

Abstract
Generation of the autacoid all-trans-retinoic acid (ATRA) from retinol (vitamin A) relies on a complex metabolon that includes retinol binding-proteins and enzymes from the short-chain dehydrogenase/reductase and aldehyde dehydrogenase gene families. Serum retinol binding-protein delivers all-trans-retinol (vitamin A) from blood to cells through two membrane receptors, Stra6 and Rbpr2. Stra6 and Rbpr2 convey retinol to cellular retinol binding-protein type 1 (Crbp1). Holo-Crbp1 delivers retinol to lecithin:retinol acyl transferase (Lrat) for esterification and storage. Lrat channels retinol directly into its active site from holo-Crbp1 by protein-protein interaction. The ratio apo-Crbp1/holo-Crbp1 directs flux of retinol into and out of retinyl esters, through regulating esterification vs ester hydrolysis. Multiple retinol dehydrogenases (Rdh1, Rdh10, Dhrs9, Rdhe2, Rdhe2s) channel retinol from holo-Crbp1 to generate retinal for ATRA biosynthesis. β-Carotene oxidase type 1 generates retinal from carotenoids, delivered by the scavenger receptor-B1. Retinal reductases (Dhrs3, Dhrs4, Rdh11) reduce retinal into retinol, thereby restraining ATRA biosynthesis. Retinal dehydrogenases (Raldh1, 2, 3) dehydrogenate retinal irreversibly into ATRA. ATRA regulates its own concentrations by inducing Lrat and ATRA degradative enzymes. ATRA exhibits hormesis. Its effects relate to its concentration as an inverted J-shaped curve, transitioning from beneficial in the “goldilocks” zone to toxicity, as concentrations increase. Hormesis has distorted understanding physiological effects of ATRA post-nataly using chow-diet fed, ATRA-dosed animal models. Cancer, immune deficiency and metabolic abnormalities result from mutations and/or insufficiency in Crbp1 and retinoid metabolizing enzymes.
Introduction
Activation of the nutrient all-trans-retinol (vitamin A, retinol) into the autacoid all-trans-retinoic acid (ATRA) relies on a complex metabolon, fueled by availability of retinol (Napoli, 2012). Cells access retinol from the serum retinol binding-protein, sRbp (encoded by rbp4) (Fig. 1). sRbp interacts with two intermembrane receptors, Stra6 (stimulated by RA6) and Rbpr2 (Rbp receptor 2). Cellular retinol binding-protein type 1 (Crbp1) and lecithin:retinol acyl transferase (Lrat) guarantee that retinol flux into cells occurs one way physiologically. Crbp1 sequesters retinol, preventing its exit from cells, and delivers retinol to Lrat. Lrat, which synthesizes retinyl esters, accesses retinol bound to Crbp1 and channels retinol directly into its active site by protein-protein interaction. A second esterifying enzyme, diacylglycerol acyltransferase type 1 (Dgat1) also can synthesize retinyl esters, especially in skin, but does not make a major contribution in most cells. The ratio apo-Crbp1/holo-Crbp1 directs flux of retinol into and out of retinyl esters, through stimulation of retinyl ester hydrolases (Reh) and inhibition of Lrat by apo-Crbp1. The latter has an inhibitory constant of ~0.2 μM for Lrat. Therefore, with saturated holo-Crbp1 (no apo-Crbp1) both Lrat and retinol dehydrogenases (Rdh) have maximum activity and generate retinyl esters for retinol storage and retinal for ATRA biosynthesis, respectively. As the concentration of apo-Crbp1 increases (deficit of retinol), holo-Crbp1 continues delivering retinol to support RA biosynthesis, but apo-Crbp1 limits storing retinol as retinyl esters.
Figure 1.
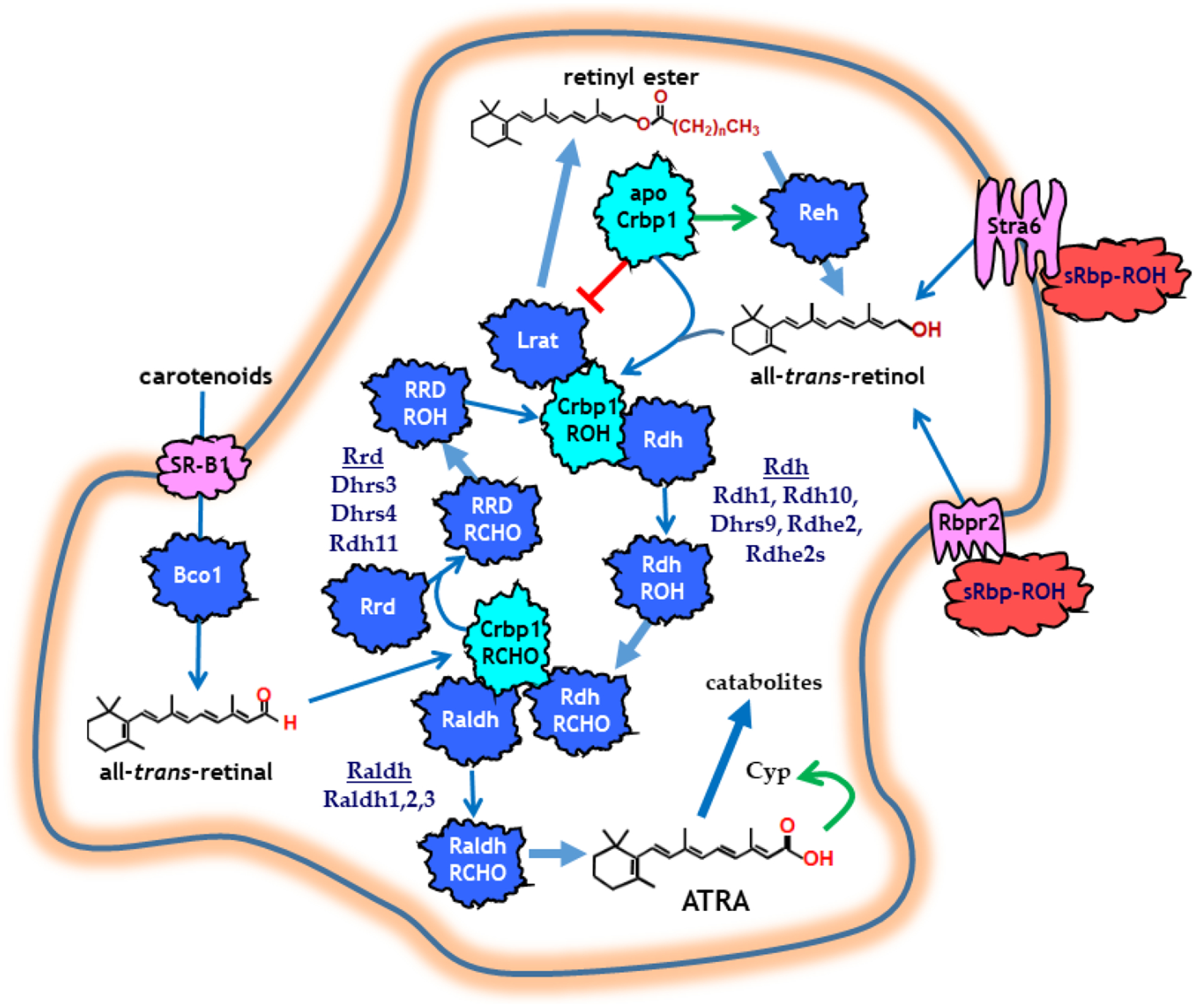
A model of the retinoid metabolon. Light blue indicates retinoid binding-proteins. Dark blue indicates enzymes. Pink indicates membrane receptors. Red indicates sRbp. The red line indicates inhibition. The green arrows show induction of activity. The wide blue arrows indicate catalytic events. The thin blue arrows indicate retinoid transfer.
Two reactions generate ATRA from all-trans-retinol. Rdh of the short-chain dehydrogenase/reductase gene (sdr) family catalyze the first reaction, the reversible dehydrogenation of retinol into all-trans-retinal (retinal). Rdh occur in microsomes (Rdh1) or associate with lipid droplets (Rdh10). The NAD+/NADH ratio and the irreversible second reaction that converts retinal into ATRA drive the reversible Rdh-catalyzed reaction forward. Multiple Rdh have been identified (Rdh1, Rdh10, Dhrs9, Rdhe2, Rdhe2s). Those that have been tested access retinol from holo-Crbp1 by channeling through protein-protein interactions, and do not rely on retinol diffusion from Crbp1.
The membrane receptor scavenger receptor-B1 (SR-B1) transports carotenoids into cells, where β-carotene oxidase type 1 (Bco1) converts those that have a β-ionone ring into retinal (Harrison, 2012). Retinal reductases (Rrd), also of the sdr gene family, catalyze reduction of retinal into retinol. Outside the retina, these include Dhrs3, Dhrs4 and Rdh11, and are either microsomal or peroxisomal. Where tested, Rrd channel retinal from Crbp1.
Retinal dehydrogenases (Raldh), of the aldehyde dehydrogenase gene family (aldh), catalyze irreversible retinal dehydrogenation into ATRA. These cytosolic enzymes include: Raldh1, Raldh2 and Raldh3, and also channel retinal directly from Crbp1.
Retinol and retinal are chaperoned intracellularly by Crbp1 (except for the intestinal enterocyte) (Ross, 1993; Napoli, 2017). Crbp1 has high affinity (≤1 nM) for retinol. Therefore, intracellular retinol occurs as holo-Crbp1 in most tissues. Ablation of Crbp1 disrupts retinol metabolism and causes metabolic disease and cancer (Kane et al., 2011; Pierzchalski, Yu, Norman, & Kane, 2013).
ATRA controls its own intracellular concentrations by inducing Lrat, thereby directing more retinol into retinyl esters, and also by inducing cytochrome P-450’s (Cyp26a1, b1 and c1) to initiate its own catabolism.
Two cytosolic enzymes of the alcohol dehydrogenase gene family (adh), also known as medium-chain alcohol dehydrogenases, convert retinol into retinal in vitro. Adh do not access retinol bound to Crbp1—the form of retinol that occurs in cells. Adh isozymes active with retinol in vitro include AdhI and AdhIV in the mouse. AdhIII has little to no activity with retinol. Adh, however, do not contribute to ATRA biosynthesis in vivo from physiological amounts of retinol. In vitro and in vivo, they access retinol concentrations in the toxic range, which exceed capacities of binding proteins, and overcome restrictions on ATRA biosynthesis. This action contributes to retinol toxicity.
ATRA and hormesis
Dosing larger amounts of retinol or ATRA reduces obesity in rodents fed high-fat diets, i.e. ameliorates diet-induced obesity (Berry, DeSantis, Soltanian, Croniger, & Noy, 2012; Mercader et al., 2006). Retinol and ATRA, however, exhibit hormesis; their effects relate to the amount dosed as an inverted J-shaped curve (Fig. 2). As concentrations increase beyond optimum, beneficial effects diminish and toxic effects ensue (Hayes, 2007). Thus, dosing high amounts of retinol or ATRA confounds physiology with toxicology. Chow diets, which contain copious amounts of vitamin A, exacerbate hormesis effects. Chow diets also contain pro-vitamin A carotenoids, which raise ATRA levels. Hormesis has distorted understanding physiological ATRA effects post-nataly, particularly with respect to energy balance, in chow-diet fed, retinol/ATRA-dosed rodents.
Figure 2.
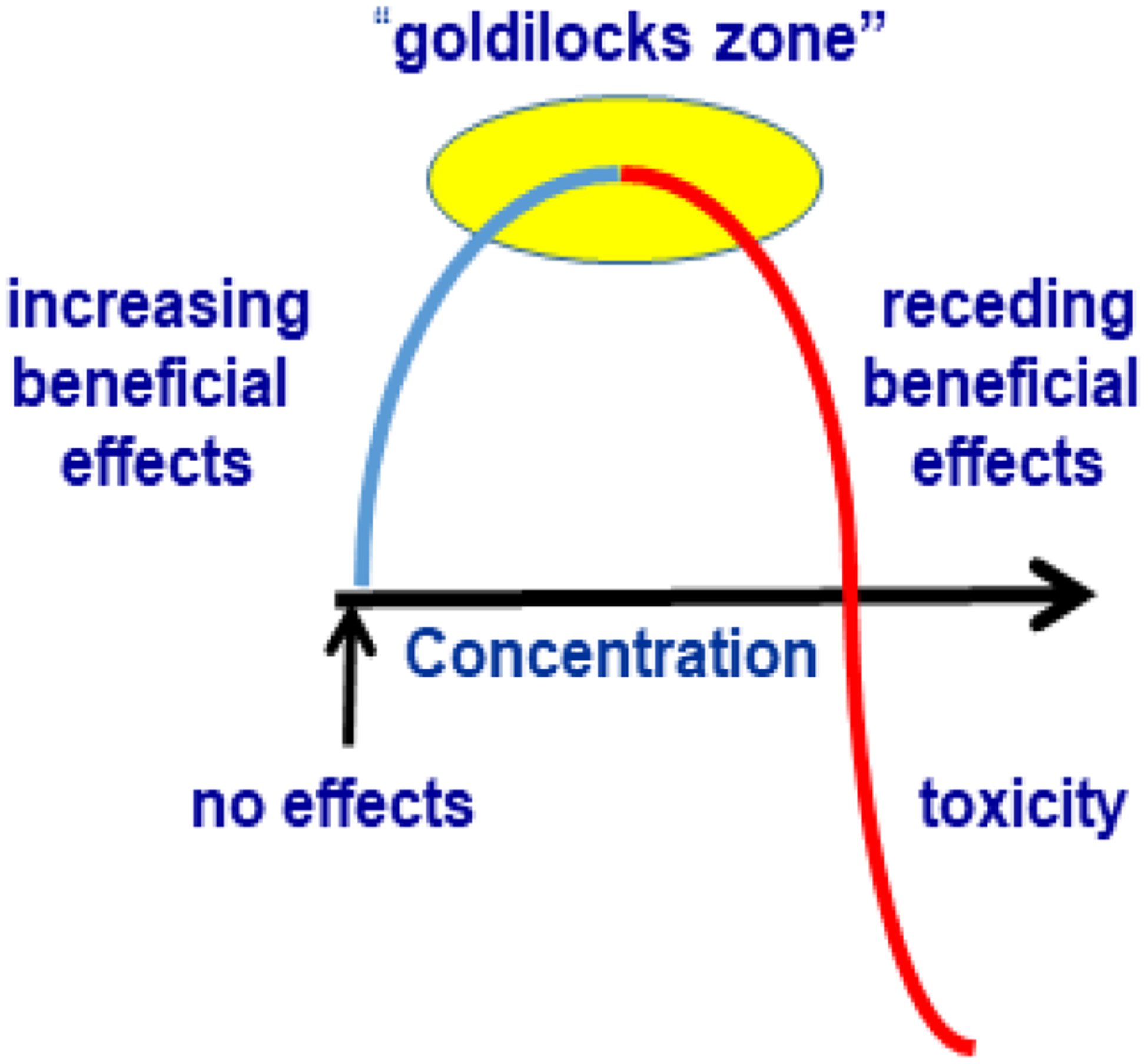
ATRA has hormesis effects. Only in the “goldilocks” zone does ATRA exert optimum physiological activity. As concentrations increase toxicity sets in.
Retinol dehydrogenases
Background
Initial detailed characterization of ATRA biosynthesis was conducted in an established cell line (the pig kidney epithelial cell line LLC-PK1) to preserve cell organization, and let the “cell speak for itself”, rather than rely on subcellular fractions. This study established retinol dehydrogenation as the rate-limiting reaction, and indicated that the retinol concentration was a primary determinant of the ATRA synthesis rate over a wide range. In other words, substrate availability helps drive retinol dehydrogenation. Retinol esterification/de-esterification and cell uptake control the retinol concentration and therefore substrate availability. In contrast to retinol dehydrogenation, retinal reduction and dehydrogenation occur 4–8-fold and 30–60-fold faster, respectively. Consistent with this, retinal generated ~11-fold more ATRA than retinol. This study also established that ATRA up to 100 nM, 2 to 20-fold higher than occurs in vivo, did not inhibit its own synthesis. The potent aldehyde oxidase inhibitor 4’-(9-acridinylamino)methanesulfon-m-anisidide did not inhibit ATRA biosynthesis, nor did 220 mM ethanol, or higher concentrations (<100 mM) of the potent AdhI and AdhII inhibitor 4-methylpyrazole. These data excluded aldehyde oxidase, AdhI, and AdhII from contributing to ATRA biosynthesis, at least in this cell line. The cytochrome P-450 (Cyp) inhibitor ketoconazole increased the ATRA concentration during biosynthesis, indicating that the ATRA concentration depends on the catabolic rate, in addition to the production rate, suggesting tight control of intracellular ATRA.
The next phase involved identifying enzymes in the physiological pathway of ATRA biosynthesis. Intracellularly, retinol occurs bound to cellular retinol binding-protein type 1 (Crbp1) with high affinity (kd ≤ 1 nM) (Ross, 1993; Newcomer, Jamison, & Ong, 1998). Crbp1 engulfs retinol buried in its binding pocket with the alcohol functional group oriented deep inside. Holo-Crbp1 resists proteolysis by Arg-C during an hour of incubation, indicating a tightly closed structure (Jamison, Newcomer, & Ong, 1994). This presents a conundrum of how metabolic enzymes would access retinol. Access could occur if select enzymes interacted with holo-Crbp1, capturing its retinol via protein-protein interaction, channeling retinol directly from binding-protein to enzyme. Kinetic experiments comparing esterification of free retinol with Crbp1-bound retinol demonstrated occurrence of channeling. Free retinol can be esterified using acyl-CoA as substrate (Arat), but bound retinol undergoes esterification using acyl moieties from the SN1 position of phosphatidylcholine, i.e. lecithin:retinol acyltransferase (Lrat) (Ong, MacDonald, & Gubitosi, 1988). Lrat has identical Km and Vmax values in presence of either free or Crbp1-bound retinol. This verifies that Lrat interacts with holo-Crbp1 and directly captures its retinol, because Crbp1 renders the free retinol concentration negligible.
In this context, a physiologically relevant Rdh should have capacity to recognize holo-Crbp1 and capture its retinol through channeling. Kinetics of retinol conversion into retinal in the presence of holo-Crbp1, with added apo-Crbp1 to ensure total retinol sequestration, revealed that rodent liver microsomes express such activity (Poach, Bowerman, Burns, & Napoli, 1991). This identification of holo-Crbp1 as the authentic substrate for converting retinol into retinal in the path of ATRA biosynthesis allowed comparison of activities in microsomes vs cytosol and classification of the activities (Bowerman & Napoli, 1996). Microsomes from liver, kidney, testis and lung have 4 to 13-fold higher capacity to generate retinal in a comparison to cytosolic activity, with 5 μM holo-Crbp1 as substrate in the presence of 2 μM apo-Crbp1, a reasonable approximation of their concentrations in liver. Neither ethanol nor the Adh inhibitor 4-methylpyrazole inhibited the microsomal reactions. In contrast, the IC50 values of ethanol and 4-methylpyrazole for the cytosolic reaction were 20 μM and 180 mM, respectively. These results exclude Adh2 and Adh3 from consideration for the cytosolic activity, because these two Adh have far different sensitivities to ethanol and 4-methypyrazole than the cytosolic retinol dehydrogenase activity. Carbenoxolone, a potent inhibitor of Sdr, inhibited both the microsomal and cytosolic activities with IC50 values of 55 and 70 μM, respectively. These data, along with previous data, exclude Adh isoforms from contributing to cytosolic retinol dehydrogenase activity and reveal that microsomal Rdh generated most retinal for ATRA biosynthesis.
An Rdh involved in ATRA biosynthesis was identified through cross-linking with holo-Crbp1, using either microsomes or an enzyme purified to two gel bands (Boerman & Napoli, 1995). Both sources of Rdh produced the same results, showing specificity of cross-linking. Cross-linking required pyridine nucleotide cofactor, consistent with an ordered bi-substrate reaction mechanism of a dehydrogenase. The ~36 kDa protein identified had a subunit molecular weight and other attributes typical of an Sdr, including the highly-conserved sequences GXXXGXG (cofactor binding), WXLVNNAG, and YCISK (active site residues), sensitivity to carbenoxolone (IC50 = 55 μM), and insensitivity to inhibition by 860 mM ethanol and the Adh inhibitor 4-methylpyrazole. The rat, mouse and human orthologs were cloned, characterized and dubbed RodhI and II, Rdh1, and Rodh4/Rodh-E/Rdh-E (Chai, Boerman, Zhai, & Napoli, 1995; Chai, Zhai, Popescu, & Napoli, 1995; Gough, VanOoteghem, Sint, & Kedishvili, 1998; Jurukovski et al., 1999; M Zhang, Chen, Smith, & Napoli, 2001). Through the peculiarities of nomenclature revision these are now known as Rdh2, Rdh1 and Rdh16, respectively.
Some concern has been raised about the purine nucleotide preferred cofactor (NAD+ vs NADP+) by Rdh, based on the perception that dehydrogenases prefer NAD+, whereas NADP+ preferring enzymes should be considered reductases that really use NADPH. This is not a biochemically sound concern, because most Rdh can use either cofactor, although the Km value usually is lower with one or the other. The “preference” depends not only on the Km value, but also on the cellular concentration of the cofactor. The function of the enzyme in an intact cell provides the answer, when presented with retinol vs retinal. All known Rdh generate ATRA in the presence of an Raldh in intact cells, as shown for Rdh1 (Zhang et al., 2001).
Functions of mRdh1/hRdh16
Rdh1 associates with the microsomal membrane with the body of the enzyme projecting into the cytoplasm (Min Zhang, Hu, & Napoli, 2004). An N-terminal sequence of 22 residues forms a hydrophobic helix that ends in a net positive charge. This leader anchors Rdh1 in the membrane bilayer. Mutating the leader sequence to place the positive charges at the beginning, and replacing the positively charged residues at the end with neutral residues, completely reverses Rdh1 orientation to face the lumen of the smooth endoplasmic reticulum. Thus, Rdh1 adheres to the “positive inside rule”, which defines the orientation of many microsomal and plasma membrane-associated proteins with respect to the cytoplasm. Simply put, the positive charges in the leader sequence occur in the cytoplasm.
The mouse embryo expresses Rdh1 by embryo day 7 (e7), with expression increasing each embryo day until parturition (M Zhang et al., 2001). Rdh1 mRNA expression relates inversely to Cyp26a1 and 26b1 expression during embryogenesis. Cyp26 catabolize ATRA. Therefore, diametrically opposed expression between Cyp and Rdh1 suggests coordination between the catabolic and biosynthetic enzymes to maximally reduce or enhance the ATRA concentration during development.
Rdh1/Rdh16 has enzymatic activity with all-trans-retinol, 9-cis-retinol, 5α-androstane-3α,17β-diol (3-adiol) and androsterone in vitro (M Zhang et al., 2001). But also accesses retinol bound with Crbp1 (Jurukovski et al., 1999). Human Rdh16 now has the designation Sdr9c8.
The rdh1 mouse knockout has a phenotype consistent with a decrease in ATRA, but serum steroids do not change. The knockout generates mice in Mendelian frequency with no obvious phenotypic differences from wild-type, when fed a chow diet (copious amount of vitamin A). Rdh1-null male mice fed a diet with 4 IU vitamin A/g (as recommended by the National Research Council) have liver retinol levels ~2-fold greater than wild type littermates, consistent with decreased ATRA biosynthesis. Feeding the 4 IU vitamin A/g diet to rdh1-null mice revealed that cyp26a1 mRNA and protein decreased by 2.5-fold. This compensatory decrease in ATRA catabolism confirms importance of Rdh1 to generating ATRA. Knockout mice fed a vitamin A-restricted diet become heavier due to increased adiposity, even when fed a low-fat diet. At 33 weeks old, male rdh1-null mice weigh 10.6 g (37%) more than wild type; females weigh 5.8 g (23%) more than WT. Increased adiposity accounts for the increase in weight. Mesentery, femoral and inguinal fat pads increase relative to body weight (% body weight) and per g/mouse. Knock-out mice did not consume more food than WT, nor reduce their activity.
The rdh1-null mouse fed a low-fat diet with recommended retinol levels offered opportunity to study physiological functions of lowering endogenous ATRA. Rdh1 has widespread tissue expression, but neither muscle or nor epidydymal white fat pads express rdh1. Brown adipose tissue (BAT) expresses rdh1 more intensely than most other tissues, and rdh1-null mice have an average body temperature ~0.8 °C lower than WT under normal vivarium conditions (Krois et al., 2019). These data indicate flawed BAT function. BAT differentiation seems unaffected, but mitochondrial function was damaged, as indicated by decreased proteins such as cytochrome C and Ucp1, a decreased oxygen consumption rate, and disrupted mitochondrial membrane potential. Knockout BAT had increased fatty acid uptake, but decreased lipolysis, resulting in increased lipid content and larger lipid droplets. RNAseq revealed 424 dysregulated BAT genes in knockout mice, which mostly segregated by differences during fasting (92 genes) or re-feeding (328 genes). The mRNA of only 4 genes differed from WT during both conditions. Sixty % of the genes had confirmed RARE. One of the genes whose mRNA had changed during both fasting and re-feeding encoded the serum Rbp (i.e. rbp4), which increased 2 to 3-fold, indicating rdh1 suppression of rbp4 and rbp4 promotion of adiposity. The mRNA of the GTPase, gbp2b, increased 200 to 400-fold. Over-expression of gbp2b in immortalized brown adipocytes recapitulated accumulation of larger lipids droplets. WT BAT cells showed a transient increase in ATRA 4 hr after re-feeding, absent in rdh1-null BAT. Rdh1-null BAT had increased dhrs9 and aldh1a1 (encodes Raldh1) mRNA during fasting, and decreased cyp26b1 mRNA, as apparent compensation for rdh1 loss. These data demonstrate a crucial purpose of Rdh1 in generating ATRA to maintain BAT function, not duplicated by other Rdh. These data also demonstrate the pervasive function of ATRA in normalizing BAT function through regulation of multiple genes in an energy status-specific manner. Rdh1 loss impairs glucose tolerance and causes insulin resistance, due to increased adiposity, reminiscent of metabolic syndrome.
Mouse kidney and skin have the highest rdh1 mRNA (Krois et al., 2019). Accordingly, the human ortholog rdh16 contributes to ATRA biosynthesis in human skin (Pavez Loriè, Chamcheu, Vahlquist, & Törmä, 2009; Pavez Loriè, Li, Vahlquist, & Törmä, 2009). Keratinocyte differentiation increased rdh16 expression, while reducing cyp26b1 expression (Jurukovski et al., 1999). ATRA treatment of organotypic epidermis reduces rdh16 mRNA expression by 80%. These data show that rdh16 contributes to keratinocyte differentiation and skin function.
Nerve growth factor (NGF) up regulates rdh2 expression in primary hepatocytes isolated from Sprague-Dawley rats and rdh1 expression in cholestatic mouse livers (Kao et al., 2018). NGF functions via an NF-κB pathway to protect against oxidative damage. These data suggest that NGF intensifies the anti-fibrotic and anticarcinogenic effects of ATRA by inducing its biosynthesis. The authors suggested that rdh16 could serve as a biomarker of cholestatic liver disease.
Alcohol-associated hepatocarcinogenesis down regulates rdh16 through promoter DNA methylation (Udali et al., 2015). Most human hepatocarcinoma samples and cell lines have down-regulated rdh16 mRNA and protein. Down-regulation associates with marginal survival, because rdh16 generates ATRA to suppress cell growth and division (Zhu et al., 2019).
Rdh10 Functions
Rdh10 was identified as an Rdh/sdr that contributes to 11-cis-retinal biosynthesis in the retinal pigment epithelium and all-trans-retinal biosynthesis in Muller cells of the retina. Rdh10 interacts with the retinoid binding-protein cellular retinal binding-protein (Cralbp) of the retinal pigment epithelium, effecting direct transfer of 11-cis-retinal (B. X. Wu et al., 2002, 2004; Farjo, Moiseyev, Takahashi, Crouch, & Ma, 2009). Subsequently, a contribution of Rdh10 to generating retinal for ATRA biosynthesis was demonstrated during mid-gestation (Cammas, Romand, Fraulob, Mura, & Dollé, 2007; Romand, Kondo, Cammas, Hashino, & Dollé, 2008; Siegenthaler et al., 2009; Ashique et al., 2012), and during pancreas organogenesis (Arregi et al., 2016). The next stage involved delineating Rdh10 postnatal functions, which involve generating retinal for ATRA biosynthesis in multiple organs (Parker & Crouch, 2010).
Post-natal, non-ocular Rdh10 functions
Spermatogenesis relies on Rdh10 in both Sertoli and germ cells (Tong, Yang, Davis, & Griswold, 2013). Knocking out rdh10 in Sertoli and germ cells causes testicular atrophy and prevents spermatogenesis in juvenile mice, due to ATRA absence. The effect is temporary. Males less than 7-weeks-old have impaired fertility, but males older than 9-weeks are fertile. This phenomenon resembles the Rdh10 contribution to embryogenesis, i.e. its total absence has a devastating effect exerted during a limited time frame. Retinal supplementation during this time (embryo day 7.5 through 11.5) allows production of mice in Mendelian frequency, but with distal limb abnormalities and atypical physical behavior (Rhinn, Schuhbaur, Niederreither, & Dollé, 2011)
A subset of mouse and human dendritic cells express rdh10 (along with aldh1a2, which encodes Raldh2) (Gyöngyösi et al., 2013). These cells generate ATRA to promote T-cell specification. These include human monocyte-derived dendritic cells and mouse CD103(+), granulocyte-macrophage colony-stimulating factor, and interleukin-4-treated bone marrow-derived dendritic cells. Thus, immune cells express both rdh10 and dhrs9 (see below).
Both visceral and subcutaneous fat express rdh10 (Sima, Manolescu, & Bhat, 2011). Mice with a heterozygote whole-body rdh10 knockout revealed the contribution of Rdh10 to adipose function (Yang et al., 2018). Although born in Mendelian frequency, these mice present with metabolic abnormalities of white fat, liver, muscle and bone, when fed a high-fat diet. By 16-weeks-old, male heterozygotes weigh >5.3 g more than controls (~14%) and females weigh >4.7 g more than controls (~18%). Fat accounted for the difference in weight. The increased weight led to decreased glucose tolerance and increased insulin resistance. Inguinal (subcutaneous), perirenal (visceral) and epidydymal (visceral) fat pads were enlarged, inflamed and fibrotic. Males suffer greater hepatic steatosis than controls, but females did not. In contrast, females have increased femur marrow adipocyte formation, clustered near the growth plate, regardless of a high or low fat diet, but males did not. These data illustrate the importance of Rdh10 to overall metabolic health, and show that modestly decreased ATRA has profound metabolic effects. This model provides opportunity to study physiological actions of moderately decreased endogenous ATRA. Studying decreases in endogenous ATRA avoids complications and artifacts caused by dosing ATRA, because of its hormesis properties.
Rdh10 has been reported as a major Rdh in skin (Lee, Belyaeva, Wu, & Kedishvili, 2011). This conclusion was based on over-expression, rather than on examination of endogenous Rdhs. It is now clear that Rdh16, Dhrs9, RdhE2 and E2S account for physiological generation of retinal for ATRA biosynthesis in skin (see below).
Rdh10 and cancer
Grade 3, stage III non-small-cell lung tumors have mutated rdh10 (Bankovic et al., 2010). This indicates a contribution of low Rdh10 and low ATRA to cancer promotion and progression. In a different approach, a computational strategy ranked large numbers (210-220) of genes vis-a-via their contribution to prostate cancer (Nim, Furtado, Ramialison, & Boyd, 2017). The results revealed that patients with an 85% decrease in rdh10, among other genes, had a mean survival rate of ~63 months compared to 150 months for patients without alterations.
Dhrs9 contributions to ATRA biosynthesis
Dhrs9 has been cloned by multiple labs and has been christened 3α-HSD, Rdh15, Rdh-TBE, eRoldh, Rodh-E2, RdhE and RdhL (Chetyrkin, Belyaeva, Gough, & Kedishvili, 2001; Soref et al., 2001; Rexer & Ong, 2002; Nedialka G. Markova, Pinkas-Sarafova, Karaman-Jurukovska, Jurukovski, & Simon, 2003). Human dhrs9 now has the designation Sdr9C4. Dhrs9 has activity with all-trans-retinol and various androgens (Soref et al., 2001; Chetyrkin et al., 2001). Early publications reported it as an androgen-related SDR without activity towards retinol, because of “low” activity with retinol. The low retinol activity, however, resulted from evaluation of dhrs9 (3α-HSD) expressed in Sf9 cells, which greatly reduced its activity, as Sf9 cells do for Rdh10. Subsequent publications showed activity with free retinol and with Crbp1-bound retinol, identifying Dhrs9 (eRoldh, Rodh-E2) as an Rdh (Rexer & Ong, 2002; Nedialka G. Markova et al., 2003).
Dhrs9, as with rdh1 and rdh10, has widespread tissue expression, especially in epithelial tissues. Research has focused on its functions in hair and skin, as a tumor suppressor, and as an immune stimulant. Significantly, Dhrs9 (hRoDH-E2) may also “moonlight” as a transcription repressor (Nelli G. Markova, Pinkas-Sarafova, & Simon, 2006). Dhrs9 interacts with Sp1, converting it from an activator to a repressor, in an ATRA independent manner. Affirmation of this ability was provided by the observation that knockdown of dhrs9 in astrocytes increased ATRA synthesis ~40%, by increasing aldh1a1 (encodes Raldh1) expression (C. Wang, Kane, & Napoli, 2011). This regulatory cross-talk provides a mechanism for controlling ATRA biosynthesis.
Dhrs9 in hair and skin
The hair follicle, sebaceous gland, and interfollicular epidermis of mice each express a complete retinoid metabolon (pathway of ATRA biosynthesis) and signaling apparatus. The components include Dhrs9 (aka eRodh), Raldh1,2,3 (aldh1a1,2,3), Crbp1, Crabp2 and Rarα (Everts, Sundberg, King, & Ong, 2007). Expression varies with the hair cycle, indicating spatial and temporal regulation of ATRA biosynthesis. Components were not always expressed in the same cell layer, consistent with epithelial-mesenchymal interactions in ATRA synthesis and actions. Focal alopecia in C57BL/6J mice resembles the human disease central centrifugal cicatrical alopecia. In these mice and humans, an increase in dhrs9 expression occurred in mild disease, but not in severe disease (Everts et al., 2013). In alopecia areata in mice (C3H/HeJ), rat and human genes of the ATRA biosynthetic metabolon increased, whereas genes of ATRA catabolism decreased (Duncan et al., 2013). Dhrs9 increased in mice, but not in humans. Overall, however, ATRA concentrations increased in the disease, but restricting vitamin A resulted in more severe disease. This phenomenon is consistent with the hormesis properties of vitamin A/ATRA; precise amounts of ATRA support vitamin A function in a restricted “goldilocks” concentration zone.
Dhrs9 and tumor progression
Human colon adenomas and carcinomas, caused by the adenomatous polyposis coli gene mutation, have reduced dhrs9 (aka RDHL) (Jette et al., 2004). Multiple colon carcinoma cell lines also have reduced dhrs9 expression. Reduced dhrs9 expression associates with reduced ATRA biosynthesis, leading to inhibited colonocyte differentiation. A second study demonstrated that human colorectal cancer samples have propensity to down-regulate dhrs9 relative to normal tissue (Hu et al., 2016). Decreased dhrs9 expression associates with increased lymph node metastasis, disease recurrence, and death.
Reduced proliferation of human renal cell carcinoma can be achieved with treatments that increase dhrs9 expression (Tavares, Nanus, Yang, & Gudas, 2008). Conversely, loss of dhrs9 expression associates with Burkitt lymphomas, T-cell acute lymphoblastic leukemia, and acute myeloid leukemia (Limongi, Curatolo, Pelliccia, & Rocchi, 2005; Meester-Smoor et al., 2008). Reduced dhrs9 expression occurs in >69% of patients with oral squamous cell carcinoma, and correlates with relatively poor prognosis (Shimomura, Sasahira, Nakashima, Shimomura-Kurihara, & Kirita, 2018).
Dhrs9-driven differentiation is not always beneficial. Differentiated epithelial cells allow lytic Epstein-Barr virus replication, but undifferentiated cells do not. Gastric carcinomas and Epstein-Barr positive Burkitt lymphomas have increased dhrs9 expression, increasing ATRA biosynthesis (Jones, Dickerson, Bhende, Delecluse, & Kenney, 2007). Enhanced ATRA-driven differentiation would enhance viral replication. Dhrs9 and immunity. Immune cells express dhrs9, although reports disagree as to the specific cells involved. One report concluded that macrophages do not express dhrs9, whereas granulocyte-macrophage colony-stimulating factor-derived dendritic cells do (Kim et al., 2019). Dendritic cells would secrete ATRA to resist pathogenesis in M. tuberculosis-infected macrophages. Contrary reports find that a subset of monocyte-derived macrophages, human regulatory macrophages, express dhrs9 specifically (Riquelme et al., 2017; Riquelme & Hutchinson, 2018). The secreted ATRA would contribute to Treg development.
RdhE2 and E2S (retinol dehydrogenases epidermal 2 and 2 similar)
RdhE2 and RdhE2S function as Rdh when expressed in HEK293 cells; RdhE2S has ~5-fold higher activity (L. Wu et al., 2019). Retinol and ATRA down-regulate RdhE2 mRNA in a skin raft culture model of stratified human epidermis (Adams, Lee, Belyaeva, Wu, & Kedishvili, 2017). A double knockout lacking both E2 and E2S produces offspring in a Mendelian ratio. Rdh activity of microsomes and mitochondria from double knockout skin decreases by ~10-fold and ~6-fold, respectively, but no phenotype develops when mice are fed a diet with 24 IU vitamin A/G—a high amount. The retinoid metabolon compensated for the dual loss by up-regulating rdh10, dhrs9, crabp2, and aldh1a2 (encodes Raldh2), and down regulating cyp26b1. When fed a vitamin A-deficient diet, the dual knockout has larger sebaceous glands and longer hair shafts, suggesting disrupted growth. These data show that RdhE2 and RdhE2s have a non-critical function in embryogenesis, but moderate the hair growth cycle post-nataly. Thus, like Rdh1, RdhE2 and E2s seem especially important for post-natal ATRA biosynthesis.
Other Rdh
SRP-35 has been reported as a Rdh of particular importance in muscle and adipose (Treves et al., 2012). The enzymatic characterization, however, does not support this claim unequivocally.
A cautionary note
The NCBI gene bank confuses Dhrs9 and RdhE2 as the same SDR. Confusion extends to declaring identity between Rdh16 and Rdh5, and Rdh16 and Rdh6 and Crad1. It is likely that there occurs further NCBI gene bank mis-identification of retinoid associated sdr.
Retinal reductases (Rrd) and retinoid metabolism
The reductase Dhrs3 (mouse and human retSDR1, hSdr16C1, hRdh17)
Dhrs3 initially was identified as retSDR1 in cone photoreceptors (Haeseleer, Huang, Lebioda, Saari, & Palczewski, 1998). It has wide-spread tissue expression, however. Knockout mice provide insight into essential extra-retinal function (Billings et al., 2013). Dhrs3 ablation increases embryonic ATRA amounts by ~40%, while decreasing retinol and retinyl esters by ~60 and 55%, respectively. Embryo death late in gestation of knockout mice, from multiple defects in heart development, illustrate Dhrs3 importance in controlling ATRA concentrations. These data show that Rrd contribute crucially to controlling ATRA concentrations, by controlling retinal produced from β-carotene and/or from retinol.
Even though Dhrs3 contributes essentially to ATRA homeostasis, it does not recognize retinal as its only substrate (Lundová et al., 2015). Dhrs3 also uses as substrates androstenedione, estrone, glyceraldehyde and multiple xenobiotics. In this respect, it resembles activities of other Rdh/sdr.
Interestingly, Dhrs3 heterodimerizes with Rdh10, when both are over-expressed in mammalian cells (Adams, Belyaeva, Wu, & Kedishvili, 2014). In contrast, Rdh10 does not form a heterodimer with the Rrd Rdh11. Dimerization of Dhrs3 and Rdh10 enhances both activities. This may seem counterintuitive, because the net effect might provide futile cycling between retinol and retinal. The net outcome, however would be determined by substrate availability. In other words, an influx of retinal, from β-carotene cleavage, would drive the reaction to retinol production. A decrease in ATRA would drive the reaction to retinal production by release of retinol from RE and irreversible retinal conversion into ATRA.
Dhrs3 and cancer
Neuroblastomas that regress spontaneously, rather than progress to fatality, overexpress dhrs3 (and cyp26a1) (Kamei et al., 2009). Ironically, over-expression of these two genes should decrease ATRA. This seems counterintuitive because ATRA generally reduces tumor proliferation and induces differentiation. Perhaps, overexpression represents compensation for enhanced expression of ATRA biosynthetic enzymes, or countermands ATRA biosynthesis in cells in which ATRA prompts proliferation by activating Pparδ. A different study concluded that dhrs3 activated by p53 and p63 suppressed tumor formation (Kirschner, Rother, Müller, & Engeland, 2010). These contradictory reports illustrate the problem with basing insight into ATRA functions by assessing only one (or two) gene(s) of the retinoid metabolon, and not quantifying ATRA.
An uncertain contribution of Rdh13 (Pan2)
Rdh13 (aka pan2) has widespread tissue expression (Belyaeva & Kedishvili, 2002). Although more active in vitro in reduction than in dehydrogenation, this is a common characteristic of dehydrogenases. Cofactor concentrations and enzymes that remove product determine the direction in vivo. Moreover, Rdh13 kinetic constants were determined with enzyme expressed in Sf9 cells, which often decreases Sdr activities extensively. Until activity is assessed in intact cells and it is ablated, its contribution to retinoid homeostasis remains uncertain.
The reductase Dhrs4 (mouse Rrd; human 2,4-dienoyl-CoA reductase)
Dhrs4 has been characterized as a peroxisomal retinal reductase, expressed by multiple tissues (Lei, Chen, Zhang, & Napoli, 2003). In contrast to Rdh1, which only generates retinal from retinol, when expressed in CHO-K1 cells, Dhrs4 only reduces retinal to retinol. This observation was reproduced in CHO cell lysates. Dhrs4 functions in the presence of a 2-fold molar excess of Crbp1/retinal, but holo-Crbp1 and apo-Crbp1 compete with each other. Thus, the ratio of holo-Crbp1/apoCrbp1 controls the amount of retinal available for ATRA biosynthesis. With higher amounts of apo-Crbp1, i.e. lower amounts of cellular retinol, retinal reduction is inhibited to direct flux of retinal into ATRA.
An enzymatic lesson was learned with Dhrs9. Assessment of only one time point or temperature indicated that Dhrs9 appeared to function as a dehydrogenase. But the amount of retinal produced did not increase with increasing temperature or time. Indeed, similar amounts of “product” were produced in a zero time control. These data indicate that “product” formation was not enzymatic in the dehydrogenation direction. In contrast, reduction of retinal was protein-, time-, and temperature-dependent. These data illustrate the importance of evaluating enzymes under varying conditions, in accordance with Michaelis-Menten kinetics.
The reductase Rdh11 (mouse and human RalR1, Psdr1 Scald and human Sdr7C1)
Rdh11 initially was identified as a Rrd in the visual cycle (Parker & Crouch, 2010). Rdh11, however, has widespread extra-ocular expression in the mouse and human. The knockout mouse has no identified phenotype, but shows lower reduction rates of retinal in liver and testes, and impaired generation of retinol from retinal derived from β-carotene. Reduced dietary vitamin A accentuate differences in tissue retinal. These data suggest Rdh11 contributes to retinoid homeostasis during vitamin A dietary insufficiency, but the lack of an identified phenotype restricts insight into its functional contribution to retinoid homeostasis.
Regulation of ATRA tissue concentrations
Disruption of FoxO1 lowers expression of dhrs9 in hepatocytes (Shin et al., 2012). This observation begged the question whether FoxO1 regulates ATRA concentrations. Based on the contribution of ATRA to controlling energy balance and adiposity, it would make sense if energy status regulated ATRA concentrations. Indeed, fasting increases FoxO1 presence in the nucleus, and FoxO1 induces transcription of rdh1 and rdh10 in liver, which leads to increased ATRA biosynthesis (Obrochta, Krois, Campos, & Napoli, 2015). Fasting also enhances glucagon and cortisol secretion, which inhibit the ATRA catabolic enzyme Cyp26a1. Inhibition of Cyp26a1 prevents futile cycling, because ATRA is a potent inducer of cyp26a1 transcription. Re-feeding decreases the liver concentration of ATRA by ~50%. Glucose gavage and insulin recapitulates this phenomenon. Insulin functions by causing phosphorylation of FoxO1, which excludes it from the nucleus. These data reveal a mechanism for regulating the opposing effects of ATRA and insulin on energy use, and suggest a nexus between ATRA and insulin-signaling related diseases (Fig. 3).
Figure 3.
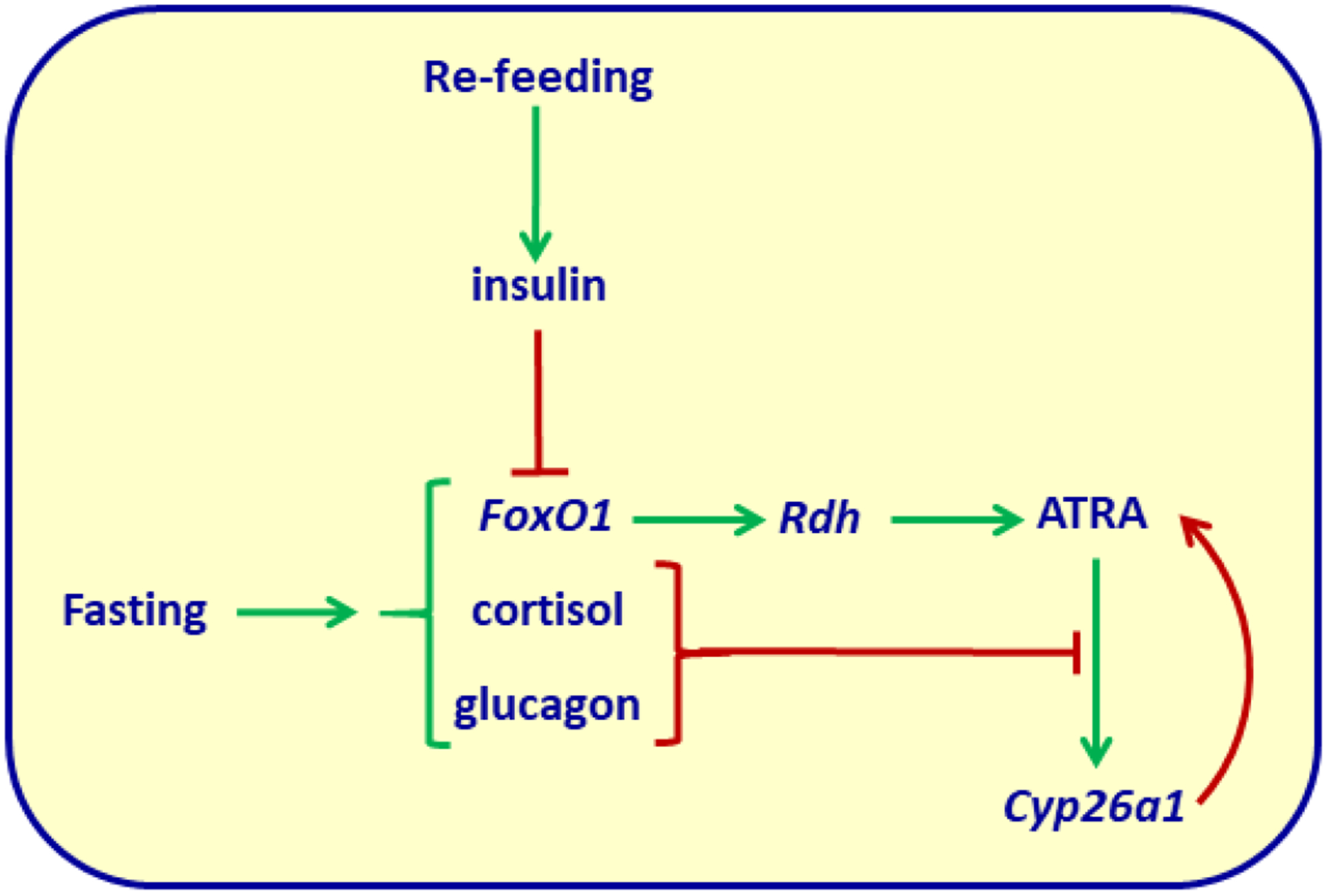
Regulation of liver ATRA concentrations by fasting and re-feeding. Green arrows indicate stimulation/increased secretion; red arrows indicate inhibition, or in the case of the red arrow extending from Cyp26a1 to ATRA, catabolism. Fasting activates FoxO1, which induces rdh transcription to increase ATRA biosynthesis. Concurrently, cortisol and glucagon ameliorate cyp26a1 transcription, which ATRA induces, to reduce ATRA catabolism. Re-feeding stimulates insulin secretion, which excludes FoxO1 from the nucleus. This reduces rdh transcription and accelerates rdh mRNA degradation. Re-feeding also reduces cortisol and glucagon secretion, allowing maximum cyp26a1 expression to catabolize ATRA. Thus, during fasting ATRA concentrations exceed those during re-feeding. ATRA concentrations inverse to insulin, optimize the contrary actions of each hormone.
Observation of insulin regulation of ATRA biosynthesis has been extended. A tissue bank of diabetic transgenic pigs (INSC94Y) and their WT littermates has been established in Munich: the Munich MIDY (mutant insulin gene-induced diabetes of youth). Livers from the MIDY pigs have a “marked” increase in rdh16 (pig designation for mouse rdh1), which associates with an ~2.5-fold increase in ATRA (Backman et al., 2019).
The importance of the reciprocal relationship between insulin and ATRA lies in their opposing actions. Whereas insulin stimulates lipogenesis, ATRA promotes fatty acid oxidation; insulin retards lipolysis, ATRA induces lipolysis; insulin arrests gluconeogenesis, ATRA stimulates gluconeogenesis. It is reasonable to conclude that the insulin-driven decrease in ATRA augments insulin actions. On the other hand, the FoxO1 increase in ATRA would augment adaptation to energy use caused by fasting.
The multiplicity of enzymes in the retinoid metabolon
Tissues, and often the same cells, co-express multiple rdh, rrd and aldh1a; gene ablations reveal different phenotypes for each, consistent with non-redundant functions. Yet, ablation of any one often prompts compensation by others, consistent with redundant functions in some locations. For example, astrocytes express rdh1, dhrs9, rdh10 and all three aldh1a (C. Wang et al., 2011). Knocking down each one decreases ATRA biosynthesis, with the exception of dhrs9 (see above). These data suggest both redundancy and differing metabolons generating ATRA for different functions. The foregoing discussion strongly supports this specificity involved in ATRA biosynthesis.
Alcohol dehydrogenases
Adh have been postulated to contribute to ATRA biosynthesis for decades, but the totality of data indicate no physiological contribution of Adh to ATRA biosynthesis (see above). The supposed Adh contribution to ATRA biosynthesis has prompted the suggestion that ethanol inhibits ATRA biosynthesis, thus accounting (in part) for ethanol toxicity and fetal alcohol spectrum disease. As discussed above, ethanol does not inhibit Rdh. In fact, the converse occurs (McCaffery et al., 2004; Kane, Folias, Wang, & Napoli, 2010; Napoli, 2011). Acute ethanol dosing increases ATRA in adult hippocampus, liver, and testis by ~2-fold. Dosing dams with 6.5% ethanol from embryonic day 13 to 19 increases ATRA in fetal hippocampus up to 20-fold and in fetal cortex up to 50-fold, depending on the dam blood alcohol content. Chronic ethanol dosing increases ATRA in adult hippocampus 20-fold, cortex 2-fold, testis 2-fold, and serum 10-fold. Tissue-specific increases in rdh mRNAs and Rdh activities, extrahepatic retinol concentrations, and ATRA catabolism combine to produce site-specific effects. Moreover, ethanol stimulates stem cell differentiation by increasing retinol cell uptake and conversion into ATRA to activate RARγ-dependent transcription (Serio, Laursen, Urvalek, Gross, & Gudas, 2019). Thus, the hypothesis has been disproved that an aspect of fetal alcohol spectrum disease involves inhibition of ATRA biosynthesis or function.
Retinal dehydrogenases
The retinal dehydrogenases (Raldh) that catalyze irreversible conversion of retinal into ATRA belong to the aldh1a gene family. They are Raldh1 (aldh1a1), Raldh2 (aldh1a2) and Raldh3 (aldh1a3).
Raldh1 was the first retinal dehydrogenase determined unequivocally as a physiological contributor to ATRA biosynthesis (Posch, Burns, & Napoli, 1992). This Raldh was identified and distinguished from other cytosolic Aldh, some of which had been postulated to serve in ATRA biosynthesis based on in vitro activity with retinal, along with many other substrates. To clarify which Aldh contributed to ATRA biosynthesis, retinal was generated in situ in microsomes (a source of most Rdh) from retinol complexed with Crbp1. Cytosolic fractions (source of Aldh) were added to determine which would generate ATRA from holo-Crbp1-produced retinal. This experiment identified the first Raldh. These data were re-enforced by experiments that showed direct access of Raldh1 to retinal bound with Crbp1 (Ottonello, Scita, Mantovani, Cavazzini, & Rossi, 1993; Penzes, Wang, & Napoli, 1997). In contrast to Raldh2, apo-Crbp1 inhibits Raldh1. This suggests primacy of other Raldh in generating ATRA. In fact, the aldh1a1 knockout has decreased ATRA and increased retinal in liver, but generally not in parametrical, epidydymal or femoral fat in either males or females (Yang et al., 2017). This can be explained by the nature of Raldh1. As an enzyme it has multiple substrates, and as a protein, it has multiple non-enzymatic functions (Grünblatt & Riederer, 2016). Thus, it is not reasonable to attribute all Raldh actions to ATRA generation.
Aldh1a1-knock out mice, in utter disagreement with ATRA actions and rdh knockout phenotypes, resist adiposity compared to WT controls. One lab attributed this to Raldh1 function as a retinal dehydrogenase, and concluded that an increase in retinal in the knockout prevented adiposity in females, and low concentrations of ATRA promote adiposity (Ziouzenkova et al., 2007). The data are overwhelming, however, that restriction of vitamin A or ATRA enhances adiposity (Berry et al., 2012; Blaner, 2019). A more reasonable interpretation would have taken into account the multiple Raldh1 functions. A re-examination of the phenotype showed that Raldh1 functions as a retinoid-independent promoter of adiposity only during adolescence in both males and females, through pre-adipocyte cell autonomous actions (Yang et al., 2017). Raldh1, therefore, acts as a retinal dehydrogenase only in select tissues and cells, not universally, and not likely in white adipose.
Upon identification as a testes Raldh, aldh1a2 (Raldh2) was cloned, expressed in E. coli and characterized, and subsequently cloned from embryonic cells (X. Wang, Penzes, & Napoli, 1996; Zhao et al., 1996). As for Raldh1, the purified recombinant enzyme produces ATRA from retinal generated in situ by microsomes from Crbp1-retinol. Equivalent rates with unbound retinal and in the presence of a 2:1 ratio of Crbp1/retinal confirm that Raldh2 can access retinal bound to Crbp1. In primary astrocytes, Raldh2 localizes most intensely in the perinuclear regions, whereas Raldh1 localizes to both cytosol and nuclei. These differences suggest that multiple Raldh exist to generate ATRA in specific areas of the cell, perhaps to support different vitamin A functions. Nuclear localization of Raldh1 also suggests activity in gene regulation.
Raldh3 (aka Aldh6), which also accesses retinal bound with Crbp1, completes the complement of known Aldh that contribute to ATRA biosynthesis (Grün, Hirose, Kawauchi, Ogura, & Umesono, 2000; Arnold et al., 2015).
Raldh and disease
Complex connections occur among Raldh and diseases. This undoubtedly reflects the multifunctional nature of Raldh1, and perhaps other Raldh. For example, over expression of Raldh1 in colon macrophages, through generating ATRA, was postulated to increase inflammation in Crohn’s disease (Sanders et al., 2014). This work did not quantify ATRA, but relied on estimation of Raldh1 activity by Aldefluor assay. This overlooks the multiple substrates metabolized by Raldh1 and its multiple functions. In other words, it does not provide any evidence of ATRA involvement in exacerbating Crohn’s disease. Raldh1 seems to confer multi-drug resistance to cancer (Kozovska et al., 2018; Chefetz et al., 2019). The latter may explain frequent association of Raldh1 with cancer progression, not its ability to generate ATRA in some loci. In contrast, Raldh2 contributes to ATRA biosynthesis to trigger an antimicrobial response in tuberculosis: low expression of Raldh2 in lung characterizes the disease (Kim et al., 2019).
References
- Adams MK, Belyaeva OV, Wu L, & Kedishvili NY (2014). The retinaldehyde reductase activity of DHRS3 is reciprocally activated by retinol dehydrogenase 10 to control retinoid homeostasis. The Journal of Biological Chemistry, 289(21), 14868–14880. 10.1074/jbc.M114.552257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams MK, Lee S-A, Belyaeva OV, Wu L, & Kedishvili NY (2017). Characterization of human short chain dehydrogenase/reductase SDR16C family members related to retinol dehydrogenase 10. Chemico-Biological Interactions, 276, 88–94. 10.1016/j.cbi.2016.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold SLM, Kent T, Hogarth CA, Griswold MD, Amory JK, & Isoherranen N (2015). Pharmacological inhibition of ALDH1A in mice decreases all-trans retinoic acid concentrations in a tissue specific manner. Biochemical Pharmacology, 95(3), 177–192. 10.1016/j.bcp.2015.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arregi I, Climent M, Iliev D, Strasser J, Gouignard N, Johansson JK, … Pera EM (2016). Retinol Dehydrogenase-10 Regulates Pancreas Organogenesis and Endocrine Cell Differentiation via Paracrine Retinoic Acid Signaling. Endocrinology, 157(12), 4615–4631. 10.1210/en.2016-1745 [DOI] [PubMed] [Google Scholar]
- Ashique AM, May SR, Kane MA, Folias AE, Phamluong K, Choe Y, … Peterson AS (2012). Morphological defects in a novel Rdh10 mutant that has reduced retinoic acid biosynthesis and signaling. Genesis (New York, N.Y.: 2000), 50(5), 415–423. 10.1002/dvg.22002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backman M, Flenkenthaler F, Blutke A, Dahlhoff M, Ländström E, Renner S, … Wolf E (2019). Multi-omics insights into functional alterations of the liver in insulin-deficient diabetes mellitus. Molecular Metabolism, 26, 30–44. 10.1016/j.molmet.2019.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankovic J, Stojsic J, Jovanovic D, Andjelkovic T, Milinkovic V, Ruzdijic S, & Tanic N (2010). Identification of genes associated with non-small-cell lung cancer promotion and progression. Lung Cancer (Amsterdam, Netherlands), 67(2), 151–159. 10.1016/j.lungcan.2009.04.010 [DOI] [PubMed] [Google Scholar]
- Belyaeva OV, & Kedishvili NY (2002). Human pancreas protein 2 (PAN2) has a retinal reductase activity and is ubiquitously expressed in human tissues. FEBS Letters, 531(3), 489–493. 10.1016/s0014-5793(02)03588-3 [DOI] [PubMed] [Google Scholar]
- Berry DC, DeSantis D, Soltanian H, Croniger CM, & Noy N (2012). Retinoic acid upregulates preadipocyte genes to block adipogenesis and suppress diet-induced obesity. Diabetes, 61(5), 1112–1121. 10.2337/db11-1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billings SE, Pierzchalski K, Butler Tjaden NE, Pang X-Y, Trainor PA, Kane MA, & Moise AR (2013). The retinaldehyde reductase DHRS3 is essential for preventing the formation of excess retinoic acid during embryonic development. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology, 27(12), 4877–4889. 10.1096/fj.13-227967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaner WS (2019). Vitamin A signaling and homeostasis in obesity, diabetes, and metabolic disorders. Pharmacology & Therapeutics, 197, 153–178. 10.1016/j.pharmthera.2019.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boerman MH, & Napoli JL (1995). Characterization of a microsomal retinol dehydrogenase: A short-chain alcohol dehydrogenase with integral and peripheral membrane forms that interacts with holo-CRBP (type I). Biochemistry, 34(21), 7027–7037. 10.1021/bi00021a014 [DOI] [PubMed] [Google Scholar]
- Boerman MH, & Napoli JL (1996). Cellular retinol-binding protein-supported retinoic acid synthesis. Relative roles of microsomes and cytosol. The Journal of Biological Chemistry, 271(10), 5610–5616. [DOI] [PubMed] [Google Scholar]
- Cammas L, Romand R, Fraulob V, Mura C, & Dollé P (2007). Expression of the murine retinol dehydrogenase 10 (Rdh10) gene correlates with many sites of retinoid signalling during embryogenesis and organ differentiation. Developmental Dynamics: An Official Publication of the American Association of Anatomists, 236(10), 2899–2908. 10.1002/dvdy.21312 [DOI] [PubMed] [Google Scholar]
- Chai X, Boerman MH, Zhai Y, & Napoli JL (1995). Cloning of a cDNA for liver microsomal retinol dehydrogenase. A tissue-specific, short-chain alcohol dehydrogenase. The Journal of Biological Chemistry, 270(8), 3900–3904. [DOI] [PubMed] [Google Scholar]
- Chai X, Zhai Y, Popescu G, & Napoli JL (1995). Cloning of a cDNA for a second retinol dehydrogenase type II. Expression of its mRNA relative to type I. The Journal of Biological Chemistry, 270(47), 28408–28412. [DOI] [PubMed] [Google Scholar]
- Chefetz I, Grimley E, Yang K, Hong L, Vinogradova EV, Suciu R, … Buckanovich RJ (2019). A Pan-ALDH1A Inhibitor Induces Necroptosis in Ovarian Cancer Stem-like Cells. Cell Reports, 26(11), 3061–3075.e6. 10.1016/j.celrep.2019.02.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetyrkin SV, Belyaeva OV, Gough WH, & Kedishvili NY (2001). Characterization of a novel type of human microsomal 3alpha -hydroxysteroid dehydrogenase: Unique tissue distribution and catalytic properties. The Journal of Biological Chemistry, 276(25), 22278–22286. 10.1074/jbc.M102076200 [DOI] [PubMed] [Google Scholar]
- Duncan FJ, Silva KA, Johnson CJ, King BL, Szatkiewicz JP, Kamdar SP, … Everts HB (2013). Endogenous retinoids in the pathogenesis of alopecia areata. The Journal of Investigative Dermatology, 133(2), 334–343. 10.1038/jid.2012.344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everts HB, Silva KA, Montgomery S, Suo L, Menser M, Valet AS, … Sundberg JP (2013). Retinoid metabolism is altered in human and mouse cicatricial alopecia. The Journal of Investigative Dermatology, 133(2), 325–333. 10.1038/jid.2012.393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everts HB, Sundberg JP, King LE, & Ong DE (2007). Immunolocalization of enzymes, binding proteins, and receptors sufficient for retinoic acid synthesis and signaling during the hair cycle. The Journal of Investigative Dermatology, 127(7), 1593–1604. 10.1038/sj.jid.5700753 [DOI] [PubMed] [Google Scholar]
- Farjo KM, Moiseyev G, Takahashi Y, Crouch RK, & Ma J (2009). The 11-cisretinol dehydrogenase activity of RDH10 and its interaction with visual cycle proteins. Investigative Ophthalmology & Visual Science, 50(11), 5089–5097. 10.1167/iovs.09-3797 [DOI] [PubMed] [Google Scholar]
- Gough WH, VanOoteghem S, Sint T, & Kedishvili NY (1998). CDNA cloning and characterization of a new human microsomal NAD+-dependent dehydrogenase that oxidizes all-trans-retinol and 3alpha-hydroxysteroids. The Journal of Biological Chemistry, 273(31), 19778–19785. [DOI] [PubMed] [Google Scholar]
- Grün F, Hirose Y, Kawauchi S, Ogura T, & Umesono K (2000). Aldehyde dehydrogenase 6, a cytosolic retinaldehyde dehydrogenase prominently expressed in sensory neuroepithelia during development. The Journal of Biological Chemistry, 275(52), 41210–41218. 10.1074/jbc.M007376200 [DOI] [PubMed] [Google Scholar]
- Grünblatt E, & Riederer P (2016). Aldehyde dehydrogenase (ALDH) in Alzheimer’s and Parkinson’s disease. Journal of Neural Transmission (Vienna, Austria: 1996), 123(2), 83–90. 10.1007/s00702-014-1320-1 [DOI] [PubMed] [Google Scholar]
- Gyöngyösi A, Szatmari I, Pap A, Dezso B, Pos Z, Széles L, … Nagy L (2013). RDH10, RALDH2, and CRABP2 are required components of PPARγ-directed ATRA synthesis and signaling in human dendritic cells. Journal of Lipid Research, 54(9), 2458–2474. 10.1194/jlr.M038984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeseleer F, Huang J, Lebioda L, Saari JC, & Palczewski K (1998). Molecular characterization of a novel short-chain dehydrogenase/reductase that reduces all-trans-retinal. The Journal of Biological Chemistry, 273(34), 21790–21799. 10.1074/jbc.273.34.21790 [DOI] [PubMed] [Google Scholar]
- Harrison EH (2012). Mechanisms involved in the intestinal absorption of dietary vitamin A and provitamin A carotenoids. Biochimica et Biophysica Acta, 1821(1), 70–77. 10.1016/j.bbalip.2011.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes DP (2007). Nutritional hormesis. European Journal of Clinical Nutrition, 61(2), 147–159. 10.1038/sj.ejcn.1602507 [DOI] [PubMed] [Google Scholar]
- Hu L, Chen H-Y, Han T, Yang G-Z, Feng D, Qi C-Y, … Gao C-F (2016). Downregulation of DHRS9 expression in colorectal cancer tissues and its prognostic significance. Tumour Biology: The Journal of the International Society for Oncodevelopmental Biology and Medicine, 37(1), 837–845. 10.1007/s13277-015-3880-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamison RS, Newcomer ME, & Ong DE (1994). Cellular retinoid-binding proteins: Limited proteolysis reveals a conformational change upon ligand binding. Biochemistry, 33(10), 2873–2879. 10.1021/bi00176a017 [DOI] [PubMed] [Google Scholar]
- Jette C, Peterson PW, Sandoval IT, Manos EJ, Hadley E, Ireland CM, & Jones DA (2004). The tumor suppressor adenomatous polyposis coli and caudal related homeodomain protein regulate expression of retinol dehydrogenase L. The Journal of Biological Chemistry, 279(33), 34397–34405. 10.1074/jbc.M314021200 [DOI] [PubMed] [Google Scholar]
- Jones RJ, Dickerson S, Bhende PM, Delecluse H-J, & Kenney SC (2007). Epstein-Barr virus lytic infection induces retinoic acid-responsive genes through induction of a retinol-metabolizing enzyme, DHRS9. The Journal of Biological Chemistry, 282(11), 8317–8324. 10.1074/jbc.M608667200 [DOI] [PubMed] [Google Scholar]
- Jurukovski V, Markova NG, Karaman-Jurukovska N, Randolph RK, Su J, Napoli JL, & Simon M (1999). Cloning and characterization of retinol dehydrogenase transcripts expressed in human epidermal keratinocytes. Molecular Genetics and Metabolism, 67(1), 62–73. 10.1006/mgme.1999.2840 [DOI] [PubMed] [Google Scholar]
- Kamei N, Hiyama K, Yamaoka H, Kamimatsuse A, Onitake Y, Sueda T, & Hiyama E (2009). Evaluation of genes identified by microarray analysis in favorable neuroblastoma. Pediatric Surgery International, 25(11), 931–937. 10.1007/s00383-009-2448-1 [DOI] [PubMed] [Google Scholar]
- Kane MA, Folias AE, Pingitore A, Perri M, Krois CR, Ryu JY, … Napoli JL (2011). CrbpI modulates glucose homeostasis and pancreas 9-cis-retinoic acid concentrations. Molecular and Cellular Biology, 31(16), 3277–3285. 10.1128/MCB.05516-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane MA, Folias AE, Wang C, & Napoli JL (2010). Ethanol elevates physiological all-trans-retinoic acid levels in select loci through altering retinoid metabolism in multiple loci: A potential mechanism of ethanol toxicity. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology, 24(3), 823–832. 10.1096/fj.09-141572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao Y-H, Lee P-H, Chiu T-C, Lin Y-C, Sun C-K, Chen P-H, & Tsai M-S (2018). Transcriptome analysis reveals a positive role for nerve growth factor in retinol metabolism in primary rat hepatocytes. Cytokine, 107, 74–78. 10.1016/j.cyto.2017.11.018 [DOI] [PubMed] [Google Scholar]
- Kim EW, De Leon A, Jiang Z, Radu RA, Martineau AR, Chan ED, … Liu PT (2019). Vitamin A Metabolism by Dendritic Cells Triggers an Antimicrobial Response against Mycobacterium tuberculosis. MSphere, 4(3). 10.1128/mSphere.00327-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner RD, Rother K, Müller GA, & Engeland K (2010). The retinal dehydrogenase/reductase retSDR1/DHRS3 gene is activated by p53 and p63 but not by mutants derived from tumors or EEC/ADULT malformation syndromes. Cell Cycle (Georgetown, Tex.), 9(11), 2177–2188. 10.4161/cc.9.11.11844 [DOI] [PubMed] [Google Scholar]
- Kozovska Z, Patsalias A, Bajzik V, Durinikova E, Demkova L, Jargasova S, … Matuskova M (2018). ALDH1A inhibition sensitizes colon cancer cells to chemotherapy. BMC Cancer, 18(1), 656 10.1186/s12885-018-4572-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krois CR, Vuckovic MG, Huang P, Zaversnik C, Liu CS, Gibson CE, … Napoli JL (2019). RDH1 suppresses adiposity by promoting brown adipose adaptation to fasting and re-feeding. Cellular and Molecular Life Sciences: CMLS, 76(12), 2425–2447. 10.1007/s00018-019-03046-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S-A, Belyaeva OV, Wu L, & Kedishvili NY (2011). Retinol dehydrogenase 10 but not retinol/sterol dehydrogenase(s) regulates the expression of retinoic acid-responsive genes in human transgenic skin raft culture. The Journal of Biological Chemistry, 286(15), 13550–13560. 10.1074/jbc.M110.181065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Z, Chen W, Zhang M, & Napoli JL (2003). Reduction of all-trans-retinal in the mouse liver peroxisome fraction by the short-chain dehydrogenase/reductase RRD: Induction by the PPAR alpha ligand clofibrate. Biochemistry, 42(14), 4190–4196. 10.1021/bi026948i [DOI] [PubMed] [Google Scholar]
- Limongi ZM, Curatolo A, Pelliccia F, & Rocchi A (2005). Biallelic deletion and loss of expression analysis of genes at FRA2G common fragile site in tumor-derived cell lines. Cancer Genetics and Cytogenetics, 161(2), 181–186. 10.1016/j.cancergencyto.2005.01.018 [DOI] [PubMed] [Google Scholar]
- Lundová T, Zemanová L, Malčeková B, Skarka A, Štambergová H, Havránková J, … Wsól V (2015). Molecular and biochemical characterisation of human short-chain dehydrogenase/reductase member 3 (DHRS3). Chemico-Biological Interactions, 234, 178–187. 10.1016/j.cbi.2014.10.018 [DOI] [PubMed] [Google Scholar]
- Markova Nedialka G., Pinkas-Sarafova A, Karaman-Jurukovska N, Jurukovski V, & Simon M (2003). Expression pattern and biochemical characteristics of a major epidermal retinol dehydrogenase. Molecular Genetics and Metabolism, 78(2), 119–135. 10.1016/s1096-7192(02)00226-3 [DOI] [PubMed] [Google Scholar]
- Markova Nelli G., Pinkas-Sarafova A, & Simon M (2006). A metabolic enzyme of the short-chain dehydrogenase/reductase superfamily may moonlight in the nucleus as a repressor of promoter activity. The Journal of Investigative Dermatology, 126(9), 2019–2031. 10.1038/sj.jid.5700347 [DOI] [PubMed] [Google Scholar]
- McCaffery P, Koul O, Smith D, Napoli JL, Chen N, & Ullman MD (2004). Ethanol increases retinoic acid production in cerebellar astrocytes and in cerebellum. Brain Research. Developmental Brain Research, 153(2), 233–241. 10.1016/j.devbrainres.2004.09.003 [DOI] [PubMed] [Google Scholar]
- Meester-Smoor MA, Janssen MJFW, Grosveld GC, de Klein A, van IJcken WFJ, Douben H, & Zwarthoff EC (2008). MN1 affects expression of genes involved in hematopoiesis and can enhance as well as inhibit RAR/RXR-induced gene expression. Carcinogenesis, 29(10), 2025–2034. 10.1093/carcin/bgn168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercader J, Ribot J, Murano I, Felipe F, Cinti S, Bonet ML, & Palou A (2006). Remodeling of white adipose tissue after retinoic acid administration in mice. Endocrinology, 147(11), 5325–5332. 10.1210/en.2006-0760 [DOI] [PubMed] [Google Scholar]
- Napoli JL (1986). Retinol metabolism in LLC-PK1 Cells. Characterization of retinoic acid synthesis by an established mammalian cell line. The Journal of Biological Chemistry, 261(29), 13592–13597. [PubMed] [Google Scholar]
- Napoli JL (2011). Effects of ethanol on physiological retinoic acid levels. IUBMB Life, 63(9), 701–706. 10.1002/iub.500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napoli JL (2012). Physiological insights into all-trans-retinoic acid biosynthesis. Biochimica Et Biophysica Acta, 1821(1), 152–167. 10.1016/j.bbalip.2011.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napoli JL (2017). Cellular retinoid binding-proteins, CRBP, CRABP, FABP5: Effects on retinoid metabolism, function and related diseases. Pharmacology & Therapeutics, 173, 19–33. 10.1016/j.pharmthera.2017.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomer ME, Jamison RS, & Ong DE (1998). Structure and function of retinoid-binding proteins. Sub-Cellular Biochemistry, 30, 53–80. 10.1007/978-1-4899-1789-8_3 [DOI] [PubMed] [Google Scholar]
- Nim HT, Furtado MB, Ramialison M, & Boyd SE (2017). Combinatorial Ranking of Gene Sets to Predict Disease Relapse: The Retinoic Acid Pathway in Early Prostate Cancer. Frontiers in Oncology, 7, 30 10.3389/fonc.2017.00030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obrochta KM, Krois CR, Campos B, & Napoli JL (2015). Insulin regulates retinol dehydrogenase expression and all-trans-retinoic acid biosynthesis through FoxO1. The Journal of Biological Chemistry, 290(11), 7259–7268. 10.1074/jbc.M114.609313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong DE, MacDonald PN, & Gubitosi AM (1988). Esterification of retinol in rat liver. Possible participation by cellular retinol-binding protein and cellular retinol-binding protein II. The Journal of Biological Chemistry, 263(12), 5789–5796. [PubMed] [Google Scholar]
- Ottonello S, Scita G, Mantovani G, Cavazzini D, & Rossi GL (1993). Retinol bound to cellular retinol-binding protein is a substrate for cytosolic retinoic acid synthesis. The Journal of Biological Chemistry, 268(36), 27133–27142. [PubMed] [Google Scholar]
- Parker RO, & Crouch RK (2010). Retinol dehydrogenases (RDHs) in the visual cycle. Experimental Eye Research, 91(6), 788–792. 10.1016/j.exer.2010.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavez Loriè E, Chamcheu JC, Vahlquist A, & Törmä H (2009). Both all-trans retinoic acid and cytochrome P450 (CYP26) inhibitors affect the expression of vitamin A metabolizing enzymes and retinoid biomarkers in organotypic epidermis. Archives of Dermatological Research, 301(7), 475–485. 10.1007/s00403-009-0937-7 [DOI] [PubMed] [Google Scholar]
- Pavez Loriè E, Li H, Vahlquist A, & Törmä H (2009). The involvement of cytochrome p450 (CYP) 26 in the retinoic acid metabolism of human epidermal keratinocytes. Biochimica Et Biophysica Acta, 1791(3), 220–228. 10.1016/j.bbalip.2008.12.004 [DOI] [PubMed] [Google Scholar]
- Penzes P, Wang X, & Napoli JL (1997). Enzymatic characteristics of retinal dehydrogenase type I expressed in Escherichia coli. Biochimica Et Biophysica Acta, 1342(2), 175–181. 10.1016/s0167-4838(97)00102-7 [DOI] [PubMed] [Google Scholar]
- Pierzchalski K, Yu J, Norman V, & Kane MA (2013). CrbpI regulates mammary retinoic acid homeostasis and the mammary microenvironment. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology, 27(5), 1904–1916. 10.1096/fj.12-219410 [DOI] [PubMed] [Google Scholar]
- Posch KC, Boerman MH, Burns RD, & Napoli JL (1991). Holocellular retinol binding protein as a substrate for microsomal retinal synthesis. Biochemistry, 30(25), 6224–6230. 10.1021/bi00239a021 [DOI] [PubMed] [Google Scholar]
- Posch KC, Burns RD, & Napoli JL (1992). Biosynthesis of all-trans-retinoic acid from retinal. Recognition of retinal bound to cellular retinol binding protein (type I) as substrate by a purified cytosolic dehydrogenase. The Journal of Biological Chemistry, 267(27), 19676–19682. [PubMed] [Google Scholar]
- Rexer BN, & Ong DE (2002). A novel short-chain alcohol dehydrogenase from rats with retinol dehydrogenase activity, cyclically expressed in uterine epithelium. Biology of Reproduction, 67(5), 1555–1564. 10.1095/biolreprod.102.007021 [DOI] [PubMed] [Google Scholar]
- Rhinn M, Schuhbaur B, Niederreither K, & Dollé P (2011). Involvement of retinol dehydrogenase 10 in embryonic patterning and rescue of its loss of function by maternal retinaldehyde treatment. Proceedings of the National Academy of Sciences of the United States of America, 108(40), 16687–16692. 10.1073/pnas.1103877108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riquelme P, Amodio G, Macedo C, Moreau A, Obermajer N, Brochhausen C, … Hutchinson JA (2017). DHRS9 Is a Stable Marker of Human Regulatory Macrophages. Transplantation, 101(11), 2731–2738. 10.1097/TP.0000000000001814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riquelme P, & Hutchinson JA (2018). Novel molecules mediate specialized functions of human regulatory macrophages. Current Opinion in Organ Transplantation, 23(5), 533–537. 10.1097/MOT.0000000000000560 [DOI] [PubMed] [Google Scholar]
- Romand R, Kondo T, Cammas L, Hashino E, & Dollé P (2008). Dynamic expression of the retinoic acid-synthesizing enzyme retinol dehydrogenase 10 (rdh10) in the developing mouse brain and sensory organs. The Journal of Comparative Neurology, 508(6), 879–892. 10.1002/cne.21707 [DOI] [PubMed] [Google Scholar]
- Ross AC (1993). Cellular metabolism and activation of retinoids: Roles of cellular retinoid-binding proteins. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology, 7(2), 317–327. 10.1096/fasebj.7.2.8440409 [DOI] [PubMed] [Google Scholar]
- Sanders TJ, McCarthy NE, Giles EM, Davidson KLM, Haltalli MLR, Hazell S, … Stagg AJ (2014). Increased production of retinoic acid by intestinal macrophages contributes to their inflammatory phenotype in patients with Crohn’s disease. Gastroenterology, 146(5), 1278–1288.e1–2. 10.1053/j.gastro.2014.01.057 [DOI] [PubMed] [Google Scholar]
- Serio RN, Laursen KB, Urvalek AM, Gross SS, & Gudas LJ (2019). Ethanol promotes differentiation of embryonic stem cells through retinoic acid receptor-γ. The Journal of Biological Chemistry, 294(14), 5536–5548. 10.1074/jbc.RA118.007153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura H, Sasahira T, Nakashima C, Shimomura-Kurihara M, & Kirita T (2018). Downregulation of DHRS9 is associated with poor prognosis in oral squamous cell carcinoma. Pathology, 50(6), 642–647. 10.1016/j.pathol.2018.06.002 [DOI] [PubMed] [Google Scholar]
- Shin D-J, Joshi P, Hong S-H, Mosure K, Shin D-G, & Osborne TF (2012). Genome-wide analysis of FoxO1 binding in hepatic chromatin: Potential involvement of FoxO1 in linking retinoid signaling to hepatic gluconeogenesis. Nucleic Acids Research, 40(22), 11499–11509. 10.1093/nar/gks932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegenthaler JA, Ashique AM, Zarbalis K, Patterson KP, Hecht JH, Kane MA, … Pleasure SJ (2009). Retinoic acid from the meninges regulates cortical neuron generation. Cell, 139(3), 597–609. 10.1016/j.cell.2009.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sima A, Manolescu D-C, & Bhat P (2011). Retinoids and retinoid-metabolic gene expression in mouse adipose tissues. Biochemistry and Cell Biology = Biochimie Et Biologie Cellulaire, 89(6), 578–584. 10.1139/o11-062 [DOI] [PubMed] [Google Scholar]
- Soref CM, Di YP, Hayden L, Zhao YH, Satre MA, & Wu R (2001). Characterization of a novel airway epithelial cell-specific short chain alcohol dehydrogenase/reductase gene whose expression is up-regulated by retinoids and is involved in the metabolism of retinol. The Journal of Biological Chemistry, 276(26), 24194–24202. 10.1074/jbc.M100332200 [DOI] [PubMed] [Google Scholar]
- Tavares TS, Nanus D, Yang XJ, & Gudas LJ (2008). Gene microarray analysis of human renal cell carcinoma: The effects of HDAC inhibition and retinoid treatment. Cancer Biology & Therapy, 7(10), 1607–1618. 10.4161/cbt.7.10.6584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong M-H, Yang Q-E, Davis JC, & Griswold MD (2013). Retinol dehydrogenase 10 is indispensible for spermatogenesis in juvenile males. Proceedings of the National Academy of Sciences of the United States of America, 110(2), 543–548. 10.1073/pnas.1214883110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treves S, Thurnheer R, Mosca B, Vukcevic M, Bergamelli L, Voltan R, … Zorzato F (2012). SRP-35, a newly identified protein of the skeletal muscle sarcoplasmic reticulum, is a retinol dehydrogenase. The Biochemical Journal, 441(2), 731–741. 10.1042/BJ20111457 [DOI] [PubMed] [Google Scholar]
- Udali S, Guarini P, Ruzzenente A, Ferrarini A, Guglielmi A, Lotto V, … Friso S (2015). DNA methylation and gene expression profiles show novel regulatory pathways in hepatocellular carcinoma. Clinical Epigenetics, 7, 43 10.1186/s13148-015-0077-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Kane MA, & Napoli JL (2011). Multiple retinol and retinal dehydrogenases catalyze all-trans-retinoic acid biosynthesis in astrocytes. The Journal of Biological Chemistry, 286(8), 6542–6553. 10.1074/jbc.M110.198382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Penzes P, & Napoli JL (1996). Cloning of a cDNA encoding an aldehyde dehydrogenase and its expression in Escherichia coli. Recognition of retinal as substrate. The Journal of Biological Chemistry, 271(27), 16288–16293. 10.1074/jbc.271.27.16288 [DOI] [PubMed] [Google Scholar]
- Wu BX, Chen Y, Chen Y, Fan J, Rohrer B, Crouch RK, & Ma J-X (2002). Cloning and characterization of a novel all-trans retinol short-chain dehydrogenase/reductase from the RPE. Investigative Ophthalmology & Visual Science, 43(11), 3365–3372. [PubMed] [Google Scholar]
- Wu BX, Moiseyev G, Chen Y, Rohrer B, Crouch RK, & Ma J-X (2004). Identification of RDH10, an All-trans Retinol Dehydrogenase, in Retinal Muller Cells. Investigative Ophthalmology & Visual Science, 45(11), 3857–3862. 10.1167/iovs.03-1302 [DOI] [PubMed] [Google Scholar]
- Wu L, Belyaeva OV, Adams MK, Klyuyeva A, Lee S-A, Goggans KR, … Kedishvili NY (2019). Mice lacking the epidermal retinol dehydrogenases SDR16C5 and SDR16C6 display accelerated hair growth and enlarged meibomian glands. The Journal of Biological Chemistry. 10.1074/jbc.RA119.010835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Krois CR, Huang P, Wang J, Min J, Yoo HS, … Napoli JL (2017). Raldh1 promotes adiposity during adolescence independently of retinal signaling. PloS One, 12(11), e0187669 10.1371/journal.pone.0187669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Vuckovic MG, Smullin CP, Kim M, Lo CP-S, Devericks E, … Napoli JL (2018). Modest Decreases in Endogenous All-trans-Retinoic Acid Produced by a Mouse Rdh10 Heterozygote Provoke Major Abnormalities in Adipogenesis and Lipid Metabolism. Diabetes, 67(4), 662–673. 10.2337/db17-0946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Chen W, Smith SM, & Napoli JL (2001). Molecular characterization of a mouse short chain dehydrogenase/reductase active with all-trans-retinol in intact cells, mRDH1. The Journal of Biological Chemistry, 276(47), 44083–44090. 10.1074/jbc.M105748200 [DOI] [PubMed] [Google Scholar]
- Zhang M, Hu P, & Napoli JL (2004). Elements in the N-terminal signaling sequence that determine cytosolic topology of short-chain dehydrogenases/reductases. Studies with retinol dehydrogenase type 1 and cis-retinol/androgen dehydrogenase type 1. The Journal of Biological Chemistry, 279(49), 51482–51489. 10.1074/jbc.M409051200 [DOI] [PubMed] [Google Scholar]
- Zhao D, McCaffery P, Ivins KJ, Neve RL, Hogan P, Chin WW, & Dräger UC (1996). Molecular identification of a major retinoic-acid-synthesizing enzyme, a retinaldehyde-specific dehydrogenase. European Journal of Biochemistry, 240(1), 15–22. 10.1111/j.1432-1033.1996.0015h.x [DOI] [PubMed] [Google Scholar]
- Zhu Y-H, Li J-B, Wu R-Y, Yu Y, Li X, Li Z-L, … Zhu X-F (2019). Clinical Significance and Function of RDH16 as a Tumor-suppressing Gene in Hepatocellular Carcinoma. Hepatology Research: The Official Journal of the Japan Society of Hepatology. 10.1111/hepr.13432 [DOI] [PubMed] [Google Scholar]
- Ziouzenkova O, Orasanu G, Sharlach M, Akiyama TE, Berger JP, Viereck J, … Plutzky J (2007). Retinaldehyde represses adipogenesis and diet-induced obesity. Nature Medicine, 13(6), 695–702. 10.1038/nm1587 [DOI] [PMC free article] [PubMed] [Google Scholar]