

SUMMARY
Bromodomain-containing protein 4 (BRD4) is a cancer therapeutic target in ongoing clinical trials disrupting primarily BRD4-regulated transcription programs. The role of BRD4 in cancer has been attributed mainly to the abundant long isoform (BRD4-L). Here we show, by isoform-specific knockdown and endogenous protein detection along with transgene expression, the less abundant BRD4 short isoform (BRD4-S) is oncogenic while BRD4-L is tumor-suppressive in breast cancer cell proliferation and migration as well as mammary tumor formation and metastasis. Through integrated RNA-seq, genome-wide ChIP-seq, and CUT&RUN association profiling, we identify Engrailed-1 (EN1) homeobox transcription factor as a key BRD4-S coregulator particularly in triple-negative breast cancer. BRD4-S and EN1 co-modulate the extracellular matrix (ECM)-associated matrisome network, including type II cystatin gene cluster, mucin 5 and cathepsin loci, via enhancer regulation of cancer-associated genes and pathways. Our work highlights the importance of targeted therapies for the oncogenic but not tumor-suppressive activity of BRD4.
Keywords: BRD4, bromodomain, BET inhibitor, transcription factor, epigenetics, enhancer, CUT&RUN, ECM, TNBC, drug resistance
Graphical Abstract
In Brief
With BRD4 isoform-specific antibodies/knockdown and mouse xenograft and transgene expression, Wu et al. demonstrate oncogenic BRD4-S and tumor-suppressive BRD4-L have opposing functions in breast cancer initiation and development in part through enhancer regulation of matrisome extracellular network modulating cancer cell migration and metastasis.
INTRODUCTION
Transcription dysregulation has been frequently implicated in cancer and disease. Targeting epigenetic regulators, including writer proteins (Lasko et al., 2017; Fedoriw et al., 2019), erasers (Dalvi et al., 2017; Wong et al., 2019), and the acetyl-lysine reader protein BRD4 (Filippakopoulos et al., 2010; Nicodeme et al., 2010) has emerged as a way to suppress cancer-associated oncogenic pathways. Nevertheless, drug resistance and cancer relapse often occur upon prolonged treatment through transcription reprogramming that bypasses targeted factor dependence or activates compensatory or independent oncogenic pathways (Fong et al., 2015; Rathert et al., 2015). Plasticity in alternative mRNA splicing and transcription complex assembly further complicates drug sensitivity and off-target concerns in cancer therapy.
The bromodomain (Brd) and extraterminal (ET) domain-containing BET family proteins are acetyl-lysine readers that bind primarily to acetylated chromatin and transcription factors and, in vertebrates, are composed of ubiquitously expressed BRD2, BRD3 and BRD4, as well as germ cell-specific BRDT (Wu and Chiang, 2007). They contain two well-structured N-terminal bromodomains (BD1 and BD2) whose association with targeted loci is regulated by a downstream acidic region containing a cluster of casein kinase II (CK2) phosphorylation sites (Wu et al., 2013). Increased phosphorylation of BRD4 in this acidic region due to decreased protein phosphatase 2A (PP2A) activity has been linked to triple-negative breast cancer (TNBC) progression (Shu et al., 2016). This phosphorylation event is also important for learning and memory as well as normal brain functioning and drug addiction as illustrated in mouse behavioral studies (Korb et al., 2015; Guo et al., 2019).
Besides BD1 and BD2, the molecular action of BRD4 also depends on the conserved ET domain, the CK2-phosphorylated region, and a unique C-terminal motif (CTM, not found in BRD2 and BRD3) that serve as interaction platforms for recruitment of various chromatin and/or transcriptional regulators (Wu and Chiang, 2007; Rahman et al., 2011; Wu et al., 2016). Enhancer/promoter-associated BRD4 controls expression of oncogenic drivers such as c-Myc (Zuber et al., 2011) and FosL1 (Lockwood et al., 2012) and immune checkpoint inhibitors, e.g., PD-L1 (Zhu et al., 2016), and has become a promising target for anticancer drug development and immunotherapy. The finding that BET bromodomain inhibitors, such as JQ1 and I-BET, are capable of blocking BET protein binding to cancer-associated enhancers/promoters and reversing cancer phenotypes in many cell and mouse models has generated excitement for targeted therapy against epigenetic regulators. Dozens of clinical trials based on BET bromodomain inhibitors are ongoing in cancer and inflammatory diseases (Xu and Vakoc, 2017). A survey of cancer patient data revealed a potential tumor suppressor role of BRD4 in tumorigenesis (Crawford et al., 2008; Fernandez et al., 2014). Oncogenic activity of a BRD4 isoform has also been reported with truncated short form-mimicking BRD4 mutants (Alsarraj et al., 2011). Given the importance of BRD4-targeted therapy, it is critical to compare the functional properties of distinct BRD4 isoforms during cancer initiation and progression.
In this study, we found a natural alternatively spliced BRD4 short isoform possesses potent oncogenic activity and the more abundant BRD4 long isoform, generally thought to be the cancer driver due to its broad coactivating function, is a tumor suppressor during breast cancer progression. Our finding unravels new genomic landscapes controlled by BRD4 short isoform and points to the importance of isoform survey before targeted therapy is conducted.
RESULTS
Opposing Functions of BRD4 Isoforms in Breast Cancer Cell Proliferation and Migration
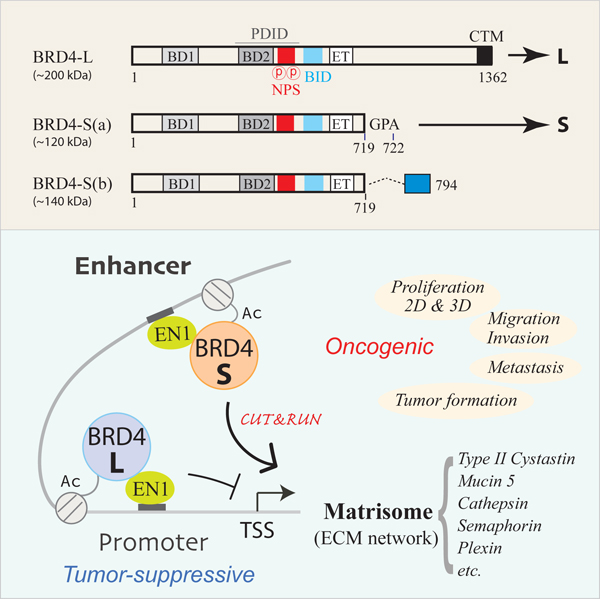
Three BRD4 isoforms, one long (BRD4-L) and two short [BRD4-S(a) and BRD4-S(b)] differing in their C-terminal amino acid residues (Floyd et al., 2013; Chiang, 2014) as a result of alternative mRNA splicing (Figure 1A), were examined for their transcript distributions in breast cancer (BCa) datasets accessible from The Cancer Genome Atlas (TCGA). By incorporating the splicing ratio (PSI, percent spliced in; TCGA SpliceSeq) and mRNA expression (TCGA RNA-seq) from 709 patient datasets, we found BRD4-L mRNA is ~2 times more abundant than BRD4-S(a) mRNA and ~56-fold higher than BRD4-S(b) mRNA (Figure 1B). Given that BRD4-S(b) protein is barely detectable with validated S(b)-specific antibody (Figure S1A) in most cancer cell lines examined (data not shown), we focus the current study on the more abundant L and S(a) isoforms (hereafter BRD4-S(a) is abbreviated as BRD4-S; see Figure 1A). BCa patients with higher L-PSI show favorable overall survival (OS), in contrast to reduced OS (i.e., higher death, indicated by hazard ratio > 1) for patients with high S-PSI (Figure S1B). Similar results were obtained when the OS analysis was conducted with isoform-specific total transcripts (Figure S1C), providing the first hint that BRD4 isoforms may have opposing functions in BCa development. When PSI and total transcripts of BRD4 isoforms were respectively analyzed according to BCa PAM50 subtypes, we found BRD4-L has higher expression in basal versus luminal A, luminal B, and HER2 subtypes by either analysis (Figure S1D and S1E). While BRD4-S transcripts were also higher in basal compared to the other subtypes (Figure S1E), its spliced ratio was somewhat reduced in the basal subtype (Figure S1D), suggesting that BRD4-S transcripts may be additionally regulated by posttranscriptional events, such as mRNA splicing, 3’-end processing, subnuclear localization, transport, and degradation, in specific BCa subtypes, making OS prediction obscure in subtype mRNA analysis (Figure S1F).
Figure 1. Opposing Functions of BRD4 Isoforms in Breast Cancer Cell Proliferation and Migration.

(A) Schematic of BRD4 domain features and antibody-targeting regions for a long (L) and two short isoforms S(a) and S(b). BRD4-S(a) is subsequently abbreviated as BRD4-S.
(B) BRD4 isoform mRNA levels in breast cancer (BCa) patients derived from RNA-seq-normalized reads (TCGA BCa) multiplying isoform PSI (TCGA SpliceSeq). Data are mean ± s.e.m. (standard error of the mean). P, two-way ANOVA.
(C) Ratio (S/L) of cellular BRD4-S and BRD4-L molecules quantified by immunoblotting (IB) with ⍺-BRD4 N antibody. Data are mean ± s.e.m. P, two-tailed t-test.
(D) MDA-MB-231 cell viability in monolayer (2D) and low-attachment sphere (3D) cultures following BRD4 pan, isoform, or control (-) siRNA knockdown. Data are mean ± s.d. (standard deviation). P, two-tailed t-test (relative to siControl).
(E) Heatmap summarizing cell viability fold change (FC) of BRD4 isoform knockdown in 10 cell lines.
(F) Representative images of invasion assay upon BRD4 isoform knockdown.
(G) Representative images of wound-healing assay performed in doxycycline (doxy)-inducible shBRD4 stable lines.
(H) Serum-induced Transwell migration assay. Data are mean ± s.e.m. Images are representative of at least three biological replicates.
Significance: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
Using validated pan- and isoform-specific BRD4 antibodies and immunoblots (Wu et al., 2016; Figure S1A), we quantified BRD4 isoform protein levels in MDA-MB-231 cells using purified recombinant BRD4 proteins as standards (Figure S1G). BRD4-L was estimated to be ~1 million and BRD4-S ~130,000 molecules per cell, which were used to measure BRD4 isoform proteins in other BCa lines selected according to their ER/PR/HER2 status (Figure S1H and S1I). BRD4-S protein typically ranges between 3–13% of BRD4-L, and the three ER-/PR-/HER2-TNBC lines with mesenchymal-like phenotypes (MDA-MB-231, MDA-MB-436 and SUM159; Lehmann et al., 2011) have the highest levels of BRD4-S (Figure 1C), further indicating that posttranscriptional regulation may play a critical role influencing BRD4 protein isoform abundance during cancer progression.
Treatment of MDA-MB-231 with BRD4-S-targeting siRNA significantly reduced cell viability in 2D monolayer and 3D low-attachment cultures, whereas BRD4-L depletion increased cell proliferation in 3D sphere culture with little effect on monolayer cell growth (Figures 1D and S2A). BRD4-Pan siRNA that reduces both isoforms had a composite effect. A general requirement of BRD4-S for cell viability was also observed in eight BCa lines and MCF10A cells, although the outcome of BRD4-L knockdown is more variable depending on cell lines and culture conditions (Figures 1E and S2B) likely reflecting cancer/cell heterogeneity and a diverse potential for growth signal-regulated factor assembly through the unique C-terminal sequence present in BRD4-L (Sabari et al., 2018; see Figure 1A). The knockdown effect could be rescued by ectopic expression of respective siRNA-resistant BRD4 isoforms as exemplified in MDA-MB-231 (Figures S3A and S3B). Collectively, these analyses indicate that BRD4-S rather than BRD4-L is required for BCa cell viability.
Invasive stellate protrusion typically observed in metastatic BCa cells grown on Matrigel (Kenny et al., 2007) mimicking extracellular matrix (ECM) components was abolished upon BRD4-S but not BRD4-L knockdown in MDA-MB-231 cells (Figure 1F). In wound-healing assay, migration of MDA-MB-231-derived doxycycline (doxy)-inducible shBRD4 stable lines (Figure S3C and S3D) was severely impaired by shBRD4-pan and shBRD4-S with little effects following BRD4-L knockdown (Figure 1G). Treatment with siBRD4-S or siBRD4-pan nearly abolished chemotaxis-induced MDA-MB-231 cell migration in a Transwell assay, whereas BRD4-L depletion significantly increased directional migration (Figure 1H) that was also observed in SUM159 but not MDA-MB-436 cells (Figure S3E). The increased migration upon BRD4-L depletion was blocked by ectopic expression of siRNA-resistant BRD4-L (Figure S3F). Likewise, impaired cell migration by BRD4-S-targeting siRNA was rescued by expressing siRNA-resistant BRD4-S (Figure S3G) with more BRD4-S protein expression further enhancing cell migration (Figure S3H). When the ECM-dependent cell growth/invasion assay was analyzed in other BCa cell lines, we found BRD4-S knockdown also reduced 3D cell proliferation in ER+/PR+ MCF-7, HER+ HCC1954 and TNBC MDA-MB-468 cells, while BRD4-L knockdown significantly enhanced SUM159 invasion (Figure S3I), consistent with the Transwell migration assay (Figure S3E). These assays together demonstrate that BRD4 isoforms have opposing functions in BCa cell proliferation and migration.
Oncogenic BRD4-S and Tumor-suppressive BRD4-L in Primary Breast Tumor Growth and Cancer Metastasis
To evaluate the in vivo function of BRD4 isoforms, we stably expressed doxy-inducible Pan- or isoform-specific shBRD4 via lentivirus delivery in MDA-MB-231 lung metastasis-prone LM2 (Minn et al., 2005; Figure S3C and S3D). Individual doxy-pretreated LM2 cells exhibiting pan or isoform-specific knockdown (Figure 2A, inset) were then injected into the 4th mammary fat pad of immunodeficient female nude mice to establish xenograft mammary tumors. The tumor growth rate of shBRD4-S LM2 was reduced compared to vector-transduced LM2, while shBRD4-L LM2 gave rise to larger tumors (Figure 2A). LM2 cells with shBRD4-Pan showed less reduced tumor growth than shBRD4-S cells. The protein levels of selective BRD4 isoforms were correspondingly reduced in the resected tumors (Figure S4A). These results suggest that low-abundant BRD4-S (~10% of BRD4-L in LM2) is the dominant isoform in mammary tumor development and that the tumor-suppressive activity of BRD4-L somewhat compromises the oncogenic activity of BRD4-S.
Figure 2. BRD4-S Enhances Mammary Tumor Growth and Metastasis While BRD4-L Suppresses Mainly Tumor Growth.

(A) Orthotopic mammary tumor growth of MDA-MB-231 LM2 cells expressing doxy-inducible BRD4 shRNA or vector. IB shows the extent of BRD4 knockdown prior to mammary fat pad injection. Data are mean ± s.e.m. P, two-way ANOVA.
(B) IB shows Cre-induced FLAG-tagged BRD4-S/L transgene (Tg) expression in MMTV-PyMT mammary tumors. Control, no Cre.
(C) Percentage of tumor-free mice with palpable mammary tumor onset in immunocompetent syngeneic mice expressing BRD4 Tg isoforms and in control mice without MMTV-Cre expression. P, Log-rank (Mantel-Cox) test.
(D) PyMT tumor images (left) and quantification of average tumor weight/number (right). Data are mean ± s.e.m. P, two-tailed t-test.
(E) Quantification of lung metastatic nodules from female mice bearing PyMT tumors with or without (control) BRD4-S Tg. Data are mean ± s.e.m. P, two-tailed t-test.
(F) BLI quantification of bone metastasis from MDA-MB-231-derived 1833 lines harboring doxy-induced BRD4 pan or isoform shRNA. Bone metastatic size is signal intensity per spot. Data are mean ± s.e.m. P, two-way ANOVA.
The opposing function of BRD4 isoforms in tumor development was examined in an MMTV-PyMT (mouse mammary tumor virus LTR-driven polyoma middle tumor-antigen)-induced syngeneic mammary tumor model by using immunocompetent knock-in mice conditionally expressing BRD4-S or BRD4-L. The coding sequence of individual BRD4 isoforms was fused with FLAG-Myc dual tags and placed downstream of a constitutive CAGGS promoter with a LoxP-STOP-LoxP sequence in between (Wu et al., 2010; Figure S4B). The entire BRD4-S or -L transgene cassette, flanked by Rosa26-targeting sequences, was inserted into the Rosa26 locus in mouse strain 129 embryonic stem (ES) cells to produce BRD4 isoform-specific transgenic mice. FLAG-Myc-tagged BRD4 transgene was ectopically expressed in female mice with MMTV-PyMT 129/B6-mixed genetic background (Figure S4C) through MMTV-Cre-mediated loxP recombination that removes the STOP cassette, thus enabling the CAGGS promoter to drive BRD4 transgene expression (Figure 2B). Consistent with the LM2 xenograft mouse study, BRD4-S transgene expression significantly enhanced PyMT-induced mammary tumor growth as evidenced by both the rate (Figure 2C) and tumor weight/number (Figure 2D), while ectopic BRD4-L expression prolonged tumor-free time and gave rise to smaller tumors. Thus, BRD4 isoforms display opposing functions in immunocompetent mice as well, with BRD4-S being oncogenic and BRD4-L tumor-suppressive for mammary tumor growth.
Since BRD4-S enhances MDA-MB-231 serum-induced directional migration and PyMT induces a high incidence of breast cancer distant metastasis to lungs (Guy et al., 1992), we analyzed H&E staining images of mouse lungs collected from control and BRD4-S transgenic mice with similar PyMT-induced tumor burden (Figure S4D) and found higher lung metastasis in the presence of BRD4-S transgene (Figure 2E). The ability of BRD4-S to enhance cancer metastasis was likewise seen in LM2 mouse xenografts in which knockdown of BRD4-S severely impaired shBRD4-S and shBRD4-Pan LM2 cell metastasis to lungs, bone, lymph node and liver as reflected by BLI (bioluminescence imaging) signals from dissected mouse organs/tissues (note that LM2 contains an integrated luciferase marker), whereas knockdown of BRD4-L enhanced shBRD4-L LM2 metastasis to lymph node and liver, but not lungs and bone (Figure S4E). These observations highlight a general dominant metastasis-promoting activity of BRD4-S but an apparent organ-specific metastasis-inhibitory activity of BRD4-L.
To confirm if BRD4 isoforms exhibit distinct metastasis tropism, we generated MDA-MB-231-derived bone-prone 1833 cells (Kang et al., 2003; Krzeszinski et al., 2014) harboring doxy-inducible pan- or isoform-specific shBRD4 by lentiviral transduction (see Figures S3C, S3D, and S4F). The doxy-pretreated lung-prone LM2- and bone-prone 1833-derived cells were then separately injected into the circulatory system of athymic female mice via retro-orbital and intracardiac route. Both BRD4 isoforms are important for LM2 lung colonization and expansion (Figure S4G) but only BRD4-S is critical for 1833 bone metastasis (Figure 2F).
BRD4-S but not BRD4-L Activates Transcription Programs Linked to ECM and ECM-Associated Matrisome Network
To identify genes and pathways uniquely regulated by individual BRD4 isoforms, we performed RNA-seq in BRD4-S/L-knockdown MDA-MB-231 cells (Figure 3A, flowchart). Using P-value, fold change and regulation (up or down) potential, we classified differentially expressed genes (DEGs) into six groups: uniquely regulated by BRD4-S (S-unique), BRD4-L (L-unique), S/L-combined (Pan-unique), or co-regulated by both (S&L; Figures 3A and S5A) with the last group further divided into three subgroups representing S-preferred, L-preferred, or no preference (equal) for specific isoforms but exhibiting either the same or opposite regulatory potential on common gene sets comparable to that seen in Pan knockdown.
Figure 3. Transcriptome Profiling Identifies Matrisome Network Genes Generally Upregulated by BRD4-S but Downregulated by BRD4-L.

(A) RNA-seq flowchart and DEG classification. RNA-seq data were from MDA-MB-231 with or without BRD4 isoform-specific knockdown.
(B) Top canonical pathways identified for BRD4-S/L DEGs highlighting S-unique-upregulated matrisome network (red) and L-preferred-downregulated core ECM (blue).
(C) Heatmap of top DEGs from MDA-MB-231 RNA-seq divided into up and downregulated (Dn) groups, each in triplicate. Scale bar is based on the Z score of CPM (counts per million).
(D) List of BRD4-S-upregulated matrisome genes with significantly higher expression in IDC vs. normal breast samples (P values from Oncomine METABRIC 2136 BCa dataset).
(E) Unique gene families identified in BRD4-S-upregulated RNA-seq from MDA-MB-231.
(F) Kaplan-Meier plot showing higher expression of a 17-matrisome-gene signature identified in S-Up predicts unfavorable DMFS in basal subtype patients. The 17 DEGs analyzed are indicated.
The six classes of genes were each separated into up- or downregulated clusters and then subjected to MSigDB pathway analysis. Major pathways identified include: S-unique-up – hemostasis, development, matrisome ECM network; S-unique-dn (down) – transcriptional regulation by p53, interferon (IFN)-stimulated antiviral genes, RNA metabolism; L-unique-up – apoptotic factor-mediated response, Rho GTPase signaling, β-catenin-independent Wnt signaling; L-unique-dn – DNA replication, cell cycle S phase; Pan-unique-up – interferon α/β signaling, innate immune system, Wnt signaling pathway; Pan-unique-dn – cytokine signaling in immune system, metabolism of lipids; and others (Figures 3B and S5B). Many matrisome genes encoding ECM and ECM-associated proteins (Socovich and Naba, 2019) are also found in the independently analyzed top 100 BRD4-S-regulated DEGs and, upon further separation into BRD4-S-upregulated (S-Up) and -downregulated (S-Dn) genes (Figures 3C and S5C), include SDC4, MUC5B, CSPG4, TNC, CTSZ/W (also identified in the S-unique-up gene cluster) and CST1, CST2, and CST4 (found in S-preferred-up; see Figure 3B). Similarly, the top 100 BRD4-L DEGs include LAMA1, WISP2, EFEMP1, and COL5A1 that are also found in the L-preferred-dn gene cluster as the core matrisome components (Figures 3B, S5B and S5C). Similar knockdown conducted in nonmetastatic ER+/PR+ MCF-7 cells did not identify the matrisome ECM network as a BRD4-S-regulated pathway (Figure S5D and S5E), highlighting its unique function in metastatic MDA-MB-231 cells.
Over half of S-Up matrisome genes (36/70) show significantly higher expression in 1,556 invasive ductal carcinoma (IDC) compared to 144 normal breast samples available in Oncomine METABRIC 2012 [Figure 3D; -log10(P-value) > 2]. The identification of multiple proteins belonging to the same family, e.g., MUC5AC/5B and SDC3/4, and pairwise family members implicated in protein-protein interaction (e.g., type II cystatin members and their interacting cathepsin proteins) and ligand-receptor binding (e.g., semaphorin-plexin) concurrently upregulated by BRD4-S (Figure 3E) further highlights the importance of matrisome in breast cancer progression. Indeed, when expression data of 1,098 patients across 8 cohorts in GOBO were analyzed, higher expression of 17 BRD4-S-upregulated matrisome genes (e.g., CST1, CST4, SEMA3B, PLXNA2, CTSB, among others) showed significantly reduced distant metastasis-free survival (DMFS) in the basal BCa subtype (Figure 3F) – about 77% of which is TNBC including MDA-MB-231 – but not in other PAM50 subtypes (data not shown). A converged function of these 17 genes lies in stroma compartment, basal-like BCa and also in early response to growth factors and steroid hormones, but has reduced immune response (Figure S5F). Patient survival analysis using a different platform (Pan Cancer Prognostics Database PROGgeneV2) also indicates the importance of BRD4-S-upregulated ECM network in breast cancer progression (Figure S5G–5I).
Engrailed-1 (EN1) Identified as a BRD4-S Coregulator in Matrisome Gene Transcription
To identify the key transcription factor (TF) dictating BRD4-S-upregulated matrisome gene transcription, we performed a TF motif screen (Cistrome MISP) focusing on regions of the top three DEG family genes (i.e., CST1/2/4, MUC5AC/5B, and CTSW) marked by H3K27ac in MDA-MB-231 (Xi et al., 2018) representing BRD4-associated active enhancers and promoters (Lee et al., 2017), and identified 70 putative TFs whose mRNAs are also enriched in IDC (Figure 4A). By comparing these 70 TF mRNA levels plus three randomly selected non-IDC-enriched mRNAs retrieved from TCGA RNA-seq databases, we found the mRNA of EN1 homeobox TF is the most enriched in basal BCa relative to other subtypes (Figure 4B).
Figure 4. EN1 functions similarly to BRD4-S in Upregulating Matrisome Gene Expression and Interacts Preferentially with BRD4-S in MDA-MB-231 Cells.

(A) 70 TF motifs identified from H3K27ac-enriched and IDC-associated BRD4-S-upregulated type II cystatin gene cluster and CTSW/MUC5 loci by MISP (Cistrome).
(B) Heatmap of 73 TF expression profiles classified according to PAM50 subtypes. Heatmap scale represents mRNA mean. Asterisks (*) mark the three TFs not enriched in IDC used as controls.
(C) Sequence logos for EN1 (MA0027.2) and EN2 (MA0642.1).
(D) RT-qPCR analysis of TF RNA. Data are mean ± s.d. P, two-tailed t-test.
(E) Knockdown of EN1 reduces S-Up CST1/2/4, MUC5AC/B and CTSW/Z RNAs as seen with BRD4-S knockdown. Data are mean ± s.d. P, two-tailed t-test.
(F) BRD4-S upregulates, while BRD4-L downregulates, matrisome gene expression in 3D culture. Data are mean ± s.d. P, two-tailed t-test.
(G) IP-IB showing EN1 preferentially interacts with BRD4-S in MDA-MB-231.
(H) EN1 interacts with both BRD4 isoforms better than EN2 using purified FLAG-tagged (f:) proteins as indicated.
(I) EN1 binds both BRD4 isoforms in SUM159 and MDA-MB-468. IP was similarly performed as in (G).
Given that EN1 was previously identified as a pro-survival TF in basal-like BCa (Beltran et al., 2014; Peluffo et al., 2019), we knocked down EN1 and related EN2 that shares a conserved core binding sequence (T/CAATTA; Figure 4C) as well as other factors by target-specific siRNAs in MDA-MB-231 cells (Figure 4D). Only knockdown of EN1, but not other homeobox TFs, reduced expression of all tested BRD4-S-upregulated matrisome genes as seen with BRD4-S, rather than BRD4-L, knockdown (Figure 4E). Similar results were obtained with BRD4 isoform knockdown performed in cells grown in 3D low-attachment culture (Figure 4F). Importantly, the level of EN1 RNA was not affected by depletion of either BRD4 isoform (see Figure 4D).
The observation that EN1 mimics BRD4-S in matrisome gene activation suggests BRD4-S may interact with EN1. Immunoprecipitation (IP) performed in MDA-MB-231 (Figure 4G) showed that α-EN1 antibody, which binds bacterially expressed/purified FLAG-tagged EN1 without cross-reaction to FLAG-tagged EN2 (lanes 1 and 2) but is unable to detect endogenous EN1 in nuclear extract (lane 3) due to its extreme low-abundance (Peluffo et al., 2019), pulled down an equivalent level of both BRD4 isoforms that were significantly reduced upon EN1 knockdown (lanes 4–6), whose targeting was efficient in depleting not only endogenous EN1 but also ectopic 3xT7-tagged EN1 protein (lanes 7 and 8). Since BRD4-S in the input lysate is ~13% of BRD4-L, this EN1-IP suggests BRD4-S interacts more efficiently with EN1 than BRD4-L in the cell (lane 3 vs. lane 5). Reciprocal IP showed only α-BRD4-S, not α-BRD4-L, antibody effectively pulled down EN1 (lanes 9–12). However, IP performed with FLAG-tagged BRD4-L and BRD4-S purified from insect Sf9 cells and FLAG-tagged EN1 and EN2 purified from bacteria (Figure 4H, lanes 1–4) showed similar interactions between EN1 and BRD4-S or BRD4-L, and both interactions were better than with EN2 (lanes 7–10). When cellular IP was performed with other TNBC lines, we found α-BRD4-L and α-BRD4-S antibodies pulled down comparable levels of EN1 in SUM159 and MDA-MB-468 nuclear extracts (Figure 4I). These results indicate the intrinsic interaction between BRD4-S/L and EN1/2 could be additionally modulated by other cellular factors, fine-tuning BRD4-S/EN1-coregulated transcription programs uniquely seen in respective basal-like TNBC cells.
Enhancer-associated EN1 and BRD4-S Upregulating CST1/2/4 Cluster Gene Transcription
The functional role of EN1 was explored by MISP motif scanning of H3K27ac-enriched regions within the 350-kb type II cystatin (CST) locus in MDA-MB-231 that encompasses six family genes on chr20p11.21, revealing scattered EN1-binding motifs around CST4 and CST2 promoters but highly clustered binding sites, ~10 kb downstream of the CST3 promoter and 60–200 kb distal to CST1/2/4 (all antisense-driven transcription), coinciding with a cluster of strong enhancers marked by ENCODE HMEC ChromHMM (Figure 5A). Since knockdown of BRD4-S significantly reduces CST1/2/4 but not CST3/5/9 expression (Figure 5B), it is likely BRD4-S acts through this distal enhancer via functional association with EN1 to activate clustered CST1/2/4 gene transcription. To test this, we designed 18 qPCR primer pairs targeting select putative EN1-binding sites with a few control regions in the CST locus and found by chromatin immunoprecipitation (ChIP) that EN1 binding to eight of these sites was diminished upon EN1 knockdown by two separate siRNAs (Figure 5C). Among these eight EN1-binding sites, BRD4-S binds specifically to #8 (enhancer) and #13 (CST4 promoter) while BRD4-L associates only with #13 (Figure 5D).
Figure 5. EN1 Recruits BRD4-S to Type II Cystatin Enhancer Critical for CST1/2/4 Cluster Gene Transcription.
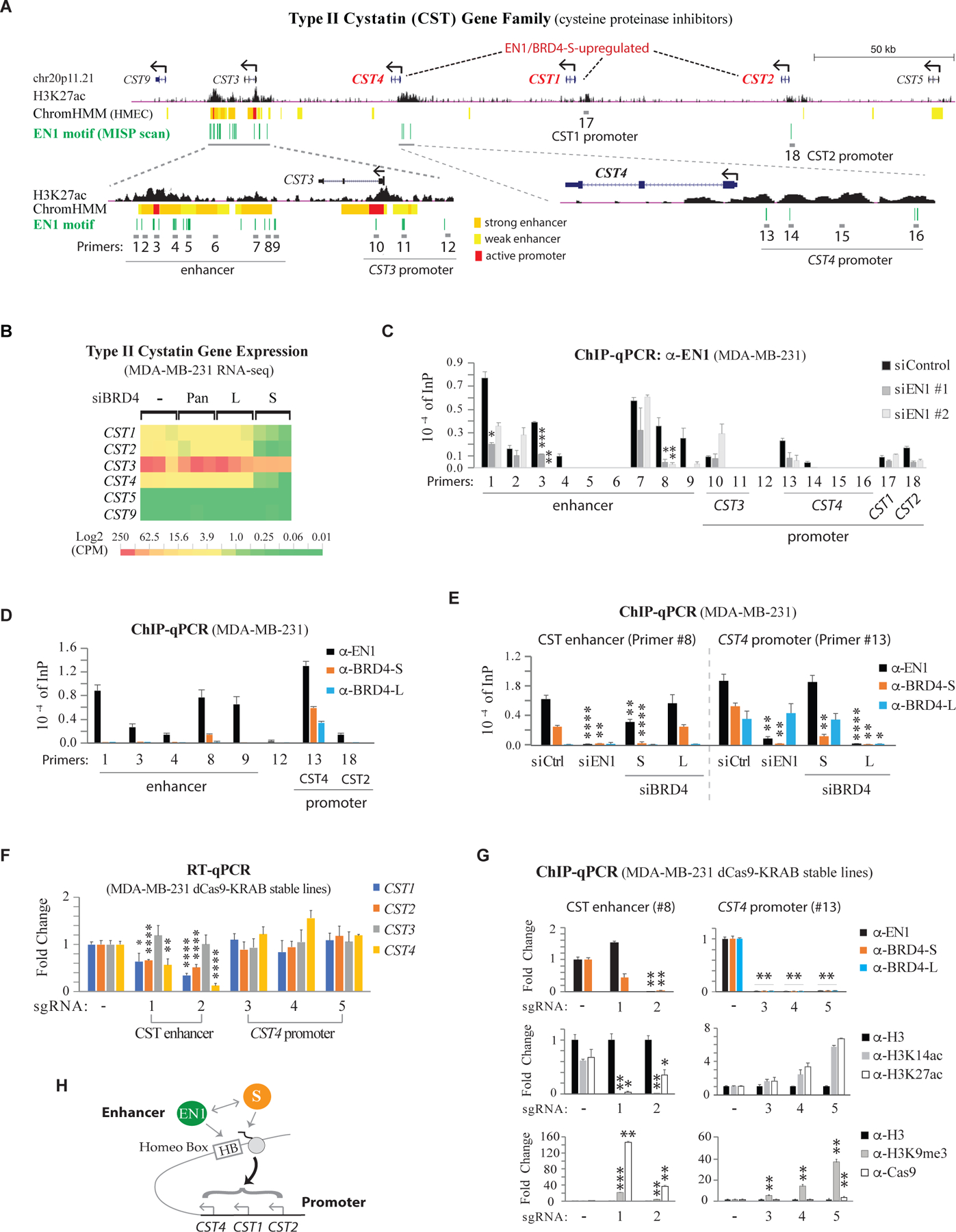
(A) Diagram of type II cystatin gene family (CST9/3/4/1/2/5) with H3K27ac ChIP-seq track (MDA-MB-231, GSE85158) and its associated enhancers/promoters identified by ChromHMM in human mammary epithelial cells (HMEC). Primer pairs (#1–18) for qPCR are indicated. Green line, predicted EN1-binding sites.
(B) Heatmap showing RNA levels of type II cystatin gene family from MDA-MB-231 RNA-seq.
(C) ChIP-qPCR of EN1 binding to 18 genomic regions in type II cystatin gene locus. Data are mean ± s.d. P, two-tailed t-test.
(D) ChIP-qPCR of BRD4 isoform occupancy in EN1-binding regions. Data are mean ± s.d.
(E) ChIP-qPCR of BRD4 isoform and EN1 binding to CST enhancer (#8 primer pair) and CST4 promoter (#13 primer pair). Data are mean ± s.d. P, two-tailed t-test.
(F) RT-qPCR of CST1/2/3/4 gene expression following sgRNA targeting in MDA-MB-231 expressing dCas9-KRAB repressor. Data are mean ± s.d. P, two-tailed t-test.
(G) ChIP-qPCR of EN1, BRD4-S, BRD4-L, histone marks, and dCas9-KRAB association. Data are mean ± s.d. P, two-tailed t-test.
(H) Model for BRD4-S/EN1-coregulated CST gene cluster.
To examine whether BRD4-S association with EN1 is critical for clustered CST1/2/4 gene transcription, we knocked down EN1, BRD4-S, or BRD4-L and found, upon EN1 depletion, BRD4-S binding to CST enhancer and CST4 promoter was abolished, whereas EN1 binding was partially reduced in the enhancer but unaffected at the CST4 promoter upon BRD4-S knockdown (Figure 5E). This result indicates EN1 recruits BRD4-S to its binding sites and BRD4-S in turn stabilizes EN1 binding in the active enhancer. While BRD4-L binding to the CST4 promoter was not affected by EN1 or BRD4-S depletion, knockdown of BRD4-L completely abolishes EN1 and BRD4-S association with CST4 promoter without altering their binding to CST enhancer, indicating a sequential entry order of BRD4-L, EN1, and BRD4-S at the CST4 promoter.
The relative importance of enhancer and promoter association with EN1 and BRD4-S was addressed by employing dCas9-KRAB-mediated silencing (Thakore et al., 2015) with sgRNAs targeting specifically to #8 CST enhancer or #13 CST4 promoter (Figure S6A) in MDA-MB-231 ectopically expressing dCas9-KRAB repressor. RT-qPCR (for RNA measurement) and ChIP-qPCR (for chromatin binding) showed that enhancer-targeting sgRNAs (1 and 2) significantly reduced CST1/2/4 RNA levels but not that of CST3 (Figure 5F), correlating with decreased binding signals for BRD4-S, EN1 (sgRNA2) and acetylated H3K14 (H3K14ac) and H3K27ac, but enhanced signals for the H3K9 trimethylation (H3K9me3) repressive mark and dCas9-KRAB occupancy (Figure 5G, left panels). Although promoter-targeting sgRNAs (3–5) blocked EN1, BRD4-S and BRD4-L binding to the CST4 promoter, coupled with enhanced H3K9me3 and dCas9-KRAB (sgRNA5), they failed to reduce H3K14 and H3K27 acetylation (Figure 5G, right panels) nor cystatin gene expression (Figure 5F), in agreement with reduced CST1 protein levels observed in cells treated with transient siRNA or doxy-induced shRNA targeting BRD4-S but not BRD4-L (Figure S6B). These results reflect a weak functional association of EN1 and BRD4 with the CST4 promoter but highlight the importance of BRD4-S-regulated enhancer action through a lineage-specific homeobox (HB) TF (Bobola and Merabet, 2017) in dictating active transcription of a distal promoter gene cluster (Figure 5H). Whether enhanced H3K9me3 and H3K27ac at the CST4 promoter upon dCas9-KRAB-triggered BRD4-L/S dissociation is attributed to relief of BRD4-inhibited H3K9 methyltransferase activity, BRD4-modulated histone acetyltransferase/deacetylase activity, or BRD4-corecruited activator/repressor in the promoter region remains to be further investigated.
EN1–BRD4-S–CST1 Regulatory Axis in Breast Cancer Cell Proliferation and Invasion
To ensure that EN1 and BRD4-S indeed co-regulate CST1/2/4 gene expression through the same regulatory axis, we performed EN1 and BRD4-S knockdown, individually or together, and found that dual knockdown of EN1 and BRD4-S did not further reduce CST1/2/4 expression, unlike the enhanced CST1/2/4 expression seen with BRD4-L knockdown alone whose effect was reversed by concurrent EN1 knockdown (Figure 6A). These results support EN1 and BRD4-S functional association through CST enhancer, not promoter occupancy (Figure 5), as well as an opposing role of activating BRD4-S and repressive BRD4-L in CST1/2/4 promoter regulation. This EN1/BRD4-cotargeted CST locus was further examined by RT-qPCR in other BCa lines. Knockdown of EN1 or BRD4-S significantly reduced CST1 expression in MCF-7, ZR-75–1, SKBR3, and MDA-MB-468 cells, contrasting the more fluctuating but reproducibly enhanced CST1 transcripts seen with BRD4-L knockdown in T47D, ZR-75–1, MDA-MB-468, MDA-MB-436, and SUM159 cells (Figure 6B). Similar knockdown effects of EN1 and BRD4-S on CST2 and CST4 expression were observed in MCF-7 and MDA-MB-468 cells, but appeared variable in other BCa lines as reflected by BRD4-L knockdown as well (Figure S6C). A heatmap summarizing EN1, BRD4-S and BRD4-L knockdown on CST1/2/4 expression across nine different BCa lines (Figure 6C) with Spearman’s correlation that identifies coexpressed pathway genes (Kumari et al., 2012) highlights positive and better coregulation between BRD4-S and EN1 than BRD4-L and EN1 (Figure 6D).
Figure 6. Knockdown of EN1 or CST1 Suppresses TNBC Cell Proliferation, Invasion and Cell Cycle Progression as Seen with BRD4-S Knockdown.

(A) RT-qPCR showing dual knockdown of EN1 and BRD4-S reduces CST1/2/4 expression comparable to individual knockdown. Data are mean ± s.d. P, two-tailed t-test.
(B) RT-qPCR of CST1 expression upon EN1 and BRD4 isoform knockdown in eight BCa cell lines. Fold difference is mean ± s.e.m. P, two-tailed t-test.
(C) Heatmap of CST1/2/4 expression following EN1, BRD4-S or BRD4-L knockdown in nine BCa cell lines.
(D) Spearman’s coefficient (rs) showing BRD4-S exhibiting positive and better correlation than BRD4-L for EN1-regulated CST1/2/4 gene expression collectively measured in nine BCa lines as analyzed in (C).
(E) Representative images of MDA-MB-231 cell invasion analyzed in 3D Matrigel culture upon BRD4-S, EN1 or CST1 knockdown.
(F) Cell cycle profiles of MDA-MB-231 following target-specific or control siRNA knockdown. P, two-tailed t-test.
(G) Cell viability of 10 cell lines in 2D culture with or without EN1 or CST1 knockdown. Data are mean ± s.e.m. P, two-tailed t-test.
Among BRD4-S-upregulated matrisome genes, CST1 shows the most significant increase in IDC [-Log10(P-value) = 297.2, see Figure 3D] and ILC (Invasive Lobular Carcinoma, data not shown) vs. normal breast samples, consistent with its essential role in BCa progression and bone metastasis (Kim et al., 2013; Dai et al., 2017) as well as in other cancers such as colorectal cancer, gastric cancer, and lung cancer (Oncomine gene disease summary; data not shown). We confirmed that knockdown of EN1 and CST1 in MDA-MB-231 reduced cell invasion (Figure 6E) and extended S phase (Figure 6F) as seen with BRD4-S knockdown, but not BRD4-L knockdown which typically prolongs cells in G1 phase (Mochizuki et al., 2008; Yang et al., 2008). The importance of EN1 and CST1 in cell proliferation was also seen in other BCa lines, particularly SUM159, MDA-MB-231, MDA-MB-436, and MDA-MB-468 TNBC lines, but with lesser effects on immortalized MCF10A, and ER+/PR+ and HER2+ BCa cells (Figure 6G). This TNBC preference for EN1-regulated cell growth is not entirely attributed to differences of EN1 RNA expression among these cell lines (Beltran et al., 2014; Peluffo et al., 2019).
When the RNA levels of EN1 and CST1 were compared with those of BRD4-S and BRD4-L in TCGA PAM50 patient datasets, we observed complete EN1-BRD4-L and modest EN1-BRD4-S positive correlations but CST1-BRD4-L/S and EN1-CST1 negative correlations (Figure S6D), with EN1 rather than CST1 RNA levels displaying a significant and high hazard ratio specifically in basal subtype (Figure S6E). This patient sample analysis argues for better survival prediction using combined expression signatures and clinical features (Figures 3D, 3F, S5G–S5I) rather than a single-gene surrogate (Zhao et al., 2011). Considering a general requirement of BRD4-S for BCa cell viability (Figure 1E) and intra/inter-tumor heterogeneity among BCa patients, these results indicate that additional pathways besides the TNBC-dominant EN1 and CST1 axis likely contribute to BRD4-S-regulated cell proliferation in other BCa subtypes and the use of an entirely RNA-based prediction may not truly reflect BRD4 isoform-regulated breast cancer and patient survival.
Genomic Landscape of BRD4-S and EN1 Association
To examine whether BRD4-S and EN1 colocalization could be observed globally, we performed ChIP followed by high-throughput sequencing (ChIP-seq) to detect factor binding to chromatin, especially in condensed regions, and conducted CUT&RUN (Cleavage Under Targets and Release Using Nuclease; Skene and Henikoff, 2017) to identify precise TF-binding sites and chromosome conformation-dependent interaction, particularly for low-abundant BRD4-S and EN1. We used BRD4 isoform-specific antibodies and chromatin samples isolated from MDA-MB-231 cells with or without ectopically expressing 3xFLAG-tagged BRD4-S (3f:S) at a level comparable to that of endogenous BRD4-L (Figure 7A, immunoblot) and also including a short-time treatment with active (+) or inactive (-) JQ1 in ChIP-seq experiments. Both ChIP-seq and CUT&RUN gave rise to reads with excellent sequencing and mapping quality (Table S1). Consistent with low abundance of BRD4-S protein (versus BRD4-L), the binding peaks for BRD4-S (1,665) from ChIP-seq is ~34-fold less than that for BRD4-L (58,353) and can be greatly enhanced by ectopic BRD4-S (3f:S) expression (47,543), with JQ1 treatment significantly dissociating chromatin-bound BRD4 (Figure 7A). Compared to ChIP-seq, CUT&RUN generates ~8-fold significant peaks for endogenous BRD4-S (13,548). That over 70% of BRD4-L and 3f:S peaks from CUT&RUN overlap with their ChIP-seq peaks indicates CUT&RUN has sensitivity similar to ChIP-seq in detecting abundant BRD4-L and 3f:S (Figure S6F).
Figure 7. Genome-wide Association of BRD4-S and EN1 by ChIP-seq and CUT&RUN.

(A) MACS2 peak numbers and IB showing BRD4-S protein level in MDA-MB-231 expressing 3xFLAG-tagged BRD4-S (3f:S).
(B) BRD4 binding in PVT1 intragenic enhancers (822E, 866E, 912E, and 919E). Signals in ChIP-seq tracks represent fold changes, relative to input, +/- JQ1 treatment. Spike-in-normalized signals are used in CUT&RUN tracks.
(C) EN1 and BRD4 co-occupy distal enhancer of BCL3, a BRD4-S-unique-up (MDA-MB-231 RNA-seq) and EN1-coregulated gene (SUM159 RNA-seq). The enhancer block (yellow) is from ENCODE HMEC chromHMM with EN1 motif sequence indicated.
(D) BRD4-L, BRD4-S, 3f:S, and EN1 peak distributions in active enhancer, primed enhancer, active promoter, and others.
(E) Heatmaps of BRD4 signals associated with EN1 peaks. SUM159 V5-tagged EN1 peaks are from GSE120957.
(F) GSEA showing S-preferred DEGs from MDA-MB-231 RNA-seq are significantly enriched in EN1 DEGs from SUM159 RNA-seq (GSE120957).
(G) Table listing TF motifs identified in enhancer-associated BRD4 peaks and heatmap showing BRD4 CUT&RUN signals associated with ChIP-seq c-Jun and TEAD4 peaks from MDA-MB-231 (GSE66081) and NF-YA peaks from K562 (ENCODE).
(H) Model for BRD4-S/EN1-coregulated enhancer modulating ECM matrisome gene expression.
Quality control of MACS2 peak calling shows BRD4-L-binding peaks have high fold change (fc; more than 500 peaks with fc > 10; Table S1) in both ChIP-seq and CUT&RUN, and endogenous BRD4-S and 3f:S have sufficient coverage in CUT&RUN according to percent fraction reads in peaks (FRiP ≥ 1%). For EN1 mapping, CUT&RUN generated better (~2-fold) genome-wide coverage and higher fc signals when compared to ChIP-seq (Table S1). Moreover, top BRD4 and EN1-binding peaks identified by CUT&RUN reside in evolutionarily conserved regions (Figure S6G) where >60% peaks overlap with DNase I hypersensitivity sites (DHS; Table S1), suggesting their preferred occupancy in TF-binding regions or in active enhancer. The signal distributions for BRD4 isoforms and EN1 are enriched in promoter and gene body (exon and intron) but reduced in intergenic regions, with endogenous BRD4-S and EN1 but not BRD4-L exhibiting binding preference for intron than exon (Figure S6H). Importantly, CUT&RUN reveals more endogenous BRD4-S promoter binding than ChIP-seq, suggesting chromatin looping-facilitated promoter contact may be better preserved by CUT&RUN. Alternatively, sonication used in ChIP-seq may partly disrupt antibody recognition of BRD4-S in its native chromatin state. The inability to efficiently detect endogenous BRD4-S binding to its chromatin targets by ChIP-seq could be improved by ectopic overexpression or by CUT&RUN, as illustrated for BRD4 isoform association with the MYC/PVT1 locus (Figure 7B; MYC/PVT1 enhancers defined in Cho et al., 2018). Similar to low-abundant BRD4-S, stronger EN1-binding peaks were detected by CUT&RUN versus ChIP-seq, with both methods identifying overrepresented sequences with common EN1 motifs (Figure S6I) potentially occupied by EN1 and BRD4 isoforms as illustrated at a BCL3-associated enhancer (Figure 7C).
With improved genome-wide detection for low-abundant proteins, we analyzed BRD4 isoform distributions and found >80% of endogenous BRD4-S and ectopic 3f:S peaks overlap with BRD4-L peaks revealed by CUT&RUN (Figure S6J). Genomic cis-element annotation with cluster analysis shows limited detection of endogenous BRD4-S at active enhancer, primed enhancer, and active promoter by ChIP-seq was significantly expanded by 3f:S expression to a binding profile analogous to that seen with BRD4-L (Figure 7D, top), whereas CUT&RUN global enhancer/promoter association for BRD4-S and 3f:S remains similar, with EN1 association likewise enhanced by CUT&RUN versus ChIP-seq (Figure 7D, bottom). Nevertheless, higher percentage (~93%) of BRD4-S shows peak overlapping with BRD4-L at active enhancer in ChIP-seq analysis (Figure S6K). In type II cystatin enhancer, both BRD4-S and EN1 could be detected at the #8 EN1-binding site by ChIP-seq and CUT&RUN with promoter association of BRD4-S, BRD4-L and EN1 at the #13 site also detected by CUT&RUN (Figure S7A), consistent with ChIP-qPCR and dCas9/KRAB analysis of EN1 and BRD4-S recruitment in regulating clustered cystatin gene transcription (Figure 5).
Association between EN1 and BRD4-S could also be detected globally in MDA-MB-231 cells by ChIP-seq and CUT&RUN, whereas BRD4-L showed only limited colocalization with EN1 detectable by CUT&RUN but not ChIP-seq (Figure 7E, left two panels). Both BRD4-S and BRD4-L, revealed by CUT&RUN in MDA-MB-231 cells, exhibited genome-wide association with EN1 ChIP-seq peaks collected in SUM159 cells (Peluffo et al., 2019; Figure 7E, right panel), in agreement with cell type-specific EN1 interaction with BRD4-S and BRD4-L (Figure 4G–4I). The regulatory potential for genome-wide EN1 association with BRD4-S and BRD4-L was analyzed by GSEA which shows significant overlap between EN1-DEGs identified in SUM159 (Peluffo et al., 2019) with BRD4-S-preferred-up and -down DEGs identified by RNA-seq performed in MDA-MB-231 (Figure 7F), but not with BRD4-L DEGs (Figure S7B), suggesting a more important role of BRD4-S than BRD4-L in regulating EN1 function in both cell lines.
To identify direct transcriptional targets (DTTs) of BRD4-S and BRD4-L, we performed integrated RNA-seq and CUT&RUN analysis using Cistrome-GO (Figure S7C, left). From classified RNA-seq DEGs (Figure S7C, right), we identified BRD4 isoform DTTs falling essentially into similar pathways identified by RNA-seq, e.g., S-unique-up DTTs in ECM organization (Figure S7D), and in general 80–95% of DEGs in each class are DTTs (compare Figures S7D and 3B).
To identify TFs potentially functioning with distinct BRD4 isoforms, we analyzed TF motifs within active enhancer/promoter-associated BRD4-S and BRD4-L peaks (see Figures 7D and S6K) and found significant association of select TF motifs in BRD4-S and/or BRD4-L peaks identified by either ChIP-seq or CUT&RUN (Figure S7E). Intriguingly, nuclear factor Y (NF-Y) motifs are overrepresented in BRD4-L but not BRD4-S CUT&RUN peaks, whereas activator protein 1 (AP-1) motifs are commonly found in both BRD4 isoform peaks (Figure 7G, left). Genome-wide association of BRD4-S and BRD4-L with the NF-YA subunit of trimeric NF-Y and the c-Jun component of dimeric AP-1 complexes, along with TEAD4 previously shown to associate with BRD4-L (Zanconato et al., 2018), illustrates NF-YA-specific BRD4-L and common c-Jun and TEAD4-associated BRD4-L and BRD4-S signal distribution (Figure 7G, right). Since NF-YA (Dolfini et al., 2019), AP-1 (Zanconato et al., 2015) and TEAD4 (Zanconato et al., 2018) have been implicated in breast cancer, the functional interplay between distinct BRD4 isoforms and EN1 (Figure 7H) as well as other cellular factors likely play crucial roles in breast cancer progression in a context-dependent and subtype-specific manner.
DISCUSSION
The function of BRD4 is regulated by posttranslational modification and interactor switch that reshape genomic landscapes, leading to rewiring of transcription programs at specific gene loci (Chiang, 2009). From detection of endogenous BRD4 protein isoforms in normal epithelial cells and distinct breast cancer subtypes, we found BRD4-S and BRD4-L each possess unique and even opposing biological and functional roles during breast cancer progression. Although typically present in less than ~13% of BRD4-L, BRD4-S is shown here to promote breast cancer growth and metastasis. BRD4-L, on the contrary, inhibits tumor progression and limits metastatic potential in certain organs/tissues, partly due to its suppressive role in growth factor-induced cell migration and cancer stem cell proliferation as seen in 3D mammosphere growth (Figures 1D, 1E, and 1H).
BRD4-S interacts with basal-like BCa-enriched EN1 to activate ECM-associated matrisome network that provides an appropriate tumor microenvironment for disease progression. The functional detection of BRD4-S association with EN1 at a unique targeting sequence (#8) situated in CST enhancer, rather than the clustered promoters, highlights the importance of enhancer-driven landscaping in transcription reprogramming. Since increasing BRD4 protein level has been linked to JQ1 resistance in several prostate cancer lines (Jin et al., 2018), alteration of genomic landscaping associated with increasing BRD4-S expression observed in 3f:S-expressing cells (Figures 7D) likely plays a critical role in cancer metastasis and drug resistance.
BRD4-S knockdown leads to S-phase arrest, similar to EN1 and CST1 knockdown. In contrast, BRD4-L knockdown delays G1 cell cycle progression (Figure 6F). These distinct effects were reflected in transcriptome profiling showing increased mitotic gene expression in BRD4-S depletion-extended S phase and increased pre-replication complex gene expression in BRD4-L depletion-prolonged G1 phase (Figure S5B). These observations indicate that each BRD4 isoform may play unique functions during cell cycle progression, genome maintenance, and DNA synthesis/repair.
While BRD4-L typically acts as a transcriptional coactivator to stimulate RNA polymerase II transcription through promoter-proximal pausing sites via CTM-dependent recruitment of P-TEFb elongation factor, a BRD4 short form (95 kDa) with molecular size smaller than that of BRD4-S (120 kDa) has been reported to act as a transcriptional corepressor in suppressing latent human immunodeficiency virus 1 (HIV-1) promoter activity (Conrad et al., 2017), consistent with our finding of the BRD4-S-inhibited HIV infection pathway uncovered by RNA-seq (Figure S5B). Given that p53 also acts as a repressor in restricting retrotransposon activity (Wylie et al., 2016) and that p53 associates with both BRD4-L (Wu et al., 2013) and BRD4-S (our unpublished data), it is likely a p53/BRD4-assembled complex may act as a genome guardian by suppressing endogenous retrotransposable element-driven transcription. If true, treating cells with BET bromodomain inhibitors may potentially relieve the repressive activity of this silencing complex, leading to reactivation of endogenous retrotransposable elements typically dormant in the human genome thus further complicating the outcome of targeted therapy.
A tumor-suppressive role of BRD4-L has been previously implicated in a transformation-resistant premature aging cell model, in which dormant oncogene-potentiated tumor progression readily occurs upon BRD4-L knockdown (Fernandez et al., 2014) and overexpressed BRD4-L significantly reduces Myc oncogene-driven mouse mammary tumor formation (Alsarraj et al., 2011). A repressive role of BRD4-L has also been reported in suppressing human papillomavirus (HPV) gene transcription in conjunction with HPV-encoded E2 protein (Wu et al., 2006) or YY1 repressor (Jha et al., 2010). It is likely that reduced cell viability upon pan-BRD4 knockdown in SUM159 partially rescued by ectopic BRD4-L expression (Shu et al., 2016) is due to BRD4-L-compensated BRD4-S function or activation of alternative cell survival pathways. Clearly, the dual function of BRD4-L and BRD4-S in transcriptional regulation and the counteracting activity of BRD4-L versus BRD4-S in tumorigenesis are context-dependent and likely cell/virus-specific.
The availability of isoform-specific antibodies in conjunction with target-specific knockdown and transgene expression in cultured cells and animal models makes it possible to perform comprehensive surveys of BRD4 protein isoform distributions and genome-wide expression and binding profiling in various cancer types and cellular systems. General use of nonselective BET inhibitors targeting both BRD4-S and BRD4-L would lead to unexpected side effects. It would be interesting to examine whether any of these BET bromodomain inhibitors in the ongoing clinical trials exhibit isoform preference. Our finding of opposing functions of BRD4 isoforms in breast carcinogenesis points to the urgency in developing more target/isoform-specific inhibitors (Chiang, 2016) that block only the oncogenic but not tumor-suppressive activity of BRD4 as well as the importance of surveying and defining isoform distributions and functions in distinct cancer cell types with molecular understanding of target selectivity by individual BET inhibitors.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Cheng-Ming Chiang (cheng-ming.chiang@utsouthwestern.edu).
Materials Availability
All plasmids, cell lines, antibodies and transgenic mice generated from this manuscript will be available upon request and approval by UT Southwestern Medical Center.
Data and Code Availability
All RNA-seq, ChIP-seq and CUT&RUN datasets, along with bigwig (bw) files used for data analysis, have been deposited in the Gene Expression Omnibus under accession code GSE136151.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial strains
DH5⍺ (Thermo Fisher Scientific) was used for regular plasmid-based DNA cloning. One Shot™ Stbl3™ Chemically Competent E. coli (Thermo Fisher Scientific) was used for constructing lentiviral delivery vectors. ArcticExpress (DE3) RIL Competent Cells from Agilent Technologies were used for BRD4-S, EN1 and EN2 protein expression.
Insect cell line
Sf9 (Expression Systems, LLC) was maintained in ESF-921 medium (Expression Systems, LCC Cat# 9600101) and used for BRD4 protein expression.
Mammalian cell lines
Human HEK Lenti-X 293T (Clontech-Takara), Phoenix-AMPHO and breast cancer cell lines MCF-7, MDA-MB-468, MDA-MB-436, MDA-MB-231, and MDA-MB-231-derived sublines (LM2, 1833, shBRD4 and sgRNA-containing stable cells, and ectopic 3f:BRD4-L/S- and dCas9-KRAB-expressing cells) were cultured in high-glucose DMEM (Sigma-Aldrich) supplemented with 10% heat-inactivated (HI) FBS, 20 mM glutamate, 10 mM sodium pyruvate, 100 units/ml penicillin (Pen) and 100 µg/ml streptomycin (Strep), with 16 µg/ml glutathione and 1 µg/ml insulin additionally included for MDA-MB-436. SUM159 was maintained in DMEM/F12 (Gibco-Invitrogen) supplemented with 10% FBS (HI) and Pen/Strep. T47D, ZR-75–1 and HCC1954 were cultured in RPMI (Sigma-Aldrich) supplemented with 10% FBS (HI) and Pen/Strep. SK-BR-3 was cultured in McCoy’s 5A (Corning Cellgro) supplemented with 10% FBS (HI) and Pen/Strep. MCF10A cells were cultured in DMEM/F12 supplemented with 5% horse serum, epidermal growth factor (EGF; 20 ng/ml), hydrocortisone (500 ng/ml), cholera toxin (100 ng/ml), insulin (10 μg/ml) and Pen/Strep. 76NF2V cells were cultured in 1:1 mix of Alpha-MEM medium (Sigma, M8042) and Ham’s F12 base medium (Fisher, MT10080CV) supplemented with 0.1 M HEPES, 2 mM L-glutamine, 1% FBS (HI), 0.035 mg/ml of bovine pituitary extract (Hammond Cell Tech, 1078NZ), 0.01 mM ascorbic acid, 2 nM β-estradiol, 2.5 ng/mL sodium selenite, 10 nM triiodothyronine (T3), ethanolamine, 1 µg/mL insulin, 1 ng/mL hydrocortisone, 0.1 mM phosphoethanolamine (PE), 0.01 mg/mL transferrin, 12.5 ng/mL EGF and Pen/Strep. All the cell lines were routinely tested for mycoplasma using MycoAlert Detection Kit (Lonza) and authenticated by STR profiling at the UT Southwestern (UTSW) Genomic core. Sources of cell lines are listed in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| BRD4 N | Chiang Lab (Covance) | aa 149–284 |
| BRD4-L | Chiang Lab (Covance) | aa 1199–1362 |
| BRD4-S(a) (or BRD4-S) | Chiang Lab (Yenzym) | CSSSSDSEDSETGPA |
| BRD4-S(b) | Chiang Lab (Yenzym) | CDVEQTAAGQPHRQS |
| pS484/488 | Chiang Lab (Yenzym) | CKVVAPP-pS-SSD-pS- SSD |
| EN-1 | Abcam | ab108598 |
| CST1 | Invitrogen | PIPA539123; AB_2555715 |
| FLAG | Sigma | F3165; AB_259529 |
| Histone H3 | Cell Signaling | 9715S; AB_331563 |
| H3K14ac | Millipore | 06–579; AB_2115283 |
| H3K27ac | Active Motif | 39133; AB_2561016 |
| H3K9me3 | Abcam | ab8898; AB_306848 |
| T7-Tag (D9E1X) | Cell Signaling | 13246; AB_2798375 |
| Cre | Millipore | 69050–3; AB_10806983 |
| Bacterial and Virus Strains | ||
| ArcticExpress (DE3) RIL | Agilent Technologies | 230193 |
| DH5⍺ | Thermo Fisher Scientific | 18258012 |
| One Shot™ Stbl3™ Chemically Competent E. coli | Thermo Fisher Scientific | C737303 |
| Biological Samples | ||
| N/A | N/A | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Doxycycline | RPI | D43020–100.0 |
| JQ1(-) | ApexBio Technology | A8181 |
| JQ1(+) | ApexBio Technology | A1910 |
| Matrigel | Corning | CB-40230C |
| Calcein AM | Corning | 354217 |
| CellTiter-Glo™ 3D | Promega | G9683 |
| Critical Commercial Assays | ||
| CytoSelect TM 24-well Wound Healing | Cell Biolabs, Inc | CBA-120 |
| TruSeq Stranded Total RNA Library Prep Kit with Ribo-Zero Human/Mouse/Rat Set A (48 samples, 12 indexes) | Illumina | RS-122–2201 |
| NEBNext® Ultra™ II DNA Library Prep Kit for Illumina® | New England (NEB) | E7645S |
| Deposited Data | ||
| RNA-seq (24 datasets), ChIP-seq (13 datasets) and CUT&RUN (11 datasets) including 21 processed data files | GSE136151 | This study |
| MDA-MB-231 histone ChIP-seq | Franco et al., 2018 | GSE85158 |
| MDA-MB-231 TEAD4 & cJun ChIP-seq | Zanconato et al., 2015 | GSE66081 |
| SUM159 EN1 RNA-seq & ChIP-seq | Peluffo et al., 2019 | GSE120957 |
| Experimental Models: Cell Lines | ||
| HEK Lenti-X 293T | Clontech-Takara | 632180 |
| MCF-7 | Weibo Luo, UT Southwestern | N/A |
| MDA-MB-436 | Weibo Luo, UT Southwestern | N/A |
| MDA-MB-231 | Weibo Luo, UT Southwestern | N/A |
| SUM159 | Weibo Luo, UT Southwestern | N/A |
| T47D | Weibo Luo, UT Southwestern | N/A |
| ZR-75–1 | Weibo Luo, UT Southwestern | N/A |
| HCC1954 | Weibo Luo, UT Southwestern | N/A |
| SK-BR-3 | Weibo Luo, UT Southwestern | N/A |
| MCF10A | Ganesh Raj, UT Southwestern | N/A |
| 76NF2V | Khandan Keyomarsi, The University of Texas M.D. Anderson Cancer Center | N/A |
| LM2 | Srinivas Malladi, UT Southwestern | N/A |
| 1833 | Yihong Wan, UT Southwestern | Krzeszinski et al., 2014 |
| MDA-MB-231 shBRD4-L/S lines | N/A | This study |
| MDA-MB-231 3f:BRD4-L/S lines | N/A | This study |
| Phoenix-AMPHO | Kai Ge | N/A |
| MDA-MB-231 T7EN1 | N/A | This study |
| Sf9 | Expression Systems, LLC | 94–001F |
| Experimental Models: Organisms/Strains | ||
| Athymic NCr-nu/nu mice | Jackson Laboratory | 553 |
| C57BL/6J | UTSW Mouse Breeding Core | N/A |
| Tg(MMTV-cre)4Mam/J | Jackson Laboratory | 003553 |
| B6.FVB-Tg(MMTV-PyVT)634Mul/LellJ | Jackson Laboratory | 022974 |
| C57BL/6J-ROSA26(BRD4-L)1Cmc | N/A | This study |
| C57BL/6J-ROSA26(BRD4-S)1Cmc | N/A | This study |
| Oligonucleotides (see Table S2 for complete oligonucleotide list) | ||
| Universal negative siRNA control | N/A | Sigma SIC001 |
| siBRD4-Pan | AAGACAAAGAAGGGAGUG | This study |
| siBRD4-L | UGUCUACACAGUAUACACA | This study |
| siBRD4-S | GGGCUAUCACCAUGGGCAA | This study |
| shBRD4-pan-(+497) | TGAACCTCCCTGATTACTATA | Pan1, this study |
| shBRD4-pan-(+1485) | CGTCCGATTGATGTTCTCCAA | Pan2, this study |
| shBRD4-L-(+4270) | CAGAGTGATCTATTGTCAATA | L1, this study |
| shBRD4-L-(+4626) | GCCAAATGTCTACACAGTATA | L2, this study |
| shBRD4-L-(+5005) | CCAACCAAAGTCAGTTCCTTC | L3, this study |
| shBRD4-S(a)-(+2394) | ATTGGACACGGACTCTTAATA | S1, this study |
| shBRD4-S(a)-(+2483) | GACAGGACTCCATCAAGTTAT | S2, this study |
| Recombinant DNA | ||
| pVL-F:BRD4-L | pVL1932 | Wu et al., 2016 |
| pVL-F:BRD4-S(a) | pVL1932 | Wu et al., 2016 |
| pVL-F:BRD4-S(b) | pVL1392 | This study |
| pF:BRD4 (1–719)-11d | pVL1932 | This study |
| pF:BRD4-S(a) | pET-11d | This study |
| pF:EN1–11d | pET-11d | This study |
| pF:EN2–11d | pET-11d | This study |
| pcDNA3–3F:BRD4-L | pcDNA3 | Wu et al., 2016 |
| pcDNA3–3F:BRD4-S(a) | pcDNA3 | This study |
| pLKO-Tet-shBRD4-Pan1 | Tet-pLKO-puro | This study |
| pLKO-Tet-shBRD4-Pan2 | Tet-pLKO-puro | This study |
| pLKO-Tet-shBRD4-L1 | Tet-pLKO-puro | This study |
| pLKO-Tet-shBRD4-L2 | Tet-pLKO-puro | This study |
| pLKO-Tet-shBRD4-L3 | Tet-pLKO-puro | This study |
| pLKO-Tet-shBRD4-S(a)1 | Tet-pLKO-puro | This study |
| pLKO-Tet-shBRD4-S(a)2 | Tet-pLKO-puro | This study |
| pLenti-LKO | Tet-pLKO-puro derivative without stuff, TetR and puromycin R | This study |
| pLKO-3F:BRD4-L | pLenti-LKO | This study |
| pLKO-3F:BRD4-S(a) | pLenti-LKO | This study |
| pLentiCRSPRv2/EGFP+/ Cas9- | plentiCRSPRv2 | This study |
| pLenti-CST-sgRNA#1 | plentiCRSPRv2/EGFP+/ Cas9- | This study |
| pLenti-CST-sgRNA#2 | plentiCRSPRv2/EGFP+/ Cas9- | This study |
| pLenti-CST4-sgRNA#1 | plentiCRSPRv2/EGFP+/ Cas9- | This study |
| pLenti-CST4-sgRNA#2 | plentiCRSPRv2/EGFP+/ Cas9- | This study |
| pLenti-CST4-sgRNA#3 | plentiCRSPRv2/EGFP+/ Cas9- | This study |
| pMSCVhyg-3xT7:EN1 | pMSCVhyg_3xT7_PA1 | This study |
| Software and Algorithm | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij |
| PRISM8 | https://www.graphpad.com | |
| FastQC v0.11.2 | Andrews, 2010 | Data.https://www.bioinformatics.babraham.ac.uk/projects/fastqc |
| FastQ Screen (v0.4.4) | Wingett, 2011 | https://www.bioinformatics.babraham.ac.uk/projects/fastq_screen |
| fastq-mcf (ea-utils/1.1.2–806) | Aronesty, 2013 | https://benthamopen.com/ABSTRACT/TOBIOIJ-7–1 |
| TopHat (v2.0.12) | Kim et al., 2013 | http://ccb.jhu.edu/software/tophat |
| FeatureCounts | Liao et al., 2013 | http://www.bioconductor.org |
| EdgeR | Robinson et al., 2010 | http://www.bioconductor.org |
| Oncomine | Rhodes et al., 2004 | www.oncomine.org |
| Cistrome-BETA | Wang et al., 2013 | http://cistrome.org/BETA |
| Cistrome-GO | Li et al., 2019 | http://go.cistrome.org |
| Cistrome-BETA-MISP | Wang et al., 2013 | http://cistrome.org/BETA |
| BWA | Li and Durbin, 2009 | http://dx.doi.org/10.1093/bioinformatics/btp324 |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| MACS2 | Zhang et al., 2008 | http://liulab.dfci.harvard.edu/MACS |
| Deeptools | Ramírez, 2016 | https://deeptools.readthedocs.io/en/develop |
| BedTools | Quinlan and Hall, 2010 | http://code.google.com/p/bedtools |
| RSAT | Nguyen et al., 2018 | http://embnet.ccg.unam.mx/rsat |
| Galaxy | Afgan et al., 2018 | https://galaxyproject.org/citing-galaxy |
| BioHPC | https://portal.biohpc.swmed.edu | |
| GSEA (v7, Broad Institute) MSigDB | Subramanian et al., 2005 | http://software.broadinstitute.org/gsea/msigdb |
| GOBO | Ringnér et al., 2011 | http://co.bmc.lu.se/gobo/gobo.pl |
| PROGgeneV2 | Goswami and Nakshatri, 2014 | http://genomics.jefferson.edu/proggene |
| Others | ||
| TCGA SpliceSeq | Ryan et al., 2016 | https://bioinformatics.mdanderson.org/TCGASpliceSeq |
| cBioPortal (TCGA) | http://www.cbioportal.org | |
| UCSC Genome Browser | Kent et al., 2002 | http://genome.ucsc.edu |
| Integrative Genomics Viewer (IGV 2.4.8) | Robinson et al., 2017 | http://www.broadinstitute.org |
| BICF ChIP-seq Analysis Workflow | Barnes et al., 2019 | https://doi.org/10.5281/zenodo.2648844 |
Mice
Xenograft mammary tumor and metastasis experiments
For xenograft tumor growth, doxycycline-inducible shBRD4 or vector-harboring LM2 cells in 2D culture were pretreated with 1 µg/ml of doxycycline for 72 h before injecting one million cells (in 25 µl 1xPBS plus 25 µl of Matrigel) into #4 mammary fat pads of athymic NCr-nu/nu mice (Jackson laboratory, strain code 553) that had been pre-fed with doxycycline (1 mg/ml)-containing drinking water 24 h ahead and continued throughout the course of the experiments. Primary tumor volume was measured by caliper twice per week for 4 weeks and calculated by Width2×Length/2. When primary tumors reached ~1500 mm3, mice were subcutaneously injected with 50 µl of luciferin (40 mg/ml in 1xPBS, Gold Biotechnology Inc.), euthanized, and dissected for metastasis to lungs, bone, liver, lymph nodes, brain, kidney, pancreas and ovary using BLI (Bioluminescence imaging; Caliper Xenogen IVIS Spectrum instrument at UTSW Small Animal Imaging Core) with sequential imaging taken at 1, 15 and 60 seconds. Luciferase signals were quantified from comparable areas of dissected organs/tissues. LM2 metastasis was observed mostly in lungs, bone, liver and lymph nodes. Independent lung metastasis and bone metastasis experiments were similarly conducted by injecting doxycycline-inducible shBRD4 or control LM2 cells (one million in 50 µl 1xPBS) via retro-orbital sinus or doxycycline-inducible shBRD4 or control 1833 cells (1x105 cells in 100 μl PBS) via intracardiac injection into the left cardiac ventricle guided by a VisualSonics Vevo 770 small-animal ultrasound device. BLI was conducted once per week for 4 weeks on both dorsal and ventral positions and quantified as described above.
Generation of BRD4 transgenic mice for primary tumor and metastasis studies
Inducible expression using Cre/LoxP with transgene knock-in at the Rosa26 locus has been previously described (Wu et al., 2010). The coding sequences of human BRD4-L and BRD4-S(a) were respectively amplified by PCR from human BRD4 cDNA (GeneCopoeia, E0102) using a BRD4 forward primer 5’-CTGACTAGTGTCGACATGTCTGCGGAGAGC-3’ (commonly found in L/S 5’ end) and a BRD4-L-specific reverse primer 5’-GATATCCTCGAGTCAGAAAAGATTTTCTTC-3’ or a BRD4-S(a)-specifc reverse primer 5’-GATATCCTCGAGTTAGGCAGGACCTGTTTC-3’), and subcloned into RfNLIII24 shuttle vector between SpeI/XhoI sites for in-frame fusion of dual FLAG/Myc tags with BRD4 sequences. The BRD4-S/L-containing shuttle vector was then digested with AseI to release FLAG/Myc-tagged BRD4-S/L along with the downstream FRT-flanked kanamycin-resistant gene. The released backbone-free BRD4-S/L-containing DNA fragment and the targeting base vector pCAGGc-LSL-Luciferase (Wu et al., 2010) were together electroporated into SW102 bacteria (National Cancer Institute, Frederick) for in vivo recombination through recombineering technology. The successfully recombined intermediate construct was transformed into 294-Flp bacteria for removal of the FRT-flanked kanamycin-resistant cassette to finalize the generation of targeting constructs that were sequenced and validated throughout the BRD4 cDNA and junction components.
For gene targeting in mouse embryonic stem (ES) cells, BRD4-S/L-targeting constructs were individually linearized by PacI and 100 µg total DNA was sent to Baylor College of Medicine Mouse Embryonic Stem Cell Core for AB2.2 (mouse strain 129S5 background) ES cell targeting. Correctly targeted ES clones were screened by their acquired puromycin resistance (the puromycin resistant gene is located between the CAGGS promoter and LoxP-STOP-LoxP) and identified by Southern blotting as previously described (Soriano, 1999; Yanagawa et al., 1999). The production of chimeras from AB2.2 ES cells was processed in the Baylor College of Medicine Genetically Engineered Mouse Core for BRD4-S transgenic mice and in the UTSW Transgenic Technology Center for BRD4-L transgenic mice. Germ line transmission of the CAG-S-hBRD4-L or CAG-S-hBRD4-S allele was determined by mouse tail genomic typing. The allele-specific primers for PCR genotyping are 5’-GCAACGTGCTGGTTATTGTG-3’ (forward) and 5’-ATTAAGGGCCAGCTCATTCC-3’ (reverse) for the knock-in allele with a product size of 0.366 kb. PCR primers for the wild-type ROSA26 locus are 5’-AAAGTCGCTCTGAGTTGTTAT-3’ (forward) and 5’-GGAGCGGGAGAAATGGATATG-3’ (reverse) with a product size of ~0.6 kb (Soriano, 1999). The genotyping PCR program includes 95°C denaturation, 15 min for one cycle, then 95°C/1 min-56°C/1 min-72°C/1 min for 35 cycles, and completed with one cycle at 72°C for 10 min.
Chimera mice harboring the BRD4-S or BRD4-L transgene were backcrossed separately with C57BL/6J (B6; purchased from UTSW Mouse Breeding Core) for three generations (BC3) before crossed together to obtain female littermates harboring both BRD4-S and BRD4-L transgenes (R26-BRD4-L/R26-BRD4-S, see Figure S4C). Female mice were then crossed with B6 males harboring dual MMTV-Cre and -PyMT transgenes. The resulting female mice expressing MMTV-PyMT transgene used for the study are all in 129/B6 genetic backgrounds containing either BRD4-S or BRD4-L transgene each with or without MMTV-Cre, allowing mammary gland-specific overexpression (OE) of respective BRD4 isoforms to be induced only in the presence but not absence (as control) of MMTV-Cre. MMTV-PyMT and MMTV-Cre mice were both in the B6 background before used for breeding. For genotyping following mouse breeding, mouse tails were cut and incubated in DirectPCR solution (Viagen) with 0.4 mg/ml Proteinase K (Fisher Bioreagents) overnight at 55°C and then spun to remove hair. Genomic DNA in the supernatant, after 5-fold dilution with autoclaved H2O, was used for PCR reactions containing FailSafe™ PCR 2x PreMix G buffer (Lucigen), GoTaq® DNA Polymerase (Promega) and primer pairs unique for ROSA26-BRD4-L, ROSA26-BRD4-S, MMTV-Cre, or MMTV-PyMT transgene amplification (see Table S2). The PCR program for transgene amplification is one cycle at 94°C for 5 min, then 35 cycles at 94°C/15 sec-60°C/30 sec-72°C/30 sec, and completed with one cycle at 72°C for 3 min. PCR products were analyzed by 1% agarose-TAE gel electrophoresis. Mammary tumor onset was monitored by palpation (~2–3 mm3). Kaplan-Meier survival plot was used to calculate the median age (days after birth) of tumor onset using log-rank (Mantel-Cox) test.
For primary tumor weight comparison, 22-week old mice with or without BRD4-S/L transgene expression were euthanized and individual tumors isolated and separated from overlying skin and surrounding adipose tissue using scissors. After weight measurement and image acquisition, tumors were cut into smaller pieces for total protein extraction or snap-frozen in liquid nitrogen and stored at −80°C. For lung metastasis analysis, mice bearing comparable size of primary tumors, with or without BRD4-S transgene expression, were euthanized and the lungs, prior to removal, were fixed by perfusion through injecting 10% buffered formalin (Fisher Scientific) into the ventral wall of trachea accessible from a small neck incision using a 27G needle-attached 5-ml syringe until lungs fully inflated. After closing the pulmonary trachea with a Serrefine clamp, the entire lungs were removed and placed in 10% formalin, shaken for 24 h at room temperature on a horizontal shaker. Lung samples after washing twice with 1xPBS were placed in tissue cassettes and submitted to UTSW Tissue Management Shared Resource Core for paraffin embedding, sectioning, and H&E staining. The H&E slides were imaged by Keyence BZ-X700 Microscope using 20x Lambda Nikon lens. Metastasis nodules and total lung areas were quantified by using ImageJ (Version 1.51). All mouse experiments were conducted according to the guidelines and protocols approved by the Institutional Animal Care and Use Committee (IACUC) at the Baylor College of Medicine and UT Southwestern Medical Center.
METHOD DETAILS
Plasmid construction
For recombinant protein expression, the protein-coding regions of BRD4(1–719), BRD4-S(a), BRD4-S(b), EN1 and EN2 were PCR-amplified respectively from pF:BRD4(1–719)-7 (Lee and Chiang, 2009), pF:BRD4(1–722)-11d (Wu et al., 2013), pCMV-F:BRD4-isoB (Floyd et al., 2013), pENTR223.1-EN1 (PlasmID #HsCD00082669) and pCR4-TOPO-EN2 (PlasmID #HsCD00341693) and subcloned individually between NdeI and BamHI into pF:TBP-11d or pVL1932-F:hBRD4 (1–722) (Wu et al., 2016), and between BamHI and XbaI into pcDNA3–3F:BRD4-L (i.e., FL), after swapping with the insert, to generate expression plasmids listed in the Key Resources Table. The BRD4 pan and isoform-specific targeting sequences used for doxycycline-inducible shBRD4 knockdown, based on Tet-pLKO-puro (Addgene #21915), are also listed in the Key Resources Table. For ectopic BRD4-L and BRD4-S(a) expression by lentiviral delivery, three FLAG-tagged (3f:) BRD4-L- and BRD4-S(a)-coding sequences were retrieved fom pcDNA3–3F:BRD4-L and pcDNA3–3F:BRD4-S(a), respectively, and inserted between SpeI and XbaI sites of Tet-pLKO-puro after prior removal of the stuffer, Tet repressor and puromycin resistance gene sequences to reduce the plasmid size. For sgRNA expression, the lentiCRSPRv2 (Addgene # 52961) plasmid was first modified by replacing the Cas9 with EGFP (from pEGFP-C1) encoding sequence between AgeI and BamHI and then used for sgRNA DNA oligo insertion at the BsmBI site. pMSCVhyg-3xT7:EN1 was constructed by swapping the PA1-coding sequence with the PCR-amplified EN1 cDNA fragment between Xho1 and Hpa1 sites in pMSCVhyg_3xT7_PA1. All the plasmids constructed and used for this study are listed in the Key Resources Table.
Protein purification
Recombinant FLAG-tagged (f:) proteins, including f:BRD4-L, f:BRD4-S(a), f:BRD4-S(b), and f:BRD4(1–719) expressed in and purified from insect Sf9 cells and f:BRD4-S(a), f:EN1, and f:EN2 expressed in and purified from bacteria, were prepared according to the established protocols (Wu et al., 2013).
siRNA transfection, and lentiviral shRNA, sgRNA and gene delivery
siRNA transfection was performed in immortalized mammary epithelial and breast cancer cells, including MDA-MB-231 and its derived 3f:BRD4-L/S-expressing lines, using BRD4 pan, BRD4-S, BRD4-L, target-specific (e.g., EN1 and other transcription factors and CST1) or control siRNA (universal negative siRNA control, Sigma SIC001) following the protocols as previously described (Wu et al., 2013). For lentiviral delivery, 3 µg of pLP1, pLP2, 2 µg of pLP/VSV-G (all from Gibco-Invitrogen), and 4 µg of a lentiviral transfer plasmid (for expressing BRD4 shRNA, type II cystatin sgRNA, dCas9-KRAB, or 3f:BRD4-L/S protein) or vector alone were mixed with 30 µl of Lipofectamine 2000 in Opti-MEM (Gibco-Invitrogen) following manufacturer’s instructions. The resulting mix was added onto ~90% confluent Lenti-X 293T cells in 6-ml DMEM growth medium. Forty-eight hours later, virus-containing supernatant was spun to remove cells and kept at 4°C overnight before virus concentration with LENTI-X CONCENTRATOR (Clontech) following manufacturer’s instructions. Approximately 2–20% of collected viruses were added to 1x105 of MDA-MB-231 or its derived cells (LM2, 1833 and dCas9-KRAB-expressing lines) grown in 0.3 ml medium. After 48-h incubation, cells were either directly expanded or pre-selected with puromycin (1 µg/ml for inducible BRD4 shRNA cells) for 7–10 days before expansion. More than 90% of BRD4 knockdown was typically observed upon treatment with 0.5–1 µg/ml of doxycycline for 72 h. The dCas9-KRAB-expressing MDA-MB-231 line was established by infecting MDA-MB-231 with Lenti-dCas9-KRAB-blast-carrying lentivirus (Addgene #89567), followed by blasticidin selection (5 µg/ml). To generate retrovirus carrying the 3xT7:EN1-coding sequence, pre-seeded Phoenix-AMPHO packaging cells (3.5x106/10-cm dish the day before) were transfected with 7 µg of pMSCVhyg-3xT7:EN1 and 21 µl of GENJET Plus DNA Reagent (Fisher) for 16 h. Virus-containing supernatant, after filtration, was used to infect MDA-MB-231 at 1:1 virus/medium ratio for 48 h. Following hygromycin selection (0.5–0.8 mg/ml, RPI) for two weeks, 3xT7:EN1 expression was confirmed by anti-T7 antibody. The stable line was then used for EN1 siRNA knockdown. Sequence information for siRNA, shRNA and sgRNA is listed in the Key Resources Table and Table S2.
Cell viability, migration, Matrigel invasion and cell cycle analysis
Breast cancer cells and immortalized mammary epithelial cells were treated with targeting or control siRNA twice for 48 hours each time or by sgRNA lentiviral transduction for 48 to 72 hours. Cells were then collected and re-seeded at 300–1000 cells per well in 384-well Black Clear Bottom plates (Corning™ 3764BC) for 2D monolayer culture or round-bottom ultra-low attachment microplates (Corning™ 4516) for 3D spheroid culture. After 5-day incubation at 37 °C in a 5% CO2 incubator, cell viability was measured by Calcein-AM (Corning) for 2D culture or CellTiter-Glo™ 3D (Promega) for 3D culture according to manufacturer’s protocols.
For Transwell migration assay, mesenchymal TNBC cells (MDA-MB-231, MDA-MB-436, and SUM159), pretreated with siRNA for 72 h, were incubated in serum-free DMEM for 24 h, collected and adjusted to 1x106 cells per 0.1 ml in serum-free DMEM (DMEM/F12 for SUM159), added into a Transwell insert (Corning™ 3422), and then placed in a receiving well containing 0.6 ml of heat-inactivated FBS-supplemented growth medium for an additional 16–40 hours at 37°C in a 5% CO2 incubator. Trypsin solution (Lonza CC-5012) containing 1 µM Calcein-AM was used to collect cells migrating to the other side of the insert and the receiving well, with collected cells quantified by fluorescence measurement per manufacturer’s instructions. For 3D on-top Matrigel culture, breast cancer cells (MCF-7, HCC1954, MDA-MB-468, MDA-MB-231, and SUM159) upon siRNA delivery were collected in DMEM/F12 supplemented with 1% FBS (HI), 2% Matrigel (Corning™ Matrigel™ Membrane Matrix GFR), EGF (20 ng/ml), insulin (10 µg/ml) and Pen/Strep, and added onto Matrigel-precoated (120 µl) 24-well plates at 2×104 cells per cm2. Cell images were taken 3–5 days later by using Keyence BZ-X700 Microscope.
For cell cycle analysis, pre-seeded MDA-MB-231 cells in 6-well plates were transfected with siRNA for 72 h, harvested, washed with PBS, fixed in 70% ethanol overnight and then incubated with a DNA-staining buffer containing 1% Triton X-100, 0.1 mg/ml RNase A (Thermo Scientific), and 10 μg/ml propidium iodide (Invitrogen) at 37°C for 30 min, followed by flow cytometry analysis by FACSCAlibur (BD Biosciences). Data were quantified by using FlowJo™ (BD Biosciences). For wound-healing assay, MDA-MB-231 cells containing doxycycline-inducible BRD4 shRNA were seeded onto 24-well plates pre-assembled with wound-healing inserts (CytoSelect TM 24-well Wound Healing, Cell Biolabs, CBA-120) at 5x105 cells/well in doxycycline (0.5 µg/ml)-supplemented growth medium. After 48-h incubation at 37 °C in a 5% CO2 incubator, inserts were removed and cells were washed twice with 1xPBS and replenished with fresh medium, followed by another 24-h incubation. Cells were then washed twice with 1xPBS, stained with 0.05% (w/v) crystal violet in 40% methanol for 30 min at RT, with subsequent wash twice with deionized H2O and air dry. Cell images were obtained by using a Keyence BZ-X700 microscope.
RNA-seq, RT-qPCR and BRD4 isoform target and pathway identification
For RNA-seq, total RNAs were extracted from BRD4 pan, BRD4-S, BRD4-L or control siRNA-treated MDA-MB-231 or MCF-7 cells, each in triplicate, by miRNeasy Mini Kit (Qiagen #217004) following manufacturer’s instructions. RNA integrity was verified using Agilent 2100 Bioanalyzer with a RIN score of 9 or higher. The library for sequencing was constructed from 1 µg of each DNase I-treated RNA sample by using the TruSeq Stranded Total RNA LT Sample Prep Kit from Illumina (Illumina #RS-122). Briefly, total RNA upon rRNA depletion was fragmented and used for strand-specific cDNA synthesis. The blunt-ended cDNA was 3’-tailed with a single A nucleotide and then ligated with indexed adapters. After adapter ligation, samples were PCR-amplified and purified with AMPURE XP beads (Beckman Coulter), then validated again on Agilent 2100 Bioanalyzer. Concentrations of the constructed cDNA libraries were quantified by Qubit, normalized, pooled, and then run on Illumina NextSeq 500. Raw reads in fastq files were analyzed by FastQC (v0.11.2) and FastQ Screen (v0.4.4) and trimmed to remove adapter sequences using fastq-mcf (ea-utils/1.1.2–806). All reads with base quality score > 30 were kept. Trimmed reads were mapped to hg19 (iGenomes) using TopHat (v2.0.12) with duplicated reads marked by Picard tools (v1.127). RNA counts were generated from FeatureCounts. After TMM normalization, differentially expressed genes (DEGs) were identified using edgeR with counts per million (CPM) > 1 in at least three samples. Statistical cutoffs based on P < 0.05 and log fold change Log2(FC) ± 0.6 were used to filter DEGs between siBRD4 and siControl (Ctrl) samples. The z scores of CPM from the top 100 DEGs filtered by false discovery rate (FDR) were used to create hierarchical clustering heatmaps using R with heatmap.2 function from the gplots package. RT-qPCR reactions were conducted as previously described (Wu et al., 2013) with total RNA collected at 48 hours (2D) or 96 hours (3D low-attachment) post siRNA transfection by NucleoSpin RNA II (Clontech) coupled to on-column DNase I treatment, and used in one-step RT-qPCR reactions composed of RevertAid Reverse Transcriptase (Thermal Fisher Scientific), SsoAdvanced™ Universal SYBR® Green Supermix (Bio-Rad) and primer pairs for matrisome genes, EN1 and other transcription factors as listed in Table S2.
For pathway analysis, BRD4-S and BRD4-L up- and downregulated DEGs from each regulatory class in the RNA-seq Venn diagram (see Figures 3A and S5D, and Tables S3 and S4 for complete DEG lists from MDA-MB-231 and MCF-7 RNA-seq) were compiled for enriched gene sets with FDR < 0.05 in MSigDB (Molecular Signature Database, 7.0) Canonical pathways (CP) using the GSEA web interface (Broad Institute). The MSigDB CP includes 2199 gene sets from BioCarta, KEGG, Matrisome Project, PID, Reactome, SigmaAldrich, Signal Transduction KE, Signaling Gateway, and SuperArray SABiosciences. The tables listing CP identified (Figures S5B and S5E) also provide values for gene ratio (# query genes found vs. # total genes collected in each pathway), P-value (% of false positives from all tests), and FDR (% of false positives from significant tests).
Analysis of patient genomic datasets for correlating cancer progression and survival rate
The expression levels of BRD4 mRNA in breast cancer (BCa) patients were retrieved from TCGA (Cancer Genome Atlas Network, 2012) RNA-seq datasets (data_RNA_Seq_v2_expression_median) with normalized TPM (Transcripts Per Million). Clinical information of patients, including months of overall survival (OS), tumor grade/stage, and cancer type, was accessible from TCGA (data_clinical_patient). Classification of PAM50 subtypes and the values of BRD4 isoform PSI (percent spliced in) were downloaded from TCGA SpliceSeq (MD Anderson; Zhao et al., 2016). A compiled list of TCGA 709 BCa datasets including BRD4 isoform PSI, normalized RNA-seq read counts of BRD4/EN1/CST1/2/4, PAM50 subtype classification and OS status is shown in Table S5 and used to calculate average PSI and/or mRNA levels of BRD4/EN1/CST1/2/4 using Prism 8 with outliers removed. PSI- or mRNA-based prognosis (according to median values) and expression correlation in all tumor and PAM50 subtypes were similarly analyzed by using Prism 8.
The prognostic impact of BRD4-S RNA-seq DEGs on disease progression and patient survival was evaluated by analyzing publicly available BCa patient datasets based on RNA-seq and microarray expression profiling. BRD4-S-upregulated (Up) matrisome genes were analyzed in Oncomine (Thermo Fisher Scientific) for enhanced expression (indicated by P-values) in invasive ductal breast carcinoma (IDC, sample size 1,556 from METABRIC; Curtis et al., 2012) or invasive breast lobular carcinoma (ILC, sample size 148) in comparison with normal breast (sample size 144). Similar analysis was conducted in other cancer types (vs. their normal tissues) using P-value less than 1E-4 and FC cutoff greater than 2. The Kaplan-Meier (KM) plots of distant metastasis-free survival (DMFS/MFS) or OS rates for BRD4-S-Up and IDC-enriched matrisome genes in tumor samples were bifurcated into higher or lower expression levels based on combined median (or mean) expression of all tested genes examined in individual cohorts by PROGgeneV2 (Pan Cancer Prognostics Database; Goswami and Nakshatri, 2014) or analyzed in multiple cohorts by GOBO (Gene expression-based Outcome for Breast cancer Online, Lund University, Sweden; Ringnér et al., 2011). The bifurcated BRD4-S-Up and IDC-enriched matrisome expression levels were compared among patients classified according to PAM50 molecular subtypes with select matrisome gene signatures further analyzed based on gene function modules using GOBO. Survival analysis in GOBO includes studies from Chin, GSE11121, GSE12093, GSE2034, GSE2603, GSE5327, GSE6532, and GSE7390, with additional incorporation of GSE1456 and GSE3494 for merged gene function modules. Three additional studies (GSE58812, NKI, and GSE22133) were included in PROGgeneV2 prediction.
RNA-seq transcription factor (TF) motif analysis
MISP (Motif-based Interval Screener with PSSM, position-specific scoring matrix; Wang et al., 2013) in Cistrome (Liu et al., 2011) was used to identify TF motifs in H3K27ac ChIP-seq peaks (GSE85158; Xi et al., 2018) proximal to the type II cystatin gene clusters and mucin 5 and CTSW loci using P < 0.001 cutoff based on TF motifs defined in TRANSFAC, Protein Binding Microarray (PBM) and Scot Wolfe’s Protein DNA Binding Database. Identified TF motifs were filtered by tolerance score cutoff > 4.5 and Oncomine TF mRNA enrichment in IDC vs. normal breast (see Table S6). The expression levels of the 70 IDC-enriched TFs identified in the above-mentioned three top BRD4-S-Up DEGs, along with three non-IDC/loci-enriched TFs (used as controls), were further analyzed according to PAM50 subtypes using TCGA RNA-seq data. The median values of individual TF expression (see Table S6) in each PAM50 subtype were then used to create a compiled TF heatmap (Figure 4B) using Prism 8.
BRD4 immunoblot (IB), protein quantification and protein-protein interaction
For IB detection of BRD4 protein isoforms, PBS-washed cells were treated with 400-mM NaCl-containing mRIPA (Wu et al., 2006) for 30 min. Cell lysates (25–50 µg) were run on an 8% SDS-PAGE or 4–20% gradient gel and then wet-transferred overnight in a cold room at 50 V (20–30 mA) for 12 h using cold transfer buffer (25 mM Tris-HCl pH 8.0, 0.192 M glycine, 0.1% SDS and 20% methanol). Highly sensitive ECL reagents (e.g., Thermo Scientific™ SuperSignal™ West Femto Chemiluminescent Substrate) were used for detection of low-abundant proteins such as BRD4-S and EN1. To quantify BRD4 protein molecules in the cell, we first determined the amount of Sf9-purified f:BRD4-L and f:BRD4-S by comparing their CBB staining signals with known amounts of BSA. IB was then performed with total lysates (10,000 or 40,000 MDA-MB-231 cells each lane) or purified BRD4 proteins (0.3–6.0 ng) with signal detection by anti-BRD4 N antibody. Quantification of cellular BRD4 (Figure S1G) was achieved by comparing IB signal intensity from ImagJ with those of purified BRD4 isoforms that were used to generate standard curves by excel trendline with R2 > 0.9. Molecular weights of BRD4-L (152,218.76 g/mol) and BRD4-S (80,463.27 g/mol) from Ensembl were used to calculate the number of BRD4 molecules in a cell. The amounts of BRD4 molecules in other cell lines (Figure S1I) were estimated by comparing each cellular BRD4 protein signal with those defined in MDA-MB-231 cells.
For cellular BRD4 and EN1 interaction analysis, BCa cells (1x107) either untreated or transfected by siCtrl or siEN1 #1 using RNAiMax (Gibco-Invitrogen) for 48–72 h were lysed in 1 ml buffer A (0.3 M sucrose, 60 mM KCl, 60 mM Tris-HCl, pH 8.0, 2 mM EDTA, and 0.5% NP-40 plus protease inhibitors). Nuclei were then collected for extract preparation by incubating pellets with 3x pellet volume of mRIPA supplemented with 400 mM NaCl at 4°C for 30 min. The salt concentration in the nuclear extract was reduced to 100 mM by adding no-salt mRIPA, followed by preclearance and subsequent immunoprecipitation (IP) with anti-EN1, anti-BRD4-S, or anti-BRD4-L antibodies (Wu et al., 2006; Wu et al., 2016). Direct BRD4-EN1 interaction was performed by in vitro pulldown using purified recombinant proteins by first mixing 0.5 µg of f:EN1 or f:EN2 with 1 µg of f:BRD4-L or 0.4 µg of f:BRD4-S (both purified from insect Sf9 cells) in BC100 buffer (Wu et al., 2013), followed by IP with anti-BRD4 N antibody (Wu et al., 2006) and IB detection with anti-FLAG antibody. All antibodies used for this study are listed in the Key Resources Table.
ChIP-qPCR and ChIP-seq
For standard ChIP-qPCR, MDA-MB-231 cells (1x107), with or without prior siRNA treatment (for 48 h), were first fixed in formaldehyde and processed as previously described (Wu et al., 2016). ChIP was conducted with 1 µg of IgG or purified antibodies against BRD4-S, EN1, H3, H3K14ac, H3K27ac, H3K9me3 and Cas9, or with 5 µl of anti-BRD4-L serum and analyzed by end-point qPCR using 4% of IP products and 0.2% of input DNA (see Table S2 for the complete primer list).
For BRD4 isoform-specific ChIP-seq, MDA-MB-231 and its derived 3f:S cells that constitutively express 3xFLAG-tagged BRD4-S established by lentivirus-mediated gene transfer were treated respectively with 1 µM of inactive JQ1(-) enantiomer (ApexBio Technology A8181) or active JQ1(+) (ApexBio Technology A1910) for 4 h and crosslinked with 1.5% formaldehyde for 10 min at room temperature. Glycine was added to a final concentration of 125 mM to stop the crosslinking reaction. Cells were washed twice with cold 1xPBS and collected in Farnham lysis buffer (5 mM PIPES pH 8.0, 85 mM KCl, 0.5% NP-40, supplemented with protease inhibitors). Pellets from 1x108 cells were resuspended in 40 ml of Farnham lysis buffer and centrifugated to remove cytosolic proteins. Resulting nuclear pellets were resuspended in 5 ml of TE buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA, supplemented with protease inhibitors) and sonicated for 5 min on ice. Detergents were then added to sonicated chromatin solutions to adjust the concentration to 1xRIPA buffer (10 mM Tris-HCl pH 7.6, 1 mM EDTA, 0.1% SDS, 0.1% sodium deoxycholate, 1% Triton X-100). For BRD4-S and EN1 ChIP-Seq, chromatin samples prepared from 1x108 cells were used. For BRD4-L ChIP-Seq, chromatin from 2x107 cells was used. For each ChIP reaction, 10–15 µg of purified antibodies were pre-incubated with Protein A-conjugated Dynabeads in 1 ml of PBS at 4°C overnight. Chromatin was mixed with antibody-Protein A complex and incubated at 4°C overnight. Bead-bound protein-DNA complexes were washed twice sequentially with each RIPA buffer, RIPA+300 mM NaCl, LiCl buffer (50 mM Tris-HCl pH 7.5, 250 mM LiCl, 0.5% NP-40, 0.5% sodium deoxycholate), and 1xPBS. Samples were reverse-crosslinked in 100 µl of elution buffer (1% SDS, 0.1 M NaHCO3, and 100 µg proteinase K) at 65°C overnight. DNA was purified by AMPure XP beads and used for sequencing library construction.
CUT&RUN (Cleavage Under Targets and Release Using Nuclease)
CUT&RUN was performed as previously described (Skene and Henikoff, 2017, Skene et al., 2018) with modification for BRD4-S and EN1 detection. Briefly, MDA-MB-231 cells with or without ectopically expressed 3f:S were scraped from unfixed plates with total 1x106 or 6x106 cells used for BRD4-L/3f:S or BRD4-S/EN1 analysis. After three rinses each with 1-ml wash buffer (20 mM HEPES, pH 7.5, 150 mM NaCl, 0.5 mM spermidine, and protease inhibitors) followed by 3 min 600 g spin at RT, cells in 1 ml wash buffer were captured by Concanavalin A-coated magnetic beads (10 µl per million cells; Bangs laboratories, Inc.) at RT for 10 min with constant rotation. Upon three more washes, captured cells were permeabilized in 100 to 300 µl of the same wash buffer plus 0.05% digitonin and 2 mM EDTA with simultaneous antibody binding (purified ⍺-BRD4-L, 1 µg; ⍺-BRD4-S or ⍺-EN1, 2 µg; and corresponding amounts of IgG control) at 4°C overnight. Permeabilized cells/nuclei retained on the beads were washed 3 times with the same digitonin-wash buffer before incubation with 150 µl of Protein A-fused Micrococcal Nuclease (pA-MNase; 700 ng/ml) at 4°C for 1 h. After washing away unbound pA-MNase, antibody-associated protein-DNA complex was released from chromatin by adding CaCl2 to digitonin-wash buffer for a final 2 mM CaCl2 concentration and incubated at 0°C (i.e., ice-water bath) for 30 min. Equal volume of 2x stop buffer (340 mM NaCl, 20 mM EDTA, 4 mM EGTA, 0.05% digitonin, 100 µg/ml RNase A, 50 µg/ml glycogen) containing spike-in yeast DNA (20 pg/ml) was then added to the reaction with an additional 30-min incubation at 37°C. The released DNA was isolated by proteinase K treatment and phenol/chloroform extraction and ethanol precipitation as described (Skene et al., 2018).
ChIP-seq and CUT&RUN Library Preparation and Sequencing
NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB) was used to prepare ChIP-seq libraries following manufacturer’s protocol and also for CUT&RUN library construction with modified procedures to preserve short DNA fragments during end-repair/dA-tailing and size selection (Liu et al., 2018) and our adjustment for BRD4-S and EN1 mapping with more isolated DNA (30–100 ng) and reduced adaptor (0.6 pmole). Barcoded libraries were pooled for single-end sequencing using NextSeq 500 High Output (75 cycles), except using Illumina HiSeq 2500 for BRD4-L ChIP sequencing.
Quality Control (QC) for ChIP-seq and CUT&RUN
Raw sequencing data from BRD4 isoform ChIP-seq and both EN1 ChIP-seq and CUT&RUN were processed by BICF ChIP-seq Analysis Workflow (Barnes et al., 2019) using BioHPC Astrocyte web interface. In general, raw reads from fastq files were trimmed by TrimGalore (Marcel, 2011), aligned to hg19 using BWA-MEM (Li and Durbin. 2009), and then converted to bam files using SAM sort/convert software (Li et al., 2009). Duplicate reads in the resulting bam files were marked with Sambamba (Tarasov et al. 2015). For BRD4 isoform CUT&RUN, FASTP (Chen et al., 2018) was used for trimming raw reads in Galaxy. Following BWA mapping to hg19 and sorting, PCR duplicates in the sorted bam files were removed by SAM/BAM RmDuP. MapQ of deduplicated reads was checked by Qualimap BamQC using Galaxy SAM/BAM tool (Table S1). Percent unique-mapped reads were defined as the ratio of deduplicated reads divided by total input reads. Sequencing read QC and library complexity were evaluated by NRF, PBC, NSC, and RSC values generated by BICF ChIP-seq Analysis Workflow using BWA-MEM and PhantomPeakQualTools (Landt et al., 2012) according to the standards set by ENCODE (Table S1). Yeast spike-in control DNA included in CUT&RUN was similarly mapped to Saccharomyces cerevisiae genome (GCF_000146045.2_R64_genomic.fna) using BWA in Galaxy.
MACS2 (Zhang et al., 2008) was used for peak calling from deduplicated bam files with appropriate input or IgG controls (Table S1). The peak-calling parameters for BRD4 CUT&RUN are the same as listed in the Galaxy default setting (effective genome size 2.45e+9, band width 300, model fold [5, 50], q cutoff 0.05, larger dataset scaled towards smaller dataset, range for calculating regional lambda 1000 bp and 10000 bp). For BRD4 ChIP-seq and both EN1 ChIP-seq and CUT&RUN, peak calling was similarly conducted with some setting changes (p cut off 0.01 --nomodel --shift 0 --extsize 165 --keep-dup all -B --SPMR) using BICF ChIP-seq Analysis Workflow. QC was performed as instructed from Cistrome ChiLin (Qin et al., 2016). Significant peak numbers with FDR (q) ≤ 0.05 and fold change (Fc) >10 or 20 for single or pooled biological samples are indicated in Table S1. FRiP (percent fraction of reads in peaks) from all significant peaks was obtained from Deeptool plotEnrichment in Galaxy (Ramírez et al., 2016). For peaks overlapping with DNase I hypersensitive sites (DHS), summits of top 5000 peaks were used to intersect ENCODE DHS sites compiled from 125 human cell types and tissues (wgEncodeAwgDnaseMasterSites.bed). For genomic distribution analysis, summits from all significant peaks were used to intersect genomic regions provided in UCSC knownGene table and including promoter (2 kb relative to TSS in both directions, processed with SlopBed), exon, intron (defined as gene regions not in exon and promoter), and intergenic regions (not found in promoter, exon, and intron). PhastCons-based conservation analysis was conducted at Cistrome using top 5000 peak summits. MACS2 peaks of BRD4 isoforms and EN1 are listed in Table S7.
Genomic Binding and TF Motif Analysis
BRD4 isoform-binding peaks were intersected using BEDTools (Quinlan and Hall, 2010) with results shown in Venn diagrams. ChIP-seq peaks for H3K27ac, H3K4me1 and H4K4me3 in MDA-MB-231 were called by MACS2 (P < 0.05) using Bowtie-aligned bam files prepared from GSE85158 datasets. Signal tracks reflecting signal fold change (sample vs. input) in ChIP-seq generated by BICF ChIP-seq Analysis Workflow or spike-in-normalized signals in CUT&RUN were displayed using Integrative Genomics Viewer (IGV 2.4.8; Robinson et al., 2017). CUT&RUN spike-in-normalized signals were generated by Deeptools bamcoverage with scaling factors determined by the ratios of spike-in DNA in the samples relative to that of 231_unt_CUT&RUN_Ig_3 control (Table S1). The ChromHMM track from HMEC (human mammary epithelial cells) used for active enhancer and promoter visualization in the type II cystatin gene clusters was obtained from the UCSC Genome Browser.
Heatmaps of BRD4-binding signals enriched in TF genome-wide binding regions were generated by plotHeatmap based on computMatrix scores calculated from ChIP-seq or CUT&RUN BRD4 fc signals within +/- 2-kb regions of TF ChIP-seq peaks. The fc signals in CUT&RUN were obtained by comparing BRD4 isoform signals (231_unt_CUT&RUN_L or 231_unt_S_CUT&RUN_2; Table S1) with that of IgG control (231_unt_CUT&RUN_Ig_1 or 231_unt_CUT&RUN_Ig_3) after spike-in normalization using Galaxy bamcompare. Genome-wide EN1 peaks used for BRD4 association heatmaps are from CUT&RUN (EN1_CUT&RUN_pooled, 16,326 regions) or ChIP-seq (231_EN1_pooled, 18,826 regions) datasets collected from MDA-MB-231 cells or downloaded from V5-tagged EN1 ChIP-seq generated in SUM159 cells (98,000 regions, GSE120957). c-Jun (24,554 regions) and TEAD4 (8,406 regions) peaks were both downloaded from MDA-MB-231 ChIP-seq datasets (GSE66081), whereas NF-YA peaks (7,647 regions) were from ENCODE ChIP-seq performed in K562 cells. Results were essentially the same when another BRD4-S biological repeat (231_unt_S_CUT&RUN_1) was used for heatmap analysis.
Correlation between BRD4-L/S-regulated DEGs identified in MDA-MB-231 (Figure S5B) with that of EN1 in SUM159 (GSE120957_combined-gene-level-counts; Peluffo et al., 2019) was analyzed by GSEA (Gene Set Enrichment Analysis, Broad Institute). Direct transcriptional targets (DTTs) were identified by integrating RNA-seq and CUT&RUN datasets using Cistrome-GO (Li et al., 2019) with enhancer mode (half decay distance = 50 kb) due to low levels (< 20%) of promoter-associated BRD4. Canonical pathway analysis was performed by MSigDB-CP as described in RNA-seq experiments.
Overrepresented TF motifs in BRD4 isoform-binding peaks were identified by RSAT (Regulatory Sequence Analysis Tools; Nguyen et al., 2018) after retrieving the interval sequences. Motif identification was performed by: 1) obtaining peak sequences +/- 500 bp on each side of peak centers, 2) finding overrepresented nucleotide patterns with position bias within 6 or 7 nucleotide length, 3) background adjustment with Markov order of background model according to sequence length, and 4) comparing identified sequences with motif databases such as JASPAR Core Nonredundant Vertebrates. Significance cutoff is -Log10(e-value) ≥ 10.
QUANTIFICATION AND STATISTICAL ANALYSIS
Biological assays are typically presented as mean ± standard error of the mean (s.e.m.) and performed with at least three independent biological samples, unless otherwise stated. RT-qPCR-based assays were presented as mean ± standard deviation (s.d.) and performed with at least three independent biological samples, whereas ChIP-qPCR was also presented as mean ± standard deviation (s.d.) and performed with at least two independent biological samples. Significance is *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 from two-way ANOVA followed by Tukey post hoc test for multiple group comparisons or from unpaired two-tailed t-test for pairwise comparison, unless otherwise specified.
Supplementary Material
Highlights.
Oncogenic BRD4-S and tumor-suppressive BRD4-L in breast cancer
Inducible BRD4-L/S transgenic mice exhibiting opposing functions of BRD4 isoforms
Genome-wide RNA-seq ChIP-seq CUT&RUN profiling of BRD4-S and BRD4-L
EN1/BRD4-S-coregulated enhancer modulating matrisome ECM network
ACKNOWLEDGMENTS
We thank Steven Henikoff for providing pA-MNase protein and spike-in yeast DNA used in CUT&RUN, Khandan Keyomarsi, Ganesh Raj, Srinivas Malladi and Weibo Luo for select breast cancer cell lines, Scott Floyd for BRD4 isoform B (i.e., short form b) cDNA, Jessica Castaneda-Gill for assistance in animal work, Venkat Malladi, Anusha Nagari and Beibei Chen for bioinformatics help, Ling Cai for guidance in patient survival and TCGA analysis, and Hongtao Yu, Jian Xu, Min Ni and Xie Wei for discussion. We also thank NGS Core for sequencing and help in RNA-seq analysis, and BioHPC for accessing genome-wide analysis platforms. We are grateful for William Chiang, Louise Chow, Thomas Wilkie, Hongtao Yu and Jian Xu for comments on the manuscript and particularly Jian Xu and Miao Cui for discussion about CUT&RUN. This work was supported in part by NIH grant 1RO1CA251698–01, CPRIT grants RP180349 and RP190077, and Welch Foundation grant I-1805 to C.-M.C. The research in Y.W.’s lab is supported by grants from NIH (RO1CA229487 and R01CA236802), DOD (W81XWH-18–1-0014), CPRIT (RP180047) and Welch Foundation (I-1751). Y.W. is Lawrence Raisz Professor in Bone Cell Metabolism and a Virginia Murchison Linthicum Scholar in Medical Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental information, including seven Figures and seven Tables, is available online.
DECLARATION OF INTEREST
The authors declare no competing interests.
REFERENCES
- Afgan E, Baker D, Batut B, van den Beek M, Bouvier D, Cech M, Chilton J, Clements D, Coraor N, Grüning BA, Guerler A, Hillman-Jackson J, Hiltemann S, Jalili V, Rasche H, Soranzo N, Goecks J, Taylor J, Nekrutenko A, and Blankenberg D (2018) The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res 46, W537–W544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsarraj J, Walker RC, Webster JD, Geiger TR, Crawford NP, Simpson RM, Ozato K, and Hunter KW (2011) Deletion of the proline-rich region of the murine metastasis susceptibility gene Brd4 promotes epithelial-to-mesenchymal transition- and stem cell-like conversion. Cancer Res 71, 3121–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronesty E (2013) Comparison of sequencing utility programs. Open Bioinf. J 7, 1–8. [Google Scholar]
- Beltran AS, Graves LM, and Blancafort P (2014) Novel role of Engrailed 1 as a pro-survival transcription factor in basal-like breast cancer and engineering of interference peptides block its oncogenic function. Oncogene 33, 4767–4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobola N, and Merabet S (2017) Homeodomain proteins in action: similar DNA binding preferences, highly variable connectivity. Curr. Opin. Genet. Dev. 43, 1–8. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network. (2012) Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Zhou Y, Chen Y, and Gu J (2018) Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C-M (2009) Brd4 engagement from chromatin targeting to transcriptional regulation: selective contact with acetylated histone H3 and H4. F1000 Biol. Rep 1, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C-M (2014) Nonequivalent response to bromodomain-targeting BET inhibitors in oligodendrocyte cell fate decision. Chem. Biol 21, 804–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C-M (2016) Phospho-BRD4: transcription plasticity and drug targeting. Drug Discov. Today Technol 19, 17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Xu J, Sun R, Mumbach MR, Carter AC, Chen YG, Yost KE, Kim J, He J, Nevins SA, Chin SF, Caldas C, Liu SJ, Horlbeck MA, Lim DA, Weissman JS, Curtis C, and Chang HY 2018. Promoter of lncRNA Gene PVT1 Is a Tumor-Suppressor DNA Boundary Element. Cell 173, 1398–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad RJ, Fozouni P, Thomas S, Sy H, Zhang Q, Zhou MM, and Ott M (2017) The short isoform of BRD4 promotes HIV-1 latency by engaging repressive SWI/SNF chromatin-remodeling complexes. Mol. Cell 67, 1001–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford NP, Alsarraj J, Lukes L, Walker RC, Officewala JS, Yang HH, Lee MP, Ozato K, and Hunter KW (2008) Bromodomain 4 activation predicts breast cancer survival. Proc. Natl. Acad. Sci. USA 105, 6380–6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, Gräf S, Ha G, Haffari G, Bashashati A, Russell R, McKinney S, METABRIC Group, Langerød A, Green A, Provenzano E, Wishart G, Pinder S, Watson P, Markowetz F, Murphy L, Ellis I, Purushotham A, Børresen-Dale AL, Brenton JD, Tavaré S, Caldas C, Aparicio S (2012) The genomic and transcriptomic architecture of 2,000 breast tumors reveals novel subgroups. Nature 486, 346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai DN, Li Y, Chen B, Du Y, Li SB, Lu SX, Zhao ZP, Zhou AJ, Xue N, Xia TL, Zeng MS, Zhong Q, and Wei WD (2017) Elevated expression of CST1 promotes breast cancer progression and predicts a poor prognosis. J. Mol. Med 95, 873–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalvi MP, Wang L, Zhong R, Kollipara RK, Park H, Bayo J, Yenerall P, Zhou Y, Timmons BC, Rodriguez-Canales J, Behrens C, Mino B, Villalobos P, Parra ER, Suraokar M, Pataer A, Swisher SG, Kalhor N, Bhanu NV, Garcia BA, Heymach JV, Coombes K, Xie Y, Girard L, Gazdar AF, Kittler R, Wistuba II, Minna JD, and Martinez ED (2017) Taxane-Platin-resistant lung cancers co-develop hypersensitivity to JumonjiC demethylase inhibitors. Cell Rep 19, 1669–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolfini D, Andrioletti V, and Mantovani R 2019. Overexpression and alternative splicing of NF-YA in breast cancer. Sci. Rep 9: 12955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedoriw A, Rajapurkar SR, O’Brien S, Gerhart SV, Mitchell LH, Adams ND, Rioux N, Lingaraj T, Ribich SA, Pappalardi MB, Shah N, Laraio J, Liu Y, Butticello M, Carpenter CL, Creasy C, Korenchuk S, McCabe MT, McHugh CF, Nagarajan R, Wagner C, Zappacosta F, Annan R, Concha NO, Thomas RA, Hart TK, Smith JJ, Copeland RA, Moyer MP, Campbell J, Stickland K, Mills J, Jacques-O’Hagan S, Allain C, Johnston D, Raimondi A, Porter Scott M, Waters N, Swinger K, Boriack-Sjodin A, Riera T, Shapiro G, Chesworth R, Prinjha RK, Kruger RG, Barbash O, and Mohammad HP (2019) Anti-tumor activity of the type I PRMT inhibitor, GSK3368715, synergizes with PRMT5 inhibition through MTAP loss. Cancer Cell 36, 100–114. [DOI] [PubMed] [Google Scholar]
- Fernandez P, Scaffidi P, Markert E, Lee JH, Rane S, and Misteli T (2014) Transformation resistance in a premature aging disorder identifies a tumor-protective function of BRD4. Cell Rep 9, 248–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, and Bradner JE (2010) Selective inhibition of BET bromodomains. Nature 468, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd SR, Pacold ME, Huang Q, Clarke SM, Lam FC, Cannell IG, Bryson BD, Rameseder J, Lee MJ, Blake EJ, Fydrych A, Ho R, Greenberger BA, Chen GC, Maffa A, Del Rosario AM, Root DE, Carpenter AE, Hahn WC, Sabatini DM, Chen CC, White FM, Bradner JE, and Yaffe MB (2013) The bromodomain protein Brd4 insulates chromatin from DNA damage signaling. Nature 498, 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong CY, Gilan O, Lam EY, Rubin AF, Ftouni S, Tyler D, Stanley K, Sinha D, Yeh P, Morison J, Giotopoulos G, Lugo D, Jeffrey P, Lee SC, Carpenter C, Gregory R, Ramsay RG, Lane SW, Abdel-Wahab O, Kouzarides T, Johnstone RW, Dawson SJ, Huntly BJ, Prinjha RK, Papenfuss AT, and Dawson MA (2015) BET inhibitor resistance emerges from leukaemia stem cells. Nature 525, 538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco HL, Nagari A, Malladi VS, Li W, Xi Y, Richardson D, Allton KL, Tanaka K, Li J, Murakami S, Keyomarsi K, Bedford MT, Shi X, Li W, Barton MC, Dent SYR, and Kraus WL (2018) Enhancer transcription reveals subtype-specific gene expression programs controlling breast cancer pathogenesis. Genome Res 28,159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami CP, and Nakshatri H (2014) PROGgeneV2: enhancements on the existing database. BMC Cancer 14, 970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Long H, Bu Q, Zhao Y, Wang H, Tian J, and Cen X (2019) Role of BRD4 phosphorylation in the nucleus accumbens in relapse to cocaine-seeking behavior in mice. Addict. Biol 30, e12808. [DOI] [PubMed] [Google Scholar]
- Guy CT, Cardiff RD, and Muller WJ (1992) Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol. Cell. Biol 12, 954–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha S, Vande Pol S, Banerjee NS, Dutta AB, Chow LT, and Dutta A (2010) Destabilization of TIP60 by human papillomavirus E6 results in attenuation of TIP60-dependent transcriptional regulation and apoptotic pathway. Mol. Cell 38, 700–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Yan Y, Wang D, Ding D, Ma T, Ye Z, Jimenez R, Wang L, Wu H, and Huang H (2018) DUB3 promotes BET inhibitor resistance and cancer progression by deubiquitinating BRD4. Mol. Cell 71, 592–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordón-Cardo C, Guise TA, and Massagué J (2003) A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 3, 537–549. [DOI] [PubMed] [Google Scholar]
- Kenny PA, Lee GY, Myers CA, Neve RM, Semeiks JR, Spellman PT, Lorenz K, Lee EH, Barcellos-Hoff MH, Petersen OW, Gray JW, and Bissell MJ (2007) The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Mol. Oncol 1, 84–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, and Haussler D (2002) The human genome browser at UCSC. Genome Res 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology, 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J-T, Lee S-J, Kang MA, Park JE, Kim B-Y, Yoon D-Y, Yang Y, Lee C-H, Yeom YI, Choe Y-K, and Lee HG (2013) Cystatin SN neutralizes the inhibitory effect of cystatin C on cathepsin B activity. Cell Death Dis 4, e974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korb E, Herre M, Zucker-Scharff I, Darnell RB, and Allis CD (2015) BET protein Brd4 activates transcription in neurons and BET inhibitor Jq1 blocks memory in mice. Nat Neurosci 18, 1464–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzeszinski JY, Wei W, Huynh H, Jin Z, Wang X, Chang TC, Xie XJ, He L, Mangala LS, Lopez-Berestein G, Sood AK, Mendell JT, and Wan Y (2014) miR-34a blocks osteoporosis and bone metastasis by inhibiting osteoclastogenesis and Tgif2. Nature 512, 431–435. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kumari S, Nie J, Chen HS, Ma H, Stewart R, Li X, Lu MZ, Taylor WM, and Wei H (2012) Evaluation of gene association methods for coexpression network construction and biological knowledge discovery. PLoS One 7, e50411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, Chen Y, DeSalvo G, Epstein C, Fisher-Aylor KI, Euskirchen G, Gerstein M, Gertz J, Hartemink AJ, Hoffman MM, Iyer VR, Jung YL, Karmakar S, Kellis M, Kharchenko PV, Li Q, Liu T, Liu XS, Ma L, Milosavljevic A, Myers RM, Park PJ, Pazin MJ, Perry MD, Raha D, Reddy TE, Rozowsky J, Shoresh N, Sidow A, Slattery M, Stamatoyannopoulos JA, Tolstorukov MY, White KP, Xi S, Farnham PJ, Lieb JD, Wold BJ, and Snyder M (2012) ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res 22, 1813–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasko LM, Jakob CG, Edalji RP, Qiu W, Montgomery D, Digiammarino EL, Hansen TM, Risi RM, Frey R, Manaves V, Shaw B, Algire M, Hessler P, Lam LT, Uziel T, Faivre E, Ferguson D, Buchanan FG, Martin RL, Torrent M, Chiang GG, Karukurichi K, Langston JW, Weinert BT, Choudhary C, de Vries P, Van Drie JH, McElligott D, Kesicki E, Marmorstein R, Sun C, Cole PA, Rosenberg SH, Michaelides MR, Lai A, and Bromberg KD (2017) Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 550, 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AY, and Chiang C-M (2009) Chromatin adaptor Brd4 modulates E2 transcription activity and protein stability. J. Biol. Chem 284, 2778–2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J-E, Park Y-K, Park S, Jang Y, Waring N, Dey A, Ozato K, Lai B, Peng W and Ge K (2017) Brd4 binds to active enhancers to control cell identity gene induction in adipogenesis and myogenesis. Nat. Commun 8, 2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, and Pietenpol JA (2011) Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest 121, 2750–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and 1000 Genome Project Data Processing Subgroup (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Wan C, Zheng R, Fan J, Dong X, Meyer CA, and Liu XS (2019) Cistrome-GO: a web server for functional enrichment analysis of transcription factor ChIP-seq peaks. Nucleic Acids Res 47, W206–W211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2013) featureCounts: an efficient general-purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liu N, Hargreaves VV, Zhu Q, Kurland JV, Hong J, Kim W, Sher F, Macias-Trevino C, Rogers JM, Kurita R, Nakamura Y, Yuan GC, Bauer DE, Xu J, Bulyk ML, and Orkin SH (2018). Direct promoter repression by BCL11A controls the fetal to adult hemoglobin switch. Cell 173, 430–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Ortiz JA, Taing L, Meyer CA, Lee B, Zhang Y, Shin H, Wong SS, Ma J, Lei Y, Pape UJ, Poidinger M, Chen Y, Yeung K, Brown M, Turpaz Y, and Liu XS (2011) Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol 12, R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockwood WW, Zejnullahu K, Bradner JE, and Varmus H (2012) Sensitivity of human lung adenocarcinoma cell lines to targeted inhibition of BET epigenetic signaling proteins. Proc. Natl. Acad. Sci. USA 109, 19408–19413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcel M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17,10–12. [Google Scholar]
- Mewes HW, Heumann K, Kaps A, Mayer K, Pfeiffer F, Stocker S, and Frishman D (1999) MIPS: a database for genomes and protein sequences. Nucleic Acids Res 27, 44–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, and Massagué J (2005) Genes that mediate breast cancer metastasis to lung. Nature 436, 518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki K, Nishiyama A, Jang MK, Dey A, Ghosh A, Tamura T, Natsume H, Yao H, and Ozato K (2008) The bromodomain protein Brd4 stimulates G1 gene transcription and promotes progression to S phase. J. Biol. Chem 283, 9040–9048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen NTT, Contreras-Moreira B, Castro-Mondragon JA, Santana-Garcia W, Ossio R, Robles-Espinoza CD, Bahin M, Collombet S, Vincens P, Thieffry D, van Helden J, Medina-Rivera A, and Thomas-Chollier M (2018) RSAT 2018: regulatory sequence analysis tools 20th anniversary. Nucleic Acids Res 46, W209–W214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H, White J, Kirilovsky J, Rice CM, Lora JM, Prinjha RK, Lee K, and Tarakhovsky A (2010) Suppression of inflammation by a synthetic histone mimic. Nature 468, 1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peluffo G, Subedee A, Harper NW, Kingston N, Jovanović B, Flores F, Stevens LE, Beca F, Trinh A, Reddy Chilamakuri CS, Papachristou EK, Murphy K, Su Y, Marusyk A, D’Santos CS, Rueda OM, Beck AH, Caldas C, Carroll JS, and Polyak K (2019) EN1 is a transcriptional dependency in triple-negative breast cancer associated with brain metastasis. Cancer Res 79, 4173–4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Q, Mei S, Wu Q, Sun H, Li L, Taing L, Chen S, Li F, Liu T, Zang C, Xu H, Chen Y, Meyer CA, Zhang Y, Brown M, Long HW, and Liu XS (2016) ChiLin: a comprehensive ChIP-seq and DNase-seq quality control and analysis pipeline. BMC Bioinformatics, 17, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman S, Sowa ME, Ottinger M, Smith JA, Shi Y, Harper JW, and Howley PM (2011) The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol. Cell. Biol 31, 2641–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016) DeepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, pW160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathert P, Roth M, Neumann T, Muerdter F, Roe JS, Muhar M, Deswal S, Cerny-Reiterer S, Peter B, Jude J, Hoffmann T, Boryń ŁM, Axelsson E, Schweifer N, Tontsch-Grunt U, Dow LE, Gianni D, Pearson M, Valent P, Stark A, Kraut N, Vakoc CR, and Zuber J (2015) Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 525, 543–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, and Chinnaiyan AM (2004) ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia 6, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringnér M, Fredlund E, Häkkinen J, Borg Å, and Staaf J (2011) GOBO: Gene expression-based outcome for breast cancer online. PLoS One 6, e17911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Wenger AM, Zehir A, and Mesirov JP (2017) Variant review with the Integrative Genomics Viewer (IGV). Cancer Res 77, 31–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010) edgeR: a bio-conductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan M, Wong WC, Brown R, Akbani R, Su X, Broom B, Melott J, and Weinstein J (2016) TCGASpliceSeq a compendium of alternative mRNA splicing in cancer. Nucleic Acids Res 44, 1018–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC, Li CH, Guo YE, Day DS, Schuijers J, Vasile E, Malik S, Hnisz D, Lee TI, Cisse II, Roeder RG, Sharp PA, Chakraborty AK, and Young RA (2018) Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu S, Lin CY, He HH, Witwicki RM, Tabassum DP, Roberts JM, Janiszewska M, Huh SJ, Liang Y, Ryan J, Doherty E, Mohammed H, Guo H, Stover DG, Ekram MB, Brown J, D’Santos C, Krop IE, Dillon D, McKeown M, Ott C, Qi J, Ni M, Rao PK, Duarte M, Wu S-Y, Chiang C-M, Anders L, Young RA, Winer E, Letai A, Barry WT, Carroll JS, Long H, Brown M, Liu XS, Meyer CA, Bradner JE, and Polyak K (2016) Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature 529, 413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, and Henikoff S (2017) An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6, e21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Henikoff JG, and Henikoff S (2018) Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat. Protoc 13, 1006–1019. [DOI] [PubMed] [Google Scholar]
- Socovich AM, and Naba A (2019) The cancer matrisome: from comprehensive characterization to biomarker discovery. Semin. Cell Dev. Biol 89, 157–166. [DOI] [PubMed] [Google Scholar]
- Soriano P (1999) Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Gene 21, 70–71. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, and Prins P (2015) Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakore PI, D’Ippolito AM, Song L, Safi A, Shivakumar NK, Kabadi AM, Reddy TE, Crawford GE, and Gersbach CA (2015) Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods 12, 1143–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Sun H, Ma J, Zang C, Wang C, Wang J, Tang Q, Meyer CA, Zhang Y, and Liu XS (2013) Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat. Protoc 8, 2502–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong M, Sun Y, Xi Z, Milazzo G, Poulos RC, Bartenhagen C, Bell JL, Mayoh C, Ho N, Tee AE, Chen X, Li Y, Ciaccio R, Liu PY, Jiang CC, Lan Q, Jayatilleke N, Cheung BB, Haber M, Norris MD, Zhang XD, Marshall GM, Wang JY, Hüttelmaier S, Fischer M, Wong JWH, Xu H, Perini G, Dong Q, George RE, and Liu T (2019) JMJD6 is a tumorigenic factor and therapeutic target in neuroblastoma. Nat. Commun 10, 3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SP, Lee DK, Demayo FJ, Tsai SY, and Tsai MJ (2010) Generation of ES cells for conditional expression of nuclear receptors and coregulators in vivo. Mol. Endocrinol 24, 1297–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S-Y, and Chiang C-M (2007) The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J. Biol. Chem 282, 13141–13145. [DOI] [PubMed] [Google Scholar]
- Wu S-Y, Lee AY, Hou SY, Kemper JK, Erdjument-Bromage H, Tempst P, and Chiang C-M (2006) Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev 20, 2383–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S-Y, Lee AY, Lai HT, Zhang H, and Chiang C-M (2013) Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol. Cell 49, 843–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S-Y, Nin DS, Lee AY, Simanski S, Kodadek T, and Chiang C-M (2016) BRD4 phosphorylation regulates HPV E2-mediated viral transcription, origin replication, and cellular MMP-9 expression. Cell Rep 16, 1733–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylie A, Jones AE, D’Brot A, Lu WJ, Kurtz P, Moran JV, Rakheja D, Chen KS, Hammer RE, Comerford SA, Amatruda JF, and Abrams JM (2016) p53 genes function to restrain mobile elements. Genes Dev 30, 64–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Y, Shi J, Li W, Tanaka K, Allton KL, Richardson D, Li J, Franco HL, Nagari A, Malladi VS, Coletta LD, Simper MS, Keyomarsi K, Shen J, Bedford MT, Shi X, Barton MC, Kraus WL, Li W, and Sharon YR (2018) Histone modification profiling in breast cancer cell lines highlights commonalities and differences among subtypes. BMC Genomics 19, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, and Vakoc CR (2017) Targeting cancer cells with BET bromodomain inhibitors. Cold Spring Harb. Perspect. Med 7, a026674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagawa Y, Kobayashi T, Ohnishi M, Kobayashi T, Tamura S, Tsuzuki T, Sanbo M, Yagi T, Tashiro F, and Miyazaki J (1999) Enrichment and efficient screening of ES cells containing a targeted mutation: the use of DT-A gene with the polyadenylation signal as a negative selection maker. Transgenic Res 8, 215–221. [DOI] [PubMed] [Google Scholar]
- Yang Z, He N, and Zhou Q (2008) Brd4 recruits P-TEFb to chromosomes at late mitosis to promote G1 gene expression and cell cycle progression. Mol. Cell. Biol 28, 967–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F, Battilana G, Forcato M, Filippi L, Azzolin L, Manfrin A, Quaranta E, Di Biagio D, Sigismondo G, Guzzardo V, Lejeune P, Haendler B, Krijgsveld J, Fassan M, Bicciato S, Cordenonsi M, and Piccolo S (2018) Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nat. Med 24, 1599–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, Rosato A, Bicciato S, Cordenonsi M, and Piccolo S (2015) Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol 17, 1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008) Model-based analysis of ChIP-seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Hoadley KA, Parker JS, and Perou CM (2016) Identification of mRNA isoform switching in breast cancer. BMC Genomics 17, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Rødland EA, Sørlie T, Naume B, Langerød A, Frigessi A, Kristensen VN, Børresen-Dale AL, and Lingjærde OC (2011). Combining gene signatures improves prediction of breast cancer survival. PLoS One 6, e17845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Bengsch F, Svoronos N, Rutkowski MR, Bitler BG, Allegrezza MJ, Yokoyama Y, Kossenkov AV, Bradner JE, Conejo-Garcia JR, and Zhang R (2016) BET Bromodomain inhibition promotes anti-tumor immunity by suppressing PD-L1 expression. Cell Rep 16, 2829–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, Taylor MJ, Johns C, Chicas A, Mulloy JC, Kogan SC, Brown P, Valent P, Bradner JE, Lowe SW, and Vakoc CR (2011) RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 478, 524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All RNA-seq, ChIP-seq and CUT&RUN datasets, along with bigwig (bw) files used for data analysis, have been deposited in the Gene Expression Omnibus under accession code GSE136151.