

Abstract
Oral cancers needs relentless research due to high mortality and morbidity associated with it. Despite of the comparable ease in accessibility to these sites, more than 2/3rd cases are diagnosed in advanced stages. Molecular/genetic studies augment clinical assessment, classification and prediction of malignant potential of oral lesions, thereby reducing its incidence and increasing the scope for early diagnosis and treatment of oral cancers. Herein we aim to review the role of apoptosis and genes associated with it in oral cancer development in order to aid in early diagnosis, prediction of malignant potential and evaluation of possible treatment targets in oral cancer. An internet-based search was done with key words apoptosis, genes, mutations, targets and analysis to extract 72 articles after considering inclusion and exclusion criteria. The knowledge of genetics and genomics of oral cancer is of utmost need in order to stop the rising prevalence of oral cancer. Translational approach and interventions at the early stage of oral cancer, targeted destruction of cancerous cells by silencing or promoting involved genes should be the ideal intervention.
Key words: Apoptosis, genes, mutations, oral cancer, target
Introduction
Cancer represents a tremendous burden on patients, their families and society, as is 2nd leading cause of death. The GLOBOCAN 2018 database reported increased global cancer burden from 8.8 million deaths (2011) to 9.6 million deaths (2018) with 18.1 million new cases in 2018. This is anticipated to escalate at 70% by 2030. Globally cancers of head and neck contribute 3.8% among all cancer cases accounting for 3.6% of deaths due to cancer. 1
Cancer development (carcinogenesis) is an intricate and multitiered process. To comprehend the molecular and genetic level changes one needs to overview and understand the basic mechanism behind the processes causing increased cell proliferation or decreased cell death.
Apoptosis is a systematic and harmonized sequential process causing cell death. This review aims to understand the role of apoptosis and genes associated with it in oral carcinogenesis to aid in early diagnosis, prediction of malignant potential and evaluation of possible treatment targets in oral cancer.
Method of data collection
An internet-based search was done with key words apoptosis, genes, mutations, targets and analysis. A total of 230 articles were displayed. On scrutinizing the title and abstracts, only 143 articles were found relevant in context to oral cancers. Out of them only the articles (72) with citation >2 and journal impact factor >1 were considered for this review.
Oral cancer
Cancer of oral cavity including lip ranks 12th most prevalent cancer in Asia, with about 16,88,50 new cases diagnosed per year. In Asia its standard incidence rate is estimated to be 3.8.2 In the Indian subcontinent, oral cancer ranks 3rd of all cancers and accounts for over 30% of all cancers in India.3
Oral cancer is a malignant neoplasm arising on lips (excluding the external surface) and/or oral cavity, but there is no uniformity in description of the term in literature. The descriptions of anatomic areas described as oral cancer sites are highly variable and hence hamper the comparison of data on oral cancers in various studies. The oral cancer comprises of cancer of the regions of oral cavity including-lips, labial and buccal mucosa, anterior two thirds of the tongue, maxillary and mandibular gingiva, retromolar region, floor of the mouth under the tongue and roof of the mouth.4 These sites are covered histologically by squamous epithelium (keratinized/non-keratinized, masticatory, specialized mucosa). Thus histologically squamous cell carcinomas with different levels of differentiation and with local and distant metastasis constitute 90% of oral cancers.5 Rest (10%) oral cancer comprises of verrucous carcinoma, minor salivary gland malignancies, mucosal melanoma, Kaposi’s sarcoma, primary intraosseous squamous cell carcinoma, osteosarcoma, odontogenic tumors, metastatic tumors and connective tissue tumors.
It is well understood that a stimulus (carcinogen) causes either genetic or epigenetic alterations that endows the cell to attain different peculiar carcinogenic traits leading to development of cancer. Also malignancy of the oral cavity may not necessarily initiate in the site analogous to the precancerous lesion and at times even the apparently normal appearing mucosa with precancerous lesion might exhibit dysplasia or molecular aberrations in another regions of oral mucosa suggesting malignant transformation. Hence cancer may develop in apparently normal tissue. This is further supported by the concept of field cancerization where a long-term exposure of a carcinogen preconditions a part of epithelium. This premalignant field extends beyond surgical margins and causes a new cancer at the same site. In response to this observation, World Health Organization under its committee on head and neck tumors recommended the term epithelial precursor lesions in its monograph and described it as altered epithelium with an increased likelihood for progression to squamous cell carcinoma.6,7
Histologically, OSCC arise from non-aberrant keratinocytes chronically exposed to a stimulus that disrupts the homeostasis causing epithelial hyperplasia, dysplasia of different grades, carcinoma in situ and an invasive carcinoma capable of metastasis, with subsequent clinical manifestations. Hence, presence of potentially malignant disorder could be first detectable clinical change in the epithelium suggesting its way progress to establishing OSCC.
But as outlined above due to difficulty in standardization of definition and identification of early pre-malignancy, its diagnosis is difficult. Therefore, various molecular and technological markers have been scrutinized to overcome this shortcoming.
Carcinogenesis: Hallmarks
Hallmarks of cancer are acquired functional capabilities that enable cancer cells to proliferate, survive, and disseminate.8 In 2017, Fouad YA defined the hallmarks more relevantly as acquired evolutionary-advantageous characteristics that facilitate transformation of phenotypically normal cells into malignant ones, and boost growth of malignant cells by exploiting host tissue (Figure 1). In different tumors, these hallmarks may be acquired by different mechanisms at various stages during carcinogenesis.9,10 The hallmarks of cancer are briefly described in Figure 2.
Apoptosis: important Hallmark and role in carcinogenesis
One of the key features acquired by cell most often though not always is capability to evade apoptosis. Apoptosis technically called as programmed cell death, is an essential element in pathogenesis of many diseases. Culprit could be either too much apoptosis like in degenerative diseases or too less apoptosis like in cancers. Too little apoptosis in cancers result in immortal malignant cells. The apoptotic machinery is intricate and comprise of several pathways. Errors at any level along the pathway can be the etiology of cancer.
Morphological and biochemical changes in apoptosis
The diverse morphological changes that ensue during apoptosis are visualized using Light and Electron microscopy are described in detail in Table 1. The main categories of biochemical changes observed in apoptosis are described in Table 2.
Table 1.
The various morphological changes that occur during apoptosis have been identified using light and electron microscopy.
| Light microscopy | Electron microscopy |
|---|---|
| Chiefly characterized by cell shrinkage and pyknosis Cell shrinkage - cells are smaller in size, with dense cytoplasm containing tightly packed organelles | Extensive plasma membrane blebbing followed by karyorrhexis and separation of cell fragments into apoptotic bodies (consisting of cytoplasm with tightly packed intact organelles with or without a nuclear fragment, enclosed within an intact plasma membrane) |
| Pyknosis - nuclear chromatin condensation | Electron-dense nuclear material characteristically aggregates peripherally under the nuclear me brane although there can also be uniformly dense nuclei |
| H&E staining - single cell or small clusters of cells appearing as a round or oval mass with dark eosinophilic cytoplasm and dense purple nuclear chromatin fragments | Apoptotic bodies are subsequently phagocytosed by macrophages, parenchymal cells, or neoplastic cells and degraded within phagolysosomes |
Table 2.
Biochemical changes in apoptosis.
| Category of changes | Observed mechanism |
|---|---|
| Activation of caspases | Inactive proenzyme form once activated often activates other procaspases allowing initiation cascade of apoptosis. Caspases have proteolytic activity and are able to cleave proteins at aspartic acid residues |
| DNA and protein breakdown | Ca2+-and Mg2+-dependent endonucleases fragments DNA into fragments of 180 to 200 base pairs (Bortner et al., 1995) along with extensive protein cross-linking through the expression and activation of tissue transglutaminase (Nemes et al., 1996) |
| Membrane changes and recognition by phagocytic cells | Expression of cell surface markers (phosphatidylserine) on the outer layers of the plasma membrane result in the early recognition and quick phagocytosis with minimal compromise to the surrounding tissue (Bratton et al., 1997). Other proteins (Annexin and calreticulin) are also exposed on the cell surface during apoptotic cell clearance. Annexin V is a recombinant phosphatidylserine-binding protein that interacts strongly and specifically with phosphatidylserine residues. Calreticulin binds to an LDL-receptor related protein on the engulfing cell and works as a recognition signal |
Pathways of apoptosis
Classical pathway
Initiation of apoptosis: i) Extrinsic/death receptor mediated pathway-Extrinsic pathway involves trans-membrane receptor mediated interactions (Figure 3); ii) Intrinsic pathway-These are mitochondrial-initiated events described in Figure 4.
Execution of apoptosis: It is a caspase - mediated event described in Figure 3.
Alternate pathway
Apoptosis can occur through alternate apoptotic pathways wherein either cytotoxic T cell directly phagocytize the cell or via Granzyme-Perforin mediated pathway/Novel pathway. The transmembrane pore forming molecules perforin is secreted which facilitates exophytic release of cytoplasmic granules through pores into the target cells.11,12
Apoptotic genes and its molecular biology
According to Jorge Finnigan A 2010, the genes participating in apoptosis can be broadly classified as: i) Pro-apoptotic genes: Includes Caspases, TNF (ligands and receptors), CARD, BCL-2, Death, CIDE domain and P53 families (Appendix Table A); ii) Anti-apoptotic genes: Comprises of different families like BCL-2, IAP, TRAF, CARD, DED and few other genes like AKT1, BRAF and BFAR (Appendix Table B).
Figure 1.
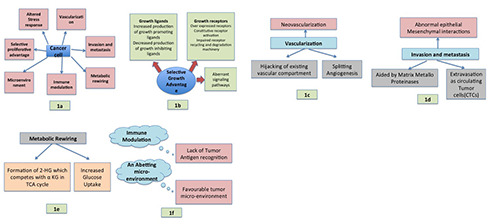
(a-f ) Depicting chief Hallmarks of cancer described by Fouad YA (2017).
Figure 2.
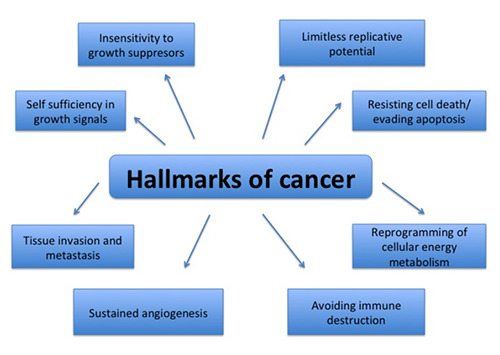
Hallmarks of cancer by Hanahan et al. (2011). Acquisition of these hallmarks of cancer by cells is crucial and interrelated in order to develop into cancerous growth. Though not always, in most of the cancers, cell needs to acquire all the hallmarks.
With an intention to comprehend the role of above -mentioned genes in oral cancer, a discussion and their current update is presented, as follows.
TNF family
TNF gene family codes for cytokines, which play vital roles in inflammation, immunity, cell proliferation, differentiation, and apoptosis.13,14 TNF gene family also codes for various ligands and their respective receptors. Majority of the TNF family members act as inducers of signaling pathways activating transcription factor NF-κB, however few ligands may cause apoptosis via binding to death receptors like Fas [CD95], TNFR1, TRAIL-R1 [DR4], TRAIL-R2 [DR5], DR6, TRAMP [DR3] and EDAR containing amino-terminal cysteine-rich domains (CRDs), to determine their specificity for ligand and death domain (DD), comprising 60-70 amino acids necessary for induction of apoptosis.13,15,16
Below is a list of genes in TNF Family:
- LTA gene (6p21.33) codes for lymphotoxin alpha, which aids in regulating innate immunity and has been shown to prevent cancer growth and destroy cancerous cell lines. LT-α, when secreted can either form a soluble homo-trimeric molecule or can heterotrimerize with lymphotoxin-beta to form a membrane bound complex (LT-α1-β2), which binds lymphotoxin-alpha with the surface of the cell.17,18 Mutations/polymorphisms in LT-α can promote disruptions in cell signaling pathways leading to cancer. In a study by Huang et al., 3 LTA polymorphisms were significantly associated with risk of having cancer.19 However such an association with oral cancer has not been reported much till date. Amongst the various polymorphisms reported, one reported by Vairaktaris et al. showed significant association with OSCC.20
- TNFRSF3 gene (12p13.31) codes for Lymphotoxin betareceptor (LTBR), a cell surface receptor for ligand lymphotoxin beta specifically LT-α1-β2 complex. LT-β receptors recruit lymphocytes to tumor cells to combat tumor growth.21 LTBR also interacts with TRAF3. The binding of LT-α1-β2 complex and LTBR induces activation of NF-κB leading to cell death. Yapijakis et al. demonstrated a strong association of TNF-α and TNF-β high expression alleles with increased risk of oral cancer.22
- TNF gene (TNFA) (6p21.3) codes for proteins which bind two receptors, TNFR1 (CD120a) and TNFR2 (CD120b). TNF receptors tri-merise upon contact with their ligand causing dissociation of SODD (inhibitory protein) thus permitting binding of adaptor protein TRADD to death domain causing: i) Activation of NF-κB factor causing transcription of proteins involved in inflammatory response, cell survival, proliferation, and anti-apoptotic factors); ii) Activation of the MAPK pathways (involved in cell proliferation, differentiation, apoptosis); and iii) Induction of death signaling via binding of TRADD and FADD to recruit caspase-8 leading to cell apoptosis. Several attempts have been made to gauze the association between tumor necrosis factor-alpha (TNFα) in oral precancers and cancers. Single nucleotide polymorphism (SNPs) in TNFα and TNF receptor 1 promoters have been significant with Oral squamous cell carcinomas thus can be used as a useful marker in high risk oral cancers.23-25 In oral premalignant lesions, significant elevation of salivary concentrations has been observed. However, in cancers not much change was observed indicating that TNFα is mainly inflammation- associated gene. Also it is believed that TNF is elevated only in premalignancies with higher component of inflammation.26-28
Figure 3.

Extrinsic pathway involves transmembrane receptor mediated interactions. It involves mainly Caspases. [ - TNFSF10 gene (3q26.31) codes for ligand TRAIL (TNFrelated apoptosis-inducing ligand). TRAIL leads to cellular death when it binds with death receptors DR4 (TRAIL-RI) and DR5 (TRAIL-RII) in normal cells.29 TRAIL also has capability to bind to decoy receptors DcR1 and DcR2 leading to transcription of antiapoptotic genes that antagonizes the death-signaling pathway. This is an observed mechanism in many cancer cells30 where it promotes inflammation.31 Owing to their anticipated role TRAIL and TRAIL receptors have been extensively evaluated for their expression in oral cancers and precancers. When compared to normal epithelium, the expression progressively diminished in oral precancers and cancers. An elevated expression of DR4, DR5 and DcR1 and a diminished DcR2 expression have been reported in oral cancer.32,33 Nagar et al. reported primary cancer cells to be more susceptible than metastatic secondary cells in TRAIL induced apoptosis but through Lysosomal Protease Cathepsin B.34 Many substances like suberoylanilide hydroxamic acid and esculetin have been observed to synergistically activate TRAIL thus promoting apoptosis demonstrated in oral cancers, cancer cell lines and animal models.35,36
- TNFRSF5 gene (20q13.12) codes for protein CD40 present on antigen presenting cells (APCs). The binding of ligand CD154 (CD40L) on TH cells to CD40 activates APCs provoking immune and inflammatory responses like expression of, chemokines cytokines, growth factors, matrix metalloproteinases, and adhesion molecules, via activation of NF kappa B.37 TNFRSF5 also interacts with TNFR-Receptor Associated Factor adaptor proteins TRAF1, TRAF2, TRAF5 and TRAF6. Gene expression profiling in Oral squamous cell carcinomas revealed that TNFRSF 5 was constantly deregulated although the increase in expression was <2 folds. Also when oral cancer cell lines were exposed to CD40 ligand apoptosis was observed revealing their vital role in tumorigenesis. 38,39
Figure 4.
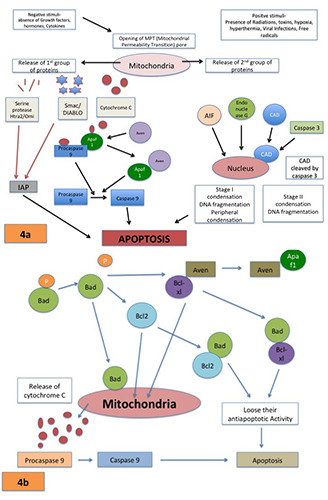
(a & b) Intrinsic pathway- Mitochondrial-initiated events having non-receptor mediated stimuli to produce intracellular signals act directly on targets within the cell causing changes in the inner mitochondrial membrane leading to opening of mitochondrial permeability transition (MPT) pore causing loss of mitochondrial trans-membrane potential along with release of proteins that causes oligo-nucleosomal DNA fragmentation followed by chromatin condensation.
- TNFRSF1 gene (12p13.31) codes for TNFR1 (tumor necrosis factor receptor 1), which acts as pervasive membrane receptor to bind TNFα also referred to as CD120a or tumor necrosis factor receptor superfamily member 1A (TNFRSF1A). TNFR1 activates the transcription factor NF-κB, regulates inflammation and reconciles apoptosis. It also interacts with adaptor proteins TRADD and TRAF2 and Anti-apoptotic protein BCL2-associated athanogene 4 (BAG4/SODD) hence regulating the receptor mediated signal transduction.40,41 Assessment of SNPs by Gupta et al. (2008) revealed lower expression in Tobacco related oral carcinomas thus qualifying TNFRSF1 SNPs as markers for high- risk groups.42
- TNFRSF25 gene (1p36.31) codes for death receptor 3 (DR3), a cell surface receptor that mediates apoptotic signaling and differentiation through TRADD adaptor molecule to enhance NF-kappa B activity or through the FADD adaptor molecule to enhance caspase activation causing apoptosis.43,44 Daigeler et al. 2008 demonstrated that combination of taurolidine and TRAIL led to upregulation of TNFRSF25 along with other proapoptotic genes on squamous carcinoma cells of the esophagus.45
- TNFRSF21 (tumor necrosis factor receptor superfamily member 21) gene (6p12.13) codes for cell surface receptor referred as death receptor 6 (DR6), which activates JNK/ MAPK8, NF-κB pathway and interacts with TRADD protein that mediates signal transduction of TNF-receptors to induce cell apoptosis. Overexpression of DR6 induces apoptosis. Daniel et al. 2001 demonstrated that Bax translocation is mandatory for DR6 triggered apoptosis, but the pathway is unknown.46 Wu et al. 2018 studied Upregulated miR-20a-5p expression in HNSCC cells and observed downregulation of TNFRSF21 expression.47
- TNFRSF7 gene (12p13.31) codes for receptor protein CD27 that binds to ligand CD70 to transduce signals activating NF-κB and MAPK8/JNK pathways. Adaptor proteins (TRAF2 and TRAF5) also mediate the signaling.48,49 CD27 can also bind to a pro-apoptotic protein (SIVA) inducing apoptosis.50 Binding of CD27 to its ligand causes T cell activation, differentiation, and survival thus suggesting its chemotherapeutic role via T cell mediated anti-tumor response.51
- TNFRSF9 gene (1p36.23) codes for receptor protein CD127 or 4-1BB and ILA (Induced by Lymphocyte Activation). The cytoplasmic domain of 4-1BB binds to TRAF proteins (TRAF1, TRAF2, and TRAF3), initiating a signaling cascade causing activation of NFκB.52,53 In cancers TNFRSF9 is upregulated due to antigen-induced activation of T cell causing their increased life expectancy and growth. Thus CD137 could be an effective biomarker which can identify antigen-stimulated T cells observed in cancers and other infections.54
BCL-2 family
Bcl-2 along with its homologs are active apoptosis regulators. In humans more than 20 members have been described. The proteins encoded can be either anti-apoptotic {BCL-2, MCL-1, BCLXL, BCL-W and Boo (Diva)} or pro-apoptotic {Bax, Bak, Bad, Bid, Bim, Bik, Hrk, Bcl-XS, Bcl-G, Nip3, APR (Noxa) and Nix (BNIP)}. The relative proportion of these anti- and pro-apoptotic proteins commands the response of cells to apoptotic stimuli.55
- BCL2 gene (18q21.33) codes for Anti-apoptotic protein bcl- 2. The bcl-2 proteins are confined to outer mitochondria, nuclear membrane and at endoplasmic reticulum, and possess hydrophobic amino acid band near C-end (trans-membrane domains) that binds bcl-2 proteins to the outer mitochondrial membrane.56,57 Bcl-2 over expression has been observed in multiple tumors including cancer of the lung, colon, glioblastomas, prostate and breast.58-60 Bcl-2 expression is also increased in OSCC and its precursor lesions.61-63 Bcl-2 is a multi- functional protein, which dimerize with another Bcl-2 proteins, binds to non- homologous proteins and forms ion- channels/pores.
- HRK gene (12q24.22) codes for Bcl-2 Interacting protein (Bip) also known as Harakiri protein. Bip localizes to membranes of intracellular organelles and differs in homology with members of Bcl-2 family apart from BH3 resembling region comprising of 8-amino acids. This BH3 domain facilitates interaction of HRK and Bcl-2 along with Bcl-XL, but not with Bax, Bak, or Bcl-XS.
- BIK gene (22q13.2) codes for Bcl-2-interacting killer protein. This protein also possesses the critical BH3 domain that interacts with Bcl-2 in order to enhance apoptosis. Bik protein is a target for anti-apoptotic proteins since its activity gets suppressed in their presence.64 Reis et al. studied genomic DNA from Primary oral cancers to reveal loss in sequences related to progression of disease. 65
- BAX gene (19q13.33) codes for pro-apoptotic bcl-2 like protein known as Bax (bcl-2-like protein 4). Bax and Bcl-2 compete with each other and Bcl-2: Bax ratio decides the relative response of cells to different apoptotic stimuli66 Bax hetero-dimerize with bcl-2 to activate apoptosis. In normal human cells, BAX is expressed in the cytosol. With onset of apoptosis, conformation of bax shifts to associate it with mitochondrial membrane67 and causes opening of voltage-dependent anion channel (VDAC), causing fall of membrane potential followed by discharge of cytochrome C and other pro-apoptotic factors from the mitochondria thus activating caspases. Bcl-2 along with p53 and Bif-1 activates BAX while VDAC2, Pin1, and IBRDC2 inactivates it. Also BAX expression is regulated by P53 and hence participates in P53-mediated apoptosis. 68-70 Bax have been extensively studied particularly in conjunction with other apoptotic proteins. Jordan et al. (1996) studied squamous cell carcinomas of the oral cavity and observed that both bcl-2 and bax were expressed differentially.71 In 2015, Kim et al. demonstrated anticancer properties of Berberine, which employed upregulation of bax and down regulation of bcl-1 and bcl-xL.72
- BCLAF1 gene (6p23.3) encodes a transcriptional repressor Bcl-2-associated transcription factor 1 which communicates with other Bcl-2 family proteins. Its overexpression induces and coexpression of BCL2 proteins suppresses apoptosis. BCLAF 1 is present all through the nucleus but in apoptotic cells it gets redistributed to a zone adjacent to the nuclear envelope.73 BCLAF1 has been observed to be suppressed in tumor cells and can promote apoptosis via disturbing the p21 mediated inhibition of caspase dependent pathway in mitochondria.74
Caspase family
The aspartate-specific cysteine protease (caspase) cascade is the main pathway orchestrating cellular death. The caspases can either be Inflammatory (involved in cytokine processing e.g. caspase- 1, 4, 5, 13, and 14) or Apoptotic like caspase-2, 3, 6, 7, 8, 9 and 10. According to role they are grouped into Initiator caspases like caspase-2, 8, 9 and 10 causing initiation of the apoptosis and Effector caspases like caspase-3, 6 and 7, causing actual cleavage of components within the cell.75
Caspases are produced as inactive zymogens (pro-caspases) that dimerise/oligomerise followed by cleavage into a small subunit and large subunit upon receiving appropriate stimulus.76 This cleavage makes favorable conformational change to expose activesite loops for enzymatic activity
- CASP1 gene (11q22.3) codes for Caspase1 protein also known as Interleukin-1 converting enzyme (ICE). It is formed as a zymogen/procaspase1which gets cleaved into subunits 20 kDa (p20) and 10 kDa (p10) to become part of the active enzyme. Caspase1 is chiefly involved in inflammation thus can play a major role in cancer associated inflammation. Tsai et al. (2004) reported dysregulation of CASP1 gene in oral cancer.77
- CASP3 gene (4q35.1) codes for protein Caspase 3. This executioner caspase gets activated by caspase 8, 9, 10 and in turn cleaves and activates caspases 6 and 7. Similar to other caspases, caspase 3 also occurs in procaspase form i.e 32 kDa and is cleaved into 17 kDa and 12 kDa (p17 and p12) subunits. Caspase-3 can be activated either by extrinsic, intrinsic or alternate pathways. Caspase 3 downregulation have been observed by many researchers in various cancers including oral cancers and correlation with degree of cellular differentiation was significantly demonstrated.78 Andressakis et al. (2008) observed reduced expression of Caspase 3 proteins in Tongue squamous cell carcinomas and that its activation induces apoptosis, a mechanism involved in many therapeutic strategies for example berberine and Ginko biloba.79-81
- CASP7 gene (10q25.3) codes for protein caspase 7 which acts as executioner for apoptosis. It exists as inactive procaspase that is proteolytically processed by upstream caspases (caspase-8, -9) to form large and small subunits, which fuses and transform into active enzyme. Caspase 7 expression has been studied in Oral cancers with a large sample size revealing a very occasional expression of caspase 7 suggesting it to be a regulator of tumorigenesis and predictor of recurrence (prognostic significance).82
- CASP9 gene (1p36.21) codes for an initiator protein caspase 9. Cytochrome c is released from mitochondria and activates apaf- 1 (apoptosome) to cleave procaspase-9 into the active dimer form of caspase 9.83 Once activated, caspase-9 cleaves caspase-3, 6 and 7, inducing the caspase cascade causing apoptosis. Caspase 9 also gets suppressed in oral cancers and therapies targeted to activate Caspase 9 tend to induce apoptosis and hence aid in chemoprevention. For example, celecoxib derivatives and Alsterpaullone, (cyclin-dependent kinase inhibitor) induces apoptosis mediated by caspase-9 activation.84,85
- CASP10 gene (2q33.1) codes for Caspase10, which exists as procaspase10. Caspase 8 processes it leading to its activation. Activated caspase 10 subsequently cleaves caspases 3 and 7 to activate them and cause apoptosis. It was demonstrated by Wang et al. (2001) that the death-effector domains of caspase-8 and 10 interact with the DED of FADD. Cengiz et al. (2007) observed loss of heterozygosity (LOH) of long arm of chromosome 2 in normal oral and cancerous tissues and elucidated that the preferentially deleted region corresponded to caspase 10 suggesting its crucial role in oral cancer development.86
CARD family
The interaction motifs called Caspase activation and recruitment domains (CARDs) are observed in many proteins involved in apoptosis. These domains/modules form a six to seven antiparallel α-helical bundle known as death domain fold that mediates the formation of larger protein complexes. CARD domains are found on kinases, helicases, caspases, mitochondrial proteins, and other cytoplasmic factors. CARDs, DEDs or DDs containing proteins exhibit homotypic interactions resulting in activation of caspase leading to apoptosis.87
- CARD9 gene (19q13.33) codes for caspase recruitment domain-containing protein 9 or TUCAN (tumor-up-regulated CARD-containing antagonist of caspase nine). Pathan et al. (2001) observed that CARD-containing protein is overexpressed in few cancer types and it binds and suppresses procaspase-9 activation, it also interferes with binding of procaspase-9 of Apaf1 and suppresses caspase activation induced by cytochrome c.88
- APAF 1 gene (12q23.1) codes for apoptotic protease activating factor 1, a cytoplasmic protein containing CARD, ATPase domain (NB-ARC), some short helical domains and many WD40 repeat domain.89 Binding of APAF 1 to cytochrome c and dATP, leads to formation of apoptosome, which activates procaspase 9 that stimulates the subsequent caspase cascade leading to apoptosis. 90 BCL-XL binds preferentially to Apaf 1 thus inhibiting its binding to caspase 9 thereby inhibiting caspase 9 mediated pathway. Lo Muzio et al. (2014) reported a decreased expression of APAF1 in oral cancer as compared to normal mucosa. Berberine has been reported to upregulate activation of Apaf 1 along with other proapoptotic factors and downregulation of antiapoptotic factors suggesting its therapeutic benefits reported in oral cancers. 72
- NOD1 gene (7p14.3) codes for a CARD containing receptor protein nucleotide-binding oligomerization domain-containing protein 1 (nod1). Stimulation of NOD1 by iE-DAP containing molecules causes transcription factor NF-κB activation.91 Wang et al. (2014) reported significantly decreased expression of NOD1 along with the progression of OSCC in terms of tumor differentiation, lymph node metastasis, and size.92
- NOL3 gene (16q22.1) codes for Nucleolar protein 3, a Caspase recruitment domain containing apoptotic repressor [ARC]. It can inhibit cell death by antagonizing both intrinsic and extrinsic pathways in contrast to most apoptosis inhibitors, which interfere with circumscribed portions of either of them.93 NOL3/ARC is found majority of cells within their cytoplasm, but in some solid tumor cell lines it was reported to be localize to the nucleus.94 Also in cancer cells ARC protein suppresses activation of NF-κB pathway and disrupts transcriptional activity of p53 by direct interaction.95,96
- CFLAR gene (2q33.1) codes for CASP8 and FADD-like apoptosis regulator protein. It is seldom known as c-FLIP (FLICElike inhibitory protein). The protein structurally resembles caspase- 8 but lacks caspase activity. Caspase 8 cleaves FLIP into 2 short peptides and thus is a regulator of apoptosis. c-FLIP has 3 splice isoforms which inhibit death receptor induced apoptosis.97,98 c-FLIP competes with procaspase-8 directly for recruitment to FADD.99 Yajima et al. (2009) performed Gene Chip microarray and qRT-PCR to reveal that the cells of the side population of human oral squamous cell carcinoma cell line exhibited significantly increased CFLAR expression.100
Death Family
- DAPK1 gene (9q21.33) codes for Death-associated protein kinase 1 which positively mediates gamma-interferon induced apoptosis. Cohen et al. (1997) demonstrated that the cytoskeletal alterations occurring during cell death causes activation of DAPkinases via Ca2/calmodulin. There are 5 DAPK1 members reported which are capable of inducing apoptosis.101 Inactivation of DAP-kinase due to aberrant methylation in the promoter region is observed in many cancers including oral cancer. DAPK1 can also be used as a molecular marker for prognosis.102-105 Zhao et al. (2015) demonstrated that DAPK1 is essential for growth of p53- mutant cancers.106
- FADD gene (11q13.3) codes for FADD (Fas-associated protein with death domain) or MORT1 adaptor protein comprising death domain (DD) and death effector domain (DED) at C terminal and N terminal respectively. During apoptosis it unites members of the TNFR superfamily (Fas-receptor) with procaspases 8 and 10 to constitute DISC (death inducing signaling complex).107 Pattje et al. (2013) reported significant up-regulated FADD expression in head and neck cancers.108 Genomic characterization sequencing in oral cancers revealed gain of function in genomic locus corresponding to FADD this is further supported by multiplex ligationdependent probe amplification (MLPA) panel directed to head and neck cancer revealing gain of genetic material in FADD chromosome loci.109-110 Lo Muzio et al. (2014) also demonstrated different expression levels of many genes including FADD in OSCC compared to normal mucosa by cDNA macroarray analysis and Real-time PCR.111-113 Squamous carcinoma cells with the expression of FADD were observed to become metastatic and to worsen survival rates.114 Another DD- containing adaptor protein named TRADD, binds to activated TNF1R, forming complex I, which gets internalized. Also FADD binds to TRADD through Death Domains of both adapter proteins, constituting complex II. This activates NFκB pathway and recruiting procaspase 8 to activate caspase cascade leading to apoptosis.115
CIDE domain Family
DNA fragmentation is a critical step in programmed cellular death and is initiated by DNA fragmentation factor (DFF) comprised of two subunits, DFF40 (a 40kDa caspase-activated nuclease) and DFF45 (45kDa inhibitor). In 1997, Liu X et al described a family of cell-death-inducing DFF45-like effectors (CIDEs). In 1998 Inohara N et al identified CIDE-A and CIDE-B, encoding proteins homologous to the N-terminal region of DFF45.55 Liang et al. in 2003 identified and described CIDE 3 that was later termed as CIDE-C.116
CIDEs possess a conserved amino acid sequence resembling CIDE-N domains in DFF40/CAD and its inhibitor DFF45/ICAD both existing as a complex from which they are released after cleavage by Caspase 3 to trigger fragmentation of DNA and nuclear condensation.
CIDEA gene (18p11.21) codes for CIDE-A and CIDEB gene (14q12.) codes for CIDE-B. These activate apoptosis inhibited by DFF45 in mammalian cells but not by caspase inhibitors. Overexpression of CIDE-B results in cell death associated with DNA fragmentation.55 Down-regulation of CIDE B has been observed in many cancers including head and neck cancers.
Tp53 family
TP53 gene (17p13.1) a tumor suppressor gene, codes for Tumor protein p53, known as p53 is popularly referred to as the guardian of the genome since it conserves genomic stability by checking mutation.117 More than 50% human cancer exhibit mutation of TP53 indicating its crucial involvement in cancer prevention as well.118 Mechanisms behind anti-carcinogenic functions includes: i) Activation of DNA-repair proteins on detecting sustained DNA damage; ii) Arrests growth by halting the cell cycle at the G1/S regulation point on recognizing DNA; damage (Enabling DNA repair proteins to mend the damage for cell cycle continuation); iii) Initiates cell death if irreparable DNA damage present; iv) It aids in shortening of telomeres due to senescence; v) It also inhibits angiogenesis.
p53 pathway
A negative regulator mdm2 binds to p53 in normal cells which dissociates upon DNA damage or other stresses causing p53 activation and cell cycle arrest or apoptosis. Cancerous cells overcome this checkpoint leading to cell survival119 (Figure 5).
There are numerous studies indicating involvement of p53 in oral cancer and precancer.
IAP family
This family comprises of proteins, which are Inhibitors of Apoptosis (IAP). They can undermine apoptosis induced via either intrinsic or extrinsic stimuli. X-linked IAP (XIAP) blocks apoptosis by directly inhibiting caspase-3, 7, and 9. The basic technique by which other IAPs inhibit apoptosis is not much known. Many IAPs can bind to caspases, but are unable to inhibit the proteolytic activity of these enzymes.
Figure 5.
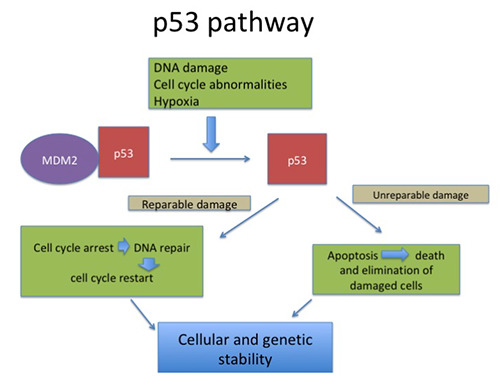
p53 pathway- In a normal cell, p53 is inactivated by its negative regulator, mdm2. Upon DNA damage or other stresses, various pathways will lead to the dissociation of the p53 and mdm2 complex. Once activated, p53 will induce a cell cycle arrest to allow either repair - survival of the cell or apoptosis to discard the damaged cell.
IAP family members contain small, zinc-coordinated domains known as 1-3 baculoviral IAP repeat (BIR) domains (essential for the anti-apoptotic activity).120
Roy et al. (1995) 1st isolated the gene, encoding neuronal apoptosis inhibitor protein (NAIP) located on 5q13.2. This gene was later designated as BIRC1 (Baculoviral IAP repeat-containing protein1). 121
- BIRC2 gene (11q22.2) codes for protein birc2 (Baculoviral IAP repeat-containing protein2) or cIAP1 (cellular inhibitor of apoptosis protein-1). BIRC2 gene has one BIR (Baculoviral IAP repeat) domain. cIAP1 is a multi-functional protein that can be found in normal cell cytoplasm and in the nucleus of tumor cells. Thus this protein has a big influence in the growth of diverse cancers including oral cancers.
- BIRC3 gene (11q22.2) codes for cIAP2 protein. It inhibits apoptosis by interfering with caspase activation. The cIAP2 protein is comprised of three BIR domains along with UBA, CARD and RING finger domains.122,123 Nagata M et al. (2011) reported down-regulation of cIAP2 and increased apoptosis using DNA microarray in parental and 5-FU-resistant OSCC cell lines, which exhibited significant increase in sensitivity of 5-FU-resistant cells to 5-FU. Also high cIAP2 tumor expression was significantly correlated with chemotherapeutic response. Thus in 5-FU resistant cases cIAP2 is a potentially useful therapeutic target, enhancing prognosis in OSCC patients.123
- XIAP gene (Xq24-25) codes for X-linked inhibitor of apoptosis protein (XIAP) popularly referred to as inhibitor of apoptosis protein 3 (IAP3) or Baculoviral IAP repeat-containing protein 4 (BIRC4). XIAP binding inhibits caspase 3, 7 and 9 leading to apoptosis inhibition. Caspase 3 and 7 are down regulated by BIR2 and caspase 9 is inhibited by BIR3 domain of XIAP.
TRAF Family
TNFR-associated factors (TRAFs) are a family of structurally related phylo-genetically conserved group of scaffold proteins that mediates transduction of signals from TNFR receptors to signaling cascades, causing activation of NF-kappa B and MAPK. TRAF proteins also aid in transcriptional and posttranslational regulation of majority of signaling pathway regulators.124,125
In mammals, earlier 6 members of TRAF family are identified but now TRAF 7 have also been identified, however it is controversial as it lacks the TRAF homology domain.
- TRAF2 gene (9q34.3) codes for TNF receptor-associated factor 2 (TRAF2). It causes TNF-alpha-mediated activation of MAPK8/JNK and NF-κB. Interaction with TNFR2 and TRADD involves C terminus whereas interaction with RIP is through N-terminus of the TRAF-C domain. Activation of NF-kB requires RING finger at the N-terminal along with adjacent two zinc fingers. 126
- TRAF3 gene (14q32.32) codes for TNF receptor-associated factor 3 (TRAF3) protein. It binds to CD40 (cytoplasmic tail) and LTbR in a ligand dependent manner to inhibit NF-kB activation. Since it participates in induction of LT-b mediated cell death and not TNF, apoptosis is inhibited by its dominant negative mutant.127
- TRAF4 gene (17q11.2) codes for TNF receptor-associated factor 4 (TRAF4) protein. TRAF4 was earlier designated CART1 due to presence of a C-rich domain associated with RING. Localisation of TRAF predominantly to the nucleus renders it unable to regulate cell surface receptors signaling.128 Jianbin Yang et al (2015) studied the expression and effect of TRAF4 on cell growth, invasion and migration in OSCC cell lines. Up-regulation in TRAF4 mRNA levels was demonstrated with an increase in TRAF4 protein levels evaluated by Western blotting analysis. TRAF4 elevation also increased cell invasion and migration.129
AKT1
AKT1 gene codes enzyme Serine/threonine-protein kinase. Akt exists in 3 mammalian isoforms (Akt1, Akt2 and Akt3). Akt becomes activated by lipid kinase phosphoinositide-3 kinase (PI3K).130,131 AKT1 phosphorylates many downstream substrates to regulate cell metabolism, proliferation, survival, growth and angiogenesis. AKT regulates NF-kappa-B-dependent gene transcription and activity of cyclic AMP response element binding protein (CREB1). The phosphorylation of CREB1 aids in binding of accessory proteins needed for transcription of anti-apoptotic genes-BCL2 and MCL1. Also AKT mediates the anti-apoptotic effects of (Insulin like Growth Factor 1) IGF-I.132 Green et al. (2013) demonstrated that p53-dependent apoptosis pathway is repressed, Puma is transcriptionally upregulated and survival of Bim- or Bad-deficient cells was prolonged by Akt1.133 Kuo et al. (2013) examined the efficacy of caffeic acid phenethyl ester (CAPE) on cellular cycle, proliferation and expression of signaling proteins in TW2.6 human oral cancer cells and demonstrated that CAPE down-regulated the proliferation and survival of cancer cells by inhibiting Akt signaling.134 Cohen et al. (2011) suggested that AKT1 dysregulation was observed in tongue squamous cell carcinoma (TSCC) and AKT1upregulation was correlated with lymph node metastasis and with reduced overall survival. Also missense mutation in the PIK3CA oncogene activated PIK3CA/AKT pathway.135 Similarly Lim et al. (2005) reported a significant correlation of p-Akt with lymph node metastasis and TNM staging in OSCC, thus aiding in prognosis of the disease.136
BRAF
It is a human gene referred as proto-oncogene B-Raf and v-Raf murine sarcoma viral oncogene homolog B coding for serine/threonine- specific protein kinase.137 It phosphorylates downstream kinase MAPK (Mitogen-activated protein kinase)/ ERK (Extracellular signal-regulated kinase). The MEK/ERK/RAS/RAF pathway identifies and transduces extracellular signals (Cytokines, hormones and growth factors) to nucleus of the cell, causing proliferation, growth, differentiation, and apoptosis.138 Mutations causing gain-of-function in BRAF is observed in cancers (about 7-8%) comprising malignant melanomas (>50%), papillary thyroid cancer (˜45%), colorectal cancer (˜10%), ovarian cancer (˜10%), and small percentage of other cancers.139 In another study, about 30 mutations of the BRAF gene have been observed. Out of all, BRAFV600E mutation is observed in about 8% of all cancers, >60% of melanomas, almost all hairy-cell leukemia and 10% of colorectal cancers.139,140 Brown et al. (2014) revealed that somatic FGFR2-RAS-BRAF mutations are frequently involved in the pathogenesis of majority of Ameloblastomas. (However Odontogenic neoplasms without ameloblastic epithelium lacks BRAF V600E mutation, suggesting its efficacy as a diagnostic marker). BRAF V600E was common in early onset and BRAF wild- type is more frequently in the maxilla and exhibited earlier recurrences. BRAF inhibitor vemurafenib inhibits proliferation of ameloblastoma cells and MAPK pathway activation therefore can be used as targeted therapy in management of ameloblastoma.141
BFAR/BAR gene
It codes for Bi-functional Apoptosis Regulator protein (a multidomain protein initially considered as inhibitor of Bax-induced cell death). BFAR inhibits apoptosis induced by both extrinsic as well as intrinsic pathway. BAR protein contains a domain (termed pseudo DEDs) resembling death effector domains, which mediates Caspase-8 binding.142,143 The SAM domain of BFAR protein interacts with Bcl-2 and Bcl-XL and suppress Bax-induced cell death while DED-like domain interacts with pro-caspases containing DED to suppress apoptosis induced by Fas. BFAR also fuses procaspase- 8 with Bcl-2 to form a protein complex. Therefore, BFAR/BAR behaves like protein with a scaffolding property so that it could link extrinsic and intrinsic apoptotic pathways.143
Conclusions
The genetics and genomics behind oral cancer is still not fully understood. However, information available with present age should immediately be placed to use in order to stop the rising prevalence of oral cancer. Translational approach using available resources and knowledge is required in solving the oral cancer problem. Interventions right at the early stage of oral cancer will be most beneficial. Similarly, targeted destruction of cancerous cells should be the ideal intervention. Regulating apoptosis seems the best way out. Various genes and pathways in apoptosis have common endpoint that is cellular death. Facilitating auto cell death of cancerous cells is achievable target and efforts in silencing or promoting involved genes is much needed.
References
- 1.Bray F, Ferlay J, Soerjomataram I, et al. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA: Cancer J Clin 2018;68:394-424. [DOI] [PubMed] [Google Scholar]
- 2.Shield KD, Ferlay J, Jemal A, et al. The global incidence of lip, oral cavity, and pharyngeal cancers by subsite in 2012. CA Cancer J Clin 2017;67:51-64. [DOI] [PubMed] [Google Scholar]
- 3.Krishna Rao SV, Mejia G, Roberts-Thomson K, Logan R. Epidemiology of oral cancer in Asia in the past decade--an update (2000-2012). Asian Pac J Cancer Prev 2013;14:5567-77. [DOI] [PubMed] [Google Scholar]
- 4. www.cancer.gov>publications>dictionaries>cancerterms>def>oral. [Google Scholar]
- 5.Lingen MW, Kalmar JR, Karrison T, Speight PM. Critical evaluation of diagnostic aids for the detection of oral cancer. Oral Oncol 2008;44:10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bremmer JF, Braakhuis BJ, Ruijter-Schippers HJ, et al. A noninvasive genetic screening test to detect oral preneoplastic lesions. Lab Invest 2005;85:1481-8. [DOI] [PubMed] [Google Scholar]
- 7.Barnes L, Eveson JW, Reichart P, Sidransky D. World Health Organization Classification of tumours: Pathology and genetics of tumours of the head and neck. Lyon: IARC Press; 2005. [Google Scholar]
- 8.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100:57-70. [DOI] [PubMed] [Google Scholar]
- 9.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646-74. [DOI] [PubMed] [Google Scholar]
- 10.Fouad YA, Aanei C. Revisiting the hallmarks of cancer. Am J Cancer Res 2017;7:1016-36. [PMC free article] [PubMed] [Google Scholar]
- 11.Brunner T, Kasibhatla S, Pinkoski MJ, et al. Expression of Fas ligand in activated T cells is regulated by c-Myc. J Biol Chem 2000;275:9767-72. [DOI] [PubMed] [Google Scholar]
- 12.Trapani JA, Smyth MJ. Functional significance of the perforin/ granzyme cell death pathway. Nat Rev Immunol 2002;2:735-47. [DOI] [PubMed] [Google Scholar]
- 13.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science 1998;281:1305-8. [DOI] [PubMed] [Google Scholar]
- 14.Wallach D, Varfolomeev EE, Malinin NL. Tumor necrosis factor receptor and Fas signaling mechanisms. Annu Rev Immunol 1999;17:331-67. [DOI] [PubMed] [Google Scholar]
- 15.Bodmer D, Brors D, Pak K, et al. Inflammatory signals increase Fas ligand expression by inner ear cells. J Neuroimmunol 2002;129:10-7. [DOI] [PubMed] [Google Scholar]
- 16.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003;114:181-90. [DOI] [PubMed] [Google Scholar]
- 17.Bauer J, Namineni S, Reisinger F. Lymphotoxin, NF-ĸB, and cancer: the dark side of cytokines. Dig Dis 2012;30:453-68. [DOI] [PubMed] [Google Scholar]
- 18.Ngo VN, Korner H, Gunn MD, et al. Lymphotoxin alpha/beta and tumor necrosis factor are required for stromal cell expression of homing chemokines in B and T cell areas of the spleen. J Exp Med 1999;189:403-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang Y, Yu X, Wang L, et al. Four Genetic Polymorphisms of Lymphotoxin-Alpha Gene and Cancer Risk: A Systematic Review and Meta-Analysis. PLoS One 2013;8:e82519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vairaktaris E, Yapijakis C, Psyrri A, et al. Loss of tumour suppressor p16 expression in initial stages of oral oncogenesis. Anticancer Res 2007;27:979-84. [PubMed] [Google Scholar]
- 21.Lukashev M, LePage D, Wilson C, et al. Targeting the lymphotoxin- beta receptor with agonist antibodies as a potential cancer therapy. Cancer Res 2006;66:9617-24. [DOI] [PubMed] [Google Scholar]
- 22.Yapijakis C, Serefoglou Z, Vylliotis A, et al. Association of Polymorphisms in Tumor Necrosis Factor Alpha and Beta Genes with Increased Risk for Oral Cancer. Anticancer Res 2009;29:2379-86. [PubMed] [Google Scholar]
- 23.Gupta R, Sharma SC, Das SN. Association of TNF-α and TNFR1 promoters and 3′ UTR region of TNFR2 gene polymorphisms with genetic susceptibility to tobacco-related oral carcinoma in Asian Indians. Oral Oncol 2008;44:455-63. [DOI] [PubMed] [Google Scholar]
- 24.Singh PK, Bogra J, Chandra G, et al. Association of TNF-α (- 238 and -308) promoter polymorphisms with susceptibility of oral squamous cell carcinoma in North Indian population. Cancer Biomark 2015;15:125-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang CM, Hou YY, Chiu YT, et al. Interaction between tumour necrosis factor-α gene polymorphisms and substance use on risk of betel quid-related oral and pharyngeal squamous cell carcinoma in Taiwan. Arch Oral Biol 2011;56:1162-9. [DOI] [PubMed] [Google Scholar]
- 26.Krishnan R, Thayalan DK, Padmanaban R, et al. Association of Serum and Salivary Tumor Necrosis Factor-α with Histological Grading in Oral Cancer and its Role in Differentiating Premalignant and Malignant Oral Disease. Asian Pac J Cancer Prev 2014;15:7141-8. [DOI] [PubMed] [Google Scholar]
- 27.Juretić M, Cerović R, Belušić-Gobić M, et al. Salivary levels of TNF-α and IL-6 in patients with oral premalignant and malignant lesions. Folia Biol (Praha) 2013;59:99-102. [DOI] [PubMed] [Google Scholar]
- 28.Brailo V, Vucicevic-Boras V, Lukac J, et al. Salivary and serum interleukin 1 beta, interleukin 6 and tumor necrosis factor alpha in patients with leukoplakia and oral cancer. Oral Med Oral Patol Oral Cir Bucal 2012;17:e10-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wiley SR, Schooley K, Smolak PJ, et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995;3:673-82. [DOI] [PubMed] [Google Scholar]
- 30.Pavet V, Shlyakhtina Y, He T, et al. Plasminogen activator urokinase expression reveals TRAIL responsiveness and supports fractional survival of cancer cells. Cell Death Dis 2014;5:e1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song JJ, Lee YJ. Differential cleavage of Mst1 by caspase-7/- 3 is responsible for TRAIL-induced activation of the MAPK superfamily. Cell Signal 2008;20:892-906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vigneswaran N, Baucum DC, Wu J, et al. Repression of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) but not its receptors during oral cancer progression. BMC Cancer 2007;7:108-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoldas B, Ozer C, Ozen O, et al. Clinical significance of TRAIL and TRAIL receptors in patients with head and neck cancer. Head Neck 2011;33:1278-84. [DOI] [PubMed] [Google Scholar]
- 34.Nagaraj NS, Vigneswaran N, Zacharias W. Cathepsin B mediates TRAIL-induced apoptosis in oral cancer cells. J Cancer Res Clin Oncol 2006;132:171-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yeh CC, Deng YT, Sha DY, et al. Suberoylanilide hydroxamic acid sensitizes human oral cancer cells to TRAIL-induced apoptosis through increase DR5 expression. Mol Cancer Ther 2009;8:2718-25. [DOI] [PubMed] [Google Scholar]
- 36.Kok SH, Yeh CC, Chen ML, Kuo YP. Esculetin enhances TRAIL-induced apoptosis through DR5 upregulation in human oral cancer SAS cells. Oral Oncol 2009;45:1067-72. [DOI] [PubMed] [Google Scholar]
- 37.Antonios C, Maria L, Gregorios C, et al. CD40/CD40L signaling and its implication in health and disease. BioFactors (Oxford, England) 2009;35:474-83. [DOI] [PubMed] [Google Scholar]
- 38.Rentoft M, Laurell G, Coates PJ, et al. Gene expression profiling of archival tongue squamous cell carcinomas provides sub-classification based on DNA repair genes. Int J Oncol 2009;35:1321-30. [DOI] [PubMed] [Google Scholar]
- 39.Sandra G, Edward JS. Analysis of apoptosis-associated genes and pathways in oral cancer cells. J Oral Pathol Med 2006;35:146-54. [DOI] [PubMed] [Google Scholar]
- 40.Baker E, Chen LZ, Smith CA, et al. Chromosomal location of the human tumor necrosis factor receptor genes. Cytogenet Cell Genet 1991;57:117-8. [DOI] [PubMed] [Google Scholar]
- 41.Schall TJ, Lewis M, Koller KJ, et al. Molecular cloning and expression of a receptor for human tumor necrosis factor. Cell 1990;61:361-70. [DOI] [PubMed] [Google Scholar]
- 42.Gupta R, Sharma SC, Das SN. Association of TNF-alpha and TNFR1 promoters and 3' UTR region of TNFR2 gene polymorphisms with genetic susceptibility to tobacco-related oral carcinoma in Asian Indians. Oral Oncol 2008;44:455-63. [DOI] [PubMed] [Google Scholar]
- 43.Bodmer JL, Burns K, Schneider P, et al. TRAMP, a novel apoptosis-mediating receptor with sequence homology to tumor necrosis factor receptor 1 and Fas (Apo-1/CD95). Immunity 1997;6:79-88. [DOI] [PubMed] [Google Scholar]
- 44.Wang EC. On death receptor 3 and its ligands. Immunology 2012;137:114-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Daigeler A, Chromik A, Geisler A, et al. Synergistic apoptotic effects of Taurolidine and TRAIL on TRAIL resistant squamous carcinoma cells of the esophagus. Arbogast R., Schackert HK, Bauer H. (eds), Chirurgisches Forum 2008. Deutsche Gesellschaft fur Chirurgie, vol 37. Berlin, Heidelberg: Springer; 2008. [Google Scholar]
- 46.Peter D, Thomas W, Isrid S, Klaus SO. The kiss of death: Promises and failures of death receptors and ligands in cancer therapy. Leukemia 2001;15:1022-32. [DOI] [PubMed] [Google Scholar]
- 47.Wu H, Pang P, Liu MD, et al. Upregulated miR-20a-5p expression promotes proliferation and invasion of head and neck squamous cell carcinoma cells by targeting of TNFRSF21. Oncol Rep 2018;40:1138-46. [DOI] [PubMed] [Google Scholar]
- 48.Yamamoto H, Kishimoto T, Minamoto S. NF-kappaB activation in CD27 signaling: involvement of TNF receptor-associated factors in its signaling and identification of functional region of CD27. J Immunol 1998;161:4753-9. [PubMed] [Google Scholar]
- 49.Akiba H, Nakano H, Nishinaka S, et al. CD27, a member of the tumor necrosis factor receptor superfamily, activates NFkappa B and stress-activated protein kinase/c-Jun N-terminal kinase via TRAF2, TRAF5, and NF-kappa B-inducing kinase. J Biol Chem 1998;273:13353-8. [DOI] [PubMed] [Google Scholar]
- 50.Prasad KV, Ao Z, Yoon Y, et al. CD27, a member of the tumor necrosis factor receptor family, induces apoptosis and binds to Siva, a proapoptotic protein. Proc Natl Acad Sci U S A 1997;94:6346-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thiemann M, Richards DM, Heinonen K, et al. A Single- Chain-Based Hexavalent CD27 Agonist Enhances T Cell Activation and Induces Anti-Tumor Immunity. Front Oncol 2018;8:387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arch RH, Gedrich RW, Thompson CB. Tumor necrosis factor receptor-associated factors (TRAFs)--a family of adapter proteins that regulates life and death. Genes Dev 1998;12:2821-30. [DOI] [PubMed] [Google Scholar]
- 53.Jang IK, Lee ZH, Kim YJ, et al. Human 4-1BB (CD137) signals are mediated by TRAF2 and activate nuclear factorkappa B. Biochem Biophys Res Commun 1998;242:613-20. [DOI] [PubMed] [Google Scholar]
- 54.Ye Q, Song DG, Poussin M, et al. CD137 accurately identifies and enriches for naturally-occurring tumor-reactive T cells in tumor. Clin Cancer Res 2014;20:44-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Inohara N, Ekhterae D, Garcia I, et al. Mtd, a novel Bcl-2 family member activates apoptosis in the absence of heterodimerization with Bcl-2 and Bcl-XL. J Biol Chem 1998;273:8705-10. [DOI] [PubMed] [Google Scholar]
- 56.Reed JC. Mechanisms of apoptosis. Am J Pathol 2000;157:1415-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krajewski S, Tanaka S, Takayama S, et al. Investigation of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res 1993;53:4701-14. [PubMed] [Google Scholar]
- 58.Tirapelli LF, Bolini PH, Tirapelli DP, et al. Caspase-3 and Bcl- 2 expression in glioblastoma: an immunohistochemical study. Arq Neuropsiquiatr 2010;68:603-7. [DOI] [PubMed] [Google Scholar]
- 59.Bray K, Chen HY, Karp CM, et al. Bcl-2 Modulation to Activate Apoptosis in Prostate Cancer. Mol Cancer Res 2009;7:1487-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu B, Sun X, Shen HY, et al. Expression of the apoptosisrelated genes BCL-2 and BAD in human breast carcinoma and their associated relationship with chemosensitivity. J Exp Clin Cancer Res 2010;29:107-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Teni T, Pawar S, Sanghvi V, Sranath D. Expression of Bcl-2 and Bax In Chewing Tobacco-Induced Oral Cancers and Oral Lesions from India. Pathol Oncol Res 2002;8:109-14. [DOI] [PubMed] [Google Scholar]
- 62.Saikrishana P, Sivapathasundharam B, Rafiuddeen IS, Krishnan B. Expression of bcl-2 oncoprotien in oral squamous cell carcinoma--an immunohistochemical study. Indian J Pathol Microbiol 2002;45:283-7. [PubMed] [Google Scholar]
- 63.Loro LL, Vintermyr OK, Liavaag PG, et al. Oral squamous cell carcinoma is associated with decreased bcl-2/bax expression ratio and increased apoptosis. Hum Pathol 1999;30:1097-105. [DOI] [PubMed] [Google Scholar]
- 64.Boyd JM, Gallo GJ, Elangovan B, et al. Bik, a novel deathinducing protein shares a distinct sequence motif with Bcl-2 family proteins and interacts with viral and cellular survivalpromoting proteins. Oncogene 1995;11:1921-8. [PubMed] [Google Scholar]
- 65.Reis PP, Rogatto SR, Kowalski LP, et al. Quantitative realtime PCR identifies a critical region of deletion on 22q13 related to prognosis in oral cancer. Oncogene 2002;21:6480-7. [DOI] [PubMed] [Google Scholar]
- 66.Oltvai ZN, Korsmeyer SJ. Checkpoints of dueling dimers foil death wishes. Cell 1994;79:189-92. [DOI] [PubMed] [Google Scholar]
- 67.Gross A, Jockel J, Wei MC, Korsmeyer SJ. Enforced dimerization of BAX results in its translocation, mitochondrial dysfunction and apoptosis. EMBO J 1998;17:3878-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 1993;74:609-19. [DOI] [PubMed] [Google Scholar]
- 69.Shi Y, Chen J, Weng C, et al. Identification of the protein-protein contact site and interaction mode of human VDAC1 with Bcl-2 family protein. Biochem Biophys Res Commun 2003;305:989-96. [DOI] [PubMed] [Google Scholar]
- 70.Westphal D, Kluck RM, Dewson G. Building blocks of the apoptotic pore: how Bax and Bak are activated and oligomerize during apoptosis. Cell Death Different 2014;21:196-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jordan RC, Catzavelos GC, Barrett AW, Speight PM. Differential expression of bcl-2 and bax in squamous cell carcinomas of the oral cavity. Eur J Cancer B Oral Oncol 1996;32B:394-400. [DOI] [PubMed] [Google Scholar]
- 72.Kim J, Oh D, Yim M, et al. Berberine induces fasl-related apoptosis through p38 activation in KB human oral cancer cell. Oncol Rep 2015;33:1775-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nagase T, Seki N, Ishikawa K, et al. Prediction of the Coding Sequences of Unidentified Human Genes. VI. The Coding Sequences of 80 New Genes (KIAA0201-KIAA0280) Deduced by Analysis of cdna Clones from Cell Line KG-1 and Brain. DNA Res 1996;3:321-9. [DOI] [PubMed] [Google Scholar]
- 74.Lee YY, Yu YB, Gunawardena HP, et al. BCLAF1 is a radiation- induced H2AX-interacting partner involved in γH2AXmediated regulation of apoptosis and DNA repair. Cell Death Dis 2012;3:e359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wong RSY. Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res 2011;30:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bao Q, Shi Y. Apoptosome: a platform for the activation of initiator caspases. Cell Death Different 2007;14:56-65. [DOI] [PubMed] [Google Scholar]
- 77.Tsai WC, Tsai ST, Ko JY, et al. The mRNA profile of genes in betel quid chewing oral cancer patients. Oral Oncol 2004;40:418-26. [DOI] [PubMed] [Google Scholar]
- 78.Hague A, Eveson JW, MacFarlane M, et al. Caspase-3 expression is reduced, in the absence of cleavage, in terminally differentiated normal oral epithelium but is increased in oral squamous cell carcinomas and correlates with tumor stage. J Pathol 2004;204:175-82. [DOI] [PubMed] [Google Scholar]
- 79.Andressakis D, Lazaris AC, Tsiambas E, et al. Evaluation of caspase-3 and caspase-8 deregulation in tongue squamous cell carcinoma, based on immunohistochemistry and computerised image analysis. J Laryngol Otol 2008;122:1213-8. [DOI] [PubMed] [Google Scholar]
- 80.Lin CC, Yang JS, Chen JT, et al. Berberine Induces Apoptosis in Human HSC-3 Oral Cancer Cells via Simultaneous Activation of the Death Receptor-mediated and Mitochondrial Pathway. Anticancer Res 2007;27:3371-8. [PubMed] [Google Scholar]
- 81.Kim KS, Rhee KH, Yoon JH, et al. Ginkgo biloba extract (EGb 761) induces apoptosis by the activation of caspase-3 in oral cavity cancer cells. Oral Oncol 2005;41:383-9. [DOI] [PubMed] [Google Scholar]
- 82.Coutinho-Camillo CM, Lourenco SV, Nishimoto IN, Kowaslski LP. Caspase expression in oral squamous cell carcinoma. Head Neck 2011;33:1191-8. [DOI] [PubMed] [Google Scholar]
- 83.Li P, Nijhawan D, Budihardjo I, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997;91:479-89. [DOI] [PubMed] [Google Scholar]
- 84.Ding H, Han C, Zhu J, et al. Celecoxib derivatives induce apoptosis via the disruption of mitochondrial membrane potential and activation of caspase 9. Int J Cancer 2005;113:803-10. [DOI] [PubMed] [Google Scholar]
- 85.Lahusen T, Siervi AD, Kunick C, Senderowicz AM. Alsterpaullone, a novel cyclin-dependent kinase inhibitor, induces apoptosis by activation of caspase-9 due to perturbation in mitochondrial membrane potential. Mol Carcinog 2003;36:183-94. [DOI] [PubMed] [Google Scholar]
- 86.Cengiz B, Gunduz M, Nagatsuka H, et al. Fine deletion mapping of chromosome 2q21-37 shows three preferentially deleted regions in oral cancer. Oral Oncol 2007;43:241-7. [DOI] [PubMed] [Google Scholar]
- 87.Bouchier-Hayes L, Martin SJ. CARD games in apoptosis and immunity. EMBO Rep 2002;3:616-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pathan N, Marusawa H, Krajewska M, et al. TUCAN, an antiapoptotic caspase-associated recruitment domain family protein overexpressed in cancer. J Biol Chem 2001;276:32220-9. [DOI] [PubMed] [Google Scholar]
- 89.Zou H, Henzel WJ, Liu X, et al. Apaf-1, a Human Protein Homologous to C. elegans CED-4, Participates in Cytochrome c-Dependent Activation of Caspase-3. Cell 1997;90:405-13. [DOI] [PubMed] [Google Scholar]
- 90.Hu Q, Wu D, Chen W, et al. Proteolytic processing of the caspase- 9 zymogen is required for apoptosome-mediated activation of caspase-9. J Biol Chem 2013;288:15142-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Inohara N, Koseki T, Lin J, et al. An induced proximity model for NF-kappa B activation in the Nod1/RICK and RIP signaling pathways. J Biol Chem 2000;275:27823-31. [DOI] [PubMed] [Google Scholar]
- 92.Wang X, Jiang W, Duan N, et al. NOD1, RIP2 and Caspase12 are potentially novel biomarkers for oral squamous cell carcinoma development and progression. Int J Clin Exp Pathol 2014;7:1677-86. [PMC free article] [PubMed] [Google Scholar]
- 93.Nam YJ, Mani K, Ashton AW, et al. Inhibition of both the extrinsic and intrinsic death pathways through nonhomotypic death-fold interactions. Mol Cell 2004;15:901-12. [DOI] [PubMed] [Google Scholar]
- 94.Wang M, Qanungo S, Crow MT, et al. Apoptosis repressor with caspase recruitment domain (ARC) is expressed in cancer cells and localizes to nuclei. FEBS Lett 2005;579:2411-5. [DOI] [PubMed] [Google Scholar]
- 95.Foo RS, Nam YJ, Ostreicher MJ, et al. Regulation of p53 tetramerization and nuclear export by ARC. Proc Natl Acad Sci U S A 2007;104:20826-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kung G, Dai P, Deng L, Kitsis RN. A novel role for the apoptosis inhibitor ARC in suppressing TNFα-induced regulated necrosis Cell Death Differ 2014;21:634-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Golks A, Brenner D, Krammer PH, Lavrik IN. The c-FLIP– NH2 terminus (p22-FLIP) induces NF-κB activation. J Exp Med 2006;203:1295-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Thome M, Tschopp J. Regulation of lymphocyte proliferation and death by FLIP. Nat Rev Immunol 2001;1:50-8. [DOI] [PubMed] [Google Scholar]
- 99.Hughes MA, Powley IR, Jukes-Jones R, et al. Co-operative and Hierarchical Binding of c-FLIP and Caspase-8: A Unified Model Defines How c-FLIP Isoforms Differentially Control Cell Fate. Mol Cell 2006;61:834-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yajima T, Ochiai H, Uchiyama T, et al. Resistance to cytotoxic chemotherapy-induced apoptosis in side population cells of human oral squamous cell carcinoma cell line Ho-1-N-1. Int J Oncol 2009;35:273-80. [PubMed] [Google Scholar]
- 101.Kogel D, Prehn JH, Scheidtmann KH. The DAP kinase family of pro-apoptotic proteins: novel players in the apoptotic game. Bioessays 2001;23:352-8. [DOI] [PubMed] [Google Scholar]
- 102.Stock RS, Roessner A, Ullrich O. DAP-kinase - Protector or enemy in apoptotic cell death. Int J Biochem Cell Biol 2005;37:1763-67. [DOI] [PubMed] [Google Scholar]
- 103.Melchers LJ, Clausen MJAM, Mastik MF, et al. Identification of methylation markers for the prediction of nodal metastasis in oral and oropharyngeal squamous cell carcinoma. Epigenetics 2015;10:850-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Noorlag R, van Kempen PM, Moelans CB, et al. Promoter hyper- methylation using 24-gene array in early head and neck cancer: better outcome in oral than in oropharyngeal cancer. Epigenetics 2014;9:1220-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ovchinnikov DA, Cooper MA, Pandit P, et al. Tumour- sup pressor gene promoter hypermethylation in saliva of head and neck cancer patients. Trans Oncol 2012;5:321-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhao J, Zhao D, Poage GM, et al. Death-associated protein kinase 1 promotes growth of p53-mutant cancers. J Clin Investig 2015;125:2707-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Huang B, Eberstadt M, Olejniczak ET, et al. NMR structure and mutagenesis of the Fas (APO-1/CD95) death domain. Nature 1996;384:638-41. [DOI] [PubMed] [Google Scholar]
- 108.Pattje WJ, Melchers LJ, Slagter-Menkema L, et al. FADD expression is associated with regional and distant metastasis in squamous cell carcinoma of the head and neck. Histopathology 2013;63:263-70. [DOI] [PubMed] [Google Scholar]
- 109.Upadhyay P, Gardi N, Desai S, et al. A Genomic characterization of tobacco/nut chewing HPV-negative early stage tongue tumors identify MMP10 asa candidate to predict metastases. Oral Oncol 2017;73:56-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ribeiro IP, Marques F, Caramelo F, et al. Genetic imbalances detected by multiplex ligation-dependent probe amplification in a cohort of patients with oral squamous cell carcinoma-the first step towards clinical personalized medicine. Tumour Biol 2014;35:4687-95. [DOI] [PubMed] [Google Scholar]
- 111.Muzio LL, Sartini D, Santarelli A, et al. Expression and prognostic significance of apoptotic genes in oral squamous cell carcinoma. Mol Carcinogen 2014;53:264-71. [DOI] [PubMed] [Google Scholar]
- 112.Sugahara K, Michikawa Y, Ishikawa K, et al. Combination effects of distinct cores in 11q13 amplification region on cervical lymph node metastases of oral squamous cell carcinoma. Int J Oncol 2011;39:761-9. [DOI] [PubMed] [Google Scholar]
- 113.Jarvinen AK, Autio R, Haapa-Paananen S, et al. Identification of target genes in laryngeal squamous cell carcinoma by highresolution copy number and gene expression microarray analyses. Oncogene 2006;25:6997-7008. [DOI] [PubMed] [Google Scholar]
- 114.Prapinjumrune C, Morita K, Kuribayashi Y, et al. DNA amplification and expression of FADD in oral squamous cell carcinoma. J Oral Pathol Med 2010;39:525-32. [DOI] [PubMed] [Google Scholar]
- 115.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003;114:181-90. [DOI] [PubMed] [Google Scholar]
- 116.Zhao LL, Xu M, Yokoyama Z, Tsaiping KL. Molecular cloning and characterization of CIDE-3, a novel member of the cell-death-inducing DNA-fragmentation-factor (DFF45)- like effector family. Biochem J 2003;370:195-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Surget S, Khoury MP, Bourdon JC. Uncovering the role of p53 splice variants in human malignancy: a clinical perspective. Onco Targets Ther 2013;7:57-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.May P, May E. Twenty years of p53 research: structural and functional aspects of the p53 protein. Oncogene 1999;18:7621-36. [DOI] [PubMed] [Google Scholar]
- 119.Chia YC, Rajbanshi R, Calhoun C, Chiu RH. Anti-neoplastic effects of gallic acid, a major component of Toona sinensis leaf extract, on oral squamous carcinoma cells. Mol 2010;15:8377-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Eckelman BP, Salvesen GS, Scott FL. Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Rep 2006;7:988-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Roy N, Deveraux QL, Takahashi R, et al. The c-IAP-1 and c- IAP-2 proteins are direct inhibitors of specific caspases. EMBO J 1997;16:6914-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Liston P, Roy N, Tamai K, et al. Suppression of apoptosis in mammalian cells by NAIP and a related family of IAP genes. Nature 1996;379:349-53. [DOI] [PubMed] [Google Scholar]
- 123.Nagata M, Nakayama H, Tanaka T, et al. Overexpression of cIAP2 contributes to 5-FU resistance and a poor prognosis in oral squamous cell carcinoma. Br J Cancer 2011;105:1322-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yang XD, Sun SC. Targeting signaling factors for degradation, an emerging mechanism for TRAF functions. Immunol Rev 2015;266:56-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Harald W, Peter S. Tumor necrosis factor receptor-associated factor (TRAF) 2 and its role in TNF signaling. Int J Biochem Cell Biol 2001;33:19-32. [DOI] [PubMed] [Google Scholar]
- 126.Robbins KM, Hambor ME, Mackey JF, et al. Assembly and Regulation of the CD40 Receptor Complex in Human B Cells. J Exper Med 1997;186:337-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.VanArsdale TL, VanArsdale SL, Force WR, et al. Lymphotoxin-beta receptor signaling complex: role of tumor necrosis factor receptor-associated factor 3 recruitment in cell death and activation of nuclear factor kappaB. Proc Natl Acad Sci U S A 1997;94:2460-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bradley JR, Pober JS. Tumor necrosis factor receptor-associated factors (TRAFs). Oncogene 2001;20:6482-91. [DOI] [PubMed] [Google Scholar]
- 129.Jianbin Y, Dongyi W, Shen WW, et al. TRAF4 enhances oral squamous cell carcinoma cell growth, invasion and migration by Wnt-beta-catenin signaling pathway. Int J Clin Exper Pathol 2015;8:11837-46. [PMC free article] [PubMed] [Google Scholar]
- 130.Burgering BM, Coffer PJ. Protein kinase B (c-Akt) in phosphatidylinositol- 3-OH kinase signal transduction. Nature 1995;376:599-602. [DOI] [PubMed] [Google Scholar]
- 131.Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol- 3,4-bisphos- phate. Science 1997;275:665-8. [DOI] [PubMed] [Google Scholar]
- 132.Rodon L, Gonzalez-Junca A, Inda Mdel M, et al. Active CREB1 Promotes a Malignant TGFβ2 Autocrine Loop in Glioblastoma. Cancer Discov 2014;4:1230-41. [DOI] [PubMed] [Google Scholar]
- 133.Green BD, Jabbour AM, Sandow JJ, et al. Akt1 is the principal Akt isoform regulating apoptosis in limiting cytokine concentrations. Cell Death Differ 2013;20:1341-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kuo YY, Lin HP, Huo C, et al. Caffeic Acid Phenethyl Ester Suppresses Proliferation and Survival of TW2.6 Human Oral Cancer Cells via Inhibition of Akt Signaling Int J Mol Sci 2013;14:8801-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cohen Y, Cohen NG, Shalmon B, et al. Mutational analysis of PTEN/PIK3CA/AKT pathway in oral squamous cell carcinoma. Oral Oncol 2011;47: 946-50. [DOI] [PubMed] [Google Scholar]
- 136.Lim J, Kim KH, Paeng JY, et al. Prognostic value of activated Akt expression in oral squamous cell carcinoma J Clin Pathol 2005;58:1199-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949-54. [DOI] [PubMed] [Google Scholar]
- 138.McCubrey JA, Steelman LS, Chappell WH, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta 2007;1773:1263-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cantwell-Dorris ER, O'Leary JJ, Sheils OM. BRAFV600E: implications for carcinogenesis and molecular therapy. Mol Cancer Ther 2011;10:385-94. [DOI] [PubMed] [Google Scholar]
- 140.Dietrich S, Glimm H, Andrulis M, et al. BRAF inhibition in refractory hairy-cell leukemia. N Engl J Med 2012;366:2038-40. [DOI] [PubMed] [Google Scholar]
- 141.Brown NA, Rolland D, McHugh JB, et al. Activating FGFR2- RAS-BRAF mutations in ameloblastoma. Clin Cancer Res 2014;20:5517-26. [DOI] [PubMed] [Google Scholar]
- 142.Roth W, Kermer P, Krajewska M, et al. Bifunctional apoptosis inhibitor (BAR) protects neurons from diverse cell death pathways. Cell Death Differ 2003;10:1178-87. [DOI] [PubMed] [Google Scholar]
- 143.Zhang H, Xu Q, Krajewski S, et al. BAR: An apoptosis regulator at the intersection of caspases and Bcl-2 family proteins. Proc Natl Acad Sci U S A 2000;97:2597-602. [DOI] [PMC free article] [PubMed] [Google Scholar]