

Abstract
Motivation
Metagenomics studies have provided key insights into the composition and structure of microbial communities found in different environments. Among the techniques used to analyse metagenomic data, binning is considered a crucial step to characterize the different species of micro-organisms present. The use of short-read data in most binning tools poses several limitations, such as insufficient species-specific signal, and the emergence of long-read sequencing technologies offers us opportunities to surmount them. However, most current metagenomic binning tools have been developed for short reads. The few tools that can process long reads either do not scale with increasing input size or require a database with reference genomes that are often unknown. In this article, we present MetaBCC-LR, a scalable reference-free binning method which clusters long reads directly based on their k-mer coverage histograms and oligonucleotide composition.
Results
We evaluate MetaBCC-LR on multiple simulated and real metagenomic long-read datasets with varying coverages and error rates. Our experiments demonstrate that MetaBCC-LR substantially outperforms state-of-the-art reference-free binning tools, achieving ∼13% improvement in F1-score and ∼30% improvement in ARI compared to the best previous tools. Moreover, we show that using MetaBCC-LR before long-read assembly helps to enhance the assembly quality while significantly reducing the assembly cost in terms of time and memory usage. The efficiency and accuracy of MetaBCC-LR pave the way for more effective long-read-based metagenomics analyses to support a wide range of applications.
Availability and implementation
The source code is freely available at: https://github.com/anuradhawick/MetaBCC-LR.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Metagenomics studies the genetic material of microorganisms directly from their natural environments (Chen and Pachter, 2005). Next-generation sequencing (NGS) technologies have enabled large-scale studies, such as the Human Microbiome Project (The Human Microbiome Project Consortium, 2012), to sequence different environments while generating large amounts of short-read data. Recovering the individual genomes present in microbial communities is a crucial step in such studies, as it aids characterization of the function and behaviour of different species. An important step in such analyses is to group the reads into individual bins representing different species. Both the number of bins and the bin to which a read belongs to have to be estimated and many binning tools have been designed for this task.
Metagenomics binning tools are broadly of two types: (i) reference-based binning (taxonomic) and (ii) reference-free binning (taxonomy-independent). Reference-based binning tools make use of features such as sequence similarity and compare them with sequences in a database of known reference genomes, e.g. LMAT (Ames et al., 2013), MetaPhlAn (Segata et al., 2012) and Kraken (Wood and Salzberg, 2014). Reference-based binning tools produce highly accurate results when the dataset consists of known microbial genomes. However, reference genomes are often unknown or incomplete and, in such cases, these tools cannot be used. In contrast, reference-free binning tools operate without the use of reference databases. These tools group sequences into unlabelled bins completely based on the genomic signatures and sequencing information. Examples include CLAME (Benavides et al., 2018), MetaProb (Girotto et al., 2016), MrGBP (Kouchaki et al., 2019) and Opal (Luo et al., 2019). Majority of these tools are designed and benchmarked using short reads with very low error rates (McIntyre et al., 2017; Pearman et al., 2019).
The use of short NGS reads is prone to ambiguous alignments or fragmented assemblies due to repeats within individual genomes and shared similar regions between different genomes (Pearman et al., 2019). To overcome these limitations, the field of metagenomics has demonstrated a rising interest towards making use of long-read data generated by third-generation sequencing technologies, such as PacBio and ONT, especially for metagenome assembly (Pearman et al., 2019). However, only a limited number of studies have been carried out to benchmark the accuracy of binning from long and error-prone reads (Pearman et al., 2019).
Among the binning tools which can handle long reads, MEGAN-LR (Huson et al., 2018), Centrifuge (Kim et al., 2016) and Kaiju (Menzel et al., 2016) perform reference-based binning where a reference database is required for k-mer matching, indexing and alignment. The tools MetaProb (Girotto et al., 2016) and BusyBee Web (Laczny et al., 2017) support reference-free binning of long reads. However, the current implementation of MetaProb (Girotto et al., 2016) does not scale well to bin large and unknown long-read datasets, while BusyBee Web (Laczny et al., 2017) is currently available as a web-based tool and has limits on the size of the input dataset. To our knowledge, there is no reference-free tool that can accurately bin large long-read datasets.
Since short reads may not contain enough species-specific signals, another approach is to assemble them into longer contigs and use contig binning tools [e.g. MetaBAT (Kang et al., 2015, 2019), MetaWatt (Strous et al., 2012), SolidBin (Wang et al., 2019), MaxBin 2.0 (Wu et al., 2016), BMC3C (Yu et al., 2018) and Autometa (Miller et al., 2019)]. However, these tools cannot be directly applied to bin long reads by treating long reads as contigs. First, contig binning tools require the coverage information for each contig, but such coverage information is usually not available for a single long read. Second, these tools use single-copy marker genes to determine the number of bins, but long reads may be too error-prone for such analysis and this may also result in an overestimated number of bins (when the coverage of a genome is larger than 1). Third, long-read datasets are much larger than contig datasets, and these tools also are unable to address the challenge of scalability. Therefore, there remains a need for a reference-free tool that can bin large long-read datasets accurately and efficiently.
In this article, we design MetaBCC-LR, a scalable reference-free binning tool to bin metagenomic long reads. MetaBCC-LR uses discriminatory features, that capture species abundance and composition information, for finding the number of bins and developing a statistical model for each bin. This is accomplished by using just a sample of the input reads, enabling MetaBCC-LR to efficiently handle large datasets. The statistical model allows us to accurately classify each long read into the bins through a maximum likelihood framework. Our experimental results demonstrate substantial improvements over state-of-the-art reference-free binning tools on both simulated and real long-read datasets, using multiple evaluation metrics. To evaluate how binning can improve downstream tasks, we applied MetaBCC-LR to pre-process large long-read datasets and bin reads into individual bins for long-read assembly. This not only reduced the running time and memory usage considerably but also improved the assembly quality.
2 Materials and methods
The input set of long reads to MetaBCC-LR is sampled and two types of feature vectors are computed for each sampled read. The first feature is a k-mer coverage histogram and the second feature is a trinucleotide composition profile. These two features are clustered, after dimension reduction, in a step-wise manner to obtain the final set of bins. Finally, the input set of long reads is classified into the bins based on a statistical model.
The complete workflow has five steps, as shown in Figure 1. The workflow accepts reads that are at least 1000 bp long (the reads shorter than 1000 bp are filtered). Step 1 performs k-mer counting for the entire dataset and builds a k-mer coverage histogram for each sampled long read. Step 2 applies dimension reduction of the histogram vectors and clusters long reads based on their k-mer coverage histograms. Step 3 computes a trinucleotide composition profile for each sampled long read. Step 4 performs dimension reduction of the composition profiles within each cluster and sub-divides each cluster into smaller bins based on the profiles. Finally, in step 5, a statistical model for each bin is built. All the long reads are classified into bins according to the models using the reads’ k-mer coverage histograms and trinucleotide composition profiles. Each of these steps will be explained in detail in the following sections.
Fig. 1.

The Workflow of MetaBCC-LR
2.1 Step 1: obtain k-mer coverage histograms for reads
Species found in metagenomic samples generally have varying abundances which would result in different sequencing coverages for the corresponding genomes. How can we infer such coverage information of a single read? All-versus-all alignments between all the reads will derive an estimated coverage of each read, but this approach is not scalable to large metagenomic datasets. We estimate the coverage of each read by breaking down the reads into k-mers (i.e. strings of length k), and use the k-mer coverage histogram of each read for binning.
A k-mer is considered as genomic if it appears in at least one genome in the metagenomic dataset, otherwise it is considered as non-genomic. Given a metagenomic dataset, the coverage of a k-mer is defined as the number of times this k-mer appears in the entire dataset. MetaBCC-LR uses the DSK (Rizk et al., 2013) tool to compute the k-mer coverage for the entire dataset and stores the k-mers and their corresponding counts as tuples in the form of , where is the count of the k-mer ki in the entire dataset. For each read, MetaBCC-LR retrieves the coverage of all its k-mers and builds a k-mer coverage histogram. We denote this feature vector by VC; in our experiments, we consider the k-mer coverage from to and thus derive a vector of size 32 using an interval size of 10. Each k-mer coverage histogram is normalized by the total number of k-mers in the corresponding read to avoid any bias due to differences in read-length. Similar to previous studies (Kolmogorov et al., 2019a; Lin et al., 2016; Ruan and Li, 2020) to process long reads, we choose the k-mer size in MetaBCC-LR to be 15 (i.e. ). This choice is sufficiently large to reduce random k-mer collisions and still fits into memory to facilitate efficient computations.
Although the set of genomic k-mers in a metagenomic dataset is unknown, the coverage of a genomic k-mer correlates to the coverage of the unknown genome which it belongs to. It has been shown that a high-coverage peak in the k-mer coverage histogram mainly consists of genomic k-mers (for a sufficiently large k, e.g. ) while a low-coverage peak in the same histogram typically corresponds to non-genomic k-mers due to sequencing errors (Kolmogorov et al., 2019a; Lin et al., 2016). For example, Figure 2a plots the k-mer coverage histograms of long reads sampled from the Zymo-1Y3B dataset (Section 3.1 has details of the dataset). It consists of long reads sampled from genomes of species with low abundance (), medium abundance () and high abundance (–). The two visible peaks around and – in Figure 2a correspond to genomic k-mers in the medium-abundance and high-abundance genomes, respectively. However, genomic k-mers from low-abundance genomes get mixed with non-genomic k-mers due to their low coverages. Figure 2a demonstrates that long reads from genomes with different coverages exhibit different k-mer coverage histograms. Thus, the histogram captures discriminatory coverage information that is effectively used in clustering in our next step.
Fig. 2.

(a) The k-mer coverage histograms of reads from species of different abundances in the Zymo-1Y3B dataset and (b) the clustering result after performing dimension reduction. Please note that colors in (a) are for illustration purposes which MetaBCC-LR is not aware of. MetaBCC-LR infers the colors (clustering result) in (b) from the mixed signals in (a)
2.2 Step 2: perform dimension reduction and clustering based on k-mer coverage histograms
Before the clustering of histogram features, dimension reduction is performed to aid visualization. Such visualization has been previously used to aid manual resolution of metagenomic data to determine the number of bins present (Laczny et al., 2015). Similar to Kouchaki et al. (2019), we use the popular Barnes-Hut t-distributed Stochastic Neighbour Embedding (BH-tSNE) (Van Der Maaten, 2014). BH-tSNE is an efficient dimension reduction technique and has been used in previous studies to bin contigs (Kouchaki et al., 2019). Each histogram vector is reduced to two dimensions, that not only aids visualization, but has been found to preserve the original local data structure (locality and density), while being sufficient for clustering (Kouchaki et al., 2019).
MetaBCC-LR uses the popular density-based clustering algorithm, DBSCAN (Ester et al., 1996), to cluster the two-dimensional data points. We utilize the variation of nearest neighbour distance to estimate the ϵ parameter in DBSCAN (Satopaa et al., 2011). The advantage of using DBSCAN is that it enables clustering based on local density and thereby discards outlier points that arise due to sequencing errors. Moreover, it automatically detects the number of clusters.
Figure 2b shows the two-dimensional clustering of the histogram vectors from reads in Zymo-1Y3B dataset after dimension reduction using BH-tSNE and three clusters of reads inferred by DBSCAN. Note that they correspond well to genomes with low, medium and high coverages in Figure 2a. As seen in Figure 2a the application of the first two steps results in long reads being well separated into clusters such that reads in the same cluster come from genomes of similar coverages. We further refine these clusters in the next two steps using an independent source of information, the trinucleotide composition of the reads.
2.3 Step 3: obtain trinucleotide composition profiles for reads
Genomic signatures of microbes display patterns which are unique to each species (Abe 2003). Hence, these genomic signatures have been used in metagenomics binning. One of the widely used genomic signatures is the oligonucleotide (k-mer for relatively small k) composition where studies have shown that oligonucleotide frequencies are conserved within a species and vary between species (Wu et al., 2016). Similar to the contigs (assembled from short reads), long reads have sufficient length to inherit oligonucleotide composition information of the underlying genome despite their high error rates. Note that this is not true for short but accurate NGS reads. Figure 3 shows the violin plots of trinucleotide composition features of (a) 100 non-overlapping short reads (150 bp) and (b) 100 non-overlapping long (PacBio) reads (10–15 kbp) simulated from the reference genome of P.aeruginosa. From Figure 3a we can see that the trinucleotide frequencies of short reads show significant deviations from that of their reference genomes due to their short lengths. On the other hand, Figure 3b shows that the trinucleotide frequencies of long reads follow a close pattern to that of the reference genome despite their high error rates. Patterns similar to PacBio reads can be seen in Nanopore (ONT) reads as well (refer to Supplementary Section S1).
Fig. 3.
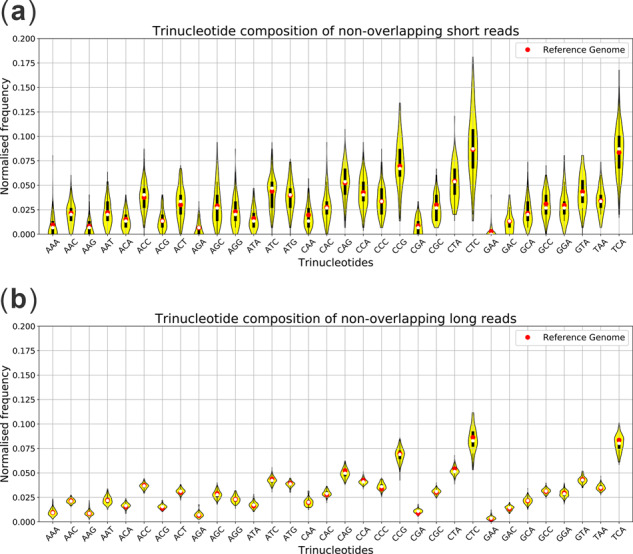
Trinucleotide composition of (a) 100 non-overlapping short reads (150 bp) and (b) 100 non-overlapping long reads (10–15 kbp) simulated from the reference genome of P.aeruginosa. Short reads were simulated modelling an Illumina MiSeq instrument with 300 bp mean read length. Error probabilities for long reads (PacBio) are; indels 0.04, insertions 0.11 and substitutions 0.01 by default for SimLoRD (Stöcker et al., 2016) simulator. For each read, the normalized frequencies are obtained by dividing the number of occurrences of each trinucleotide in the read by the total number of trinucleotides observed in the read
For each long read, MetaBCC-LR builds a profile vector VT consisting of trinucleotide (3-mer or trimer) occurrences. Since there are 32 unique trinucleotides (combining reverse complements as well), the resulting vectors will have 32 dimensions. The vector coordinates are then normalized by the total number of trinucleotides in the long read to avoid any read-length bias. For example, Figure 4a shows the violin plots of trinucleotide composition profiles of long reads obtained from the high abundance cluster in the Zymo-1Y3B dataset from Figure 2b. Despite the high error rates, long reads sampled from the same genome show similar trinucleotide composition patterns whereas reads sampled from different genomes show varying trinucleotide composition patterns. Such discriminatory patterns are utilized to further sub-divide the clusters obtained from Step 2.
Fig. 4.

(a) The trinucleotide composition of reads in the high-abundance cluster of Zymo-1Y3B dataset and (b) the binning result after performing dimension reduction. Please note that colors in (a) are for illustration purposes which MetaBCC-LR is not aware of. Colors (clustering result) are inferred in (b) from the mixed signals in (a)
2.4 Step 4: perform dimension reduction and binning based on trinucleotide composition
Similar to Step 2, MetaBCC-LR first uses BH-tSNE (Van Der Maaten, 2014) to map the trinucleotide composition profiles to two-dimensional vectors. Clusters of long reads derived from Step 2 are further divided into bins according to their trinucleotide composition profiles using DBSCAN. For example, Figure 4b shows the two-dimensional plot of reads in the high-abundance cluster of Zymo-1Y3B dataset after dimension reduction using BH-tSNE and the two clusters inferred by DBSCAN that correspond to two genomes with distinct trinucleotide composition in Figure 4a. All the clusters obtained at this step form the final set of bins B. As shown in Figure 1, MetaBCC-LR then bins the entire input set of reads, not just the sampled reads, into these bins. This classification is done by building a statistical model for each bin as explained in Step 5.
2.5 Step 5: build models for bins and bin all the reads according to these models
For the ith bin Bi, MetaBCC-LR builds a statistical model by calculating the mean and standard deviation for the vectors VC (i.e. k-mer coverage histograms) and the mean and standard deviation for the vector (i.e. trinucleotide composition profile). For each read vector , MetaBCC-LR computes the multivariate probability that it belongs to a bin with mean vector and standard deviation vector using the following Gaussian distribution:
| (1) |
Here, stands for the size of each vector that we compare (i.e. 32). More specifically, a long read r, with a k-mer coverage histogram and trinucleotide frequency vector , is assigned into a bin in a maximum likelihood framework:
| (2) |
Finally, after assigning all the reads to the identified bins, MetaBCC-LR will output a bin identifier for each read.
3 Experimental setup
3.1 Datasets
We used the following long-read datasets for our evaluation.
Oxford Nanopore GridION mock community dataset from the ZymoBIOMICS Microbial Community Standards (Nicholls et al., 2019) (denoted by Zymo-All). The original Zymo-All dataset contains Nanopore (ONT) reads. Five additional PacBio datasets (denoted by Zymo-1Y2B, Zymo-1Y3B, Zymo-2Y2B, Zymo-2Y3B and Zymo-2Y4B) and five additional Nanopore datasets (refer to Supplementary Section S2) are simulated using the species found in the Zymo-All dataset.
Artificial Skin Microbiome datasets (NCBI BioProject number PRJNA533970) consisting of four different mixes of five common skin bacteria with various noise levels (denoted by ASM-0, ASM-5, ASM-10 and ASM-15). Each dataset consists of 10 000 reads which are of length 1000 bp or longer.
The Preborn infant gut metagenome (NCBI Accession No. SRA052203) (Sharon et al., 2013). A PacBio dataset is simulated using the five most abundant species with their corresponding coverages (denoted by Sharon).
Simulated metagenome with 100 species (Wu et al., 2014). A PacBio dataset is simulated using the 100 species found (denoted as 100-genomes).
Note that simulated datasets were generated by the long-read simulators SimLoRD (Stöcker et al., 2016) using default parameters for PacBio reads (error probabilities: indels 0.04, insertions 0.11 and substitutions 0.01) and DeepSimulator (Li et al., 2018) using default parameters for ONT reads. Further details about each dataset can be found in Supplementary Section S2.
3.2 Tools compared
We compared MetaBCC-LR with two recent reference-free binning tools which support long reads, MetaProb (Girotto et al., 2016) and BusyBee Web (Laczny et al., 2017). However, current implementations of MetaProb and BusyBee cannot process the entire long-read dataset at once. Hence, we subsampled 10 000 reads from all the datasets except for the ASM datasets to provide acceptable inputs to MetaProb and BusyBee Web.
3.3 Evaluation criteria
Since the reference genomes for Zymo-All and ASM datasets are available, we mapped the reads to these reference genomes using Minimap2.1 (Li, 2018). The reads which had over 90% of the bases mapped to a reference genome were considered for evaluation. For the simulated datasets from known reference genomes, we used their ground-truth label to evaluate the binning result. We determined the precision, recall, F1 score and Adjusted Rand Index (ARI) for the binning results of each dataset. The equations used to calculate these evaluation metrics can be found in Supplementary Section S3.
4 Results
4.1 Binning results
We recorded the number of bins identified by each tool for all the datasets and the values can be found in Table 1. It was observed that MetaProb and BusyBee Web tend to produce more bins than the actual number of species present. In comparison, MetaBCC-LR was able to identify a number closer to the actual number of species present in all the datasets. Results of the five additional Zymo Nanopore (ONT) datasets can be found in Supplementary Section S4.
Table 1.
Comparison of the actual number of species present and the number of bins identified by the tools for different datasets. The numbers of bins closest to the actual number of species present are highlighted in bold text.
| Dataset | Actual no. of species present | No. of bins identified by MetaProb | No. of bins identified by BusyBee Web | No. of bins identified by MetaBCC-LR |
|---|---|---|---|---|
| Zymo-1Y2B | 3 | 6 | 23 | 3 |
| Zymo-1Y3B | 4 | 7 | 16 | 4 |
| Zymo-2Y2B | 4 | 8 | 21 | 4 |
| Zymo-2Y3B | 5 | 8 | 18 | 5 |
| Zymo-2Y4B | 6 | 9 | 18 | 6 |
| Zymo-All | 10 | 13 | 26 | 8 |
| Sharon | 5 | 8 | 123 | 4 |
| ASM-0 | 5 | 11 | 10 | 5 |
| ASM-5 | 5 | 13 | 8 | 4 |
| ASM-10 | 5 | 14 | 14 | 4 |
| ASM-15 | 5 | 22 | 10 | 5 |
| 100-genomes | 100 | 22 | 74 | 89 |
Figure 5 compares the precision, recall, F1 score and ARI of the binning results of all the datasets obtained from MetaBCC-LR with MetaProb and BusyBee Web. On average, MetaBCC-LR has the best precision, recall, F1 score and ARI for binning long reads. Note that MetaProb and BusyBee Web perform poorly on the ASM datasets when no sub-sampling is applied. This is because BusyBee Web utilizes a pre-trained model based on a selected set of reference genomes, whereas MetaProb requires overlapping k-mers to form read groups which are used for clustering reads into bins. Note that in the five simulated Zymo PacBio datasets, MetaBCC-LR improves on precision (e.g. 6.1% average increase) with a bit of compromise on recall (e.g. 0.5% average decrease) compared to other binning tools.
Fig. 5.
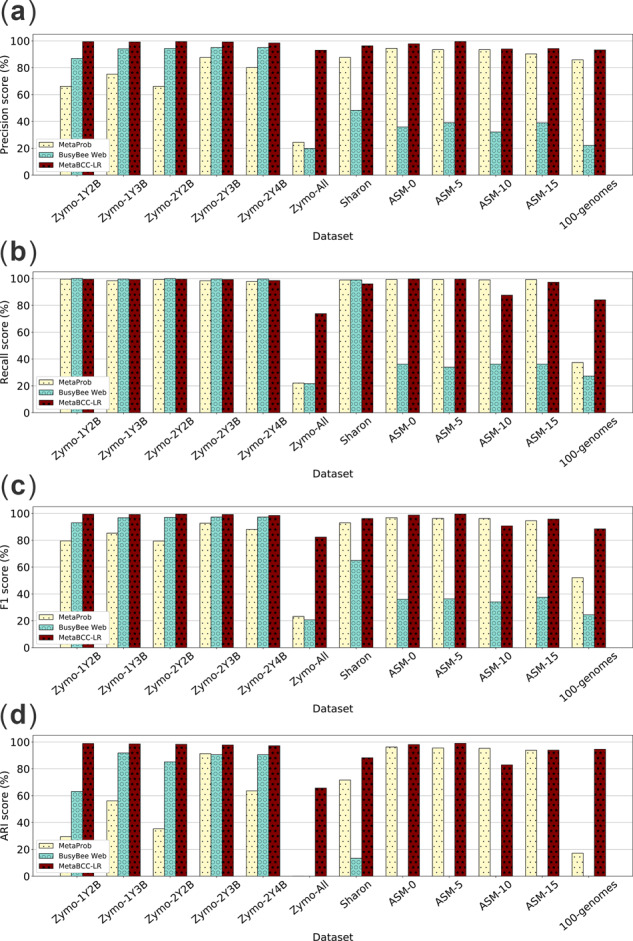
Binning precision, recall, F1 score and ARI over different datasets
Table 2 compares the mean and standard deviation of each evaluation metric averaged over all the datasets for each tool. MetaBCC-LR outperforms the other tools in all metrics. The increase in F1 score and ARI is ∼13% and ∼30% respectively over the best baseline method, MetaProb. MetaBCC-LR also has the most consistent performance with the lowest standard deviation values (less than 10%) in all metrics.
Table 2.
Comparison of mean and standard deviation (STD) of each evaluation metric averaged over all the datasets for each tool. The best values are highlighted in bold text.
| Tool | Precision (%) |
Recall (%) |
F1 score (%) |
ARI (%) |
||||
|---|---|---|---|---|---|---|---|---|
| Mean | STD | Mean | STD | Mean | STD | Mean | STD | |
| MetaProb | 78.77 | 19.77 | 87.35 | 27.11 | 81.42 | 22.19 | 62.13 | 34.42 |
| BusyBee Web | 58.46 | 31.52 | 65.75 | 35.58 | 61.27 | 32.64 | 36.24 | 43.15 |
| MetaBCC-LR | 96.66 | 2.40 | 94.12 | 8.08 | 95.27 | 5.35 | 92.28 | 9.61 |
4.2 Metagenome assembly results
To demonstrate the effect of MetaBCC-LR on metagenomics assembly, we assembled all the Zymo datasets (simulated and real) and the 100-genomes dataset individually using two popular long-read assemblers metaFlye (Kolmogorov et al., 2019b) (available in Flye v2.4.2) and Canu v1.8 (Koren et al., 2017) (denoted as complete assembly) and also assembled the partitioned reads of the individual bins from MetaBCC-LR using metaFlye and Canu (denoted as partitioned assembly). We evaluated all the assemblies using MetaQUAST (Mikheenko et al., 2016). Please note that binning results from MetaProb and BusyBee Web were not assembled as they were not able to bin the entire dataset at once.
Table 3 shows the comparison between genome fraction (total number of aligned bases in the reference/the genome size) of complete and partitioned assemblies. Applying MetaBCC-LR to bin long reads prior to assembly (i.e. partitioned assemblies) improves the genome fraction over the complete assemblies. One possible reason for such improvement is that partitioned assemblies allow assemblers to estimate more appropriate parameters for reads in each bin rather than applying the same parameters to the entire dataset. This may help to recover more genomic sequences, especially for low-abundance species.
Table 3.
Comparison of the assembled genome fraction of the different assemblies for different datasets. The best values are highlighted in bold text.
| Dataset | metaFlye assembly |
Canu assembly |
||
|---|---|---|---|---|
| Complete (%) | Partitioned (%) | Complete (%) | Partitioned (%) | |
| Zymo-1Y2B | 93.12 | 99.60 | 78.74 | 98.69 |
| Zymo-1Y3B | 93.97 | 99.65 | 78.20 | 98.79 |
| Zymo-2Y2B | 93.90 | 97.78 | 57.28 | 97.18 |
| Zymo-2Y3B | 97.35 | 93.44 | 63.53 | 95.66 |
| Zymo-2Y4B | 94.55 | 97.59 | 71.35 | 86.69 |
| Zymo-All | 86.47 | 88.68 | 68.79 | 85.42 |
| 100-genomes | 96.99 | 98.33 | 63.93 | 92.33 |
Table 4 shows the comparison of resource usage (assembly time and peak memory) for complete assembly and partitioned assembly. Assembly time of partitioned assemblies includes the CPU time elapsed for binning using MetaBCC-LR (refer to Table 6) and metagenome assembly. As expected, partitioned assemblies have consumed lesser time and memory than the complete assemblies. The reduction in resource usage is more significant in Canu assemblies than in metaFlye assemblies because Canu is a generic assembler whereas metaFlye is a dedicated metagenomics assembler. Table 5 shows the average improvements of the partitioned assemblies after using MetaBCC-LR, compared to the complete assemblies. We observed improvements in genome fraction with significant reduction in time and memory usage. These results show that MetaBCC-LR can be used to improve metagenomics assembly by binning long reads before assembly.
Table 4.
Comparison of resource usage between complete assembly and partitioned assembly for the Zymo datasets. The best values are highlighted in bold text.
| Dataset | Performance metric | metaFlye assembly |
Canu assembly |
||
|---|---|---|---|---|---|
| Complete assembly | Partitioned assembly | Complete assembly | Partitioned assembly | ||
| Zymo-1Y2B | Assembly time (h) | 12.15 | 9.25 | 74.61 | 58.01 |
| Memory usage (GB) | 35.34 | 24.21 | 13.18 | 8.01 | |
| Zymo-1Y3B | Assembly time (h) | 15.40 | 11.96 | 86.51 | 78.33 |
| Memory usage (GB) | 36.53 | 22.16 | 18.43 | 7.85 | |
| Zymo-2Y2B | Assembly time (h) | 13.41 | 10.43 | 75.20 | 61.12 |
| Memory usage (GB) | 35.41 | 23.42 | 19.45 | 11.46 | |
| Zymo-2Y3B | Assembly time (h) | 16.51 | 13.49 | 87.22 | 82.93 |
| Memory usage (GB) | 35.81 | 23.81 | 22.15 | 7.94 | |
| Zymo-2Y4B | Assembly time (h) | 20.74 | 15.63 | 102.13 | 101.16 |
| Memory usage (GB) | 54.84 | 21.65 | 25.78 | 12.44 | |
| Zymo-All (Nicholls et al., 2019) | Assembly time (h) | 45.95 | 36.45 | 437.99 | 258.00 |
| Memory usage (GB) | 129.36 | 16.19 | 108.50 | 21.76 | |
Note: Assembly time for partitioned assemblies includes the CPU time elapsed for binning using MetaBCC-LR and metagenomics assembly.
Table 6.
Memory and time taken by MetaBCC-LR to bin different datasets.
| Dataset | Size (GB) | Wall time (h) |
|---|---|---|
| Zymo-1Y2B | 4.2 | 0.24 |
| Zymo-1Y3B | 5.45 | 0.32 |
| Zymo-2Y2B | 4.35 | 0.25 |
| Zymo-2Y3B | 5.65 | 0.33 |
| Zymo-2Y4B | 7.15 | 0.40 |
| Zymo-All | 14.24 | 0.86 |
Note: Wall times are recorded for the complete binning process using 8 CPUs.
Table 5.
Average improvement in Genome Fraction (GF) due to MetaBCC-LR considering only the Zymo datasets.
| Assembly | Complete |
Partitioned with MetaBCC-LR |
||
|---|---|---|---|---|
| GF (%) | GF (%) | Memory saved (%) | Time saved (%) | |
| metaFlye | 93.23 | 96.12 | 87.48 | 24.64 |
| Canu | 69.65 | 93.74 | 79.94 | 41.09 |
4.3 Running times and memory usage
Table 6 shows the times taken by MetaBCC-LR to bin the datasets Zymo-1Y2B, Zymo-1Y3B, Zymo-2Y2B, Zymo-2Y3B, Zymo-2Y4B and Zymo-All. Please note that the running times for MetaProb and BusyBee Web were not recorded. They were not able to bin the entire dataset at once and running times for BusyBee Web could not be measured since it is a web application. These factors make it challenging to conduct a fair comparison of running time.
Please note that all the steps of MetaBCC-LR are performed using under 5 GB of peak memory. Further information about memory usage can be found in Supplementary Section S6.
5 Discussion
Our approach, MetaBCC-LR, is a two-phased binning approach to bin noisy long reads without the use of reference databases. The first phase uses k-mer coverage histograms and the second phase uses trinucleotide composition to separate reads. The two phases are executed sequentially. The two phases of MetaBCC-LR and the final step of building a statistical model for each bin, they all use a sample of the input dataset. Finally, a bin is assigned to all the reads in the input data.
We conducted experiments to see how alternative approaches and read assignment would affect the final binning results. Moreover, we conducted experiments to see how subsampling improves the scalability of MetaBCC-LR without compromising the binning quality. Furthermore, we conducted experiments to show the importance of MetaBCC-LR in solving real-world metagenomics binning problems.
5.1 Alternative approaches for binning long reads
We conducted experiments on two alternative approaches: (i) separate long reads first by their trinucleotide composition and then by their coverage (Composition first) and (ii) combining the k-mer coverage histograms and trinucleotide composition profiles and apply dimension reduction to bin reads (k-mer Coverage + Composition). Table 7 shows the comparison of results obtained for the Zymo-2Y4B dataset using each of these methods with MetaBCC-LR.
Table 7.
Comparison of alternative approaches with MetaBCC-LR using the Zymo-2Y4B dataset. The best values are highlighted in bold text.
| Method | No. of bins identified | Precision (%) | Recall (%) | F1 score (%) | ARI (%) |
|---|---|---|---|---|---|
| Composition first | 5 | 92.71 | 90.14 | 91.41 | 79.78 |
| k-mer Coverage + Composition | 2 | 99.59 | 47.80 | 64.60 | 0.43 |
| Coverage first (MetaBCC-LR) | 6 | 98.46 | 98.46 | 98.64 | 97.21 |
We observed that even though we obtained very high precision values with the combined approach (k-mer Coverage + Composition), it resulted in poor recall and ARI values. Overall, MetaBCC-LR has outperformed other alternative methods and produced better evaluation scores.
5.2 Read assignment
We conducted experiments to compare the performance of doing dimension reduction and binning on the entire dataset with our method where we assign reads based on the models obtained from doing dimension reduction and binning on the subsample of reads from a dataset. Our method took 1 min and 19 s of wall time (on a Linux system with Ubuntu 18.04.1 LTS, 16 GB memory and Intel(R) Core(TM) i7-7700 CPU @ 3.60 GHz with 4 CPU cores and 8 threads) to bin and assign the reads of the Zymo-2Y4B dataset (with a sample of 8620 reads) whereas when we performed dimension reduction and binning on the entire dataset at once, the process did not finish even after 3 hours of wall time.
We observed that the precision values of clustering of sampled reads and read assignment afterwards remain similar. Initial uniform sampling was done to obtain a smaller representation of the dataset considering the computational complexity of BH-tSNE. Read assignment is labelling the unsampled reads to the bins identified from the sampled set of reads. Table 8 shows the precision and recall of the initial sampled binning and final binning after read assignment for the Zymo-1Y2B, Zymo-1Y3B, Zymo-2Y2B, Zymo-2Y3B, Zymo-2Y4B and Zymo-All datasets. Even though the precision has reduced slightly after read assignment, we obtained a significant performance gain by using this method over doing dimension reduction for the entire dataset.
Table 8.
Comparison of the precision and recall of sampled reads and after assigning all the reads. The best values are highlighted in bold text.
| Dataset | No. of sampled reads | Precision of sampled reads (%) | Recall of sampled reads (%) | Total no. of reads available | Precision after read assignment (%) | Recall after read assignment (%) |
|---|---|---|---|---|---|---|
| Zymo-1Y2B | 5028 | 99.42 | 99.42 | 502 890 | 99.47 | 99.47 |
| Zymo-1Y3B | 6576 | 98.97 | 98.97 | 657 610 | 99.27 | 99.27 |
| Zymo-2Y2B | 5259 | 99.13 | 99.13 | 525 922 | 99.51 | 99.51 |
| Zymo-2Y3B | 6806 | 98.81 | 98.81 | 680 642 | 99.24 | 99.24 |
| Zymo-2Y4B | 8620 | 98.46 | 98.46 | 862 021 | 98.46 | 98.46 |
| Zymo-All (Nicholls et al., 2019) | 34 910 | 97.64 | 71.92 | 3 491 078 | 93.09 | 73.84 |
5.3 Effect of initial sample size
We conducted experiments to check the effect of varying initial sample sizes on the final binning results. We selected sample sizes 0.5, 1 and 1.5% of reads from each of the complete simulated Zymo datasets to determine the number of bins and build their corresponding statistical profiles. Then, we calculated the precision, recall, F1-score and ARI for the binned sample of reads and the values can be found in Supplementary Section S5. The average results obtained for the simulated Zymo datasets for 0.5, 1 and 1.5% are shown in Table 9 as we observed the most significant gain in performance when the sampling size was increased from 0.5 to 1%.
Table 9.
Comparison of average evaluation metrics for varying sample sizes of the simulated Zymo datasets. The best values are highlighted in bold text.
| Sample size (%) | Precision (%) | Recall (%) | F1 score (%) | ARI (%) |
|---|---|---|---|---|
| 0.5 | 98.49 | 98.49 | 98.49 | 97.03 |
| 1 | 99.19 | 99.19 | 99.19 | 98.14 |
| 1.5 | 99.04 | 99.04 | 99.04 | 97.87 |
We observed that the final binning precision and recall after performing read assignment remains very similar for each sample size. Finally, 1% was chosen to retain reads of very low abundant species during the clustering process.
5.4 Separate coral genomes from its symbiont genomes
Many adult reef-building corals are known to be supplied through a mutually beneficial relationship with its microbial symbionts that live inside the coral cells. However, this symbiotic relationship makes it very challenging to separate adult coral from its microbial symbionts as they live inside the coral cells (Ying et al., 2018 and personal communication). To evaluate the utility of MetaBCC-LR in coral studies, we simulated a PacBio dataset using the Coral P.lutea and its microbial symbiont Cladocopium C15 found from the Coral and its microbial symbiont communities from the Orpheus Island, Australia (Robbins et al., 2019) (denoted by Coral+Symbio). Details about the dataset can be found in Supplementary Section S2. We used MetaBCC-LR to separate the reads of coral from its microbial symbiont, to show the importance of our tool in solving real-world metagenomics binning problems.
Figure 6 denotes the two-dimensional plot of the read clusters obtained from the subsample of the Coral+Symbio dataset, (a) after separating by coverage and (b) after separating by composition. Figure 6a shows that using coverage (k-mer coverage histograms) has failed to separate P.lutea and Cladocopium C15 as expected due to their similar abundance in the Coral+Symbio dataset. However, the trinucleotide composition allows us to identify two clearly separated clusters corresponding to P.lutea and Cladocopium C15 (Fig. 6b) because they have different trinucleotide composition. Moreover, MetaBCC-LR resulted in precision, recall, F1 score and ARI of 98.21, 98.21, 98.21 and 92.97% for the final binning of the entire Coral+Symbio dataset. Hence, it can be seen that MetaBCC-LR can be used to solve real-world metagenomics binning problems. However, if two species have similar abundances and similar composition, it would be more challenging to perform binning.
Fig. 6.
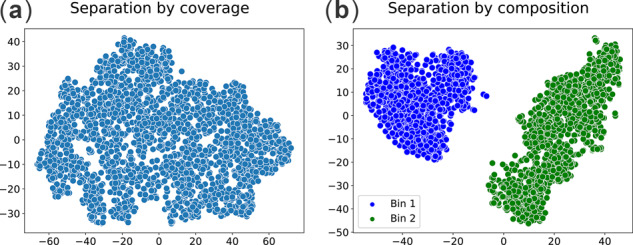
Read clusters obtained from simulated PacBio reads from the Coral P.lutea and its microbial symbiont Cladocopium C15 found from the Coral and its microbial symbiont communities from the Orpheus Island, Australia (Robbins et al., 2019) (denoted by Coral+Symbio). MetaBCC-LR achieved the final binning precision, recall, F1 score and ARI as 98.21, 98.21, 98.21 and 92.97%, respectively
6 Conclusion
In this article, we design and evaluate MetaBCC-LR, a scalable reference-free binning tool to cluster large long-read datasets. MetaBCC-LR uses k-mer coverage histograms and oligonucleotide composition of the reads to estimate the number of bins and classify the input reads to the bins. Our extensive experimental results, on several datasets with varying coverage and error rates, show that MetaBCC-LR outperforms state-of-the-art reference-free binning tools by a substantial margin.
Typically, assemblers erroneously assume uniform coverages and low abundance species tend to be ignored—that can be ameliorated by binning reads before assembly. We indeed observe that binning long reads using MetaBCC-LR prior to assembly improves assembled genome fractions. Further, this is achieved along with a considerable reduction in time and memory usage. Binning is a crucial step in many metagenomics studies and the efficiency and accuracy of MetaBCC-LR can potentially lead to improved characterization of microbial communities to provide valuable biological insights. In future, we intend to extend this work to bin reads across multiple samples to improve the overall binning result.
Supplementary Material
Acknowledgements
The authors thank Dr Hua Ying from the Research School of Biology, the Australian National University for her suggestions. Vaibhav Rajan was supported by the Singapore Ministry of Education Academic Research Fund [R-253-000-138-133]. Furthermore, this research was undertaken with the assistance of resources and services from the National Computational Infrastructure (NCI), Australia.
Financial Support: none declared.
Conflict of Interest: none declared.
References
- Abe T. (2003) Informatics for unveiling hidden genome signatures. Genome Res., 13, 693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames S.K. et al. (2013) Scalable metagenomic taxonomy classification using a reference genome database. Bioinformatics, 29, 2253–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benavides A. et al. (2018) CLAME: a new alignment-based binning algorithm allows the genomic description of a novel Xanthomonadaceae from the Colombian andes. BMC Genomics, 19, 858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K., Pachter L. (2005) Bioinformatics for whole-genome shotgun sequencing of microbial communities. PLoS Comput. Biol., 1, e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ester M. et al. (1996) A density-based algorithm for discovering clusters a density-based algorithm for discovering clusters in large spatial databases with noise. In: KDD’96 AAAI Press, Portland, Oregon. pp. 226–231.
- Girotto S. et al. (2016) MetaProb: accurate metagenomic reads binning based on probabilistic sequence signatures. Bioinformatics, 32, i567–i575. [DOI] [PubMed] [Google Scholar]
- Huson D.H. et al. (2018) MEGAN-LR: new algorithms allow accurate binning and easy interactive exploration of metagenomic long reads and contigs. Biol. Direct, 13, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang D.D. et al. (2015) MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ, 3, e1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang D.D. et al. (2019) MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ, 7, e7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. et al. (2016) Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res., 26, 1721–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolmogorov M. et al. (2019. a) Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol., 37, 540–546. [DOI] [PubMed] [Google Scholar]
- Kolmogorov M. et al. (2019. b) metaFlye: scalable long-read metagenome assembly using repeat graphs. bioRxiv. [DOI] [PMC free article] [PubMed]
- Koren S. et al. (2017) Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res., 27, 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouchaki S. et al. (2019) A signal processing method for alignment-free metagenomic binning: multi-resolution genomic binary patterns. Sci. Rep., 9, 2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laczny C.C. et al. (2015) Alignment-free visualization of metagenomic data by nonlinear dimension reduction. Sci. Rep., 4, 4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laczny C.C. et al. (2017) BusyBee Web: metagenomic data analysis by bootstrapped supervised binning and annotation. Nucleic Acids Res., 45, W171–W179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. (2018) Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics, 34, 3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. et al. (2018) DeepSimulator: a deep simulator for Nanopore sequencing. Bioinformatics, 34, 2899–2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y. et al. (2016) Assembly of long error-prone reads using de Bruijn graphs. Proc. Natl. Acad. Sci. USA, 113, E8396–E8405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y. et al. (2019) Metagenomic binning through low-density hashing. Bioinformatics, 35, 219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre A.B.R. et al. (2017) Comprehensive benchmarking and ensemble approaches for metagenomic classifiers. Genome Biol., 18, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzel P. et al. (2016) Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun., 7, 11257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikheenko A. et al. (2016) MetaQUAST: evaluation of metagenome assemblies. Bioinformatics, 32, 1088–1090. [DOI] [PubMed] [Google Scholar]
- Miller I.J. et al. (2019) Autometa: automated extraction of microbial genomes from individual shotgun metagenomes. Nucleic Acids Res., 47, e57–e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls S.M. et al. (2019) Ultra-deep, long-read nanopore sequencing of mock microbial community standards. GigaScience, 8, giz043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearman W.S. et al. (2019) The advantages and disadvantages of short- and long-read metagenomics to infer bacterial and eukaryotic community composition. bioRxiv.
- Rizk G. et al. (2013) DSK: k-mer counting with very low memory usage. Bioinformatics, 29, 652–653. [DOI] [PubMed] [Google Scholar]
- Robbins S.J. et al. (2019) A genomic view of the reef-building coral Porites lutea and its microbial symbionts. Nat. Microbiol., 4, 2090–2100. [DOI] [PubMed] [Google Scholar]
- Ruan J., Li H. (2020) Fast and accurate long-read assembly with wtdbg2. Nat. Methods, 17, 155–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satopaa V. et al. (2011) Finding a “kneedle” in a haystack: detecting knee points in system behavior. In: 2011 31st International Conference on Distributed Computing Systems Workshops, IEEE, Minneapolis, MN, USA, pp. 166–171.
- Segata N. et al. (2012) Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods, 9, 811–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon I. et al. (2013) Time series community genomics analysis reveals rapid shifts in bacterial species, strains, and phage during infant gut colonization. Genome Res., 23, 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöcker B.K. et al. (2016) SimLoRD: simulation of long read data. Bioinformatics, 32, 2704–2706. [DOI] [PubMed] [Google Scholar]
- Strous M. et al. (2012) The binning of metagenomic contigs for microbial physiology of mixed cultures. Front. Microbiol., 3, 410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Human Microbiome Project Consortium. (2012) Structure, function and diversity of the healthy human microbiome. Nature, 486, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Maaten L. (2014) Accelerating t-SNE using tree-based algorithms. J. Mach. Learn. Res., 15, 3221–3245. [Google Scholar]
- Wang Z. et al. (2019) SolidBin: improving metagenome binning with semi-supervised normalized cut. Bioinformatics, 35, 4229–4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood D.E., Salzberg S.L. (2014) Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol., 15, R46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.-W. et al. (2014) Maxbin: an automated binning method to recover individual genomes from metagenomes using an expectation–maximization algorithm. Microbiome, 2, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.-W. et al. (2016) MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics, 32, 605–607. [DOI] [PubMed] [Google Scholar]
- Ying H. et al. (2018) Comparative genomics reveals the distinct evolutionary trajectories of the robust and complex coral lineages. Genome Biol., 19, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G. et al. (2018) BMC3C: binning metagenomic contigs using codon usage, sequence composition and read coverage. Bioinformatics, 34, 4172–4179. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.