

Abstract
The features that stabilize the structures of membrane proteins remain poorly understood. Polar interactions contribute modestly, and the hydrophobic effect contributes little to the energetics of apolar side-chain packing in membranes. Disruption of steric packing can destabilize the native folds of membrane proteins, but is packing alone sufficient to drive folding in lipids? If so, then membrane proteins stabilized by this feature should be readily designed and structurally characterized—yet this has not been achieved. Through simulation of the natural protein phospholamban and redesign of variants, we define a steric packing code underlying its assembly. Synthetic membrane proteins designed using this code and stabilized entirely by apolar side chains conform to the intended fold. Although highly stable, the steric complementarity required for their folding is surprisingly stringent. Structural informatics shows that the designed packing motif recurs across the proteome, emphasizing a prominent role for precise apolar packing in membrane protein folding, stabilization, and evolution.
As membrane proteins (MPs) exit the translocon, they complete folding and assembly in the lipid milieu. However, the forces that stabilize proteins in membranes are less well understood for MPs than for water-soluble proteins. The hydrophobic effect provided by packing apolar side chains in the protein interior represents the predominant driving force for protein folding in water, yet it is negligible in lipid membranes. In MPs, it is unclear whether analogous side-chain packing in the native state can be the primary source of structural stabilization. On the one hand, those same apolar moieties pack similarly with lipid tails in the exposed unfolded state; on the other hand, structural informatics suggests that side chains pack more efficiently in MPs (1–4) and thus stabilize folding via favorable van der Waals (vdW) interactions and possibly also lipid-specific effects, such as solvophobic exclusion (5–9). Mutations to MPs that strongly disrupt vdW packing in the protein interior, either by introducing voids or steric clashes, have been shown to destabilize their native state to various degrees (8, 10–12). However, it has proven difficult to determine whether apolar packing can play a dominant role in MP folding, or whether this feature is secondary to other more-stabilizing interactions, namely hydrogen bonding (13–17), topology (18), and weakly polar interactions (19, 20) (Fig. 1). If tight, sterically compatible apolar packing is strongly stabilizing, then it should be possible to design and structurally characterize folded MPs stabilized by this feature alone. However, this has not been accomplished, despite many attempts (14, 17, 21–24), and all successfully designed MPs have relied on hydrogen bonding (17, 25), metal-ligand interactions (26, 27), small residue motifs (28, 29), or templating of their folds with extramembrane loops and water-soluble domains (30). In this study, we used a multipronged approach to design MPs that show that apolar side-chain packing plays a major role throughout MP folding.
Fig. 1. Noncovalent forces in MP folding.
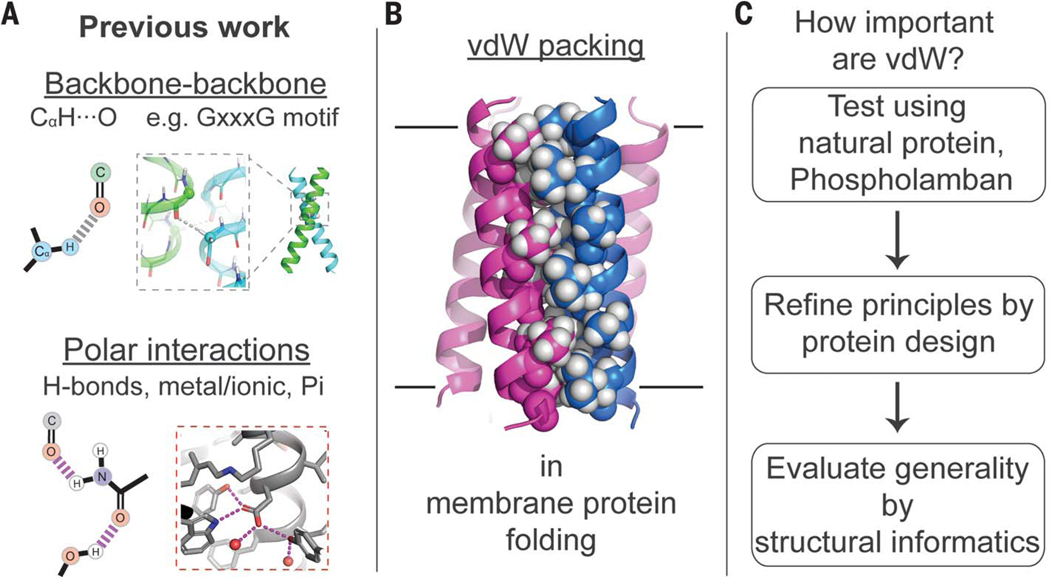
(A) Polar interactions are known to stabilize MP structures. (B) vdW packing is abundant in the folded state, but similar interactions with membrane lipids occur in the unfolded state; it is unknown whether packing alone can drive MP folding. (C) Overview of the multipronged approach used.
To isolate the role of packing in MP stabilization, we focus on the self-association of transmembrane (TM) α helices from single-span MPs, chosen for their pervasive biological importance and technical advantages. More than 50% of all MPs are single-spanning, yet these are the least structurally characterized class of MPs. Lateral interactions between single-span TM helices play vital roles in processes such as signaling and ion conduction (31, 32), and the aberrant assembly of these helices is central to diseases ranging from cancer to Alzheimer’s disease (33, 34). Additionally, unlike complex multipass proteins whose folds are subject to constraining loops and extramembrane domains, single-span TM bundles allow investigation of unconstrained interhelical interactions with a clear unfolded state—a monomeric α helix—where conformational specificity and thermodynamics can be evaluated by the oligomeric distribution.
We began by analyzing the homopentameric assembly of the small single-span MP phospholamban (PLN), a regulator of the sarcoplasmic reticulum Ca2+ pump. PLN’s TM helix contains an LxxIxxx sequence repeat (Fig. 2), which has been extensively studied in water-soluble coiled coils (35, 36). Given the canonical labeling for α-helical seven-residue repeats (abcdefg), a and d side chains (Leu and Ile of LxxIxxx, respectively) project toward the bundle core, whereas e and g residues face the intersubunit interface (37). Recent water-soluble peptide studies have demonstrated that it is possible to design four-to eight-stranded bundles by manipulating the physicochemical properties of the residues at the e and g positions (35). Previous strategies to design MP assemblies by apolar interactions focused primarily on a and d positions. Thus, we explored the role of both core and interfacial packing interactions, focusing on both PLN and rationally designed peptides. We show that when apolar packing is used as the sole stabilizing principle, folding requires side-chain geometric complementarity that is surprisingly stringent. Nevertheless, the optimization of steric packing alone can determine the thermodynamics and the three-dimensional (3D) architecture of these highly stable TM protein bundles. These synthetic MPs provide comprehensible and robust model systems for future investigations of apolar packing in MP folding, including the relative contributions of dispersion forces and entropy.
Fig. 2. All-atom MD simulation of PLN, a pentameric TM α-helical bundle, illuminates critical apolar packing interactions.
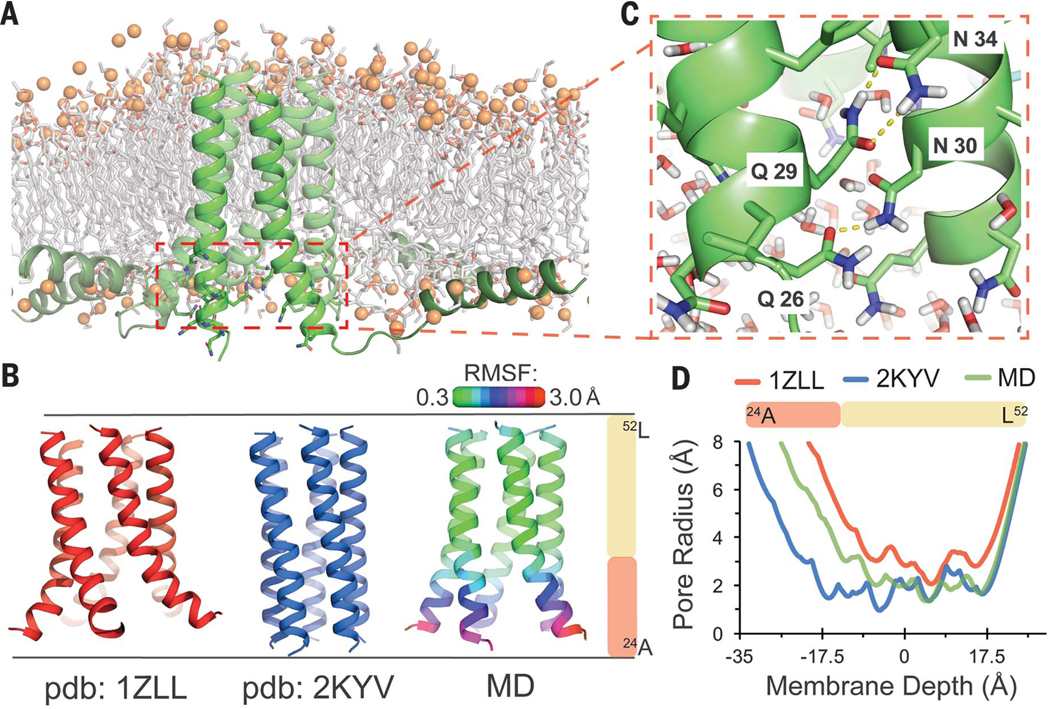
(A) Final simulation frame for full-length PLN in a POPC bilayer. Water molecules are hidden. (B) The TM domains of published NMR structures of PLN (PDB IDs: 1ZLL, red; 2KYV, blue) compared with our MD simulation. RMSF of backbone atoms versus the simulation medoid is displayed as ribbon color. (C) Snapshot showing the water-filled central cavity and rapidly fluctuating polar side-chain interactions at the splayed N-terminal third of the TM-spanning α helices. (D) Central cavity size within each helical bundle versus membrane depth. Single-letter abbreviations for the amino acid residues are as follows: A, Ala; C, Cys; D, Asp; E, Glu; F, Phe; G, Gly; H, His; I, Ile; K, Lys; L, Leu; M, Met; N, Asn; P, Pro; Q, Gln; R, Arg; S, Ser; T, Thr; V, Val; W, Trp; and Y, Tyr.
MD simulations of PLN identify a rigid pentameric domain stabilized by vdW packing
The membrane-spanning segment (residues 24 to 52) of PLN forms a homopentameric helical bundle. The N-terminal third of this bundle houses several strongly polar residues, whereas the C-terminal two-thirds is highly hydrophobic, containing 2.5 copies of the critical LxxIxxx motif (Fig. 2) (38). The interactions stabilizing the pentameric structure remain unclear, particularly the role of the polar sector. Two early nuclear magnetic resonance (NMR) structures of the PLN helical bundle derived from relatively sparse experimental restraints differ substantially [backbone root mean square deviation (RMSD) = 4.8 Å; residues 24 to 52]—most notably at the polar TM sector (39, 40). To obtain a refined model of PLN’s TM bundle, we performed atomistic molecular dynamics (MD) simulations of the full-length protein in a fluid bilayer [1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC)], beginning with the more recent NMR model [Protein Data Bank (PDB) ID 2KYV].
The initial model rapidly rearranged to a stable conformational ensemble (Fig. 2B and fig. S1B) similar to a previous 15-ns simulation using the alternative NMR structure (PDB ID 1ZLL) (41). The apolar C-terminal LxxIxxx-domain was tightly packed and very rigid [backbone root mean square fluctuation (RMSF) to medoid = 0.53 Å; residues 33 to 51], indicating that it plays an important role in stabilizing the structure.
By contrast, the polar N-terminal TM sector was more dynamic (1.53 Å) and became widely splayed (fig. S1B)—likely making it less critical for stabilization. Water rushed into this end of the bundle, accompanied by rapidly fluctuating hydration and interhelical side-chain hydrogen bonds (fig. S1, D to F). These findings are consistent with previous mutational studies of PLN (38, 42, 43) and with other MPs in which polar TM residues were found to modulate functional dynamics rather than stability (44). In PLN, tuning of stability and dynamics is likely important for functional interactions with cyclic adenosine monophosphate–dependent protein kinase and the Ca2+ adenosine triphosphatase.
Design and structural stabilization of apolar variants of the PLN pentamer
We next asked whether the well-packed apolar region of PLN, when free of strongly polar residues, can drive pentameric assembly of a PLN-like peptide, designated PL5. PL5 was designed using an idealized model built de novo by mathematical parameterization of PLN’s rigid C-terminal domain from simulation. For the C-terminal half of PL5, PLN’s native sequence was retained, changing only lipid-facing β-branched residues to Leu or Phe to facilitate synthesis. By contrast, the N-terminal TM portion of PLN was redesigned. Asparagines in the splayed polar sector of the bundle were converted to less-polar residues that were more sterically compatible with the idealized bundle (Fig. 3A). The remaining lipid-facing and terminal residues were chosen to facilitate synthesis, purification, and positioning within the bilayer. To determine whether the weak polarity of the interfacial Ser9, Cys11, and Cys16 of PL5 contributed to assembly, these residues were converted to the isosteric and fully hydrophobic S-Cα-ethyl-Gly (EtGly) in PL5EtG and PL5EtG3 (Fig. 3A) (45).
Fig. 3. Design and structural characterization of PLN-like pentameric protein PL5.
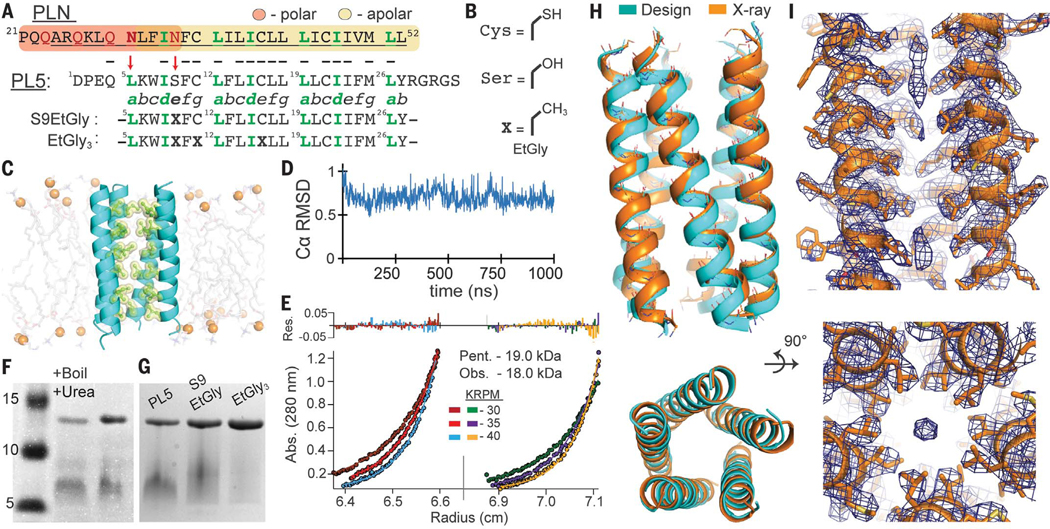
(A) Sequence of PLN and PL5. PLN’s polar region (orange) and apolar region (yellow) highlighted; membrane-spanning α helix underlined (residues 24 to 52). The seven-residue α-helical repeat is labeled “abcdefg”; LxxIxxx motif, green. Red arrows indicate polar-to-apolar mutations. Bold Xs denote EtGly residues. (B) EtGly is approximately isosteric to Ser and Cys. (C) Snapshot from a 1.0-μs MD simulation of PL5 in a POPC bilayer. (D) PL5 shows conformational rigidity by Cα RMSD versus medoid frame (residues 5 to 29). (E) Equilibrium analytical ultracentrifugation, PL5 at 58 μM and 79 μM in 33 mM myristyl sulfobetaine micelles, globally fit to a single species model. Apparent molecular weight = 4.74 monomers. Pent., pentamer; Obs., observed. (F) SDS-PAGE of PL5 in reducing conditions shows a single oligomeric state, resistant to heating (95°C, 30 min) in 2% LDS, 8 M urea. Similar to PLN, PL5 exhibits aberrant gel migration. Figure S2 confirms that the slower band is pentameric. (G) PL5, PL5EtG, and PL5EtG3 have similar oligomeric distributions. (H) The pentameric x-ray structure of PL5 (PDB ID 6MQU) closely matches the MD-refined design model (cyan). (I) Well-packed side chains are well-resolved in the 2Fo-Fc electron density map (σ = 1.0). Elongated density is present only at divergent ends of the bundle.
PL5 was shown to form stable pentamers by MD and solution measurements (Fig. 3, C to I). In all-atom MD, PL5’s initial conformation was stable over 1.0 μs (0.71 Å mean RMSD versus medoid). Analytical ultracentrifugation of PL5 in myristyl dimethyl-3-ammonio-1-propanesulfonate micelles gave an apparent molecular weight of 18 kDa (Fig. 3E), which is within the experimental error of the pentamer (19 ± 1.5 kDa, based on propagation of a 3% error for the computed partial specific volume). PL5, PL5EtG, and PL5EtG3 migrate as a single oligomeric state, confirmed to be pentameric, by SDS–polyacrylamide gel electrophoresis (PAGE) (fig. S2). Additionally, the PL5 pentamer is highly stable even after heating (95°C) in lithium dodecyl sulfate (LDS) and 8 M urea (Fig. 3D).
The 3.17-Å-resolution crystallographic structure of micelle-solubilized PL5 was in excellent agreement with the guiding MD model (1.1-Å backbone RMSD) (Fig. 3, figs. S3 and S4, and table S2). This structure can be mathematically described to within 0.6 Å as either a parallel left-handed coiled coil or a bundle of straight, tilted α helices (Fig. 4). The bundle appears to maximize interhelical packing at the expense of packing the central core, resulting in a small pore. Throughout most of the bundle, the pore is too small to accommodate even a single water molecule and was dry during MD. However, diffuse density observed near the ends of the bundle might represent portions of a detergent tail (Fig. 3I). The radius of the bundle expands by up to 1 Å as the steric bulk of the interfacial e and g residues progressively increases: Ser9, Cys11, Cys16, Leu18, Ile23, and Met25 (fig. S5). As expected from previous experiments with PLN (46) and our simulations (fig. S6), the side chains of Ser9, Cys11, and Cys16 donate intrahelical hydrogen bonds to preceding carbonyls (residue i-4), rather than forming interhelical hydrogen bonds.
Fig. 4. Side-chain steric packing at PL5’s symmetric helix-helix interface.
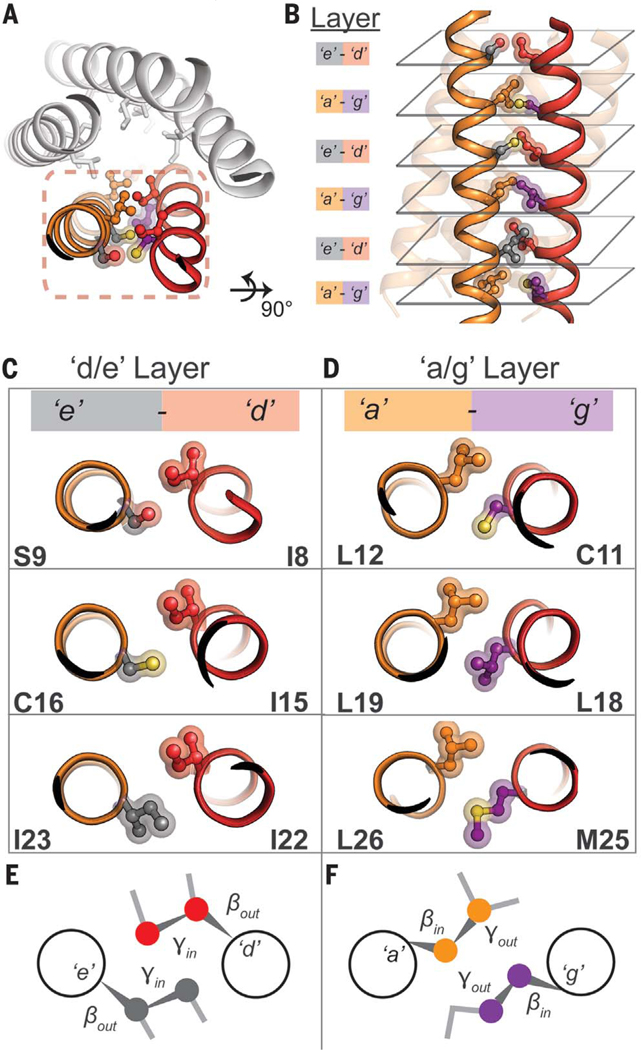
(A) The pairwise interaction of helices, symmetrically repeated, provides the primary stabilization for PL5. (B) High geometric complementarity of interacting residues across the helix-helix interface, roughly in layers: the a/g and e/g layers. (C and D) Axial view of side-chain packing of individual layers. (E) A potential stereochemical code required for pentameric assembly. At the e/d layer, the Cα-Cβ bond vector of each amino acid points outward from the helix-helix interface (βout), whereas the Cβ-Cγ bond vector faces inward (γin). This suggests that a heavy atom (e.g., N, C, O, S) at the gauche+ position is required for tight interhelical packing. (F) In the a/g layer, the opposite is true; the Cα-Cβ bond vector points inward (βin) and the Cβ-Cγ bond vector faces outward (γout).
A steric code for specific assembly and design of TM five-helix bundles
Inspection of PL5’s x-ray structure revealed knobs-into-holes packing (Fig. 4, C and D), as in multistranded coiled coils (36, 37). Side chains at d and e positions of opposing helices interact intimately along a d/e interface; side chains at a and g positions form the a/g interface (Fig. 4, E and F). At the a/g interface, the Cα-Cβ bond vector of each side chain is directed inward toward its helical neighbor, whereas the Cβ-Cγ bond is directed outward to avoid a steric clash (Fig. 4F). The close approach of opposing Cβ atoms provides evidence against placement of β-branched amino acids at g positions in the context of PL5’s sequence (Leu, a). The reverse is seen at the d/e interface; the Cα-Cβ bond vector is directed outward and the Cβ-Cγ bond points inward. Here, β-branched amino acids are sterically poised for tight packing. Although not β-branched, Ser and Cys also pack well at e, because they can position a heavy atom (O, S) in place of Cγ when in a gauche+ rotamer (i.e., −60°); this rotamer is further stabilized by intrahelical side-chain–main-chain hydrogen bonding. Interestingly, in contrast with the aforementioned packing code, water-soluble LxxIxxx-repeating peptides containing non-β-branched Glu at e and β-branched Ile at g specifically assembled into pentameric bundles (35), perhaps reflecting differences in forces between water-soluble versus membrane proteins.
To test our steric model, we first prepared a single-site variant of PL5, in which the g position Leu18 was replaced by its β-branched isomer Ile, resulting in a steric clash between this Ile18 Cγ methyl and Leu19 (a) Cβ methylene in models. In water-soluble coiled coils, similar conservative single-site mutations at noncore positions are generally tolerated or, at worst, alter the stoichiometry of association (35, 36). However, this replacement entirely destabilized the PL5 pentamer (fig. S7 and table S3). Thus, the requirement for steric complementarity is so stringent that misplacement of a single methyl group per helix eliminates detectable pentamer formation.
Next, we designed peptides to test the proposed rules for steric packing at the e position in pentamer assembly (Fig. 5). The peptide sequences repeated a minimalist heptad LaxxIdXexxLg, systematically varying the e position (Xe) to either one of the β-branched amino acids Ile, Val, Thr, or Cys (designated e-Ile, e-Val, e-Thr, and e-Cys, respectively). As stringent controls, we also prepared variants with Ala or Leu at e, whose side chains would not pack as favorably but would not necessarily clash either (Fig. 5B).
Fig. 5. Design of pentameric MPs from first principles of steric packing.
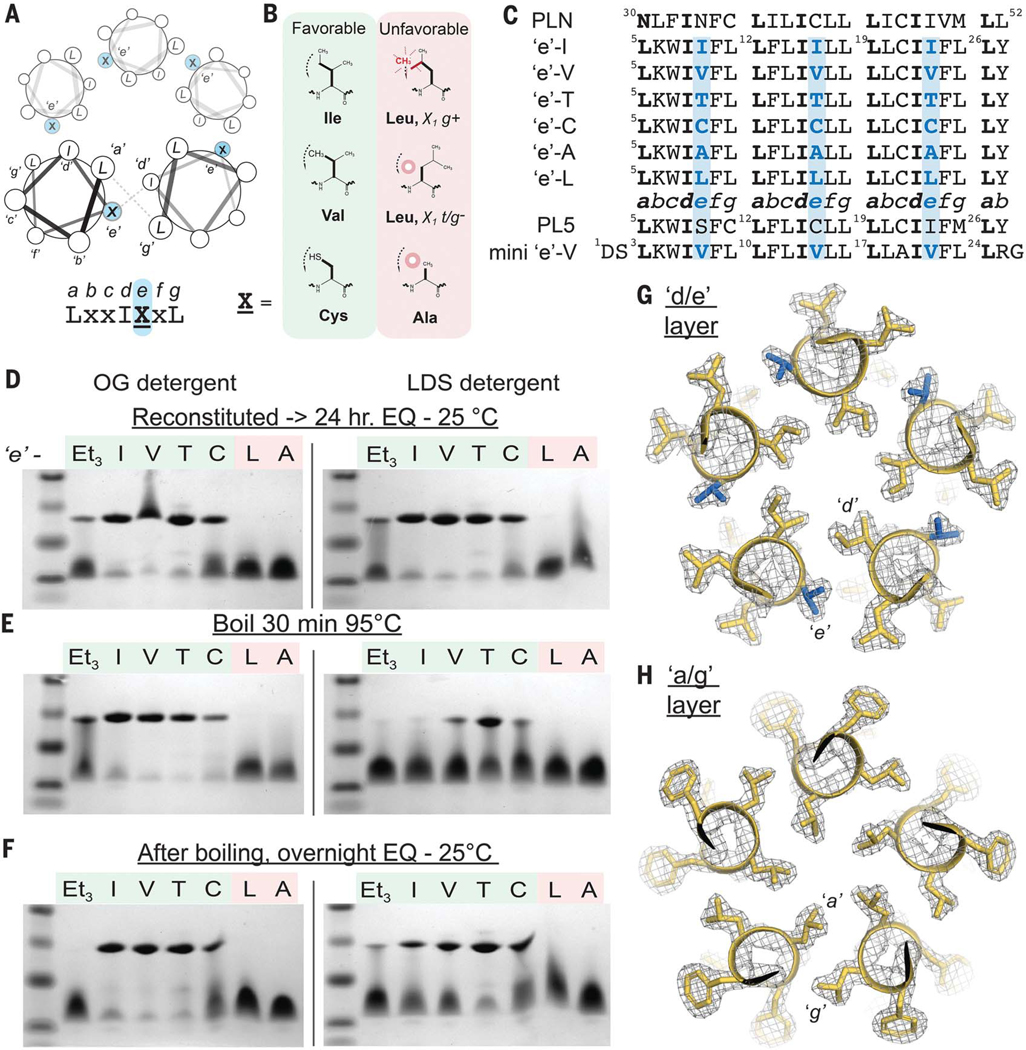
(A) Helical wheel diagram of a pentameric bundle, the identity of the e position at the intersubunit interface. (B) Given the proposed steric code in Fig. 4E, Ile, Val, and Cys should facilitate favorable packing at position e, whereas Leu and Ala are expected to be unfavorable. (C) Aligned sequences of e-series peptides, PLN, and PL5. Leu is fixed at each g, whereas e is systematically varied. (D to F) SDS-PAGE of peptides under different detergent, temperature, and incubation conditions; the PL5EtG3 peptide is abbreviated as Et3. After incubating in the indicated micelle and temperature in the presence of 4 mM of reducing agent tris(2-carboxyethyl)phosphine (TCEP), 2 μg was loaded. After heating, fresh TCEP was added before electrophoresis to 4 mM final. EQ, equilibrium. (G and H) The pentameric x-ray crystal structure of the mini e-Val peptide (1.9-Å resolution, gold). The 2Fo-Fc electron density map (σ = 1.5) highlights layers of e/g (G) and a/g (H) packing layers in the central LxxIVxL repeat; Val at e, blue.
SDS-PAGE showed that the designed peptides e-Ile, e-Val, e-Thr, and e-Cys formed pentamers, whereas the negative controls (e-Ala and e-Leu) were entirely monomeric in both mild and harsh detergents, octyl glucoside (OG) and LDS, respectively (Fig. 5D). Thus, small modifications in the amino acid structure magnify to large changes in stability. Only very low intensity bands were observed between the monomer and pentamer, showing that the assembly is highly cooperative and specific.
The thermal stability of the assemblies was assessed after 95°C incubation and again after cooling to 25°C (24 hours). In OG, the pentamers are essentially unperturbed by heating. In LDS micelles, heating partially dissociates the pentamers, with e-Thr being more stable than both e-Val and e-Cys. After cooling, the original distribution was largely recovered, demonstrating that assembly is enthalpically favorable and slowly reversible. Densitometric tracing (tables S4 and S5) allowed extraction of apparent equilibrium constants and associated energetics. The apparent free energy differences between e-Ile and e-Val relative to e-Thr are 3.4 and 2.8 kcal per mol of pentamer in LDS (25°C), respectively. Furthermore, the apparent free energy difference between e-Ile and e-Leu isomers is at least 9.8 kcal per mol of pentamer (OG, 25°C), given that e-Leu formed less than 0.5% pentamer under conditions where e-Ile was predominantly pentameric.
To confirm the structural basis for association, we determined x-ray structures of a slightly shortened variant of e-Val in three crystal forms between 1.90- and 2.50-Å resolution (fig. S8 and tables S2 and S6). The pentameric structure, detailed side-chain packing, and rotamer distribution of the core residues were exactly as designed (0.63-Å backbone RMSD) (Fig. 5, G and H). Each residue had clear density, and all interhelical contacts were between apolar side chains. Thus, accurate design of stable TM domain architectures mediated entirely by apolar side chains is indeed feasible.
Packing motifs in designed assemblies are observed across the membrane proteome
The constitutive helix-helix packing interactions in coiled coils such as PL5 and e-Val are often structurally analogous to those found at diverse tertiary contexts in numerous globular protein folds. We therefore asked whether the intersubunit helix-helix geometries seen in e-Val and PL5 occur frequently within the structures of natural MPs. Successive pairs of nine-residue stretches (18 total) (Fig. 6A) from e-Val and PL5 were compared with the 3D structures of helical pairs from a nonredundant database of experimental MP structures at the main-chain level (table S7) (47). For each stretch of e-Val, a large number of matches (RMSD < 0.85 Å) were found (50 to 91 matches; mean = 79), demonstrating that structurally similar helix-helix geometries occur frequently in many MP architectures (Fig. 6B). This geometry encompasses the most common parallel left-handed interhelical packing motifs used by MPs (3, 48). Variations of this geometry were much less frequent (fig. S9).
Fig. 6. Helix-helix packing motifs stabilizing the designed TM bundles are common across the membrane proteome.
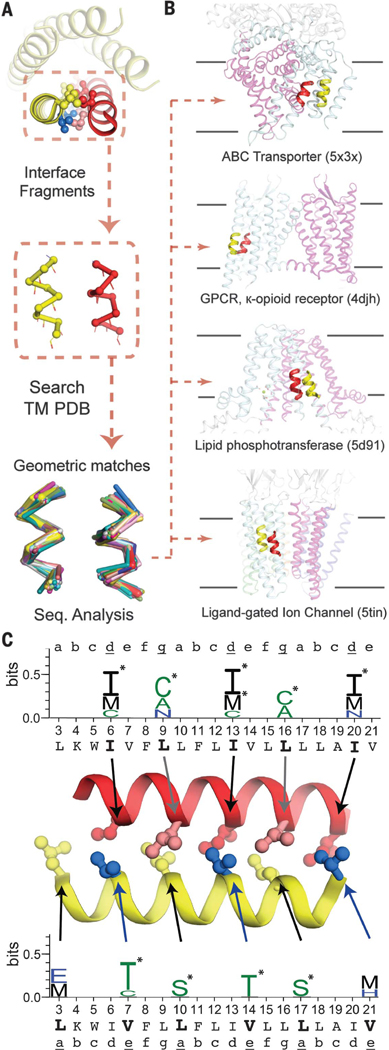
(A) Adjacent helices that make up the repeated helix-helix interface in mini e-Val were decomposed into successive 18-residue fragments. Backbone atoms were used to search for close structural matches (<0.85 Å RMSD) within a nonredundant database of MP experimental structures. (B) The helix-helix geometries in mini e-Val are found frequently in nature, within MPs of diverse architectures and functions. GPCR, G protein–coupled receptor. (C) Amino acids enriched in the fragments (P < 0.05) at equivalent position in e-Val are plotted in WebLogo format (55), with asterisks denoting amino acids enriched at >3 standard deviations (P < 0.003). Steric bulk of enriched amino acids at d, e, and g are similar to those in e-Val, and all are consistent with our proposed steric code. A total of nine positions have at least one amino acid enriched with P < 0.003. The binomial probability associated with finding even one position with this level of enrichment is 0.03, whereas the probability for finding nine positions thusly enriched is 1.1 × 10−21.
We calculated the degree of enrichment of specific amino acid types in the aligned sequences compared with the background amino acid distribution in the MP database (Fig. 6C and table S9), including the probability of finding each enrichment to denote statistical significance (p-value, P). We found that the pattern of amino acids significantly enriched were also in agreement with the packing analysis (Fig. 4). A perfect correspondence was not expected, because the sequence and structural contexts vary in different natural proteins. Nevertheless, β-branched amino acids were highly enriched at the d/e layer, with a clear preference for Ile at d, precisely matching Ile6, Ile13, and Ile20 in the LxxIxxx motif. Furthermore, the observed preference for Thr at e—rather than Val—matches the thermal stability seen for e-Thr and is likely explained by the additional stabilization of its gauche+ rotamer by the intrahelical main-chain hydrogen bonding of Thr (49). In the final heptad, where the bundle radius has widened (Val21), the larger Met is enriched instead. Similar results were obtained probing PL5 (fig. S10).
Using e-Val as the search template, the amino acid enrichments observed at the a/g interface are also consistent with the steric pattern (Fig. 4). No β-branched amino acids were found enriched at the a/g interface, although they are also not strictly forbidden (table S9). Cys was strongly enriched at g (P = 0.0001) for e-Val, matching evolutionary conservation seen in PLN (Cys36, g). For g at the equivalent PL5 helix-helix interface (fig. S10), Cys was highly enriched at Cys11, Cys at Leu18, and Leu at Met25 (P = 0.0001, 0.013, and 0.004, respectively). The enriched amino acids also progressively increase in size as the bundle widens (fig. S10). At a, the leucine side chains project toward the bundle center, where their terminal atoms appear to be important for stabilizing the pentamer’s core, rather than only maximizing pairwise interhelical interactions. In our analysis, a number of non-β-branched residues are accommodated at a, as expected, including the enriched Ser. Although slightly polar residues are commonly enriched in this and other positions, our data suggest that the steric properties of these residues, rather than their interhelical hydrogen bonding capability, are responsible. Moreover, in those natural examples containing Ser, Thr, or Cys, interhelical hydrogen bonding was rare (7.3% of cases).
Position-dependent propensities within the database of natural MPs are in accord with our steric model (Fig. 4). Using this approach, we separate the trends general to the constitutive pairwise helix-helix packing motif—recurring in diverse contexts of natural MPs—from features distinct to pentameric bundles and the LxxIxxx repeat, such as core packing.
Finally, we asked how strict packing restraints are in MPs versus water-soluble proteins by instead searching a nonredundant database of water-soluble proteins. Interestingly, bulky hydrophobic Leu and Ile residues are enriched at essentially all interfacial positions (fig. S11). The distinct amino acid distributions seen in the two classes of protein likely reflect differences in the stabilizing forces at play. A strong hydrophobic driving force dominates folding in water, so natural proteins need not achieve stringent packing to fold, although further optimization of packing is advantageous and can produce stabilities not seen in natural proteins (50, 51). Without a hydrophobic force in bilayers, it appears that geometric complementarity must be more strictly optimized to achieve folding in MPs.
Outlook
This work places on firm experimental ground what had been long hypothesized: apolar side-chain packing can provide a sufficient driving force to determine an MP’s fold. The design of highly stable TM assemblies can be encoded solely by steric side-chain complementarity. Furthermore, the interhelical packing motifs of these pentamers are frequently found in diverse MP families, including transporters, ion channels, enzymes, and G protein–coupled receptors. Previous studies suggest that only a small number of interhelical motifs, such as this one, constitute most tertiary building blocks used within MPs of distinct architectures (3, 52). The helix-helix interfaces of these commonly occurring motifs are also rich in well-packed apolar side chains (3). Thus, analogous steric principles likely encode these different motif geometries and are used to direct a wide diversity of folds and stoichiome-tries throughout nature.
The observed sequence-specific stringency indicates that cooperativity of the distributed vdW interactions across a large interface is crucial, particularly in the absence of alternative stabilizing interactions, such as hydrogen bonds. Local defects propagate over a large area, disrupting the precise geometric complementarity required for TM helix association. Thus, small changes in protein-protein interactions can impart strict specificity. Notably, others have also recently observed single Leu-to-Ile substitutions abolishing select TM helix-helix interactions (53). Furthermore, in the very common GxxxG motif, steric properties of apolar side chains distant from the motif sequence strongly modulate stability and specificity, which is consistent with our findings (7, 11).
Our study, together with other recent work (26, 30, 54), demonstrates that the principles for MP design have progressed substantially, and the engineering of more-complex structures and functions within lipid bilayers is on the horizon.
Supplementary Material
ACKNOWLEDGMENTS
We thank H. T. Kratochvil, J. M. Nicoludis, N. Joh, M. Grabe, R. Jung, and T. Kortemme for careful reading of the manuscript.
Funding: We acknowledge support from NIH (NIGMS R35–122603 and R01GM117593) as well as the NSF (CHE-1413295). M.M. is supported by the Howard Hughes Medical Institute Gilliam Fellowship. This research used resources of the Advanced Light Source, a DOE Office of Science User Facility, under contract DE-AC02–05CH11231. Beamline 8.3.1 at the Advanced Light Source is operated by the University of California Office of the President, Multicampus Research Programs and Initiatives grant MR-15–328599 and NIGMS grants P30 GM124169 and R01 GM124149.
Footnotes
Competing interests: The authors have identified no conflicts of interest.
Data and materials availability: X-ray structures have been submitted to the Protein Data Bank under IDs 6MQU, 6MCT, 6MPW, and 6M2Q.
REFERENCES AND NOTES
- 1.Oberai A, Joh NH, Pettit FK, Bowie JU, Proc. Natl. Acad. Sci. U.S.A 106, 17747–17750 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eilers M, Shekar SC, Shieh T, Smith SO, Fleming PJ, Proc. Natl. Acad. Sci. U.S.A 97, 5796–5801 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang SQ et al. , Structure 23, 527–541 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adamian L, Liang J, J. Mol. Biol 311, 891–907 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Langosch D, Heringa J, Proteins 31, 150–159 (1998). [DOI] [PubMed] [Google Scholar]
- 6.Hong H, Arch. Biochem. Biol 564, 297–313 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Anderson SM, Mueller BK, Lange EJ, Senes A, J. Am. Chem. Soc 139, 15774–15783 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Joh NH, Oberai A, Yang D, Whitelegge JP, Bowie JU, J. Am. Chem. Soc 131, 10846–10847 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yano Y, Kondo K, Kitani R, Yamamoto A, Matsuzaki K, Biochemistry 54, 1371–1379 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Baker RP, Urban S, Nat. Chem. Biol 8, 759–768 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doura AK, Kobus FJ, Dubrovsky L, Hibbard E, Fleming KG, J. Mol. Biol 341, 991–998 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Guo R. et al. , Nat. Chem. Biol 12, 353–360 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joh NH et al. , Nature 453, 1266–1270 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choma C, Gratkowski H, Lear JD, DeGrado WF, Nat. Struct. Biol 7, 161–166 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Teese MG, Langosch D, Biochemistry 54, 5125–5135 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Senes A, Ubarretxena-Belandia I, Engelman DM, Proc. Natl. Acad. Sci. U.S.A 98, 9056–9061 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou FX, Merianos HJ, Brunger AT, Engelman DM, Proc. Natl. Acad. Sci. U.S.A 98, 2250–2255 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cymer F, von Heijne G, White SH, J. Mol. Biol 427, 999–1022 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson RM, Hecht K, Deber CM, Biochemistry 46, 9208–9214 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Langosch D, Arkin IT, Protein Sci. 18, 1343–1358 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gurezka R, Laage R, Brosig B, Langosch D, J. Biol. Chem 274, 9265–9270 (1999). [DOI] [PubMed] [Google Scholar]
- 22.Yano Y. et al. , Biochemistry 41, 3073–3080 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Johnson RM, Heslop CL, Deber CM, Biochemistry 43, 14361–14369 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Whitley P, Nilsson I, von Heijne G, Nat. Struct. Biol 1, 858–862 (1994). [DOI] [PubMed] [Google Scholar]
- 25.Tatko CD, Nanda V, Lear JD, DeGrado WF, J. Am. Chem. Soc 128, 4170–4171 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Joh NH et al. , Science 346, 1520–1524 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Korendovych IV et al. , J. Am. Chem. Soc 132, 15516–15518 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yin H. et al. , Science 315, 1817–1822 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Mravic M. et al. , Protein Eng. Des. Sel 31, 181–190 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu P. et al. , Science 359, 1042–1046 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lomize AL, Lomize MA, Krolicki SR, Pogozheva ID, Nucleic Acids Res. 45, D250–D255 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kirrbach J. et al. , Bioinformatics 29, 1623–1630 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Partridge AW, Therien AG, Deber CM, Proteins 54, 648–656 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Schlebach JP, Sanders CR, Q. Rev. Biol 48, 1–34 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thomson AR et al. , Science 346, 485–488 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Harbury PB, Zhang T, Kim PS, Alber T, Science 262, 1401–1407 (1993). [DOI] [PubMed] [Google Scholar]
- 37.Crick FH, Acta Crystallogr. 6, 689–697 (1953). [Google Scholar]
- 38.Arkin IT et al. , EMBO J. 13, 4757–4764 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Verardi R, Shi L, Traaseth NJ, Walsh N, Veglia G, Proc. Natl. Acad. Sci. U.S.A 108, 9101–9106 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oxenoid K, Chou JJ, Proc. Natl. Acad. Sci. U.S.A 102, 10870–10875 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim T, Lee J, Im W, Proteins 76, 86–98 (2009). [DOI] [PubMed] [Google Scholar]
- 42.Fujii J, Maruyama K, Tada M, MacLennan DH, J. Biol. Chem 264, 12950–12955 (1989). [PubMed] [Google Scholar]
- 43.Simmerman HK, Kobayashi YM, Autry JM, Jones LR, J. Biol. Chem 271, 5941–5946 (1996). [DOI] [PubMed] [Google Scholar]
- 44.Bowie JU, Curr. Opin. Struct. Biol 21, 42–49 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karim CB et al. , J. Biol. Chem 276, 38814–38819 (2001). [DOI] [PubMed] [Google Scholar]
- 46.Arkin IT et al. , J. Membr. Biol 155, 199–206 (1997). [DOI] [PubMed] [Google Scholar]
- 47.Zhou J, Grigoryan G, Protein Sci. 24, 508–524 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walters RF, DeGrado WF, Proc. Natl. Acad. Sci. U.S.A 103, 13658–13663 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.MacKenzie KR, Prestegard JH, Engelman DM, Science 276, 131–133 (1997). [DOI] [PubMed] [Google Scholar]
- 50.Huang PS et al. , Science 346, 481–485 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moll JR, Ruvinov SB, Pastan I, Vinson C, Protein Sci. 10, 649–655 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Feng X, Barth P, Nat. Chem. Biol 12, 167–173 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.He L. et al. , eLife 6, e27701 (2017). [Google Scholar]
- 54.Young M. et al. , Proc. Natl. Acad. Sci. U.S.A 115, 7051–7056 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Crooks GE, Hon G, Chandonia JM, Brenner SE, Genome Res. 14, 1188–1190 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.