged variants from multiple samples, 97.78% of them contained exactly the same sets of variants after the simulation. The added noise to the variant boundaries caused some previously merged variants to exceed the distance threshold. Also, some originally close variants in the same sample traded places during the merging process. This analysis shows that the method is highly robust to variation in the positions and lengths of structural variants across samples.
MAS2.0 Genome Assembly
We established de novo genome assemblies and associated gene and repeat annotations for a subset of the 100 accessions sequenced for SV analysis. This included the PAS014479 (SP), BGV006775 (SP), BGV006865(SLC), BGV007989 (SLC), BGV007931 (SLC), PI303721 (SLL), PI169588 (SLL), EA00990 (SLL), LYC1410 (SLL), Floradade (SLL), EA00371 (SLL), M82 (SLL), Fla.8924 (SLL), and Brandywine (SLL) accessions. Collectively, we refer to these assemblies and annotations as “MAS2.0”, and they are freely available to download at the Sol Genomics Network (https://solgenomics.net/projects/tomato100).
A diagram describing the assembly pipeline is depicted in Figure S2A. A hybrid assembly was performed for each accession using the MaSuRCA assembler (v3.3.3 or v3.3.4) (Zimin et al., 2017). Sequencing data used for assembly are described in “Short-read DNA sequencing” and “Long-read DNA sequencing” and Table S2D. M82 and Fla.8924 were not sequenced in-house for this study, but rather come from a previous publication (Alonge et al., 2019). As is recommended by the MaSuRCA documentation, no preprocessing was done on any of the sequencing data. For the ONT reads, we used the longest 35X coverage of reads with an average Phred quality score of at least 7. Library insert sizes for all Illumina data was set to 500 +/− 50. All assemblies employed the Flye unitigger during the final stage of MaSuRCA, except M82, which used default unitigging settings. All other MaSuRCA parameters were set to default values. The MaSuRCA draft assembly stats are found in Table S2A.
Each set of initial draft contigs underwent two rounds of short-read polishing with POLCA (MaSuRCA v3.3.4)(Zimin and Salzberg, 2019). As input for each of the two rounds of polishing, we used seqtk to randomly sample ⅔ of the Illumina data used during assembly (https://github.com/lh3/seqtk). After polishing, we screened each set of contigs for bacterial contamination by aligning them to the tomato SL4.0 reference and a bacterial reference genome. Every RefSeq bacterial genome, downloaded on October 1st, 2019, comprised our bacterial reference. Contigs were mapped to both references with Minimap2 (-k19 -w19) (Li, 2018). Any contig covered more by bacterial alignments than by tomato alignments were deemed contaminated and removed from the assembly. Only the BGV006865 and PI303721 accessions contained contaminated contigs. Finally, polished and screened contigs were scaffolded according to the SL4.0 reference genome using RaGOO (v1.1) (-T corr) (Alonge et al., 2019). The MaSuRCA mega-reads associated with the initial assemblies were used for misassembly correction. “Chromosome 0” of the SL4.0 was not considered during RaGOO scaffolding (-e). We generated dotplots for each assembly by aligning the final pseudomolecules to the SL4.0 reference genome using nucmer (-l 100 -c 500) and finally plotting with mummerplot (--fat --layout) (Figures S2C–S2P) (Kurtz et al., 2004). Finally, we used BUSCO to assess genome completeness (v3.0.2, -l solanaceae_odb10 -m genome -c 10 -sp tomato)(Table S2B) (Simão et al., 2015).
To observe SV concordance between our panSV-genome and the MAS2.0 assemblies, we called SVs from the assemblies using two techniques. First, we aligned the MAS2.0 assemblies to the SL4.0 reference genome using Nucmer (v3.1, -maxmatch -l 100 -c 500) and called SVs with Assemblytics (unique_length_required=500 min_size=15, max_size=100500)(Nattestad et al., 2016). Additionally, we simulated 60X coverage of perfect 25 kbp reads from the MAS2.0 assemblies and called SVs with NGMLR (v0.2.7, -x ont –bam-fix) and Sniffles (v1.0.11, -s 2 -l 15 –cluster –min_homo_af 0.7 -n 1000) with respect to the SL4.0 reference genome. Combining the Assemblytics and Sniffles MAS2.0 SV sets, we observed the pairwise SV concordance with the corresponding 14 accessions in our panSV-genome. The % SV overlap for each of the 14 accessions is as follows: BGV006775: 95.5571, BGV006865: 94.5002, BGV007931: 95.8251, BGV007989: 91.8735, Brandywine: 91.1921, EA00371: 87.8088, EA00990: 86.9073, Fla.8924: 89.4226, Floradade: 84.7832, LYC1410: 93.3863, M82: 90.3600, PAS014479: 92.8686, PI169588: 88.5430, PI303721: 70.9839.
We note that we do not expect perfect overlap between the read-mapping and assembly-based SV calls, since both have unique fallibilities and biases. For example, larger variants found with one approach may be broken into multiple smaller variants found by the other approach. Or, the exact position of variants may shift within genomic repetitive elements. Also, SVs in regions of the genome that fail to assemble may still be detected by aligning reads to a reference genome. Furthermore, expected variability in nanopore sequencing, along with other factors, likely contributes to the between accession variation that we observe. Broadly, an average overlap of 90% is a positive indication of SV accuracy and data quality.
MAS2.0 Gene Annotation
We used a “lift-over” approach to annotating the MAS2.0 assemblies with gene models. Along with the tomato reference ITAG4.0 gene models, our reference gene model set included previously published “pan-genome” genes which may be missing from ITAG4.0 but present in our assemblies (Gao et al., 2019). Gene models were lifted-over onto each of the 14 MAS2.0 assemblies with geneLift (v1.1, -c 90 -i 95) (https://github.com/srividya22/geneLift). Briefly, geneLift maps reference cDNA sequences to target assemblies using GMAP and Minimap2 and retains alignments with at least 90% coverage and 95% identity (Wu et al., 2016). The remaining non-overlapping GMAP alignments constitute the initial gene models, which are then supplemented by Minimap2 alignments to unannotated regions providing additional non-redundant gene models. Gene IDs reported by geneLift match the reference gene IDs and any gene duplications reported have an added suffix “-c” followed by the respective copy number of the gene to make them unique. Annotated “pan-genome” genes can be distinguished by a “TomatoPan” gene ID prefix. The geneLift statistics for each assembly can be found in Table S2C.
MAS 2.0 and SV Repeat Annotation
We used REPET to annotate MAS2.0 assemblies and panSV-genome insertion/deletion sequences with repeats (Flutre et al., 2011). From each MAS2.0 genome assembly, we built a sub-genome by selecting the longest contigs up to a cumulative size ranging 360–380 Mbp. This allowed us to sample a large portion of the genome while keeping the downstream computation tractable (Jouffroy et al., 2016). Each sub-genome was used to generate libraries of consensus sequences that are representative of repeats present therein using the TEdenovo pipeline from the REPET package v2.4 (parameters were set to consider repeats with at least 5 copies). The libraries produced were filtered to keep only those sequences that are found at least once as a full-length copy in the respective sub-genomes. Each resulting library of consensus sequences was then used as query for annotation of respective whole genomes using the TEannot pipeline from the REPET package v2.4. The library of consensus sequences was classified using PASTEC followed by semi-manual curation (Hoede et al., 2014).
For the annotation of insertions and deletions, the filtered consensus libraries obtained from ten of the 14 MAS2.0 assemblies (the first 10 to be completed) were pooled and appended to those from SL4.0 which were generated previously using the protocol described above. This combined library was then used as query for whole genome annotation by TEannot using default settings.
PI129033 NSGT Local Assembly
None of our 14 MAS2.0 assemblies contained the NSGT deletion allele described in “New Reference Genomes Resolve Multiple Haplotypes for the “Smoky” Volatile Locus”. Therefore, we performed a local assembly of the NSGT locus in PI129033, a sample known to carry this deletion allele. Using the same long-read alignments as described in “Long-read Structural Variant Calling, Filtering, and Merging”, we extracted PI129033 reads that aligned to the NSGT locus (SL4.0ch09:65168601–65653800) using samtools view. These reads were then error corrected with Canu (corOutCoverage=999, genomeSize=475k) and assembled with Flye (--nano-corr, --genome-size 475k) (Kolmogorov et al., 2019; Koren et al., 2017). Flye produced a single contig 534,847 bp in length representing the NSGT locus in PI129033. We next sought to polish this contig with short reads to produce an accurate representation of the locus. To do this, we first placed the contig into the SL4.0 reference genome in order to provide a suitable reference genome for short-read mapping. This avoids the potential poor quality of mapping when aligning WGS reads to a small segment of the genome. To create this pseudo-reference genome, we first started with the SL4.0 genome and replaced the NSGT locus (SL4.0ch09:65168601–65653800) with our local assembly. We also added 100bp gaps to the flanks of the inserted contig so that we could identify and retrieve it after polishing. We aligned short reads to this pseudo-reference using bwa and performed two rounds of short-read polishing with Racon (-u). Finally, we removed the local assembly from the pseudo-reference using samtools faidx and aligned it with Minimap2 (-ax asm5) to the SL4.0 reference genome to precisely define the deletion coordinates.
SV Hotspot and Introgression Analysis
For each accession, we counted the number of SVs in non-overlapping 1Mpb windows of the reference genome. Bins with a relatively large number of SVs are informally referred to as “SV hotspots”. An example distribution of SV frequency in 1 Mbp bins for M82 is shown in Figure S3M. SV frequency, shown in heatmap and circos form, is depicted in Figure 2A and S3A–S3L (http://omgenomics.com/circa/). Our observation of “hotspots” usually results from visual interpretation of these plots. SV hotspot heatmap rows are ordered within each phylogenetic group (GAL, CHE, SP, SLC, SLL) by the R “heatmap.2” default row ordering. These ordered groups were then concatenated to produce the final heatmap.
Since we hypothesized that introgression from wild donors could account for many of the observed SLL hotspots, we developed a technique to compare accessions to look for genomic regions of SV similarity. The custom Python code used for this task can be found in a GitHub repository (https://github.com/malonge/CallIntrogressions). The script “get_distances.py” compares SLL accessions to one or many accessions from any other “comparison” group (SP, SLC, GAL, or CHE). The algorithm considers successive 1Mpb windows of the reference genome. For each SLL accession, its set of SVs in a given window is compared to the set of SVs in all accessions in the comparison group in the same window. To compare two sets of SVs, we calculate the Jaccard similarity, requiring at least 5 SVs in both SV sets. The script then outputs, for each 1 Mpb window and for each SLL accession, the maximum Jaccard Similarity with any other comparison accession. If all comparisons for a given window had fewer than 5 SVs in either SV set, an “NA” value is reported.
We calculated similarity for all 45 SLL accessions at the same time by comparing each accession to each non-SLL accession. Comparisons against GAL and CHE did not yield any candidate introgressions from these groups, so we did not display those results. Comparisons against SP and SLC, which both show many regions of putative admixture/introgression from donors of these groups, are shown in Figure S3A–S3L. Tables S3A–S3L report the comparison accessions which yielded the maximum Jaccard similarity for each window depicted in Figures S3A–S3L. In Figures 2D and 2E, we also show an instance where we compare SLL accessions against a single SP comparison accession (LA1589).
SV Genomic Feature Annotation
Throughout the manuscript, we describe various relationships between SVs and other genomic features such as genes. Generally, we annotated our panSV-genome with genomic features using vcfanno (Pedersen et al., 2016). We define an “annotation” as the association of a particular SV with particular feature IDs (such as a gene ID) based on some relationship. vcfanno annotates SVs by finding their intersection (overlap) with genomic feature intervals. Accordingly, some of the annotations reported in the manuscript can be directly interpreted from vcfanno, such as “Insertions in exons”, or “Deletions overlapping 5 kbp upstream”, since these can be directly interpreted from feature intersection. Other annotations, such as SV containment of genes, required some combination of intersection calculations. For example, to detect genes contained by SVs, we first checked if the gene start and end positions intersected a given SV. If that SV intersected both the start and end of a gene, it contains that gene.
We ultimately produced many SV/feature annotation classes which are explained in more detail here. In any applicable annotation, “upstream” or “downstream” refers to the 5’ or 3’ flanking regions of genes, respectively. In supplemental material, these “upstream” and “downstream” regions may also be referred to as “5’ UTR” and “3’ UTR” respectively. “Insertions in exons”, “Insertions in introns”, “Insertions in 5 kbp downstream”, “Insertions in 5 kbp upstream”, “Deletions overlapping 5 kbp upstream”, and “Deletions overlapping 5 kbp downstream” are self-explanatory. “Duplications” are duplications that contain entire genes. “Deletions of exons” are deletions that delete at least one entire CDS exon of a gene, but do not delete the entire gene. Finally, “Deletions of CDS start” are deletions that contain 50 bp upstream and downstream of a CDS start site.
The Impact of SVs on Gene Expression
Data analysis was performed in R using custom scripts. In each tissue (apex, cotyledon and root), gene expression was averaged over the biological replicates in each accession (23 accessions with 3 replicates each in apex and root, and 22 accessions with 4 replicates each in cotyledon), and the genes with average expression count of at least 1 across the accessions were retained for further analysis. We averaged read counts across replicates to effectively treat the replicate expression as estimating a fixed effect. These gene expression averages within each accession/tissue were ranked and standardized so that the values were constrained between 0 and 1. While most of our analyses operate on these rank data, in order to provide estimates of fold change, we used the average expression profiles across replicates directly. These values were normalized by division of total read count of each accession and then fold changes were calculated across these normalized values between accessions with and without the SV.
Are SV-associated genes differentially expressed?
We first defined a list of SV-gene pairs based on SV annotations (see SV Genomic Feature Annotation). We filtered this list to only include SV-gene pairs which had the SV present in at least 5 and absent in at least 5 of the accessions for which we had RNA-seq data. For each of the SV-gene pairs, the accessions were split into two groups: with and without the SV. The extent of differential expression of the associated gene was calculated using a two-sided Mann-Whitney U test across the accession split. The Mann Whitney U test is a rank-based test that is very robust to underlying distributions in the expression values. The p-values among a specific annotation and tissue type were adjusted by applying Benjamini-Hochberg procedure (Benjamini and Hochberg, 1995). The adjusted p-values for each annotation and tissue type were aggregated using two methods: Fisher’s method and a harmonic mean estimate (Sitgreaves and Haggard, 1960; Wilson, 2019), and are listed in Table S4A.
At least half of the SV-associated genes in each SV type were common to all three tissues, exhibiting different levels of differential expression across the same accession split. In order to determine an average differential expression across the tissues, we used Fisher’s method to aggregate p-values across the three tissues for each SV-associated gene, and subsequently applied Benjamini-Hochberg method to limit the number of false positives (Table S4B).
Can we predict SV-associated genes from their differential expression?
For this analysis, we formulated a prediction task: Using the SV annotations as a “ground truth” labeled feature set (the gene associated with the SV is positively labeled and all other genes are negatively labeled), we measured how well we could predict the presence of an associated SV (positive label) given differential expression. A diagram depicting the workflow of this analysis is shown in Figure S4B. We used AUROC (Area under the ROC) scores as a measure of the performance of this task, which is calculated as follows: For each SV of a given annotation type, the p-values corresponding to the differential expression across the accession split (with or without the SV) was calculated for all genes in a given tissue via a two-sided Mann-Whitney U test, and the list of p-values was ranked (highest rank corresponds to the most significant p-value). For each SV, AUROC scores were analytically calculated by determining the positively labeled gene’s position in the ranked list of all gene p-values (high AUROC score corresponds to a near-perfect identification of the SV-associated gene). In other words, genes are predicted to be associated with a variant if they exhibit excess differential expression when comparing accessions with vs. without the SV. Conceptually, this can also be described as our classifier choosing a series of cutoff positions in this list, generating a ROC curve (and associated AUROC) by calculating the true and false positive rate associated with each cutoff (Figure S4B). Since all genes are affected by the underlying phylogenetic structure in the data, successful prediction of the true SV-associated gene in the list of all genes only occurs when predictions are robust to confounding population structure.
We have thus far described our prediction task when considering a single SV-gene pair. To assess the broad impact of SVs on expression, we combined all SV-gene pairs in a given annotation and tissue type. This is conceptually the same as for single SV-gene pairs, except the gene labels are combined into an aggregated labeled set where there is one positive gene label for each SV-gene pair (Figure S4B). The resulting ROC curve and associated AUROC effectively measures the average performance of the classifier over all SV-gene pairs. A high AUROC would indicate SVs globally have a significant impact on associated gene expression.
Our aggregated classifier’s performance can be measured by computing an overall p-value as follows. For a given variant and tissue type, the ranks of p-values of all SV-associated genes are removed from the list of sequential ranks of all expressed genes in a given tissue (for example, the ranks of 17 genes associated with duplications in apex tissue are removed from the sequence of ranks 1:20029 of the 20029 expressed apex genes). A One-tailed Mann-Whitney U test was performed to evaluate if the median of the ranks of SV-gene pair p-values was lower than the median of ranks of p-values of all other expressed genes. The resulting p-value is depicted by the size of the circle in Figure 3E. It is important to note that the overall p-values (circle size) are influenced by the number of SV-associated genes used in classification in each case, as well as the fold change in expression. For instance, duplications in apex have a larger p-value (p <4.06×10−4, with 17 variants used in classification) than insertions in 5 kbp downstream (p <1.72×10−16, with 1129 variants used in classification). Lists of AUROC scores of all SV-associated genes for each tissue and variant type are provided in Tables S4C–S4L.
Plant phenotyping
To quantify floral organ size, lengths of sepals and anther cones of closed yellow flower buds just before opening were measured. Inflorescence complexity was measured by counting the number of branching events per inflorescence. Flowering time was quantified by counting the number of leaves before the first inflorescence.
NSGT haplotype analyses
Thirteen of the fourteen MAS2.0 genome assemblies filled the gaps at the chromosome 9 “guaiacol” GWAS locus. To annotate this region, the full-length protein sequence of NSGT1 was used for BLAST search against the Heinz SL4.0 reference genome and the 14 MAS2.0 assemblies. We used the protein sequence as the query for BLAST to achieve more sensitive and more contiguous alignments while still allowing for the discrimination of NSGT alleles. Based on the BLAST results and sequence differences, four coding sequence variants including NSGT1, NST2, nsgt1 and nsgt2 are annotated in these genomes (Tikunov et al., 2013). We observed several accessions missing sequencing coverage at this locus, suggesting a deletion. We selected one such accession (PI129033) for a local assembly of the deletion haplotype (see “PI129033 NSGT Local Assembly”). The local assembly revealed the large deletion haplotype V.
Short-read based genotyping
NSGT locus coding sequence variants genotyping
From short-read alignments to the SL4.0 reference genome, we extracted reads overlapping with NGST locus (SL4.0ch09:65390765–65417476) using samtools view. In addition, we included previously unmapped reads. These mapped and unmapped read sets were converted back to a fastq files using samtools bam2fq. Subsequently, the reads were mapped to the unique portion of nsgt1 (117bp, GTTAGGTTTTAGGGTTTCAATTATGCTTGGAAATTTGGAagaagccatttgaaaggcttgaataaggttt aggtaccATCTTTAACAACTACCTCCAAAATTATAAACCTTTTTCTT), nsgt2 (86bp, CCAATACTTGAATGgttcaaaattagactttgtactttcaagaaaaccttgtGGAACCATTTCTTCAATTGTTT TGTTCACCCCTT), NSGT1 (100bp, ATATAATAGCTTCAACAACTTTTTAACCCCTTcatcaatagctttcaattttatcttctcactcaattgCATTG CCTTCAAATGAATTTGTTTCCTAGGC) and NSGT2(123bp, CAAAGGCTTTCTCATCGCGTGGTTTTATTGGTTTCATATCTAATTTCTTGatctcatagtcatga agaaaaggAAAAGATGTAAGGCTTGAACTCCCATAAAGAAATTGGTGGTAAAGGTAGG) simultaneously using bwa mem (-M). After mapping, reads with edit distance (NM tag) smaller than 15 and a minimum mapping quality of 20 were extracted. We used samtools depth to compute the coverage of the filtered reads across only the core of the unique regions (lower case sequences above) for nsgt1, nsgt2, NSGT1 and NSGT2. If more than 4 core bp had 0 coverage, we discarded the total mapped read counts for the sequence. If there was read count support for any of the nsgt1, nsgt2, NSGT1 or NSGT2 haplotypes, we report as them as “presence”. Since the “unique” sequence of NSGT1 is also present in nsgt1, if both nsgt1 and NSGT1 were genotyped as “presence”, we only labeled nsgt1 as “presence”. This is based on the observation that no sequencing resolved haplotypes have both nsgt1 and NSGT1 together. This genotyping was consistent with the observed haplotypes in our MAS2.0 assemblies.
NSGT locus deletion variant genotyping
From the short-read alignments to SL4.0, we counted the reads with a mapping quality of at least 20 in the middle region of the haplotype V deletion: SL4.0ch09:65401889–65404136. Accessions with less than 5 mapped reads were genotyped as “deletion”. The pipeline was benchmarked against PCR genotyped samples including 138 accessions with no deletion and 17 accession with deletions. Results from our pipeline were 100% consistent with PCR genotyping results.
sb1 duplication genotyping
From the short-read alignments to SL4.0, we extracted the reads mapped to a broad region that contained the sb1 duplication locus: SL4.0ch01:77727550–77765153. For each sample, we also extracted the unmapped reads. Mapped and unmapped read sets were converted to fastq files using samtools. Subsequently, we aligned the extracted reads to a portion of the sb1 locus (SL4.0ch01:77737550–77745153), which avoided high copy number TEs and represented a unique sequence of this locus. This was done with bwa mem (-M). We counted the number of reads mapped to this locus using samtools idxstats. The raw counts were normalized based on the total number of reads mapped for each sample. We manually checked the read alignments to SL4.0 and verified 22 single-copy accessions and eight duplication accessions. Accessions with normalized coverage lower than mean (verified single-copy accessions) – 1 standard deviation were genotyped as “single-copy” and accessions with normalized coverage greater than mean (verified duplication accessions) + 1 standard deviation were genotyped as “duplication”.
Tissue collection, RNA extraction and quantification
For 3’ RNA-sequencing (3’ RNA-seq), seeds were treated with 50% bleach for 20 minutes to homogenize germination and were germinated in petri dishes with moistened filter paper in the dark at 28 °C. Whole root tissues were collected 3 days after germination with a mixture of several seedlings as one biological replicate and three such replicates for each of a total of 23 accessions. For cotyledon tissues, seedlings after germination at similar stages were transplanted to soil in 96-cell flats and grown in the greenhouse. Cotyledons of seedlings were collected when two true leaves start to visibly emerge (10~11 days after sowing). Four biological replicates each with several seedlings combined for each of a total of 22 accessions were collected. For apex tissue, seedlings after germination at similar stages were transplanted to soil in 96-cell flats and grown in the greenhouse. For apex tissue collection, seeds were germinated, and seedlings were transplanted as above. Vegetative apical meristem together with the two youngest/smallest leaf primordia were collected 4 days after transplanting (Park et al., 2012). Eight to twelve apices were combined as one biological replicate and three replicates were collected for each of a total of 23 accessions. Total RNA was extracted using the RNeasy Plant Mini Kit (Qiagen) and treated with the RNase Free DNase Set (Qiagen) according to the manufacturer’s instructions. Total RNA samples were sent to the Genomic Diversity Facility at Cornell University for high-throughput 3’ RNA (single-end, read length=75bp) as described (Kremling et al., 2018).
For quantitative RT-PCR, seeds were germinated on moistened filter paper at 28 °C in dark. After germination, seedlings at similar stages were transferred to soil in 96-cell plastic flats and grown in the greenhouse. Shoot apices were collected at the transition and floral meristem stage of meristem maturation (Park et al., 2012), and immediately flash-frozen in liquid nitrogen. Total RNA was extracted as described above. 100 ng to 1 μg of total RNA was used for cDNA synthesis using the SuperScript III First-Strand Synthesis System (Invitrogen). qPCR was performed with gene-specific primers using the iQ SYBR Green SuperMix (Bio-Rad) reaction system on the CFX96 Real-Time system (Bio-Rad). Primer sequences are available in Table S6.
NSGT1/2 expression analysis
Published RNA-seq data of tomato fruit pericarp tissue from 405 accessions were downloaded from SRA PRJNA396272. Reads were trimmed by quality using Trimmomatic (ILLUMINACLIP:TruSeq3-PE-2.fa:2:40:15:1:FALSE LEADING:30 TRAILING:30 MINLEN:100) and aligned to the cDNA annotation of reference genome sequence of tomato (SL4.0) using kallisto quant (Bray et al., 2016). The output of kallisto generates normalized transcripts per million reads (TPM) which was used for quantifying NSGT1/2 expression. Because only one copy of NSGT1/2 is annotated in the SL4.0 and sequences of NSGT1 and NSGT2 are highly similar, we used the TPM of the annotated copy of NSGT (Solyc09g089585) to represent the expression level of both NSGT1 and NSGT2. TMPs are in Table S5C.
Metabolite profiling
Published fruit guaiacol contents were obtained from (Tieman et al., 2017). To minimize environmental effects, only data from one field season (2015) were used (Table S5D).
Fruit guaiacol and methylsalicylate contents in our new GWAS panel were quantified as previously described (Tieman et al., 2017). Briefly, at least six fruits (two fruits for each replicate) of red ripe stage were collected from each variety. Volatile compound identification was determined by gas chromatography-mass spectrometry and co-elution with known standards (Sigma-Aldrich, St. Louis MO). Metabolite contents are in Table S5E and S5F.
3’ RNA-seq data processing and gene expression analysis for individual duplication locus
3’ RNA-seq reads were trimmed by quality using Trimmomatic (v0.36, ILLUMINACLIP:TruSeq3-SE.fa:2:30:10 LEADING:30 TRAILING:30 MINLEN:30 HEADCROP:12) and mapped to SL4.0 reference genome using STAR with default parameters (Dobin et al., 2013). Bam files generated by STAR were sorted by read name and gene expression was quantified as uniquely mapped reads to annotated gene features in the ITAG4.0 reference annotation using HTSeq-count (--format=bam --order=name --stranded=no --type=exon --idattr=Parent) (Anders et al., 2015). Gene counts were processed in R for visualization. First, we filtered expressed genes by only keeping genes with sum of counts across all samples greater than the sum of replicates. Then the count table was imported into R package “DESeq2” (Love et al., 2014) and normalized counts were used for making box plots.
Generation of F2 populations segregating for the fw3.2 duplication or promoter SNP
The fw3.2 duplication and the derived allele of the promoter SNP are highly, but not completely associated. From our collection of accessions, we carefully selected four pairs of accessions carrying either single or double copies of fw3.2 but fixed at the promoter SNP (M9) of KLUH and all other known fruit weight QTL genes (Figure S5D). Four bi-parental F2 populations were developed from each pair of accessions, so that the duplication of fw3.2 would segregate. We genotyped the F2 plants by fw3.2 duplication markers and markers flanking the entire duplicated region. Similarly, six bi-parental F2 populations that segregated for the promoter SNP but fixed as the single-copy of fw3.2 and other known fruit weight QTL genes were developed. We genotyped F2 plants using M9 markers. In each population, ten homozygous F2 plants carrying each of the contrasting genotype were grown in the field. At harvest, we selected 15 to 20 large fruits after mature green stage and recorded their average weight to represent the potential of largest fruit from a single plant. Poor fruit setting was observed in population 19S313 so only about 10 representative fruits were used for each plant. In extreme cases, the fruit weight of three plants were represented by less than 5 fruits. Fruit weight data are in Table S5G.
CRISPR-Cas9 mutagenesis, plant transformation, and selection of mutant alleles
CRISPR-Cas9 mutagenesis and generation of transgenic tomato was performed following our standard protocol (Brooks et al., 2014). Briefly, guide RNAs (gRNAs) were designed using the CRISPRdirect tool (https://crispr.dbcls.jp/) (Naito et al., 2015). Binary vectors for gRNAs and Cas9 were assembled using the Golden Gate cloning system as described (Rodríguez-Leal et al., 2017; Soyk et al., 2017; Werner et al., 2012). Final binary vectors were transformed into the tomato cultivar M82 by Agrobacterium tumefaciens-mediated transformation through tissue culture (Gupta and Van Eck, 2016). Transplanting of first generation transgenic (T0) plants and genotyping of CRISPR-generated mutations were performed as (Soyk et al., 2017). Briefly, CRISPR-targeted region was PCR amplified and wild type (WT) size products were sequenced for T0 plants and those with mutations were selfed or crossed to WT M82 plants for further characterization of mutant alleles. All gRNA sequences are listed in Table S6.
Generation of hybrid plants for different KLUH dosages
To test the dosage-dependent effect of KLUH in an isogenic background with uniform “cherry” fruit type, the fertile T0 plant with CRISPR-Cas9 targeting SlKLUH (slkluhCR T0–1) was crossed with the SP accession LA1589. About half of F1 plants carried the Cas9 transgene (1:1 segregation of transgene). Analyses were focused on F1 plants that did not inherit the Cas9 transgene, because they are a fixed, uniform genotype. In contrast, plants with the Cas9 transgene would be genetically intractable for dosage analyses, because of the random chimerism that occurs within individual plants carrying the Cas9 transgene. From eight individual F1 plants without the Cas9 transgene (genotypic group B), KLUH gene PCR products were cloned and eight individual clones were sequenced. All eight plants were confirmed to have only mutant slkluh alleles and a WT SpKLUH allele (Figure S5F). Sepal length, flower length and fruit weight were quantified from these plants. Most of the F1 plants with the Cas9 transgene showed slightly smaller floral organs, and several of these plants had extremely small floral organs and no fruit set. From four individual F1 plants with the Cas9 transgene that showed tiny floral organs (genotypic group C), sepal length and flower size were quantified. To determine whether this effect was due to trans-targeting of SpKLUH, two plants with extremely small floral organs were randomly selected and sequenced for multiple PCR-cloned KLUH alleles. Consistently, sequencing of the two plants showed only mutant alleles for SlKLUH and SpKLUH (mutant alleles and their combinations are shown in Figure S5F and S5G), consistent with the CRISPR-Cas9 trans-targeting the SpKLUH gene copy. WT M82 was crossed with LA1589 and the F1 plants were used as controls. Quantification data of sepal, flower length and fruit weight are in Table S5H and S5I.
STM3 Phylogenetic analyses and sequence analyses
Sequences of homologous proteins of STM3 and TM3 were obtained from tomato and Arabidopsis genome and aligned using the ClustalW2.1 program in Geneious 11.1.5. Phylogenetic tree was constructed using “Geneious Tree Builder” with Jukes-Cantor genetic distance model and Neighbor-Joining method with 1,000 bootstrap replicates. STM3 and TM3 fell in the same clade with Arabidopsis flowering time regulator SOC1 (Lee and Lee, 2010).
Delta SNP index plot and genome coverage plot
Mapping of genomic position of sb1 was reported in (Soyk et al., 2019). Briefly, F2 segregation population was generated from crosses between a branched M82 j2TE ej2W double mutant with an unbranched j2TE ej2W double mutant (Fla.8924). A group of excessively branched inflorescences (6–36 branches) and a group of clearly suppressed plants (1–4 branches) were selected. An equal amount of tissue from each plant (~0.2 g) was pooled for DNA extraction for the two groups using standard protocols. Libraries were prepared with the Illumina TruSeq DNA PCR-free prep kit from 2 μg genomic DNA sheared to 550 bp insert size and sequenced on an Illumina NextSeq platform at the CSHL Genome Center. After aligning reads to reference genome (SL3.0), SNPs were called with samtools/bcftools (Li, 2011; Li et al., 2009) using read alignments for the two genomic DNA sequencing pools in addition to the M82 (Bolger et al., 2014a) and Fla.8924 (Lee et al., 2018) parents. Called SNPs were then filtered for bi-allelic high-quality SNPs at least 100 bp from a called indel using bcftools (Li, 2011). Read depth for each allele at segregating bi-allelic SNPs in 100-kb sliding windows (by 10 kb) was summed for the various sequencing pools and allele frequencies were calculated. Finally, the difference in allele frequency (SNP index) between the branched and unbranched pools was calculated and plotted across the 12 tomato chromosomes. One of the two regions that exceeded a genome-wide 95% cut-off in SNP index was located on chromosomes 1 and was named sb1. The candidate interval based on SL3.0 is SL3.0ch01:80006250–86570024.
To show the genome coverages at the sb1 locus in M82, M82 j2TEej2W, Fla.8924 and S. pimpinellifolium, we calculated the coverage from Illumina data using bedtools multicov only counting properly paired reads in 10-kb windows across chromosome 1. Depths in the four genotypes were normalized by dividing by the average depth using R.
Generation of F2 populations segregating for sb1 CRISPR alleles, j2TE and ej2W
Homozygous sb1CR−1 and sb1CR-del plants were each crossed with M82 j2TEej2W, respectively, to construct two F2 populations segregating at those three loci. In the F2 generation, plants were first genotyped for j2TE and ej2W mutations at seedling stage in flats. All double mutants were transplanted and further genotyped for CRISPR alleles and quantified for inflorescence complexity/branching. Genotyping primer sequences are in Table S6. Phenotype related to sb1 are in Table S5J, S5K and S5L.
QUANTIFICATION AND STATISTICAL ANALYSES
“n” is defined in all relevant figure legends. All statistical tests were performed in R. Significance is only ever defined for the SV differential expression analysis (Figure 3C) (Table S4A and S4B) and it is defined as a p-value less than 0.05. Two-sided Mann-Whitney U tests were used for analysis in Figures 3C–F. The Mann-Whitney U test provides a robust estimate to compute the significance of the expression change that does not depend on any assumption of underlying distributions. The p-values for these tests underwent FDR correction with the Benjamini-Hochberg procedure. Adjusted p-values were aggregated using Fisher’s method and a harmonic mean estimate. Detailed methods for these analyses can be found in “The Impact of SVs on Gene Expression”. For expression analysis in Figures 4E, 5C, 6E and S5A, numbers of accessions for each genotype are presented in the figures and differences between groups were compared using two-tailed, two-sample t-tests. Fruit guaiacol and methylsalicylate contents were compared between genotypes using two-tailed, two-sample t-tests. For quantitative analysis in sepal length, flower length, fruit weight and inflorescence complexity n= number of flowers and inflorescences quantified was used for two-tailed, two-sample t-tests. The number of plants (n =) used for each genotype is also labeled in the figures. For above analysis, all data points were plotted as single dots in the box plots. For expression analysis with qRT-PCR, three biological replicates of pooled meristems were used for each genotype and two technical replicates were performed for each biological replicate. Mean values of normalized expression were compared using two-tailed, two samples t-tests. For flowering time quantification, number of plants of each genotype is labeled in the figure. Means ± s.d. were shown and mean values between groups were compared by two-sample t-tests.
Supplementary Material
Figure S1. Sample Selection and Long-Read Sequencing, Related to Figure 1. (A and B) Two SNP-based phylogenetic trees for distinct collections of tomato accessions (see STAR Methods) (Razifard et al., 2020; Soyk et al., 2019). Accessions represented in these trees and selected for long-read sequencing are marked with circles or asterisks along the circumference.
(C) Overview of the SV calling pipeline. The process involves aligning ONT long-reads to the SL4.0 reference genome and then calling, filtering and annotating SVs (see STAR Methods).
(D) Histograms of long-read coverage across the SL4.0 genome. Two representative accessions from each taxonomic group are displayed. Average read depths range from 50X to 60X and coverage falls along a Poisson distribution, indicative of genome wide even coverage.
(E) Euler diagrams (http://eulerr.co) depicting the concordance of SV calls derived from short vs. long-reads. A total of four accessions are examined: 2 SLL (top) and 2 SLC (bottom).
(F) Distance matrices depicting pairwise hamming distances between SLL, SLC and SP accessions, graphically represented as heatmaps. The dendrograms represent a hierarchical clustering of the accessions. The distribution of hamming distances is depicted in the heatmap color legends. The larger hamming distances in SLC and SP relative to SLL indicate greater SV diversity in these two groups. Clustering was done using the heatmap.2 function in R with default parameters.
Figure S2. MAS2.0 Assembly and Annotation, Related to STAR Methods. (A) Diagram depicting the MAS2.0 assembly and annotation pipeline.
(B) “Nchart” showing the contiguity of all 14 MAS2.0 assemblies compared to the SL4.0 reference genome. An “Nchart” is a generalization of the N50 metric (50% on the x-axis) in graphical form.
(C-P) Dotplots with respect to the SL4.0 reference for each of the 14 MAS2.0 assemblies.
(Q) The percentage of ITAG4.0 genes that are anchored in each of the 14 MAS2.0 assemblies.
(R) The total number of pan-genome genes anchored in each of the 14 MAS2.0 assemblies.
Figure S3. SV Hotspot and Introgression Analysis, Related to Figure 2. (A-L) Twelve panels are presented, one for each of the 12 chromosomes in the SL4.0 reference genome. Blue heatmaps at left depict the number of SVs in 1 Mbp non-overlapping windows along the reference chromosome. Rows of green heatmaps at top right correspond to the top 45 rows (all SLL accessions) of the blue heatmap. The green heatmap shows the Jaccard Similarity for each SLL accession and for each 1Mbp window to the SP accessions (see STAR Methods). Green heatmap at bottom right (same row order as the top right green heatmap) also shows SV Jaccard Similarity but with respect to all SLC accessions. Specifically, dark green heatmap cells show 1 Mbp windows that show high SV similarity with at least one SP (top) or SLC (bottom) accession. Grey cells in the green heatmap represent “NA” values for regions of the genome that did not have enough SVs to perform the similarity calculations.
(M) The distribution of SV counts in 1 Mbp windows (blue heatmaps from A-L) for the M82 genome.
Figure S4. Differential Expression ROCs, Related to Figure 3. (A) Distribution of the absolute value of the log fold changes of mean gene expression, separated by SV annotation and tissue types. All SV-gene pairs are shown on top, while only significant pairs are shown on the bottom. Numbers beneath boxplots are the number of SV-gene pairs. Fold changes above 27 are not shown.
(B) Diagram of the classifier predicting the presence of a nearby SV given differential gene expression. The diagram depicts a simplified scenario wherein there is a total of three expressed genes, three SVs, and three SV-gene pairs. (I) One of the example SV-gene pairs, with SV-1 overlapping the upstream region of gene-1. (II) Accessions are split into two groups: with and without SV-1. Differential expression of gene-1 between these two groups is tested with a Mann-Whitney U test, yielding a p-value. (III) The test is performed across the same accession split for the two other expressed genes (gene-2 and gene-3). The original gene (gene-1) is positively labeled (+, cyan shade), while the other genes are negatively labeled (−). (IV) The p-values for all tests are ranked, from the most (1) to the least significant (3). (V) Steps I - IV are repeated for all SV-gene pairs using their respective SV accession splits. (VI - VIII) The p-value ranks and gene labels are combined across all SV-gene pairs. The classifier iteratively considers all p-value rank cutoffs and predicts “+” or “-” for all genes at/above or below the cutoff, respectively. The associated ROC curve is built by calculating the true and false-positive rates for each cutoff.
(C-E) ROCs for the nine SV annotation types for cotyledon, root, and apical meristem tissue respectively. “del”, “ins” and “dup” are deletions, insertions and duplications respectively. “5utr” and “3utr” refer to 5 kbp up and downstream of genes respectively. “del CDS” refers to deletions of the CDS start site, whereas “insCDS” refers to insertions in CDS exons. “int” refers to introns.
(F) Volcano plots for specific SV-gene pair examples (extension of Figure 3F). Gene diagrams are not drawn to scale.
Table S1. Tomato accession information and long-read sequencing statistics, related to Figure 1 and Figure S1.
Table S2. Statistics for MAS2.0 genome assemblies and gene annotation, related to Figure S2.
Table S3. Accessions with highest similarity for each window for introgression heatmap analysis, related to Figure 2 and Figure S3.
Table S4. Lists of SV-associated genes and statistics of differential expression, related to Figure 3 and Figure S4.
Table S5. Raw data of genotyping and phenotyping experiments, related to STAR Methods.
Figure S5. A Tandem Duplication and Not a Promoter SNP of the Slkluh Gene Is Responsible for the Fw3.2 Fruit Size Domestication QTL, Related to Figure 5. (A) Expressions of genes within the fw3.2 duplication are approximately two-fold higher in accessions carrying the fw3.2 duplication. Genes within the duplication are in red font. RNA-seq box plots of duplicated and flanking genes from cotyledons (left) and roots (right) are shown. Each point is one biological replicate from one accession (see STAR Methods). n, number of accessions.
(B) Fruit weight quantification in three additional F2 populations segregating for the fw3.2 duplication but fixed for the promoter SNP. Increased fruit weight is associated with the duplication in all three families. n: number of plants measured for each genotype.
(C) Fruit weight quantification in six F2 populations segregating for the promoter SNP (M9) of SlKLUH but fixed for the fw3.2 duplication. There is no consistent association between the M9 SNP and fruit weight. n, number of plants measured for each genotype.
(D) Table showing accessions and genotypes at five known fruit weight QTLs in the 10 F2 populations used to test associations between fruit weight and the fw3.2 duplication or the promoter SNP.
(E) Table showing CRISPR-Cas9 generated mutant alleles of SlKLUH in three independent T0 plants. g1 and g4 refer to guide RNA1 and guide RNA4, respectively.
(F) Mutated spkluh alleles in genotypic group C. Red font, guide RNA targets. Cyan font, mutations. An LA1589 SNP (blue font) permits distinction of KLUH allele parent-of-origin.
(G) Summary of Sanger sequencing resolved alleles of slkluh and spkluh in individual F1 plants of genotypic group C. Allele names in the table refer to Figure 5G and Figure S5G.p-values in (A, B and C) are based on two-tailed, two-sample t-tests.
(H) Summary of Sanger sequencing resolved alleles of slkluh and SpKLUH in individual F1 plants of genotypic group B. These plants were used for quantifying floral organ size and fruit weight. Allele names in the table refer to Figure 5G.
Figure S6. Expression, Flowering, and Inflorescence Branching Phenotypes of the sb1CR Mutation and Allele Distribution of STM3 Duplication, Related to Figure 6. (A) Phylogenetic tree of STM3-TM3 paralogous proteins in tomato and Arabidopsis. Bootstrap values (%) from 1,000 replicates are shown.
(B) Quantification of relative expressions of STM3 and TM3 in transition and floral meristems in Fla.8924 j2TE ej2W and M82 j2TE ej2W genotypes by quantitative reverse transcription PCR (qRTPCR). UBIQUITIN is used as internal control. Expressions are normalized to those in Fla.8924 and are shown as means ± s.d. Three biological pooled meristem samples and two technical replicates of each were assayed.
(C) Genomic short-read resequencing coverage map showing an increase in short reads at the STM3 locus in accessions with the SB1 duplication compared to those with single copy sb1.
(D) Schematic showing positions of guide RNAs used and mutant alleles recovered in the two CRISPR-Cas9 targeting of the STM3-TM3 locus. Small indel alleles of stm3 and tm3 in sb1CR−1 (top) and a large deletion allele removing two copies of STM3 and TM3 in sb1CR-del (bottom) are shown. Orange box shows the common exon 2 between STM3 and TM3 and black box and thinner black box show exons and UTRs, respectively. Red font, guide RNA targets. The cyan font shows mutations.
(E) Flowering time measured by number of leaves to first inflorescence in wild type control (M82) and the two CRISPR alleles of sb1. n, number of plants phenotyped.
(F) Schematic depicting the sb1CR1−1 CRISPR allele at the STM3-TM3 locus (top). Representative inflorescences from the indicated genotypes showing suppression of branching in j2TE ej2w by sb1CR−1 (bottom). Arrowheads mark branch points.
(G) STM3 duplication allele frequency in distant wild tomato species, the closest wild relatives of domesticated tomato (S. galapagense, S. cheesmaniae, S. pimpinellifolium), “admixtures”, early tomato domesticates (S. lyc. var. cerasiforme), and cultivated tomatoes (S. lycopersicum). The number of accessions in each group is indicated.
p-values in (B and E) are based on two-tailed, two-sample t-tests.
ACKNOWLEDGEMENTS
We thank members of the Schatz and Lippman labs at J.H.U. and CSHL for helpful discussions. We thank Aleksey Zimin at J.H.U for helpful discussions about genome assembly. We thank T. Mulligan, A. Krainer, S. Qiao and K. Schlecht from CSHL for assistance with plant care. We thank S. Muller, R. Wappel, S. Mavruk-Eskipehlivan, and E. Ghiban from the CSHL Genome Center for sequencing support. We thank Sanwen Huang at the Chinese Academy of Agricultural Sciences and Dani Zamir at the Hebrew University for sharing sequencing data and germplasm. We thank Prashant Hosmani, Susan Strickler, Naama Menda, and Lukas Mueller at the Boyce Thompson Institute for Plant Research for providing early access to the SL4.0 reference genome and hosting data on the Solanaceae Genomics Network. We thank Surya Saha and Lukas Mueller for early access to Heinz 1706 PacBio data. This work was supported by the Howard Hughes Medical Institute, and grant support from: the US National Institutes of Health for their support of the CSHL Cancer Center Next Generation Shared Resource (5P30CA045508-31) and (R01LM012736 and R01MH113005) to J.G. and (UM1 HG008898) to F.J.S., the USDA National Institute of Food and Agriculture (AFRI no. 2018-67013-27896, SCRI no. 2015-51181-24312) to S.H., (AFRI no. 2016-67013-24452) to S.H. and Z.B.L., the BARD (United States-Israel Binational Agricultural Research and Development Fund, IS-5120-18C) to Y.E. and Z.B.L., the ISF (Israel Science Foundation, 1913/19) to Y.E., and the National Science Foundation Plant Genome Research Program (IOS 1855585) to D. M. T. and H.K., (IOS 1564366) to A.L.C., D.M.T. and E.v.K., (IOS-1350041) to M.C.S., and (IOS-1732253) to J.V.E., E.v.K., M.C.S. and Z.B.L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
W.R.M. is a founder and shareholder of Orion Genomics, a plant genetics company. Z.B.L. is a consultant for and a member of the Scientific Strategy Board of Inari Agriculture. Orion Genomics and Inari Agriculture had no role in the planning, execution or analysis of the experiments described here.
REFERENCES
- Abyzov A, Urban AE, Snyder M, and Gerstein M (2011). CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 21, 974–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aflitos S, Schijlen E, De Jong H, De Ridder D, Smit S, Finkers R, Wang J, Zhang G, Li N, Mao L, et al. (2014). Exploring genetic variation in the tomato (Solanum section Lycopersicon) clade by whole-genome sequencing. Plant J. 80, 136–148. [DOI] [PubMed] [Google Scholar]
- Aflitos SA, Sanchez-Perez G, de Ridder D, Fransz P, Schranz ME, de Jong H, and Peters SA (2015). Introgression browser: high-throughput whole-genome SNP visualization. Plant J. 82, 174–182. [DOI] [PubMed] [Google Scholar]
- Aguet F, Brown AA, Castel SE, Davis JR, He Y, Jo B, Mohammadi P, Park YS, Parsana P, Segrè AV, et al. (2017). Genetic effects on gene expression across human tissues. Nature 550, 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonge M, Soyk S, Ramakrishnan S, Wang X, Goodwin S, Sedlazeck FJ, Lippman ZB, and Schatz MC (2019). RaGOO: fast and accurate reference-guided scaffolding of draft genomes. Genome Biol. 20, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasiou E, Kenz S, Gerstung M, MacLean D, Timmer J, Fleck C, and Lenhard M (2007). Control of Plant Organ Size by KLUH/CYP78A5-Dependent Intercellular Signaling. Dev. Cell 13, 843–856. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audano PA, Sulovari A, Graves-Lindsay TA, Cantsilieris S, Sorensen M, Welch AME, Dougherty ML, Nelson BJ, Shah A, Dutcher SK, et al. (2019). Characterizing the Major Structural Variant Alleles of the Human Genome. Cell 176, 663–675.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belhaj K, Chaparro-Garcia A, Kamoun S, and Nekrasov V (2013). Plant genome editing made easy: targeted mutagenesis in model and crop plants using the CRISPR/Cas system. Plant Methods 9, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y (1995). Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 57, 289–300. [Google Scholar]
- Beyter D, Ingimundardottir H, Eggertsson HP, Bjornsson E, Kristmundsdottir S, Mehringer S, Jonsson H, Hardarson MT, Magnusdottir DN, Kristjansson RP, et al. (2019). Long read sequencing of 1,817 Icelanders provides insight into the role of structural variants in human disease. BioRxiv. 10.1101/848366. [DOI] [PubMed] [Google Scholar]
- Bohn GW, and Tucker CM (1939). Immunity to fusarium wilt in the tomato. Science 89, 603–604. [DOI] [PubMed] [Google Scholar]
- Bolger A, Scossa F, Bolger ME, Lanz C, Maumus F, Tohge T, Quesneville H, Alseekh S, Sørensen I, Lichtenstein G, et al. (2014a). The genome of the stress-tolerant wild tomato species Solanum pennellii. Nat. Genet 46, 1034–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014b). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Brooks C, Nekrasov V, Lipppman ZB, and Van Eck J (2014). Efficient gene editing in tomato in the first generation using the clustered regularly interspaced short palindromic repeats/CRISPR-associated9 system. Plant Physiol. 166, 1292–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti M, Zhang N, Sauvage C, Muños S, Blanca J, Cañizares J, Diez MJ, Schneider R, Mazourek M, McClead J, et al. (2013). A cytochrome P450 regulates a domestication trait in cultivated tomato. Proc. Natl. Acad. Sci. U. S. A 110, 17125–17130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Wang Y, Zhang R, Zhang H, and Gao C (2019). CRISPR/Cas Genome Editing and Precision Plant Breeding in Agriculture. Annu. Rev. Plant Biol 70, 667–697. [DOI] [PubMed] [Google Scholar]
- Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, Cox AJ, Kruglyak S, and Saunders CT (2016). Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222. [DOI] [PubMed] [Google Scholar]
- Chiang C, Scott AJ, Davis JR, Tsang EK, Li X, Kim Y, Hadzic T, Damani FN, Ganel L, Montgomery SB, et al. (2017). The impact of structural variation on human gene expression. Nat. Genet 49, 692–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Coster W, and Van Broeckhoven C (2019). Newest Methods for Detecting Structural Variations. Trends Biotechnol. 37, 973–982. [DOI] [PubMed] [Google Scholar]
- Danilevicz MF, Tay Fernandez CG, Marsh JI, Bayer PE, and Edwards D (2020). Plant pangenomics: approaches, applications and advancements. Curr. Opin. Plant Biol 54, 18–25. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flutre T, Duprat E, Feuillet C, and Quesneville H (2011). Considering Transposable Element Diversification in De Novo Annotation Approaches. PLoS One 6, e16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foolad MR, and Panthee DR (2012). Marker-Assisted Selection in Tomato Breeding. CRC. Crit. Rev. Plant Sci 31, 93–123. [Google Scholar]
- Frary A, Nesbitt TC, Frary A, Grandillo S, Van Der Knaap E, Cong B, Liu J, Meller J, Elber R, Alpert KB, et al. (2000). fw2.2: A quantitative trait locus key to the evolution of tomato fruit size. Science 289, 85–88. [DOI] [PubMed] [Google Scholar]
- Fuentes RR, Chebotarov D, Duitama J, Smith S, De la Hoz JF, Mohiyuddin M, Wing RA, McNally KL, Tatarinova T, Grigoriev A, et al. (2019). Structural variants in 3000 rice genomes. Genome Res. 29, 870–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Gonda I, Sun H, Ma Q, Bao K, Tieman DM, Burzynski-Chang EA, Fish TL, Stromberg KA, Sacks GL, et al. (2019). The tomato pan-genome uncovers new genes and a rare allele regulating fruit flavor. Nat. Genet 51, 1044–1051. [DOI] [PubMed] [Google Scholar]
- Gupta S, and Van Eck J (2016). Modification of plant regeneration medium decreases the time for recovery of Solanum lycopersicum cultivar M82 stable transgenic lines. Plant Cell. Tissue Organ Cult 127, 417–423. [Google Scholar]
- Ho SS, Urban AE, and Mills RE (2020). Structural variation in the sequencing era. Nat. Rev. Genet 21, 171–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoede C, Arnoux S, Moisset M, Chaumier T, Inizan O, Jamilloux V, and Quesneville H (2014). PASTEC: An Automatic Transposable Element Classification Tool. PLoS One 9, e91929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosmani PS, Flores-Gonzalez M, Geest H. van de, Maumus F, Bakker LV, Schijlen E, Haarst J. van, Cordewener J, Sanchez-Perez G, Peters S, et al. (2019). An improved de novo assembly and annotation of the tomato reference genome using single-molecule sequencing, Hi-C proximity ligation and optical maps. BioRxiv. 10.1101/767764. [DOI] [Google Scholar]
- Hutton SF, Scott JW, and Vallad GE (2014). Association of the Fusarium wilt race 3 resistance gene, I-3, on chromosome 7 with increased susceptibility to bacterial spot race T4 in tomato. J. Am. Soc. Hortic. Sci 139, 282–289. [Google Scholar]
- Jeffares DC, Jolly C, Hoti M, Speed D, Shaw L, Rallis C, Balloux F, Dessimoz C, Bähler J, and Sedlazeck FJ (2017). Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat. Commun 8, 14061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouffroy O, Saha S, Mueller L, Quesneville H, and Maumus F (2016). Comprehensive repeatome annotation reveals strong potential impact of repetitive elements on tomato ripening. BMC Genomics 17, 624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakatsu T, Huang S shan C, Jupe F, Sasaki E, Schmitz RJJ, Urich MAA, Castanon R, Nery JRR, Barragan C, He Y, et al. (2016). Epigenomic Diversity in a Global Collection of Arabidopsis thaliana Accessions. Cell 166, 492–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Knaap E, Chakrabarti M, Chu YH, Clevenger JP, Illa-Berenguer E, Huang Z, Keyhaninejad N, Mu Q, Sun L, Wang Y, et al. (2014). What lies beyond the eye: the molecular mechanisms regulating tomato fruit weight and shape. Front. Plant Sci 5, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolmogorov M, Yuan J, Lin Y, and Pevzner PA (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol 37, 540–546. [DOI] [PubMed] [Google Scholar]
- Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, and Phillippy AM (2017). Canu: Scalable and accurate long-read assembly via adaptive κ-mer weighting and repeat separation. Genome Res. 27, 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremling KAG, Chen SY, Su MH, Lepak NK, Romay MC, Swarts KL, Lu F, Lorant A, Bradbury PJ, and Buckler ES (2018). Dysregulation of expression correlates with rare-allele burden and fitness loss in maize. Nature 555, 520–523. [DOI] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, and Salzberg SL (2004). Versatile and open software for comparing large genomes. Genome Biol. 5, R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layer RM, Chiang C, Quinlan AR, and Hall IM (2014). LUMPY: A probabilistic framework for structural variant discovery. Genome Biol. 15, R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, and Lee I (2010). Regulation and function of SOC1, a flowering pathway integrator. J. Exp. Bot 61, 2247–2254. [DOI] [PubMed] [Google Scholar]
- Lee TG, Shekasteband R, Menda N, Mueller LA, and Hutton SF (2018). Molecular markers to select for the j-2–mediated jointless pedicel in tomato. HortScience 53, 153–158. [Google Scholar]
- Li H (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2018). Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Chitwood J, Menda N, Mueller L, and Hutton SF (2018). Linkage between the I-3 gene for resistance to Fusarium wilt race 3 and increased sensitivity to bacterial spot in tomato. Theor. Appl. Genet 131, 145–155. [DOI] [PubMed] [Google Scholar]
- Lin T, Zhu G, Zhang J, Xu X, Yu Q, Zheng Z, Zhang Z, Lun Y, Li S, Wang X, et al. (2014). Genomic analyses provide insights into the history of tomato breeding. Nat. Genet 46, 1220–1226. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lye ZN, and Purugganan MD (2019). Copy Number Variation in Domestication. Trends Plant Sci. 24, 352–365. [DOI] [PubMed] [Google Scholar]
- Meyer RS, and Purugganan MD (2013). Evolution of crop species: Genetics of domestication and diversification. Nat. Rev. Genet 14, 840–852. [DOI] [PubMed] [Google Scholar]
- Miyoshi K, Ahn B-O, Kawakatsu T, Ito Y, Itoh J-I, Nagato Y, and Kurata N (2004). PLASTOCHRON1, a timekeeper of leaf initiation in rice, encodes cytochrome P450. Proc. Natl. Acad. Sci 101, 875–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu Q, Huang Z, Chakrabarti M, Illa-Berenguer E, Liu X, Wang Y, Ramos A, and van der Knaap E (2017). Fruit weight is controlled by Cell Size Regulator encoding a novel protein that is expressed in maturing tomato fruits. PLoS Genet. 13, e1006930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muños S, Ranc N, Botton E, Bérard A, Rolland S, Duffé P, Carretero Y, Paslier MC Le, Delalande C, Bouzayen M, et al. (2011). Increase in tomato locule number is controlled by two single-nucleotide polymorphisms located near WUSCHEL. Plant Physiol. 156, 2244–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito Y, Hino K, Bono H, and Ui-Tei K (2015). CRISPRdirect: Software for designing CRISPR/Cas guide RNA with reduced off-target sites. Bioinformatics 31, 1120–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattestad M, Bioinformatics MS-, and 2016, U. (2016). Assemblytics: a web analytics tool for the detection of variants from an assembly. Bioinformatics 32, 3021–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen KM, and Wendel JF (2013). A Bountiful Harvest: Genomic Insights into Crop Domestication Phenotypes. Annu. Rev. Plant Biol 64, 47–70. [DOI] [PubMed] [Google Scholar]
- Paaby AB, and Rockman MV (2014). Cryptic genetic variation: Evolution’s hidden substrate. Nat. Rev. Genet 15, 247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SJ, Jiang K, Schatz MC, and Lippman ZB (2012). Rate of meristem maturation determines inflorescence architecture in tomato. Proc. Natl. Acad. Sci. U. S. A 109, 639–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen BS, Layer RM, and Quinlan AR (2016). Vcfanno: Fast, flexible annotation of genetic variants. Genome Biol. 17, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch T, Zichner T, Schlattl A, Stutz AM, Benes V, and Korbel JO (2012). DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333–i339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razifard H, Ramos A, Della Valle AL, Bodary C, Goetz E, Manser EJ, Li X, Zhang L, Visa S, Tieman D, et al. (2020). Genomic Evidence for Complex Domestication History of the Cultivated Tomato in Latin America. Mol. Biol. Evol 1–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Leal D, Lemmon ZH, Man J, Bartlett ME, and Lippman ZB (2017). Engineering Quantitative Trait Variation for Crop Improvement by Genome Editing. Cell 171, 470–480.e8. [DOI] [PubMed] [Google Scholar]
- Sackton TB, and Hartl DL (2016). Genotypic Context and Epistasis in Individuals and Populations. Cell 166, 279–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Tabata S, Hirakawa H, Asamizu E, Shirasawa K, Isobe S, Kaneko T, Nakamura Y, Shibata D, Aoki K, et al. (2012). The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485, 635–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott JW (1999a). University of Florida tomato breeding accomplishments and future directions. Annu. Proc. Soil Crop Sci. Soc. Florida 58, 8–11. [Google Scholar]
- Scott JW (1999b). Tomato Plants Heterozygous for Fusarium Wilt Race 3 Resistance Develop Larger Fruit Than Homozygous Resistant Plants. Proc. Fla. State Hort. Soc 112, 305–307. [Google Scholar]
- Scott JW, and Jones JP (1989). Monogenic resistance in tomato to Fusarium oxysporum f. sp. lycopersici race 3. Euphytica 40, 49–53. [Google Scholar]
- Sedlazeck FJ, Rescheneder P, Smolka M, Fang H, Nattestad M, Von Haeseler A, and Schatz MC (2018a). Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 15, 461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlazeck FJ, Lemmon Z, Soyk S, Salerno WJ, Lippman Z, and Schatz MC (2018b). SVCollector: Optimized sample selection for validating and long-read resequencing of structural variants. BioRxiv. 10.1101/342386. [DOI] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, and Zdobnov EM (2015). BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. [DOI] [PubMed] [Google Scholar]
- Sitgreaves R, and Haggard EA (1960). Intraclass Correlation and the Analysis of Variance. J. Am. Stat. Assoc 55, 384. [Google Scholar]
- Song J-M, Guan Z, Hu J, Guo C, Yang Z, Wang S, Liu D, Wang B, Lu S, Zhou R, et al. (2020). Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus. Nat. Plants 6, 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soyk S, Lemmon ZH, Oved M, Fisher J, Liberatore KL, Park SJ, Goren A, Jiang K, Ramos A, van der Knaap E, et al. (2017). Bypassing Negative Epistasis on Yield in Tomato Imposed by a Domestication Gene. Cell 169, 1142–1155.e12. [DOI] [PubMed] [Google Scholar]
- Soyk S, Lemmon ZH, Sedlazeck FJ, Jiménez-Gómez JM, Alonge M, Hutton SF, Van Eck J, Schatz MC, and Lippman ZB (2019). Duplication of a domestication locus neutralized a cryptic variant that caused a breeding barrier in tomato. Nat. Plants 5, 471–479. [DOI] [PubMed] [Google Scholar]
- Strobel JW, Hayslip NC, Burgis DS, and Everett PH (1969). Walter, a determinate tomato resistant to races 1 and 2 of the Fusarium wilt pathogen. Circ. Fla. Agric. Exp. Stn; 1969 (S-202)9 Pp. [Google Scholar]
- Sun S, Zhou Y, Chen J, Shi J, Zhao H, Zhao H, Song W, Zhang M, Cui Y, Dong X, et al. (2018). Extensive intraspecific gene order and gene structural variations between Mo17 and other maize genomes. Nat. Genet 50, 1289–1295. [DOI] [PubMed] [Google Scholar]
- Team, R.C. (2017). R Core Team (2017). R: A language and environment for statistical computing. R Found. Stat. Comput. Vienna, Austria. URL Http//Www. R-Project. Org/., Page R Found. Stat. Comput. URL http://www.R-project.org/.
- Tieman D, Zhu G, Resende MFR, Lin T, Nguyen C, Bies D, Rambla JL, Beltran KSO, Taylor M, Zhang B, et al. (2017). A chemical genetic roadmap to improved tomato flavor. Science 355, 391–394. [DOI] [PubMed] [Google Scholar]
- Tikunov YM, Molthoff J, de Vos RCH, Beekwilder J, van Houwelingen A, van der Hooft JJJ, Nijenhuis-de Vries M, Labrie CW, Verkerke W, van de Geest H, et al. (2013). Non-smoky GLYCOSYLTRANSFERASE1 prevents the release of smoky aroma from tomato fruit. Plant Cell 25, 3067–3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaser R, Sović I, Nagarajan N, and Šikić M (2017). Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veitia RA, Bottani S, and Birchler JA (2013). Gene dosage effects: Nonlinearities, genetic interactions, and dosage compensation. Trends Genet. 29, 385–393. [DOI] [PubMed] [Google Scholar]
- Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, Burovski E, Peterson P, Weckesser W, Bright J, et al. (2020). SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voichek Y, Weigel D (2020). Identifying genetic variants underlying phenotypic variation in plants without complete genomes. Nat. Genet 10.1038/s41588-020-0612-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace JG, Rodgers-Melnick E, and Buckler ES (2018). On the Road to Breeding 4.0: Unraveling the Good, the Bad, and the Boring of Crop Quantitative Genomics. Annu. Rev. Genet 52, 421–444. [DOI] [PubMed] [Google Scholar]
- Walter JM, and Kelbert DGA (1953). Manalucie: A Tomato with Distinctive New Features (University of Florida, Agricultural Experiment Stations; ). [Google Scholar]
- Weber E, Engler C, Gruetzner R, Werner S, and Marillonnet S (2011). A Modular Cloning System for Standardized Assembly of Multigene Constructs. PLoS One 6, e16765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner S, Engler C, Weber E, Gruetzner R, and Marillonnet S (2012). Fast track assembly of multigene constructs using golden gate cloning and the MoClo system. Bioeng. Bugs 3, 38–43. [DOI] [PubMed] [Google Scholar]
- Wilson DJ (2019). The harmonic mean p-value for combining dependent tests. Proc. Natl. Acad. Sci. U. S. A 116, 1195–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TD, Reeder J, Lawrence M, Becker G, and Brauer MJ (2016). GMAP and GSNAP for genomic sequence alignment: Enhancements to speed, accuracy, and functionality In Methods in Molecular Biology, (Humana Press Inc.), pp. 283–334. [DOI] [PubMed] [Google Scholar]
- Xu C, Liberatore KL, Macalister CA, Huang Z, Chu YH, Jiang K, Brooks C, Ogawa-Ohnishi M, Xiong G, Pauly M, et al. (2015). A cascade of arabinosyltransferases controls shoot meristem size in tomato. Nat. Genet 47, 784–792. [DOI] [PubMed] [Google Scholar]
- Yang N, Liu J, Gao Q, Gui S, Chen L, Yang L, Huang J, Deng T, Luo J, He L, et al. (2019). Genome assembly of a tropical maize inbred line provides insights into structural variation and crop improvement. Nat. Genet 51, 1052–1059. [DOI] [PubMed] [Google Scholar]
- Yoon S, Xuan Z, Makarov V, Ye K, and Sebat J (2009). Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 19, 1586–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate S, Carroll A, Krashenina O, Sedlazeck FJ, Jun G, Salerno W, Boerwinkle E, and Gibbs R (2018). Parliament2: Fast Structural Variant Calling Using Optimized Combinations of Callers. BioRxiv. 10.1101/424267. [DOI] [Google Scholar]
- Zhou Y, Minio A, Massonnet M, Solares E, Lv Y, Beridze T, Cantu D, and Gaut BS (2019). The population genetics of structural variants in grapevine domestication. Nat. Plants 5, 965–979. [DOI] [PubMed] [Google Scholar]
- Zhu G, Wang S, Huang Z, Zhang S, Liao Q, Zhang C, Lin T, Qin M, Peng M, Yang C, et al. (2018). Rewiring of the Fruit Metabolome in Tomato Breeding. Cell 172, 249–261.e12. [DOI] [PubMed] [Google Scholar]
- Zimin AV, and Salzberg SL (2019). The genome polishing tool POLCA makes fast and accurate corrections in genome assemblies. BioRxiv. 10.1101/2019.12.17.864991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimin AV, Puiu D, Luo MC, Zhu T, Koren S, Marçais G, Yorke JA, Dvořák J, and Salzberg SL (2017). Hybrid assembly of the large and highly repetitive genome of Aegilops tauschii, a progenitor of bread wheat, with the MaSuRCA mega-reads algorithm. Genome Res. 27, 787–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Sample Selection and Long-Read Sequencing, Related to Figure 1. (A and B) Two SNP-based phylogenetic trees for distinct collections of tomato accessions (see STAR Methods) (Razifard et al., 2020; Soyk et al., 2019). Accessions represented in these trees and selected for long-read sequencing are marked with circles or asterisks along the circumference.
(C) Overview of the SV calling pipeline. The process involves aligning ONT long-reads to the SL4.0 reference genome and then calling, filtering and annotating SVs (see STAR Methods).
(D) Histograms of long-read coverage across the SL4.0 genome. Two representative accessions from each taxonomic group are displayed. Average read depths range from 50X to 60X and coverage falls along a Poisson distribution, indicative of genome wide even coverage.
(E) Euler diagrams (http://eulerr.co) depicting the concordance of SV calls derived from short vs. long-reads. A total of four accessions are examined: 2 SLL (top) and 2 SLC (bottom).
(F) Distance matrices depicting pairwise hamming distances between SLL, SLC and SP accessions, graphically represented as heatmaps. The dendrograms represent a hierarchical clustering of the accessions. The distribution of hamming distances is depicted in the heatmap color legends. The larger hamming distances in SLC and SP relative to SLL indicate greater SV diversity in these two groups. Clustering was done using the heatmap.2 function in R with default parameters.
Figure S2. MAS2.0 Assembly and Annotation, Related to STAR Methods. (A) Diagram depicting the MAS2.0 assembly and annotation pipeline.
(B) “Nchart” showing the contiguity of all 14 MAS2.0 assemblies compared to the SL4.0 reference genome. An “Nchart” is a generalization of the N50 metric (50% on the x-axis) in graphical form.
(C-P) Dotplots with respect to the SL4.0 reference for each of the 14 MAS2.0 assemblies.
(Q) The percentage of ITAG4.0 genes that are anchored in each of the 14 MAS2.0 assemblies.
(R) The total number of pan-genome genes anchored in each of the 14 MAS2.0 assemblies.
Figure S3. SV Hotspot and Introgression Analysis, Related to Figure 2. (A-L) Twelve panels are presented, one for each of the 12 chromosomes in the SL4.0 reference genome. Blue heatmaps at left depict the number of SVs in 1 Mbp non-overlapping windows along the reference chromosome. Rows of green heatmaps at top right correspond to the top 45 rows (all SLL accessions) of the blue heatmap. The green heatmap shows the Jaccard Similarity for each SLL accession and for each 1Mbp window to the SP accessions (see STAR Methods). Green heatmap at bottom right (same row order as the top right green heatmap) also shows SV Jaccard Similarity but with respect to all SLC accessions. Specifically, dark green heatmap cells show 1 Mbp windows that show high SV similarity with at least one SP (top) or SLC (bottom) accession. Grey cells in the green heatmap represent “NA” values for regions of the genome that did not have enough SVs to perform the similarity calculations.
(M) The distribution of SV counts in 1 Mbp windows (blue heatmaps from A-L) for the M82 genome.
Figure S4. Differential Expression ROCs, Related to Figure 3. (A) Distribution of the absolute value of the log fold changes of mean gene expression, separated by SV annotation and tissue types. All SV-gene pairs are shown on top, while only significant pairs are shown on the bottom. Numbers beneath boxplots are the number of SV-gene pairs. Fold changes above 27 are not shown.
(B) Diagram of the classifier predicting the presence of a nearby SV given differential gene expression. The diagram depicts a simplified scenario wherein there is a total of three expressed genes, three SVs, and three SV-gene pairs. (I) One of the example SV-gene pairs, with SV-1 overlapping the upstream region of gene-1. (II) Accessions are split into two groups: with and without SV-1. Differential expression of gene-1 between these two groups is tested with a Mann-Whitney U test, yielding a p-value. (III) The test is performed across the same accession split for the two other expressed genes (gene-2 and gene-3). The original gene (gene-1) is positively labeled (+, cyan shade), while the other genes are negatively labeled (−). (IV) The p-values for all tests are ranked, from the most (1) to the least significant (3). (V) Steps I - IV are repeated for all SV-gene pairs using their respective SV accession splits. (VI - VIII) The p-value ranks and gene labels are combined across all SV-gene pairs. The classifier iteratively considers all p-value rank cutoffs and predicts “+” or “-” for all genes at/above or below the cutoff, respectively. The associated ROC curve is built by calculating the true and false-positive rates for each cutoff.
(C-E) ROCs for the nine SV annotation types for cotyledon, root, and apical meristem tissue respectively. “del”, “ins” and “dup” are deletions, insertions and duplications respectively. “5utr” and “3utr” refer to 5 kbp up and downstream of genes respectively. “del CDS” refers to deletions of the CDS start site, whereas “insCDS” refers to insertions in CDS exons. “int” refers to introns.
(F) Volcano plots for specific SV-gene pair examples (extension of Figure 3F). Gene diagrams are not drawn to scale.
Table S1. Tomato accession information and long-read sequencing statistics, related to Figure 1 and Figure S1.
Table S2. Statistics for MAS2.0 genome assemblies and gene annotation, related to Figure S2.
Table S3. Accessions with highest similarity for each window for introgression heatmap analysis, related to Figure 2 and Figure S3.
Table S4. Lists of SV-associated genes and statistics of differential expression, related to Figure 3 and Figure S4.
Table S5. Raw data of genotyping and phenotyping experiments, related to STAR Methods.
Figure S5. A Tandem Duplication and Not a Promoter SNP of the Slkluh Gene Is Responsible for the Fw3.2 Fruit Size Domestication QTL, Related to Figure 5. (A) Expressions of genes within the fw3.2 duplication are approximately two-fold higher in accessions carrying the fw3.2 duplication. Genes within the duplication are in red font. RNA-seq box plots of duplicated and flanking genes from cotyledons (left) and roots (right) are shown. Each point is one biological replicate from one accession (see STAR Methods). n, number of accessions.
(B) Fruit weight quantification in three additional F2 populations segregating for the fw3.2 duplication but fixed for the promoter SNP. Increased fruit weight is associated with the duplication in all three families. n: number of plants measured for each genotype.
(C) Fruit weight quantification in six F2 populations segregating for the promoter SNP (M9) of SlKLUH but fixed for the fw3.2 duplication. There is no consistent association between the M9 SNP and fruit weight. n, number of plants measured for each genotype.
(D) Table showing accessions and genotypes at five known fruit weight QTLs in the 10 F2 populations used to test associations between fruit weight and the fw3.2 duplication or the promoter SNP.
(E) Table showing CRISPR-Cas9 generated mutant alleles of SlKLUH in three independent T0 plants. g1 and g4 refer to guide RNA1 and guide RNA4, respectively.
(F) Mutated spkluh alleles in genotypic group C. Red font, guide RNA targets. Cyan font, mutations. An LA1589 SNP (blue font) permits distinction of KLUH allele parent-of-origin.
(G) Summary of Sanger sequencing resolved alleles of slkluh and spkluh in individual F1 plants of genotypic group C. Allele names in the table refer to Figure 5G and Figure S5G.p-values in (A, B and C) are based on two-tailed, two-sample t-tests.
(H) Summary of Sanger sequencing resolved alleles of slkluh and SpKLUH in individual F1 plants of genotypic group B. These plants were used for quantifying floral organ size and fruit weight. Allele names in the table refer to Figure 5G.
Figure S6. Expression, Flowering, and Inflorescence Branching Phenotypes of the sb1CR Mutation and Allele Distribution of STM3 Duplication, Related to Figure 6. (A) Phylogenetic tree of STM3-TM3 paralogous proteins in tomato and Arabidopsis. Bootstrap values (%) from 1,000 replicates are shown.
(B) Quantification of relative expressions of STM3 and TM3 in transition and floral meristems in Fla.8924 j2TE ej2W and M82 j2TE ej2W genotypes by quantitative reverse transcription PCR (qRTPCR). UBIQUITIN is used as internal control. Expressions are normalized to those in Fla.8924 and are shown as means ± s.d. Three biological pooled meristem samples and two technical replicates of each were assayed.
(C) Genomic short-read resequencing coverage map showing an increase in short reads at the STM3 locus in accessions with the SB1 duplication compared to those with single copy sb1.
(D) Schematic showing positions of guide RNAs used and mutant alleles recovered in the two CRISPR-Cas9 targeting of the STM3-TM3 locus. Small indel alleles of stm3 and tm3 in sb1CR−1 (top) and a large deletion allele removing two copies of STM3 and TM3 in sb1CR-del (bottom) are shown. Orange box shows the common exon 2 between STM3 and TM3 and black box and thinner black box show exons and UTRs, respectively. Red font, guide RNA targets. The cyan font shows mutations.
(E) Flowering time measured by number of leaves to first inflorescence in wild type control (M82) and the two CRISPR alleles of sb1. n, number of plants phenotyped.
(F) Schematic depicting the sb1CR1−1 CRISPR allele at the STM3-TM3 locus (top). Representative inflorescences from the indicated genotypes showing suppression of branching in j2TE ej2w by sb1CR−1 (bottom). Arrowheads mark branch points.
(G) STM3 duplication allele frequency in distant wild tomato species, the closest wild relatives of domesticated tomato (S. galapagense, S. cheesmaniae, S. pimpinellifolium), “admixtures”, early tomato domesticates (S. lyc. var. cerasiforme), and cultivated tomatoes (S. lycopersicum). The number of accessions in each group is indicated.
p-values in (B and E) are based on two-tailed, two-sample t-tests.
Data Availability Statement
All sequencing data generated in this study have been deposited at the Sequence Read Archive (http://ncbi.nlm.nih.gov/sra) under BioProject PRJNA557253. Github repositories for software presented in this work are listed as follows: https://github.com/malonge/DupCheck, https://github.com/mkirsche/Jasmine, https://github.com/srividya22/geneLift, https://github.com/malonge/CallIntrogressions. All genome assemblies/annotations and SV VCF files are available at the Solanaceae Genomics Network (https://solgenomics.net/projects/tomato100).