

Summary
Metabolites, phytohormones, and genes involved in dehydration responses/tolerance have been predicted in several plants. However, metabolite/phytohormone–gene regulatory networks in soybean organs under dehydration conditions remain unclear. Here, we analyzed the organ specificity of metabolites, phytohormones, and gene transcripts and revealed the characteristics of their regulatory networks in dehydration‐treated soybeans. Our metabolite/phytohormone analysis revealed the accumulation of raffinose, trehalose, and cis‐zeatin (cZ) specifically in dehydration‐treated roots. In dehydration‐treated soybeans, raffinose, and trehalose might have additional roles not directly involved in protecting the photosynthetic apparatus; cZ might contribute to root elongation for water uptake from the moisture region in soil. Our integration analysis of metabolites–genes indicated that galactinol, raffinose, and trehalose levels were correlated with transcript levels for key enzymes (galactinol synthase, raffinose synthase, trehalose 6‐phosphate synthase, trehalose 6‐phosphate phosphatase) at the level of individual plants but not at the organ level under dehydration. Genes encoding these key enzymes were expressed in mainly the aerial parts of dehydration‐treated soybeans. These results suggested that raffinose and trehalose are transported from aerial plant parts to the roots in dehydration‐treated soybeans. Our integration analysis of phytohormones–genes indicated that cZ and abscisic acid (ABA) levels were correlated with transcript levels for key enzymes (cytokinin nucleoside 5′‐monophosphate phosphoribohydrolase, cytokinin oxidases/dehydrogenases, 9‐cis‐epoxycarotenoid dioxygenase) at the level of individual plants but not at the organ level under dehydration conditions. Therefore, processes such as ABA and cZ transport, among others, are important for the organ specificity of ABA and cZ production under dehydration conditions.
Keywords: soybean, dehydration, metabolites, phytohormones, gene transcripts
Significance Statement
We analyzed the characteristics of metabolite/phytohormone–gene regulatory networks in dehydration‐treated soybean plants. Our analysis revealed the accumulation of raffinose, trehalose, and cZ specifically in dehydration‐treated roots, and our integration analysis of metabolite/phytohormone–genes indicated that several metabolite/phytohormone levels, including those of raffinose, trehalose, and cZ, were correlated with transcript levels for these key enzymes at the level of individual plants but not at the organ level under dehydration conditions.
INTRODUCTION
According to recent reports and simulation studies, climate change is expected to adversely affect the yields of all major crops worldwide (Varshney et al., 2011; Wheeler and von Braun, 2013; Rosenzweig et al., 2014; Zhao et al., 2017). Under dehydration conditions, several crops showed reduced early seedling growth, root and shoot dry weight, and vegetative growth. These adverse effects led to a decrease in crop yields (Fahad et al., 2017). As soybean is cultivated by using mainly rainfed agricultural practices in the major soybean‐producing countries (e.g. USA, Brazil, and Argentina), yields might be reduced by even short periods of soil water deficit (Sinclair, 2017). Hence, genes that are isolated based on analyses of the metabolite/phytohormone–gene regulatory networks underlying the dehydration response in soybeans are important for molecular breeding under dehydration conditions.
An essential step in elucidating these metabolite/phytohormone–gene regulatory networks is to identify dehydration‐responsive metabolites, phytohormones, and gene transcripts. To date, such studies have been conducted for the most part in Arabidopsis thaliana. Studies of metabolomes and phytohormones based on mass spectrometry (MS) have led to the characterization of dehydration‐responsive metabolites and phytohormones. The profiles of several types of dehydration‐responsive metabolites have been reported. For instance, the levels of oligosaccharides, proline, branched‐chain amino acids (BCAAs), γ‐aminobutyrate, saccharopine, and agmatine are significantly higher in dehydration‐treated plants than in untreated plants (Urano et al., 2009; Skirycz et al., 2010; Verslues and Juenger, 2011; Maruyama et al., 2014a; Todaka et al., 2017). Analyses of phytohormone profiles have revealed that abscisic acid (ABA) is the main dehydration‐responsive phytohormone (Müller et al., 2002; Chiwocha et al., 2003; Yamaguchi‐Shinozaki and Shinozaki, 2006; Hirayama and Shinozaki, 2007; Cutler et al., 2010; Raghavendra et al., 2010; Weiner et al., 2010; Maruyama et al., 2014a; Todaka et al., 2017; Urano et al., 2017).
Transcriptome studies involving microarray analysis and next‐generation sequencing have led to the characterization of dehydration‐responsive genes in several plant species, including genes encoding metabolic enzymes, late embryogenesis‐abundant (LEA) proteins, detoxification enzymes, chaperones, protein kinases, and transcription factors (Kilian et al., 2007; Shinozaki and Yamaguchi‐Shinozaki, 2007; Cutler et al., 2010; Maruyama et al., 2012, 2014a; Todaka et al., 2017). The transcript levels of many genes in dehydration‐treated plants are correlated with those in ABA‐treated plants (Seki et al., 2002; Rabbani et al., 2003). Mutant analysis has revealed that ABA plays an essential role in the response to dehydration and that an increase in endogenous ABA is required for activating ABA‐dependent signalling networks (Iuchi et al., 2001; Urano et al., 2009, 2017).
ABA is recognized by pyrabactin resistance1/PYR1‐like/regulatory components of ABA receptor (PYR/PYL/RCAR) proteins (Ma et al., 2009; Nishimura et al., 2009; Park et al., 2009; Santiago et al., 2009). In plants with low levels of ABA, PYR/PYL/RCAR proteins and group‐A protein phosphatase 2C (PP2C) complexes inhibit the kinase activity of subclass III sucrose non‐fermenting 1‐related protein kinase 2 (SnRK2). By contrast, in plants with high levels of ABA, ABA–PYR/PYL/RCAR–PP2C complexes form, the inhibition of SnRK2 activity is relieved, and ABA signal transduction is activated. Transcription factors, such as abscisic acid‐responsive element (ABRE)‐binding protein/ABRE‐binding factors (AREB/ABFs), are phosphorylated by SnRK2, and genes downstream of these transcription factors are upregulated (Cutler et al., 2010; Raghavendra et al., 2010). ABREs are highly conserved in the promoters of dehydration‐ and ABA‐inducible genes and of genes that function downstream of the AREB family, suggesting that ABRE/AREB family‐dependent gene expression plays a major role in dehydration‐inducible gene expression (Maruyama et al., 2012). Cytokinins (CKs), which are involved in various processes of plant growth and development, have also been reported to respond to abiotic stresses such as dehydration and high salinity (Kieber and Schaller, 2014). Among CKs, the levels of trans‐zeatin (tZ) and N 6‐(∆2‐isopentenyl) adenine (iP) decrease in xylem sap under dehydration (Alvarez et al., 2008). CKs are generally thought to have antagonistic actions relative to ABA and function in opposition to ABA in stomatal movement, seed germination, etc. (Hare et al., 1997; Peleg and Blumwald, 2011). The functions of tZ and iP under abiotic stress conditions have been studied in detail, but the role of cis‐zeatin (cZ) is not well understood (Schäfer et al., 2015). We previously described an increase in ABA signalling and a decrease in CK signalling in the aerial parts of rice under cold and dehydration conditions (Maruyama et al., 2014). However, the role of cZ in rice under cold and dehydration conditions was unclear based on our integration analysis of phytohormones and gene transcripts.
Studies of the changes in metabolites, phytohormones, and gene transcripts in various species of plants subjected to dehydration stress have revealed many dehydration‐responsive molecules, but studies of their organ‐specific accumulation have been limited. In the current analysis, we surveyed the molecular responses of specific soybean organs (leaves, stems, and roots) to dehydration during vegetative growth. Using two types of MS, liquid chromatography coupled with MS (LC‐MS) and gas chromatography coupled with time‐of‐flight MS (GC‐TOF‐MS), we identified and characterized representative dehydration‐responsive metabolites and phytohormones. We also analyzed gene transcripts differentially expressed under dehydration conditions via oligo microarray analysis and cap analysis of gene expression (CAGE). Oligo microarray analysis is suitable for comprehensive gene expression analysis of a species whose genome has been sequenced. CAGE uses cap trap technology based on biotinylation of the 7‐methylguanosine cap of mRNA transcribed by RNA polymerase II and large‐scale parallel sequencing of the 5 ends of cDNAs. Therefore, CAGE can be used not only for high‐throughput gene expression analysis but also for identifying transcription start sites (TSSs) (Carninci et al., 2005; Murata et al., 2014). Finally, we analyzed the regulatory networks of metabolites–genes and phytohormones–genes and predicted several genes encoding metabolic enzymes that might play key roles in the dehydration response in soybeans.
RESULTS
Responses of soybeans to dehydration stress
To investigate the metabolites, phytohormones, and gene transcripts in various soybean organs in response to dehydration, we used our cultivation system to grow soybean plants to the V3 growth stage (the full opening of the 3rd trifoliate leaf) (Figure 1a) and then subjected them to dehydration stress by withholding water for 4 days (Figure 1b–k). Our cultivation system utilizing capillary phenomena can maintain a soil moisture content of ~20% under normal growth conditions (Figure 1l). The soil moisture content slowly decreased during the 4‐day dehydration treatment (Figure 1m). We measured the photosynthetic rate, transpiration rate, and H2O conductance of soybean plants after exposure to dehydration (Figure 1n–p) and found that these values were close to zero after 3 days of dehydration. These results indicated that the plants closed their stomata in response to dehydration. We collected five different organs (1st [L1], 2nd [L2], and 3rd [L3] trifoliate leaves; stems [S]; and roots [R]) from soybean plants exposed to dehydration for 3 or 4 days (D3 or D4) or watered normally (untreated for 3 [U3] or 4 days [U4]) and analyzed their metabolite, phytohormone, and gene transcript levels.
Figure 1.
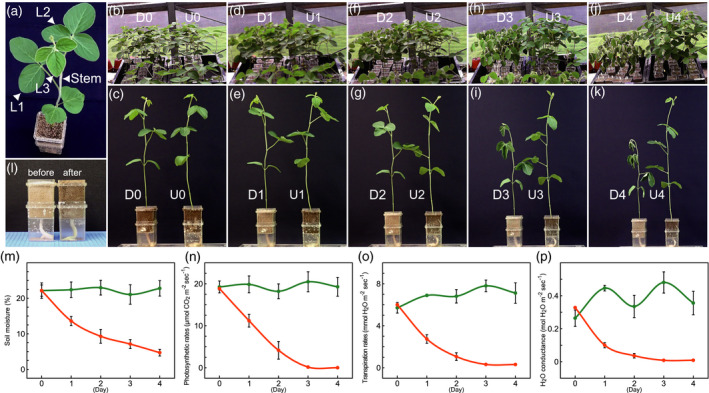
Dehydration treatment of soybean seedlings using our cultivation system. Soybean plant at the V3 growth stage (a). L1, L2, and L3 indicate 1st, 2nd, and 3rd trifoliate leaves, respectively. Soybean seedlings before (b, c) and 1 day (d, e), 2 days (f, g), 3 days (h, i), and 4 days (j, k) after exposure to dehydration. D and U indicate dehydration and untreated, respectively. Soil in pots before and after water supply (l). Graphs show soil moisture content (m) in pots and photosynthetic rate (n), transpiration rate (o), and H2O conductance (p) in soybean plants after exposure to dehydration. Orange and green indicate dehydration and untreated, respectively. Error bars indicate standard deviation (SD) from 36 measurements (m–p).
Metabolite profiles of soybean organs exposed to dehydration
We used GC‐TOF‐MS to measure dehydration‐responsive metabolites in the five types of soybean organs (L1, L2, L3, S, and R) in response to the four different treatments (U3, U4, D3, and D4) and identified 197 independent metabolites according to their retention‐time indices and specific mass fragments. Compared with those in the corresponding 3‐day and 4‐day untreated plants, the levels of 59 (L1‐D3), 113 (L1‐D4), 55 (L2‐D3), 111 (L2‐D4), 78 (L3‐D3), 121 (L3‐D4), 124 (S‐D3), 135 (S‐D4), 123 (R‐D3), and 110 (R‐D4) metabolites were significantly higher in dehydration‐treated plants (Benjamini and Hochberg false discovery rate [FDR]: P < 0.05; fold change [FC] >2), whereas the levels of 12 (L1‐D3), 7 (L1‐D4), 14 (L2‐D3), 25 (L2‐D4), 18 (L3‐D3), 19 (L3‐D4), 5 (S‐D3), 3 (S‐D4), 10 (R‐D3), and 7 (R‐D4) metabolites were significantly lower (FDR, P < 0.05; FC < 0.5; Table S1).
We compared the metabolite profiles via principal component analysis (PCA) (Figure 2a). The cumulative contribution ratio of the PCA was 72% up to the second principal component (PC2). The PCA had two key features. The first principal component (PC1) reflected increases in the levels of metabolites after exposure to dehydration. The PC1 values for dehydration‐treated organs were higher than those for untreated organs. The PC2 values reflected differences in the variety of metabolites in organs among dehydration‐treated organs. The PC2 values were highest for dehydration‐treated roots, whereas dehydration‐treated leaves had the lowest PC2 values. These results indicated that the organ specificity of metabolite accumulation was higher in dehydration‐treated organs than in untreated organs. We displayed PCA loadings using scatter plots (Figure 2b). PCA loadings correspond to the coefficients of each principal component. In the metabolite profiles, the highest PC1 loading was for pinitol. Pinitol levels were significantly higher in dehydration‐treated organs than in untreated organs (Figure 2c) and were higher in leaves than in stems and roots after exposure to dehydration. The 3rd leaves of plants subjected to the 4‐day dehydration treatment had the highest pinitol levels. Asparagine had the highest PC2 loading (Figure 2b). Asparagine levels were higher in stems and roots than in leaves after exposure to dehydration. The highest asparagine levels were detected in the roots of plants subjected to the 4‐day dehydration treatment (Figure 2c).
Figure 2.
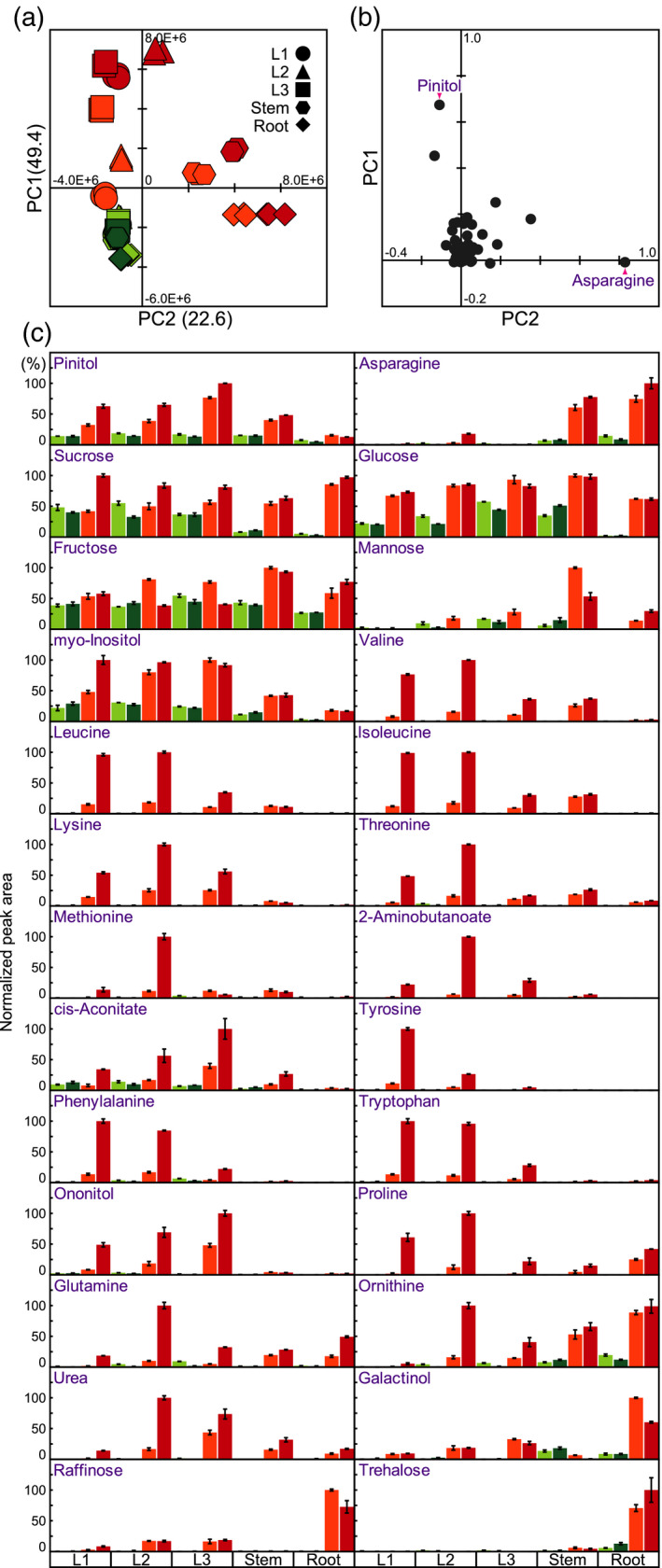
Statistical analysis of the metabolite profiles of soybean organs (1st [L1], 2nd [L2], and 3rd [L3] trifoliate leaves; stems [S]; and roots [R]) after exposure to dehydration. Metabolite levels were measured in various soybean organs subjected to four treatments: dehydration (water withheld) for 3 days (D3) or 4 days (D4) and untreated for 3 or 4 days (U3 and U4, respectively). (a) Principal component analysis (PCA) of metabolite levels. Values on the y and x axes indicate PC1 and PC2, respectively. (b) PCA loadings for metabolites. (c) Representative metabolites under water‐deficit stress conditions. In each case, the maximum level of metabolite was set to 100. Values are means (n = 3 experiments); error bars show SDs.
We previously reported dehydration‐inducible sugars, amino acids, and oligosaccharides in Arabidopsis and rice (Maruyama et al., 2009; Urano et al., 2009; Maruyama et al., 2014a). In the current study, metabolite analyses of soybean organs revealed that the levels of several organic acids, sugars, amino acids, oligosaccharides, and polyols were also significantly higher in dehydration‐treated organs than in untreated organs, and pinitol was the most highly dehydration‐inducible metabolite in various soybean organs (Figure 2b,c). Dehydration‐inducible changes in pinitol levels have also been observed in Mesembryanthemum crystallinum (ice plant) (Paul and Cockburn, 1989), Porteresia coarctata (halophytic wild rice) (Sengupta et al., 2008), and Medicago sativa (alfalfa) (Aranjuelo et al., 2011).
In addition, our metabolite analyses revealed the organ specificity of several types of dehydration‐inducible metabolites (Figure 2c). Both sucrose and glucose levels were higher in leaves and stems than in roots under untreated conditions. The levels of both metabolites were significantly higher in dehydration‐treated organs than in untreated organs, and they dramatically increased in roots after exposure to dehydration. Both metabolites had a high fold change in roots after dehydration treatment (Figure 2c). The highest levels of both fructose and mannose were detected in stems subjected to the 3‐day dehydration treatment. In the 2nd and 3rd leaves and in the stems, both fructose and mannose levels increased after dehydration treatment, peaking on the third day of dehydration treatment, and then decreased on the fourth day (Figure 2c). The levels of myoinositol, BCAAs (valine, leucine, and isoleucine), lysine, threonine, methionine, 2‐aminobutanoate, and cis‐aconitate were higher in leaves and stems than in roots under dehydration conditions (Figure 2c). Greater increases in tyrosine, phenylalanine, tryptophan, and ononitol levels were detected in leaves than in stems and roots after exposure to dehydration (Figure 2c). The highest levels of proline, glutamine, ornithine, and urea were found in 2nd leaves subjected to the 4‐day dehydration treatment. The accumulation of these metabolites also increased in stems and roots after dehydration (Figure 2c). Galactinol, raffinose, and trehalose levels were higher in roots than in leaves and stems under dehydration conditions (Figure 2c).
Phytohormone profiles of soybean organs exposed to dehydration
We identified 13 phytohormones in dehydration‐treated or untreated soybean organs using LC‐MS (Table S2) and performed PCA to compare dehydration‐responsive phytohormone levels (Figure 3a). The cumulative contribution ratio of the PCA was 77.5% up to PC2. Two key features emerged from the PCA. PC1 reflected increases in the levels of phytohormones after exposure to dehydration. The highest PC1 value was for the 3rd leaves subjected to the 4‐day dehydration treatment. The PC2 value reflected differences in the variety of phytohormones in different organs. The PC2 values were positive for stems, negative for roots, and near zero for leaves. These results indicate that some phytohormones exhibit high organ specificity in soybeans under both untreated and dehydration conditions.
Figure 3.
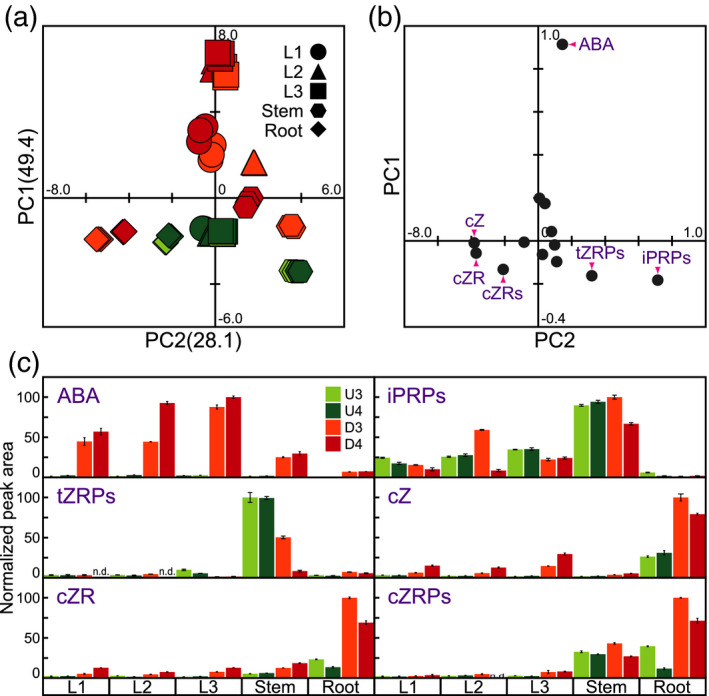
Statistical analysis of the phytohormone profiles of soybean organs after exposure to dehydration. Phytohormone levels were measured in various soybean organs subjected to four treatments: U3, U4, D3, and D4. (a) Principal component analysis (PCA) of phytohormones. Values on the y and x axes indicate PC1 and PC2, respectively. (b) PCA loadings for phytohormones. (c) Relative levels of representative phytohormones. n.d. indicates that the phytohormone could not be detected. In each case, the maximum phytohormone level was set to 100. Values are the mean ± SD (n = 3 experiments). ABA, abscisic acid; iPRPs, N 6‐(Δ2‐isopentenyl) adenine ribosides; tZRPs, trans‐zeatin riboside‐5′‐phosphates; cZ, cis‐zeatin; cZR, cis‐zeatin riboside; cZRPs, cis‐zeatin 5′‐phosphates.
We focused on representative phytohormones by analyzing PCA loadings (Figure 3b). The highest PC1 loading was for ABA. ABA levels were significantly higher in dehydration‐treated organs than in untreated organs and were higher in leaves than in stems and roots after exposure to dehydration. The highest ABA levels were found in 3rd leaves subjected to the 4‐day dehydration treatment (Figure 3c). The first‐ and second‐highest PC2 loadings were for iP riboside phosphates (iPRPs) and tZ riboside phosphates (tZRPs), respectively; iPRPs and tZRPs are precursors of iP and tZ, respectively. The levels of both precursors were higher in stems than in leaves and roots (Figure 3c). The lowest PC2 loading was for cZ. cZ levels were higher in roots than in leaves and stems, and cZ levels in untreated roots were similar to those in dehydration‐treated leaves and stems. The highest cZ levels were detected in the roots of plants subjected to the 3‐day dehydration treatment (Figure 3c). Both cZ riboside (cZR) and cZR phosphates (cZRPs) are precursors of cZ; these compounds had the second‐ and third‐lowest PC2 loading, respectively. The highest levels of both precursors were also found in the roots of plants subjected to the 3‐day dehydration treatment (Figure 3c).
Transcript profiles of soybean organs exposed to dehydration
We performed oligo microarray analysis to identify dehydration‐responsive genes in these soybean organs. Overall, 2736 (L1‐D3), 3630 (L1‐D4), 2904 (L2‐D3), 3956 (L2‐D4), 3497 (L3‐D3), 4666 (L3‐D4), 2910 (S‐D3), 3797 (S‐D4), 2589 (R‐D3), and 2881 (R‐D4) genes were significantly upregulated (FDR: P < 0.05; FC > 2), and 3461 (L1‐D3), 3862 (L1‐D4), 3982 (L2‐D3), 4476 (L2‐D4), 4176 (L3‐D3), 5067 (L3‐D4), 4058 (S‐D3), 8238 (S‐D4), 4497 (R‐D3), and 4409 (R‐D4) genes were significantly downregulated (FDR: P < 0.05; FC < 0.5; Table S3) in response to dehydration relative to those in the corresponding untreated plants. In addition, to confirm the dehydration‐responsive genes, we performed CAGE using L3‐U4, L3‐D4, S‐U4, S‐D4, R‐U4, and R‐D4. We detected 27 233 transcripts by CAGE. Correlation analysis showed that the two kinds of transcriptome analyses (oligo microarray and CAGE) were highly correlated. The correlation coefficients were 0.89 (L3‐D4), 0.89 (S‐D4), and 0.77 (R‐D4) (Figure 4a).
Figure 4.
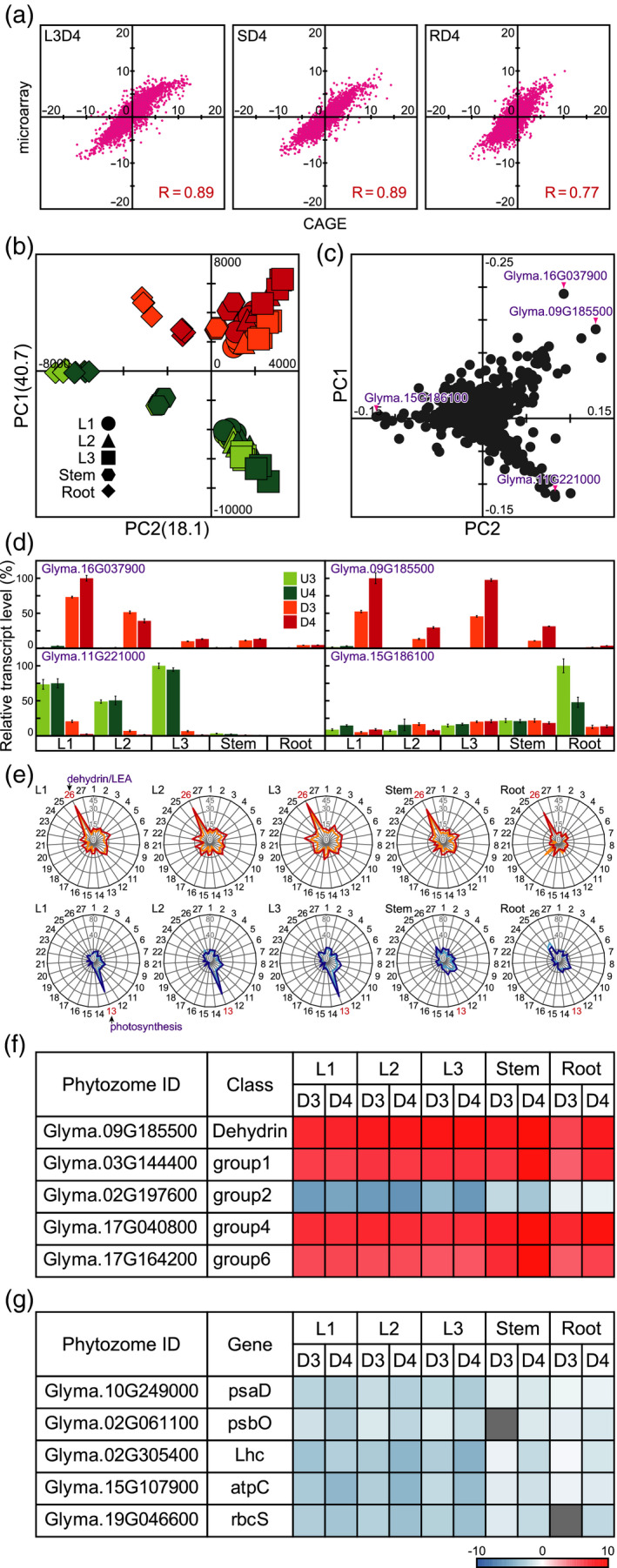
Statistical analysis of the transcript profiles of various soybean organs after exposure to dehydration. (a) Correlation analysis of gene transcript levels. Values on the y and x axes indicate fold change in microarray and CAGE analyses, respectively. (b) Principal component analysis (PCA) of gene transcript levels. Values on the y and x axes indicate PC1 and PC2, respectively. (c) PCA loadings for gene transcripts. (d) Expression levels of representative genes under dehydration conditions. In each case, the maximum gene transcript level was set to 100. Values are the mean ± SD (n = 3 experiments). (e) Molecular functions of dehydration‐responsive genes in various soybean organs. Radar charts of 27 upregulated and downregulated molecular function classes indicating the frequency of each class in dehydration‐treated soybean organs. 1, Carbohydrate metabolism; 2, Energy metabolism; 3, Lipid metabolism; 4, Nucleotide metabolism; 5, Amino acid metabolism; 6, Metabolism of other amino acids; 7, Glycan biosynthesis and metabolism; 8, Metabolism of cofactors and vitamins; 9, Metabolism of terpenoids and polyketides; 10, Biosynthesis of other secondary metabolites; 11, Xenobiotic biodegradation and metabolism; 12, Biosynthesis of plant hormones; 13, Photosynthesis; 14, Signal transduction; 15, Transcription; 16, Translation; 17, Epigenetics; 18, Molecular chaperone; 19, DNA replication and repair; 20, Helicase; 21, Ribonuclease; 22, Ubiquitination; 23, Protease/peptidase; 24, Transport; 25, Detoxification enzyme; 26, dehydrin/LEA proteins; 27, Other. Orange and light blue indicate the 3‐day dehydration treatment. Red and blue indicate the 4‐day dehydration treatment. (F, G) Heatmaps illustrate the log ratios of dehydration‐responsive genes in representative molecular classes of dehydrin/LEA proteins (f) and photosynthesis (g). psaD, photosystem I subunit II; psbO, photosystem II oxygen‐evolving enhancer protein 1; Lhc, chlorophyll a/b‐binding protein; atpC, CF1‐ATPase γ‐subunit; rbcS, ribulose 1,5‐bisphosphate carboxylase/oxygenase small subunit.
We compared the transcript profiles of the samples via PCA (Figure 4b). The cumulative contribution ratio of the PCA was 58.8% up to PC2. Two key features emerged from the PCA. The PC1 value reflected increases in transcript levels after exposure to dehydration. The PC1 values were higher for dehydration‐treated organs than for untreated organs. The PC2 value reflected differences in the variety of transcripts in these organs. The PC2 values for untreated and dehydration‐treated 3rd leaves were similar and were the highest among the organs. Untreated roots had the lowest PC2 values. These results indicated that the organ specificity of transcript accumulation was lower in dehydration‐treated plants than in untreated plants.
We selected representative dehydration‐responsive genes for PCA loading (Figure 4c). The transcript levels of four selected genes were confirmed by reverse‐transcription quantitative PCR (RT‐qPCR) and are displayed as bar charts (Figure 4d). Among the transcript profiles, Glyma.16G037900 and Glyma.09G185500 showed the first‐ and second‐highest PC1 loadings, respectively. The levels of both transcripts were significantly higher in dehydration‐treated organs than in untreated organs and were higher in leaves than in stems and roots under dehydration conditions (Figure 4d). The lowest PC1 loading was for Glyma.11G221000. Glyma.11G221000 transcript levels were significantly lower in dehydration‐treated leaves than in untreated leaves and were higher in leaves than in stems and roots (Figure 4c). The lowest PC2 loading was for Glyma.15G186100. Untreated roots showed the highest Glyma.15G186100 transcript levels (Figure 4d).
We used our in‐house gene ontology (GO) database (Maruyama et al., 2012) to annotate the molecular functions of all identified dehydration‐responsive genes (Figure 4e). The GO profiles of the upregulated genes were similar among the five organs examined, revealing dehydrin/LEA genes as representative genes whose transcript levels increased under dehydration conditions. Transcript levels of dehydrin/LEA genes upregulated under dehydration conditions increased by 35.8% (L1‐D3), 39.5% (L1‐D4), 35.8% (L2‐D3), 37.0% (L2‐D4), 30.9% (L3‐D3), 42.0% (L3‐D4), 30.9% (S‐D3), 35.8% (S‐D4), 24.7% (R‐D3), and 32.1% (R‐D4) relative to those in the corresponding untreated plants (Figure 4e). As dehydrin/LEA genes are classified into seven groups in the Pfam database, we constructed heatmaps to illustrate the log ratios of expression of these dehydration‐responsive genes (Figure 4f; Figure S1). We analyzed 74 dehydrin/LEA genes by oligo microarray analysis. Five genes belong to the dehydrin group, and four genes, 53 genes, six genes, three genes, two genes, and one gene belong to groups 1, 2, 3, 4, 5, and 6 of LEA genes, respectively. All genes in the dehydrin group and in LEA gene groups 1, 4, 5, and 6 were upregulated in all five organs under dehydration conditions. Among group 2 LEA genes, a few were upregulated under dehydration conditions, and several genes (e.g. Glyma.02G197600) were downregulated. We selected genes involved in photosynthesis to represent downregulated genes under dehydration conditions (Figure 4e). The transcript levels of photosynthesis‐related genes downregulated by dehydration decreased by 45.3% (L1‐D3), 56.8% (L1‐D4), 40.5% (L2‐D3), 60.8% (L2‐D4), 48.0% (L3‐D3), 65.5% (L3‐D4), 8.8% (S‐D3), 25.7% (S‐D4), 14.2% (R‐D3), and 21.6% (R‐D4) relative to those in the corresponding untreated plants. Photosynthesis‐related genes include psa and psb, which encode subunits of photosystems I and II, respectively. psaD, psaE, psaF, psaG, psaH, psaK, psbO, psbP, psbR, psbW, and psbY were significantly downregulated in dehydration‐treated leaves. Lhc (chlorophyll a/b‐binding protein), atpC (CF1‐ATPase γ‐subunit), atpD (CF1‐ATPase δ‐subunit), and rbcS (ribulose 1,5‐bisphosphate carboxylase/oxygenase small subunit) were also significantly downregulated in dehydration‐treated leaves (Figures 4g and S2).
Expression of genes involved in carbohydrate and amino acid metabolism in soybeans
Our metabolite and gene transcript analyses revealed the organ specificity of dehydration‐induced metabolites and gene transcripts in soybeans, respectively (Figures 2 and 3). We investigated the expression of genes involved in carbohydrate and amino acid metabolism in dehydration‐treated soybean organs using KEGG, Kappa‐view, and Phytozome. Among the genes involved in sucrose metabolism, transcript levels of the gene encoding alkaline/neutral invertase (INV) (Glyma.01G211000) were upregulated in leaves and stems after dehydration treatment (Figure 5a,b). Glyma.01G211000 transcript levels were >30‐fold higher in dehydration‐treated 1st and 2nd leaves than in untreated 1st and 2nd leaves and were higher on the third day of dehydration treatment than on the fourth day. Branched‐chain amino acid aminotransferase (BCAT) is important not only for BCAAs but also for 2‐aminobutanoate biosynthesis (Figure 5c). The highest transcript levels of the gene encoding BCAT (Glyma.06G050100) were found in 2nd leaves subjected to the 4‐day dehydration treatment (Figure 5a). Among glutamine and methionine biosynthesis, the transcript levels of the genes encoding glutamine synthetase (GS) (Glyma.02G244000) and homocysteine S‐methyltransferase (HMT) (Glyma.20G148900) were upregulated after exposure to dehydration. The 1st leaves and the stems subjected to the 4‐day dehydration treatment had the highest transcript levels of Glyma.02G244000 and Glyma.20G148900, respectively (Figure 5a,c). Among the genes in the glyoxylate cycle, the transcript levels of genes encoding isocitrate lyase (ICL) (Glyma.12G100500) and malate synthase (Glyma.17G128000) were higher in leaves and stems than in roots under dehydration conditions (Figure 5a,d). Of the genes associated with asparagine metabolism, the transcript levels of the gene encoding glutamine‐dependent asparagine synthetase (ASNS) (Glyma.18G061100) were higher in leaves and stems than in roots under dehydration conditions (Figure 5a,d). Among the genes involved in proline metabolism, the transcript levels of genes encoding Δ1‐pyrroline‐5‐carboxylate dehydrogenase (P5CD) (Glyma.05G029200) and Δ1‐pyrroline‐5‐carboxylate synthase (P5CS) (Glyma.01G099800) were significantly higher in all dehydration‐treated organs than in untreated organs (Figure 5a,e). The highest transcript levels of both Glyma.05G029200 and Glyma.01G099800 were found in 2nd leaves subjected to the 4‐day dehydration treatment (Figure 5a). Among the genes involved in mannose metabolism, we detected high transcript levels of Glyma.19G223000, which encodes mannan endo‐1,4‐β‐mannosidase (MAN), in only stems under dehydration conditions (Figure 5a,f). Among the genes involved in trehalose metabolism, the transcript levels of the genes encoding trehalose 6‐phosphate phosphatase (TPP) (Glyma.18G018100) and trehalose 6‐phosphate synthase (TPS) (Glyma.17G067800) were upregulated in the dehydration‐treated leaves and stems but were downregulated in dehydration‐treated roots (Figure 5a,f). We investigated the expression levels of the dehydration‐inducible galactinol (GolS) and raffinose synthase (RS) genes in soybeans via oligo microarray and CAGE analysis and confirmed their transcript levels by RT‐qPCR (Figure 5h). Probes corresponding to several GolS genes were not included in our oligo microarray slide. The transcript levels of Glyma.19g227800 (GolS) and Glyma.03g137900 (RS) were significantly higher in dehydration‐treated organs than in untreated organs (Figure 5h). The transcript levels of Glyma.19g227800 were higher in leaves and stems than in roots under dehydration conditions. Stems subjected to the 3‐day dehydration treatment showed the highest transcript levels of Glyma.19g227800. The transcript levels of Glyma.03g137900 were higher in leaves than in stems and roots after exposure to dehydration. The highest transcript levels of Glyma.03g137900 were detected in 1st leaves subjected to the 4‐day dehydration treatment.
Figure 5.
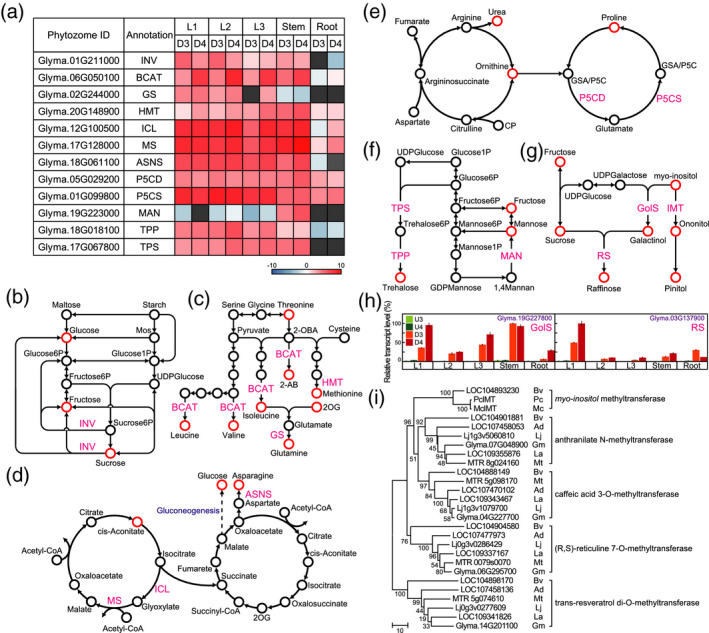
Amino acid and carbohydrate metabolic pathways. (a) Heatmaps illustrate the log ratios of genes involved in amino acid and carbohydrate biosynthesis and metabolism. INV, alkaline/neutral invertase; BCAT, branched‐chain amino acid aminotransferase; GS, glutamine synthetase; HMT, homocysteine S‐methyltransferase; ASNS, glutamine‐dependent asparagine synthetase; ICL, isocitrate lyase; MS, malate synthase; P5CD, Δ1‐pyrroline‐5‐carboxylate dehydrogenase, P5CS, Δ1‐pyrroline‐5‐carboxylate synthetase; MAN, mannan endo‐1[,]4‐β‐mannosidase, TSP, trehalose 6‐phosphate synthase; TPP, trehalose 6‐phosphate phosphatase. (b) Starch degradation and sucrose metabolism. Mos, malto‐oligosaccharides. (c) BCAAs, glutamine, and methionine biosynthesis. 2‐OBA, 2‐oxobutyrate; 2‐AB, 2‐aminobutanoate; 2OG, 2‐oxoglutarate. (d) Glyoxylate cycle and tricarboxylic acid cycle. (e) Arginine and proline metabolism. CP, carbamoyl phosphate. P5C, Δ1‐pyrroline‐5‐carboxylate. GSA, l‐glutamate γ‐semialdehyde. (f) Mannose and trehalose metabolism. (g) Raffinose and pinitol biosynthesis. GolS, galactinol synthase; RS, raffinose synthase; IMT, myoinositol methyltransferase. (h) Transcript levels of galactinol synthase and RS genes, as determined by RT‐qPCR. Values are shown as the mean ± SD (n = 3 experiments). (i) Phylogenetic trees of SAM‐MTases. Sequence sets are listed in Table S4. Ad, Arachis duranensis; Bv, Beta vulgaris; Gm, Glycine max; La, Lupinus angustifolius; Lj, Lotus japonicus; Mt, Medicago truncatula; Mc, Mesembryanthemum crystallinum; Pc, Porteresia coarctata.
Our metabolite analysis revealed that the increased levels of polyols (ononitol and pinitol) were higher in leaves than in stems and roots after exposure to dehydration (Figure 2). High levels of ononitol and/or pinitol have been reported in halophytic wild rice, legumes, and ice plants (Quemener and Brillouet, 1983; Adams et al., 1992; Sengupta et al., 2008), and there is a correlation between the expression of genes encoding myoinositol methyltransferase 1 (McIMT1) and both ononitol and pinitol levels in ice plants (Ishitani et al., 1996). McIMT1, which belongs to the S‐adenosyl‐l‐methionine‐dependent methyltransferase (SAM‐MTase) family, is a key enzyme in both ononitol and pinitol biosynthesis in ice plants (Ishitani et al., 1996). Transgenic tobacco plants that overexpress McIMT1 accumulate more endogenous ononitol and exhibit improved dehydration tolerance compared with those of wild‐type plants (Vernon et al., 1993; Sheveleva et al., 1997). In addition, a cDNA for a McIMT1 homologue (PcIMT1) was reported in only a halophytic wild rice accession (Sengupta et al., 2008). To predict the myoinositol methyltransferase (IMT) genes in soybeans, we performed a similarity search for McIMT1 against the Phytozome and GenBank databases and constructed phylogenetic trees of representative members of the SAM‐MTase family from several species, including soybean (Figures 5i and S3 and Tables S4 and S5). The known IMTs (McIMT1 and PcIMT1) are highly conserved and are most similar to LOC104893230 in Beta vulgaris, forming a single group. The level of homology between McIMT1 and PcIMT1 is quite high. Surprisingly, McIMT1 and PcIMT1 differ by only 4 of 1095 bases, with this small number of differences between the two genes at nucleotide positions 6, 9, 47, and 709 from the initiation codon (Figure S4). We recognized several identifiable clusters in our phylogenetic trees, which suggest that members of the SAM‐MTase family are classified into groups of proteins with the same function, such as anthranilate N‐methyltransferase, caffeic acid 3‐O‐methyltransferase, (R,S)‐reticuline 7‐O‐methyltransferase, and trans‐resveratrol di‐O‐methyltransferase (Figure 6i). This classification was not related to species. These results suggested that there are no genes with high levels of amino acid sequence similarity to McIMT1 in legumes, including in soybeans. As ononitol is present in soybeans, SAM‐MTases other than myoinositol methyltransferase might be involved in ononitol biosynthesis in soybeans. We then searched for dehydration‐inducible SAM‐MTase genes but found no SAM‐MTase genes that were expressed at high levels under dehydration conditions (Figure S5). Ononitol levels were significantly increased in 35S:McIMT1 transgenic soybean plants, whereas pinitol levels in these plants were not significantly different from those in wild‐type plants (Chiera et al., 2006). Taken together, these results suggested that the roles of SAM‐MTases in ononitol and pinitol accumulation in plants under dehydration conditions differ between soybeans and ice plants.
Figure 6.
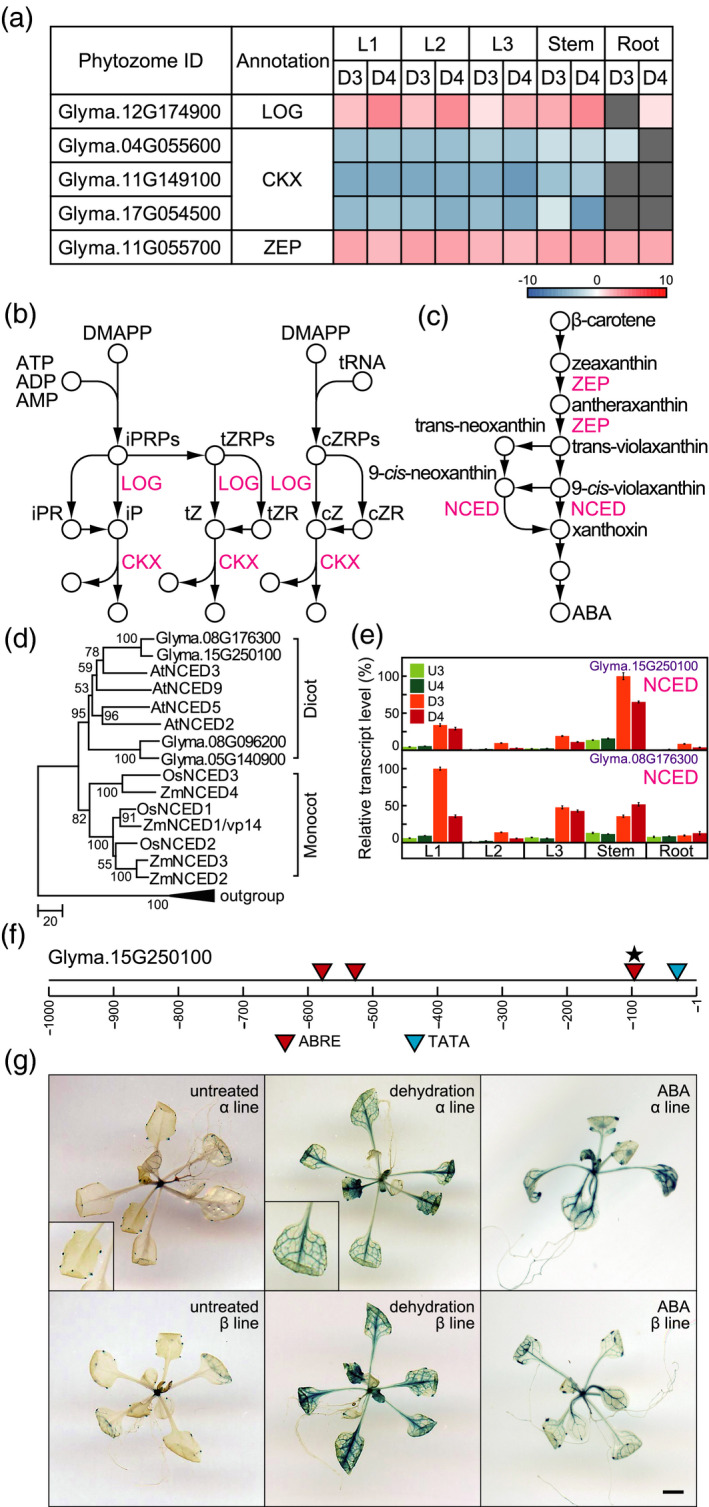
Pathways for CK and ABA biosynthesis and metabolism. (a) Heatmaps illustrate the log ratios of genes involved in cZ and ABA biosynthesis and metabolism. (b) CK biosynthesis and metabolism. (c) ABA biosynthesis. LOG, cytokinin nucleoside 5'‐monophosphate phosphoribohydrolase; CKX, cytokinin oxidase/dehydrogenase; ZEP, zeaxanthin epoxidase; NCED, 9‐cis‐epoxycarotenoid dioxygenase. (d) Phylogenetic trees of NCEDs. The phylogenetic tree was constructed using the neighbour‐joining method with MEGA6 software (Tamura et al., 2013). At3g63520, Glyma.13g202200, GRMZM2G057243, and Os12g0640600 encode carotenoid cleavage dioxygenases and were used as an outgroup. (e) Transcript levels of genes encoding NCED, as determined by RT‐qPCR. Values are shown as the mean ± SD (n = 3 experiments). (f) Schematic diagram of promoters of Glyma.15g250100. Stars indicate duplication of ABREs. (g) Glyma.15g250100 promoter−driven GUS expression in dehydration‐ and ABA‐treated Arabidopsis plants. Left, centre, and right panels show untreated, dehydration‐treated, and ABA‐treated Arabidopsis plants, respectively. The upper and lower panels show two independently isolated lines, α and β. Bar = 0.5 mm.
Expression of genes involved in phytohormone biosynthesis and metabolism
Our phytohormone profiling indicated that cZ and its riboside levels increased in soybean roots after dehydration treatment (Figure 3c). ABA levels under dehydration conditions were higher in leaves than in stems and roots (Figure 3c). We screened dehydration‐responsive genes involved in cZ and ABA biosynthesis and metabolism. Among the genes involved in cZ biosynthesis and metabolism, the transcript levels of the gene encoding LONELY GUY (LOG) (Glyma.12G174900) were increased, and those of the genes encoding CK oxidases/dehydrogenases (CKXs) (Glyma.04G055600, Glyma.11G149100, Glyma.17G054500) were decreased in leaves and stems after exposure to dehydration (Figure 6a,b). We also investigated the expression of genes involved in ABA biosynthesis under dehydration in soybean organs. Among the genes involved in ABA biosynthesis, the transcript levels of the gene encoding zeaxanthin epoxidase (ZEP) (Glyma.11G055700) were increased in all organs after dehydration treatment (Figure 6a,c). The enzyme 9‐cis‐epoxycarotenoid dioxygenase (NCED), which converts 9‐cis‐neoxanthin and 9‐cis‐violaxanthin into xanthoxin, plays a key role in ABA biosynthesis (Figure 6c). We searched for coding sequences (CDSs) of soybean NCEDs and constructed a phylogenetic tree of representative NCEDs from Arabidopsis, rice, maize, and soybean (Figure 6d). Phylogenetic analysis of NCEDs suggested that they can be divided into monocot and dicot plant proteins. Among soybean NCEDs, Glyma.15g250100 and Glyma.08g176300 are most similar to AtNCED3 and form a single group (Figure 6d and Table S6). We investigated the expression levels of the dehydration‐inducible NCED genes in soybeans via oligo microarray and CAGE analysis and confirmed their transcript levels by RT‐qPCR. Probes corresponding to several NCED genes were not included in our oligo microarray slide. Both Glyma.15g250100 and Glyma.08g176300 showed markedly higher transcript levels in dehydration‐treated organs than in the untreated organs (Figure 6e), and the levels of transcripts for both genes were higher in leaves and stems than in roots under dehydration conditions. The highest transcript levels of Glyma.15g250100 and Glyma.08g176300 were found in stems and 1st leaves subjected to the 3‐day dehydration treatment, respectively (Figure 6e).
In addition, we analyzed the promoter for Glyma.15g250100. This promoter contains four ABREs within 1 kb upstream from the TSS (Figure 6f), whereas the regulatory region of the AtNCED3 promoter in Arabidopsis is located 3 kb upstream from the TSS (Behnam et al., 2013). To predict Glyma.15g250100 promoter activity in dehydration‐ and ABA‐treated transgenic plants, we transformed Arabidopsis plants with a fragment of the Glyma.15g250100 promoter fused to the β‐glucuronidase gene (GUS) and analyzed GUS expression. In untreated transgenic plants, hydathodes exhibited Glyma.15g250100 promoter activity. By contrast, dehydration and ABA treatment enhanced Glyma.15g250100 promoter activity in the vascular tissues of leaves and stems of the transgenic plants (Figure 6g). These results support our transcript analyses of Glyma.15g250100 in soybeans. The Glyma.15g250100 promoter is likely to be functional in the vascular tissue of leaves and stems in dehydration‐treated plants and is likely to be regulated by ABA via a positive feedback regulation loop.
DISCUSSION
There is a close relationship between the level of dehydration stress in plants and ABA concentration (Beardsell and Cohen, 1975; Bensen et al., 1988). In plants with mild dehydration stress levels, the endogenous ABA levels are low, and changes in gene transcript and metabolite levels are minimal. By contrast, in plants with severe dehydration stress levels, the endogenous ABA levels are high, and gene transcript and metabolite levels undergo dynamic changes (Todaka et al., 2017; Urano et al., 2017). In Arabidopsis, ethylene signalling is involved under mild osmotic stress conditions (Skirycz et al., 2010). The metabolite/phytohormone–gene regulatory networks under dehydration stress with low levels of ABA might also be different from ABA‐dependent metabolite/phytohormone–gene regulatory networks in soybean. In addition, ABA is related to not only dehydration responses but also senescence, seed maturation, etc. (Lim et al., 2007). The metabolite/phytohormone–gene regulatory networks in reproductive stages of plant organs under dehydration conditions are considered to be more complex than those of vegetative stages due to effects such as senescence, seed maturation, etc. In the present study, we conducted an integration analysis to understand the metabolite/phytohormone–gene regulatory networks involved in the dehydration responses in soybean organs of the vegetative growth stages at dehydration stress levels during which stomatal closure occurs.
An integration analysis enabled us to predict important genes encoding metabolic enzymes for carbohydrates, amino acids, and phytohormones that are involved in the response of soybean plants to dehydration. Based on our results, INV (Glyma.01G211000) is important for sucrose metabolism in dehydration‐treated soybean plants. High levels of glucose and fructose were correlated with transcript levels of Glyma.01G211000 in the aerial parts of soybeans under dehydration conditions (Figures 2 and 5). In the glyoxylate cycle, ICL and MS are important enzymes, and the expression levels of the genes that encode them play a role in activating gluconeogenesis (Eastmond et al., 2000; Dunn et al., 2009). Among the genes in the glyoxylate cycle, the transcript levels of ICL (Glyma.12G100500) and MS (Glyma.17G128000) were upregulated significantly after exposure to dehydration (Figure 5). High levels of monosaccharide were correlated with transcript levels of both Glyma.12G100500 and Glyma.17G128000 in the aerial parts of plants under dehydration conditions (Figures 2 and 5). We suggest that both genes have important roles in increasing the accumulation of monosaccharides under dehydration conditions in soybeans. Our findings also indicate that MAN (Glyma.19G223000) is important for mannose metabolism in dehydration‐treated stems. Increased mannose levels were correlated with high transcript levels of Glyma.19G223000 in stems under dehydration conditions (Figures 2 and 5). Consistently, the highest levels of mannose were found in stems subjected to dehydration treatment. The highest transcript levels of Glyma.19G223000 were also found in stems subjected to dehydration treatment.
BCAT (Glyma.06G050100) is an important gene in both BCAA and 2‐aminobutanoate biosynthesis. Increased BCAAs and 2‐aminobutanoate levels were correlated with high transcript levels of Glyma.06G050100 in dehydration‐treated soybean plants (Figures 2 and 5). The highest levels of both BCAAs and 2‐aminobutanoate were found in 2nd leaves subjected to the 4‐day dehydration treatment. The highest transcript levels of Glyma.06G050100 were also found in 2nd leaves subjected to the 4‐day dehydration treatment. P5CS (Glyma.01G099800) is an important gene in proline metabolism. Increased proline levels were correlated with high transcript levels of Glyma.01G099800 in all organs under dehydration conditions (Figures 2 and 5). The highest levels of proline were found in 2nd leaves subjected to the 4‐day dehydration treatment. The highest transcript levels of Glyma.01G099800 were also found in 2nd leaves subjected to the 4‐day dehydration treatment. Levels of asparagine were correlated with transcript levels of ASNS (Glyma.18G061100) at the level of the individual plants but not at the organ level under dehydration conditions. Asparagine levels increased in mainly stems and roots under dehydration conditions (Figure 2), but Glyma.18G061100 was expressed in mostly the aerial parts of soybeans under dehydration conditions (Figure 5). Asparagine is a typical compound for nitrogen transport and storage (Lam et al., 1994; Lea et al., 2007). Our integration analysis suggested that asparagine is biosynthesized in the aerial parts of soybean plants and is transported to the roots under dehydration conditions.
Trehalose is a compatible solute that functions under various abiotic stresses in rice. Transgenic plants with increased trehalose accumulation show improved photosystem II function under dehydration conditions (Garg et al., 2002). Increased trehalose levels were correlated with transcript levels of TSP (Glyma.17G067800) and TPP (Glyma.18G018100) at the level of the individual plants but not at the organ level under dehydration conditions. Trehalose levels increased in mainly roots under dehydration conditions (Figure 2), but both Glyma.17G067800 and Glyma.18G018100 were expressed in mostly the aerial parts of soybeans under dehydration conditions (Figure 5). Our integration analysis suggested that trehalose is biosynthesized in the aerial parts of soybean plants and is transported to the roots under dehydration conditions. Raffinose is thought to function in processes such as translocation (Haritatos et al., 2000) and polymer trapping (Turgeon and Wolf, 2009). In addition, raffinose functions as an osmoprotectant to stabilize cellular membranes, as a scavenger of reactive oxygen species to protect the photosynthetic apparatus in chloroplasts under dehydration conditions (Nishizawa et al., 2008), and as a stabilizer of photosystem II to protect cold‐acclimated leaf cells from damage during freezing (Knaupp et al., 2011). Our integration analysis revealed that GolS (Glyma.19g227800) and RS (Glyma.03g137900) were expressed in mainly the aerial parts of soybeans, but galactinol and raffinose accumulated to the greatest extent in the roots of soybeans under dehydration conditions (Figures 2 and 5). Our integration analyses indicated that galactinol and raffinose levels were correlated with transcript levels of Glyma.19g227800 and Glyma.03g137900, respectively, at the level of the individual plant but not at the organ level under dehydration conditions. Therefore, along with transcriptional regulation of Glyma.19g227800 and Glyma.03g137900, transport of their resulting metabolites may also be important for the accumulation of galactinol and raffinose in roots under dehydration conditions. In addition, the specific accumulation of trehalose and raffinose in dehydration‐treated soybean roots detected in our study suggests that trehalose and raffinose have additional roles not directly involved in protecting the photosynthetic apparatus of soybeans under dehydration conditions.
Our integration analyses indicated that cZ levels were correlated with transcript levels of LOG (Glyma.12G174900) and CKX (Glyma.04G055600, Glyma.11G149100, Glyma.17G054500) at the level of individual plants but not at the organ level under dehydration conditions. In general, cZ is resistant to CKX, but overexpression of an Arabidopsis cytosolic isoenzyme, CKX7, reduces levels of cZ and its conjugates (Köllmer et al., 2014). cZ accumulated most notably in roots after exposure to dehydration (Figure 3), but transcriptional changes in LOG and CKX genes were detected in the aerial parts under dehydration conditions. In addition, among the genes involved in the biosynthesis and metabolism of cZ, genes expressed specifically in roots were not detected under dehydration conditions (Table S3). As cZR is relatively abundant in phloem sap (Hirose et al., 2008), cZ detected in roots might be translocated from aerial organs. Elevation of cZ in roots has been reported under high‐salt conditions in maize (Vyroubalová et al., 2009) and under dehydration conditions in tobacco (Nicotiana tabacum) (Havlová et al., 2008; Macková et al., 2013). In general, cZ is a CK species with little activity. It was suggested that cZ maintains a minimal level of CK activity under abiotic stress conditions (Schäfer et al., 2015). The increase in cZ and its riboside in dehydration‐treated soybean roots might be involved in the maintenance of certain physiological functions of CKs under water‐deficit conditions. Arabidopsis mutants for cZ biosynthesis, which show chlorotic phenotypes, have a shortened primary root, which likely results from a reduced root meristem size and ectopic protoxylem formation (Miyawaki et al., 2006; Köllmer et al., 2014). These results support the possibility that cZ accumulation in soybean roots might contribute to root elongation for water uptake from the moisture region in soil. Our integration analyses indicated that ABA levels were correlated with transcript levels of NCED (Glyma.15g250100 and Glyma.08g176300) at the individual level but not at the organ level under dehydration conditions. These results suggested that processes such as ABA transport and others were important for the accumulation of ABA in the youngest leaves of soybeans under dehydration conditions as well as for the transcriptional regulation of NCED genes. The ABA exporter AtABCG25 has been isolated from Arabidopsis (Kuromori et al., 2010). The accumulation of ABA under dehydration conditions might be attributed to the activity of an ABA exporter localized to stems. High levels of ABA are important for plant growth and dehydration tolerance, as shown in several plant species (Yamaguchi‐Shinozaki and Shinozaki, 2006; Hirayama and Shinozaki, 2007; Cutler et al., 2010). Our phytohormone profiling of various soybean organs revealed that ABA is the most strongly dehydration‐inducible phytohormone. Among the dehydration‐treated organs, the youngest leaves (3rd leaves) had the highest ABA levels, and the roots had the lowest (Figure 3). These results suggested that the effects of ABA on plant growth and dehydration tolerance are greater in the aerial parts of plants, especially the youngest leaves, than in the underground parts of plants under dehydration conditions.
Experimental procedures
Plant materials and growth conditions
Soybean seedlings (Glycine max [L.] Merr. cultivar Williams 82) were grown in plastic pots filled with nutrient soil (Kureha) for 3 weeks under a 12‐h light (28°C)/12‐h dark (25°C) photoperiod (ca. 1500 μmol of photons m−2 sec−1). Soybean seedlings were subjected to dehydration treatment by withholding watering for the indicated number of days, after which they were allowed to recover by rehydration treatment (Figure S6). Transgenic Arabidopsis seedlings (Arabidopsis thaliana ecotype Columbia) were grown in plates containing germination medium in agar at 22°C under a 16‐h light/8‐h dark photoperiod (50 ± 10 μmol of photons m−2 sec−1). Three‐week‐old plants were removed from the germination medium and transferred to empty Petri dishes that were subsequently sealed with Parafilm for dehydration treatment, as described (Liu et al., 1998).
GC‐TOF‐MS analysis
Metabolites were extracted from five different organs (1st [L1], 2nd [L2], and 3rd [L3] trifoliate leaves; stems [S]; and roots [R]) of soybean plants at the V3 growth stage (25 mg/sample) using extraction medium (3:1:1 [v/v/v] methanol/chloroform/water). Extraction and derivatization were performed as described (Kusano et al., 2007, 2011). Metabolites were detected using a GC instrument (Model 6890, Agilent Technologies, Palo Alto, CA, USA) fitted with an Rxi‐5Sil MS column (0.25‐mm i.d., 0.25‐µm film; Restek) coupled to a TOF mass spectrometer (Leco, St. Joseph, MI, USA). Ten stable isotope reference compounds were used as internal standards (Kusano et al., 2007). All raw data in netCDF format were pre‐processed by hyphenated data analysis (Jonsson et al., 2005, 2006). The obtained data matrix was normalized and summarized using the cross‐contribution‐compensating multiple standard normalization method (Redestig et al., 2009). For metabolite identification, we cross‐referenced the obtained mass spectra with GC‐EI‐MS mass spectral and RI libraries (Schauer et al., 2005) in the Golm Metabolome Database (Kopka et al., 2005) and our own in‐house libraries. The reproducibility of GC‐TOF‐MS analysis was assessed with three biological replicates per experiment.
LC‐MS analysis
Phytohormone levels in soybean tissue (100 mg) were quantified as described previously (Kojima and Sakakibara, 2012) using an LC‐MS system (AQUITY UPL System/Xevo‐TQS; Waters, MA, USA) fitted with an octadecylsilyl column (AQUITY UPLC BEH C18, 1.7 μm, 2.1 × 100 mm; Waters). The reproducibility of LC‐MS analysis was assessed with three biological replicates per experiment.
Oligo microarray analysis
Total RNA was isolated from soybean plants and labelled using Low RNA Input Linear Amplification/Labelling kit reagents (Agilent). Cy5‐labelled cRNA experimental samples and Cy3‐labelled cRNA control samples were hybridized to the oligo microarray chips. Biological and technical (dye‐swap) replicates of the sample sets were analyzed as described (Maruyama et al., 2014b). After hybridization, the oligo microarray slides were scanned (scanner model G2505C with scan control software version A.8.5.1; Agilent), and the data were analyzed using Feature Extraction software version 10.10.1.1 (Agilent), as described (Maruyama et al., 2014b). Raw data were analyzed using GeneSpring GX software version 12.0 (Agilent). Expression log ratios and Benjamini and Hochberg FDR P‐values were calculated using GeneSpring GX as described (Maruyama et al., 2014b). The oligo microarray design and data were deposited at ArrayExpress (accession number E‐MTAB‐7010).
CAGE analysis
CAGE TSS clustering was performed using the protocol from the third stage of the Functional Annotation of Mouse project (Carninci et al., 2005). CAGE TSS clusters were created by summing the number of CAGE tags at each genome position, which required at least one CAGE tag for at least one experimental condition. The TSSs within 20 bp of each other on the same chromosomal strand were clustered into a single TSS cluster. The tags per million (tpm) values for the TSS clusters were calculated by dividing the number of CAGE tags in each TSS cluster for each experimental condition by the total number of mapped CAGE tags for that condition and multiplying by 1 000 000. TSS clusters with values <10 tpm over the experimental condition were discarded. The FASTQ files of the sequencing reads were submitted to the DDBJ Sequence Read Archive under accession number DRA007142.
RT‐qPCR analysis
RT‐qPCR was performed as described (Urano et al., 2017). Total RNA was extracted from soybean plants using RNAiso (TaKaRa, Kusatsu, Shiga, Japan). First‐strand cDNA was synthesized from total RNA with SuperScript III Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA). RT‐qPCR was performed on a LightCycler (Roche, Basel, Switzerland) with TB Green Premix Ex Taq II (TaKaRa). Primer sets are listed in Table S7. The reproducibility of RT‐qPCR was assessed with three biological replicates per experiment.
AUTHOR CONTRIBUTIONS
KM and KU designed the research; HS, K. Saito, and K. Shinozaki supervised the experiments; KM, KU, M. Kusano, TS, HT, M. Kojima, KY, M. Kobayashi, and M. Kishimoto performed the research; KM, M. Kusano, and KY analyzed the data; and KM wrote the paper.
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
Open Research Badges
This article has earned an Open Data Badge for making publicly available the digitally shareable data necessary to reproduce the reported results.
This article has earned an Open Materials Badge for making publicly available the components of the research methodology needed to reproduce the reported procedure and analysis.
Supporting information
Figure S1. Expression of genes encoding dehydrin/LEA proteins.
Figure S2. Expression of genes encoding photosynthesis‐related proteins.
Figure S3. Phylogenetic tree of SAM‐MTases.
Figure S4. Nucleotide sequences of PcIMT1 and McIMT1.
Figure S5. Expression of SAM‐MTase genes.
Figure S6. Soybean seedlings 1 day (Re1), 2 days (Re2), 3 days (Re3), and 4 days (Re4) after rehydration treatment.
Table S1. Metabolite levels in various soybean organs.
Table S2. Phytohormone levels in various soybean organs.
Table S3. Transcript levels in various soybean organs.
Table S4. Genes encoding representative SAM‐MTases.
Table S5. Genes encoding SAM‐MTases in soybean.
Table S6. Genes encoding representative NCEDs.
Table S7. Primers used for RT‐qPCR.
Acknowledgements
This work was supported by grants from the Ministry of Agriculture, Forestry and Fisheries (MAFF); the Program for the Promotion of Basic and Applied Research for Innovations in Bio‐oriented Industry (BRAIN); the Science and Technology Research Partnership for Sustainable Development (SATREPS) of the Japan Science and Technology Agency (JST)/Japan International Cooperation Agency (JICA); Grants‐in‐Aid for Scientific Research by the Ministry of Education, Culture, Sports, Science and Technology (MEXT); and Japan Society for the Promotion of Science (JSPS).
Data Availability
All relevant data can be found within the manuscript and its supporting materials.
References
- Adams, P. , Thomas, J.C. , Vernon, D.M. , Bohnert, H.J. and Jensen, R.G. (1992) Distinct cellular and organismic responses to salt stress. Plant Cell Physiol. 33, 1215–1223. [Google Scholar]
- Alvarez, S. , Marsh, E.L. , Schroeder, S.G. and Schachtman, D.P. (2008) Metabolomic and proteomic changes in the xylem sap of maize under drought. Plant Cell Environ. 31, 325–340. [DOI] [PubMed] [Google Scholar]
- Aranjuelo, I. , Molero, G. , Erice, G. , Avice, J.C. and Nogués, S. (2011) Plant physiology and proteomics reveals the leaf response to drought in alfalfa (Medicago sativa L.). J. Exp. Bot. 62, 111–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beardsell, M.F. and Cohen, D. (1975) Relationships between leaf water status, abscisic acid levels, and stomatal resistance in maize and sorghum. Plant Physiol. 56, 207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnam, B. , Iuchi, S. , Fujita, M. , Fujita, Y. , Takasaki, H. , Osakabe, Y. , Yamaguchi‐Shinozaki, K. , Kobayashi, M. and Shinozaki, K. (2013) Characterization of the promoter region of an arabidopsis gene for 9‐cis‐epoxycarotenoid dioxygenase involved in dehydration‐inducible transcription. DNA Res. 20, 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensen, R.J. , Boyer, J.S. and Mullet, J.E. (1988) Water deficit‐induced changes in abscisic acid, growth, polysomes, and translatable RNA in soybean hypocotyls. Plant Physiol. 88, 289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carninci, P. , Kasukawa, T. , Katayama, S. et al . (2005) The transcriptional landscape of the mammalian genome. Science, 309, 1559–1563. [DOI] [PubMed] [Google Scholar]
- Chiera, J.M. , Streeter, J.G. and Finer, J.J. (2006) Ononitol and pinitol production in transgenic soybean containing the inositol methyl transferase gene from Mesembryanthemum crystallinum . Plant Sci. 171, 647–654. [Google Scholar]
- Chiwocha, S.D.S. , Abrams, S.R. , Ambrose, S.J. , Cutler, A.J. , Loewen, M. , Ross, A.R.S. and Kermode, A.R. (2003) A method for profiling classes of plant hormones and their metabolites using liquid chromatography‐electrospray ionization tandem mass spectrometry: an analysis of hormone regulation of thermodormancy of lettuce (Lactuca sativa L.) seeds. Plant J. 35, 405–417. [DOI] [PubMed] [Google Scholar]
- Cutler, S.R. , Rodriguez, P.L. , Finkelstein, R.R. and Abrams, S.R. (2010) Abscisic acid: emergence of a core signaling network. Annu. Rev. Plant Biol. 61, 651–679. [DOI] [PubMed] [Google Scholar]
- Dunn, M.F. , Ramirez‐Trujillo, J.A. and Hernandez‐Lucas, I. (2009) Major roles of isocitrate lyase and malate synthase in bacterial and fungal pathogenesis. Microbiology, 155, 3166–3175. [DOI] [PubMed] [Google Scholar]
- Eastmond, P.J. , Germain, V. , Lange, P.R. , Bryce, J.H. , Smith, S.M. and Graham, I.A. (2000) Postgerminative growth and lipid catabolism in oilseeds lacking the glyoxylate cycle. Proc. Natl Acad. Sci. USA, 97, 5669–5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahad, S. , Bajwa, A.A. , Nazir, U. et al . (2017) Crop production under drought and heat stress: plant responses and management options. Front. Plant Sci. 8, 1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg, A.K. , Kim, J.K. , Owens, T.G. , Ranwala, A.P. , Choi, Y.D. , Kochian, L.V. and Wu, R.J. (2002) Trehalose accumulation in rice plants confers high tolerance levels to different abiotic stresses. Proc. Natl Acad. Sci. USA, 99, 15898–15903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare, P.D. , Cress, W.A. and van Staden, J. (1997) The involvement of cytokinins in plant responses to environmental stress. Plant Growth Regul. 23, 79–103. [Google Scholar]
- Haritatos, E. , Medville, R. and Turgeon, R. (2000) Minor vein structure and sugar transport in Arabidopsis thaliana . Planta, 211, 105–111. [DOI] [PubMed] [Google Scholar]
- Havlová, M. , Dobrev, P.I. , Motyka, V. , Štorchová, H. , Libus, J. , Dobrá, J. , Malbeck, J. , Gaudinová, A. and Vanková, R. (2008) The role of cytokinins in responses to water deficit in tobacco plants over‐expressing trans‐zeatin O‐glucosyltransferase gene under 35S or SAG12 promoters. Plant Cell Environ. 31, 341–353. [DOI] [PubMed] [Google Scholar]
- Hirayama, T. and Shinozaki, K. (2007) Perception and transduction of abscisic acid signals: keys to the function of the versatile plant hormone ABA. Trends Plant Sci. 12, 343–351. [DOI] [PubMed] [Google Scholar]
- Hirose, N. , Takei, K. , Kuroha, T. , Kamada‐Nobusada, T. , Hayashi, H. and Sakakibara, H. (2008) Regulation of cytokinin biosynthesis, compartmentalization and translocation. J. Exp. Bot. 59, 75–83. [DOI] [PubMed] [Google Scholar]
- Ishitani, M. , Majumder, A.L. , Bornhouser, A. , Michalowski, C.B. , Jensen, R.G. and Bohnert, H.J. (1996) Coordinate transcriptional induction of myo‐inositol metabolism during environmental stress. Plant J. 9, 537–548. [DOI] [PubMed] [Google Scholar]
- Iuchi, S. , Kobayashi, M. , Taji, T. , Naramoto, M. , Seki, M. , Kato, T. , Tabata, S. , Kakubari, Y. , Yamaguchi‐Shinozaki, K. and Shinozaki, K. (2001) Regulation of drought tolerance by gene manipulation of 9‐cis‐epoxycarotenoid dioxygenase, a key enzyme in abscisic acid biosynthesis in Arabidopsis . Plant J. 27, 325–333. [DOI] [PubMed] [Google Scholar]
- Jonsson, P. , Johansson, A.I. , Gullberg, J. , Trygg, J. , A, J. , Grung, B. , Marklund, S. , Sjöström, M. , Antti, H. and Moritz, T. (2005) High‐throughput data analysis for detecting and identifying differences between samples in GC/MS‐based metabolomic analyses. Anal. Chem. 77, 5635–5642. [DOI] [PubMed] [Google Scholar]
- Jonsson, P. , Johansson, E.S. , Wuolikainen, A. , Lindberg, J. , Schuppe‐Koistinen, I. , Kusano, M. , Sjöström, M. , Trygg, J. , Moritz, T. and Antti, H. (2006) Predictive metabolite profiling applying hierarchical multivariate curve resolution to GC−MS data a potential tool for multi‐parametric diagnosis. J. Proteome Res. 5, 1407–1414. [DOI] [PubMed] [Google Scholar]
- Kieber, J.J. and Schaller, G.E. (2014) Cytokinins. Arabidopsis Book, 12, e0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilian, J. , Whitehead, D. , Horak, J. , Wanke, D. , Weinl, S. , Batistic, O. , D’Angelo, C. , Bornberg‐Bauer, E. , Kudla, J. and Harter, K. (2007) The AtGenExpress global stress expression data set: protocols, evaluation and model data analysis of UV‐B light, drought and cold stress responses. Plant J. 50, 347–363. [DOI] [PubMed] [Google Scholar]
- Knaupp, M. , Mishra, K.B. , Nedbal, L. and Heyer, A.G. (2011) Evidence for a role of raffinose in stabilizing photosystem II during freeze–thaw cycles. Planta, 234, 477–486. [DOI] [PubMed] [Google Scholar]
- Kojima, M. and Sakakibara, H. (2012) Highly sensitive high‐throughput profiling of six phytohormones using MS‐probe modification and liquid chromatography‐tandem mass spectrometry. Methods Mol. Biol. 918, 151–164. [DOI] [PubMed] [Google Scholar]
- Köllmer, I. , Novák, O. , Strnad, M. , Schmülling, T. and Werner, T. (2014) Overexpression of the cytosolic cytokinin oxidase/dehydrogenase (CKX7) from Arabidopsis causes specific changes in root growth and xylem differentiation. Plant J. 78, 359–371. [DOI] [PubMed] [Google Scholar]
- Kopka, J. , Schauer, N. , Krueger, S. et al . (2005) GMD@CSB.DB: the golm metabolome database. Bioinformatics, 21, 1635–1638. [DOI] [PubMed] [Google Scholar]
- Kuromori, T. , Miyaji, T. , Yabuuchi, H. , Shimizu, H. , Sugimoto, E. , Kamiya, A. , Moriyama, Y. and Shinozaki, K. (2010) ABC transporter AtABCG25 is involved in abscisic acid transport and responses. Proc. Natl Acad. Sci. USA, 107, 2361–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusano, M. , Fukushima, A. , Arita, M. , Jonsson, P. , Moritz, T. , Kobayashi, M. , Hayashi, N. , Tohge, T. and Saito, K. (2007) Unbiased characterization of genotype‐dependent metabolic regulations by metabolomic approach in Arabidopsis thaliana . BMC Syst. Biol. 1, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusano, M. , Tabuchi, M. , Fukushima, A. et al . (2011) Metabolomics data reveal a crucial role of cytosolic glutamine synthetase 1;1 in coordinating metabolic balance in rice. Plant J. 66, 456–466. [DOI] [PubMed] [Google Scholar]
- Lam, H.M. , Peng, S.S.Y. and Coruzzi, G.M. (1994) Metabolic regulation of the gene encoding glutamine‐dependent asparagine synthetase in Arabidopsis thaliana . Plant Physiol. 106, 1347–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lea, P.J. , Sodek, L. , Parry, M.A.J. , Shewry, P.R. and Halford, N.G. (2007) Asparagine in plants. Ann. Appl. Biol. 150, 1–26. [Google Scholar]
- Lim, P.O. , Kim, H.J. and Nam, H.G. (2007) Leaf senescence. Annu. Rev. Plant Biol. 58, 115–136. [DOI] [PubMed] [Google Scholar]
- Liu, Q. , Kasuga, M. , Sakuma, Y. , Abe, H. , Miura, S. , Yamaguchi‐Shinozaki, K. and Shinozaki, K. (1998) Two transcription factors, DREB1 and DREB2, with an EREBP/AP2 DNA binding domain separate two cellular signal transduction pathways in drought‐ and low‐temperature‐responsive gene expression, respectively, in Arabidopsis . Plant Cell, 10, 1391–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, Y. , Szostkiewicz, I. , Korte, A. , Moes, D. , Yang, Y. , Christmann, A. and Grill, E. (2009) Regulators of PP2C phosphatase activity function as abscisic acid sensors. Science, 324, 1064–1068. [DOI] [PubMed] [Google Scholar]
- Macková, H. , Hronková, M. , Dobrá, J. et al . (2013) Enhanced drought and heat stress tolerance of tobacco plants with ectopically enhanced cytokinin oxidase/dehydrogenase gene expression. J. Exp. Bot. 64, 2805–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama, K. , Takeda, M. , Kidokoro, S. et al . (2009) Metabolic pathways involved in cold acclimation identified by integrated analysis of metabolites and transcripts regulated by DREB1A and DREB2A. Plant Physiol. 150, 1972–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama, K. , Todaka, D. , Mizoi, J. et al . (2012) Identification of Cis‐acting promoter elements in cold‐ and dehydration‐induced transcriptional pathways in Arabidopsis, rice, and soybean. DNA Res. 19, 37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama, K. , Urano, K. , Yoshiwara, K. et al . (2014a) Integrated analysis of the effects of cold and dehydration on rice metabolites, phytohormones, and gene transcripts. Plant Physiol. 164, 1759–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama, K. , Yamaguchi‐Shinozaki, K. and Shinozaki, K. (2014b) Gene expression profiling using DNA microarrays. Methods Mol. Biol. 1062, 381–391. [DOI] [PubMed] [Google Scholar]
- Miyawaki, K. , Tarkowski, P. , Matsumoto‐Kitano, M. , Kato, T. , Sato, S. , Tarkowska, D. , Tabata, S. , Sandberg, G. and Kakimoto, T. (2006) Roles of Arabidopsis ATP/ADP isopentenyltransferases and tRNA isopentenyltransferases in cytokinin biosynthesis. Proc. Natl Acad. Sci. USA, 103, 16598–16603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller, A. , Düchting, P. and Weiler, E. (2002) A multiplex GC‐MS/MS technique for the sensitive and quantitative single‐run analysis of acidic phytohormones and related compounds, and its application to Arabidopsis thaliana . Planta, 216, 44–56. [DOI] [PubMed] [Google Scholar]
- Murata, M. , Nishiyori‐Sueki, H. , Kojima‐Ishiyama, M. , Carninci, P. , Hayashizaki, Y. and Itoh, M. (2014) Detecting expressed genes using CAGE. Transcr. Factor Regul. Netw. Methods Protoc. 1164, 67–85. [DOI] [PubMed] [Google Scholar]
- Nishimura, N. , Hitomi, K. , Arvai, A.S. , Rambo, R.P. , Hitomi, C. , Cutler, S.R. , Schroeder, J.I. and Getzoff, E.D. (2009) Structural mechanism of abscisic acid binding and signaling by dimeric PYR1. Science, 326, 1373–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizawa, A. , Yabuta, Y. and Shigeoka, S. (2008) Galactinol and raffinose constitute a novel function to protect plants from oxidative damage. Plant Physiol. 147, 1251–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, S.Y. , Fung, P. , Nishimura, N. et al . (2009) Abscisic acid inhibits type 2C protein phosphatases via the PYR/PYL family of START proteins. Science, 324, 1068–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul, M.J. and Cockburn, W. (1989) Pinitol, a compatible solute in Mesembryanthemum crystallinum L.? J. Exp. Bot. 40, 1093–1098. [Google Scholar]
- Peleg, Z. and Blumwald, E. (2011) Hormone balance and abiotic stress tolerance in crop plants. Curr. Opin. Plant Biol. 14, 290–295. [DOI] [PubMed] [Google Scholar]
- Quemener, B. and Brillouet, J.‐M. (1983) Ciceritol, a pinitol digalactoside form seeds of chickpea, lentil and white lupin. Phytochemistry, 22, 1745–1751. [Google Scholar]
- Rabbani, M.A. , Maruyama, K. , Abe, H. , Khan, M.A. , Katsura, K. , Ito, Y. , Yoshiwara, K. , Seki, M. , Shinozaki, K. and Yamaguchi‐Shinozaki, K. (2003) Monitoring expression profiles of rice genes under cold, drought, and high‐salinity stresses and abscisic acid application using cDNA microarray and RNA Gel‐Blot analyses. Plant Physiol. 133, 1755–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavendra, A.S. , Gonugunta, V.K. , Christmann, A. and Grill, E. (2010) ABA perception and signalling. Trends Plant Sci. 15, 395–401. [DOI] [PubMed] [Google Scholar]
- Redestig, H. , Fukushima, A. , Stenlund, H. , Moritz, T. , Arita, M. , Saito, K. and Kusano, M. (2009) Compensation for systematic cross‐contribution improves normalization of mass spectrometry based metabolomics data. Anal. Chem. 81, 7974–7980. [DOI] [PubMed] [Google Scholar]
- Rosenzweig, C. , Elliott, J. , Deryng, D. et al . (2014) Assessing agricultural risks of climate change in the 21st century in a global gridded crop model intercomparison. Proc. Natl Acad. Sci. USA, 111, 3268–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago, J. , Dupeux, F. , Round, A. , Antoni, R. , Park, S.‐Y. , Jamin, M. , Cutler, S.R. , Rodriguez, P.L. and Márquez, J.A. (2009) The abscisic acid receptor PYR1 in complex with abscisic acid. Nature, 462, 665–668. [DOI] [PubMed] [Google Scholar]
- Schäfer, M. , Brütting, C. , Meza‐Canales, I.D. , Großkinsky, D.K. , Vankova, R. , Baldwin, I.T. and Meldau, S. (2015) The role of cis‐zeatin‐type cytokinins in plant growth regulation and mediating responses to environmental interactions. J. Exp. Bot. 66, 4873–4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauer, N. , Steinhauser, D. , Strelkov, S. et al . (2005) GC‐MS libraries for the rapid identification of metabolites in complex biological samples. FEBS Lett. 579, 1332–1337. [DOI] [PubMed] [Google Scholar]
- Seki, M. , Narusaka, M. , Ishida, J. et al . (2002) Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold and high‐salinity stresses using a full‐length cDNA microarray. Plant J. 31, 279–292. [DOI] [PubMed] [Google Scholar]
- Sengupta, S. , Patra, B. , Ray, S. and Majumder, A.L. (2008) Inositol methyl tranferase from a halophytic wild rice, Porteresia coarctata Roxb. (Tateoka): regulation of pinitol synthesis under abiotic stress. Plant Cell Environ. 31, 1442–1459. [DOI] [PubMed] [Google Scholar]
- Sheveleva, E. , Chmara, W. , Bohnert, H.J. and Jensen, R.G. (1997) Increased salt and drought tolerance by D‐ononitol production in transgenic Nicotiana tabacum L. Plant Physiol. 115, 1211–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinozaki, K. and Yamaguchi‐Shinozaki, K. (2007) Gene networks involved in drought stress response and tolerance. J. Exp. Bot. 58, 221–227. [DOI] [PubMed] [Google Scholar]
- Sinclair, T.R. (2017) Soybean In Water‐Conservations Traits to Increase Crop Yields in Water‐Deficit Environments: Case Studies (Sinclair T.R., ed.). Switzerland: Springer. [Google Scholar]
- Skirycz, A. , de Bodt, S. , Obata, T. et al . (2010) Developmental stage specificity and the role of mitochondrial metabolism in the response of arabidopsis leaves to prolonged mild osmotic stress. Plant Physiol. 152, 226–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura, K. , Stecher, G. , Peterson, D. , Filipski, A. and Kumar, S. (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todaka, D. , Zhao, Y. , Yoshida, T. et al . (2017) Temporal and spatial changes in gene expression, metabolite accumulation and phytohormone content in rice seedlings grown under drought stress conditions. Plant J. 90, 61–78. [DOI] [PubMed] [Google Scholar]
- Turgeon, R. and Wolf, S. (2009) Phloem transport: cellular pathways and molecular trafficking. Annu. Rev. Plant Biol. 60, 207–221. [DOI] [PubMed] [Google Scholar]
- Urano, K. , Maruyama, K. , Ogata, Y. et al . (2009) Characterization of the ABA‐regulated global responses to dehydration in Arabidopsis by metabolomics. Plant J. 57, 1065–1078. [DOI] [PubMed] [Google Scholar]
- Urano, K. , Maruyama, K. , Jikumaru, Y. , Kamiya, Y. , Yamaguchi‐Shinozaki, K. and Shinozaki, K. (2017) Analysis of plant hormone profiles in response to moderate dehydration stress. Plant J. 90, 17–36. [DOI] [PubMed] [Google Scholar]
- Varshney, R.K. , Bansal, K.C. , Aggarwal, P.K. , Datta, S.K. and Craufurd, P.Q. (2011) Agricultural biotechnology for crop improvement in a variable climate: hope or hype? Trends Plant Sci. 16, 363–371. [DOI] [PubMed] [Google Scholar]
- Vernon, D.M. , Tarczynski, M.C. , Jensen, R.G. and Bohnert, H.J. (1993) Cyclitol production in transgenic tobacco. Plant J. 4, 199–205. [Google Scholar]
- Verslues, P.E. and Juenger, T.E. (2011) Drought, metabolites, and Arabidopsis natural variation: a promising combination for understanding adaptation to water‐limited environments. Curr. Opin. Plant Biol. 14, 240–245. [DOI] [PubMed] [Google Scholar]
- Vyroubalová, Š. , Václavíková, K. , Turečková, V. , Novák, O. , Šmehilová, M. , Hluska, T. , Ohnoutková, L. , Frébort, I. and Galuszka, P. (2009) Characterization of new maize genes putatively involved in cytokinin metabolism and their expression during osmotic stress in relation to cytokinin levels. Plant Physiol. 151, 433–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner, J.J. , Peterson, F.C. , Volkman, B.F. and Cutler, S.R. (2010) Structural and functional insights into core ABA signaling. Curr. Opin. Plant Biol. 13, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler, T. and von Braun, J. (2013) Climate change impacts on global food security. Science, 341, 508–513. [DOI] [PubMed] [Google Scholar]
- Yamaguchi‐Shinozaki, K. and Shinozaki, K. (2006) Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Annu. Rev. Plant Biol. 57, 781–803. [DOI] [PubMed] [Google Scholar]
- Zhao, C. , Liu, B. , Piao, S. et al . (2017) Temperature increase reduces global yields of major crops in four independent estimates. Proc. Natl Acad. Sci. USA, 114, 9326–9331. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Expression of genes encoding dehydrin/LEA proteins.
Figure S2. Expression of genes encoding photosynthesis‐related proteins.
Figure S3. Phylogenetic tree of SAM‐MTases.
Figure S4. Nucleotide sequences of PcIMT1 and McIMT1.
Figure S5. Expression of SAM‐MTase genes.
Figure S6. Soybean seedlings 1 day (Re1), 2 days (Re2), 3 days (Re3), and 4 days (Re4) after rehydration treatment.
Table S1. Metabolite levels in various soybean organs.
Table S2. Phytohormone levels in various soybean organs.
Table S3. Transcript levels in various soybean organs.
Table S4. Genes encoding representative SAM‐MTases.
Table S5. Genes encoding SAM‐MTases in soybean.
Table S6. Genes encoding representative NCEDs.
Table S7. Primers used for RT‐qPCR.
Data Availability Statement
All relevant data can be found within the manuscript and its supporting materials.