

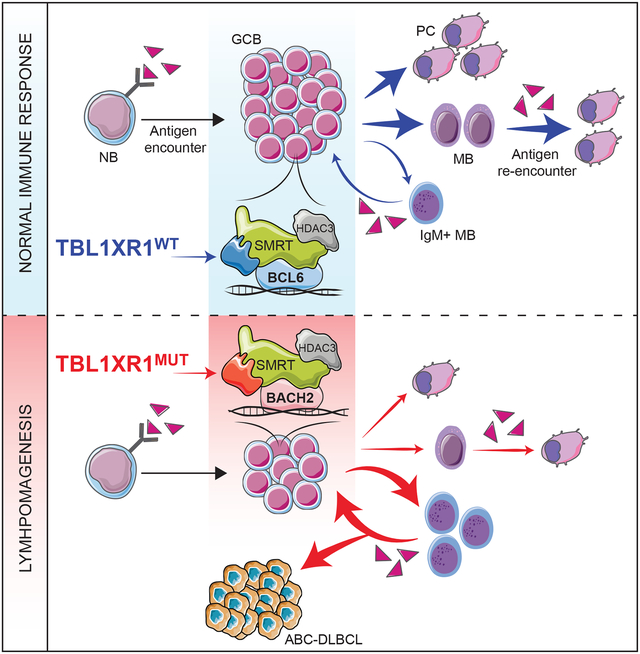
SUMMARY
The most aggressive B-cell lymphomas frequently manifest extranodal distribution, and carry somatic mutations in the poorly-characterized gene TBL1XR1. Here, we show that TBL1XR1 mutations skew the humoral immune response towards generating abnormal immature memory B-cells (MB), while impairing plasma cell differentiation. At the molecular level, TBL1XR1 mutants co-opt SMRT/HDAC3 repressor complexes towards binding the MB cell transcription factor (TF) BACH2, at the expense of the germinal center (GC) TF BCL6, leading to pre-memory transcriptional reprogramming and cell-fate bias. Upon antigen recall, TBL1XR1 mutant MB cells fail to differentiate into plasma cells, and instead preferentially reenter new GC reactions, providing evidence for a cyclic reentry lymphomagenesis mechanism. Ultimately, TBL1XR1 alterations lead to a striking extranodal immunoblastic lymphoma phenotype that mimics the human disease. Both human and murine lymphomas feature expanded MB-like cell populations, consistent with a MB-cell origin and delineating an unforeseen pathway for malignant transformation of the immune system.
In brief
A subset of B cell lymphomas are driven by mutations that impair plasma cell differentiaton and instead bias cell fate toward immature memory B cells, which preferentially re-enter germinal center reactions to drive lymphomagenesis
Graphical Abstract
INTRODUCTION
Diffuse large B-cell lymphomas (DLBCLs) are the most common class of lymphomas (Beham-Schmid, 2017), and likely represent a heterogeneous group of diseases arising from different immune system processes. Gene expression profiling studies classified DLBCLs into two major subtypes: Germinal Center B-cell (GCB) and Activated B-cell (ABC)-DLBCLs. GCB-DLBCLs reflect the transcriptional signature of B-cells that are transiting through the germinal center (GC) reaction (Wright et al., 2003). GCs form transiently in response to T-cell dependent antigens, and are composed of rapidly proliferating B-cells that simultaneously undergo immunoglobulin somatic hypermutation (SHM). After transiting to a T-cell rich GC “light zone”, GCB expressing the highest affinity immunoglobulins undergo plasma cell (PC) differentiation, while others transition to forming memory B-cells (reviewed in (Cyster and Allen, 2019)). ABC-DLCBLs are described as manifesting a “post-GC” transcriptional signature, and evidence of chronic active B-cell receptor signaling (BCR) and NF-kB activation, linked to somatic mutations in the BCR and toll-like receptor (TLR) signaling pathways (Phelan et al., 2018; Wright et al., 2003). However, the cell of origin (COO) and mechanisms of malignant transformation of ABC-DLBCLs remain poorly understood. From a clinical perspective this is problematic, since these are among the most incurable and aggressive lymphomas (Beham-Schmid, 2017).
DLBCLs were more recently classified into putative distinct entities based on constellations of genetic lesions (Chapuy et al., 2018; Reddy et al., 2017; Schmitz et al., 2018). Two independent studies identified a novel genetically defined ABC-DLBCL subtype (“MCD” or “Cluster 5 (C5)” lymphomas) associated with unfavorable clinical outcomes, somatic activating mutations of MYD88, CD79B, and recurrent mutation of the poorly characterized gene TBL1XR1 (Chapuy et al., 2018; Schmitz et al., 2018). TBL1XR1 alleles are likely founder mutations, based on variant allele frequency (Chapuy et al., 2018). From the clinical and biological perspective, a striking feature of these lymphomas is their unusual extranodal distribution (Chapuy et al., 2018; Schmitz et al., 2018), invading sites like the central nervous system and testes (Chapuy et al., 2016; Gonzalez-Aguilar et al., 2012). Neither GCB nor PC normally home to these tissues, raising questions about the pathogenesis and origin of these tumors, within the complex milieu of the immune system.
Congenital missense mutations in TBL1XR1 similar to those in lymphoma have also been linked to early-childhood developmental disorders, such as Pierpont Syndrome (Heinen et al., 2016; Laskowski et al., 2016), suggestive of TBL1XR1’s broad biological relevance. From the biochemical standpoint, TBL1XR1 is a core component of the SMRT/NCOR1 transcriptional repressor complexes (Yoon et al., 2003). These complexes also contain HDAC3, which enables the complex to repress transcription (Hatzi et al., 2013). Little is known about the exact role of TBL1XR1, which has been alternatively described as: a) enhancing the functionality (Tomita et al., 2004), or b) driving the disassembly (Perissi et al., 2004) of these complexes. Whichever the case, it is notable that in GCB the majority of SMRT/NCOR1 complexes are bound and recruited by the transcriptional repressor BCL6 (Hatzi et al., 2013), which is essential for GC formation. Indeed, most recurrent mutations in DLBCL directly or indirectly enhance BCL6 functionality (reviewed in (Hatzi and Melnick, 2014)), a driving force on which most of these tumors rely for their survival (Cardenas et al., 2016; Cerchietti et al., 2010). TBL1XR1 mutations are thus highly intriguing, given their potential to affect SMRT/NCOR1 complexes, as well as BCL6, and the complete lack of information on how this might play out from the molecular and biological perspectives. Herein, we explore the mechanism of action of TBL1XR1 mutations, their impact on the humoral immune response, and whether they provide critical insight into the origin and immunological nature of aggressive extranodal MCD/C5 DLBCLs.
RESULTS
Tbl1xr1 mutation impairs germinal center development
TBL1XR1 somatic mutations occur in ~5–10% of DLBCL and follicular lymphoma (FL) cases, mainly as heterozygous missense alleles (Figure 1A). In line with recent studies (Chapuy et al., 2018; Schmitz et al., 2018), our analysis of three independent DLBCL cohorts confirmed that mutations occur more frequently in ABC-DLBCL (Data S1A and Table S1), are highly enriched in the MCD subtype (Data S1B), and often co-occur with MYD88 mutations (Data S1C). TBL1XR1 mutations largely occur within its WD40 domain (Figures 1A–B), and affect aromatic residues exposed on the same surface of the barrel structure (Figure 1B), that are predicted to mediate protein-protein interactions (PPI, (Wang et al., 2015)).
Figure 1. Tbl1xr1 mutation impairs GC development.
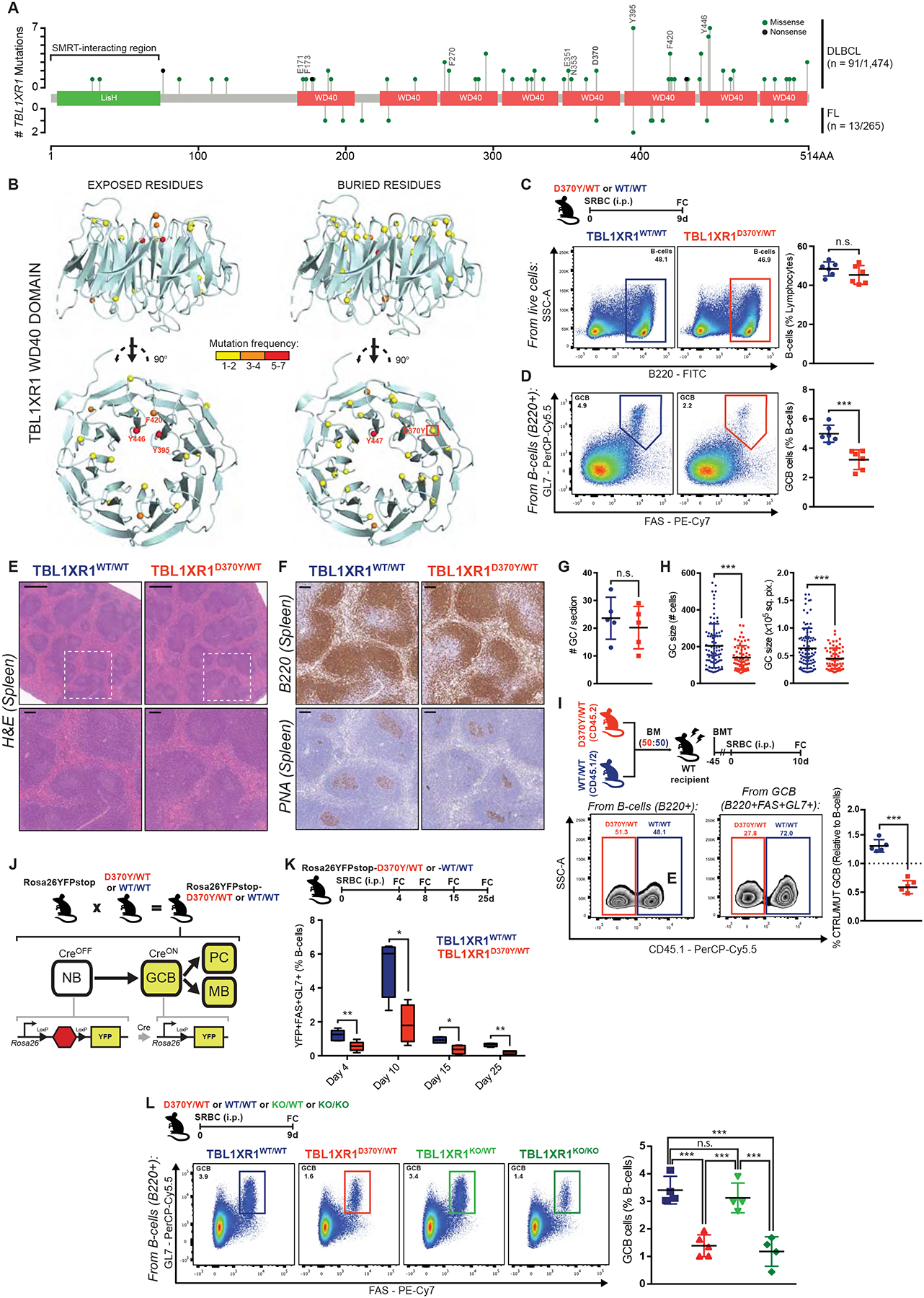
A, TBL1XR1 mutations in DLBCL (Arthur et al., 2018; Ma et al., 2019; Reddy et al., 2017) and FL (Krysiak et al., 2017; Ma et al., 2019; Ortega-Molina et al., 2015). TBL1XR1-SMRT interacting region (Zhang et al., 2002), and PPI mutated positions are indicated. See also Data S1A–C and Table S1.
B, Exposed (>25% accessible surface) or buried residues affected by missense DLBCL mutations in TBL1XR1 WD40 domain (4LG9; DOI: 10.2210/pdb4lg9/pdb).
C-D, FC analysis of splenic (C) total B-cells or (D) GCB. See also Data S1D–F.
E, Spleen sections H&E from animals treated as in C. Insets show zoom of outlined areas. Scale = 500μm (top), 100μm (bottom).
F, B220 or PNA IHC in consecutive spleen sections from E. Scale = 100μm.
G-H, (G) Number of GC per spleen section or (H) GC size as (left) number of cells or (right) area, based on PNA staining. Dots represent individual (G) animals or (H) GCs. Results for 5 animals per genotype.
I, FC analysis of D370Y/WT and WT/WT relative contribution to total B-cells and GCB, based on CD45 allelic frequencies. See also Data S1H–J.
J, Use of the Rosa26YFPstop reporter.
K, FC analysis of splenic GCB. Left to right: n = 4, 5, 5, 4 per genotype. See also Data S1K–L.
L, FC analysis of splenic GCB.
Values represent mean ± SEM. Data reproducible with three repeats. NS, not significant; *P < 0.05; **P < 0.01; ***P < 0.001, using unpaired (C,D,K) or paired (I) two-tailed Student’s t-test; or Mann-Whitney U-test (G,H); or one-way ANOVA with Tukey’s post-test (L).
Faced with the lack of a clear hotspot, we decided to model the D370Y allele in mice given it occurs both in lymphomas and congenital developmental disorders (Laskowski et al., 2016), suggestive of its biological relevance. The Tbl1xr1 locus was engineered for conditional expression of D370Y (Figure S1A–B). Since TBL1XR1MUT lymphomas are derived from GC or post-GC B-cells (Chapuy et al., 2018), we crossed Tbl1xr1D370Y mice to the Cγ1Cre strain (Casola et al., 2006), to restrict D370Y expression to (pre)GCB and GC-derived cells. Sorted GCB manifested the correct Tbl1xr1 genotype (Figure S1C), and showed no difference in overall Tbl1xr1 mRNA or protein abundance (Figures S1D–E).
Given a previous report suggesting that TBL1XR1 might limit SMRT functionality (Perissi et al., 2004), we hypothesized that TBL1XR1MUT would further enhance the activity of BCL6-SMRT complexes. We immunized Tbl1xr1D370Y/WT (D370Y/WT) or Tbl1xr1WT/WT (WT) mice with the T-cell dependent antigen sheep red blood cells (SRBC), and sacrificed them at the peak of the GC reaction. Unexpectedly, while B-cell abundance remained unaltered in D370Y/WT mice (Figure 1C and Data S1D–E), these showed significant decrease in the absolute number (p value = 0.0008; Data S1E) and proportion (p value = 0.0006; Figure 1D) of FAS+GL7+ GCB. Similar results were observed gating GCB as FAS+CD38− (Data S1F). Animals showed no disruption of splenic architecture (Figure 1E). Immunohistochemistry (IHC) staining for B-cells (B220) and GCB (peanut agglutinin, PNA) revealed normal follicular structures with smaller GCs in D370Y/WT mice, but conserved GC numbers per spleen (Figures 1F–H). Dark zone (DZ) centroblasts (CB) and light zone (LZ) centrocytes (CC) ratios were similar to WT (Data S1G).
Although D370Y/WT GC were smaller, it is possible that these GCB could be fitter when in direct competition. We performed mixed chimera experiments where equal numbers of Cd45.2;Tbl1xr1D370Y/WT or Cd45.1/2;Tbl1xr1WT/WT bone marrow (BM) cells were transplanted into lethally irradiated syngeneic recipients. Following immunization, the relative proportions of Cd45.1 and Cd45.1/2 B220+ cells were roughly equal, but D370Y/WT GCB manifested significant disadvantage compared to WT (Figure 1I), regardless of the initial ratio of transferred cells (Data S1H). Immunization with hapten-protein conjugates yielded similar results (Data S1I). Observations were also not dependent on the Cd45 allelic variant, as identical results were obtained with Cd45.1;Tbl1xr1D370Y/WT (Data S1J).
To study the possibility of a premature/delayed GC burst as explanation for the reduced GC size, we crossed D370Y/WT mice to include a Rosa26lox-stop-loxYFP (Rosa26YFP) allele, which reports on Cre activity (Figure 1J and Data S1K). D370Y/WT mice presented reduced GCB abundance at all timepoints (Figure 1K and Data S1L), and GCs completely resolved by day 25. Collectively, these data indicate that Tbl1xr1 mutation impairs, rather than enhances, the GC reaction (perhaps by hindering BCL6 function), while maintaining normal kinetics.
Tbl1xr1 mutations phenocopy TBL1XR1 complete loss
The absence of a clear hotspot suggests that TBL1XR1 mutations behave as loss of function (LOF). Along these lines, focal TBL1XR1 deletions are also detected in DLBCL patients (Data S1M; (Schmitz et al., 2018)), albeit at lower frequency than missense mutations. We then generated a Cγ1Cre-inducible Tbl1xr1 knockout mouse (Figures S1F–I), and again observed that while total B-cell abundance was unchanged (Figures S1J and S1L), there was significant GCB reduction in immunized Tbl1xr1KO/KO (KO/KO) mice (Figures S1K–L and Data S1N). There was no disruption of splenic architecture or lymphoid follicles (Figure S1M), although there was significant reduction in GC size, but not GC number (Figures S1N–P). Mixed chimera experiments confirmed that KO/KO GCB are at disadvantage against WT (Figure S1Q). The kinetics of the GC reaction in KO/KO mice were similar to WT, but were again consistently hypoplastic (Figure S1R). Finally, KO/KO GCs showed normal ratios of LZ-DZ GCB (Data S1O).
The fact that that KO/KO phenocopies D370Y/WT, supports the idea that Tbl1xr1 mutations confer LOF, and suggests mutations may have a dominant negative (DN) character. Along these lines, GC formation appeared normal in heterozygous Tbl1xr1KO/WT mice (Figure 1L), and did not recapitulate the phenotype induced by D370Y/WT, which instead was virtually identical to KO/KO. This strongly suggests that TBL1XR1 mutants function as DN LOF alleles.
Tbl1xr1 mutation impairs GC proliferation
Apoptosis is prevalent in the GC, with up to half GCBs dying every 6h (Mayer et al., 2017). D370Y/WT manifested similar frequency of apoptotic/dead cells to WT GCB, by AnnexinV/DAPI FC (Figure 2A and Data S1P) or TUNEL staining (Figure 2B). Similar findings were observed for KO/KO (Figure S2A). On the other hand, there was significant reduction in D370Y/WT GCB positive for the proliferation marker PCNA (Figure 2C). Abundance of T follicular cells (TFH), which express CD40 ligand and induce GCB proliferation, was comparable in mutant and WT mice (Figure S2B). To assess for intrinsic defects in response to CD40 stimulation, we performed ex vivo activation of mixed D370Y/WT and WT naïve B (NB)-cells in organoid cultures (Beguelin et al., 2017) (Figure S2C). D370Y/WT and WT B-cells acquired GC surface markers to approximately the same extent (Figure S2D), without significant differences in proliferation rate (Figure S2E). Hence, proliferation of D370Y/WT B-cells is not impaired when given unrestricted CD40 stimulation.
Figure 2. Tbl1xr1 mutation impairs GC proliferation.
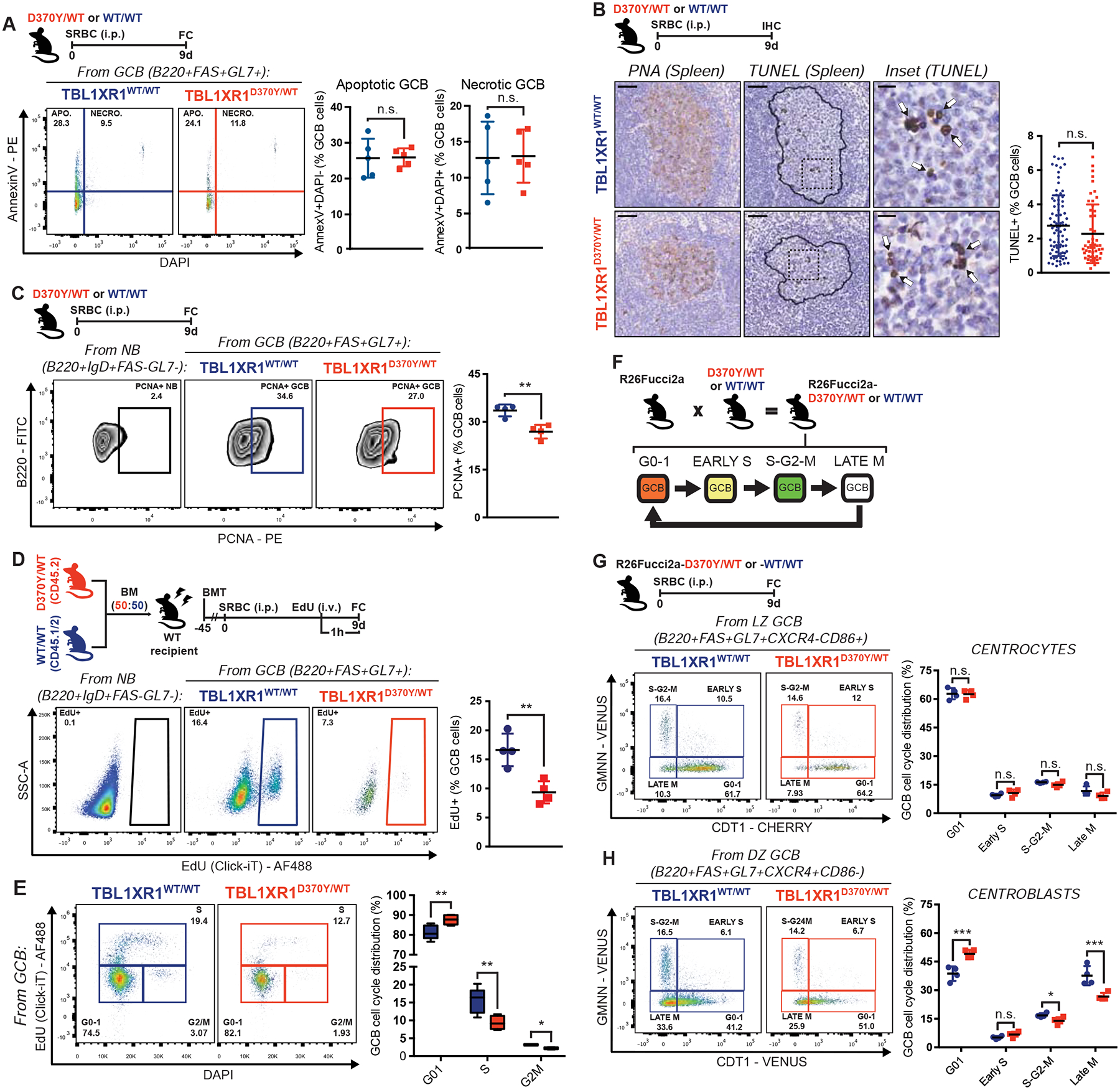
A, FC analysis of AnnexinV/DAPI staining of splenic GCB. See also Data S1P.
B, IHC staining in consecutive spleen sections. Contour delineates GC area based on PNA stain. Insets show zoom of outlined areas. Arrows point representative TUNEL+ GCB. Graph shows frequency of TUNEL+ GCB per GC, where each dot is a GC. Results for 5 animals per genotype. Scale = 50μm (left and center), 10μm (right).
C, FC analysis of splenic PCNA+ GCB. NB from a WT mouse illustrate non-proliferating cells.
D, FC analysis of EdU incorporation by splenic GCB. NB from a WT mouse illustrate non-proliferating cells.
E, FC analysis of splenic GCB cell cycle in D, based on EdU/DAPI staining. Data for n=5 per genotype.
F, Use of the R26-Fucci2aR reporter.
G-H, FC analysis of cell cycle distribution of (G) CC or (H) CB. See also Data S1Q.
Values represent mean ± SEM. Data reproducible with two repeats. P-values calculated using unpaired (A,C,D,G,H) or paired (D,E) two-tailed Student’s t-test; or Mann-Whitney U-test (B).
To assess cell cycle kinetics, we tested EdU incorporation in immunized D370Y/WT or WT mice. In line with PCNA staining, EdU+ GCB were significantly depleted in D370Y/WT (Figure 2D), with a significant increase in GCB at the G0–1 stage, and reciprocal reduction in S and G2/M (Figure 2E). Similar results were observed for KO/KO (Figures S2F–G). As an orthogonal approach, D370Y/WT animals were crossed with R26-Fucci2aR reporter mice (Figure 2F and Data S1Q). In line with previous studies (Stewart et al., 2018), most CC in WT mice were at G0–1, and D370Y/WT had no significant impact on cell cycle distribution (Figure 2G). As expected, WT CB showed high abundance of G2/M cells and fewer cells in G0–1 (Figure 2H). In contrast, D370Y/WT CB manifested significant reduction in the proportion of G2/M and late M cells, as well as increase in G0–1 (Figure 2H). BCL6 represses cell cycle checkpoint genes in GCB (Hatzi and Melnick, 2014), but D370Y/WT or KO/KO CC showed no induction of these genes (Figure S2H), suggesting that proliferation effects are driven by a different mechanism. Impairment in proliferation largely explains the reduced abundance of GCB in Tbl1xr1 mutant and KO/KO mice.
Tbl1xr1 mutation induces ABC-DLBCL-like signatures suggestive of antagonism with BCL6
While most DLBCL mutations engineered in mice induce GC hyperplasia (Beguelin et al., 2013; Dominguez et al., 2018; Hashwah et al., 2017; Hatzi et al., 2019), the Tbl1xr1 phenotype was opposite and counter-intuitive as a lymphoma lesion. To gain mechanistic insight, we performed RNA-seq in sorted D370Y/WT GCB. Unsupervised analysis revealed that D370Y/WT induces a distinct transcriptome (Figures S3A–B). Supervised analysis identified a D370Y/WT specific signature (|log2FC|>1.5; FDR<0.05), skewed towards gene de-repression (Figure 3A and Table S2). Pathway analysis revealed enrichment for ABC-DLBCL associated signatures (Figure 3B and Table S2), as well as pathways involved in chemotaxis, immune cytokine, and NF-kB signaling, all of which are repressed in GCB by BCL6-SMRT (Figure 3B). Accordingly, we observed significant enrichment for genes repressed through BCL6-SMRT enhancer binding, genes induced by BCL6 siRNA and CREBBP target genes (CREBBP normally reverses BCL6-SMRT effects in the LZ; (Jiang et al., 2017)) (Figure 3B). This suggests that TBL1XR1 plays a critical role in supporting the function of the BCL6-SMRT complex. Repression of EBI2 (GPR183) and S1PR1 by BCL6 is largely responsible for GCB confinement to lymphoid follicles (Arnon et al., 2013; Pereira et al., 2009), whereas they reactivate in cells exiting the GC. D370Y/WT GCB upregulated Gpr183 and S1pr1 (Figures 3A and S3C–D), raising the possibility that TBL1XR1MUT drive GCB to manifest post-GC phenotypes.
Figure 3. Tbl1xr1 mutation induces preMB expansion.
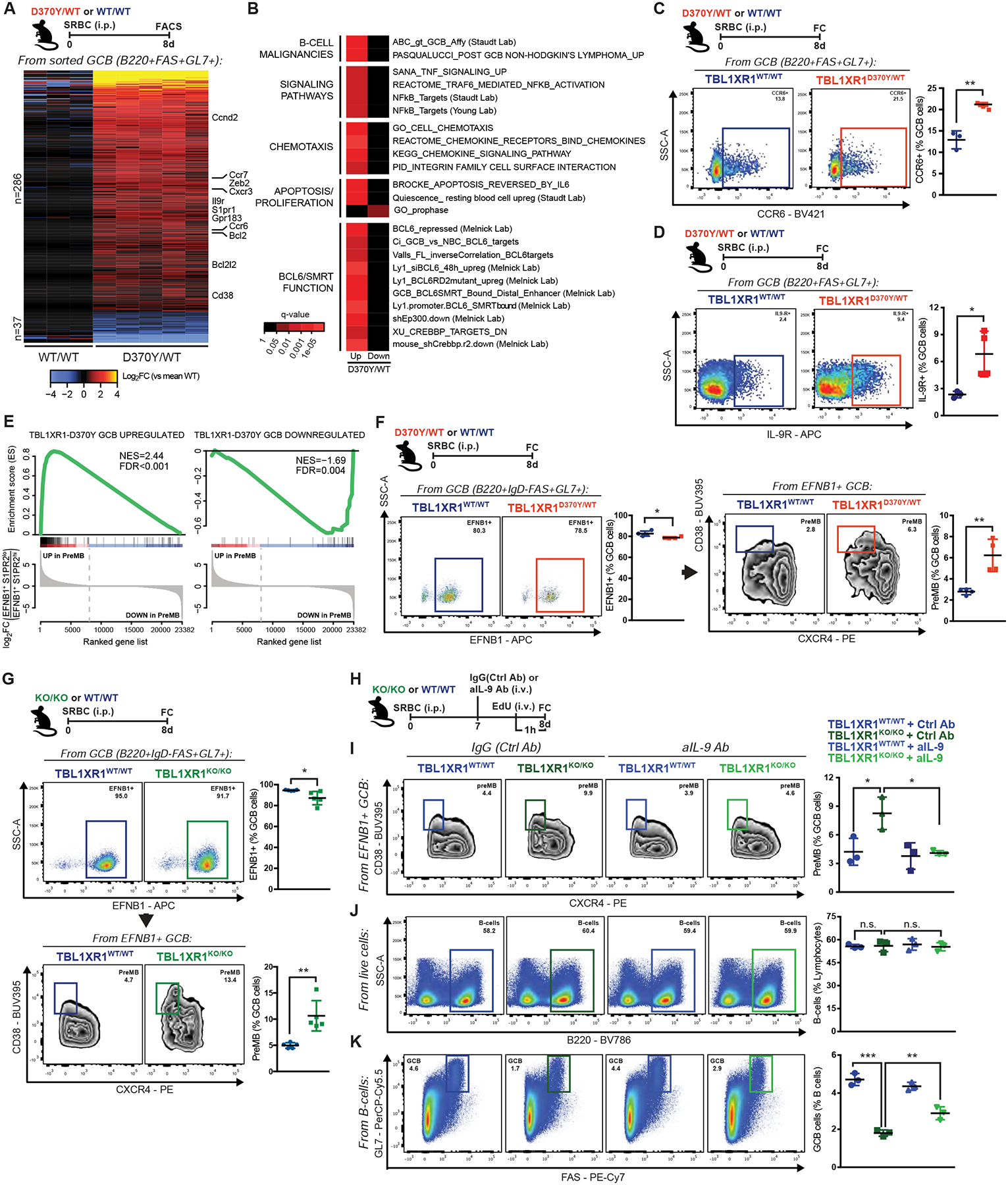
A, Differentially expressed genes in splenic D370Y/WT GCB. Genes of interest are highlighted. See also Figures S3A–C and Table S2.
B, Pathway enrichment for genes in A. See also Table S2.
C-D, FC analysis of (C) CCR6+ or (D) IL-9R+ splenic GCB.
E, GSEA of D370Y/WT GCB against preMB (EFNB1+S1PR2lo) GCB (GSE89897). See also Figures S3G–H.
F-G, FC analysis of EFNB1+ and preMB populations in splenic GCB. See also Figures S3I–K and Data S1R.
H, Experimental scheme and timeline for I-K.
I, FC analysis of preMB in splenic GCB.
J-K, FC analysis of splenic (J) total B-cells or (K) GCB.
Values represent mean ± SEM. Data reproducible with two repeats. P-values calculated using unpaired two-tailed Student’s t-test (C,D,F,G), or one-way ANOVA with Tukey’s post-test (I-K).
Tbl1xr1 mutation induces expansion of the preMB compartment
D370Y/WT GCB showed upregulation of Cd38 (Figures 3A and S3C), a gene which in mice is upregulated in MB, but not in PC (Oliver et al., 1997; Ridderstad and Tarlinton, 1998). Upregulation of CD38 is also a feature of precursor MBs (preMB) in the GC (Laidlaw et al., 2017). PreMB were alternatively defined as LZ GCB with augmented expression of CCR6 (Suan et al., 2017), a receptor upregulated in D370Y/WT and KO/KO GCB (Figures 3A, 3C, S3C and S3E). A third study defined preMB as cell cycle arrested IL-9R+ GCB (Wang et al., 2017b), traits present in D370Y/WT and KO/KO mice (Figures 3A, 3D, S2G, S3C and S3F). Accordingly, we observed enrichment of the D370Y/WT signature among transcriptional profiles of preMB cells in these reports (Figures 3E and S3G–H).
Immunophenotypic analysis of D370Y/WT GCs revealed marked and significant increase in preMB at all studied timepoints (p value=0.0045, Figures 3F and S3I), and also with independent antigens (Figure S3J). PreMB expansion was also seen for KO/KO (Figure 3G and Data S1R), but not for Tbl1xr1KO/WT (Data S1S), supporting that TBL1XR1 mutations act as DN LOF. Given that D370Y/WT and KO/KO GCB showed significant Il9r upregulation, which plays a role in MB formation (Wang et al., 2017b), we tested whether IL-9 blockade would rescue the observed phenotype (Figure 3H). Strikingly, blockade of IL-9R function reverted the KO/KO-driven preMB expansion (Figure 3I). While total B-cells were not affected by IL-9 blockage (Figure 3J), there was partial rescue of GC impairment in KO/KO mice (Figure 3K), concomitant with increased GCB proliferation (Data S1T). Hence, preMB expansion downstream of TBL1XR1 deficiency remains dependent on key MB signaling pathways.
Tbl1xr1 deficiency biases GC fate towards MB cells, and away from the PC lineage
We next explored whether preMB expansion translated into bias towards MB. D370Y/WT GCB transcriptome showed strong enrichment for MB gene signatures, but no enrichment for PC genes (Figures 4A–B; (Luckey et al., 2006)). In agreement, GCB expression of the master PC TF BLIMP1 was not affected by Tbl1×1 mutation or loss (Figures S4A–B). To confirm these findings, we used a mixed chimera approach with Rosa26YFP;Tbl1xr1D370Y/WT vs. Rosa26YFP;Tbl1xr1WT/WT cells, to track GC and post-GC cells (Figure 4C; additional mice were administered CD40-blocking antibodies). D370Y/WT splenocytes revealed a lower percentage of YFP+ cells (Figure 4D). As expected, most YFP+ cells corresponded to GCB (B220+CD138−GL7+FAS+), which were reduced in the D370Y/WT compartment (Figures 4E and S4C, and Data S1U). On the other hand, D370Y/WT cells showed significant increase in MB (B220+CD138-FAS−/loGL7−CD38+IgD−), and a concomitant reduction in plasmablasts (PB; B220+CD138+) and PC (B220−CD138+, Figures 4E and S4C). Experiments with Rosa26YFP;Tbl1xr1KO/KO mice yielded the same phenotype (Figures S4D–E).
Figure 4. Tbl1xr1 mutations bias cell fate towards MB.
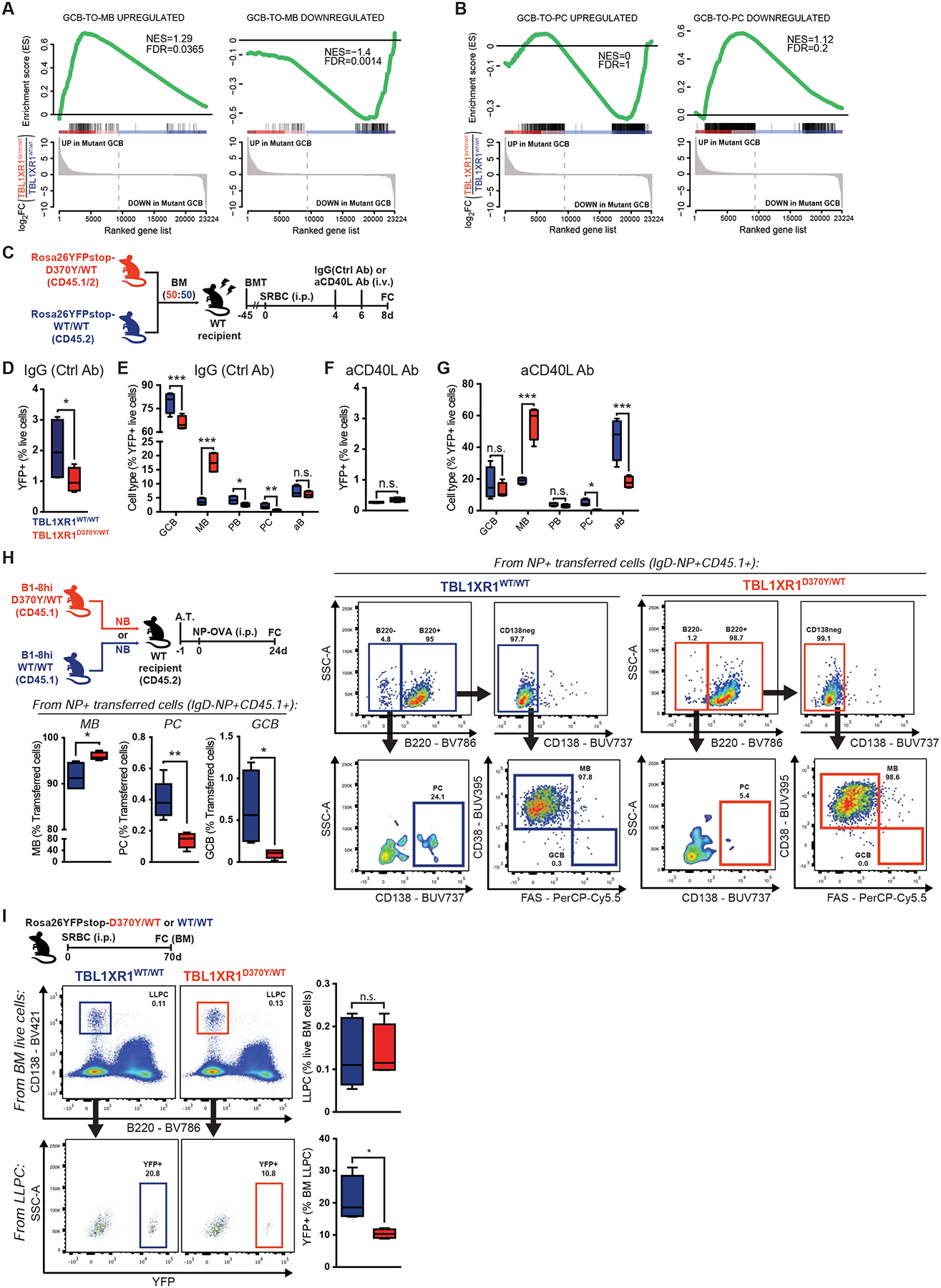
A-B, GSEA of antigen-specific (A) MB or (B) PC, relative to GCB (GSE4142), against D370Y/WT GCB.
C, Experimental scheme and timeline for D-G.
D-E, FC analysis of (D) total YFP+ splenocytes in IgG(Ctrl Ab) treated mice, and (E) the relative cell type composition of this population. Results for 4 animals. See also Figure S4C and Data S1U.
F-G, FC analysis of (F) total YFP+ splenocytes in anti-CD40L treated mice, and (G) the relative cell type composition of this population. Results for 4 animals.
H, FC analysis of cell type composition of antigen-specific splenocytes. Results for 5 animals. See also Data S1V.
I, FC analysis of (top) total or (bottom) GC-derived BM LLPC.
Values represent mean ± SEM. Data reproducible with two repeats. P-values calculated using unpaired (H,I) or paired (D-G) two-tailed Student’s t-test.
GCB with intermediate affinity BCR’s tend to become MB cells after T-cell help, whereas high affinity GCBs receive strong T-cell help, causing GCBs to differentiate into PCs or recycle to the DZ (Shinnakasu et al., 2016). CD40-blocking antibodies in mixed chimeras (Figure 4C) led to profound reduction in GCBs, as expected (Baumjohann et al., 2013), to the point where differences in YFP+ cell abundance among D370Y/WT and WT were lost (Figures 4F and 4G). Reduction of CD40 input drove the majority of the remaining D370Y/WT GC-derived B-cells to the MB compartment. In contrast, the majority of residual WT cells were IgD+ activated B-cells (“aB”), that presumably had not yet entered the GC reaction. No PCs were identified in this setting from D370Y/WT, suggesting that the MB cell fate is hardwired into the mutant B-cells (Figure 4G).
Since PC formation is a rather late event in the GC (Weisel et al., 2016), we evaluated whether commitment bias was also detectable at later time-points. To this end, used an adoptive transfer system to quantify antigen-specific MB and PC 24d after immunization (Figure 4H and Data S1V). Assessment of NP-specific populations, again showed that D370Y/WT biased cell fate towards MB, at the expense of PC (Figure 4H). Finally, assessment of long-lived PC (LLPC) in the BM, 70d after immunization of Rosa26YFP;Tbl1xr1D370Y/WT mice, revealed that while the abundance of total LLPC was unaltered, D370Y/WT had less GC-derived LLPC than WT mice (Figure 4I).
TBL1XR1 mutations cause a BCL6-to-BACH2 molecular switch
TBL1XR1 is a core component of the SMRT/NCOR1 complex, which in GCB is recruited to chromatin by BCL6 (Hatzi et al., 2013). We found that TBL1XR1 is also present in chromatin-bound BCL6 complexes, by RIME-LC-MS/MS (Figure S5A and Table S3). Interaction of endogenous TBL1XR1 with the SMRT/HDAC3 complex and BCL6 was confirmed by Co-IPs (Figure S5B). In other systems, TBL1XR1 can have SMRT-independent functions (Li and Wang, 2008). We therefore performed proximity ligation-mass spectrometry (BioID) for TBL1XR1WT interacting partners in GC-derived cell lines (Figure S5C). The majority of significantly interacting proteins for TBL1XR1WT were restricted to components of the SMRT complex (Figure 5A and Table S3). In addition to these, only BCL6 and another BTB-domain containing TF (BACH2), appeared as significant hits (Figure 5A), largely ruling out alternative functions for TBL1XR1WT in GC derived B-cells.
Figure 5. TBL1XR1 mutations introduce a BCL6-to-BACH2 switch.
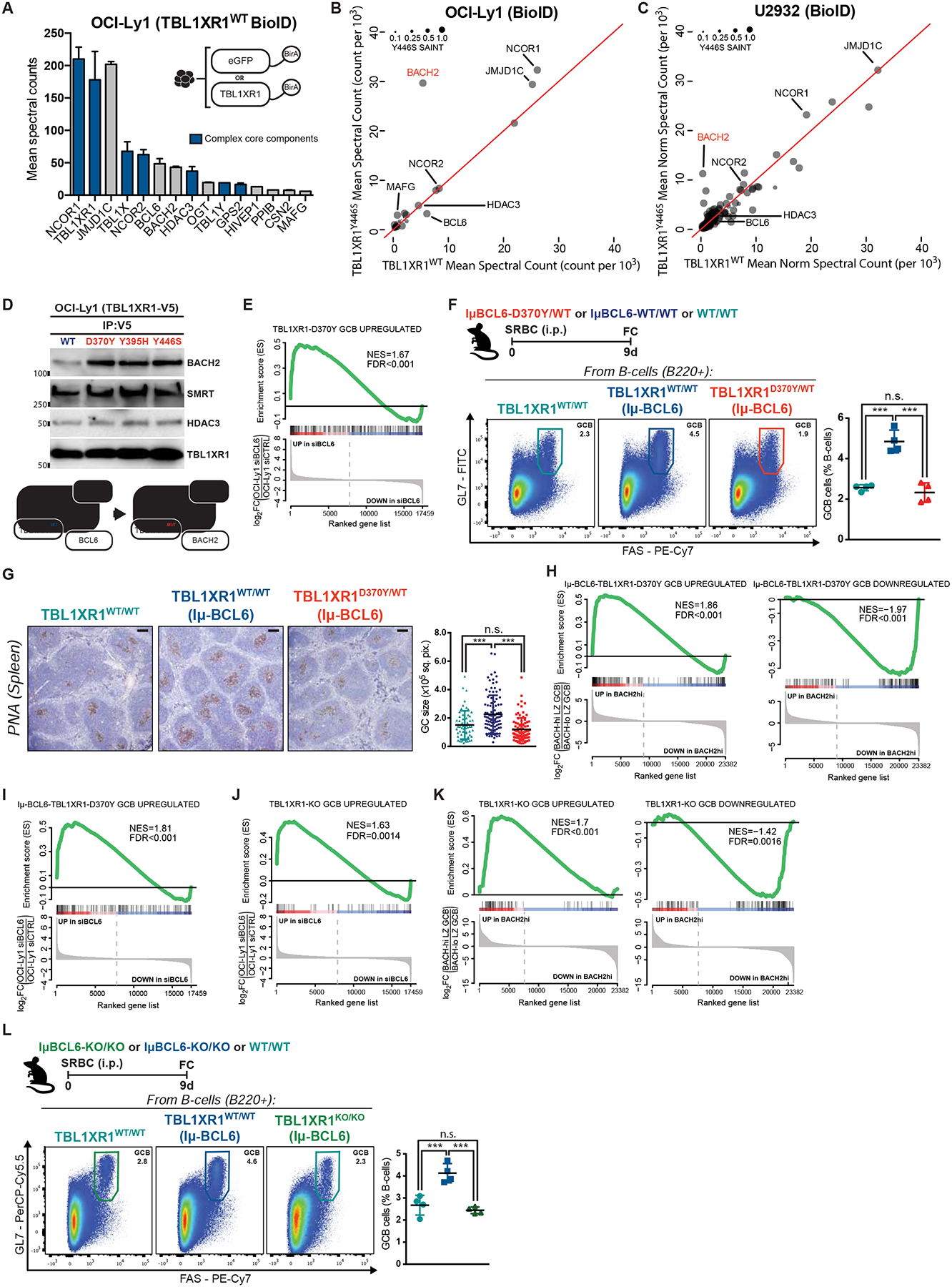
A, TBL1XR1WT interacting proteins. Shown are hits with SAINT>0.75 and 2-fold spectral counts (averaged between biological duplicates) over eGFP-BirA. See also Figure S5C and Table S3.
B-C, Comparison between TBL1XR1 WT and Y446S BioID. Results as average of biological duplicates for each condition.
D, Co-IP results for V5 pulldown in cells inducibly expressing WT or mutant TBL1XR1-V5 fusion proteins. See also Figures S5D–E.
E, GSEA of D370Y/WT GCB against siBCL6-treated OCI-Ly1 (GSE29282).
F, FC analysis of splenic GCB. See also Data S1W.
G, IHC staining of spleen sections from F. Graph shows GC area based on PNA staining. Dots are individual GCs. Results for 4 animals per genotype. Scale = 100μm.
H-I, GSEA of IμBcl6-Tbl1xr1D370Y/WT GCB against (H) BACH2hi GCB (GSE77319) or (I) siBCL6-treated OCI-Ly1.
J-K, GSEA of KO/KO GCB against (J) siBCL6-treated OCI-Ly1 or (K) BACH2hi GCB.
L, FC analysis of splenic GCB.
Values represent mean ± SEM. Data reproducible with two repeats. P-values calculated using one-way ANOVA with Tukey’s post-test (F,L), or Kruskal–Wallis H test with Dunn’s post-test (G).
To assess whether TBL1XR1 mutations alter its interactome, we conducted additional assays modeling one of the most recurrent DLBCL mutations (TBL1XR1Y446S; Figure 1A). Surprisingly, while the interaction with SMRT/HDAC3 complex was preserved, the only significant change in the TBL1XR1MUT interactome was ~6-fold gain of interaction with BACH2, at the expense of BCL6 (Figure 5B). Similar results were obtained in an ABC-DLBCL cell line (Figures 5C and S5C, and Table S3), and in HEK293 cells (not shown). Assessment of three independent TBL1XR1 mutants by Co-IPs confirmed gain of interaction with BACH2 (Figure 5D), and loss of interaction with BCL6 (Figure S5D), without alteration of total protein levels (Figure S5D–E).
In accordance with these data, genes de-repressed in D370Y/WT GCB in vivo, matched those repressed by BCL6 (Figure 5E). Since Tbl1xr1 mutation did not affect BCL6 expression levels (Figure S5F), this suggested interference at the functional level. To test this, we crossed D370Y/WT mice to the IμBcl6 strain, which constitutively expresses BCL6 in GCB, and manifests GC hyperplasia (Cattoretti et al., 2005). Indeed, IμBcl6 mice manifested GC hyperplasia after immunization (Figures 5F–G and Data S1W), which in the presence of the Tbl1xr1D370Y was rescued back to the levels of the WT control (Figures 5F–G). As reported above in a WT background, IμBcl6-Tbl1xr1D370Y/WT GCB manifested a preMB-like transcriptome (Data S1X and Table S4), and actual preMB expansion (Data S1Y). In agreement with our biochemical data, we observed enrichment for a BACH2-driven signature in IμBcl6-Tbl1xr1D370Y/WT GCB (Figure 5H) and significant depletion of the BCL6 program (Figure 5I). BACH2 is reported to be a key TF in MB formation (Shinnakasu et al., 2016), and our data suggest that TBL1XR1 mutations mistarget the SMRT complex to BACH2, thus inducing preferential transcriptional rewiring towards MB, while preventing PC formation by maintaining BACH2 repression of BLIMP1 (Data S1Z; (Huang et al., 2014; Ochiai et al., 2006)).
Transcriptional profiling of KO/KO GCB likewise enriched both preMB and IμBcl6-Tbl1xr1D370Y/WT signatures (Figures S5G–H and Table S4). More importantly, it recapitulated the upregulation of BCL6-targets (Figure 5J), and engagement of BACH2-driven program (Figure 5K). KO/KO did not affect BCL6 expression in GCB (Figure S5I), but was able to rescue BCL6-induced hyperplasia in IμBcl6 GCs (Figure 5L). To assess SMRT/HDAC3 shuttling at the biochemical level, we developed TBL1XR1-KO variants of a GC-derived lymphoma cell line using CRISPR.Cas9 (Figure S5J). Similar to primary GCB, these clones showed impaired proliferation (Figure S5K). Notably, TBLXR1-KO resulted in loss of BCL6-SMRT/HDAC3 interaction, and gain in BACH2-SMRT/HDAC3 Co-IP (Figure S5L). TBL1XR1 is thus critical for the preferential association of SMRT with BCL6 in GCBs, and its LOF induces preferential interaction with BACH2 (Data S1AA), thus explaining the GC impairment and MB cell fate bias.
Tbl1xr1 mutant MB cells manifest traits associated with preferential reentry to the GC reaction
MB reentry into the GC has been proposed as a putative mechanism of GC-derived lymphomagenesis (Sungalee et al., 2014). Given our findings, we postulated that Tbl1xr1 mutation might generate aberrant MBs with capacity for preferential reentry into new GCs. To explore whether D370Y/WT MBs persisted over time, we immunized mice with NP-CGG, and sacrificed long after GC resolution, which revealed long-lasting expansion of NP-specific MB in spleen and BM (Figures 6A–B and Data S1BB–CC). A subset of non-class switched MB cells in humans are proposed to re-seed GCs upon recall, while switched MB differentiate into Ig-secreting cells (Dogan et al., 2009). D370Y/WT and KO/KO GCB showed significant restriction in class-switch recombination (CSR), with relative increase in IgM+ and reduction in IgG1+ cells (Figures 6C and S6A). This was not due to delayed kinetics, since later timepoints showed similar isotype representation (Figure S6B), nor due to an impairment in Aicda induction (Figures S6C–E). The CSR restriction was also evident in GC-derived MB, tracked with the Rosa26YFP reporter (Figures 6D and S6F, and Data S1EE). In accordance with surface Ig profiles, NP-OVA immunized D370Y/WT and KO/KO mice showed impaired IgG1 antibody responses (Figures S6G–H). Still, affinity maturation was not significantly affected (Figures S6I–J), nor were there differences in Ig SHM burden between D370Y/WT and WT GCB (Figure S6K), further suggesting that AID activity was not compromised.
Figure 6. Tbl1xr1 mutant MB cells preferentially become GCB upon recall.
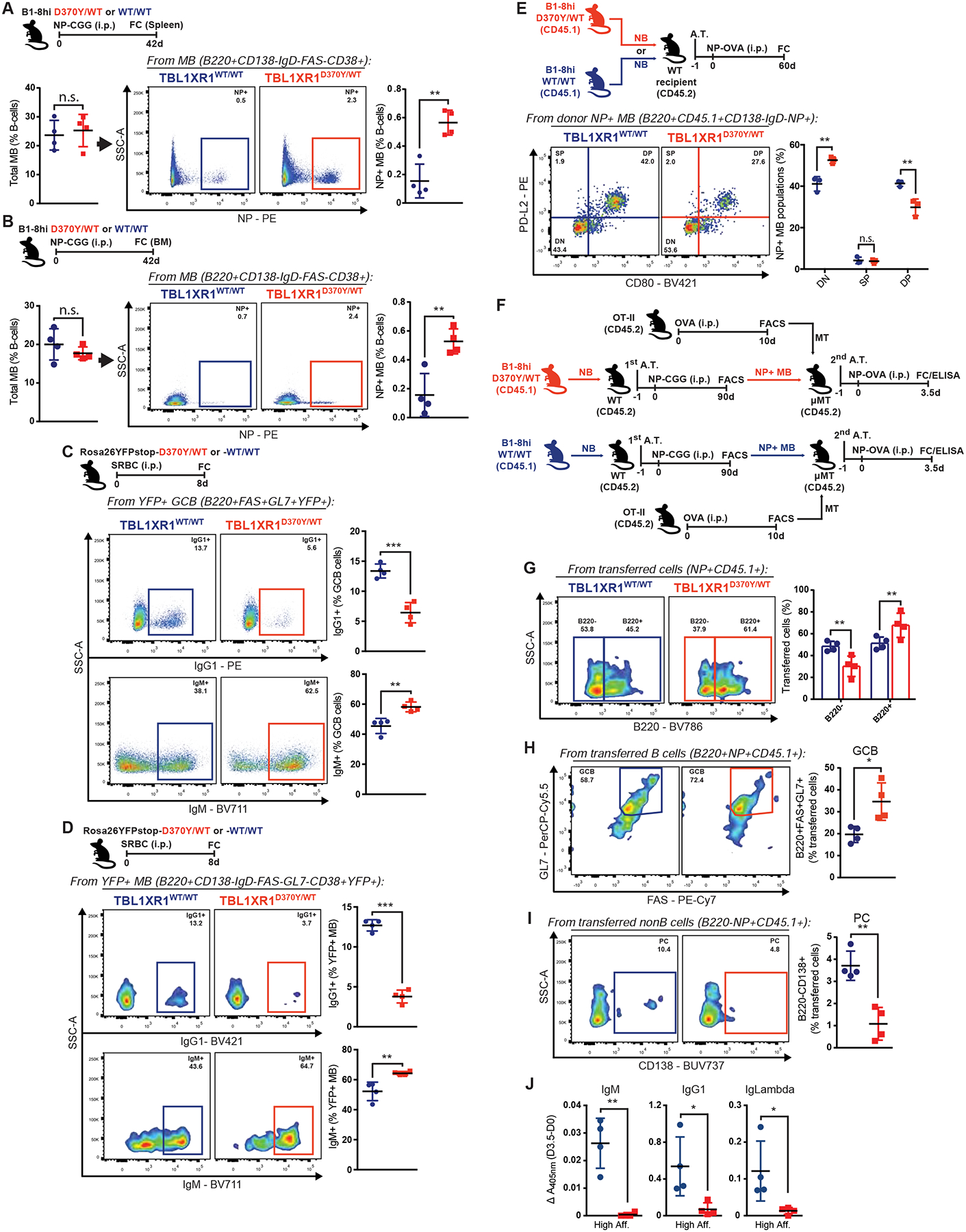
A-B, FC analysis of total or antigen-specific MB population in (A) spleen or (B) BM. See also Data S1BB–CC.
C-D, FC analysis of IgG1+ or IgM+ splenic (C) GCB or (D) MB. See also Figure S6B and Data S1DD.
E, FC profiling of donor-derived antigen-specific splenic MB. See also Figure S6L.
F, Experimental scheme and timeline for G-J.
G-I, FC analysis of (G) total B220+, (H) GCB or (I) PC donor-derived antigen-specific cells, in the spleens of MB-recipient animals. See also Figure S6M.
J, ELISA for NP-specific serum Ig in MB-recipients in F. Graphs show difference between 0 and 3.5d post-immunization.
Values represent mean ± SEM. Data reproducible with two repeats. P-values calculated using unpaired two-tailed Student’s t-test.
In mice, CD80−PDL2− MBs, which are mostly IgM+, show higher capacity for GC reentry, while CD80+PDL2+ MB are generally class-switched, and tend to differentiate into PC (Zuccarino-Catania et al., 2014). Profiling of donor-derived antigen-specific MB cells (CD45.1+CD138−B220+IgD−NP+) from an adoptive transfer system, revealed that D370Y/WT MB were significantly over-represented in the CD80−PDL2− population (Figure 6E). Most D370Y/WT MB were also IgM+, further confirming a CSR restriction (Figure S6L).
Tbl1xr1 mutant MB cells preferentially become GCB-cells upon antigen recall
Results above suggest that TBL1XR1MUT might predispose MB for GC reentry upon recall. To test this, we conducted secondary adoptive transfer experiments (Figure 6F), where sorted D370Y/WT or WT antigen-specific MB were transferred in equal numbers into μMT recipient mice, along with OT-II memory T-cells to boost MB recall (Zuccarino-Catania et al., 2014). μMT mice lack mature B-cells and antigen-specific antibodies (Kitamura et al., 1991), allowing specific detection of those coming from transferred cells. Recall was assessed 3.5d after immunization of recipients, to exclude confounding factors such as differences in GC expansion, that might occur later on. D370Y/WT transferred cells (NP+CD45.1+, Figure S6M) persisted largely as B220+ (Figure 6G), and over 70% of these were FAS+GL7+ (Figure 6H). The relative total abundance of GCB was almost 2-fold higher in animals transferred with mutant MB, than those receiving WT MB (Figure 6H), indicating that D370Y/WT favors MB reentry into new GC reactions. In contrast, ~50% of the WT cells downregulated B220 (Figure 6G), consistent with PC differentiation, as evidenced by CD138 upregulation in some of these (Figure 6I). Terminal differentiation of D370Y/WT MB into PC was practically null at the assessed timepoint (Figure 6I). In agreement, only WT MB lead to a detectable secretory recall response, in the form of high affinity IgG1/Lambda Ig (Figure 6J). These data suggest that TBL1XR1MUT causes aberrant expansion of persisting MB, and provide the first evidence that a lymphoma mutation can drive preferential reentry of putative clonal precursor cells into subsequent immune reactions.
Tbl1xr1 alterations lead to extranodal immunoblastic-like lymphomas
Since TBL1XR1-driven transformation may require GC reentry over an extended period of time, incompatible with mice lifespan (e.g. (Sungalee et al., 2014)) we used CD19cre to increase the frequency of mature B-cells carrying Tbl1xr1 alterations. This did not affect B-cell development (Data S1EE–GG), and recapitulated the Cγ1Cre phenotype (Figures S7A–D). Since C5/MCD carry near-uniform 18q gain and high BCL2 levels (Chapuy et al., 2018; Wright et al., 2020), we further crossed our animals with VavP-Bcl2 mice (Ogilvy et al., 1999), to phenocopy this effect. We generated cohorts of VavP-Bcl2;CD19Cre;Tbl1xr1KO/KO and VavP-Bcl2;CD19Cre;Tbl1xr1WT/WT mice, and immunized them periodically to make sure they generated GCs (Figure 7A). Mice were sacrificed at a timepoint when VavP-Bcl2 animals show pre-tumoral lymphoproliferation (Egle et al., 2004). While all KO/KO animals had developed macroscopic tumors at necropsy, most with extranodal localization such as kidney, lung, liver and intestines (Figure 7B), no masses were found in controls. Splenic hyperplasia was evident in all animals, but was exacerbated in KO/KO (Figure 7C). Splenocytes in the control group had the expected representation of T-cells (CD3+) and B-cells (B220+) (Figure 7D), but a massive overrepresentation of GCB (Figure 7E). In contrast, KO/KO manifested an expansion of B220− cells (Figure 7D). The few remaining B220+ KO/KO cells lacked GCB markers for the most part, but showed relative expansion of (pre)MB populations (Figures 7F–G and Data S1HH–II). B220− KO/KO cells, in turn, expressed higher, albeit variable, levels of CD138 than the controls (Figure 7H). Both groups showed comparable levels of mature B-cells in their BM, suggesting the phenotype was not linked to an early-stage developmental impairment (Data S1JJ–KK).
Figure 7. Tbl1xr1 alterations lead to extranodal immunoblastic-like lymphomas.
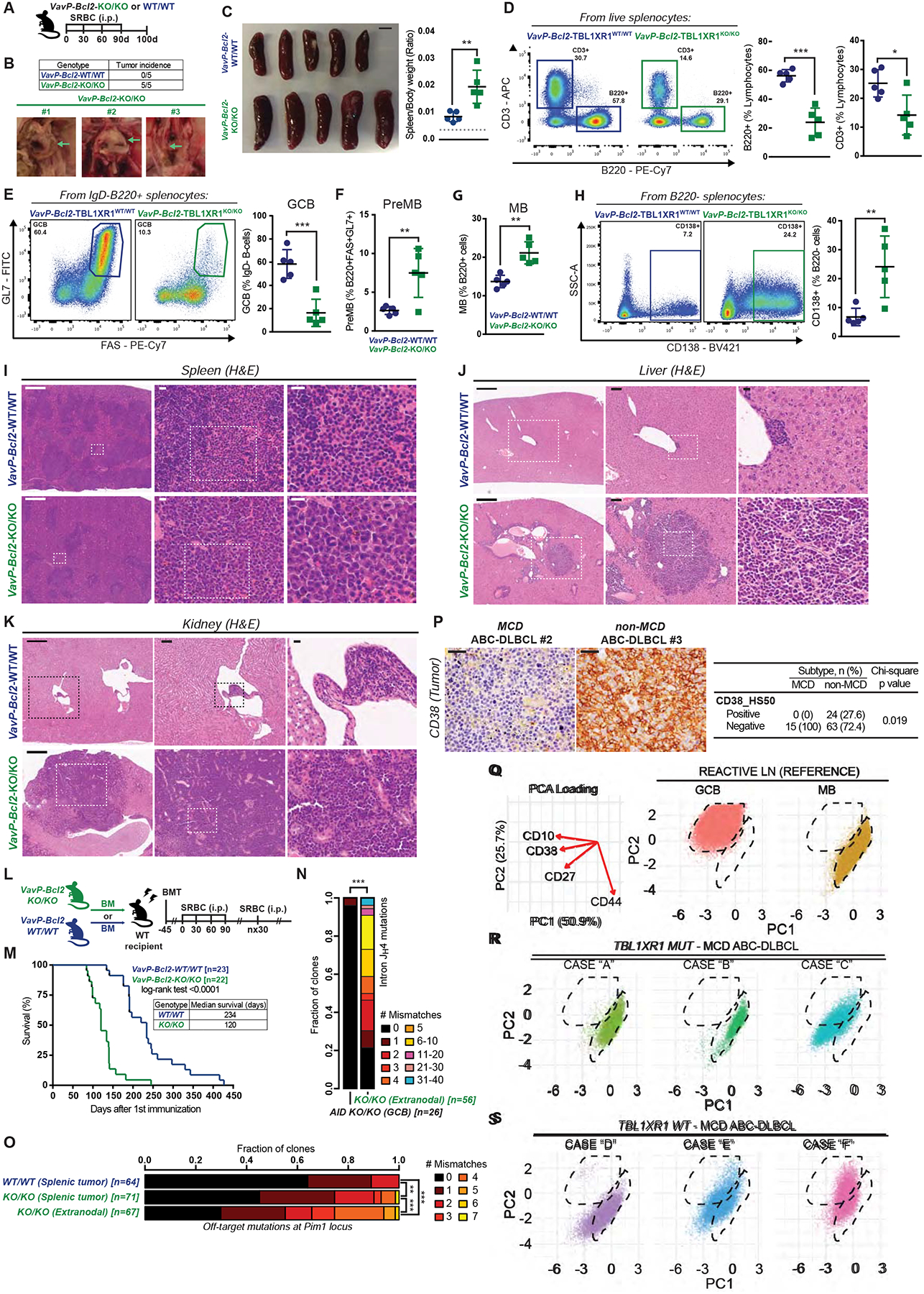
A, Experimental scheme and timeline for B-K and N-O.
B, Incidence and representative images of macroscopic tumors found in VavP-Bcl2-CD19Cre-Tbl1xr1KO/KO mice. Left to right: tumors associated to intestine, lungs and para-aortic tissues. Numbers represent different animals.
C, Spleens at time of necropsy; scale = 500mm. Graph shows spleen to body weight ratios. Dashed line represents average for non-VavP-Bcl2 age-matched controls (n=10).
D-H, FC analysis of splenic (D) B/T-cell populations, (E) GCB, (F) preMB, or (G) MB (B220+CD138−IgD−FAS−/loGL7−CD38+), or (H) CD138+ cells. See also Data S1HH–II.
I, H&E of spleen sections from animals in A. Scale = 500μm (left) or 15μm (center and right). See also Figure S7M.
J-K, H&E of (J) liver or (K) kidney sections from animals in A. Scale = 500μm (left), 100μm (center), 15μm (right).
L, Experimental scheme and timeline for M.
M, Survival curve for VavP-Bcl2;CD19Cre-Tbl1xr1KO/KO and VavP-Bcl2;CD19Cre-Tbl1xr1WT/WT mice.
N, SHM burden at Ig intron JH4 in extranodal tumors from A. Numbers in square brackets are total numbers of clones, pooled for 3 animals. AicdaKO/KO GCB were used as control.
O, Mutation burden at Pim1 locus in tumors from A. Numbers in square brackets represent total clones, pooled for n=5 per genotype.
P, Representative images and quantification of CD38 IHC for MCD/C5 and non-MCD/C5 ABC-DLBCL human specimens. Scale = 20μm.
Q, (Left) Surface marker contribution to PCA loading and (right) PCA plots for MB and GC-B from reactive LN, used as reference in CyTOF experiments.
R-S, PCA for CyTOF profiling of human DLBCL specimens carrying canonical MCD/C5 lesions and (R) TBL1XR1 mutations or (S) TBL1XR1 WT alleles. See also Table S5.
Values represent mean ± SEM. Data reproducible with two repeats. P-values calculated using unpaired two-tailed Student’s t-test (D-H), or log-rank test (M), or Wilcoxon rank sum test with (O) or without (N) BH correction, or two-sided Chi-square test (P).
Histological analysis of lymphoid and other tissues in KO/KO mice revealed predominantly large and highly atypical immunoblasts, often with large and irregularly shaped nuclei and a moderate amount of cytoplasm, mimicking the appearance of human extranodal ABC-DLBCLs (Figure S7E). In the spleen, these cells were seen predominantly outside the follicles in the red pulp, disrupting the splenic architecture (Figure 7I). These cells also extensively infiltrated and distorted many tissues including liver (Figure 7J) and kidneys (Figure 7K). Consistent with this extranodal scenario, lymph nodes (LN) were only focally involved. Immunoblastic cells in KO/KO specimens were virtually all Ki67+, and many stained for CD138 (Figures S7F–G), consistent with the notion that TBL1XR1 loss results in a post-GC lymphoma phenotype. In comparison, the overall architecture of the liver, spleen, kidneys and lung was relatively intact for Tbl1xr1WT/WT (Figures 7I–K), although LNs showed variable sinusoidal expansion by smaller infiltrating lymphocytes. Analysis of tissues yielded sporadic, small, focal lymphocytic infiltrates linked to blood vessels (Figure 7J and 7K) in the LN, spleen and BM that rarely affected tissue architecture (Figure 7I and not shown). IHC revealed infiltrates were composed of T- and B-cells, as well as PC (Figures S7F–G). KI67 stain was low for small plasmacytoid cells/plasma cells and lymphoid cells, and higher in the few larger cells (Figures S7F–G). To assess whether the disease translated into accelerated lethality, we conducted a survival study in an additional cohort of mice, immunized to induce GC formation (Figure 7L). The study revealed that KO/KO mice died of their disease significantly sooner than WT (Figure 7M).
Human C5/MCD DLBCLs show the highest AID footprint (Chapuy et al., 2018), suggesting they originate from GC-transitioned cells. We examined Ig SHM in our KO/KO mouse lymphomas, to exclude that these derived from extrafollicular responses. Sequencing of intron JH4 revealed significant SHM burden in tumors, that was absent in AcidaKO/KO GCB used as controls (Figure 7N). We then performed targeted sequencing of a region in the Pim1 locus frequently impacted by off-target AID activity (Liu et al., 2008), since PIM1 mutations are a hallmark of C5/MCD tumors, and frequently co-occur with TBL1XR1 mutations (Data S1C). Notably, KO/KO tumors accumulated more mutations than VavP-Bcl2 controls (Figure 7O). Strikingly, the highest rate of off-target mutations was detected in extranodal tumors from KO/KO mice (Figure 7O). To further confirm that these lymphomas could arise from GCB, we generated Cγ1Cre;VavP-Bcl2;Tbl1xr1KO/KO mice. These mice again showed extensive extranodal infiltrates, composed of a heterogeneous mixture of lymphocytic and immunoblastic populations (Figure S7H), with higher percentage of B220+ cells than the CD19Cre variant (Figure S7I). Hence, the more homogeneous plasmacytic-like presentation in CD19Cre mice is not required for transformation or extranodal dissemination of malignant B-cells, but might instead represent a later or alternative stage in disease progression. These data are consistent with TBL1XR1 driving MCD/C5 pathogenesis by inducing MB cell reentry into successive GC reactions.
Human MCD/C5 tumors show evidences of MB origin
If MB served as COO for C5/MCD lymphomas, we would expect to find phenotypic evidence in clinical specimens. In humans, CD38 is expressed in GCB and PC, but lost in MB (Arpin et al., 1995). Performing IHC in a DLBCL cohort (Arthur et al., 2018), we observed that while ~40% of all TBL1XR1WT DLBCLs were CD38+, only ~10% of TBL1XR1MUT cases were CD38+ (p=0.03; Figure S7J). We further subclassified (Wright et al., 2020) tumors in this cohort, and observed significant and striking differences between MCD cases (0% CD38+) and non-MCD ABC-DLBCL cases (~30% CD38+) (p=0.01; Figure 7P).
To further support the notion that C5/MCD reflect a MB COO, we conducted CyTOF on patients from an independent DLBCL cohort (Table S6 and (Nissen et al., 2019)). Cells from reactive LN specimens were used to define B-cell subpopulations. In these samples, GCB were identified as CD10hiCD38hi, while MB showed lower levels of these markers, and distinctive CD27 upregulation (Figure 7Q; (Agematsu et al., 2000)). GCB-DLBCL cases, included as controls, mapped to GCB reference populations (not shown). In contrast, ABC-DLBCL tumors carrying TBL1XR1 and MCD/C5 canonical mutations narrowly recapitulate the surface marker profile of MB cells (Figure 7R). The MB profile was also observed in MCD DLBCLs without TBL1XR1 mutations (Figure 7S), consistent with the notion that MCD tumors arise from MB, albeit through distinct mutational trajectories. Collectively, we find that TBL1XR1 LOF drives expansion of aberrant MB cells with enhanced tendency to reenter GC reactions, ultimately resulting in canonical post-GC extranodal ABC-DLBCL with MB-like features, revealing the manner in which these tumors arise naturally from the humoral immune response.
DISCUSSION
Herein, we show how TBL1XR1 mutations result in transcriptional re-wiring of GCB cells to drive lymphomagenesis. A previous report noted TBL1XR1 interaction with BCL6 in a lymphoma cell line, using mass spectrometry (Miles et al., 2005). We observed that TBL1XR1 forms complexes with BCL6 at the chromatin level, and that its interactome is largely confined to the SMRT complex, and the TFs BCL6 and BACH2. In GCB, BCL6-SMRT complexes transiently poise enhancers controlling terminal differentiation, immune synapse signaling and cell cycle checkpoint genes, thus enabling proliferative bursts (Hatzi et al., 2013; Jiang et al., 2017). Accordingly, the GC reaction in Tbl1xr1 LOF mice was significantly impaired, and GCB showed upregulation of BCL6 targets. Whereas Bcl6-KO completely abrogates GC formation (Hatzi and Melnick, 2014), Tbl1xr1 LOF effects are reminiscent of the milder effects observed in mice carrying NCOR/SMRT loss-of-function mutations that disrupt interaction with HDAC3 (Jiang et al., 2017), or Hdac3 conditional deletion (Stengel et al., 2019). The difference in severity between BCL6 vs. TBL1XR1/SMRT complex deficiency phenotype is likely due to SMRT/HDAC3-independent BCL6 functions, such as interaction with BCOR and LSD1 (Hatzi et al., 2019).
TBL1XR1’s lack of enzymatic activity has largely hindered efforts to elucidate its function. Still, one study proposed that TBL1XR1 acts as a repressor/coactivator exchange factor for hormone-receptors (Perissi et al., 2004). In this model, TBL1XR1 recruited UbcH5 and the 19S proteasome, leading to SMRT degradation. This finding was not reproduced by latter studies (Huang et al., 2009; Yoon et al., 2005; Zhang et al., 2006), and our own data show that in B-cells, TBL1XR1MUT had no effect on SMRT protein levels. Furthermore, our analysis of TBL1XR1 interactome failed to identify ubiquitin or proteasome machinery. In agreement with this, acute CD40L signaling (a differentiation cue for GCB) evicts SMRT from chromatin, without affecting its stability (Polo et al., 2008). Rather than altering SMRT turnover, TBL1XR1 mutation or KO instead shift SMRT complex from BCL6 to BACH2, resulting in transcriptional reprogramming.
WD40 domains function as PPI or protein-DNA interaction motifs, mainly through aromatic residues on the surface of the β-propeller (reviewed in (Jain and Pandey, 2018)), Notably, these TBL1XR1 residues are the most frequently mutated in DLBCL. Structural studies show that TBL1XR1 forms tetramers, and binds SMRT through its N-terminal domain (Oberoi et al., 2011; Zhang et al., 2002), which is largely spared by mutations. Hence, WD40 mutations would not be predicted to disrupt TBL1XR1-SMRT interaction. Accordingly, a study on Pierpont Syndrome found that the interaction between TBL1XR1Y446C (a mutant also found in DLBCL) and SMRT/HDAC3, was maintained in vitro (Heinen et al., 2016). The fact that TBL1XR1MUT remains within the SMRT complex, suggests that the WD40 plays a critical role in facilitating preferential association with BCL6, since TBL1XR1 KO yields a similar effect, both from the biological and biochemical standpoints. Importantly, the overall structure of the SMRT complex has not been resolved, but will be necessary to understand how the holo-complex associates with BCL6 in the chromatin context, and why TBL1XR1 LOF switches the conformation of the complex to favor binding to BACH2.
During the humoral immune response, B-cells can transition to MB and PC phenotypes, with implications for formation of clonal precursor cells. Previous studies suggested a role for IL-9R signaling (Wang et al., 2017b), or the relative level of BCR affinity for antigen (Shinnakasu et al., 2016) as factors driving GCB into the (pre)MB compartment. In the case of TBL1XR1 LOF, preMB expansion is caused by switching the SMRT complex to BACH2, and away from BCL6. This situation is different from simple de-repression of BCL6 target genes, which can enable differentiation to either PC or MB cell fate (Diehl et al., 2008; Ise et al., 2018; Kuo et al., 2007; Laidlaw et al., 2017). The reason for this is explained by the critical role of BACH2 in driving B-cells towards the MB lineage (Shinnakasu et al., 2016), and the fact that PC master TF BLIMP1 is repressed both by BCL6 and BACH2 (Ochiai et al., 2006; Tunyaplin et al., 2004). Thus, in the TBL1XR1 LOF setting, BLIMP1 and the PC fate remain suppressed through the actions of BACH2-SMRT complexes. Notably, Il9r is a direct target gene of BLIMP1, and BLIMP1 LOF results in upregulation of Il9r transcripts (Minnich et al., 2016). Furthermore, transcriptional profiling of T-cells from Bach2-KO mice revealed reduced levels of Il9r, and increased levels of Prdm1 (Geng et al., 2019). Accordingly, we found that Il9r plays an important role in mediating TBL1XR1 LOF-induced acquisition of preMB phenotypes. Beyond IL-9R, our findings provide a mechanistic explanation for how a switch to BACH2-driven programs might normally take place during the GC. In this view, somatic mutations of TBL1XR1 simply exaggerate this effect, to lock GC cell fate into the preMB pathway and expand the population of pre-malignant MB cells, to set the stage for post-GC extranodal DLBCL lymphomagenesis.
Mutations affecting TBL1XR1 associate with activating mutations in the BCR and TLR pathways (Chapuy et al., 2018; Schmitz et al., 2018), to define a group of aggressive extranodal post-GC lymphomas. Our finding that Tbl1xr1 mutations disrupt canonical BCL6 transcriptional programs to drive GCB towards a post-GC/preMB stage, are consistent with a MB COO for MCD/C5 DLBCLs. In agreement with this, lymphomas in KO/KO animals recapitulated canonical histological features of the human tumors, such as immunoblastic morphology, diffuse infiltration of extranodal tissues, and sparing of lymphoid follicles (Kashyap et al., 2011). The extrafollicular pattern may be linked to expression of MB-associated migratory chemokines induced by TBL1XR1MUT, as part of the MB cell program. Indeed, the transcriptional profile of D370Y/WT GCB showed enrichment for post-GC derived lymphoma transcriptomes and associated features, such as NF-kB/CD40 activation. An additional canonical MCD/C5 trait observed in our models was restriction in CSR (Lenz et al., 2007), with Tbl1xr1 mutant and KO/KO cells remaining largely as IgM+. The selective preference for IgM expression could reflect possible benefits for the growth of post-GC lymphomas. We hypothesize that the increase in IgM+ GC-derived populations is due to the strong bias introduced by TBL1XR1 alterations towards the generation/expansion of immature MB with high capacity to reenter the GC reaction. Most strikingly, both our Tbl1xr1 deficient murine lymphomas and primary human ABC-DLBCLs manifest an increased fraction of malignant cells with MB phenotype and a heavy burden of AID-induced on-target and off-target SHM. Importantly, this MB phenotype is observed in ABC-DLBCLs beyond those with TBL1XR1 mutations, suggesting that there are additional mutational trajectories that favor this novel MB-cell associated malignant transformation pathway.
Tbl1xr1 mutations not only skew GC output towards MB, but also suppressed formation of Ab-secreting cells upon recall. Along these lines, it is notable that extinction of the BACH2 program is normally required for PC differentiation of MB (Kometani et al., 2013). Persistent BACH2-driven transcriptional programming in TBL1XR1MUT MB could then explain the impairment in forming PC. Most strikingly, TBL1XR1MUT MB cells showed an increased tendency to reenter new GC reactions. Cyclic reentry of BCL2-overexpressing MB into the GC reaction has been proposed to originate FLs (Sungalee et al., 2014). Presumably, these BCL2-overexpressing cells self-sustain and persist for long periods of time. However, we find that TBL1XR1MUT expressly skew GC cell fate to produce the class of MB cells that are most prone to reenter the GC reaction, thus serving as clonal precursor cells to aggressive C5/MCD DLBCLs. The fact that TBL1XR1 mutations in these lymphomas appear as early clonal events (Chapuy et al., 2018), further supports a model where TBL1XR1MUT MB engage in successive rounds of GC reactions, eventually acquiring further hits in other genes (as shown here for Pim1), that further accelerate malignant transformation. This would likely explain the canonical complex phenotype of these tumors, where they manifest an aberrant and heterogenous composite of features reflecting aspects of MB, PB and GCB. These results point to the need for surveillance studies in humans to identify precursor clonal cells at risk for transformation to these aggressive and incurable forms of DLBCL, much in the way current studies are attempting to predict which patients with clonal hematopoiesis of indeterminate prognosis are at risk for development of acute leukemia. The fact that this process is dependent on the SMRT/HDAC3 complex, points to the potential for newly reported selective HDAC3 inhibitors (Mondello et al., 2020) as potential therapeutic agents for these clonal disorders. Perhaps combining such compounds with blockade of IL-9R signaling could further potentiate their activity. The recent discovery that auto-immune disorders like Sjogren’s disease are driven by MB cells harboring canonical MCD mutations (Singh et al., 2020; Wang et al., 2017a) point to the further potential medical impact of such therapeutic interventions.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ari M. Melnick (amm2014@med.cornell.edu).
Materials Availability
Unique reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
RNA sequencing data from this paper have been deposited in the Gene Expression Omnibus (GEO) database, under accession number GSE139059. All raw mass spectrometry files have been deposited at the MassIVE archive (massive.ucsd.edu). Code used for analysis is available upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse models
Animal care was in strict compliance with institutional guidelines established by the Weill Cornell Medical College, the Guide for the Care and Use of Laboratory Animals (National Academy of Sciences 1996), and the Association for Assessment and Accreditation of Laboratory Animal Care International. The Research Animal Resource Center of the Weill Cornell Medical College of Medicine approved all mouse procedures.
The following strains were obtained from The Jackson Laboratory (Ben Harbor, ME, USA): C57Bl/6J (CD45.2, stock 000664), Cγ1-Cre (stock 010611), CD19-Cre (stock 006785), μMT (stock 002288), B6.SJL-PtprcaPepcb/Boy (CD45.1, stock 002014), Rosa26-lox-stop-lox-YFP (stock 006148), OT-II (stock 004194; (Barnden et al., 1998)), R26:FLPe knock in strain (stock 003946) and B1–8hi (stock 007594). The R26-Fucci2aR model (Mort et al., 2014) was developed by I.J. Jackson (University of Edinburgh, Scotland). IμBcl6 mice (Cattoretti et al., 2005) were obtained from R. Dalla-Favera (Columbia University, NY, USA). The VavP-Bcl2 (Ogilvy et al., 1999) model was developed by J.M. Adams (Walter and Eliza Hall Institute of Medical Research, Australia). The Aicda-KO mouse model (Muramatsu et al., 2000) was a generous gift from T. Honjo (Kyoto University Graduate School of Medicine, Kyoto, Japan).
Conditional Tbl1xr1-D370Y mice were generated by inGenious Targeting Laboratory Inc. (Ronkonkoma, NY, USA), using an inversion and deletion approach (Figure S1A). To achieve the conditional activation of the D370Y mutation, WT LoxP and mutant Lox66/Lox71 (RE/LE mutants) sites were used. In brief, the genomic sequence from Tbl1xr1 exon 11 to exon 12, and their flanking sequences, were duplicated and flanked by Lox66 and Lox71 sites. The specific point mutation GAC>TAC (D>Y) was introduced into the duplicated exon 12. Then, the Lox66/71 flanked sequence was inserted into intron 12, in the reverse direction. A 5’ WT LoxP site was inserted upstream of exon 11. An FRT-flanked Neomycin selection cassette was inserted immediately upstream of the Lox66/71 flanked inversion sequence. Embryonic stem (ES) cells were electroporated with this homologous targeting construct, and stable G418 clones were derived. Targeted iTL IC1 (C57Bl/6) ES cells were microinjected into BALB/c blastocysts. Resulting chimeras with a high percentage of black coat were further screened by PCR, and mated to C57Bl/6J R26:FLPe mice, to remove the Neomycin cassette. Founder mice were further back-crossed for at least 10 generations into the C57Bl/6J background.
Conditional Tbl1xr1-KO mice were obtained by microinjection of targeted JMB8A3.N1 Tbl1xr1tm1a(EUCOMM)Hmgu ES cells (clone HEPD0744_3_E01; EUCOMM program) into C57BL/6-albino blastocytes, and implantation into pseudo-pregnant foster C57BL/6-albino recipients (stock 000058; The Jackson Laboratory). Procedures were carried out by the Mouse Genetic - Transgenic Core Facility at Memorial Sloan-Kettering Cancer Center (MSKCC, New York, NY, USA). Resulting chimeras with a high percentage of black coat were further screened by PCR, and mated to C57Bl/6J R26:FLPe mice, to remove the FRT-flanked lacZ-Neomycinresistance cassette. Founder mice were further back-crossed for at least 10 generations into the C57Bl/6J background.
All mouse experiments were conducted using aged and sex-matched specimens. Experiments were designed to include male and female specimens in all groups, and no sex-based influence/bias was detected in the observations made in this work. Unless stated otherwise in the text, all animals were 8 to 12 weeks of age at the time of experimentation.
Cell lines
The DLBCL cell line OCI-Ly1 (CVCL_1879; male origin) was grown in Iscove Modified Dulbecco Media (12440061; ThermoFisher Scientific) , supplemented with 10% FBS and penicillin G/streptomycin; U2932 (CVCL_1896; female origin) cells were grown in Roswell Park Memorial Institute medium (10–040-CV; Corning; Corning, NY, USA), supplemented with 10% FBS, penicillin G/streptomycin, L-glutamine, and HEPES; HEK293T cells (CVCL_0063; female origin) were maintained in Dulbecco’s Modified Eagle Medium (11965–092; ThermoFisher Scientific), supplemented with 10% FBS and penicillin G/streptomycin. Murine 40LB cells ((Nojima et al., 2011); male origin) were grown in DMEM media with 10% FBS. All cells were grown in incubators at 37C, in a 5% CO2 atmosphere. Cell line authentication testing was performed at IDEXX BioResearch (http://www.idexxbioresearch.com/cellcheck), using methods recommended by the American National Standards Institute (ANSI ASN-0002-2011). The cell lines were confirmed to be of human origin and tested for evidence of cross-species contamination (mouse, rat, Chinese hamster and African Green monkey). Short tandem repeat (STR) testing was performed and the genetic profile obtained was compared to the established cell line profile to confirm the cell lines are consistent with the established profile. These cell lines were also routinely tested for Mycoplasma contamination in the laboratory.
Bacteria strains
TOP10, Stbl3 and DH5α E. Coli cells (ThermoFisher Scientific) were cultured in autoclave-sterilized Luria Bertani broth (BP1426-2; Fisher Scientific), in a MaxQ™ 8000 orbital shaker (ThermoFisher Scientific) set at 200rpm and 37°C.
Human subjects
Information on TBL1XR1 mutation status in human lymphoma specimens was retrieved from publicly available datasets. DLBCL cases (Arthur et al., 2018; Ma et al., 2019; Reddy et al., 2017; Schmitz et al., 2018); FL cases: (Krysiak et al., 2017; Ma et al., 2019; Ortega-Molina et al., 2015). Clinic-pathological characteristics of these cases are detailed in the original publications. Further information on TBL1XR1 mutation status is summarized in Table S1.
All DLBCL human samples used for immunoprofiling studies were obtained with informed consent, and according to protocols approved by the BCCA Research Ethics Board. CD38 IHC on human specimens was performed on formalin-fixed paraffin-embedded tissue (FFPE) biopsies of 341 DLBCL (Arthur et al., 2018). Clinic-pathological characteristics of these cases are detailed in the original publication. For CyTOF immunoprofiling, diagnostic pre-treatment LN biopsies from patients with DLBCL, or non-malignant “reactive” LN (rLN) were acquired from the lymphoma tumor bank at the BC Cancer Agency (as described in (Nissen et al., 2019)), and were selected based on sufficient numbers of viable cells for CyTOF analysis (2 million per sample). Clinic-pathological characteristics of these cases are detailed in the original publication. Further details on somatic mutations in these cases are summarized in Table S5.
METHOD DETAILS
Germinal center assessment in mice
To induce GC formation, age- and sex-matched mice were immunized intraperitoneally at 8 to 12 weeks of age with either 0.5ml of a 2% sheep SRBC suspension in PBS (Cocalico Biologicals; Reamstown, PA, USA), or 100 μg of the highly substituted hapten NP (NP16 to NP32) conjugated to the carrier protein ovalbumin (OVA), or CGG (Chicken Gamma Globulin), or KLH (Keyhole Limpet Hemocyanin; all from Biosearch Technologies; Novato, CA, USA) absorbed to Imject™ Alum Adjuvant (77161; ThermoFisher Scientific; Waltham, MA, USA) at a 1:1 ratio. In the experiments where TFH interactions were blocked in vivo, mice received and i.v. injection of 100μg anti-CD40L antibody (clone MR-1, BE0017; BioXCell; West Lebanon, NH, USA) or control IgG antibody (BE0091; BioXCell) 4 days after SRBC immunization, and a second dose of antibody 2 days later. In the experiments where IL-9 was blocked in vivo, mice received a single i.v. injection of 200μg anti-IL-9 antibody (clone 9C1, BE0181; BioXCell) or control IgG2a antibody (BE0085; BioXCell) 7 days after SRBC immunization. In these experiments, littermates of the same sex were randomly assigned to experimental groups. In experiments where GC cell cycle distribution was assessed, animals received an i.v. injection of 1mg EdU (E10187; ThermoFisher Scientific), 1h before euthanasia.
BM transplantations and lymphomagenesis studies
Bone marrow cells were harvested from the tibia and femur of 8–12 weeks old donor mice. After treatment with red blood cell lysis solution (158904; QIAGEN; Germantown, MD, USA), cells were mixed at the indicated ratios, and 1–2 million cells were injected into the tail vein of lethally irradiated C57Bl/6J host mice (2 doses of 450rad, on a Rad Source Technologies RS 2000 Biological Research X-ray Irradiator). Transplanted mice were used for experiments 6–8 weeks after transplant, to allow for full engraftment. With the exception of mice euthanized at specific time points, all mice involved in lymphomagenesis studies were monitored until any one of several criteria for euthanizing were met, including severe lethargy, more than 10% body weight loss, and palpable splenomegaly that extended across the midline, in accordance with our Weill Cornell Medicine Institutional Animal Care and Use Committee–approved animal protocol (protocol #2011–0031).
NB and MB adoptive transfers
For NB cell adoptive transfers, total splenocytes were harvested from 8–12 weeks old B1–8hi donor mice (Cd45.1 or Cd45.1/2), and mature B-cells were isolated using negative selection with CD43 magnetic beads (130-049-801; Miltenyi Biotec; Somerville, MA, USA). The percentage of NP-binding cells in this population was determined by FC (see below), and a number of mature B-cells corresponding to 2–3×105 NP-binding B-cells was injected i.v. into C57Bl/6J recipient mice (Cd45.2). Recipient animals were immunized with an NP-conjugate 16h after cell transfer, and euthanized for analysis at the stated timepoints.
Antigen-specific MB cells for transfer experiments were generated following the protocol above, and splenic NP-binding MB cells were FACS-sorted three months, or more, after immunization of recipient mice with NP-CGG or NP-KLH. Splenic memory T-cells (MT; CD3+CD4+CD44+CD62L−) were FACS-sorted from OT-II mice, 10 days after immunization with 100μg OVA peptide (#A5503, Sigma-Aldrich; San Luis, MO, USA). For re-call experiments, 8×103 MB and 4×104 MT cells were injected i.v. into μMT recipient mice (Cd45.2). Recipient animals were immunized with NP-OVA 16h after transfer, and euthanized for analysis 3.5 days after immunization.
Flow cytometry analysis and cell sorting
Single-cell suspensions from mouse spleens or bone marrow were stained using the following fluorescent-labeled anti-mouse antibodies: from eBioscience ThermoFisher Scientific: APC anti-CD38 (17–0381, dilution 1:500), PE anti-CXCR4 (12–9991, dilution 1:400), PerCP-Cy5.5 anti-CD45.1 (45–0453, dilution 1:500), FITC anti-PD-1 (11–9985, dilution 1:200), PE-Cy7 streptavidin (25–4317, dilution 1:1000); from BD Biosciences (San Jose, CA, USA): PE-Cy7 anti-CD44 (560569, dilution 1:500), APC, PE-Cy7 and BV786 anti-B220 (553087, 552772 and 563894, dilution 1:500), PE-Cy7 anti-FAS (557653, dilution 1:500), BV395 anti-CD38 (740245, dilution 1:500), PE-Cy7 and BV421 anti-CD86 (560582 and 564198, dilution 1:300), biotin anti-CXCR4 (551968, dilution 1:200), biotin anti-CXCR5 (551960, dilution 1:200), BV421 and BV510 anti-IgD (744291 and 563110, dilution 1:500), PE and BV421 anti-IgG1 (550083 and 562580, dilution 1:500), BUV737 anti-CD138 (564430, dilution 1:500), FITC and BV711 anti-IgM (553437 and 743327, dilution 1:500), BV421 anti-CCR6 (564736, dilution 1:500), APC anti-CD117 (561074, dilution 1:500), FITC anti-CD19 (553785, dilution 1:500), PE-Cy7 anti-CD25 (561780, dilution 1:500), FITC and AF647 anti-GL7 (553666 and 561529, dilution 1:500); from BioLegend (San Diego, CA, USA): BV510 anti-CD62L (104441, dilution 1:500), PE anti-PCNA (307908, dilution 1:50), APC-Cy7 anti-CD4 (100414, dilution 1:500), PE anti-PD-L2 (107206, dilution 1:300), PerCP-Cy5.5 anti-CD45.2 (109828, dilution 1:500) BV421 anti-CD80 (104725, dilution 1:200) APC anti-CD3 (100235, dilution 1:500), PE anti-BCL6 (648304, dilution 1:500), AF647 anti-BLIMP1 (150004, dilution 1:300), BV421, APC-Cy7 and PE anti-B220 (103240, 103224 and 103208, dilution 1:500), APC-Cy7 anti-CD38 (102728, dilution 1:500), PerCP-Cy5.5 anti-GL7 (144610, dilution 1:500), PerCP-Cy5.5 anti-FAS (152610, dilution 1:500), PE-Cy7 and BV421 anti-CD138 (142514 and 142508, dilution 1:500), APC streptavidin (405207, dilution 1:1000), APC-Cy7 streptavidin (405208, dilution 1:1000); from R&D Systems (Minneapolis, MN, USA): biotin anti-EFNB1 (BAF473, dilution 1:60) and anti-mS1P1 (MAB7089, dilution 1:50); from Biosearch Technologies: PE NP (N-5070–1, dilution 1:500); from Life Technologies: anti-AID (1AID-2E11; dilution 1:100). NIP-haptenated FITC was obtained from M.J. Shlomchik (University of Pittsburg, PA, USA) (Anderson et al., 2007). For S1PR1 staining, a biotinylated goat anti-rat secondary antibody was used (112-065-167; dilution 1:1000, Jackson Immunoresearch; West Grove, PA, USA). For AID staining, a goat anti-mouse IgG cross-adsorbed secondary antibody conjugated to Alexa Fluor 647, was used (A21235, dilution 1:300; Thermo Scientific). To detect surface IL-9R expression, cells were incubated with 8 ng/μl recombinant IL-9 (219–19; Peprotech, Rocky Hill, NJ) on ice for 60min, washed and fixed in 1% paraformaldehyde for 30min, and then stained with APC anti-IL-9 (RM9A4, dilution 1:250; BioLegend) on ice. As the negative control, incubation with IL-9 was omitted. Ghost Dye Violet 510 (13–0870; Tonbo Biosciences; San Diego, CA, USA) or DAPI (D1306; ThermoFisher Scientific) were used for the exclusion of dead cells. For intracellular markers, cells were fixed and permeabilized with the eBioscience Foxp3/Transcription Factor Staining Buffer Set (00-5523-00; ThermoFisher Scientific). PE-conjugated AnnexinV (556421, dilution 1:100; BD Biosciences) in AnnexinV binding buffer (BD Biosciences) was used to identify apoptotic cells. For cell cycle assessment, EdU incorporation was detected using the Click-iT Plus AF488 Flow Cytometry Assay Kit (C10633; ThermoFisher Scientific). Proliferation rates were assessed in cell lines using the CellTrace™ CFSE Cell Proliferation Kit (C34554; ThermoFisher Scientific), and in primary cultures using the eBioscience™ Cell Proliferation Dye eFluor™ 670 (65-0840-85; ThermoFisher Scientific), according to the vendor’s protocol. Data were acquired on BD FACS Canto II or BD Fortessa flow cytometer analyzers, and analyzed using FlowJo software package (TreeStar).
When B-cell populations were sorted, single-cell suspensions of splenocytes were pre-enriched in B-cells using positive selection with anti-B220 magnetic microbeads (130-049-501; Miltenyi Biotec), or negative selection with the EasySep™ Mouse B-Cell Isolation Kit (19854; StemCell Technologies,). The stated populations were then isolated using a BD FACSAria II or a BD Influx sorter (BD Biosciences).
ELISA
For analysis of T-cell dependent antibody production, mice were immunized intraperitoneally with 100μg NP-CGG or NP-OVA or NP-KLH, ratio 16–32, in alum (1:1). Serum from each animal was collected before immunization, and on the stated days after immunization. Titers of low and high-affinity isotype-specific antibodies to NP were measured in plates coated with NP28-BSA or NP8-BSA, respectively, using the SBA Clonotyping System (5300–05; Southern Biotechnology; Birmingham, AL, USA), according to the manufacturer’s protocol. Results were assessed by spectrophotometric measurement of absorbance at 405nm using a Biotek Synergy Neo Alpha Plate Reader (BioTek; Winooski, VT, USA). Background readings of absorbance in negative control wells were A450<0.050.
Histology and immunohistochemistry
Mice organs were fixed in 4% formaldehyde and embedded in paraffin. Tissue processing and staining were done by the Laboratory of Comparative Pathology (MSKCC). Briefly, five micron-sections were deparaffinized and heat antigen-retrieved in citrate buffer pH=6.4, and endogenous peroxidase (HRP) activity was blocked by treating the sections with 3% hydrogen peroxide in methanol. Indirect immunohistochemistry was performed with anti-species-specific biotinylated secondary antibodies followed by avidin–horseradish peroxidase or avidin-AP, and developed by Vector Blue or DAB color substrates (Vector Laboratories; Burlingame, CA USA). Sections were counterstained with hematoxylin. The following primary antibodies were used: biotin-conjugated anti-B220 (550286; BD Biosciences), anti-CD3 (ab16669; Abcam; Cambridge, UK), anti-PNA (B1075; Vector Laboratories), anti-CD138 (553712; BD Biosciences), and anti-KI67 (12202; Cell Signaling Technology; Danvers, MA, USA). TUNEL staining was carried out as previously described (Gavrieli et al., 1992). IHC on human specimens was performed on formalin-fixed paraffin-embedded tissue (FFPET) biopsies of 341 DLBCL cases within the cohort as described previously (Ennishi et al., 2017; Scott et al., 2015). Briefly, IHC staining on 4μm slides of TMAs was performed for CD38 (SPC32; MA5–14413; ThermoFisher Scientific) on the Benchmark XT platform (Ventana, AZ, USA), according to the previously described method (Ennishi et al., 2017; Scott et al., 2015). Slides were scanned using a Zeiss Mirax Slide Scanner and photomicrographs were examined using Aperio eSlide Manager (Leica Biosystems; Wetzlar, Germany). Fiji software (Schindelin et al., 2012; Schneider et al., 2012) was used to quantify GC and TUNEL+ cell/areas. CD38 staining was semi-quantitatively assessed on tumor cells using HistoScore (HS = IxP): intensity (I = [1–3]) and percentage of positive cells (P = [0–100]). Specimens with HS>50 were defined as CD38 positive.
3D B cell follicular organoid
Experiments using the organoid culture system were performed as previously described (Beguelin et al., 2017). In brief, splenic B-cells were obtained from naïve animals through negative selection, using the EasySep™ Mouse B-Cell Isolation Kit in accordance with manufacturer’s protocol. Feeder 40LB cells (Nojima et al., 2011) were grown in DMEM media with 10% FBS and, at the time of experiment, were mitotically inhibited through incubation in cell culture complete medium containing 0.01 mg/ml Mitomycin C (MO503, Sigma-Aldrich,) at 37 °C for 55 min before the encapsulation. For organoid fabrication, gelatin stock solution was freshly prepared by mixing gelatin powder (Sigma-Aldrich) in RPMI-1640 medium followed by sterilization using syringe filter. Cells were mixed with warmed 5% gelatin stock solution and diluted accordingly using cell culture medium. Silicate nanoparticles (SiNP) with 25–30 nm in diameter and 1 nm in thickness were obtained from Southern Clay Products Inc. (Gonzales, TX, USA). A 3% hydrogel SiNP suspension was freshly prepared before the encapsulation procedure by mixing SiNP powder with deionized water and vortexing the resulting solution, followed by filtration through 0.22μm syringe filters immediately before use. Organoids were fabricated in 96-well plates by first adding 10 μl of 3% hydrogel SiNP followed by injecting 10 μl cell-containing gelatin solution into the initial SiNP droplet, and then mixing the entire hydrogel through repeated pipetting. Each organoid contained 50,000 B cells and 80,000 40LB cells. Organoids were cured for ~ 10 min before the addition of RPMI media with 10% FBS and penicillin G/streptomycin, containing 50 ng/ml murine recombinant IL-4 (404-ML; R&D Systems) and 25ng/ml murine recombinant IL-21 (594-ML; R&D Systems) and were incubated at 37 °C with 5% CO2. Cell culture medium was renewed every 3 days.
Generation of TBL1XR1-KO cell lines
To generate TBL1XR1-KO clones using CRISPR, cell lines were electroporated using an Amaxa Nucleofector Unit and the SF Cell Line 4D-Nucleofector X Kit (PBC2–22500; Lonza; Basel, Switzerland), to incorporate a recombinant Cas9 nuclease (Alt-R® S.p. Cas9 Nuclease V3, 1081058), a TBL1XR1-targeting Alt-R® CRISPR-Cas9 crRNA (TBL1XR1_1 crRNA: 5’-GAU AUG GCU UUC UAU ACC AAG UUU UAG AGC UAU GCU-3’; TBL1XR1_2 crRNA: 5’-UAU UGG UCG ACC AUC AAA CAG UUU UAG AGC UAU GCU-3’), and an Alt-R® CRISPR-Cas9 tracrRNA (1075927; all from Integrated DNA Technologies; Coralville, IA, USA), following vendor’s recommendations. Forty-eight hours after electroporation, cells were single-cell plated into 96 well plates, and allowed to grow for at least 2 weeks. Resulting clones were screened by WB, and positive clones were further verified by Sanger sequencing of genomic DNA. Clones expressing parental-like levels of TBL1XR1, and WT DNA sequences, were used as congenic controls for experiments.
Generation of TBL1XR1 mutant cell lines
TBL1XR1 open reading frame (ORF) was amplified from HEK293T cells, and a Linker-His_Tag-V5_Tag sequence was added downstream by PCR (5’-GGCAGCAGCGGCCATCATCACCATCACCACGGTAAGCCTATCCCTAACCCTCTCCTCGGTCTCGATTCTACG-3’). The TBL1XR1-V5 insert was cloned into the pSBtet-GP backbone (#60495; Addgene; Watertown, MA, USA; (Kowarz et al., 2015) using the SfiI restriction enzyme (New England Biolabs; Ipswich, MA, USA). TBL1XR1 point mutations (D370Y, Y395H and Y446S) were introduced using the QuikChange II XL Site-directed mutagenesis kit (200521; Agilent Technologies; Santa Clara, CA, USA), and verified by Sanger sequencing. OCI-Ly1 cells were nucleofected to incorporate the different pSBtet-GP-TBL1XR1-V5 plasmids, along with a construct coding for the SB100X transposase [pCMV(CAT)T7-SB110; #34879, Addgene; (Mates et al., 2009)]. Cells were selected with Puromycin for 7 days, and incorporation of the construct was validated by FC for GFP. To induce TBL1XR1-V5 expression, cells were stimulated with Doxycycline (DOX) 1μg/ml for 48h in complete growth media.
Generation of cell lines for BioID experiments
TBL1XR1 or eGFP ORF were cloned into the pLIX_403 backbone (#41395; Addgene; David Root (Broad Institute, Cambridge, MA, USA), downstream of FLAG-BirA(R118G) ORF (#36047; Addgene; (Roux et al., 2012), using the Gateway™ LR Clonase™ II Enzyme mix (11791020; ThermoFisher Scientific). TBL1XR1-Y446S mutation was introduced using the QuikChange II Site-directed mutagenesis kit, and verified by Sanger sequencing. Lentivirus were produced in HEK293T cells co-transfected with pCMV-VSV-G and pCMV-dR8.91 plasmids, and then concentrated using PEG-it (LV810A-1; System Biosciences; Palo Alto, CA, USA). OCI-Ly1 and U2932 cells were infected and cultured for 48h, and then selected with Puromycin for 7 days. To induce expression of the FLAG-BirA-fusion proteins, cells were stimulated with DOX 1μg/ml for 24h. Incorporation of the construct was validated by WB for BirA (Figure S5C), or FC for eGFP.
BioID experiments
Expression of FLAG-BirA-fusion proteins in OCI-Ly1 and U2932 cells was induced by treatment with Doxycycline 1μg/ml for 24h, after which biotin 50μM (B4501; Sigma-Aldrich) was added to the media for additional 24h. Cell pellets were washed twice with PBS, and lysed in modified RIPA lysis buffer (50mM Tris-HCl pH=7.5, 150mM NaCl, 1mM EDTA, 1mM EGTA, 1% Triton X-100, 0.1% SDS, 1:500 protease inhibitor cocktail (11873580001; Sigma-Aldrich), 1:100 Benzonase nuclease (E1014-5KU; Sigma-Aldrich), at 4°C for 1h, and sonicated for 30s at 35% power to disrupt visible aggregates. Lysates were then centrifuged at 16,000rpm for 30min, and clarified supernatants were incubated with 30μL packed pre-equilibrated streptavidin-Sepharose beads (GE17-5113-01; GE Healthcare Life Sciences; Chicago, IL, USA) at 4°C for 3h. Beads were collected by centrifugation, washed six times with 50mM ammonium bicarbonate pH=8.3, and treated with TPCK trypsin (V5111; Promega Corporation; Madison, WI, USA). The supernatant, containing tryptic peptides, was collected and lyophilized. Peptides were resuspended in 0.1% formic acid.
LC-MS/MS was conducted using a 120min reversed-phase buffer gradient running at 150nL/min (column heated to 40°C) on a Proxeon EASY-nLC pump in-line with a hybrid LTQ-Orbitrap Velos mass spectrometer (ThermoFisher Scientific). A parent ion scan was performed in the Orbitrap, using a resolving power of 60000. Simultaneously, up to twenty of the most intense peaks were selected for MS/MS (minimum ion count of 1000 for activation) using standard CID fragmentation. Fragment ions were detected in the LTQ. Dynamic exclusion was activated such that MS/MS of the same m/z (within a 10ppm window, exclusion list size 500) detected three times within 45s were excluded from analysis for 30s. For protein identification, Proteowizard was used to convertraw files to .mzXML, and searched using X!Tandem (Craig and Beavis, 2004; Kessner et al., 2008) against Human RefSeq Version 45. Search parameters specified a parent MS tolerance of 15ppm and an MS/MS fragment ion tolerance of 0.4Da, with up to two missed cleavages allowed for trypsin. Oxidation of methionine and ubiquitylation of lysine residues were allowed as variable modifications. Data were analyzed using a trans-proteomic pipeline via the ProHits software suite (Liu et al., 2010). Proteins identified with a ProteinProphet cut-off value 0.80 (corresponding to FDR < 1%) were analyzed with SAINT Express (Choi et al., 2011). Four control runs of Flag-BirA-eGFP were used for comparative purposes. The control runs were collapsed to the two highest spectral counts for each hit. Prey identified with ≥2 spectral counts in each of the two replicates with a SAINT ≥ 75% were considered significant. Known common contaminants [CRAPome Database v1.1 for Jurkart cells; (Mellacheruvu et al., 2013)] were removed from the final protein lists. All raw mass spectrometry files have been deposited at the MassIVE archive (massive.ucsd.edu).
Cell lysis, immunoblotting and immunoprecipitation
Whole cell protein lysates from cell lines or GCB were obtained using a buffer containing Tris-HCl 50mM (pH=7.4), NaCl 150mM, EDTA 1mM, EGTA 1mM, NP-40 1%, SDS 0.1%, glycerol 10%, and complete protease inhibitor cocktail. Protein extracts for IP experiments were obtained using the Nuclear Complex Co-IP Kit (54001; Active Motif; Carlsbad, CA, USA). For IP experiments, 500–1000ug of nuclear protein extracts were incubated ON with V5-Tag magnetic beads (M16711; MBL International Corporation; Woburn, MA, USA), or with the indicated primary antibodies [BCL6 (sc-858) and HDAC3 (sc-11417) from Santa Cruz Biotechnology (Dallas, TX, USA); NCOR2 (06–891) from Sigma Aldrich; BACH2 (ARP39513_P050) from Aviva Systems Biology (San Diego, CA, USA); Rabbit anti-mouse control IgG (ab46540) from Abcam], followed by a 2h incubation with Protein A Dynabeads (10001D; ThermoFisher Scientific). Whole cell lysates or IP products were resolved by SDS-PAGE, transferred to PVDF membrane (1620177; Bio-Rad Laboratories; Hercules, CA, USA), and probed with the indicated primary antibodies [BCL6 (sc-7388), HDAC3 (sc-376957) and ß-ACTIN (sc-47778) from Santa Cruz Biotechnology; NCOR2 (PA1–843) from ThermoFisher Scientific; BACH2 (ARP39513_P050); TBL1XR1 (H00079710-M01) from Abnova (Taipei City, Taiwan); BirA (BID-CP-100) from BioFront Technologies (Tallahassee, FL, USA)]. Membranes were then incubated with a corresponding peroxidase-conjugated secondary antibody (Jackson Immunoresearch). Protein signals were detected using enhanced chemiluminescence (RPN2232; GE Healthcare Life Sciences).
Rapid immunoprecipitation mass spectrometry of endogenous proteins
RIME experiments (Mohammed et al., 2016) in OCI-Ly1 cells were performed by Active Motif, using an anti-BCL6 antibody (sc-858), or an isotype-matched IgG control (ab46540). Biological duplicates were prepared for each immunoprecipitation. Known common contaminants (CRAPome Database v1.1 for Jurkart cells) were removed from the final protein list.
RNA and genomic DNA and extraction
Genomic DNA was extracted using: a) the QuickExtract™ DNA Extraction Solution (QE09050; Epicentre; Madison, WI, USA), for human cell lines; b) the Puregene Gentra cell kit (158388; QIAGEN), for mouse cell suspensions; c) the DirectPCR Lysis Reagent (101-T; Viagen Biotech; Los Angeles, CA, USA), for mouse tails. Total RNA was extracted from cell suspensions using Trizol LS (10296010; ThermoFisher Scientific) and RNeasy Mini Kit (74106; QIAGEN), with DNase treatment. RNA concentration was determined using Qubit Fluorometric Quantification (ThermoFisher Scientific) and integrity was verified using Agilent 2100 Bioanalyzer (Agilent Technologies).
Genotyping PCR
PCR-based genotyping of the Tbl1xr1-D370Y allele was done on genomic DNA extracted from mouse tail tissues, using the GoTaq® DNA Polymerase (M3001; Promega Corporation). Primers were designed to flank the distal LoxP site located between Tbl1xr1 exons 10 and 11, in the targeted allele (Figures S1A–B) (Forward: 5’-AAC GAG AAG CCA AGG TTC CAG TTC-3’; Reverse: 5’-GAT GCA AAG GTG TTG TTG CTC TGC-3’). PCR program: [2’: 95C] x1, [30”: 95C, 30”: @55C, 45”: 72C] x35, [5’: 72C] x1. PCR-based validation of exon 5 excision in Tbl1xr1KO/KO mice was done on genomic DNA from sorted GCB. Primers were designed to target Tbl1xr1 exon 5 (Figure S1G) (Forward: 5’-TAT CCT CAG CGG TGG GTT AC-3’; Reverse: 5’-GCC AGT AGG GGT TGC TGA TA-3’). PCR program: [2’: 95C] x1, [1’: 95C, 1’: @52C, 2’: 72C] x35, [5’: 72C] x1. PCR products were resolved by agarose gel electrophoresis, and visualized using SYBR Safe DNA stain (S33102; ThermoFisher Scientific).
Quantitative real-time PCR
cDNA synthesis from total RNA extracts was performed using the Verso cDNA Synthesis kit (AB1453B; ThermoFisher Scientific). Expression of the genes of interest was detected using the Fast SYBR Green Master Mix (4385614; ThermoFisher Scientific) on a QuantStudio6 Flex Real-Time PCR System (ThermoFisher Scientific). Gene expression was normalized to Actin or GAPDH levels, using the ΔΔC(t) method, and results were represented as mRNA expression. See Table S6 for primer sequences.
Sanger sequencing
cDNA from FACS-sorted Tbl1xr1WT/WT or Tbl1xr1D370Y/WT GCB, or from the indicated cells, was generated as described above. Sanger sequencing was performed by GENEWIZ (South Plainfield, NJ, USA).
Assessment of Pim1 somatic mutations
Pim1 locus was amplified from genomic DNA using the primers from Liu et al. (Liu et al., 2008): Fwd: 5’-TTC GGC TCG GTC TAC TCT G-3’; Rev: 5’-GGA GGG AAA AGT GGG TCA TAC-3’. PCR program: [2’: 95C] x1, [1’: 95C, 1’: @65C, 1’: 72C] x25, [15’: 72C] x1. PCR products were resolved by agarose gel electrophoresis, and extracted using the QIAquick Gel Extraction Kit (28704; QIAGEN). Purified products were then cloned using the TOPO TA cloning kit for sequencing (K457501; ThermoFisher Scientific), following manufacturer instructions. Plates were grown ON at 37C, and bacteria colony sequencing was performed by GENEWIZ, using the T7 universal sequencing primer (5’-TAA TAC GAC TCA CTA TAG GG-3’). Only good-quality sequence was considered, as determined by inspection of the chromatograms. Mutation mismatch counts were calculated from Sanger sequencing using the sangerseqR ((Hill et al., 2014)) and Biostrings (R package version 2.54.0) packages. As a negative control for SHM, genomic DNA extracted from sorted AcidaKO/KO GCB was used as PCR template.
JH4 intron sequencing
Sorted GCB or tumor cells were collected by centrifugation and resuspended in 500μL DNA cell lysis buffer (100mM Tris pH 8.8, 200mM NaCl, 5mM EDTA, 0.2% SDS) containing 0.2mg/mL proteinase K and incubated at 56°C overnight. 500μL isopropanol and 40μg glycogen was added to precipitate DNA. The mixture was incubated at 4°C for 20 minutes followed by centrifugation for 20 minutes at 20,000 × g and 4°C. The pellet was washed with 1mL 70% ethanol and centrifuged for 10 minutes at 20,000xg and 4°C. The pellet was resuspended with 20μL DEPC-H2O and incubated at 37°C for 40 minutes. JH4 intron sequences were amplified from germinal center B cell genomic DNA by PCR using JH4 forward primer (5’-GGA ATT CGC CTG ACA TCT GAG GAC TCT GC-3’), JH4 reverse primer (5’-GAC TAG TCC TCT CCA GTT TCG GCT GAA TCC-3’) ((Jolly et al., 1997)), and Phusion High-Fidelity DNA Polymerase (M0530L: New England BioLabs). PCR program: [3’: 98C] x1, [30”: 98C, 1’: 72C] x39, [10’: 72C] x1. PCR products were resolved by agarose gel electrophoresis, and the 1.2kb band extracted using the QIAquick Gel Extraction Kit. The amplicon was ligated into the pCR-Blunt II-TOPO vector and transformed into One Shot TOP10 chemically competent E. coli (K280002; ThermoFisher Scientific) according to the manufacturer’s protocol. Clones were sequenced using the bacterial colony Sanger sequencing service provided by Eton Bioscience (Union, NJ, USA) and JH4 sequencing primer (5’-CCA TAC ACA TAC TTC TGT GTT CC-3’). As a negative control for SHM, genomic DNA extracted from sorted AcidaKO/KO GCB was used as PCR template. Sequences were aligned using MUSCLE (EMBL-EBI) included in Biopython package (Cock et al., 2009) and analyzed for mutations using Python 3 script.
RNA sequencing and analysis
Library preparation, sequencing and post-processing of the raw data was performed at the Epigenomics Core at Weill Cornell Medicine. Samples that passed the quality control (RNA Integrity Number ≥8) were subjected to library preparation using the Illumina TruSeq stranded-mRNA sample kits (Illumina; San Diego, CA, USA), according to the manufacturer. Briefly, poly A+ RNA was purified from 100 ng of total RNA with oligo-dT beads. Purified mRNA was fragmented with divalent cations at elevated temperature, to ~200 bp. Following dscDNA synthesis, the double stranded products are end repaired, followed by addition of a single ‘A’ base and then ligation of the Illumina TruSeq adaptors. The resulting product was amplified with 15 cycles of PCR. Libraries were validated using the Agilent Technologies 2100 Bioanalyzer and Quant-iT™ dsDNA HS Assay (Q33120; ThermoFisher Scientific). Each library was made with a unique Index sequence and libraries were pooled for sequencing. The pool was clustered at 6.5pM on a single end read flow cell and sequenced for 50 cycles on an Illumina HiSeq 2500 to obtain ~50 M reads per sample. Single-end sequencing (SE50), for the Tbl1xr1D370Y/WT GCB profiling, or paired end-sequencing (PE50×2) for the IμBcl6-Tbl1xr1D370Y/WT and Tbl1xr1KO/KO GCB profiling, were performed on an Illumina HiSeq2500 sequencer. Primary processing of sequencing images was done using Illumina’s Real Time Analysis software (RTA) as suggested by Illumina. CASAVA 1.8.2 software was used to perform image capture, base calling, demultiplexing samples and generation of raw reads and respective quality scores. Sequencing results were aligned to mm10 using STAR (Dobin et al., 2013) and annotated to RefSeq using the R subread package (Liao et al., 2014). Differentially expressed genes were identified using the EdgeR package GLM (Robinson et al., 2010) with thresholds of fold-change >1.5 and p<0.05, adjusted for multiple testing using Benjamini-Hochberg correction. Hierarchical clustering was performed using Euclidean distance of log FPKM values of genes within the top 5th percentile of standard deviation across replicates and Ward’s minimum variance. Gene set enrichment analysis was performed using the GSEA algorithm, as described in (Subramanian et al., 2005). Pathway analysis was performed using PAGE algorithm (Goodarzi et al., 2009).
CyTOF immunophenotyping of DLBCL specimens
Diagnostic pre-treatment LN biopsies from patients with DLBCL, or non-malignant “reactive” LN (rLN) were acquired from the lymphoma tumor bank at the BC Cancer Agency. All samples were obtained with informed consent and according to protocols approved by the BCCA Research Ethics Board, and were selected based on sufficient numbers of viable cells for CyTOF analysis (2 million per sample).
Vials of frozen cells were rapidly thawed from liquid nitrogen to 37°C using a water bath, and cell suspensions were washed with RPMI supplemented with 10% fetal bovine serum. Cells were used immediately after thawing and were incubated in cisplatin for 15 minutes to label dead cells, then washed thoroughly in PBS+2%FCS. Cell pellets were then incubated with Fc Receptor Blocking Solution (422302; BioLegends) for 10 minutes to inhibit non-specific binding of antibodies, before the addition of a master mix of metal-conjugated antibodies at optimal concentrations for 30 minutes. The relevant antibodies used for the current analysis are; CD10–156Nd (HI10a; Fluidigm, South San Francisco, CA, USA), CD27–167Er (L128; Fluidigm), CD38–172Yb (HIT2; Fluidigm) and CD44–171Yb (IM7; Fluidigm).
Samples were bar-coded with the Cell-ID 20-plex Pd kit (#201060; Fluidigm) as per manufacturer’s instructions before being pooled together for analysis to ensure consistent acquisition. Batch effects were accounted for using a spike-in staining control of rLN cells. Pooled suspensions were then spiked with DVS normalization beads to account for technical drift during acquisition, and acquired on a Fluidigm CyTOF 2 instrument. After acquisition, samples were normalized based on bead signal intensity and de-multiplexed to distinguish individual samples using published tools (https://github.com/nolanlab/bead-normalization, https://github.com/nolanlab/single-cell-debarcoder).
Viable cells were gated in FlowJo based on negative staining for cisplatin, and all subsequent analysis was performed using R. Malignant tumor cells were identified as previously described (Nissen et al., 2019). Isolated malignant cells and manually-defined germinal center B cells and memory B cells were then pooled together for principal component analysis.
Exome sequencing on samples (Table S6) was carried out using genomic DNA extracted from DLBCL samples using QIAGEN reagents. Exome libraries were created using IDT exome capture kits, and samples were sequenced using an Illumina HiSeq 2500 (PE_2×125). Somatic SNV/indel variants were identified using the intersection of calls predicted by VarScan (version 2.3.6) (Koboldt et al., 2012), Strelka (version 1.0.13) (Saunders et al., 2012) and MuTect (version 1.1.4) (Cibulskis et al., 2013). All variants were annotated using SnpEff (version 4.2) (Cingolani et al., 2012) and filtered for effects predicted to have an impact at the protein level (nonsynonymous, stop-gained, splice site, UTR). Variants were further filtered to remove potential germline SNPs if the variant: (i) had a GMAF score > 1% or (ii) was present in dbSNP (version 137) (Sherry et al., 2001) and not COSMIC (version 68) (Tate et al., 2019). All tumors were paired against a pooled control that was constructed from 10 normal samples, to account for sequencing noise.
Graphical abstract
Cartoons used in the graphical abstract were adapted from images in the SERVIER MEDICAL ART repository (https://smart.servier.com/). Use of these images falls within the terms of the Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical parameters including the exact value and definition of n, precision measures (mean ± SEM or SD), and statistical significance are reported in the Figures and Figure Legends. Statistical analysis was conducted using GraphPad Prism 6.00 (GraphPad Software, San Diego, CA, USA), or R statistical language scripts and packages specified under Method Details. Data was judged to be statistically significant when p<0.05. Asterisks in figures denote statistical significance (*, p<0.05; **, p<0.01; ***, p<0.001). Statistical analysis of Sequencing data is described under Method Details section and in Figure Legends.
Supplementary Material
Table S2. Related to Figure 3. Differentially expressed genes in Tbl1xr1- D370Y/WT GCB (|log2FC|≥1.5 and FDR<0.05). Significantly enriched pathways for the Tbl1xr1-D370Y/WT GCB transcriptional signature.
Table S3. Related to Figure 5. Spectral counts for BCL6 RIME assay in the OCI-Ly1 cell line. Spectral counts for TBL1XR1 BioID assays in OCI-Ly1 and U2932 cell lines.
Table S4. Related to Figure 5. Differentially expressed genes in IμBcl6-Tbl1xr1-D370Y/WT GCB (|log2FC|≥1.5 and FDR<0.05). Differentially expressed genes in Tbl1xr1-KO/KO GCB (|log2FC|≥1.5 and FDR<0.05).
Table S5. Related to Figure 7. Somatic mutation profiles of DLBCL specimens used for CyTOF immunophenotyping.
HIGHLIGHTS.
TBL1XR1 mutation skews the humoral immune response towards producing memory B-cells
TBL1XR1 mutant memory cells feature aberrant cyclic reentry to new germinal centers
Mutant TBL1XR1 acts by triggering aberrant targeting of SMRT complex to BACH2
TBL1XR mutation gives rise to extranodal ABC-DLBCLs derived from memory B-cells
ACKNOWLEDGEMENTS
We thank all members of the Melnick Lab for thoughtful discussions and suggestions, and Hao Shen for his support with mice colonies. We acknowledge the Epigenomics, Genomics, and Flow Cytometry cores (WCM), the Molecular Cytology core (MSKCC) and the Center of Comparative Medicine and Pathology (WCM/MSKCC). LV is a Career Development Program Fellow from the Leukemia & Lymphoma Society (Grant 5469-18). AMM is supported by NCI-R35-CA220499, LLS-TRP-6572-19, LLS-SCOR-7012-16, the Chemotherapy Foundation, and the Follicular Lymphoma Consortium.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
AM receives research funding for Janssen, is on the scientific board of KDAC pharmaceuticals and has consulted for Constellation and Epizyme. MG consults for Verastem Oncology.
REFERENCES
- Agematsu K, Hokibara S, Nagumo H, and Komiyama A (2000). CD27: a memory B-cell marker. Immunol Today 21, 204–206. [DOI] [PubMed] [Google Scholar]
- Anderson SM, Tomayko MM, Ahuja A, Haberman AM, and Shlomchik MJ (2007). New markers for murine memory B cells that define mutated and unmutated subsets. J Exp Med 204, 2103–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnon TI, Horton RM, Grigorova IL, and Cyster JG (2013). Visualization of splenic marginal zone B-cell shuttling and follicular B-cell egress. Nature 493, 684–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arpin C, Dechanet J, Van Kooten C, Merville P, Grouard G, Briere F, Banchereau J, and Liu YJ (1995). Generation of memory B cells and plasma cells in vitro. Science 268, 720–722. [DOI] [PubMed] [Google Scholar]
- Arthur SE, Jiang A, Grande BM, Alcaide M, Cojocaru R, Rushton CK, Mottok A, Hilton LK, Lat PK, Zhao EY, et al. (2018). Genome-wide discovery of somatic regulatory variants in diffuse large B-cell lymphoma. Nat Commun 9, 4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnden MJ, Allison J, Heath WR, and Carbone FR (1998). Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol 76, 34–40. [DOI] [PubMed] [Google Scholar]
- Baumjohann D, Preite S, Reboldi A, Ronchi F, Ansel KM, Lanzavecchia A, and Sallusto F (2013). Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity 38, 596–605. [DOI] [PubMed] [Google Scholar]
- Beguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, Shen H, Yang SN, Wang L, Ezponda T, et al. (2013). EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 23, 677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beguelin W, Rivas MA, Calvo Fernandez MT, Teater M, Purwada A, Redmond D, Shen H, Challman MF, Elemento O, Singh A, et al. (2017). EZH2 enables germinal centre formation through epigenetic silencing of CDKN1A and an Rb-E2F1 feedback loop. Nat Commun 8, 877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beham-Schmid C (2017). Aggressive lymphoma 2016: revision of the WHO classification. Memo 10, 248–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas MG, Yu W, Beguelin W, Teater MR, Geng H, Goldstein RL, Oswald E, Hatzi K, Yang SN, Cohen J, et al. (2016). Rationally designed BCL6 inhibitors target activated B cell diffuse large B cell lymphoma. J Clin Invest 126, 3351–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casola S, Cattoretti G, Uyttersprot N, Koralov SB, Seagal J, Hao Z, Waisman A, Egert A, Ghitza D, and Rajewsky K (2006). Tracking germinal center B cells expressing germ-line immunoglobulin gamma1 transcripts by conditional gene targeting. Proc Natl Acad Sci U S A 103, 7396–7401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattoretti G, Pasqualucci L, Ballon G, Tam W, Nandula SV, Shen Q, Mo T, Murty VV, and Dalla-Favera R (2005). Deregulated BCL6 expression recapitulates the pathogenesis of human diffuse large B cell lymphomas in mice. Cancer Cell 7, 445–455. [DOI] [PubMed] [Google Scholar]
- Cerchietti LC, Ghetu AF, Zhu X, Da Silva GF, Zhong S, Matthews M, Bunting KL, Polo JM, Fares C, Arrowsmith CH, et al. (2010). A small-molecule inhibitor of BCL6 kills DLBCL cells in vitro and in vivo. Cancer Cell 17, 400–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuy B, Roemer MG, Stewart C, Tan Y, Abo RP, Zhang L, Dunford AJ, Meredith DM, Thorner AR, Jordanova ES, et al. (2016). Targetable genetic features of primary testicular and primary central nervous system lymphomas. Blood 127, 869–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, Lawrence MS, Roemer MGM, Li AJ, Ziepert M, et al. (2018). Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med 24, 679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H, Larsen B, Lin ZY, Breitkreutz A, Mellacheruvu D, Fermin D, Qin ZS, Tyers M, Gingras AC, and Nesvizhskii AI (2011). SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat Methods 8, 70–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, and Getz G (2013). Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, and Ruden DM (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cock PJ, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, et al. (2009). Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig R, and Beavis RC (2004). TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467. [DOI] [PubMed] [Google Scholar]
- Cyster JG, and Allen CDC (2019). B Cell Responses: Cell Interaction Dynamics and Decisions. Cell 177, 524–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl SA, Schmidlin H, Nagasawa M, van Haren SD, Kwakkenbos MJ, Yasuda E, Beaumont T, Scheeren FA, and Spits H (2008). STAT3-mediated upregulation of BLIMP1 Is coordinated with BCL6 down-regulation to control human plasma cell differentiation. J Immunol 180, 4805–4815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogan I, Bertocci B, Vilmont V, Delbos F, Megret J, Storck S, Reynaud CA, and Weill JC (2009). Multiple layers of B cell memory with different effector functions. Nat Immunol 10, 1292–1299. [DOI] [PubMed] [Google Scholar]
- Dominguez PM, Ghamlouch H, Rosikiewicz W, Kumar P, Beguelin W, Fontan L, Rivas MA, Pawlikowska P, Armand M, Mouly E, et al. (2018). TET2 Deficiency Causes Germinal Center Hyperplasia, Impairs Plasma Cell Differentiation, and Promotes B-cell Lymphomagenesis. Cancer Discov 8, 1632–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egle A, Harris AW, Bath ML, O’Reilly L, and Cory S (2004). VavP-Bcl2 transgenic mice develop follicular lymphoma preceded by germinal center hyperplasia. Blood 103, 2276–2283. [DOI] [PubMed] [Google Scholar]
- Ennishi D, Mottok A, Ben-Neriah S, Shulha HP, Farinha P, Chan FC, Meissner B, Boyle M, Hother C, Kridel R, et al. (2017). Genetic profiling of MYC and BCL2 in diffuse large B-cell lymphoma determines cell-of-origin-specific clinical impact. Blood 129, 2760–2770. [DOI] [PubMed] [Google Scholar]
- Gavrieli Y, Sherman Y, and Ben-Sasson SA (1992). Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol 119, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng J, Wei H, Shi B, Wang YH, Greer BD, Pittman M, Smith E, Thomas PG, Kutsch O, and Hu H (2019). Bach2 Negatively Regulates T Follicular Helper Cell Differentiation and Is Critical for CD4(+) T Cell Memory. J Immunol 202, 2991–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Aguilar A, Idbaih A, Boisselier B, Habbita N, Rossetto M, Laurenge A, Bruno A, Jouvet A, Polivka M, Adam C, et al. (2012). Recurrent mutations of MYD88 and TBL1XR1 in primary central nervous system lymphomas. Clin Cancer Res 18, 5203–5211. [DOI] [PubMed] [Google Scholar]
- Goodarzi H, Elemento O, and Tavazoie S (2009). Revealing global regulatory perturbations across human cancers. Mol Cell 36, 900–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashwah H, Schmid CA, Kasser S, Bertram K, Stelling A, Manz MG, and Muller A (2017). Inactivation of CREBBP expands the germinal center B cell compartment, down-regulates MHCII expression and promotes DLBCL growth. Proc Natl Acad Sci U S A 114, 9701–9706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzi K, Geng H, Doane AS, Meydan C, LaRiviere R, Cardenas M, Duy C, Shen H, Vidal MNC, Baslan T, et al. (2019). Histone demethylase LSD1 is required for germinal center formation and BCL6-driven lymphomagenesis. Nat Immunol 20, 86–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzi K, Jiang Y, Huang C, Garrett-Bakelman F, Gearhart MD, Giannopoulou EG, Zumbo P, Kirouac K, Bhaskara S, Polo JM, et al. (2013). A hybrid mechanism of action for BCL6 in B cells defined by formation of functionally distinct complexes at enhancers and promoters. Cell Rep 4, 578–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzi K, and Melnick A (2014). Breaking bad in the germinal center: how deregulation of BCL6 contributes to lymphomagenesis. Trends Mol Med 20, 343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinen CA, Jongejan A, Watson PJ, Redeker B, Boelen A, Boudzovitch-Surovtseva O, Forzano F, Hordijk R, Kelley R, Olney AH, et al. (2016). A specific mutation in TBL1XR1 causes Pierpont syndrome. J Med Genet 53, 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JT, Demarest BL, Bisgrove BW, Su YC, Smith M, and Yost HJ (2014). Poly peak parser: Method and software for identification of unknown indels using sanger sequencing of polymerase chain reaction products. Dev Dyn 243, 1632–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Geng H, Boss I, Wang L, and Melnick A (2014). Cooperative transcriptional repression by BCL6 and BACH2 in germinal center B-cell differentiation. Blood 123, 1012–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Ghisletti S, Perissi V, Rosenfeld MG, and Glass CK (2009). Transcriptional integration of TLR2 and TLR4 signaling at the NCoR derepression checkpoint. Mol Cell 35, 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ise W, Fujii K, Shiroguchi K, Ito A, Kometani K, Takeda K, Kawakami E, Yamashita K, Suzuki K, Okada T, et al. (2018). T Follicular Helper Cell-Germinal Center B Cell Interaction Strength Regulates Entry into Plasma Cell or Recycling Germinal Center Cell Fate. Immunity 48, 702–715 e704. [DOI] [PubMed] [Google Scholar]
- Jain BP, and Pandey S (2018). WD40 Repeat Proteins: Signalling Scaffold with Diverse Functions. Protein J 37, 391–406. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Ortega-Molina A, Geng H, Ying HY, Hatzi K, Parsa S, McNally D, Wang L, Doane AS, Agirre X, et al. (2017). CREBBP Inactivation Promotes the Development of HDAC3-Dependent Lymphomas. Cancer Discov 7, 38–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly CJ, Klix N, and Neuberger MS (1997). Rapid methods for the analysis of immunoglobulin gene hypermutation: application to transgenic and gene targeted mice. Nucleic Acids Res 25, 1913–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashyap R, Rai Mittal B, Manohar K, Balasubramanian Harisankar CN, Bhattacharya A, Singh B, Malhotra P, and Varma S (2011). Extranodal manifestations of lymphoma on [(1)(8)F]FDG-PET/CT: a pictorial essay. Cancer Imaging 11, 166–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessner D, Chambers M, Burke R, Agus D, and Mallick P (2008). ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 24, 2534–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura D, Roes J, Kuhn R, and Rajewsky K (1991). A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature 350, 423–426. [DOI] [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, and Wilson RK (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kometani K, Nakagawa R, Shinnakasu R, Kaji T, Rybouchkin A, Moriyama S, Furukawa K, Koseki H, Takemori T, and Kurosaki T (2013). Repression of the transcription factor Bach2 contributes to predisposition of IgG1 memory B cells toward plasma cell differentiation. Immunity 39, 136–147. [DOI] [PubMed] [Google Scholar]
- Kowarz E, Loscher D, and Marschalek R (2015). Optimized Sleeping Beauty transposons rapidly generate stable transgenic cell lines. Biotechnol J 10, 647–653. [DOI] [PubMed] [Google Scholar]
- Krysiak K, Gomez F, White BS, Matlock M, Miller CA, Trani L, Fronick CC, Fulton RS, Kreisel F, Cashen AF, et al. (2017). Recurrent somatic mutations affecting B-cell receptor signaling pathway genes in follicular lymphoma. Blood 129, 473–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo TC, Shaffer AL, Haddad J Jr., Choi YS, Staudt LM, and Calame K (2007). Repression of BCL-6 is required for the formation of human memory B cells in vitro. J Exp Med 204, 819–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laidlaw BJ, Schmidt TH, Green JA, Allen CD, Okada T, and Cyster JG (2017). The Eph-related tyrosine kinase ligand Ephrin-B1 marks germinal center and memory precursor B cells. J Exp Med 214, 639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, Tyagi N, Johnson D, Joss S, Kinning E, McWilliam C, Splitt M, Thornton JM, Firth HV, Study DDD, et al. (2016). Integrating population variation and protein structural analysis to improve clinical interpretation of missense variation: application to the WD40 domain. Hum Mol Genet 25, 927–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz G, Nagel I, Siebert R, Roschke AV, Sanger W, Wright GW, Dave SS, Tan B, Zhao H, Rosenwald A, et al. (2007). Aberrant immunoglobulin class switch recombination and switch translocations in activated B cell-like diffuse large B cell lymphoma. J Exp Med 204, 633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, and Wang CY (2008). TBL1-TBLR1 and beta-catenin recruit each other to Wnt target-gene promoter for transcription activation and oncogenesis. Nat Cell Biol 10, 160–169. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liu G, Zhang J, Larsen B, Stark C, Breitkreutz A, Lin ZY, Breitkreutz BJ, Ding Y, Colwill K, Pasculescu A, et al. (2010). ProHits: integrated software for mass spectrometry-based interaction proteomics. Nat Biotechnol 28, 1015–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, and Schatz DG (2008). Two levels of protection for the B cell genome during somatic hypermutation. Nature 451, 841–845. [DOI] [PubMed] [Google Scholar]
- Luckey CJ, Bhattacharya D, Goldrath AW, Weissman IL, Benoist C, and Mathis D (2006). Memory T and memory B cells share a transcriptional program of self-renewal with long-term hematopoietic stem cells. Proc Natl Acad Sci U S A 103, 3304–3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma MCJ, Tadros S, Bouska A, Heavican TB, Yang H, Deng Q, Moore D, Akhter A, Hartert K, Jain N, et al. (2019). Pathognomonic and epistatic genetic alterations in B-cell non-Hodgkin lymphoma. bioRxiv, 674259. [Google Scholar]
- Mates L, Chuah MK, Belay E, Jerchow B, Manoj N, Acosta-Sanchez A, Grzela DP, Schmitt A, Becker K, Matrai J, et al. (2009). Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat Genet 41, 753–761. [DOI] [PubMed] [Google Scholar]
- Mayer CT, Gazumyan A, Kara EE, Gitlin AD, Golijanin J, Viant C, Pai J, Oliveira TY, Wang Q, Escolano A, et al. (2017). The microanatomic segregation of selection by apoptosis in the germinal center. Science 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellacheruvu D, Wright Z, Couzens AL, Lambert JP, St-Denis NA, Li T, Miteva YV, Hauri S, Sardiu ME, Low TY, et al. (2013). The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat Methods 10, 730–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles RR, Crockett DK, Lim MS, and Elenitoba-Johnson KS (2005). Analysis of BCL6-interacting proteins by tandem mass spectrometry. Mol Cell Proteomics 4, 1898–1909. [DOI] [PubMed] [Google Scholar]
- Minnich M, Tagoh H, Bonelt P, Axelsson E, Fischer M, Cebolla B, Tarakhovsky A, Nutt SL, Jaritz M, and Busslinger M (2016). Multifunctional role of the transcription factor Blimp-1 in coordinating plasma cell differentiation. Nat Immunol 17, 331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed H, Taylor C, Brown GD, Papachristou EK, Carroll JS, and D’Santos CS (2016). Rapid immunoprecipitation mass spectrometry of endogenous proteins (RIME) for analysis of chromatin complexes. Nat Protoc 11, 316–326. [DOI] [PubMed] [Google Scholar]
- Mondello P, Tadros S, Teater M, Fontan L, Chang AY, Jain N, Yang H, Singh S, Ying HY, Chu CS, et al. (2020). Selective Inhibition of HDAC3 Targets Synthetic Vulnerabilities and Activates Immune Surveillance in Lymphoma. Cancer Discov 10, 440–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mort RL, Ford MJ, Sakaue-Sawano A, Lindstrom NO, Casadio A, Douglas AT, Keighren MA, Hohenstein P, Miyawaki A, and Jackson IJ (2014). Fucci2a: a bicistronic cell cycle reporter that allows Cre mediated tissue specific expression in mice. Cell Cycle 13, 2681–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, and Honjo T (2000). Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 102, 553–563. [DOI] [PubMed] [Google Scholar]
- Nissen MD, Kusakabe M, Wang X, Simkin G, Gracias D, Tyshchenko K, Hill A, Meskas J, Hung S, Chavez EA, et al. (2019). Single Cell Phenotypic Profiling of 27 DLBCL Cases Reveals Marked Intertumoral and Intratumoral Heterogeneity. Cytometry A. [DOI] [PubMed] [Google Scholar]
- Nojima T, Haniuda K, Moutai T, Matsudaira M, Mizokawa S, Shiratori I, Azuma T, and Kitamura D (2011). In-vitro derived germinal centre B cells differentially generate memory B or plasma cells in vivo. Nat Commun 2, 465. [DOI] [PubMed] [Google Scholar]
- Oberoi J, Fairall L, Watson PJ, Yang JC, Czimmerer Z, Kampmann T, Goult BT, Greenwood JA, Gooch JT, Kallenberger BC, et al. (2011). Structural basis for the assembly of the SMRT/NCoR core transcriptional repression machinery. Nat Struct Mol Biol 18, 177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochiai K, Katoh Y, Ikura T, Hoshikawa Y, Noda T, Karasuyama H, Tashiro S, Muto A, and Igarashi K (2006). Plasmacytic transcription factor Blimp-1 is repressed by Bach2 in B cells. J Biol Chem 281, 38226–38234. [DOI] [PubMed] [Google Scholar]
- Ogilvy S, Metcalf D, Print CG, Bath ML, Harris AW, and Adams JM (1999). Constitutive Bcl-2 expression throughout the hematopoietic compartment affects multiple lineages and enhances progenitor cell survival. Proc Natl Acad Sci U S A 96, 14943–14948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver AM, Martin F, and Kearney JF (1997). Mouse CD38 is down-regulated on germinal center B cells and mature plasma cells. J Immunol 158, 1108–1115. [PubMed] [Google Scholar]
- Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang Y, Zhao C, Jiang M, Hu D, Agirre X, Niesvizky I, et al. (2015). The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med 21, 1199–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira JP, Kelly LM, Xu Y, and Cyster JG (2009). EBI2 mediates B cell segregation between the outer and centre follicle. Nature 460, 1122–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perissi V, Aggarwal A, Glass CK, Rose DW, and Rosenfeld MG (2004). A corepressor/coactivator exchange complex required for transcriptional activation by nuclear receptors and other regulated transcription factors. Cell 116, 511–526. [DOI] [PubMed] [Google Scholar]
- Phan RT, and Dalla-Favera R (2004). The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature 432, 635–639. [DOI] [PubMed] [Google Scholar]
- Phelan JD, Young RM, Webster DE, Roulland S, Wright GW, Kasbekar M, Shaffer AL 3rd, Ceribelli M, Wang JQ, Schmitz R, et al. (2018). A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature 560, 387–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo JM, Ci W, Licht JD, and Melnick A (2008). Reversible disruption of BCL6 repression complexes by CD40 signaling in normal and malignant B cells. Blood 112, 644–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranuncolo SM, Polo JM, Dierov J, Singer M, Kuo T, Greally J, Green R, Carroll M, and Melnick A (2007). Bcl-6 mediates the germinal center B cell phenotype and lymphomagenesis through transcriptional repression of the DNA-damage sensor ATR. Nat Immunol 8, 705–714. [DOI] [PubMed] [Google Scholar]
- Ranuncolo SM, Wang L, Polo JM, Dell’Oso T, Dierov J, Gaymes TJ, Rassool F, Carroll M, and Melnick A (2008). BCL6-mediated attenuation of DNA damage sensing triggers growth arrest and senescence through a p53-dependent pathway in a cell context-dependent manner. J Biol Chem 283, 22565–22572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, Leppa S, Pasanen A, Meriranta L, Karjalainen-Lindsberg ML, et al. (2017). Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 171, 481–494 e415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridderstad A, and Tarlinton DM (1998). Kinetics of establishing the memory B cell population as revealed by CD38 expression. J Immunol 160, 4688–4695. [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Raida M, and Burke B (2012). A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol 196, 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, and Cheetham RK (2012). Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28, 1811–1817. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, Roulland S, Kasbekar M, Young RM, Shaffer AL, et al. (2018). Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J Med 378, 1396–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DW, Mottok A, Ennishi D, Wright GW, Farinha P, Ben-Neriah S, Kridel R, Barry GS, Hother C, Abrisqueta P, et al. (2015). Prognostic Significance of Diffuse Large B-Cell Lymphoma Cell of Origin Determined by Digital Gene Expression in Formalin-Fixed Paraffin-Embedded Tissue Biopsies. J Clin Oncol 33, 2848–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, and Sirotkin K (2001). dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29, 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinnakasu R, Inoue T, Kometani K, Moriyama S, Adachi Y, Nakayama M, Takahashi Y, Fukuyama H, Okada T, and Kurosaki T (2016). Regulated selection of germinal-center cells into the memory B cell compartment. Nat Immunol 17, 861–869. [DOI] [PubMed] [Google Scholar]
- Singh M, Jackson KJL, Wang JJ, Schofield P, Field MA, Koppstein D, Peters TJ, Burnett DL, Rizzetto S, Nevoltris D, et al. (2020). Lymphoma Driver Mutations in the Pathogenic Evolution of an Iconic Human Autoantibody. Cell 180, 878–894 e819. [DOI] [PubMed] [Google Scholar]
- Stengel KR, Bhaskara S, Wang J, Liu Q, Ellis JD, Sampathi S, and Hiebert SW (2019). Histone deacetylase 3 controls a transcriptional network required for B cell maturation. Nucleic Acids Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart I, Radtke D, Phillips B, McGowan SJ, and Bannard O (2018). Germinal Center B Cells Replace Their Antigen Receptors in Dark Zones and Fail Light Zone Entry when Immunoglobulin Gene Mutations are Damaging. Immunity 49, 477–489 e477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9(4):493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suan D, Krautler NJ, Maag JLV, Butt D, Bourne K, Hermes JR, Avery DT, Young C, Statham A, Elliott M, et al. (2017). CCR6 Defines Memory B Cell Precursors in Mouse and Human Germinal Centers, Revealing Light-Zone Location and Predominant Low Antigen Affinity. Immunity 47, 1142–1153 e1144. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sungalee S, Mamessier E, Morgado E, Gregoire E, Brohawn PZ, Morehouse CA, Jouve N, Monvoisin C, Menard C, Debroas G, et al. (2014). Germinal center reentries of BCL2-overexpressing B cells drive follicular lymphoma progression. J Clin Invest 124, 5337–5351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takatsuka S, Yamada H, Haniuda K, Saruwatari H, Ichihashi M, Renauld JC, and Kitamura D (2018). IL-9 receptor signaling in memory B cells regulates humoral recall responses. Nat Immunol 19, 1025–1034. [DOI] [PubMed] [Google Scholar]
- Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, et al. (2019). COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res 47, D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita A, Buchholz DR, and Shi YB (2004). Recruitment of N-CoR/SMRT-TBLR1 corepressor complex by unliganded thyroid hormone receptor for gene repression during frog development. Mol Cell Biol 24, 3337–3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunyaplin C, Shaffer AL, Angelin-Duclos CD, Yu X, Staudt LM, and Calame KL (2004). Direct repression of prdm1 by Bcl-6 inhibits plasmacytic differentiation. J Immunol 173, 1158–1165. [DOI] [PubMed] [Google Scholar]
- Wang JQ, Jeelall YS, Humburg P, Batchelor EL, Kaya SM, Yoo HM, Goodnow CC, and Horikawa K (2017a). Synergistic cooperation and crosstalk between MYD88(L265P) and mutations that dysregulate CD79B and surface IgM. J Exp Med 214, 2759–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Hu XJ, Zou XD, Wu XH, Ye ZQ, and Wu YD (2015). WDSPdb: a database for WD40-repeat proteins. Nucleic Acids Res 43, D339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Shi J, Yan J, Xiao Z, Hou X, Lu P, Hou S, Mao T, Liu W, Ma Y, et al. (2017b). Germinal-center development of memory B cells driven by IL-9 from follicular helper T cells. Nat Immunol 18, 921–930. [DOI] [PubMed] [Google Scholar]
- Weisel FJ, Zuccarino-Catania GV, Chikina M, and Shlomchik MJ (2016). A Temporal Switch in the Germinal Center Determines Differential Output of Memory B and Plasma Cells. Immunity 44, 116–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright G, Tan B, Rosenwald A, Hurt EH, Wiestner A, and Staudt LM (2003). A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc Natl Acad Sci U S A 100, 9991–9996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright GW, Huang DW, Phelan JD, Coulibaly ZA, Roulland S, Young RM, Wang JQ, Schmitz R, Morin RD, Tang J, et al. (2020). A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell 37, 551–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon HG, Chan DW, Huang ZQ, Li J, Fondell JD, Qin J, and Wong J (2003). Purification and functional characterization of the human N-CoR complex: the roles of HDAC3, TBL1 and TBLR1. EMBO J 22, 1336–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon HG, Choi Y, Cole PA, and Wong J (2005). Reading and function of a histone code involved in targeting corepressor complexes for repression. Mol Cell Biol 25, 324–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Kalkum M, Chait BT, and Roeder RG (2002). The N-CoR-HDAC3 nuclear receptor corepressor complex inhibits the JNK pathway through the integral subunit GPS2. Mol Cell 9, 611–623. [DOI] [PubMed] [Google Scholar]
- Zhang XM, Chang Q, Zeng L, Gu J, Brown S, and Basch RS (2006). TBLR1 regulates the expression of nuclear hormone receptor co-repressors. BMC Cell Biol 7, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccarino-Catania GV, Sadanand S, Weisel FJ, Tomayko MM, Meng H, Kleinstein SH, Good-Jacobson KL, and Shlomchik MJ (2014). CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat Immunol 15, 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. Related to Figure 3. Differentially expressed genes in Tbl1xr1- D370Y/WT GCB (|log2FC|≥1.5 and FDR<0.05). Significantly enriched pathways for the Tbl1xr1-D370Y/WT GCB transcriptional signature.
Table S3. Related to Figure 5. Spectral counts for BCL6 RIME assay in the OCI-Ly1 cell line. Spectral counts for TBL1XR1 BioID assays in OCI-Ly1 and U2932 cell lines.
Table S4. Related to Figure 5. Differentially expressed genes in IμBcl6-Tbl1xr1-D370Y/WT GCB (|log2FC|≥1.5 and FDR<0.05). Differentially expressed genes in Tbl1xr1-KO/KO GCB (|log2FC|≥1.5 and FDR<0.05).
Table S5. Related to Figure 7. Somatic mutation profiles of DLBCL specimens used for CyTOF immunophenotyping.
Data Availability Statement
RNA sequencing data from this paper have been deposited in the Gene Expression Omnibus (GEO) database, under accession number GSE139059. All raw mass spectrometry files have been deposited at the MassIVE archive (massive.ucsd.edu). Code used for analysis is available upon request.