

Summary
In multiple sclerosis and experimental autoimmune encephalomyelitis (EAE), myeloid cells comprise a major part of the inflammatory infiltrate in the central nervous system (CNS). We previously described that motile sperm domain‐containing protein 2 (MOSPD2) is expressed on human myeloid cells and regulates monocyte migration in vitro. The role of MOSPD2 in EAE pathogenesis was studied by generating MOSPD2 knock‐out (KO) mice and monoclonal antibodies directed against MOSPD2. We found that EAE development in MOSPD2 KO mice was significantly suppressed. While frequency representation of leukocyte subsets in lymphoid tissues was comparable, the ratio of inflammatory monocytes in the blood was markedly reduced in MOSPD2 KO mice. In addition, T cells from MOSPD2 KO mice displayed reduced secretion of proinflammatory cytokines and increased production of interleukin (IL)‐4. Prophylactic and post‐onset treatment using monoclonal antibodies (mAbs) generated against MOSPD2 abrogated development and reduced EAE severity. These results suggest that MOSPD2 is key in regulating migration of inflammatory monocytes, and that anti‐MOSPD2 mAbs constitute a potential therapy for the treatment of CNS inflammatory diseases.
Keywords: EAE, migration, MOSPD2, Myeloid
We explored the role of motile sperm domain‐containing protein 2 (MOSPD2), a newly characterized protein expressed by monocytes, in experimental autoimmune encephalomyelitis (EAE) pathogenesis. We show that EAE was significantly suppressed in MOSPD2 KO mice or after treatment of wild‐type mice with anti‐MOSPD2 monoclonal antibodies, with pathological evidence demonstrating restricted monocyte and T cell infiltration into the CNS. The study substantiates a key role for MOSPD2 in regulating migration of inflammatory monocytes and suggests that anti‐MOSPD2 antibodies constitute a potential therapy for the treatment of CNS inflammatory diseases such as multiple sclerosis via a mechanism that interferes with monocyte accumulation in the CNS.

Introduction
Multiple sclerosis (MS) is an inflammatory disease of the central nervous system (CNS). It is characterized by neurological impairment resulting from demyelination ensuing from an autoimmune response against myelin [1]. In experimental autoimmune encephalomyelitis (EAE), the well‐studied model for MS, disease pathogenesis involves infiltration of T helper type 1 (Th1)‐ and Th17‐differentiated T cells into the CNS [2, 3], followed by recruitment of blood‐borne monocytes [4]. The mobility of these leukocytes from the circulation to the CNS is dependent upon the interaction between chemokine receptors and their cognate ligands [5, 6, 7]. Several lines of evidence indicate that monocytes are essential for EAE development and progression. Studies have shown that in EAE, an increase in circulating inflammatory monocytes coincides with relapses [8] and obstructing penetration of circulating monocytes into the CNS prevented disease progression [9]. Moreover, a recent study found that infiltrating monocytes account for the initiation of demyelination at disease onset [10]. Monocyte involvement in EAE is not, however, limited to myelin degradation, but also includes recruitment of pathogenic T cells [11, 12], antigen presentation and generation of oxidative stress and inflammatory mediators [11, 13, 14, 15, 16, 17]. Inflammatory circulating monocytes that migrate into the CNS were identified as CD11b+Ly6Chi, rendering a subset of chemokine receptor (CCR2+) within Ly6Chi monocytes instrumental for disease progression [18]. Nevertheless, no drugs are available that directly target chemokines or chemokine receptors to specifically attenuate infiltration of circulating monocytes into the CNS of MS patients. Motile sperm domain‐containing protein 2 (MOSPD2) is a surface protein expressed on human monocytes. Using specific sh‐RNA and monoclonal antibodies (mAb), MOSPD2 was found to regulate monocyte migration regardless of the activating ligand [19]. Given the paramount role of circulating monocytes in EAE, we set out to explore the effect of MOSPD2 on disease pathogenesis using knock‐out (KO) mice and blocking antibodies. Characterization of the immune system of MOSPD2 KO mice revealed normal lymphoid tissue development, where the number of leukocytes and subsets frequency was comparable to those of wild‐type (WT) mice. Activated T cells from KO mice proliferated normally, whereas the cytokine profile indicated a reduced proinflammatory and propensity towards Th2 differentiation. Frequency profiling in the periphery showed that the percentage of CD11b+ Ly6Chi inflammatory monocytes was cut by half. Further examination demonstrated that the release of monocytes from the bone marrow (BM) to the circulation was attenuated in MOSPD2 KO mice, and that a lower percentage of peripheral monocytes represents steady state during homeostasis in these mice. Upon immunization with myelin oligodendrocyte glycoprotein peptide pMOG35‐55, EAE development and progression in MOSPD2 KO mice was suppressed and a pathological examination revealed that infiltration of monocytes and T cells into the CNS was markedly reduced. Moreover, mAbs generated to target MOSPD2 profoundly suppressed EAE development. Collectively, the study indicates that MOSDP2 is essential for EAE pathogenesis and that targeting MOSPD2 using mAbs holds promise as a remedy for MS via a mechanism that interferes with monocyte accumulation in the CNS.
Materials and methods
Mice
Female 8–10‐week‐old C57BL/6 mice were purchased from Envigo (Rihovot, Israel) (cat. no. 2BL/626). The T cell receptor (TCR) transgenic mice C57BL/6‐Tg (Tcra2D2, Tcrb2D2)1Kuch/J (2D2) (cat. no. 006912) and B6.Cg‐Tg (TcraTcrb)425Cbn/J (OT‐II) (cat. no. 004194) which, respectively, recognize myelin oligodendrocyte glycoprotein peptide (pMOG35–55) and peptide ovalbumin (pOVA323–339), were purchased from Jackson Laboratories (Bar Harbor, ME, USA). The health status of the animals in those studies was examined on arrival. Only animals in good health were acclimatized to laboratory conditions and were used in this study. Animals were housed within a limited‐access rodent specific‐pathogen‐free (SPF) facility, kept in groups of up to 10 mice in polypropylene cages fitted with solid bottoms and wood shavings as bedding material. Animals were provided with a commercial rodent diet (Harlan/Envigo) ad libitum and were allowed free access to drinking water, which was supplied to each cage via a polyethylene bottle with stainless steel sipper tubes. When monitoring weakness in the hind limbs, wet food was placed within the cage in order to allow access to food and water. Environmental conditions were targeted to maintain temperature at 20–24°C, with a 12‐h light/12‐h dark cycle. At the end of the study, animals were euthanized by inhalation of CO2.
Generation of MOSPD2 KO mice
MOSPD2 KO mice were generated in collaboration with geneOway (Lyon, France). The MOSPD2‐targeting construct was introduced into embryonic stem cells on a C57BL background by homologous recombination. Two genes that belong to the same family, MOSPD1 and MOSPD3, seemed to be implicated in development. The neomycin selection cassette introduced in the targeting vector is an independent transcription unit driven by a strong promoter (phosphoglycerate kinase: pGK) that can interfere with the MOSPD2 endogenous promoter. To avoid any MOSPD2 gene expression deregulation in embryonic stem cells (ES cells) that could potentially jeopardize the generation of chimeras and/or the germline transmission from these chimeras, the selection cassette was bordered with flippase recognition target (FRT) sites. After Southern blotting, validated recombined ES cell clones were transfected with a plasmid expressing Flp‐recombinase to remove the neo‐cassette and generate the neo‐deleted recombined ES cell clones. These clones were then injected into the blastocysts of albino C57BL mice, resulting in coat color chimerism. To generate heterozygous mice carrying the KO allele, highly chimeric (> 50% coat color chimerism rate) males derived from the Flp‐mediated neo‐excised ES cell clones were mated with C57BL/6 Cre deleter females. After reaching sexual maturity, the MOSPD2‐heterozygous constitutive KO animals were interbred to generate the homozygous constitutive KO mouse line. For Southern blot validation of homozygous constitutive KO mice, the blotted genomic Allium sativum protease inhibitor (ASPI) digestion was tested with a 5' probe.
For mice genotyping, a polymerase chain reaction (PCR) reaction was conducted with the following primers:
Set 1: GGGACAGTTTATGACAAATTCTTAGGAGGC hybridizes close to locus of X‐over P1 (loxP) site adjacent to exon 3 and TGGTTTAGTCCAATCAAATGGGTACTGG hybridizes close to the loxP site adjacent to exon 5. WT mice putatively show a product of 4278 base pairs (bp), but this oversized PCR product was not expected to be amplified under the assay conditions, while KO mice yield a product of 768 bp.
Set 2: CGCTATCTGATGAGTTTTCAACAGACTCAC and TGGTTTAGTCCAATCAAATGGGTACTGG, which hybridizes to a site between exon 5 and the adjacent loxP site. WT mice yield a product of 260 bp, while no product is expected from the KO mice.
Reagents
Pertussis toxin (PT) (cat. no. P7208), peptides corresponding to residues 35–55 of rat MOG (MEVGWYRSPFSRVVHLYRNGK) and residues 323–339 (ISQAVHAAHAEINEAGR) of OVA were purchased from Sigma (Rehovot, Israel). Anti‐mouse CD4‐fluorescein isothiocyanate (FITC) (cat. no. 11‐0042‐85; clone RM4‐5), CD4‐phycoerythrin (PE) (cat. no. 12‐0042‐82; clone RM4‐5), CD8a‐PE‐cyanine 7 (Cy7) (cat. no. 25‐0081‐81; clone 53‐6.7), interferon (IFN)‐γ‐PE, interleukin (IL)‐17‐Alexa‐Fluor 647, CD25‐PE (cat. no. 12‐0251‐82; clone PC61.5), CD16/32 (cat. no. 14‐0161‐85; clone 93), forkhead box protein 3 (FoxP3)‐peridinin chlorophyll (PerCP)‐Cy5.5 (cat. no. 45‐5773‐80; clone FJK‐16s), CD3 (cat. no. 16‐0031‐85; clone 145‐2C11), CD28 (cat. no. 16‐0281‐85; clone 37.51), anti‐Ki‐67‐PE (cat. no. 12‐5698‐80; clone SoIA15), anti‐lymphocyte antigen 6C (Ly6C) Alexa‐Fluor 488 (cat. no. 53‐5932‐82; clone HK1.4), anti‐Ly6G‐allophycocyanin (APC) (cat no. 17‐9668‐82; clone 1A8‐Ly6g), anti‐CD11b‐PE (cat. no. 12‐0112‐82; clone M1/70), anti‐CD115‐APC (cat. no. 17‐1152‐82; clone AFS98) and anti‐B220‐APC (cat. no. 17‐0452‐82; clone RA3‐6B2) were all purchased from Thermo Fisher Scientific (Fremont, CA, USA.
Isolation of mouse and human immune cell subsets
Mouse monocytes and T cells were isolated from the spleen using CD11b (cat. no. 130‐049‐601) and CD90.2 (cat. no. 130‐049‐101) microbeads, respectively. Venous blood samples were obtained from healthy male donors. Peripheral blood mononuclear cells (PBMCs) were isolated on Ficoll‐Paque Plus (cat. no. 17‐1440‐03; GE Healthcare, Chicago, IL, USA) using 50‐ml Leucosep tubes (cat. no. 227290; Greiner Bio‐One, Kremsmünster, Austria). Cells were washed in phosphate‐buffered saline (PBS) (Biological Industries, Beit HaEmek, Israel), and incubated at 4°C for 15 min in buffer containing PBS and 0·5% bovine serum albumin (BSA) with human CD14 microbeads (cat. no. 130‐050‐201; Miltenyi Biotec, Bergisch Gladbach, Germany).
T cell stimulation and cytokine analysis
For APC‐free stimulation, T cells (2 × 106/ml) were seeded in 24‐well flat‐bottomed plates and activated with 5 μg/ml plate‐bound anti‐CD3 and anti‐CD28. Cells were then collected, spun down for supernatant collection and analyzed by flow cytometry. Supernatants were tested by enzyme‐linked immunosorbent assay (ELISA) for the secretion of IL‐17A (cat. no. DY421), IFN‐γ (cat. no. DY485), IL‐10 (cat. no. DY417) and IL‐4 (cat. no. DY404) (all from R&D Systems, Minneapolis, MN, USA). For in‐vitro pOVA323–339 antigen‐dependent proliferation, OT‐II CD4+ T cells (5 × 105) were labeled with carboxyfluorescein succinimidyl ester (CFSE) (cat. no. 21888; Sigma, St Louis, MO, USA), cultured in 24‐well flat‐bottomed plates in the presence of CD11b splenic monocytes (5 ×105) and pOVA323–339 for 3 days. Cells were then collected, spun down, stained for CD4 and analyzed by flow cytometry. Supernatants were collected and analyzed using Milliplex MAP mouse cytokine/chemokine multiplex assay (cat no. MCYTOMAG‐70K). For ex‐vivo stimulation, mice were immunized subcutaneously at two sites in the flank with 100 μl inoculums per site of pMOG35–55 (1·5 mg/ml) emulsified in complete Freund’s adjuvant (CFA) (cat. no. BD231141; BD Biosciences, San Jose, CA, USA) (2·5 mg/ml). Seven to 10 days later, lymph node cells were excised, a single‐cell suspension was prepared and cells were seeded at a concentration of 4 × 106/ml in the presence of 30 μg/ml pMOG35–55 or 2 μg/ml anti‐CD3 for 3 days. Supernatants were collected and analyzed by ELISA kits (R&D Systems).
Western blots
Cells were washed and resuspended in lysis buffer containing 1 : 100 dithiothreitol (DTT, cat. no. 457779), phosphatase (cat. no. P0044) and protease inhibitors (cat. no. P8340), all purchased from Sigma. Samples were loaded onto a precast Criterion TGX gel (cat. no. 5678124; Bio‐Rad, Hercules, CA, USA) and transferred onto nitrocellulose membrane. Blots were blocked with 5% milk or BSA in Tris‐buffered saline and Tween 20 (TBST) for 1 h, followed by incubation with mouse anti‐human MOSPD2 monoclonal antibody (mAb) developed in‐house (1 : 1000) and secondary antibody horseradish peroxidase (HRP) goat anti‐mouse (cat. no. 115‐035‐071) (1 : 3000) from Jackson ImmunoResearch (West Grove, PA, USA). Heat shock protein (HSP) 90 (cat. no. SC‐13119) (1 : 1000) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
In‐vitro cell migration
To test for chemotaxis, RANTES (CCL5, 100 ng/ml) (cat. no. 300‐06), MCP‐1 (CCL2, 100 ng/ml) (cat. no. 300‐04) or SDF‐1 (CXCL12, 25 ng/ml) (cat. no. 300‐28; all from PeproTech, Rocky Hill, NJ, USA) were dissolved in 0.5% FBS/RPMI‐1640 and placed in the lower chamber of a QCM 24‐well migration assay plate (5‐mm pores) (cat. no. CA‐3421; Corning‐Costar, Cambridge, MA, USA). Cells (3 × 105) were seeded in the upper chamber, followed by incubation for 3 h, after which the number of cells that migrated to the lower compartment was determined by fluorescence activated cell sorter (FACS).
IF staining
Staining was performed on six 4‐μm paraffin sections (three duplicates) from different spinal cord segments (thoracic, lumbar and sacral). Sections were deparaffinized and rehydrated. For CD3 and macrophage antigen‐2 (Mac‐2) staining, antigen retrieval was performed in 10 mM citric acid pH6 for 10 min in a microwave, using a low boiling program to break protein cross‐links and unmask the antigens. After preincubation with 20% normal horse serum and 0·2% Triton X‐100, slides were incubated with the primary antibodies at room temperature (RT) for 24 h, followed by incubation at 4ºC for 48 h. The primary antibodies used were: anti‐CD3 for T cells (cat. no. Ab5690) and anti‐MAC‐2 (cat. no. CL8942AP; clone M3/38) for activated macrophages. The second antibody step involved labeling with highly cross‐absorbed Cy2 (cat. no. 712‐225‐150) or Cy3 (cat. no. 711‐165‐152) conjugated antibodies for 30–60 min. Stained sections were examined and photographed using a fluorescence microscope (Eclipse Ni‐U; Nikon, Tokyo, Japan) equipped with Plan Fluor objectives (×20) connected to a monochrome camera (DS‐Qi1; Nikon). The number of infiltrating cells per mouse was determined by counting the cells in the most populated infiltrate duplicate of each spinal section and then calculating the average number of infiltrating cells for the three sections. The counted area of the infiltrate was 0·16 mm2.
For myelin basic protein (MBP) staining, antigen retrieval was performed for 10 min using a low boiling program in the microwave to break protein cross‐links and unmask antigens and epitopes. Sections were preincubated with 20% normal horse serum and 0·2% Triton X‐1000 for 1 h followed by incubation with primary antibody (cat. no. MAB386; 1 : 100) at RT for 24 h and for an additional 48 h at 4ºC. To enhance the signal, a secondary antibody, biotinylated anti‐rat (cat. no. 712‐065‐150), was added for 90 min followed by Cy2 (cat. no. 016‐220‐084) for 1 h r. The fluorescence intensity, measured in sections taken from healthy control mice, was determined as 100% MBP coverage area. Fluorescence intensity was then measured for all six spinal cord segments of the experimental mice. The average intensity of the most affected sections from each segment was calculated. The percentage of total MBP coverage area was calculated as follows:
EAE induction
For active immunization, mice were immunized subcutaneously at two sites in the flank with 100 μl inoculum per site of pMOG35–55 (1·5 mg/ml) emulsified in CFA (2·5 mg/ml). PT (500 ng/500 μl PBS) was injected intraperitoneally (i.p.) immediately and 48 h after immunization.
For adoptive transfer, 2D2 mice were immunized as described above without PT. One week later, spleen and lymph nodes were excised to prepare cell suspension, which was activated with pMOG35–55 (20 μg/ml) for 3 days in the presence of recombinant mouse IL‐12 (10 ng/ml) (cat. no. 210‐12‐10; PeproTech). Cells were then collected, washed and injected i.p. (10–15 × 106/mouse) into irradiated (400 rad) naive recipients.
EAE severity was assessed as follows: 0 = normal, 1 = limp tail, 2 = weakness of hind limbs, 3 = hind leg paralysis, 4 = hind and fore leg paralysis, 5 = moribund or death.
Bone marrow transplant
Recipient mice were irradiated (1000 rad) using a clinical linear accelerator (Varian 2110C, 6MV, 240 mU/min). The next day, bone marrow cells (5 × 106) isolated from the donor’s femur and tibia were administered intravenously (i.v.), and 3 weeks later recipient mice were induced for EAE as described above.
Depletion of circulating monocytes
To deplete circulating monocytes, mice were injected with 200 μl clodronate liposomes (cat. no. LIP‐01; Liposoma, Amsterdam, the Netherlands). CD115+ levels in the periphery were monitored to assess depletion and recovery of peripheral monocytes.
Generation of anti‐human MOSPD2 monoclonal antibodies
Mouse anti‐human MOSPD2 mAbs were generated in collaboration with Proteogenix (Schiltigheim, France). Gene synthesis was performed to include a ×6 His‐tagged extracellular region (amino acids 1–496) of human MOSPD2. The insert was subcloned and the expression vector was transfected into Chinese hamster ovary (CHO)‐S cells. The protein was purified on a nickel resin and AKTA protein purification system (Marlborough, MA, USA). The final product was evaluated by sodium dodecyl sulphate‐polyacrylamide gel electrophoresis (SDS‐PAGE) and Western blot. Mice were injected with the extracellular region of human MOSPD2. Spleens were fused with mouse myeloma cell line Sp2/0. Serum and hybridoma supernatants were screened by ELISA against the immunizing antigen and flow cytometry against cells transfected with a construct of green fluorescent protein (GFP)‐His‐tagged MOSPD2. Antibodies for in‐vivo experiments were produced under animal‐free, serum‐free cell culture conditions.
Linear epitope mapping of anti‐MOSPD2 mAbs
Linear epitope mapping of anti‐MOSPD2 mAbs was performed against the extracellular region of human MOSPD2 and was translated into linear 15‐amino acid peptides with a peptide–peptide overlap of 14 amino acids. Human MOSPD2 peptide microarrays were incubated with anti‐MOSPD2 mAb at a concentration of 1 μg/ml followed by staining with secondary goat anti‐mouse IgG Fc DyLight680 antibody (cat. no. NBP1‐72880; Novus Biological, Littleton, CO, USA) and were read out using a LI‐COR Odyssey Imaging System.
Prevention and treatment of EAE with anti‐MOSPD2 mAb
For prevention experiments, 500 μg of anti‐MOSPD2 mAbs or isotype‐matched controls [mouse immunoglobulin (Ig)G1, cat. no. BP0083; clone MOPC‐21 and mouse IgG2a, cat. no. BP0085; clone C1.18.4 from BioXcell, Upper Heyford, UK] were injected i.p. 1 day before disease induction and five more times, once every 3 days, for a total of six times. For treatment regiment, when the disease reached an average score of 2, mice were assigned into two groups: one group received 500 μg of anti‐MOSPD2 mAb and the other group was given an isotype‐matched control antibody every other day, for a total of seven times.
Statistics
Statistical analyses were performed using SigmaPlot version 14.0 (SigmaPlot Software, Chicago, IL, USA). Statistical significance of differences between groups was determined by two‐tailed Student’s t‐test or by analysis of variance (ANOVA) with multiple comparisons test as indicated. All data are expressed as mean ± standard error (s.e.) or as indicated. The criterion for statistical significance was a P‐value of less than 0·05.
Study approval
All animal experiments described in this paper were approved by the Institutional Animal Care and Use Committee of the Sheba Medical Center, Ramat Gan, Israel. Venous blood samples were obtained from healthy male donors in compliance with the Institutional Review Board at Sheba Medical Center, Ramat Gan, Israel. Written informed consent was received from participants prior to sample donation.
Results
Generation of MOSPD2 KO mice
To assess the effect of MOPSD2 on the development of inflammation, we generated mice with a targeted deletion of exons 4–5 in the MOSPD2 gene using the targeting construct described in Fig. 1a. The homozygous constitutive KO mice were identified by PCR (Fig. 1b) and further verified by Southern blot analysis (Fig 1c). Ablation of protein was verified by Western blots performed on cells isolated from spleen, BM and thymus (Fig. 1d). MOSPD2 KO mice were born at expected Mendelian ratios and developed normally.
Fig. 1.
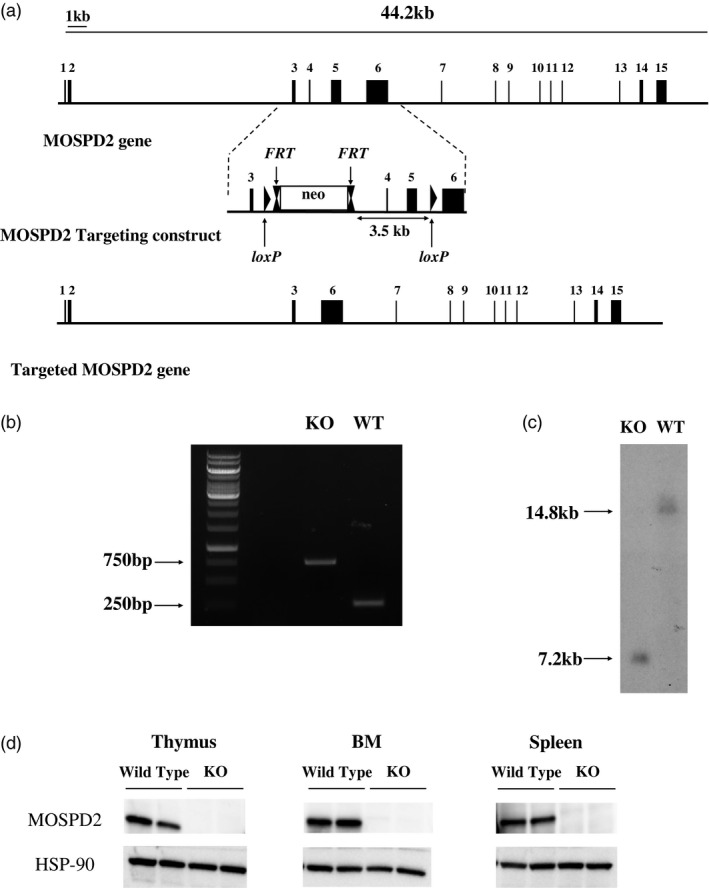
Targeted disruption of the mouse motile sperm domain‐containing protein 2 (MOSPD2) gene. (a) The exons of the MOSPD2 gene are represented by solid black rectangles. The middle panel shows the MOSPD2‐targeting construct with exons 3–6 (solid black rectangles), neo‐cassette (blank rectangle), locus of X‐over P1 (loxP) and flippase recognition target (FRT) elements (indicted by arrows). The targeted MOSPD2 gene after homologous recombination is shown in the bottom panel. (b) Polymerase chain reaction (PCR) products of tail biopsies from homozygous wild‐type (WT) and MOSPD2 knock‐out (KO) mice. The expected 768 base pairs (bp) and 260 bp DNA fragments were detected in both MOSPD2 KO and WT mice, respectively. (c) Southern blot genotyping results for MOSPD2 KO and WT mice. The Allium sativum protease inhibitor (AspI)‐digested DNAs were blotted and hybridized with a 5' probe. (d) Immunoblot analysis using anti‐MOSPD2 monoclonal antibodies (mAb) for detection of MOSPD2 protein expression in organs enriched with immune cells from WT and MOSPD2 KO mice. Two mice are shown for each organ.
Lymphoid tissues and their cell populations in MOSPD2 KO mice are intact
Our previous study indicated that MOSPD2 is expressed in humans on immune system cells of myeloid origin [19]. We assessed the effect of MOSPD2 deficiency on the development of lymphoid tissues and cell subsets thereof. The total number of spleen cells was the same for WT and MOSPD2 KO mice (Supporting information, Fig. S1a), and the ratios of splenic and lymph node B cells, monocytes, CD4 T cells, as well as regulatory T cells, were also comparable (Supporting information, Fig. S1b–e,i). In addition, the total number of thymocytes and ratios of thymic SP CD4 and SP CD8 cells were similar in WT and MOSPD2‐deficient mice (Supporting information, Fig. S1f–h).
MOSPD2 KO mice demonstrate reduced ratio of peripheral inflammatory monocytes
Next, we sought to determine whether MOSPD2 deficiency affects the frequency of cell subsets in the peripheral blood. We found that in MOSPD2 KO mice the total white blood cell (WBC) count was moderately reduced compared with WT mice (Fig. 2a). Moreover, ratios of CD11b+Ly6C myeloid cells and CD11b+Ly6Chi inflammatory monocytes as well as of CD11b+Ly6G neutrophils were decreased significantly in MOSPD2 KO mice (Fig. 2b–e). Previous reports indicated that a subset of cells within CD11b+Ly6Chi monocytes, which express chemokine receptor CCR2, is crucial for advancing inflammation [18, 20, 21]. Analysis of this subset showed that among CD11b+Ly6Chi monocytes, CCR2+ cells were significantly reduced in MOSPD2 KO mice (Fig. 2f). Nonetheless, testing the frequency of lymphocyte population in the periphery revealed similar ratios of B cells and a moderate increase in T lymphocytes in MOSPD2 KO mice (Fig. 2g,h). As the pool of circulating inflammatory monocytes is maintained by their release from the BM, we examined the number of total cells and the percentage of CD11b+Ly6Chi in the BM. The total number of cells and CD11b+Ly6Chi ratio in the BM was similar for MOSPD2 KO and WT mice (Fig. 2i–k). These results indicate that MOSPD2 may regulate the motility of monocytes and neutrophils from the BM to the circulation. CD115 is a macrophage‐CSF (M‐CSF) receptor, and is commonly used as a marker for circulating monocytes; i.v. injection of clodronate liposomes results in rapid elimination of all peripheral CD115+ cells, followed by their gradual reappearance. We depleted blood monocytes in WT and MOSPD2 KO mice and then tracked their return to the periphery. During steady state, CD115+ cells in the periphery of MOSPD2 KO mice were significantly reduced compared with WT mice. Twenty‐four h after clodronate administration, peripheral monocytes were ablated from blood in both KO and WT mice. Three days later, a notable increase in WT peripheral monocytes was detected, but only a minor replenishment of monocytes was observed for KO mice. At subsequent time‐points, a steady‐state monocyte level in the periphery was attained for both WT and MOSPD2 KO mice, but the difference in blood monocyte ratio persisted throughout (Fig. 2l). These data suggest that the release of monocytes from the BM to the periphery, rather than their differentiation from HSCs, is impaired in MOSPD2‐deficient mice.
Fig. 2.
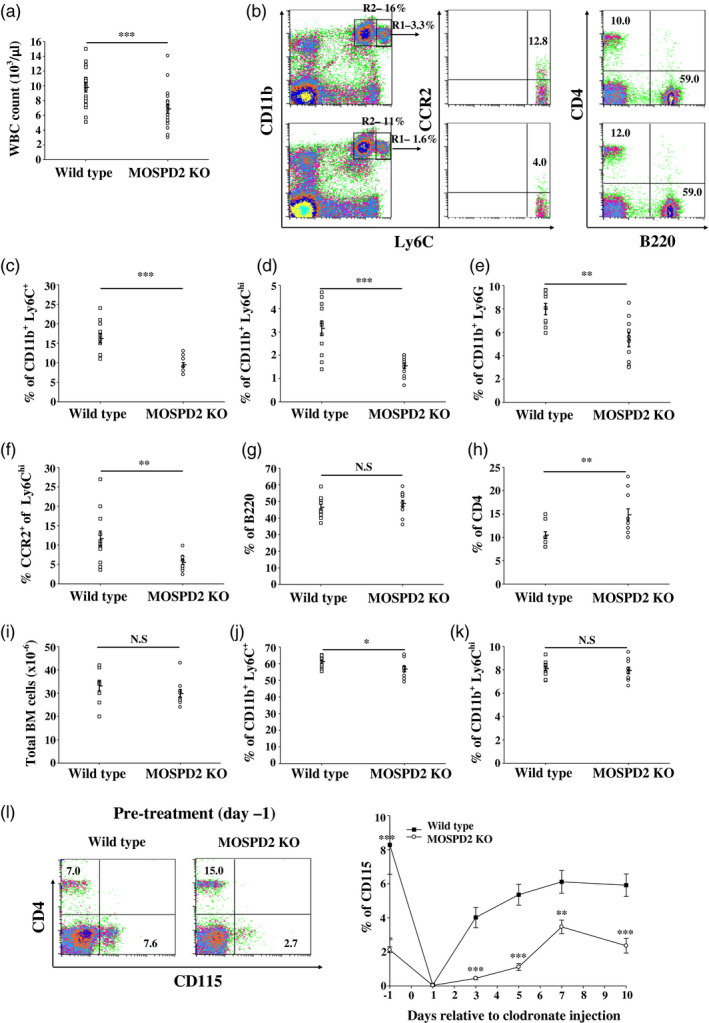
Motile sperm domain‐containing protein 2 (MOSPD2) knock‐out (KO) mice demonstrate significantly reduced peripheral blood monocytes. (a) Total white blood cell count (n = 18 per group). (b–h) Flow cytometric evaluation of peripheral blood (c–f) myeloid subsets (n = 12 per group), (g) B lymphocyte (n = 12 per group) and (h) CD4+ T cells (n = 12 per group). (h) Total number of bone marrow (BM) cells (n = 10 per group). (i–k) Flow cytometric evaluation of BM monocyte subsets (n = 10 per group). P‐values were calculated by Student’s t‐test. Data are presented as mean ± standard error (s.e.). *P ≤ 0·05; **P ≤ 0·01; ***P ≤ 0·001. (l) Flow cytometric evaluation monitoring CD115+ total blood monocytes at different time‐points following intravenous injection of clodronate liposomes (n = 12 per group). One of two experiments is shown. P‐values were calculated by analysis of variance (ANOVA) with multiple comparisons test. **P ≤ 0·01; ***P ≤ 0·001.
MOSPD2 deficiency does not affect myeloid cell effector function
To answer whether the absence of MOSPD2 affects myeloid effector functions, CD11b cells were isolated from the spleens of WT and MOSPD2 KO mice. After 72 h of incubation with CFSE‐labeled CD4+ TCR transgenic T cells specific to the ovalbumin peptide pOVA323–339 and antigen, cells were tested for CFSE dilution. CFSE dilution of pOVA323–339‐specific CD4 T cells was similar when activated with APCs from both WT and MOSPD2 KO mice, indicating that MOSPD2 is not essential for the presentation of antigens that induce T cell activation (Supporting information, Fig. S2a). Analysis of supernatants showed no significant differences in cytokine secretion, although a propensity towards a Th2 phenotype with lower Th1 and Th17 cytokine production was noted (Supporting information, Fig. S2b). An ex‐vivo challenge of lymph nodes explanted from mice immunized with pOVA323–339 demonstrated comparable rates of T cell proliferation (Supporting information, Fig. S2c). In addition, phagocytosis by neutrophils and cytokine production from splenic myeloid cells in response to lipopolysaccharide (LPS) was not perturbed by MOSPD2 deficiency (Supporting information, Fig. S2d,e).
EAE induction is suppressed in MOSPD2 KO mice
T cells and monocytes were shown to be involved in the pathogenesis of the autoimmune inflammatory disease MS and its animal model EAE. Given the attenuated presence of inflammatory monocytes in the periphery of MOSPD2 KO mice, we explored how MOSPD2 deficiency affects EAE induction and progression. The results, presented in Fig. 3a, show that MOSPD2 KO mice were protected against the development of EAE. Infiltrations of immune cells into the CNS, along with demyelination, are the main features in EAE pathology. Accordingly, we tested whether MOSPD2 deficiency impairs infiltration of immune cells into the CNS. To that end, spinal cord sections were stained by immunofluorescence with anti Mac‐2 and CD3 to indicate the presence of activated macrophages and T cells, and were analyzed for MBP to evaluate myelin integrity. Infiltration of peripheral monocytes and T cells was severely impaired in MOSPD2 KO mice (Fig. 3b–d) and the ratio of demyelinated regions was profoundly lower in spinal cords taken from MOSPD2 KO mice (Fig. 3b,e). Isolation of mononuclear cells from the CNS of WT and MOSPD2 KO mice after induction of EAE further demonstrated reduced infiltration of inflammatory monocytes in the latter (Supporting information, Fig. S3). To assess the significance of MOSPD2 expression on radio‐sensitive hematopoietic cells and radio‐resistant compartments for EAE pathogenesis, MOSPD2 KO and WT mice were irradiated with a lethal dose and subsequently transplanted with BM from WT or KO mice. Disease course and severity were similar for KO and WT recipient mice reconstituted with BM from WT mice (Fig. 3f), but when MOSPD2 expression was absent in hematopoietic cells, disease progression was suppressed compared with mice that received BM cells from WT donors. These data indicate that MOSPD2 presence in non‐hematopoietic cells is dispensable for EAE development and that its expression on hematopoietic cells, most probably myeloid cells, is necessary for disease progression. To provide supporting evidence of a role for MOSPD2 in monocytes in disease pathogenesis, 2D2 T cells were adoptively transferred into WT and KO recipient mice. In the first phase, when the disease is mediated by T cells, clinical severity was comparable among the groups of mice. In later stages, however, when monocytes are implicated, clinical symptoms persisted and worsened in WT mice, whereas KO mice exhibited a decrease in disease severity (Fig. 3g).
Fig. 3.
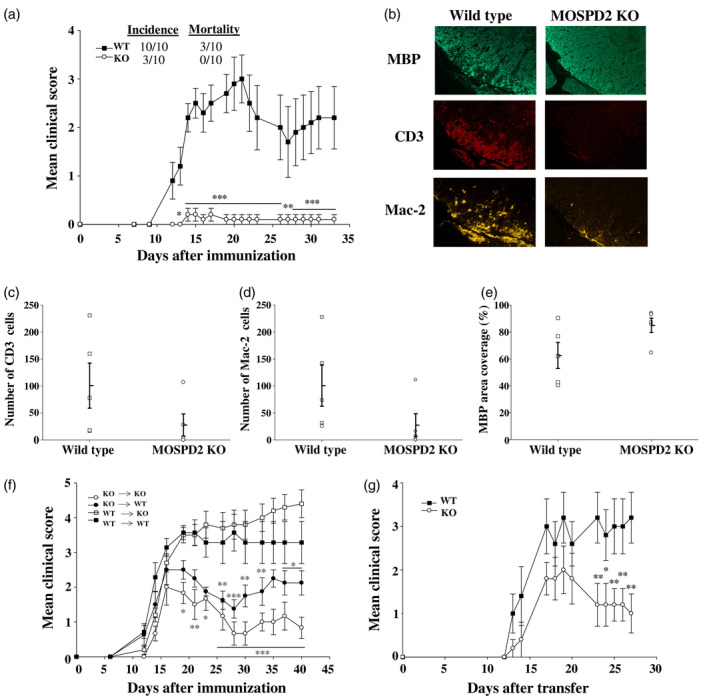
Experimental autoimmune encephalomyelitis (EAE) is suppressed in motile sperm domain‐containing protein 2 (MOSPD2) knock‐out (KO) mice. (a) Wild‐type and MOSPD2 KO mice were immunized with pMOG35‐55/complete Freund’s adjuvant (CFA). Pertussis toxin (PT) was injected on day 0 and 48 h later (n = 10 per group). One of four experiments is shown. (b) Spinal cords were stained by immunofluorescence with anti‐CD3, anti‐macrophage antigen‐2 (Mac‐2) and anti‐myelin basic protein (MBP) for the detection of infiltrating T cells, monocytes/macrophages and proportion of myelin degradation, respectively. (c,d) Number of infiltrating CD3 T cells and Mac‐2 monocytes/macrophages in the most populated infiltrate. The counted area of the infiltrate was 0·16 mm2 (n = 5 per group). (e) Mean MBP coverage area. The fluorescence intensity measured in sections taken from healthy control mice was determined as 100% coverage area (n = 5 per group). For (c–e), one of two experiments is shown. (f) Bone marrow (BM) chimeric mice were immunized as in (a), 3 weeks after BMT was performed (n = 7–10 per group). One of two experiments is shown. (g) 2D2 T cells were adoptively transferred into irradiated recipients (n = 5 per group). One of two experiments is shown. Data are presented as mean ± standard error (s.e.). P‐values were calculated by analysis of variance (ANOVA) with multiple comparisons test. *P ≤ 0·05; **P ≤ 0·01; ***P ≤ 0·001.
T cells from MOSPD2 KO mice display propensity for a Th2‐like phenotype
MOSPD2 is expressed on myeloid cells in mice, as in humans, and could not be detected in resting or activated T cells (Fig. 4a, Supporting information, Fig. S4). To test whether the effect on EAE pathogenesis in MOSPD2 KO mice involves indirect attenuation of T cell effector function, WT and MOSPD2 KO mice were immunized with pMOG35–55 and the secretion of the proinflammatory cytokines was measured after ex‐vivo challenge. The results, presented in Fig. 4b,c, demonstrate that immunization of MOSPD2 KO mice generated pMOG35–55 T cells with attenuated secretion of the proinflammatory cytokines IFN‐γ and IL‐17. This observation was evidently not antigen‐specific, as polyclonal stimulation with anti‐CD3 showed similar differences in cytokine secretion between WT and KO mice T cells. To further determine whether this feature is acquired inherently, T cells were isolated from the spleens of WT and MOSPD2 KO mice and activated with anti‐CD3 and anti‐CD28 antibodies. After 24, 48 and 72 h, cells were collected, stained with Ki‐67 and the ratio of proliferating cells was analyzed. Figure 4d shows that proliferation of T cells derived from MOSPD2 KO mice was not impeded. Analysis of supernatants collected 48 and 72 h after activation showed, however, that the secretion of IL‐4 in T cells purified from MOSPD2 KO mice increased more than twofold (Fig. 4e), whereas IL‐17 and IFN‐γ dropped by ~40% (Fig. 4f,g). MOSPD2 deficiency did not hamper the production of the anti‐inflammatory cytokine IL‐10 (Fig. 4h).
Fig. 4.
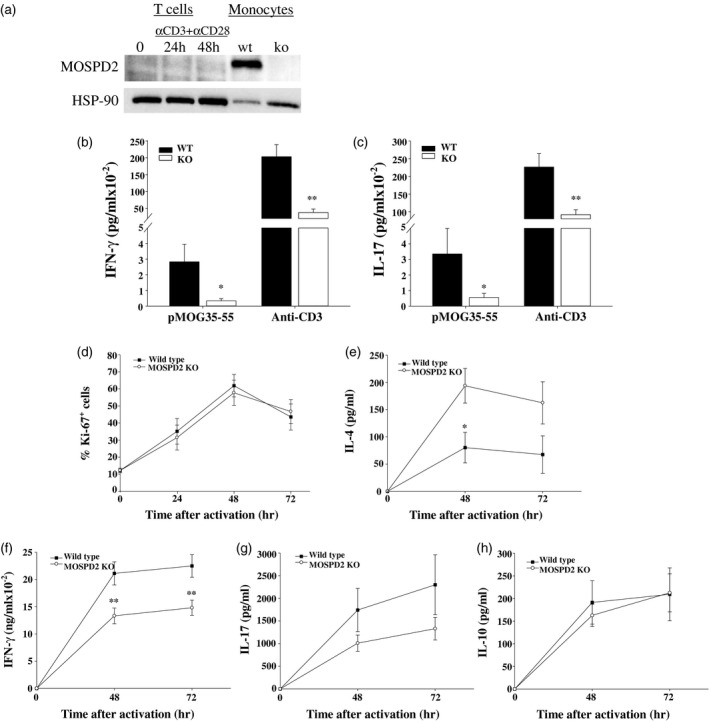
T cells from motile sperm domain‐containing protein 2 (MOSPD2) knock‐out (KO) mice secrete reduced level of proinflammatory cytokines. (a) Relative protein expression of MOSPD2 in resting and activated T cells and monocytes isolated from wild‐type (WT) mouse spleen. Purified T cells were activated with plate‐bound anti‐CD3 and anti‐CD28 for the indicated time‐points. (b,c) Lymph node cells were excised from WT and MOSPD2 KO mice immunized subcutaneously with pMOG35–55 in complete Freund’s adjuvant (CFA) and stimulated for 3 days with pMOG35–55 (30 μg/ml) or anti‐CD3 (2 μg/ml). Supernatant was collected and tested for (b) IFN‐γ (c) and interleukin (IL)‐17 (n = 9 per group). P‐values were calculated by Student’s t‐test. Experiments were performed in triplicate and data are presented as mean ± standard error (s.e.). *P < 0·05; **P < 0·01. (d) Proliferation of purified T cells activated with plate‐bound anti‐CD3 and anti‐CD28 (5 μg/ml) analyzed by 3‐day staining for Ki‐67+ cells (n = 5 per group). Supernatant from T cells described in (d) was collected pre‐, 48 and 72 h after activation and tested by enzyme‐linked immunosorbent assay (ELISA) for (e) IL‐4, (f) interferon (IFN)‐γ, (g) IL‐17 and (h) IL‐10 (n = 12–13 per group). P‐values for (d–h) were calculated by analysis of variance (ANOVA) with multiple comparisons. *P < 0·05; **P < 0·01.
Monocyte release from BM in MOSPD2 KO is enhanced in EAE
In EAE, the frequency of peripheral CD11b+Ly6Chi inflammatory monocytes increases during preclinical and peak disease [8]. Following their impaired exit from the BM in MOSPD2 KO mice, we monitored for changes in circulating inflammatory monocytes during peak disease and at euthanasia. We found that even though EAE was suppressed in MOSPD2 KO mice throughout the experiment (Fig. 5a), the frequency alteration pattern in total and inflammatory CD11b+Ly6Chi monocytes was similar for WT and KO mice, but remained lower in the latter (Fig. 5b–d).
Fig. 5.
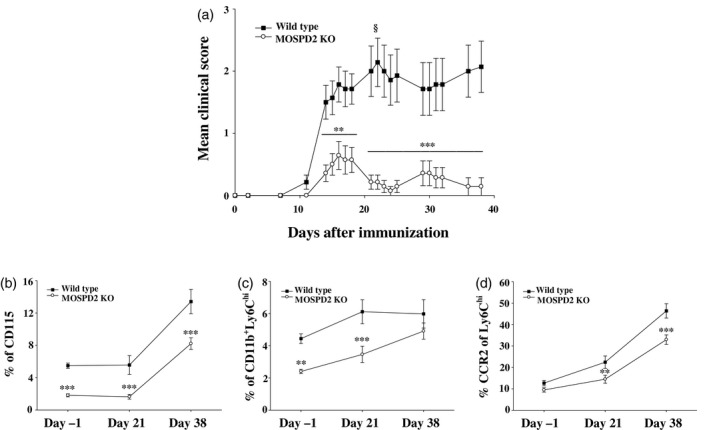
Monocyte release from bone marrow (BM) in motile sperm domain‐containing protein 2 (MOSPD2) knock‐out (KO) mice is enhanced during experimental autoimmune encephalomyelitis (EAE). (a) Wild‐type and MOSPD2 KO mice were immunized with pMOG35‐55/complete Freund’s adjuvant (CFA). Pertussis toxin (PT) was injected on day 0 and 48 h later (n = 14 per group). §Two mice died (b–d). Mice were bled 1 day before disease induction, on day 20 and upon euthanasia. Peripheral blood was stained for (a) total monocytes, (b) inflammatory monocytes and (c) Chemokine receptor (CCR2+) inflammatory monocytes (n = 12 per group). One of three experiments is shown. Data are presented as mean ± standard error (s.e.). P‐values were calculated by analysis of variance (ANOVA) with multiple comparisons test. **P ≤ 0·01; ***P ≤ 0·001.
Generation and selection of lead anti‐MOSPD2 mAbs
Recognizing the potential of MOSPD2 as a therapeutic target in MS and possibly additional monocyte‐affected inflammatory diseases, we set out to develop anti‐MOSPD2 mAbs that would show a beneficial effect on EAE pathogenesis. Mouse mAbs were generated by immunizing mice with the extracellular portion of human MOSPD2. To support selection of lead antibody, all mAbs were tested by flow cytometry for binding to plasma membrane expressing full‐length human MOSPD2 (amino acids 1–518) and subsequently examined for their effect on the migration of human monocytes in vitro. Two of the tested clones, clones‐1 and ‐3, profoundly inhibited the migration of human monocytes but not of T cells (Fig. 6a). Antibodies can bind to their antigen at different affinity rates, with equilibrium dissociation constant (KD) values ranging from 10−6 M for low‐affinity antibodies to 10−12 for very high‐affinity antibodies. To assess the binding strength of clones‐1 and ‐3 to human MOSPD2, affinity tests were run using Biacore. Results presented in Table 1 show that KD values for both antibodies were in the lower range (10−9–10−10 M), indicating a strong antibody/antigen interaction. Between the two, clone‐3 was the preferred binder to MOSPD2, with a lower KD value than clone‐1. To delineate the epitopes in human MOSPD2 recognized by the two mouse clones that displayed biological activity, a microarray containing 15‐amino acid‐long peptides with a peptide–peptide overlap of 14 amino acids demonstrated that clones‐1 and ‐3 display a clear response against a single similar epitope. In agreement with its KD, clone‐3 also exhibited a stronger response to the identified epitope than did clone‐1 (Fig. 6b). MOSPD2 is a conserved protein, with 90% sequence identity between human and mouse (Fig. 6c). Moreover, the binding regions of clones‐1 and ‐3 are completely identical in human and mouse MOSPD2. Accordingly, we anticipated that clones‐1 and ‐3 would cross‐react with mouse MOSPD2. Western blots and FACS analysis shown in Fig. 6d,e confirmed binding of both clones to mouse MOSPD2. When anti‐MOSPD2 mAb clones‐1 and ‐3 were employed on myeloid cells isolated from mouse spleen, they profoundly inhibited their migration (Fig. 6f).
Fig. 6.
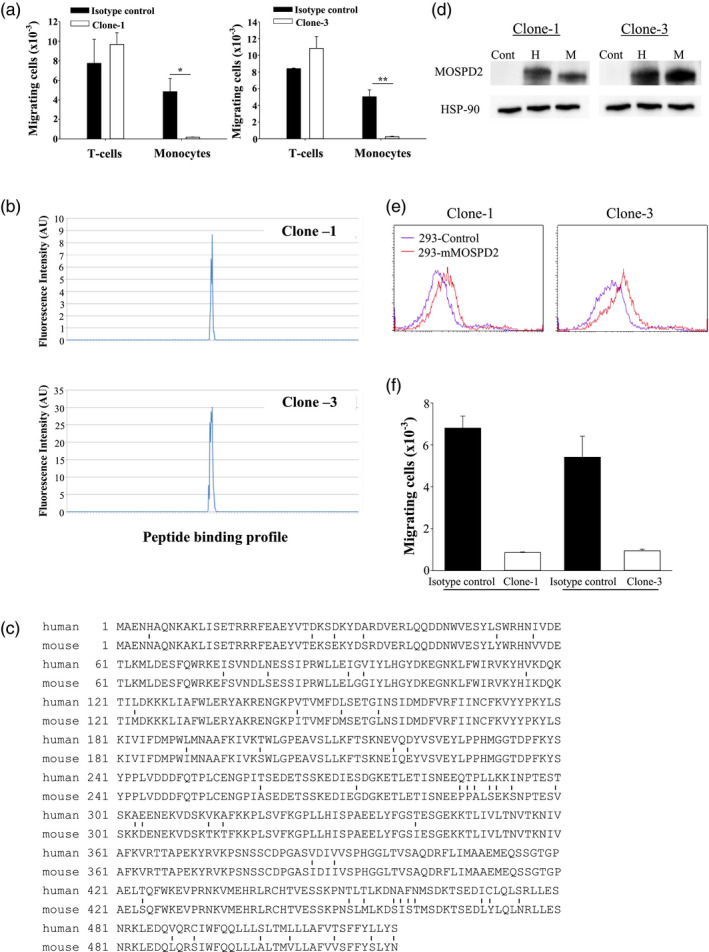
Selection of lead anti‐motile sperm domain‐containing protein 2 (MOSPD2) monoclonal antibodies (mAbs). (a) Migration of primary CD14+ human monocytes towards stromal derived factor‐1 alpha + monocyte chemotactic factor 1 (SDF‐1+MCP‐1) and CD3 T cells towards SDF‐1 + regulated upon activation, normal T cell expressed and secreted (RANTES) in the presence of 10 μg/ml anti‐MOSPD2 mAb Clone‐s1 or ‐3 compared with matched isotype controls. One of three experiments is shown. P‐values were calculated by Student’s t‐test. Data are of triplicates and shown as mean ± standard error (s.e.). *P < 0·005; **P < 0·001. (b) Linear epitope mapping of clones‐1 and ‐3 against the extracellular region of human MOSPD2. (c) Alignment of human and mouse MOSPD2. Differences are indicated by vertical lines. (d) Western blots of human embryonic kidney 293 (HEK) 293 cells transfected with human and mouse MOSPD2 and detected with anti‐human MOSPD2 mAbs clones‐1 or ‐3. One of two experiments is shown. (e) Fluorescence activated cell sorter (FACS) analysis for the recognition of anti‐human MOSPD2 mAbs clones‐1 and ‐3 of surface‐expressed mouse MOSPD2. One of two experiments is shown. (f) Migration of mouse splenic CD11b cells towards SDF‐1 + MCP‐1 in the presence of 10 μg/ml anti‐MOSPD2 mAb clone‐1 or clone‐3 compared with matched isotype controls. One of three experiments is shown. P‐values were calculated by Student’s t‐test. Data are of triplicates and shown as mean ± s.e. *P < 0·005; **P < 0·001.
Table 1.
Kinetic parameters and affinity of anti‐MOSPD2 mAb/antigen interaction
| Antibody | Ka (1/Ms) | Kd (1/s) | KD (M) |
|---|---|---|---|
| Clone‐1 | 1·603 × 105 | 5·071 × 10−4 | 3·163 × 10−9 |
| Clone‐3 | 4·555 × 105 | 3·803 × 10−4 | 8·35 × 10−10 |
MOSPD2 = motile sperm domain‐containing protein 2; mAb = monoclonal antibody.
Anti‐MOSPD2 mAbs display therapeutic effect against EAE
The essential role demonstrated for MOSPD2 in EAE development using KO mice suggests that clones‐1 and ‐3 mAbs may have a therapeutic effect in EAE. To put this to the test, we administered the antibodies in a prophylactic regimen, starting 1 day before immunization of WT mice for EAE induction with subsequent antibody injections every other day. We found that clones‐1 and ‐3 significantly inhibited disease development, with clone‐3 exhibiting superior efficacy over clone‐1 (Fig. 7a,b). Similar to the pathology observed for MOSPD2 KO mice immunized for EAE induction, administration of anti‐MOSPD2 antibodies markedly reduced the number of infiltrating T cells and monocytes and restricted myelin damage (Fig. 7c–f). We then investigated whether targeting MOSPD2 with mAbs in advanced stages of the disease can ameliorate EAE. To that end, EAE was first induced in WT mice, and when mean clinical severity reached a score of 2, mice were divided into two groups and treated with isotype control or anti‐MOSPD2 mAb clone‐3. The results presented in Fig. 7g show that targeting MOSPD2 at peak disease with specific mAbs significantly reduced disease severity.
Fig. 7.
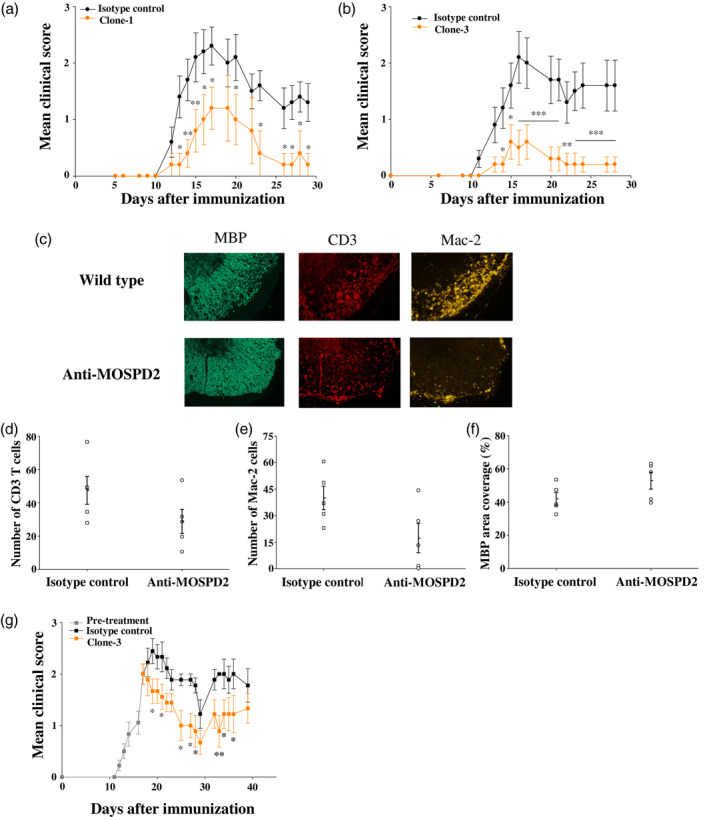
Treatment with anti‐human motile sperm domain‐containing protein 2 (MOSPD2) monoclonal antibodies (mAb) ameliorates experimental autoimmune encephalomyelitis (EAE). (a,b) Prevention of EAE with anti‐human MOSPD2 mAbs clones‐1 and ‐3. Clone‐1 or isotype control [mouse immunoglobulin (Ig)G2a] (500 μg/injection) were given on days −1, 3, 6 and 9 to disease induction. Clone‐3 or isotype control (mouse IgG1) (500 μg/injection) was given on days −1, 3, 6, 9, 13 and 16 (n = 6‐10 per group). One of two experiments is shown. Data are presented as mean ± standard error (s.e.). (c) Spinal cords from (b) were stained by immunofluorescence with anti‐CD3, anti‐macrophage antigen‐2 (Mac‐2) and anti‐myelin basic protein (MBP) for the detection of infiltrating T cells, monocytes/macrophages and proportion of myelin degradation, respectively (d,e). Number of infiltrating CD3 T cells and Mac‐2 monocytes/macrophages in the most populated infiltrate. The counted area of the infiltrate was 0·16 mm2 (n = 5 per group). (f) Mean MBP coverage area. The fluorescence intensity measured in sections taken from healthy control mice was determined as 100% coverage area (n = 5 per group). (g) EAE treatment regimen. When mean severity reached a score of 2 (day 17), mice were divided into two groups and treated with clone‐3 or isotype control by injection given up to day 34 every 2–3 days (500 μg/injection) (n = 9 per group). Data are presented as mean ± s.e. P‐values were calculated by analysis of variance (ANOVA) with multiple comparisons test. *P ≤ 0·05; **P ≤ 0·01; ***P ≤ 0·001.
Discussion
In both MS and its animal model EAE, lymphocytes and monocytes are implicated in disease initiation and progression. However, whereas some current MS drugs are designed to attenuate the adaptive immune reaction, none directly target monocytes/macrophages. These cells, which are heavily associated with ongoing demyelination [22, 23] and are found in the inflamed CNS of MS patients, were thought to originate from self‐maintained CNS microglia seeded in the yolk sac stage and/or from circulating monocytes. Until recently, no functional distinction could be made between microglia and circulating monocytes with regard to their contribution to EAE pathogenesis. In their pivotal paper, however, Yamasaki et al. [10] demonstrated that monocyte‐derived macrophages coming from the circulation are involved in demyelination, whereas microglia are more likely to clear debris. These data suggest that blocking the migration of circulating monocytes to the CNS of MS patients could have therapeutic potential. Chemokines and their cognate receptors play an important role in regulating monocyte migration, facilitating their release from the BM to the blood and their arrival to tissues in steady state and during injury. However, monocyte chemotaxis is controlled by a range of different chemokines and various cognate receptors. Overcoming this redundancy for therapeutic purposes of inflammatory diseases such as MS is therefore highly important.
In this study, we demonstrate that MOSPD2, a protein shown to regulate chemokine‐induced human monocyte migration regardless of the activating ligand, is a potential target for treating MS. For this purpose, we undertook two different approaches. The first was to generate MOSPD2‐deficient mice, enabling the study of clinical manifestation, histopathology and mechanism, and the second included the use of mAbs that were developed against MOSPD2. Data base (BioGPS) and our study indicate that MOSPD2 expression is circumscribed to immune cells from myeloid origin. We therefore sought to investigate the effect of MOSPD2 deficiency on immune system development and function in the gene‐targeted mice. Unlike the thymus, lymph nodes and spleen, which developed normally in the KO mice and presented comparable frequencies of immune cell subsets to WT mice, the profile drawn from the periphery demonstrated a decrease in the frequency of CD11b+Ly6C+ inflammatory monocytes. Moreover, expression of CCR2+ among CD11b+Ly6C+ monocytes, which is required for their egress from the BM and entry into inflamed tissue, was also reduced. In most tissues, macrophage population is seeded before birth and self‐renewed during steady state [24, 25], while peripheral monocytes are constantly replenished from the BM [26, 27]. Accordingly, we reasoned that the disparity observed in monocyte frequency between the circulation and the spleen could be related to the role MOSPD2 plays in regulating monocyte migration later in ontogeny. Indeed, following peripheral ablation, the frequency of blood monocytes in MOSPD2 KO mice returned to its baseline and never equaled that of WT mice. In this respect, it would be interesting to determine whether tissues that are constantly replenished with HSC‐derived monocytes, such as the intestine, will present a reduced number of tissue macrophages. King et al. [8] demonstrated that during the course of EAE, CD11b+Ly6C+ monocytes accumulate in the blood. Even though MOSPD2 deficiency reduced steady‐state frequency of blood CD11b+Ly6C+ monocytes, it did not impede frequency increase following disease induction. These results suggest that, to some extent, conditions created during inflammation overcome MOSPD2 regulation of monocyte release from BM. However, penetration of blood‐borne monocytes into the CNS of MOSPD2 KO mice was dramatically reduced, possibly due to tighter MOSPD2 regulation of the migration properties of monocytes from the periphery to the CNS. Nonetheless, we could not exclude the possibility that restricted infiltration of monocytes to the CNS in actively immunized KO mice is, at least partially, the outcome of a restrained proinflammatory milieu instituted during the initial phase of EAE by encephalitogenic T cells. Regarding T cell effector function, it is currently not clear what causes T cells from MOSPD2 KO mice to produce decreased amounts of Th1/Th17 proinflammatory cytokines and increase IL‐4 secretion, given that MOSPD2 could not be detected in T cells. RNA‐Seq studies to assess regulation on gene expression by MOSPD2 in monocytes, which may also affect T cell differentiation, are under way.
In contrast to the results seen with KO mice, in BM chimera experiments, the induction phase in EAE mediated by T cells was unimpaired when HSCs from KO mice were engrafted into KO or WT recipients. Nonetheless, subsequent stages in disease pathogenesis, which involve migration of peripheral monocytes to the CNS, were presumably curbed, as disease progression was suppressed in mice engrafted with BM from KO mice. This finding was further supported by adoptive transfer experiments to induce disease using MOG TCR‐transgenic mice, in which altered disease pathogenesis was exhibited in the KO recipients.
The profound effect that MOSPD2 deficiency had on EAE pathogenesis prompted the development of mAbs with the prospect of perturbing monocyte infiltration into the CNS through specific binding and neutralization of MOSPD2. Accordingly, we developed an array of mAbs against human MOSPD2 and tested for their effect on human monocyte migration. Among these mAbs, two clones had a significant impact on monocyte chemotaxis. Both mAbs have high affinity KD values, and epitope mapping revealed a common core binding sequence on MOSPD2. Although the mAbs described in this study were directed against human MOSPD2, high general homology and total identity of the antibody binding regions to human and murine MOSPD2 enabled in‐vivo studies to evaluate their efficacy on EAE induction and progression. The results strongly suggest that anti‐MOSPD2 antibodies have a potential to treat CNS inflammatory diseases, such as MS, in which monocyte migration and accumulation are implicated. Among all approved MS medications, none is directly aimed at the innate immune cells. Restricting the entry of blood‐borne monocytes into the CNS of MS patients could attenuate disease deterioration in several ways, as monocytes are not only instrumental in the demyelination process, but also produce inflammation mediators that foment recruitment of leukocytes and can become monocyte‐derived dendritic cells that proficiently present newly exposed antigens (epitope spreading). We therefore conclude that targeting MOSPD2 may offer a remedy with a new action mechanism for the treatment of MS.
Disclosures
The authors are employees and stock option holders of VBL Therapeutics.
Author contributions
N. Y., P. K., Y. S., O. P. M. and B. F. performed and guided experimental work. E. B. and I. M. supervised the design and performance of the experiments. I. M. wrote the manuscript.
Supporting information
Fig. S1. Cell subset characterization in lymphoid tissues of MOSPD2 KO mice. Total spleen cell count (a) Flow cytometric ratio evaluation in the spleen of (b) B‐lymphocytes (c) myeloid (d) CD4 T cells and (e) regulatory CD4+ T cells. (f) Total thymus cell count. Flow cytometric ratio evaluation in the thymus of (g) SP CD4 and (H) SP CD8 T cells. (n = 5‐10 per group). P values were calculated by Student’s t test. Results are mean ± SE. *P ≤ 0·05. (I) Flow cytometric ratio evaluation in lymph nodes. Mean ± SE of two experiments is shown. Each experiment includes a pool of five mice.
Fig. S2. Effector function in MOSPD2‐deficient myeloid cells. (a) Purified TCR Tg CD4+ T cells specific for pOVA323‐339 were labelled with CFSE, co‐cultured with monocytes isolated from wild‐type (purple) or MOSPD2‐deficient (red) mice, and activated for 72 hr in the presence of 0·1 μg/ml or 1 μg/ml peptide. CFSE dilution was analyzed by FACS. Each experiment was performed in triplicate. One of three experiments is shown. (b) OT‐II T cells were co‐cultured for 3 days with CD11b+ myeloid cells isolated from spleens of wild‐type and MOSPD2 KO mice in the presence of the antigen at the indicated concentration and supernatants were tested for the indicated cytokines (n = 5‐6 per group). Results are mean ± SE. (c) Wild‐type and MOSPD2 KO mice were immunized with pOVA323‐339 in CFA and 7‐10 days later lymph nodes were excised. Cells were challenged for 3 days and then tested for proliferation by flow cytometry using Ki‐67 (n = 6 per group). Results are mean ± SE. (e) Whole blood from wild‐type and MOSPD2 KO mice was treated to deplete RBC. Leukocytes were incubated with PE‐labeled E. coli for 2 hr and the ratio of PE‐labeled cells was measured. Results are on gated neutrophils based on SSC and FSC (n = 5‐6). Results are mean ± SE. (e) CD11b+ myeloid cells were isolated from spleens of wild‐type and MOSPD2 KO mice in the presence of LPS (100 ng/ml) for 24 hr. Supernatants were collected and tested by ELISA for TNF‐α and IL‐6 (n = 8 per group). Results are mean ± SE.
Fig. S3. Infiltration of inflammatory monocytes into the CNS. Mononuclear cells were isolated from the brains of wild‐type and MOSPD2 KO mice on day 38 after EAE induction and stained for CD45 and Ly6C. CD45 is used to distinguish between CNS‐resident microglia (CD45low) and invading leukocytes (CD45high). Results are of pooled mononuclear cells from five mice from each group.
Fig. S4. Expression of MOSPD2 in mouse immune cell subsets. B cells and monocytes were isolated from the spleen of wild‐type mice using CD45R and CD11b microbeads, respectively. Neutrophils were isolated from the BM using mouse Neutrophil Isolation Kit.
References
- 1. Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol 2005; 23:683–747. [DOI] [PubMed] [Google Scholar]
- 2. Langrish CL, Chen Y, Blumenschein WM et al IL‐23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 2005; 201:233–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sriram S, Solomon D, Rouse RV, Steinman L. Identification of T cell subsets and B lymphocytes in mouse brain experimental allergic encephalitis lesions. J Immunol 1982; 129:1649–51. [PubMed] [Google Scholar]
- 4. Mishra MK, Yong VW. Myeloid cells – targets of medication in multiple sclerosis. Nat Rev Neurol 2016; 12:539–51. [DOI] [PubMed] [Google Scholar]
- 5. Holman DW, Klein RS, Ransohoff RM. The blood‐brain barrier, chemokines and multiple sclerosis. Biochim Biophys Acta 2011;1812:220–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Proudfoot AE, de Souza AL, Muzio V. The use of chemokine antagonists in EAE models. J Neuroimmunol 2008; 198:27–30. [DOI] [PubMed] [Google Scholar]
- 7. Cheng W, Chen G. Chemokines and chemokine receptors in multiple sclerosis. Mediat Inflamm 2014; 2014:659206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. King IL, Dickendesher TL, Segal BM. Circulating Ly‐6C+ myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. Blood 2009; 113:3190–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FM. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci 2011; 14:1142–9. [DOI] [PubMed] [Google Scholar]
- 10. Yamasaki R, Lu H, Butovsky O et al Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med 2014; 211:1533–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Benveniste EN. Role of macrophages/microglia in multiple sclerosis and experimental allergic encephalomyelitis. J Mol Med 1997; 75:165–73. [DOI] [PubMed] [Google Scholar]
- 12. Bauer J, Huitinga I, Zhao W, Lassmann H, Hickey WF, Dijkstra CD. The role of macrophages, perivascular cells, and microglial cells in the pathogenesis of experimental autoimmune encephalomyelitis. Glia 1995; 15:437–46. [DOI] [PubMed] [Google Scholar]
- 13. Mossakowski AA, Pohlan J, Bremer D et al Tracking CNS and systemic sources of oxidative stress during the course of chronic neuroinflammation. Acta Neuropathol 2015; 130:799–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. van Horssen J, Witte ME, Schreibelt G, de Vries HE. Radical changes in multiple sclerosis pathogenesis. Biochim Biophys Acta 2011; 1812:141–50. [DOI] [PubMed] [Google Scholar]
- 15. Levesque SA, Pare A, Mailhot B et al Myeloid cell transmigration across the CNS vasculature triggers IL‐1beta‐driven neuroinflammation during autoimmune encephalomyelitis in mice. J Exp Med 2016; 213:929–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wolf Y, Shemer A, Polonsky M et al Autonomous TNF is critical for in vivo monocyte survival in steady state and inflammation. J Exp Med 2017; 214:905–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jordao MJC, Sankowski R, Brendecke SM et al Single‐cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 2019; 363:eaat7554. [DOI] [PubMed] [Google Scholar]
- 18. Mildner A, Mack M, Schmidt H et al CCR2+Ly‐6Chi monocytes are crucial for the effector phase of autoimmunity in the central nervous system. Brain 2009; 132:2487–500. [DOI] [PubMed] [Google Scholar]
- 19. Mendel I, Yacov N, Salem Y, Propheta‐Meiran O, Ishai E, Breitbart E. Identification of motile sperm domain‐containing protein 2 as regulator of human monocyte migration. J Immunol 2017; 198:2125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mahad DJ, Ransohoff RM. The role of MCP‐1 (CCL2) and CCR2 in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Semin Immunol 2003; 15:23–32. [DOI] [PubMed] [Google Scholar]
- 21. Tacke F, Alvarez D, Kaplan TJ et al Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest 2007; 117:185–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute multiple sclerosis lesions. Brain 1997; 120(Pt 3):393–9. [DOI] [PubMed] [Google Scholar]
- 23. Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med 1998; 338:278–85. [DOI] [PubMed] [Google Scholar]
- 24. Hashimoto D, Chow A, Noizat C et al Tissue‐resident macrophages self‐maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013; 38:792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mass E, Ballesteros I, Farlik M et al Specification of tissue‐resident macrophages during organogenesis. Science 2016; 353:aaf4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Varol C, Mildner A, Jung S. Macrophages: development and tissue specialization. Annu Rev Immunol 2015; 33:643–75. [DOI] [PubMed] [Google Scholar]
- 27. Patel AA, Zhang Y, Fullerton JN et al The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J Exp Med 2017; 214:1913–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Cell subset characterization in lymphoid tissues of MOSPD2 KO mice. Total spleen cell count (a) Flow cytometric ratio evaluation in the spleen of (b) B‐lymphocytes (c) myeloid (d) CD4 T cells and (e) regulatory CD4+ T cells. (f) Total thymus cell count. Flow cytometric ratio evaluation in the thymus of (g) SP CD4 and (H) SP CD8 T cells. (n = 5‐10 per group). P values were calculated by Student’s t test. Results are mean ± SE. *P ≤ 0·05. (I) Flow cytometric ratio evaluation in lymph nodes. Mean ± SE of two experiments is shown. Each experiment includes a pool of five mice.
Fig. S2. Effector function in MOSPD2‐deficient myeloid cells. (a) Purified TCR Tg CD4+ T cells specific for pOVA323‐339 were labelled with CFSE, co‐cultured with monocytes isolated from wild‐type (purple) or MOSPD2‐deficient (red) mice, and activated for 72 hr in the presence of 0·1 μg/ml or 1 μg/ml peptide. CFSE dilution was analyzed by FACS. Each experiment was performed in triplicate. One of three experiments is shown. (b) OT‐II T cells were co‐cultured for 3 days with CD11b+ myeloid cells isolated from spleens of wild‐type and MOSPD2 KO mice in the presence of the antigen at the indicated concentration and supernatants were tested for the indicated cytokines (n = 5‐6 per group). Results are mean ± SE. (c) Wild‐type and MOSPD2 KO mice were immunized with pOVA323‐339 in CFA and 7‐10 days later lymph nodes were excised. Cells were challenged for 3 days and then tested for proliferation by flow cytometry using Ki‐67 (n = 6 per group). Results are mean ± SE. (e) Whole blood from wild‐type and MOSPD2 KO mice was treated to deplete RBC. Leukocytes were incubated with PE‐labeled E. coli for 2 hr and the ratio of PE‐labeled cells was measured. Results are on gated neutrophils based on SSC and FSC (n = 5‐6). Results are mean ± SE. (e) CD11b+ myeloid cells were isolated from spleens of wild‐type and MOSPD2 KO mice in the presence of LPS (100 ng/ml) for 24 hr. Supernatants were collected and tested by ELISA for TNF‐α and IL‐6 (n = 8 per group). Results are mean ± SE.
Fig. S3. Infiltration of inflammatory monocytes into the CNS. Mononuclear cells were isolated from the brains of wild‐type and MOSPD2 KO mice on day 38 after EAE induction and stained for CD45 and Ly6C. CD45 is used to distinguish between CNS‐resident microglia (CD45low) and invading leukocytes (CD45high). Results are of pooled mononuclear cells from five mice from each group.
Fig. S4. Expression of MOSPD2 in mouse immune cell subsets. B cells and monocytes were isolated from the spleen of wild‐type mice using CD45R and CD11b microbeads, respectively. Neutrophils were isolated from the BM using mouse Neutrophil Isolation Kit.