

Abstract
The underlying neurological events accompanying dog domestication remain elusive. To reconstruct the domestication process in an experimental setting, silver foxes (Vulpes vulpes) have been deliberately bred for tame vs. aggressive behaviors for more than 50 generations at the Institute for Cytology and Genetics in Novosibirsk, Russia. The hypothalamus is an essential part of the hypothalamic-pituitary-adrenal axis and regulates the fight-or-flight response, and thus, we hypothesized that selective breeding for tameness/aggressiveness has shaped the hypothalamic transcriptomic profile. RNA-seq analysis identified 70 differentially expressed genes. Seven of these genes, DKKL1, FBLN7, NPL, PRIMPOL, PTGRN, SHCBP1L, and SKIV2L, showed the same direction expression differences in the hypothalamus, basal forebrain, and prefrontal cortex. The genes differentially expressed across the three tissues are involved in cell division, differentiation, adhesion, and carbohydrate processing, suggesting an association of these processes with selective breeding. Additionally, 159 transcripts from the hypothalamus demonstrated differences in abundances of alternative spliced forms between the tame and aggressive foxes. WGCNA analyses also suggested gene modules in hypothalamus were significantly associated with tame vs. aggressive behavior. Pathways associated with these modules include signal transduction, interleukin signaling, cytokine-cytokine receptor interaction, and peptide ligand-binding receptors (e.g., G-protein coupled receptor [GPCR] ligand binding). Current studies reveal selection for tameness vs. aggressiveness in foxes is associated with unique hypothalamic gene profiles partly shared with other brain regions and highlight differentially expressed genes involved in such biological processes as development, differentiation, and immunological responses. The role of these processes in fox and dog domestication remains to be determined.
Keywords: Domestication, Canine, Evolutionary Selection, Genetics, Breeding, Dog, Brain, Hypothalamus, RNA-seq, Behavior
1. INTRODUCTION
Domestication of various animal species has been occurring historically for at least the last 10,000 years1. Domesticated species tend to show enhanced social affiliation but reduced fear and aggression towards humans and individuals within their own species2–4. Such changes are likely multifaceted in origin but may be accompanied by physiological and anatomical alterations, collectively referred to as “domestication syndrome”5,6. Domesticated dogs (Canis familiaris) evolved approximately 15,000 years ago from wolf ancestors (Canis lupus)7,8. In the process, stress responses became blunted, coat color pellage began to acquire unusual morphs, such as spotting, the tail went from being exclusively straight to sometimes curled, and the cranium became shortened with overall more rounding of the skull9,10. Additionally, differences in human-directed behavior of wolf and dog pups are observed. For instance, communication patterns began to diverge relative to ancestral wolves, such that domesticated dog puppies display a greater number of communication signals to stimulate social interactions with human caregivers, including distress vocalizations, tail wagging, and direct gazing at the face of humans11,12. While domestication of diverse species occurred at different times throughout evolutionary history and was presumably driven in each case by unique extrinsic factors, in all species, such alterations were most likely due to changes in gene networks that affected neurobehavioral, hormonal, and structural responses13. In support of the idea that common gene changes drive behaviors associated with domestication, the increased human-directed hypersociability demonstrated by dogs but not wolves was shown to be associated with structural variations in GTF2I and GTF2IRD114, the genes implicated in Williams-Beuren Syndrome (WBS) in humans15,16, which is characterized by hyper-social behavior.
The hypothalamus is an essential organ in orchestrating social and stress responses with the latter being through the hypothalamic-pituitary-adrenal (HPA) axis. Thus, it is one brain region that has been heavily sculpted by domestication in varying species, including domesticated dogs compared to wolf ancestors and coyotes (Canis latrans)17, as well as domesticated chickens (Gallus gallus domesticus) relative to ancestral red junglefowl (Gallus gallus). Experimental selection of red junglefowl for tameness also leads to changes in hypothalamic gene expression18–21. Comparison of wild and laboratory “domesticated” strains of rats (Rattus rattus) and mice (Mus musculus) reveal that laboratory strains possess higher densities of oxytocin (OXT)- immunoreactive (ir) and vasopressin (AVP)-ir neurons in the medial preoptic area (MPOA) of the hypothalamus22, consistent with laboratory strains being easier to handle and less aggressive and fearful compared to wild counterparts.
The most longstanding and well-known domestication experiment involves farmed silver foxes (Vulpes vulpes) that have been deliberately bred for over 50 generations in an attempt to better understand the molecular and physiological mechanisms contributing to dog domestication23. Initially, one group of foxes was selectively bred based on those who exhibited reduced fear of and increased sociableness to humans. At the other end of the spectrum, another group of foxes were selectively bred for increased aggression to humans13,24. Comparison of behavioral, biochemical, and molecular responses in the two fox populations representing the extreme ends of the continuum have provided critical insight into how these factors are altered as foxes become more or less tame. Initial studies showed that features of the HPA axis began to diverge between the two groups, as has also been documented to occur in tame vs. aggressive rats and domesticated guinea pigs compared to their wild ancestors13,24. Tame foxes show a muted stress response compared to their aggressive counterparts, as evidenced by reductions in ACTH levels, decreased ACTH response to stress, and reduced glucocorticoids in tame foxes24,25. However, gene expression of corticotrophin releasing factor (CRF) by the hypothalamus did not differ between the two fox groups25,26. Differences in serotonergic and catecholaminergic systems have been reported in the hypothalamus of tame compared to aggressive foxes, with the latter having reduced density of serotonin 1A (5-HT1A) receptors in this brain region, but demonstrating greater amounts of serotonin and noradrenaline in the anterior hypothalamus27–30. Recently, global transcriptomic changes have been identified in the prefrontal cortex, basal forebrain, and anterior pituitary gland of tame vs. aggressive foxes26,31. To determine how selection for behavior affected hypothalamic gene expression differences, RNA-seq analysis was used to examine the transriptomic profile of this brain region and compare it with transriptomic profiles of the prefrontal cortex, basal forebrain and anterior pituitary gland of the same tame and aggressive foxes.
2. Materials and Methods
2.1. Animals, sample collection, and RNA isolation
Foxes were bred at the experimental farm of the Institute of Cytology and Genetics (ICG) in Novosibirsk, Russia. All animal procedures at the ICG were performed in accordance with standards for humane care and use of laboratory animals by foreign institutions. The current study was approved by the Institutional Animal Care and Use Committees (IACUC) of the University of Illinois at Urbana-Champaign. Hypothalamic tissue was dissected from twelve tame and twelve aggressive sexually naive 1.5-yr-old male foxes. The foxes were selected based on their relatedness and behavioral scores24,32. All tame foxes used in the study had the highest behavioral score (score 4); the behavior of foxes in tame population is scored on a scale from 1 to 432. The behavior of aggressive foxes ranged from −1.5 to −3, with the later score corresponding to the most aggressive foxes; the behavior of foxes in aggressive population is scored on a scale from −1 to −432. Most of the selected foxes did not have the same parents and grandparents, with the exception being two aggressive foxes that had one common relative (a parent of one fox and a grandparent of another fox), and two pairs of tame foxes, each of which shared one grandparent. All of the selected foxes from the tame population belonged to the “elite” group of domesticated foxes, i.e., had the highest behavioral scores for tameness, while all selected foxes from aggressive population had low (most aggressive) behavioral scores33,34. Foxes were euthanized with sodium thiopental. The skull was immediately incised with a saw and the whole brain removed. The brains were cut in the sagittal plane into right and left halves. The left half of brain of each fox was fixed in formalin and used for imaging studies (Supplementary Information Appendix S1: Imaging studies for fox brain samples). The hypothalamus was immediately dissected from the right half of the brain and placed into RNAlater (Life Technologies, Grand Island, NY). The sampling was done with unaided eye and by using a set of scalpels and tissue scissors. The sample collection was video recorded, and anatomical location of the sequenced brain regions is presented in Figure S1–S3. The samples were stored at −70°C. For extraction, samples were minimally defrosted, and ~ 100 mg of hypothalamic tissue was dissected out. In accordance with the manufacture’s protocol, total mRNA was extracted using RNeasy Mini Kit (Qiagen, Valencia, CA).
2.2. RNA sequencing and quality analysis
One microgram of high quality RNA from each sample was used for sequencing. Stranded RNAseq libraries were prepared using TruSeq SBS Sequencing kit version 3 (Illumina, San Diego, CA). Libraries were barcoded and pooled and sequenced on two lanes on a HiSeq2500. Reads were single-end, stranded, and 100 base pairs in length. Sequencing results were processed by CASAVA 1.8 (Illumina, San Diego, CA). Data quality, including base quality per position across reads, GC content, and distribution of sequence length, was initially assessed with FastQC35.
2.3. Bioinformatics analyses of RNA-seq data
The raw sequences (FastQ) were checked for quality using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), followed by adapter removal by cutadapt36 and quality control using windowed adaptive quality trimming by fqtrim (https://ccb.jhu.edu/software/fqtrim/). The quality trimming was performed for phred score >30 by a sliding window scan for 6 nucleotides. Finally, reads of 30 nucleotides or longer were selected and mapped to the reference genome. In the absence of a complete fox genome assembly37–39, closely related dog genome assembly (Canis familiaris 3.1 genome) was used as the reference for mapping. This reference genome was also used in previous studies comparing the prefrontal cortex, basal forebrain, and pituitary gland of tame vs. aggressive foxes26,31. The reference genome used is available at ftp://ftp.ensembl.org/pub/release-79/fasta/canis_familiaris/dna/. The Hisat2 mapper (https://ccb.jhu.edu/software/hisat2/), which is a fast and sensitive alignment program of next-generation sequencing data40, was used for alignment of reads to the genome. The program FeatureCounts41 was then used to quantify read counts that mapped to Ensembl annotation genes (CanFam3.1). The differentially expressed genes (DEGs) between aggressive and tame foxes were determined by edgeR-robust42 (Supplementary Information Appendix S2: DEGs and all other genes list). A false discovery rate (FDR) < 0.05 was used as a significance threshold.
We also compared the expression pattern of the genes DE of the HYP from the current study with that of the PFC and BFB regions from the previous study31. For this analysis, the read count data of a total of 185 genes that were exclusively DE in each of the three regions (PFC, BFB, HYP) were used by PCA. Each gene was labeled to include metadata if it was DE to HYP or BFB or HYP. The PCA was conducted seprately to compare how the gene expression of the 185 genes vary in one brain region relative to the other two regions. We also conducted PCA to compare upregulated vs. downregulated genes within HYP. For this analysis, a subset of genes differentially expressed in HYP was used. This included genes that were DE in HYP but not PFC or BFB, as well as genes that were commonly DE among the three brain regions. Thus, this analysis was conducted to determine the pattern of upregulation and downregulation of genes within HYP which may also be linked to differential regulation of genes in the two regions of the fox brain. The count data included metadata specifying which gene was either upregulated or downregulated in HYP. All the analysis was done using the R package ‘ggfortify.’
2.4. Description of PCA analysis.
The read count data of all identified genes across the 24 tame and aggressive fox samples was used to perform PCA of gene expression. The calculation of the principal components and visualization of the plot was performed in R (package pca3d).
2.5. Functional Analysis
An information approach43 was adopted to infer gene expression network analysis based on genes identified as DE based on edgeR-robust anlaysis. In this method, mutual information (MI) of expression variation was calculated in pairwise manner between genes across samples. The MI measures the information content between two genes, and determines how much knowing one gene would predict variability of the other. The Maximum Relevance Minimum Redundancy (MRMR) method44 was then used from the mutual information matrix to infer gene expression networks. Degree centrality and prediction of key players were inferred from the MI networt as described earlier45.
Functional differences in DEGs were also analyzed with DAVID Bioinformatics Resources 6.846 with the Gene Ontology (GO) terms database and the dog genome as the background database.
Finally, the ClueGO app47 in Cytoscape48 was used for pathway enrichment analyses based on DEG upregulated in tame foxes (decreased in aggressive foxes) and those upregulated in aggressive foxes. After a gene list is imported into this app, the ones meeting set criteria, GO level, # genes, % genes, are selected. A p value is then determined based on the Fisher exact test. Terms are connected based on shared genes, i.e. a kappa score, which is used to define groups. The edges of the diagram indicate the kappa score with thicker lines delineating the terms have more genes in common than others. Size of the colored nodes indicates the enrichment significance with bigger terms being the most significance ones. Color of each node indicates that proportion of genes from each cluster that are associated with a given term.
2.6. Alternative splicing analysis
Differences in alternative splicing frequencies between tame and aggressive hypothalamic reads were further analyzed by replicate Multivariate Analysis of Transcript Splicing (rMATS)49 from bam files generated by TopHat. Because rMATS requires reads of equal lengths, post-filtered reads were further filtered using SAMtools to remove all reads < 100 nucleotides in length. Ensembl annotation, version 1.93, was used with rMATS to investigate reads crossing splicing junctions. Transcripts that showed skipped exon differences were further analyzed based on human orthologs and all human encoding protein genes with WebGestalt for functional enrichment in diseases (Disgenet database), phenotype (Human_Phenotype _Orthology), and pathways (KEGG).
2.7. Weighted gene coexpression network analysis
Weighted gene coexpression network analysis (WGCNA) describes network relationships among a collection of genes based on the pattern of gene expression correlations. We utilized the WGCNA package in R50 to find unsigned weighted gene coexpression modules. The blockwiseModules function was run with a soft thresholding power of 18 to indicate the gene cluster. In order to identify significant gene modules, we calculated the correlation score between genes and trait to rank the gene cluster, which utilized the eigengene network methodology by using Pearson Correlation/Bicor. The trait data include the behavioral score of each sample (−3 to 4), as detailed in24,32 (Supplementary Information Appendix S3: Behavioral scores for tame and aggressive foxes). For each module, we calculated the MM, which describes the correlation of the modules and genes. We applied the bicor correlation function in MM to calculate the biweight mid-correlations of the ME, expression matrix, and corresponding p-value. Functional enrichment analysis also known as over-representation analysis (ORA) or gene set enrichment analysis for GO Terms was performed by g:Go st on input gene lists. This application maps genes to known functional information sources and detects statistically significantly enriched terms. The statistical domain scope was all hypothalamic genes identified in tame and aggressive foxes, and the significance threshold was Benjamini-Hochberg FDR ≤ 0.05. For those significant gene modules, potential interaction with queried genes in the module and associated genes were identified with GeneMANIA app51 in Cytoscape48. With this program, black node color represent queried genes, and grey node color indicates associated genes. The size of the nodes for the associated genes indicates the strength of the correlation. For this app, we limited it to searching for co-expressed genes (designated with violet edges) and co-localized genes (designated with blue edges). Finally, the ClueGO app47 in Cytoscape48 was used for pathway enrichment analyses. Details of this program and significance of size and color of nodes is detailed above. We use the exportNetworkToCytoscape function in WGCNA package to export the module of interest, midnight blue, into Cytoscape48. This function exports the edge and node list files into Cytoscape48. In Cytoscape48, we select the gene with the largest number of connection to other genes in the network as a hub gene.
2.8. qPCR analysis
The quantitative polymerase chain reaction (qPCR) procedure was performed on the Applied Biosystems 7500 Real-Time PCR System (Carlsbad, CA) using the Applied Biosystems PowerUP SYBR Green Master Mix kit (catalogue number A25741). The procedures for this kit were followed. Primer sequences for the candidate genes examined are listed in Table S1, and primers were purchased from IDT (Coralville, IA). All samples were run in duplicate or triplicate. The qPCR conditions employed were 1) 15 minutes at 95°C for polymerase activation 2) 40 cycles of: denaturation, 40 seconds at 94°C; annealing, 40 seconds at 56°C; and extension, 1.50 minutes at 72°C 3) dissociation melt curve analysis from 60°C to 90°C. Gene expression for each of the test genes was normalized to the expression pattern of the housekeeping gene, succinate dehydrogenase complex, subunit A (SDHA), a gene that previously has not shown DE between tame and aggressive foxes in the anterior pituitary gland26 nor in the current studies for the hypothalamus. Mean ΔCt values for each test gene relative to SDHA expression were analyzed by using SAS (version 9.4, SAS, Cary, NC) and ANOVA analyses with treatment as main effects and individual fox as statistical unit. Graphs for these data represent 2−ΔΔCt values where mean gene expression values of aggressive foxes was set to 1 (by subtracting mean ΔCT of aggressive fox group from all individual tame ΔCT values to obtain ΔΔCT values, and then calculating the 2−ΔΔCt values). Thus, the expression pattern (fold changes of individual genes) in the tame group of foxes was considered relative to those of the results obtained for the aggressive foxes.
3. Results
3.1. General Characterizations of Fox Hypothalamus Transcriptome
To identify gene expression differences in brains of tame vs. aggressive foxes, we performed hypothalamic RNA-seq analysis with 12 replicates in each group and compared hypothalamic transcriptome with previously analysed transcriptomes of pre-frontal cortex and basal forebrain of same foxes31. The anatomical location and connectivity of each sampled region are shown in Figure S1–S3. Table S2 summarizes the alignment of hypothalamic RNA-seq reads to the Canis familiaris 3.1 reference genome. For tame and aggressive foxes analyzed together, the average number of QC reads was ~30 million, average number of mapped reads was ~23 million, and average mapping rate was 75%. In both groups of foxes, 14,976 genes were identified altogether (Supplementary Information Appendix S2: DEGs and all other genes list). Some genes were highly expressed in both tame and aggressive foxes, though the expression level of the top 10 most abundant genes in tame foxes was different in rank order than those in the aggressive foxes (Table 1). Principal component analysis (PCA) of hypothalamic transcripts did not show clear separation between tame and aggressive individuals, which might be due to heterogeneity within hypothalamic samples (Figure 1).
Table 1.
The top expressed genes in the hypothalamus of tame and aggressive foxes. The gene expression rank is shown as (GER#). The mean RPKM values are indicated in parentheses. The top 10 GER are italicized or bolded for aggressive and tame foxes, respectively.
| Gene Symbol | Gene Name | Aggressive Foxes | Tame Foxes |
|---|---|---|---|
| MBP | Myelin basic protein | GER# 1 (5467.567) | GER# 1 (4576.599) |
| PLP1 | Proteolipid protein 1 | GER# 2 (4581.421) | GER# 2 (3556.126) |
| PTGDS | Prostaglandin D2 synthase | GER# 8 (1204.963) | GER# 3 (1476.062) |
| SPARC | Secreted protein acidic and cysteine rich | GER# 4 (1538.644) | GER# 4 (1440.66) |
| APOE | Apolipoprotein E | GER# 6 (1453.506) | GER# 5 (1385.807) |
| ENSCAFG00000005106 | GER# 3 (1982.721) | GER# 6 (1302.408) | |
| SPARCL1 | SPARC like 1 | GER# 7 (1392.475) | GER# 7 (1299.786) |
| GFAP | Glial fibrillary acidic protein | GER# 5 (1500.543) | GER# 8 (1232) |
| CLU | Clusterin | GER# 15 (965.258) | GER# 9 (1189.477) |
| MT3 | Metallothionein 3 | GER# 12 (1015.945) | GER# 10 (1124.97) |
| CPE | Carboxypeptidase E | GER# 10 (1057.1) | GER# 11 (1080.682) |
| FTH1 | Ferritin Heavy Chain 1 | GER# 9 (1108.135) | GER# 17 (868.742) |
Figure 1.
PCA analysis of all genes. A) No clear clustering is evident in the hypothalamic transcriptome results between tame vs. aggressive foxes.
PCA analysis and hierarchical cluster analysis show that the gene expression profile in the fox hypothalamus (HYP) is distinct from that previously identified in the basal forebrain (BFB) and prefrontal cortex (PFC)31 of the same foxes examined in the current study (Figures 2A and B, respectively).
Figure 2.
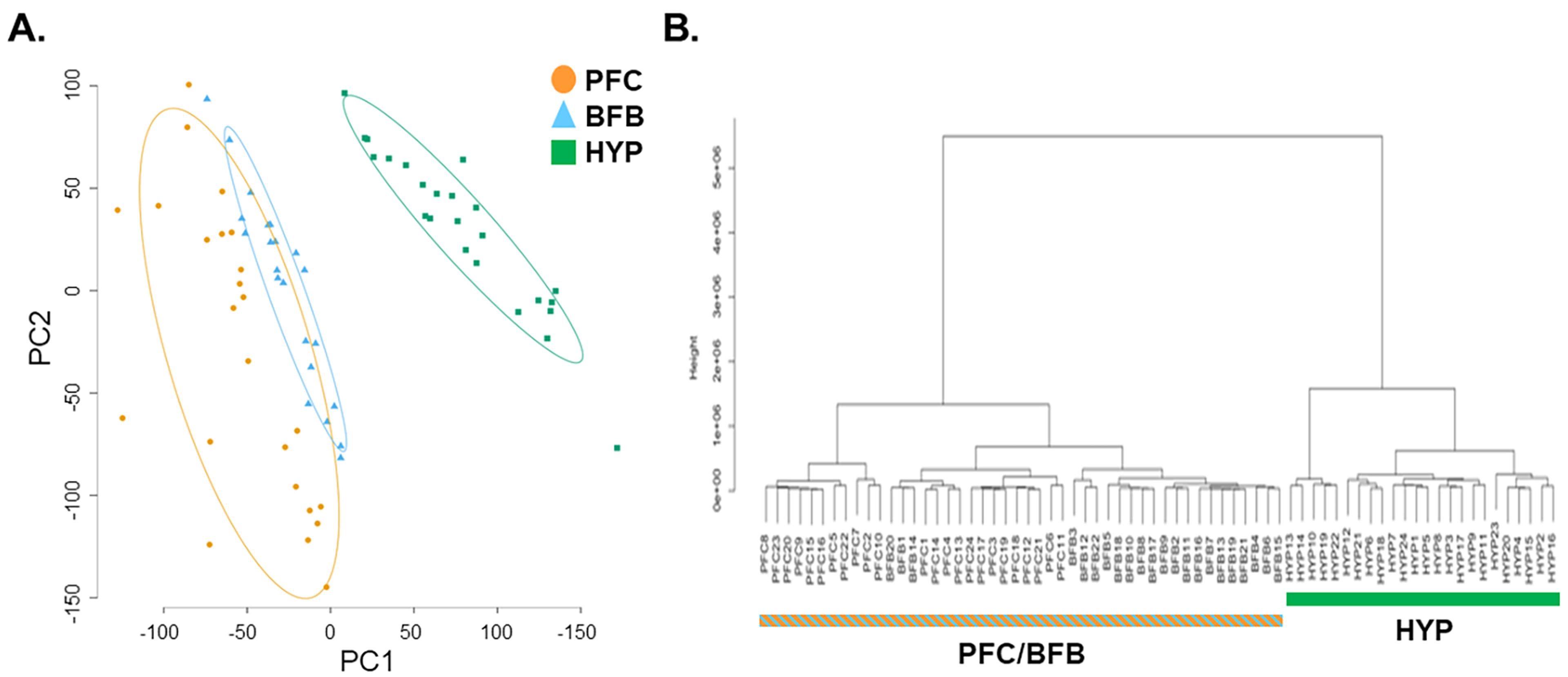
PCA plot and hierarchical clustering of current hypothalamic results and previous basal forebrain (BFB) and prefrontal cortex (PFC) gene expression results. A) The PCA plot using expression of all genes shows that the fox hypothalamic transcriptome is distinct from that of PFC (cortex) and BFB (forebrain). B) Similarly, hierarchical cluster analysis using expression of all genes also shows that the fox hypothalamus transcriptome is distinct from that of cortex and forebrain.
3.2. Gene Expression Differences
Comparison of hypothalamic gene expression between tame and aggressive foxes identified 70 differentially expressed genes (DEGs) at an FDR q-value of 0.05 (Figure 3A; Supplementary Information Appendix S2: DEGs and all other genes list). Predictably, a hierarchical clustering heatmap based on the DEGs in this analysis revealed relatively distinct separation between tame vs. aggressive foxes (Figure 3B), however, as with the PCA analysis, heterogeneity of hypothalamic samples within the tame and aggressive groups was observed.
Figure 3.
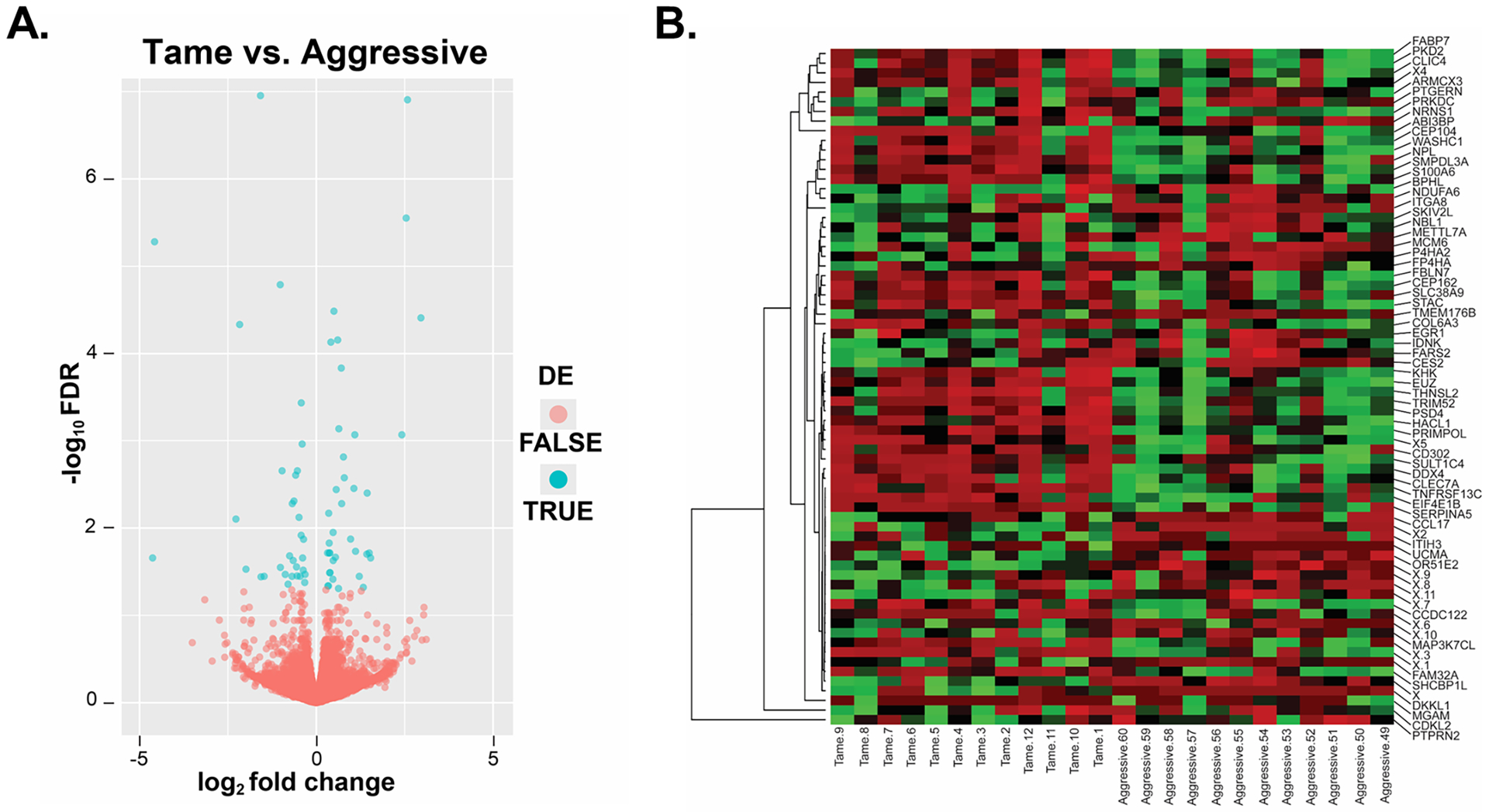
Volcano plot and hierarchical clustering heatmap analyses of genes differentially expressed in tame vs. aggressive foxes. A) The volcano plot represents the relationship of each gene’s log2 fold change vs -log10 FDR. Red points represent those genes that are not differentially expressed in the hypothalamus of tame vs. aggressive foxes; whereas those in blue are significantly different (FDR < 0.05, and log2 fold change > 0 or log2 fold change < 0). B) Consideration of only those hypothalamic genes that were DE based on EdgeR analyses revealed overall separate clustering between tame vs. aggressive foxes with heatmap analysis. The heatmap was generated with http://heatmapper.ca/137.
Among the 70 DEGs, 33 genes showed increased expression in HYP of tame foxes with log2 fold changes ranging from −4.6 to −0.31 (mean −1.17; SD 1.18) and 37 showed increased expression in HYP of aggressive foxes with log2 fold changes ranging from 0.31 to 2.96 (mean 0.93; SD 0.71) (Figure 3A). The genes up- and down-regulated in tame foxes vs aggressive foxes at FDR ≤0.01 are presented in Tables 2 and 3, respectively.
Table 2.
Genes downregulated in aggressive vs. tame foxes with a FDR ≤ 0.01.
| GeneID (CanFam3.1) | Gene Symbol | Gene Name | q-Value | Log2 Fold Change (aggressive vs. tame) | Fold Change (aggressive vs. tame) |
|---|---|---|---|---|---|
| ENSCAFG00000004656 | ITGA8 | integrin subunit alpha 8 | 1.11e−7 | −1.58 | 0.33 |
| ENSCAFG00000003681 | DKKL1 | dickkopf like acrosomal protein 1 | 1.63e−5 | −1.02 | 0.49 |
| ENSCAFG00000002070 | FAM32A | family with sequence similarity 32 member A | 4.66e−5 | −2.17 | 0.22 |
| ENSCAFG00000023837 | PTGFRN | prostaglandin F2 receptor inhibitor | 0.0004 | −0.42 | 0.74 |
| ENSCAFG00000009483 | FARS2 | phenylalanyl-tRNA synthetase 2, mitochondrial | 0.001 | −0.40 | 0.76 |
| ENSCAFG00000004673 | KHK | ketohexokinase | 0.002 | −0.53 | 0.69 |
| ENSCAFG00000001254 | EGR1 | early growth response 1 | 0.002 | −0.97 | 0.51 |
| ENSCAFG00000000824 | P4HA2 | prolyl 4-hydroxylase subunit alpha 2 | 0.002 | −0.58 | 0.67 |
| ENSCAFG00000028566 | NRSN1 | neurensin 1 | 0.005 | −0.63 | 0.65 |
| ENSCAFG00000032583 | METTL7A | methyltransferase like 7A | 0.005 | −0.67 | 0.62 |
| ENSCAFG00000029948 | IDNK | IDNK, gluconokinase | 0.007 | −0.48 | 0.71 |
| ENSCAFG00000015068 | ITIH3 | inter-alpha-trypsin inhibitor heavy chain H3 | 0.008 | −2.27 | 0.21 |
| ENSCAFG00000015137 | NBL1 | neuroblastoma suppressor of tumorigenicity 1 | 0.01 | −0.42 | 0.74 |
| ENSCAFG00000017704 | ARMCX3 | armadillo repeat containing X-linked 3 | 0.01 | −0.36 | 0.78 |
Table 3.
Genes upregulated in aggressive vs. tame foxes with a FDR ≤ 0.01.
| GeneID (CanFam3.1) | Gene Symbol | Gene Name | q-Value | Log2 Fold Change (aggressive vs. tame) | Fold Change (aggressive vs. tame) |
|---|---|---|---|---|---|
| ENSCAFG00000006875 | DDX4 | DEAD-box helicase 4 | 2.81e−6 | 2.54 | 5.82 |
| ENSCAFG00000007408 | THNSL2 | threonine synthase like 2 | 3.28e−5 | 0.50 | 1.41 |
| ENSCAFG00000029000 | SERPINA5 | plasma serine protease inhibitor | 3.91e−5 | 2.96 | 7.78 |
| ENSCAFG00000004585 | TMEM176B | transmembrane protein 176B | 7.01e−5 | 0.60 | 1.52 |
| ENSCAFG00000015743 | WASHC1 | WASH complex subunit 1 | 7.42e−5 | 0.41 | 1.33 |
| ENSCAFG00000006851 | SLC38A9 | solute carrier family 38 member 9 | 0.0001 | 0.71 | 1.63 |
| ENSCAFG00000007746 | PRIMPOL | primase and DNA directed polymerase | 0.0007 | 0.64 | 1.56 |
| ENSCAFG00000003841 | MGAM | maltase-glucoamylase | 0.0008 | 2.42 | 5.35 |
| ENSCAFG00000009506 | ABI3BP | ABI family member 3 binding protein | 0.0008 | 1.09 | 2.13 |
| ENSCAFG00000004768 | STAC | SH3 and cysteine rich domain | 0.001 | 0.77 | 1.70 |
| ENSCAFG00000023655 | SULT1C4 | sulfotransferase family 1C member 4 | 0.002 | 0.79 | 1.73 |
| ENSCAFG00000012226 | COL6A3 | collagen type VI alpha 3 chain | 0.003 | 1.06 | 2.09 |
| ENSCAFG00000013160 | NPL | N-acetylneuraminate pyruvate lyase | 0.004 | 0.56 | 1.48 |
| ENSCAFG00000017553 | S100A6 | S100 calcium binding protein A6 | 0.005 | 0.72 | 1.64 |
| ENSCAFG00000006555 | PRKDC | protein kinase, DNA-activated, catalytic polypeptide | 0.007 | 0.35 | 1.28 |
| ENSCAFG00000016848 | acyl-coenzyme A thioesterase 1 | 0.01 | 0.47 | 1.39 | |
| ENSCAFG00000013504 | CLEC7A | C-type lectin domain containing 7A | 0.01 | 0.97 | 1.96 |
| ENSCAFG00000019498 | CEP104 | centrosomal protein 104 | 0.01 | 0.37 | 1.29 |
While a different number of genes were differentially expressed (DE) among HYP, PFC31 , and BFB31, as shown in the Venn diagram (Figure 4A), there were selected genes that were shared between one or more brain regions (Figures 4B and C). Overall, out of 70 DE genes identified in the HYP, 7 genes (10 %) showed the same pattern of differential expression in the HYP, PFC and BFB31 (Figure 4D; Table S3). Two of these genes (PRIMPOL and SHCBP1L) were also DE in anterior pituitary26 (Table S3). In all three brain regions (HYP, PFC, and BFB), DKKL1, FBLN7, PTGFRN, and SKIV2L were upregulated in tame foxes, whereas NPL, PRIMPOL, and SHCBP1L were upregulated in aggressive foxes.
Figure 4.
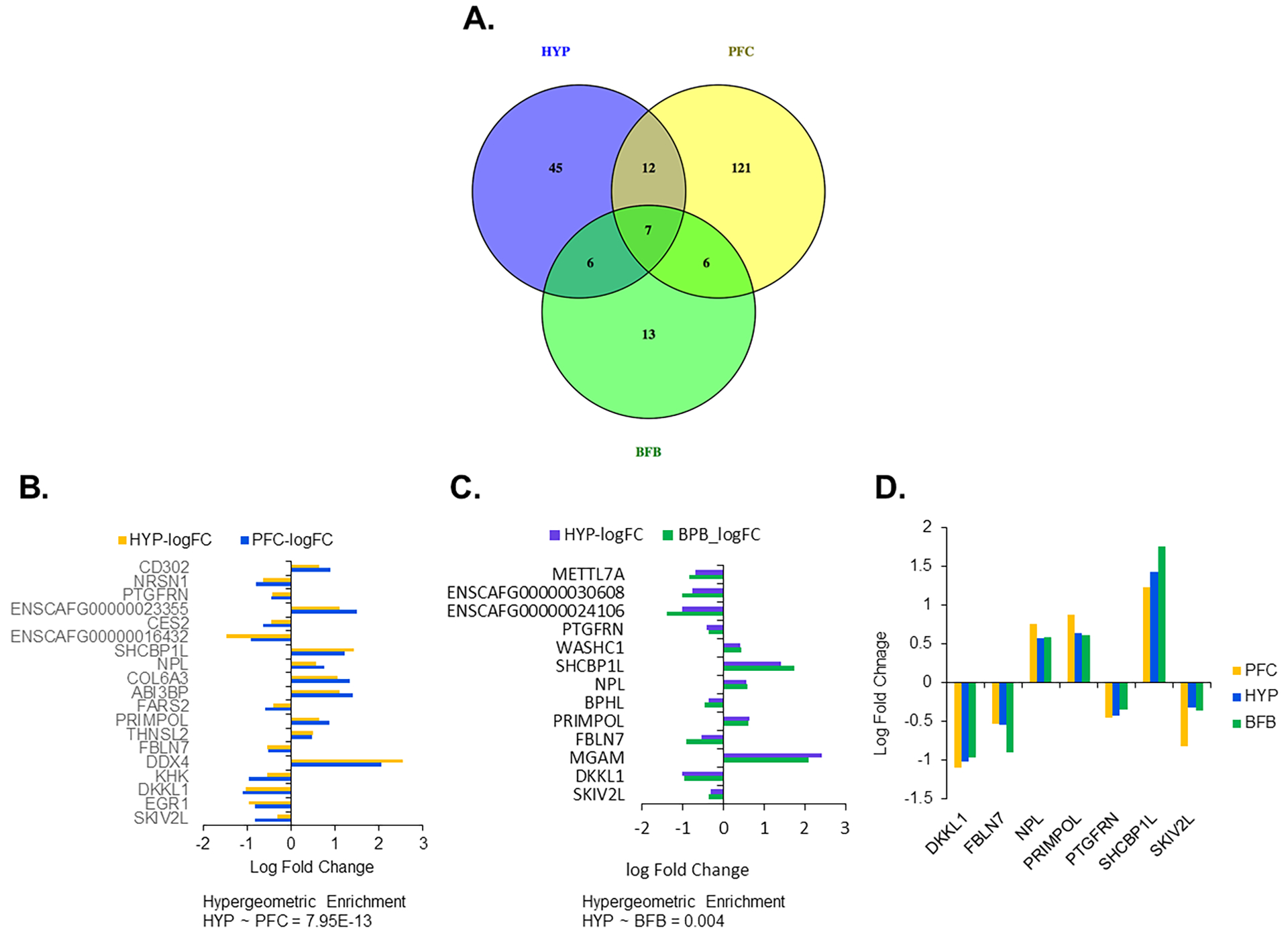
Comparison of DEG identified in hypothalamus compared to those previously identified in the prefrontal cortex (PFC) and basal forebrain (BFB). A) Venn diagram showing number of DEG common or specific to hypothalamus, frontal cortex or forebrain. B and C) The expression (log fold changes between tame and aggressive) of the common genes between hypothalamus and PFC (B) and between hypothalamus and BFB (C). The enrichment was determined by calculating p-value of hypergeometric test from the number of common genes, specific genes to each and the total number of genes for each comparison. D) A column graph showing the expression (log fold changes between tame and aggressive) of the common genes among hypothalamus, PFC and BFB.
We also compared expression patterns of DEGs that were DE in each of the three brain regions (PFC, BFB, HYP) and accounted in total for 185 genes. The read counts of those 185 genes extracted from the current study (for HYP) and the previous study31 (for PFC and BFB) were analyzed by PCA (Figure S4). The dataset included samples of the three brain regions from both tame and aggressive animals (total 70 samples, this number is due to the fact that two samples were dropped from the analysis of BFB31). In addition, the dataset included a metadata information column specifying which gene was DE in which brain region. Each dot in the PCA plot represents a brain sample, and the samples representing the three brain regions have been color coded. Thus, in each plot within this figure, a comparison was made of specific brain region with the other two regions. This analysis revealed that although these 185 genes show distinct groupings in expression in different regions, PC1 explains a lower level of variance (85.4%) in the hypothalamus (HYP) compared to that of the PFC (96.79%) and BFB (93.88%). Thus, based on the difference in variance for PC1 for the HYP , it is possible that these 185 genes are differentially regulated in the hypothalamus than in PFC and BFB, although additional studies would be needed to confirm this potential differential regulation. In addition to comparing brain regions, we also compared how the DE genes (upregulated vs. downregulated) varied within HYP (Figure S5). For this purpose, genes were selected if they were DE in HYP but not PFC or BFB (n = 45) along with genes that were commonly DE in all three brain regions (n =7). The purpose of using this subset of genes was to identify the differential expression pattern of genes in HYP whose regulation may be linked to differential regulation of genes in PFC and BFB. The count data included metadata specifying whether each gene was up-regulated or downregulated in HYP. This analysis showed that the PC1 explains a relatively higher level of variance in the BFB (90.81%) compared to that of the PFC (88.23%) and HYP (88.8%). Thus, based on this variance within PC1, these genes may be regulated in the HYP in a similar manner to PFC but regulation of these genes in both of these brain regions differ from the potential regulation in the BFB.
Comparison of 70 hypothalamic DEGs with genomic regions associated with selection for behavior37 identified 10 DEGs located in such regions (CDKL2, CLEC7A, FBLN7, PSD4, PTGFRN, STAC, THNSL2, TRIM52, WASHC1, and ENSCAFG00000009852), three of which were also DEGs in PFC (THNSL2), BFB (WASHC1) or both PFC and BFB (FBLN7 and PTGFRN).
3.3. Functional Annotation
3.3.1. GO and Pathway Analysis
Functional analysis for GO terms and pathways (KEGG and Reactome) was performed with ClueGO47, a Cytoscape app48. Groups of genes analyzed included genes upregulated in tame foxes and, separately, genes upregulated in aggressive foxes, as determined by edgeR-robust analysis. No significant GO terms were identified in these groups, but both sets of DEGs were associated with functional enrichment for KEGG/Reactome pathways (Figure 5). Primary pathways enriched in upregulated genes in tame foxes included phase I functionalization of compounds (including biological oxidations and metabolism), neutrophil degranulation (including innate immune system), metabolism of proteins, and extracellular matrix organization (Figure 5A). Those pathways enriched in upregulated genes in aggressive foxes included mTOR signaling pathway, innate immune system, endocytosis, and cilium assembly (Figure 5B).
Figure 5.
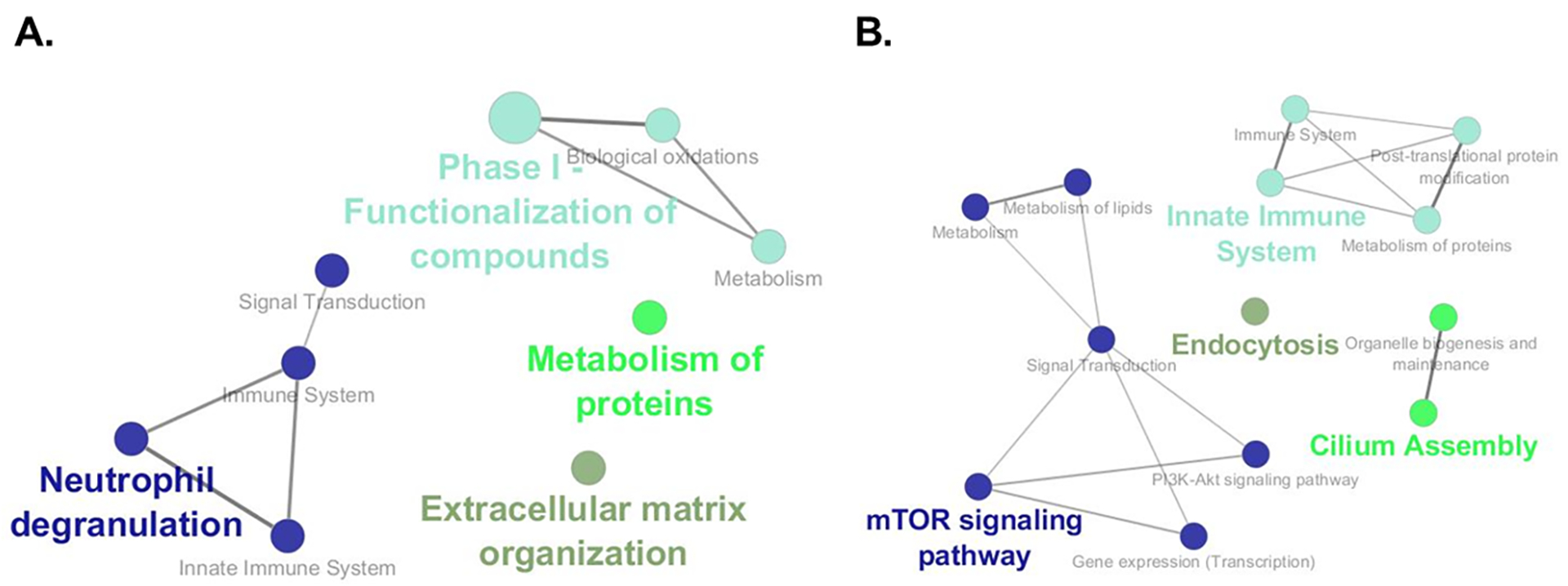
Pathways enriched based on upregulated genes in tame and those upregulated in aggressive foxes. ClueGo App47 was used to examine for enriched KEGG and Reactome pathways based on DEG upregulated in tame (A) and those upregulated in aggressive (B). Primary pathways enriched based on upregulated genes in tame foxes included phase I functionalization of compounds, including biological oxidations and metabolism, neutrophil degranulation, including innate immune system, metabolism of proteins, and extracellular matrix organization. Those pathways enriched based on upregulated genes in aggressive foxes include mTOR signaling pathway, innate immune system, endocytosis, and cilium assembly.
3.3.2. Network Analysis and Prediction of Key Players
Gene expression network analysis of 70 hypothalamic DEGs predicted several transcriptional interactions between genes that are downregulated in aggressive foxes (those listed in blue on Figure 6A) and those upregulated in aggressive foxes (those listed in red on Figure 6A). This network analysis, specifically the degree centrality of the interacting genes (as detailed in the Methods) was also used to identify the top 10 genes that are considered key players (Table 4). In this list, most are upregulated in aggressive foxes and involved in a variety of processes. The only key player gene that was downregulated in aggressive foxes is ITGA8 (the most significant upregulated gene in tame foxes), which mediates varied cellular processes, including cell adhesion, cytoskeleton rearrangement, and activation of cell signaling pathways. The other 9 key player genes are upregulated in aggressive foxes: CD302, CEP104, FUZ, PRIMPOL, NPL, PRKDC, S100A6, SMPDL3A, and TRIM52. Using the GTEx Portal (a human based database located at https://gtexportal.org/home/), a heatmap was generated based on key player genes that were DE in tame vs. aggressive foxes. This heatmap revealed that S100A6 and FUZ are the two most abundant genes in the hypothalamus and other brain regions of humans (Figure 6B). The expression levels of these two genes in foxes are S100A6: 221.54 and 125.71 RPKM values in aggressive and tame foxes, respectively (FDR = 0.00525), and FUZ: 11.30 and 8.95 RPKM values in aggressive and tame foxes, respectively (FDR = 0.04605). Two of these genes (PRIMPOL and NPL) are also DE in PFC and BFB31 and one gene (CD302) is DE in PFC31; all three genes showed differential expression in these bran regions in the same direction as in hypothalamus samples of tame and aggressive foxes.
Figure 6.
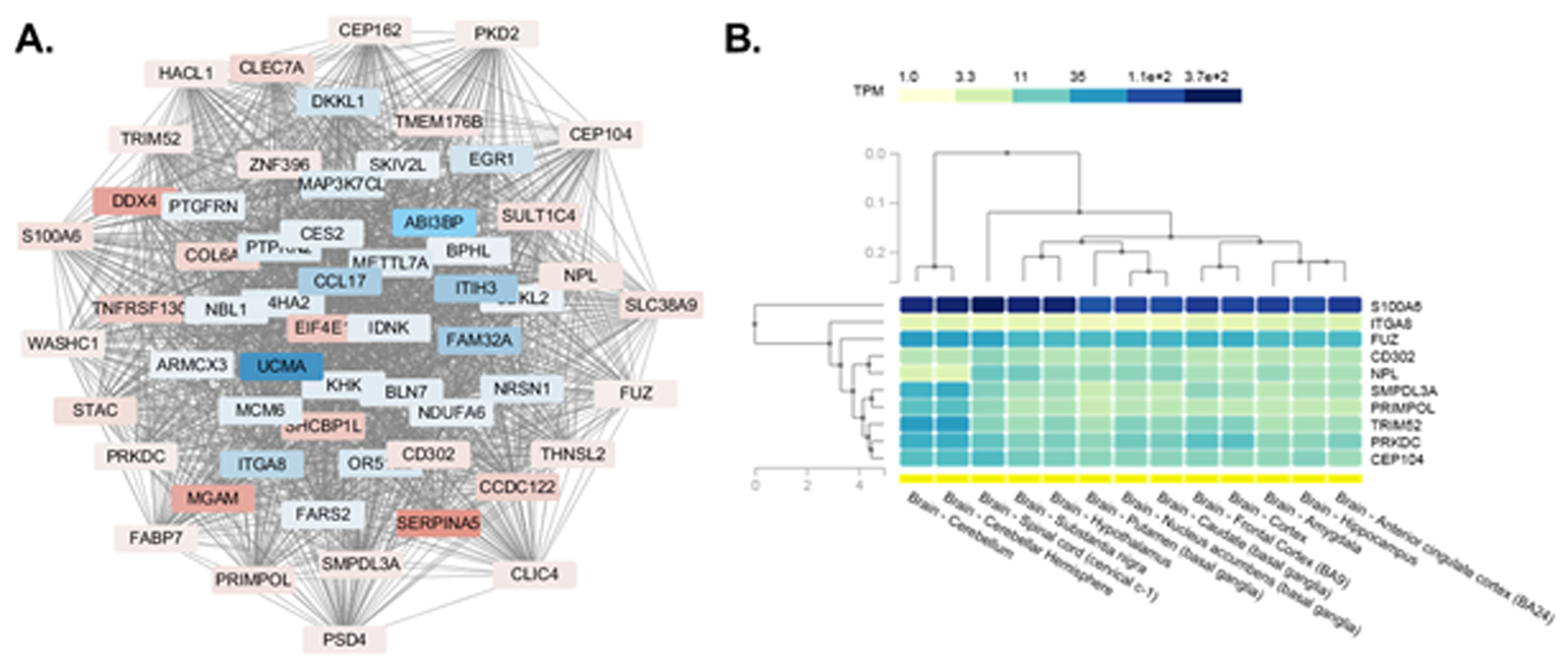
Gene expression network to identify ten hub or “key player” genes and their expression pattern in human brain regions. A) The diagram shows the inter-relationships of the gene to be DE based on edgeR robust. As shown, there are connections between almost all 70 genes that are DE. This network analysis was used to identify ten key player genes within the 70 DEG. Those in blue represent genes that are downregulated in aggressive relative to tame foxes; whereas, those in red indicate genes that are upregulated in aggressive relative to tame foxes. B). Analysis of the expression pattern for the ten key player genes in various human brain regions. The GTEx Portal site (https://gtexportal.org/home/) was screened to determine the expression pattern of the identified ten key player genes that were DE in tame vs. aggressive foxes. As shown, S100A6 followed by FUZ has greatest expression in all human brain regions, including the hypothalamus.
Table 4.
The top 10 DEGs that are considered to be key players in aggressive vs. tame foxes.
| GeneID (CanFam3.1) | Gene Symbol | Gene Name | q-Value | Log2 Fold Change (aggressive to tame) | Directionality in terms of expression in tame foxes |
|---|---|---|---|---|---|
| ENSCAFG00000030080 | CD302 | CD302 molecule | 0.04 | 0.62 | ↓ |
| ENSCAFG00000019498 | CEP104 | centrosomal protein 104 | 0.01 | 0.36 | ↓ |
| ENSCAFG00000003502 | FUZ | fuzzy planar cell polarity protein | 0.05 | 0.33 | ↓ |
| ENSCAFG00000004656 | ITGA8 | integrin subunit alpha 8 | 1.11e−7 | −1.58 | ↑ |
| ENSCAFG00000007746 | PRIMPOL | primase and DNA directed polymerase | 0.0007 | 0.64 | ↓ |
| ENSCAFG00000013160 | NPL | N-acetylneuraminate pyruvate lyase | 0.003 | 0.56 | ↓ |
| ENSCAFG00000006555 | PRKDC | protein kinase, DNA-activated, catalytic polypeptide | 0.007 | 0.35 | ↓ |
| ENSCAFG00000017553 | S100A6 | S100 calcium binding protein A6 | 0.005 | 0.72 | ↓ |
| ENSCAFG00000001002 | SMPDL3A | sphingomyelin phosphodiesterase acid like 3A | 0.02 | 0.54 | ↓ |
| ENSCAFG00000012697 | TRIM52 | tripartite motif containing 52 | 0.03 | 0.38 | ↓ |
The hypothalamus, similar to other brain regions, includes a heterogenous mix of cells, and thus, we searched each of these 10 genes with the Brain RNA-Seq database (https://www.brainrnaseq.org/)52 to define the potential neuronal cells expressing these genes in mouse brain. As shown in Figure 7, Cd302 and Itga8 are expressed in highest amounts in oligodendrocyte precursor cells (OPC). Oligodendrocytes myelinate axons within the CNS. Microglia (resident macrophages), astrocytes (support glial cells), and endothelial cells (lining blood vessels within the CNS) also expressed high amounts of Cd302. Fuz and Primpol are expressed in relatively similar amounts in all nervous tissue cells screened, including astrocytes, neurons, OPC, newly formed oligodendrocytes, myelinating oligodendrocytes, microglia, and endothelial cells. Prkdc and Smpdl3a are abundantly expressed in astrocytes with the latter also widely prevalent in endothelial cells. S100a6 expression is primarily confined to various stages of oligodendrocytes (OPC, newly formed oligodendrocytes, and myelinating oligodendrocytes). Taken together, the 10 key player genes likely originate from a variety of nervous tissue cells, including microglial or immune cells.
Figure 7.
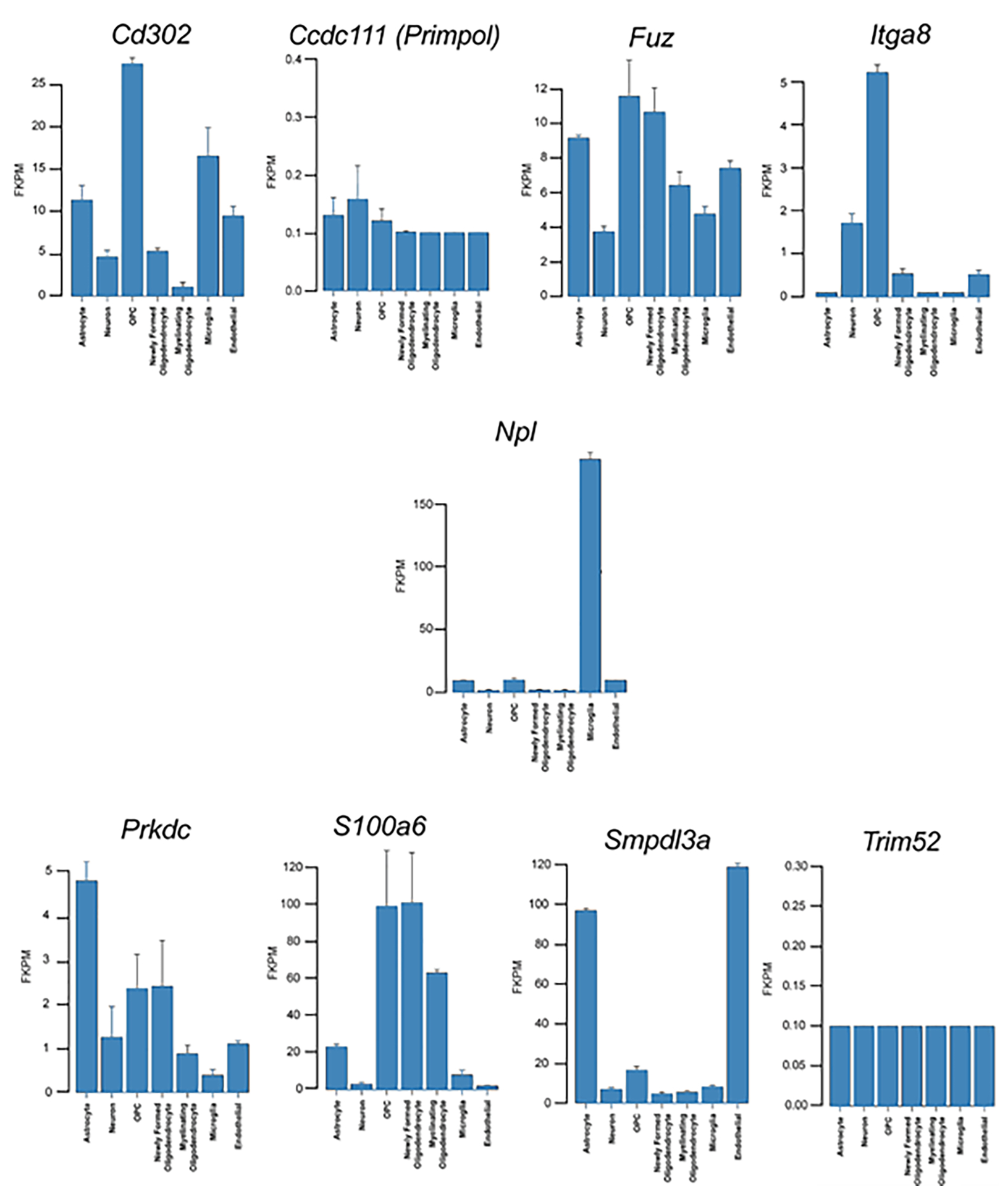
Neuronal cellular analysis of the ten key player genes. To pinpoint which neuronal cells might be contributing to the difference in expression for nine of the ten key player genes, were analyzed with the Brain RNA-Seq (http://www.brainrnaseq.org/) mouse database. No information was available for Cep104. As shown, differences in DEG might arise from various neuronal cell type and depends upon the gene. More details on individual genes are provided in the Results section.
3.4. qPCR validation
Validation by qPCR analyses revealed that NADH:Ubiquinone Oxidoreductase Subunit A6 (NDUFA6), neurensin 1 (NRSN1), integrin subunit α 8 (ITGA8), cyclin dependent kinase like 2 (CDKL2), and olfactory receptor family 51 Subfamily E Member 2 (OR51E2) were significantly upregulated in tame vs. aggressive foxes; whereas protein kinase, DNA-activated, catalytic polypeptide (PRKDC), and DNA directed primase/polymerase protein (PRIMPOL) were strongly upregulated in aggressive vs. tame foxes (p < 0.05, Figure 8). These qPCR results are consistent with the RNA-seq data. Based on the importance of OXT and AVP in modulating social behaviors and the potential linkage of these neuropeptides with dog and other species’ domestication53–56, their expression pattern was also examined with qPCR. However, no differences in expression were detected for these two genes, again consistent with our RNA-seq results (Figure S6). Expression results for other genes that did not differ significantly based on qPCR analyses between tame and aggressive foxes are shown in Table S4.
Figure 8.
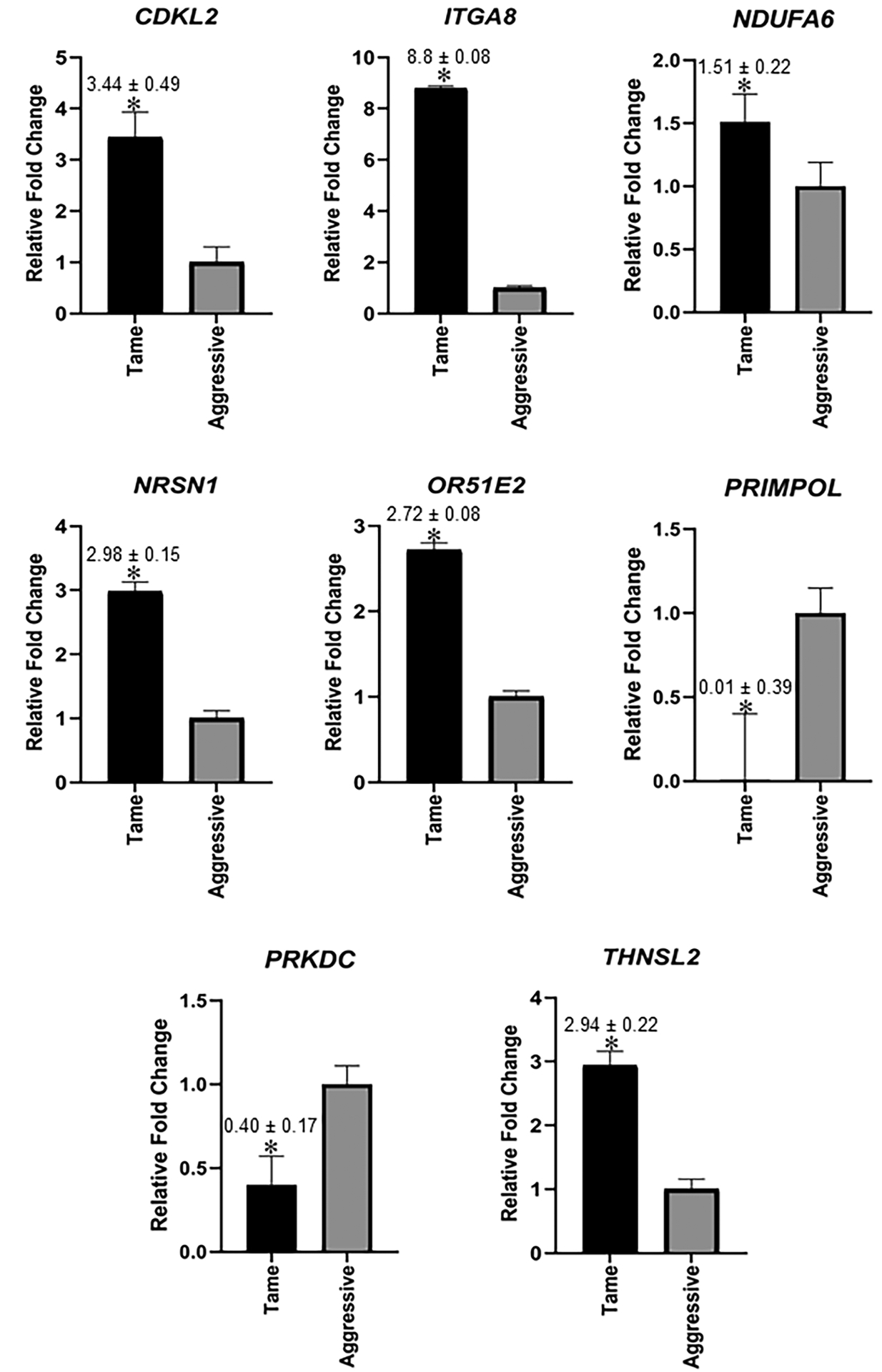
qPCR validation of select hypothalamic genes that were shown to be DE based on RNA-seq results. Graphs represent average 2−ΔΔCT ± SEM with aggressive foxes used as the reference and mean value for this group set to 1. As detailed, statistical analyses were based on ΔCT values. N= 12 individuals for tame and 12 individuals for aggressive foxes, i.e. same samples used for RNA-seq were used for qPCR analysis.
3.5. Alternative Splicing and Pathway Differences
rMATS49 was used to identify 159 different genes with exons skipped at different frequencies in tame vs. aggressive fox hypothalamic reads at FDR < 0.05 (Supplementary Information Appendix S4: rMATS splice form analysis) and 25 transcripts at FDR < 0.0001 (Table 5). Six of these genes (FBRSL1, MFAP5, MTHFSD, SPICE1, TRIM65, UNC5D) were also found among genes with skipped exons in anterior pituitary samples of tame and aggressive foxes26. WebGestalt57 was then used to examine human-associated diseases, human phenotype ontology, and KEGG pathways associated with transcript variants between tame and aggressive foxes (Figure 9). Many of these relate to central nervous system disorders, including dementia, neurodegenerative disease, learning and memory disorders, anxiety disorders, Alzheimer’s disease, repetitive compulsive behavior, dyskinesia, agitation, restlessness, stereotypy (repetitive behaviors), which may also be influenced by the fact that nervous tissue is being screened. The KEGG pathways affected include fatty acid biosynthesis, fatty acid metabolism, glutathione metabolism, glycerolipid metabolism, arachidonic acid metabolism, the glucagon signaling pathway, drug metabolism (cytochrome P450), the calcium signaling pathway, tight junction, and cell adhesion molecules.
Table 5.
Genes containing exons skipped at different frequencies in aggressive vs. tame fox hypothalamic reads (FDR < 0.0001), including conserved domains overlapping the skipped exon.
| Gene | Exon Location | Percent Reads Skipping Exon (Tame) | Percent Reads Skipping Exon (Aggressive) | FDR | Skipped in Isoforms* | In Conserved Domains |
|---|---|---|---|---|---|---|
| GSTA4 | chr12: 20433452–20433585 | 0.253 | 1.773 | 0 | X1, X2, X3 | Thioredoxin_like super family |
| GSTA4 | chr12: 20435194–20435246 | 1.162 | 6.964 | 0 | X1, X2, X3 | None |
| GSTM4 | chr6: 42206292–42206393 | 0.391 | 4.652 | 0 | X1, X2, X3, X4 (Canis forms) | GST_C_family super family |
| GSTM4 | chr6: 42206493–42206575 | 0.562 | 5.977 | 0 | X1, X2, X3, X4 (Canis forms) | Thioredoxin_like super family |
| TMA16 | chr15: 59743149–59743234 | 0.204 | 5.519 | 4.92E-11 | X1, X2, X3 (Canis forms) | Tma16 super family |
| MTHFSD | chr5: 66312701–66312815 | 0.000 | 0.929 | 6.00E-11 | None | None |
| NA | Chr23: 27215997–27216139 | 4.983 | 0.715 | 5.33E-10 | X5, X6, X7 | None |
| CDKL2 | chr32: 168420–168527 | 3.841 | 14.218 | 5.62E-10 | X1, X2, X5 | None |
| DNAJC15 | chr22: 7795378–7795430 | 3.804 | 4.762 | 1.89E-08 | X1, X2 (Canis forms) | None |
| LRP12 | chr13: 5710069–5710126 | 14.973 | 28.571 | 2.53E-08 | X1 | None |
| MFAP5 | chr27: 37034668–37034704 | 8.654 | 0.921 | 4.55E-08 | X1 | None |
| MIOS | chr14: 23205051–23205181 | 4.888 | 4.211 | 8.67E-08 | X1, X2, X3 | RING_Ubox super family |
| UNC5D | chr16: 29308689–29308857 | 12.844 | 0.895 | 7.93E-08 | None | TSP1 |
| BCAS1 | chr24: 39703194–39703265 | 2.025 | 18.421 | 1.32E-07 | X1, X2, X3, X4, X5, X6 | None |
| FAM71E1 | chr1: 106261638–106261757 | 37.838 | 7.264 | 2.04E-07 | X1, X2 | None |
| DNAJC15 | chr22: 7792280–7792354 | 1.429 | 5.000 | 1.05E-06 | X1, X2 (Canis forms) | None |
| MPZL1 | chr7: 30684041–30684144 | 1.124 | 0.413 | 3.62E-06 | None | None |
| PTGDS | chr9: 48649471–48649611 | 0.724 | 10.638 | 7.45E-06 | None | Lipocalin |
| FBRSL1 | chr26: 542234–542303 | 2.769 | 2.542 | 8.98E-06 | X1, X2, X3, X4, X7, X8, X9, X12, X13, X15, X16, X17, X18, X19, X20, X21 | None |
| KIAA0556 | chr6: 19001226–19001494 | 5.252 | 10.294 | 1.70E-05 | X1, X2, X3 | None |
| TRIM65 | chr9: 4716736–4716911 | 19.811 | 4.594 | 2.92E-05 | X1, X2 | None |
| PECAM1 | chr9: 12181163–12181220 | 21.071 | 2.933 | 3.50E-05 | X1 | None |
| TADA2A | chr9: 37232686–37232758 | 7.849 | 2.616 | 3.96E-05 | X1, X2 | None |
| NA | chr17: 48250060–48250102 | 0.880 | 12.000 | 6.17E-05 | X1, X2, X3, X4, X5, X6, X7 | None |
| EPB41L5 | chr19: 29940406–29940471 | 27.778 | 3.826 | 0.000136 | X1, X2, X3, X4, X5 | None |
If not specified, the listed isoforms represent those in Vulpes vulpes.
Figure 9.

Functional analyses based on splice variant data. Transcript isoforms shown to be differentially expressed in the hypothalamus of tame vs. aggressive foxes was analyzed with WEB-based Gene SeT AnaLysis Toolkit (WebGestalt, http://www.webgestalt.org/option.php)57. To perform this analyses, the canine genes were converted to their human orthologs, and three functional analyses were then considered: A) Diseases based on Disgenet database, which reveals that several human neurological diseases, including neurodegenerative, anxiety, and learning and memory disorders, and Alzheimer Disease; B) Phenotype based on Human_Phenotype _Orthology. This functional analysis revealed the splice form differences between the two groups of foxes are associated with human phenotypic conditions, including repetitive compulsive behavior, memory impairment, restlessness, and stereotypy. C) KEGG pathway functional analysis showed that these transcript differences are linked to various types of carbohydrate and lipid biosynthesis and metabolism, glucagon signaling pathway, drug metabolism, calcium signaling pathway, and cell adhesion molecules (CAM).
3.6. Integrative Correlation Analyses
All genes identified in tame and aggressive foxes (14,976 genes ) were further analyzed with a Weighted Gene Correlation Network Analysis (WGCNA58) to identify modules of co-expressed genes. Individual modules were represented by different colors (Figure S7) and module eigengenes (ME) were correlated to quantitative measures of tameness vs. aggressiveness. This approach identified 34 distinct color modules. Significant correlations were found for four modules: dark orange, midnight blue, dark turquoise, and pale turquoise (Figure 10). All of the modules were negatively associated with tameness, indicating the genes within these modules showed reduced expression in tame foxes. However, within the significant modules, only one gene (BPHL) in the midnight blue module overlapped with DE genes (Supplementary Information Appendix S5: gene module membership for those that are significantly correlated with tameness vs. aggressiveness). We further analyzed the hub genes in the significant modules listed above. Of these, only midnight blue contained a significant hub gene, which was BPHL. As shown in Figure S8, 135 different nodes came off the hub gene, BPHL. Of these, 70 have identified gene names and corresponding symbols. Supplementary Information Appendix S6: distribution of the other 69 DEG within the various color modules provides information on the distribution of the other 69 DEG within the various color modules. The two modules that contained most of the DEG were Grey (n = 34) and Turquoise (n = 18). The grey module correponds to sets of genes that do not cluster in any of the other modules. In other words, the grey module is reserved for genes that are not part of a co-expressed module. Since it is a random collection of genes, we have not included further details on the grey module. The rest of the genes within the Turquoise module are listed in Supplementary Information Appendix S7: DEG and other genes within the Turquoise module.
Figure 10.
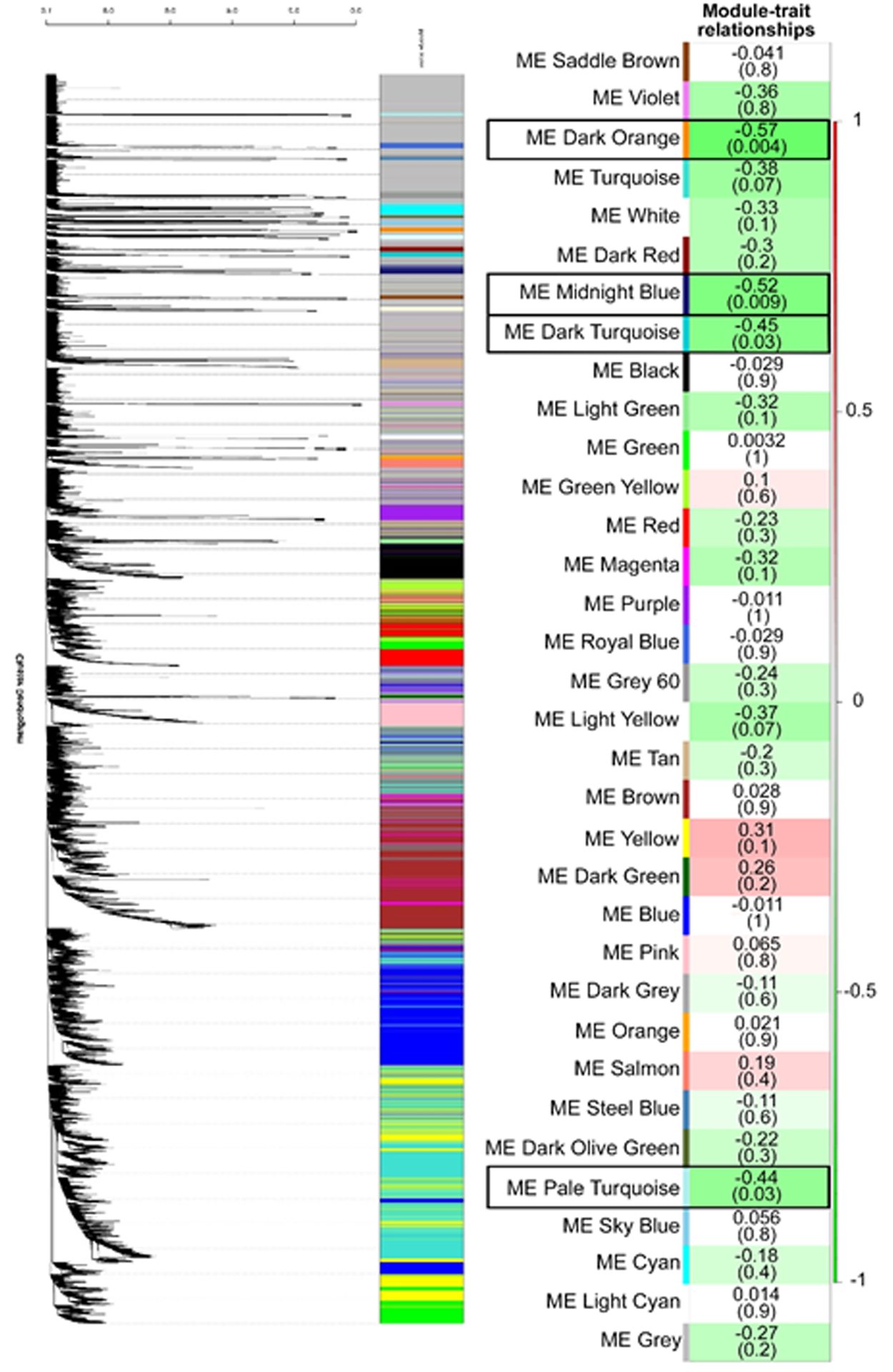
Relationship of WGCNA results and tameness vs. aggressiveness. The modules identified in Figure S7 were then correlated with tameness vs. aggressiveness for the same foxes whose hypothalamus was analyzed with RNA-seq. As detailed in33,34, the foxes were assigned a tameness score of −3 to 4 with 4 being the highest degree of tameness. Each row corresponds to a Module Eigengene (ME) and colors represent the correlation coefficient between the ME and tameness vs aggressiveness. There are two numbers on the right of each row, the number at the top of each row represents the degree of correlation, and those values with a negative integer indicate an inverse correlation with degree of tameness, numbers on the bottom, in parentheses represent the p value associated with the ME~tameness correlation. There are black boxes around the four ME that were significantly associated with degree of tameness: ME dark orange, ME midnight blue, ME dark turquoise, ME pale turquoise (p ≤ 0.05). All four ME were negatively correlated with this behavioral trait.
As determined by the GeneMANIA app51 in Cytoscape48, extensive interactions that included co-expressed and co-localized genes within each of these four significant modules were evident (Figures S9 and S10). Pathway analysis with ClueGO47 reveals that signal transduction, immune system, Cho transports from the extracellular space to the cytosol, developmental biology, signaling by interleukins, cytokine-cytokine receptor interaction, and peptide ligand-binding receptors (e.g., G-protein coupled receptor [GPCR] ligand binding) were enriched in pale turquoise and midnight blue modules, Figure 11). Overall, these modules highlighted the importance of cell membrane processes associated with immune response and development, which were also identified in the KEGG/Reactome pathway analysis of hypothalamic DEGs (Figure 5). The novel finding included choline (Cho) transports pathway. Choline is implicated in the synthesis of the phospholipid components of the cell membrane, serves as a methyl-group donor in methionine metabolism, and it is a precursor of the neurotransmitter acetylcholine59. Choline is essential for brain development and the loss of cholinergic neurons is associated with neurodegenerative disorders60,61 further suggesting the relevance of this pathway to brain processes. The dark turquoise and dark orange modules were also enriched for pathways related to cell membrane processes and signal transduction: cGMP-PKG signaling pathway, regulation of gene expression, cellular response to cytokine stimulus, and cell surface receptor signaling pathway (Figure S11). The GO terms significantly enriched for module genes (Supplementary Information Appendix S8: GO terms associated with genes in modules that are significantly correlated with tameness vs. aggressiveness) further highlighted processes associated with acetylcholine, neurotransmitter transporter activity, cognition, learning and memory, vocalization behavior, forebrain neuron fate commitment, and immune responses in the pale turquoise module; extracellular region, chemotaxis, and immune response in the midnight blue module; hormones, receptor ligand/regulator activity and neuropeptide receptor binding in the dark turquoise module; and thyroid hormone generation, NAD(P)H oxidase activity, and metabolic processes in the dark orange module.
Figure 11.
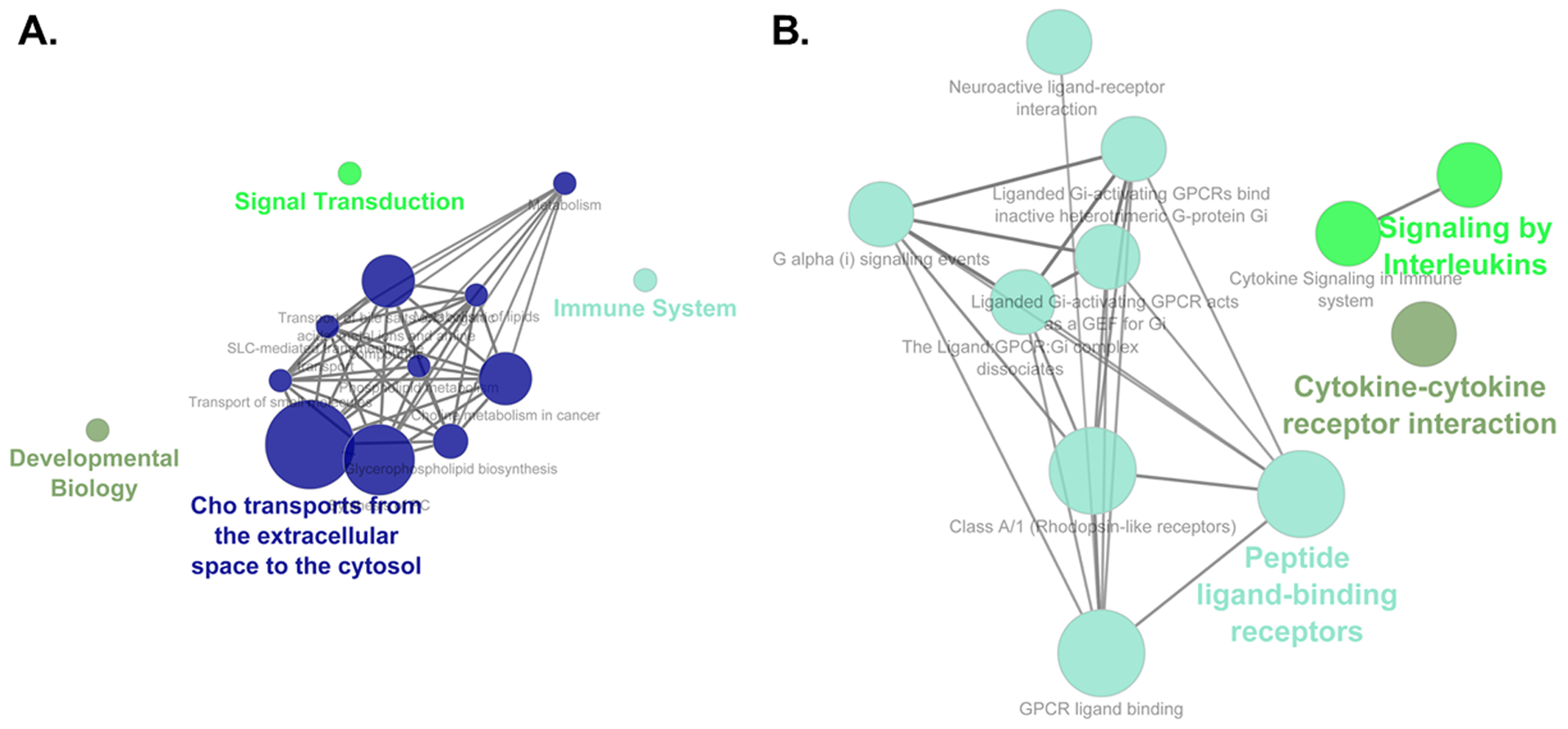
Functional enrichment pathway analyses for the pale turquoise and the midnight blue modules, as determined by the ClueGo App47 in Cytoscape48. A) For the pale turquoise module, signal transduction, immune system, Cho transports from the extracellular space to the cytosol, and developmental biology were enriched. B) In the midnight blue module, signaling by interleukins, cytokine-cytokine receptor interaction, and peptide ligand-binding receptors were enriched.
4. DISCUSSION
Changes in complex neurological processes presumably accompanied the domestication of dogs and other species. As the hypothalamus is a key brain region involved in regulation of fear and aggression62,63, we sought to compare the transcriptome profiles in tame vs. aggressive silver foxes, hypothesizing that differences in gene expression between the fox strains may provide insight into genes and pathways associated with domestication. We identified 70 DE genes in the hypothalamus of tame and aggressive foxes. Of these, 10 DEGs with the highest number of connections to other DEGs were considered to be key players (Table 4; Figure 6B). The ITGA8 gene, encoding the alpha8 subunit of the integrin alpha8beta1, is a key player upregulated in tame foxes and the most significant DEG (Table 2; Supplementary Information Appendix S2: DEGs and all other genes list). Integrins are heterodimeric transmembrane receptors which mediate binding extracellular matrix proteins and interactions between cells64,65. The GO term extracellular matrix organization was also found to be enriched for genes upregulated in tame foxes (Figure 5A). Although none of the integrin subunits is neural specific, the alpha8 subunit is expressed in several brain regions,66 and it is implicated in neuronal development, differentiation, migration, and synaptic plasticity67–69. Mouse mutants for alpha8 were found to exhibit a specific impairment of long term potentiation (LTP) at CA1 synapses in hippocampus70 and beta1 conditional knockouts show deficiencies in the cortical neuron layer formation and the development and function of central excitatory synapses71. Integrins are likely to contribute to imbalanced synaptic function in neurological diseases72 and a missense mutation in the ITGA8 gene was found to be protective against schizophrenia in Japanese female patients73. Although ITGA8 functions in hypothalamus remain to be investigated, its importance for brain development, neuron outgrowth, and signal transduction suggests that differences in expression of ITGA8 may be implicated in behavioral differences between the tame and aggressive foxes.
The remaining nine key player genes are all upregulated in aggressive foxes. These genes are involved in diverse processes in the body, but their functions in hypothalamus remain to be investigated. S100A6 belongs to the family of low-molecular-weight calcium-binding proteins74, and it is implicated in cell proliferation, differentiation, migration, cytoskeletal dynamics, and ubiquitination of β-catenin75, which acts as a signal transducer in the Wnt signaling pathway. It is also suggested that S100A6 transduction through integrin beta1 can increase adhesion and inhibit cell proliferation76. In the mouse brain, the level of S100A6 decreases in response to chronic mild stress, suggesting that this protein may modify stress responses77. Two of the key player genes: CEP104 (Centrosomal Protein 104) and FUZ (homolog of Fuzzy in Drosophila) are involved in ciliogenesis. Cilia function as sensors of extracellular cues and represent a critical part of signaling pathways such as Hedgehog (Hh), PDGF and Wnt,78,79 pathways that are critical in neuronal development. In humans, mutations in CEP104 cause Joubert syndrome, a developmental disorder characterized by a distinctive mid-hindbrain and cerebellar malformation80. FUZ plays a crucial role in embryonic development and it is involved in planar cell polarity, ciliogenesis and directional cell movement81. FUZ is implicated in both Hh and Wnt/b-catenin signaling82 and mutations in this gene are associated with neural tube defects in humans83.
Two key player genes, NPL (N-Acetylneuraminate Pyruvate Lyase) and CD302 (C-type lectin receptor) are associated with carbohydrate modification and carbohydrate binding. NPL regulates cellular concentrations of N-acetyl-neuraminic acid (sialic acid) which typically occupy the terminal position of glycan chains84. The brain has the highest concentration of N-acetylneuraminic acid, which serves as an integral part of ganglioside structure in synaptogenesis and neural transmission85. CD302 binds specific types of carbohydrates, but a ligand for CD302 has yet to be identified. CD302 is colocalized with F-actin–rich filopodia and lamellopodia, suggesting a potential role of this gene in cell adhesion and cell migration86. In addition to the key player genes, four other hypothalamus DEGs (EIF4E1B, IDNK, KHK, MGAM) are involved in carbohydrate/glucose metabolism87. The differences in glucose level were observed between strains of tame and aggressive rats (Rattus norvegicus) with higher glucose levels in the blood of aggressive rats88. Differences in brain expression of genes involved in glucose metabolism were also shown in populations of honey bees with different levels of aggression89,90. Taking into account the role of hypothalamus in glucose homeostasis maintenance91, it is possible to suggest that changes in glucose matabolism were associated with selection of foxes for tame vs. aggressive behavior.
Four other key player genes display significant differences between tame and aggressive foxes, but there is no clear mechanism that connects these genes and their functions to the behavioral differences we see in the foxes. Two key player genes are associated with DNA replication and DNA double strand break repair and recombination. PRIMPOL (Primase and DNA Directed Polymerase) catalyzes the synthesis of short RNA primers that serve as starting points for DNA synthesis and DNA polymerase activity, and facilitates DNA damage tolerance92,93. PRKDC (Protein Kinase, DNA-Activated, Catalytic Subunit) acts as a molecular sensor for DNA damage. It functions with the Ku70/Ku80 heterodimer protein in DNA double strand break repair and recombination94. Another key player gene, SMPDL3A (Sphingomyelin Phosphodiesterase Acid Like 3A) hydrolyzes nucleotide tri- and diphosphates and their derivatives95. Finally, the TRIM52 (Tripartite Motif Containing 52) is a novel noncanonical antiviral TRIM gene which is involved in NF-κB pathway activation and plays an important role in antiviral innate immunity96,97.
The functions of key player genes are in line with the GO terms identified in the enrichment analysis of hypothalamus DE genes: cilium assembly, innate immune system and extracellular matrix organization (Figure 5). Overall, the functions of the key player genes and GO terms identified in the enrichment analysis of hypothalamus DEGs correspond to a broad spectrum of biological processes, indicating that differential expression in hypothalamus of tame vs. aggressive foxes involves development, cell differentiation, cell interaction, and migration, rather than specific neurotransmitter pathways.
Comparison of expression patterns of DEGs that were DE in each of the three brain regions (PFC, BFB, HYP) reveales that selected genes are differentially regulated in each of these three brain regions (Figures S4 and S5). These findings highlight that expression of certain genes is important in regulating specific brain regions. On the other hand, the comparison of DEGs in the three brain regions highlighted seven genes which showed differential expression in the same direction in all three tissues (Figure 4). Two of these genes (NPL and PRIMPOL) are also the key player genes in hypothalamus. Little is known about the functions of the remaining five genes. DKKL1 (Dickkopf Like Acrosomal Protein 1) is one of the least investigated members of the Dickkopf family98. It is abundantly expressed in testis and likely involved in testicular development and spermatogenesis. In mouse brain, DKKL1 was shown to be expressed in embryonic dorsal root ganglia neurons99 and in cortical neurons of the adult brain100. Previously, we demonstrated that DKKL1 is one of the most significant DEGs in PFC of tame vs. aggressive foxes31. DKKL1 is most closely related to DKK3, which acts as antagonist of Wnt signaling101 and is involved in many cellular processes including cell proliferation, differentiation, adhesion, and apoptosis102. FBLN7 (Fibulin 7) is a member of the fibulin protein family, cell-secreted glycoproteins that function as cell adhesion molecules and interact with other extracellular matrix proteins as well as cell receptors103. First identified as an extracellular matrix molecule in developing teeth104, FBLN7 was found to be expressed in a variety of tissues and upregulated in several cancers, with the highest expression in glioblastomas105. PTGFRN (Prostaglandin F2 Receptor Inhibitor) is a glycosylated type 1 integral membrane protein with immunoglobulin domains that interacts with tetraspanins CD9 and CD81106, integral membrane proteins present on the plasma membrane of many cells107,108. Tetraspanins are involved in regulation of cell interaction and cell migration and implicated in numerous physiological processes including immune response, reproduction, development, angiogenesis, and tumorogenesis109–112. It was also shown that expression of PTGFRN in COS-1 cells inhibits the binding of prostaglandin F2-alpha (PGF2-alpha) to its specific FP receptor113. The functions in brain tissues of the two other genes from Figure 4D in brain tissues are largely unknown. SHCBP1L (SHC Binding And Spindle Associated 1 Like) maintains stability of the spindle integrity during meiosis and implicated in spermatogenesis114. SKIV2L (Ski2 Like RNA Helicase) encodes a DEAD box protein which shares homology with yeast exosome components. Based on this similarity it was suggested that SKIV2L functions as an RNA helicase115,116. SKIV2L has a protective effect against the age-related macular degeneration117.
Analysis of the genes which show gene expression differences between tame and aggressive foxes in three brain regions showed consistent differences in expression of genes involved in broad biological processes rather than in processes specific to CNS. It would be interesting to test if expression of these genes is also altered in other tissues of tame and aggressive foxes and particularly in neural crest cells. Given the importance of these genes for such processes as cell division, differentiation, and adhesion they may potentially have an effect on neural crest cell migration and differentiation, which was proposed to be involved in the domestication syndrome6.
The functions of both key player genes and genes from Figure 4D are in agreement with GO terms identified in the current study as well as in the analysis of genomic regions which differentiate fox populations37. Comparison of genomic regions which differentiate tame and aggressive foxes also identified GO terms related to damaged DNA binding, carbohydrate binding, and immunity (interleukin-1 receptor binding and cytokine activity)37. Tame and aggressive foxes are maintained under the same farm conditions, and thus, it is more likely that differences in genomic regions containing immune genes are directly or indirectly associated with selection for behavior. The link between behavior and immunity has been demonstrated in a large body of literature (reviewed in118–120).
Although finding consistent gene expression differences across brain tissues increases confidence that our experimental and analytical methods detect DEGs between sampled foxes, we cannot conclude that all identified DEGs genes are implicated in behavioral differences between tame and aggressive fox strains. While sufficient replicates were used in the current study, it is possible that some alleles could drift in frequency in these outbred populations sufficiently to give the observed changes in expression by chance alone. Further studies including different sets of tame and aggressive foxes and comparisons with transcriptomic differences between pairs of domesticated vs. wild species need to be performed to pinpoint genes whose expression affects behavior of these foxes.
WGCNA identified four eigengene modules with significant gene expression differences between tame and aggressive foxes (Figure 10). Integrative correlation analyses revealed that all four modules that were significantly different were negatively associated with tameness. Pathway analyses of these modules identified biological processes similar to the process identified in the enrichment analysis of hypothalamus DEG. However, the enrichment analysis of significant WGCNA modules also highlighted GO terms more specific to neural system such as acetylcholine, neurotransmitter transporter activity cognition, learning and memory, and forebrain neuron fate commitment (Supplementary Information Appendix S8: GO terms associated with genes in modules that are significantly correlated with tameness vs. aggressiveness). We found only one DEG (BPHL) in the significant WGCNA modules.This gene is also the only hub gene identified in all four significant modules, with connections to 135 other genes in this significant module (Midnight Blue). As a hub gene, BPHL, might regulateother genes connected to it. Some of the genes directly connected to it include keratin 36 (KRT36), AVP, myosin light chain 3 (MYL3), solute carrier family 5 member 1 (SLC5A1), tissue factor pathway inhibitor 2 (TFPI2), growth factor independent 1B transcriptional repressor (GFI1B), T cell receptor beta variable 3–1 (TRBV3–1), and vasoactive intestinal peptide (VIP). Of these, we did not detect any differences in AVP mRNA expression with RNA-seq (Supplementary Information Appendix S2: DEGs and all other genes list) or qPCR analysis (Figure S6).
Finding GO terms more specific to neural system in WGCNA open the possibility that differences in expression of genes related to brain-specific pathways take place in particular cell types or nuclei in hypothalamus. Because our hypothalamus samples include heterogeneous populations of cells and nuclei, differences in expression of these genes between tame and aggressive samples can be masked and may not reach statistical significance.
Analysis of alternative splicing identified 159 genes with differentially skipped exons in reads from tame and aggressive fox hypothalamus. Six of these genes were also differentially spliced in anterior pituitary samples of tame and aggressive foxes26. Comparison of DEGs (Supplementary Information Appendix S1: DEGs and all other genes list) with genes with differentially skipped exons (Supplementary Information Appendix S3: rMATS splice form analysis) identified only a small overlap, three genes (CDKL2, CES2, and SLC38A9) were found to be significant in both datasets. These results indicate that differential expression is not the only mechanism leading to transcriptomic differences and detailed analysis of gene transcription and processing is needed to identify a more complete picture of transcriptomic differences between samples. Functional enrichment analysis of skipped exon data based on rMATS analysis suggests that primary KEGG pathways affected include those regulating fatty acid biosynthesis, fatty acid metabolism, glutathione metabolism, glycerolipid metabolism, and glucagon signaling pathways, providing evidence that selection pressures as foxes were bred for tameness also acted on the individual transcript level for these metabolic pathways. Further screening of this database also revealed differential expression of transcripts associated with various neurobehavioral disorders in humans, including dementia, neurodegenerative disorders, Alzheimer’s disease, learning and memory dysfunction, anxiety disorders, and repetitive behaviors. As foxes were selected for tameness, they showed reduced anxiety and cognitive alterations24,25,121.
We hypothesized that primary differences that would emerge between tame and aggressive foxes would be found in neuropeptides, such as OXT and AVP, and other genes associated with social affiliation. These neuropeptides and their receptors have been proposed as facilitators of domestication in the dog and other species53–56. However, neither OXT, AVP, nor any of their splice forms were altered with RNA-seq or qPCR analyses. This could be due to the area of the hypothalamus sampled, as we did not selectively screen the paraventricular (PVN) and supraoptic (SON) nuclei. Alternatively, differences might emerge in tame and aggressive foxes in the release of these neuropeptides from the posterior pituitary gland. Previous work only examined the anterior pituitary gland from these fox populations26.
We compared DEGs and GO terms identified in hypothalamus of tame and aggressive foxes with genes and pathways identified in transcriptomes of domesticated and wild species. A study by Albert et al.,122 used frontal cortex tissues obtained from dogs and wolves, pigs and wild boars, domesticated and wild guinea pigs, and domesticated and wild rabbits and identified a large number of GO terms differentiating wild and domestic species. Similar to our findings, the identified GO terms largely belonged to a broad spectrum of biological processes such as immunity, development, reproduction, and metabolism; as well as cellular processes such as cell adhesion, cell-cell interaction, motility, and signal transduction. Comparably, the immune-related processes were enriched in the comparison of frontal cortex transcriptomes of wild boar and domestic pig in a study by Long et al.123 and in a study of blood transcriptomes between wolves and dogs124. The comparative studies of transcriptomes from thalamus/hypothalamus of wild Red Junglefowl and Red Junglefowl selected for tameness for five generations largely identified genes related to spermatogenesis and immunity21, but the comparative analysis of cerebral hemisphere transcriptomes in these populations suggested an enrichment for genes associated with behavioral processes125. Furthermore, genes that were DE between the two groups of foxes include those regulating the metabolism of fats and carbohydrates, such as MGAM, KHK, IDNK, FABP7, and CES2. Differences in fat metabolism have also been identified in the adrenal glands of these two fox populations (Hekman et al., In Preparation). A previous study on dog domestication also found enrichment in genes for fat and other metabolism, e.g., CCRN4L, SCP2D1, and PDXC1126. The findings led the authors to speculate that domestication impacted dietary selection as proto-dogs hunted and fed alongside hunter-gatherers. Overall, the results of transcriptomic studies between domesticated species and their wild progenitors indicate importance of genes whose functions in adult brain are not well understood. These findings may suggest that genes involved in neural functions are less important for domestication than genes involved in immunity and development, e.g. affecting sizes of brain regions implicated in fear processing127; alternatively, these results may indicate current limitations of transcriptomic studies of domestication. Changes in expression of genes with neurological functions may be specific to particular cell types and therefore not found in the heterogeneous tissue samples used in conventional RNA-seq experiments.
Comparison of hypothalamus DEGs with the gene content of genomic regions differentiating fox populations37 identified 10 genes in common. Two genes highlighted in our study, PTGRFN and FBLN7, are both located on fox chromosome 8 in genomic regions (regions 50 and 52, respectively) which show increased divergence between tame and aggressive fox populations37. Because PTGRFN and FBLN7 are two out of many genes located in these regions, it is impossible to conclude without additional experiments whether PTGRFN and FBLN7 are genes targeted by selection for behavior or their differential expression in the two fox strains is a result of a tight linkage of these genes with genes under selection. However, finding DEGs within genomic regions targeted by selection allows us to prioritize positional candidate genes for screening. A hypothalamus DEG, STAC (SH3 And Cysteine Rich Domain), is located in a genomic region (region 61) which includes only seven genes32. In neurons, STAC modulates Ca2+ entry via l-type, but not via non-l-type, Ca2+ channels. Expression of the major neuronal isoform (STAC2) is increased in postnatal forebrain and cerebellum, which could provide developmental regulation of l-type channel Ca2+ signaling in these brain regions. In skeletal muscle, STAC protein is essential for proper trafficking and function of the l-type Ca2+ channel and mutations in STAC cause severe myopathy128. STAC will be prioritized for screening as a positional candidate gene in region 61.
One limitation of the current study is that there was likely cellular heterogeneity in the hypothalamus tissue samples, as shown in the PCA analysis (Figure 1) and hierarchical heatmap clustering analysis (Figure 2B). In foxes, as in dogs, the hypothalamus is a substantial brain region, and since we used only a small amount of dissected tissue for RNA extraction, we inevitably introduced heterogeneity across sequenced samples. However, in these initial studies, the initial aim was to broadly analyze the hypothalamus in these two fox populations with the idea that further work can examine discrete nuclei, including the PVN and SON. Future studies will employ single cell RNA-seq to pinpoint which cells have been affected to the greatest extent by the selective breeding scheme as evidenced by the cell-specific transcript changes. Our abilities to collect cell-specific information in undisturbed landscapes in solid tissues is limited but growing arsenal of omics techniques129 and precise brain mapping approaches130 provide a framework for these future studies. Revealing brain cell-specific activity is critical for understanding complex processes underlying cognitive and behavioral functions.
Domesticated dogs demonstrate increased sociability to humans and a greater ability to use human directed cues relative to their wolf ancestors, traits that appear to have been selected for during the domestication process131–136. Besides showing reduced fear and aggression towards humans, similar socio-cognitive traits have evolved in this silver fox population bred for tameness121, providing robust evidence that these foxes have potential to provide valuable insight into the underlying neurological events promoting canine domestication. The current studies reveal that selection for tameness vs. aggressiveness in foxes is associated with unique hypothalamic gene and transcript signature profiles. Functional analyses of genes altered between these two groups reveals that processes affected by this selective breeding include those regulating development, immunity, lipid and carbohydrate metabolism, and genes involved in extracellular matrix organization, cell interaction, and several signaling pathways.
Supplementary Material
Table S1. Sequence of primers used for qPCR analyses.
Table S2. Sequence reads and mapping rate of samples.
Table S3. Hypothalamus DEGs that are DE in one or more of the three other brain regions: PFC 31, BFB 31, and anterior pituitary 26.
Table S4. qPCR expression for other hypothalamic genes that did not differ between aggressive vs. tame foxes. Average ± SEM for 2−ΔΔCt values.
Figure S2. Anatomical location (red) and connectivity (blue) of the basal forebrain (BFB) dissection samples. Connectivity includes the olfactory tubercule, piriform lobe, stria terminalis, amygdala, medial temporal lobe, and lateral temporal cortex (ectosylvian and suprasylvian gyri).
Figure S3. Anatomical location (red) and connectivity (blue) of the medial prefrontal cortex (PFC) dissection samples. Connectivity includes the olfactory tubercule, stria terminalis, caudate, cingulum bundle, and medial temporal lobe.
Figure S1. Anatomical location (red) and connectivity (blue) of the hypothalamus (HYP) dissection samples. Connectivity includes the olfactory tubercule, piriform lobe, amygdala, brainstem, medial prefrontal cortex, and medial temporal lobe.
Figure S4. Expression pattern of three groups of DEG in the different brain regions. A) Expression pattern of DEG within PFC (cortex) in the three brain regions, cortex, forebrain, and hypothalamus. B) Expression pattern of DEG within BFB (forebrain) within the three brain regions. C) Expression pattern of DEG within hypothalamus within the three brain regions.
Figure S6. qPCR analysis for AVP and OXT. No differences in gene expression were detected for these genes between tame and aggressive foxes.
Figure S5. Expression pattern of hypothalamic DEG (between tame and aggressive foxes) within the hypothalamus for the three brain regions. A) Expression pattern of hypothalamic DEG in the PFC (cortex). B) Expression pattern of hypothalamic DEG in the BFB (forebrain). C) Expression pattern of hypothalamic DEG in the hypothalamus. The count data included a meta data information specifying which gene was either up-regulated or downregulated in HYP.
Figure S9. Gene interactions as determined by the GeneMANIA app 51 in Cytoscape 48,for the pale turquoise and midnight blue modules. A) Pale Turquoise. B) Midnight blue. Extensive interactions for genes within both of these modules is evident. Legends for the colors of the nodes and edges are detailed in the figure.
Figure S8. Diagram showing BPHL as a hub gene with 135 genes within the midnight blue module connecting to this central gene. This figure though only shows 70 genes that have corresponding gene symbols of the 135 genes based on Ensembl IDs.
Figure S7. Identification and characterization of weighted gene co-expression network analyses (WGCNA) and clustering dendrogram of genes. The clustering dendrogram is based on variation in topological overlap and assigned module colors are shown above and to the side of the heatmap.
Figure S10. Gene interactions as determined by the GeneMANIA app 51 in Cytoscape 48, for the dark turquoise and the dark orange modules. A) Dark Turquoise. B) Dark Orange. Extensive interactions for genes within both of these modules is evident. Legends for the colors of the nodes and edges are detailed in the figure.
Figure S11. Functional enrichment pathway analyses for the dark turquoise and dark orange modules, as determined by the ClueGo App 47 in Cytoscape 48. A) For the dark turquoise module, the cGMP-PKG signaling pathway was the primary enriched pathway. B) In the dark orange module, regulation of gene expression, cellular response to cytokine stimulus, and cell surface receptor signaling pathway were enriched in the dark orange module.
Supplementary Information Appendix S1: Imaging studies for fox brain samples
Supplementary Information Appendix S3. Behavioral scores for aggressive and tame foxes.
Supplementary Information Appendix S4. rMATS splice form analysis.
Supplementary Information Appendix S5. Gene module membership for those that are significantly correlated with tameness vs. aggressiveness.
Supplementary Information Appendix S6. Distribution of the other 69 DEG within the various color modules. The two modules that contained most of the DEG were Grey (n = 34 DEG), which is a module generally made up of genes that do not fit into any of the modules, and Turquoise (n = 18).
Supplementary Information Appendix S7. DEG and other genes within the Turquoise module. Highlighted genes within this module are DEG.
Supplementary Information Appendix S8. GO terms associated with genes in modules that are significantly correlated with tameness vs. aggressiveness.
Supplementary Information Appendix S2. Differentially expressed genes (DEGs) and all other genes.
ACKNOWLEDGEMENTS
We thank Irina V. Pivovarova, Tatyana I. Semenova, Eugene A. Martinov, and all the animal keepers at the Institute of Cytology and Genetics (ICG) experimental farm for research assistance. This project was supported by NIH R01 ES025547 (to C.S.R.), NIH GM120782 (to A.V.K.), USDA Federal Hatch Project (to A.V.K.), a Campus Research Board grant from the University of Illinois at Urbana-Champaign (to A.V.K.), the Institute of Cytology and Genetics of the Siberian Branch of the Russian Academy of Sciences (grant number 0324-2018-0016; animal maintenance), the Russian Scientific Foundation (grant number 16-14-10216; data collection and analysis), the Jonathan Baldwin Turner Scholarship Program (to J.P.H.), and IMSD-EXPRESS program (R25GM056901, to M.T.O.). We appreciate Donald L. Connor’s assistance with figure preparation and Dr. Wes Warren’s input for the informatics analyses. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from: GTEx Multi Gene Query on the GTEx Portal on 03/19/19 and used to generate Figure 6B.
Footnotes
CONFLICT OF INTEREST
The authors have no conflicts of interest to report.
REFERENCES
- 1.Evolution Diamond J., consequences and future of plant and animal domestication. Nature. 2002;418(6898):700–707. [DOI] [PubMed] [Google Scholar]
- 2.Price EO. Principles and Applications of Domestic Animal Behavior: An Introductory Text. Wallingford, United Kingdom: CABI; 2008. [Google Scholar]
- 3.Campler M, Jongren M, Jensen P. Fearfulness in red junglefowl and domesticated White Leghorn chickens. Behav Processes. 2009;81(1):39–43. [DOI] [PubMed] [Google Scholar]
- 4.Driscoll CA, Macdonald DW, O’Brien SJ. From wild animals to domestic pets, an evolutionary view of domestication. Proc Natl Acad Sci U S A. 2009;106 Suppl 1:9971–9978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hare B, Wobber V, Wrangham R. The self-domestication hypothesis: evolution of bonobo psychology is due to selection against aggression. Anim Behav. 2012;83(3):573–585. [Google Scholar]
- 6.Wilkins AS, Wrangham RW, Fitch WT. The “domestication syndrome” in mammals: a unified explanation based on neural crest cell behavior and genetics. Genetics. 2014;197(3):795–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vonholdt BM, Pollinger JP, Lohmueller KE, et al. Genome-wide SNP and haplotype analyses reveal a rich history underlying dog domestication. Nature. 2010;464(7290):898–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Savolainen P, Zhang YP, Luo J, Lundeberg J, Leitner T. Genetic evidence for an East Asian origin of domestic dogs. Science. 2002;298(5598):1610–1613. [DOI] [PubMed] [Google Scholar]
- 9.Schoenebeck JJ, Ostrander EA. Insights into morphology and disease from the dog genome project. Annu Rev Cell Dev Biol. 2014;30:535–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shearin AL, Ostrander EA. Canine morphology: hunting for genes and tracking mutations. PLoS Biol. 2010;8(3):e1000310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gacsi M, Gyori B, Miklosi A, et al. Species-specific differences and similarities in the behavior of hand-raised dog and wolf pups in social situations with humans. Dev Psychobiol. 2005;47(2):111–122. [DOI] [PubMed] [Google Scholar]
- 12.Hall NJ, Lord K, Arnold AM, Wynne CD, Udell MA. Assessment of attachment behaviour to human caregivers in wolf pups (Canis lupus lupus). Behav Processes. 2015;110:15–21. [DOI] [PubMed] [Google Scholar]
- 13.Trut L, Oskina I, Kharlamova A. Animal evolution during domestication: the domesticated fox as a model. Bioessays. 2009;31(3):349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.vonHoldt BM, Shuldiner E, Koch IJ, et al. Structural variants in genes associated with human Williams-Beuren syndrome underlie stereotypical hypersociability in domestic dogs. Science Advances. 2017;3(7):e1700398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Porter MA, Dobson-Stone C, Kwok JB, Schofield PR, Beckett W, Tassabehji M. A role for transcription factor GTF2IRD2 in executive function in Williams-Beuren syndrome. PLoS One. 2012;7(10):e47457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edelmann L, Prosnitz A, Pardo S, et al. An atypical deletion of the Williams-Beuren syndrome interval implicates genes associated with defective visuospatial processing and autism. J Med Genet. 2007;44(2):136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saetre P, Lindberg J, Leonard JA, et al. From wild wolf to domestic dog: gene expression changes in the brain. Brain Res Mol Brain Res. 2004;126(2):198–206. [DOI] [PubMed] [Google Scholar]
- 18.Belteky J, Agnvall B, Bektic L, Hoglund A, Jensen P, Guerrero-Bosagna C. Epigenetics and early domestication: differences in hypothalamic DNA methylation between red junglefowl divergently selected for high or low fear of humans. Genet Select Evol. 2018;50(1):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lotvedt P, Fallahshahroudi A, Bektic L, Altimiras J, Jensen P. Chicken domestication changes expression of stress-related genes in brain, pituitary and adrenals. Neurobiology of stress. 2017;7:113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fallahsharoudi A, de Kock N, Johnsson M, et al. Genetic and Targeted eQTL Mapping reveals strong candidate genes modulating the stress response during chicken domestication. G3. 2017;7(2):497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Belteky J, Agnvall B, Johnsson M, Wright D, Jensen P. Domestication and tameness: brain gene expression in red junglefowl selected for less fear of humans suggests effects on reproduction and immunology. Royal Soc. 2016;3(8):160033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruan C, Zhang Z. Laboratory domestication changed the expression patterns of oxytocin and vasopressin in brains of rats and mice. Anat Sci Intl. 2016;91(4):358–370. [DOI] [PubMed] [Google Scholar]
- 23.Trut L, Dugatkin LA. How to build a dog. Sci Am. 2017;316(5):68–73. [DOI] [PubMed] [Google Scholar]
- 24.Trut LN, Pliusnina IZ, Os’kina IN. An experiment on fox domestication and debatable issues of evolution of the dog. Genetika. 2004;40(6):794–807. [PubMed] [Google Scholar]
- 25.Gulevich RG, Oskina IN, Shikhevich SG, Fedorova EV, Trut LN. Effect of selection for behavior on pituitary-adrenal axis and proopiomelanocortin gene expression in silver foxes (Vulpes vulpes). Physiol Behav. 2004;82(2–3):513–518. [DOI] [PubMed] [Google Scholar]
- 26.Hekman JP, Johnson JL, Edwards W, et al. Anterior pituitary transcriptome suggests differences in ACTH release in tame and aggressive foxes. G3. 2018;8(3):859–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Popova NK, Kulikov AV, Avgustinovich DF, Voitenko NN, Trut LN. Effect of domestication of the silver fox on the main enzymes of serotonin metabolism and serotonin receptors. Genetika. 1997;33(3):370–374. [PubMed] [Google Scholar]
- 28.Osadchuk LV, Voitenko NN. [Sex steroid hormones and brain serotonin in the estrous cycle of silver foxes]. Fiziol Zh SSSR Im I M Sechenova. 1992;78(4):118–123. [PubMed] [Google Scholar]
- 29.Popova NK, Voitenko NN, Kulikov AV, Avgustinovich DF. Evidence for the involvement of central serotonin in mechanism of domestication of silver foxes. Pharmacol Biochem Behav. 1991;40(4):751–756. [DOI] [PubMed] [Google Scholar]
- 30.Nikulina EM. [The brain catecholamines during domestication of the silver fox Vulpes fulvus]. Zh Evol Biokhim Fiziol. 1990;26(2):156–160. [PubMed] [Google Scholar]
- 31.Wang X, Pipes L, Trut LN, et al. Genomic responses to selection for tame/aggressive behaviors in the silver fox (Vulpes vulpes). Proc Natl Acad Sci U S A. 2018;115(41):10398–10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kukekova AV, Trut LN, Acland GM. Genetics of domesticated behavior in dogs and foxes. Chapter 10 In: Grandin T, Deesing M, eds. Genetics and the Behavior of Domestic Animals, 2nd Edition.2014:361–396. [Google Scholar]
- 33.Trut LN, Pliusnina IZ, Os’kina IN. [An experiment on fox domestication and debatable issues of evolution of the dog]. Genetika. 2004;40(6):794–807. [PubMed] [Google Scholar]
- 34.Kukekova AV, Temnykh SV, Johnson JL, Trut LN, Acland GM. Genetics of behavior in the silver fox. Mamm Genome. 2012;23(1–2):164–177. [DOI] [PubMed] [Google Scholar]
- 35.Andrews S FastQC: a quality control tool for high throughput sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc. 2010.
- 36.Martin M Cutadapt removes adaptor sequences from high-throughput sequencing reads. EMBnetjournal. 2011;17:10. [Google Scholar]
- 37.Kukekova AV, Johnson JL, Xiang X, et al. Red fox genome assembly identifies genomic regions associated with tame and aggressive behaviours. Nat Ecol Evol. 2018;2(9):1479–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rando HM, Farre M, Robson MP, et al. Construction of Red Fox Chromosomal Fragments from the Short-Read Genome Assembly. Genes (Basel). 2018;9(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rando HM, Stutchman JT, Bastounes ER, et al. Y-Chromosome Markers for the Red Fox. J Hered. 2017;108(6):678–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–930. [DOI] [PubMed] [Google Scholar]
- 42.Zhou X, Lindsay H, Robinson MD. Robustly detecting differential expression in RNA sequencing data using observation weights. Nucleic Acids Res. 2014;42(11):e91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Budden DM, Crampin EJ. Information theoretic approaches for inference of biological networks from continuous-valued data. BMC Syst Biol. 2016;10(1):89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ding C, Peng H. Minimum redundancy feature selection from microarray gene expression data. J Bioinform Comput Biol. 2005;3(2):185–205. [DOI] [PubMed] [Google Scholar]
- 45.Azuaje FJ. Selecting biologically informative genes in co-expression networks with a centrality score. Biol Direct. 2014;9:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiao X, Sherman BT, Huang da W, et al. DAVID-WS: a stateful web service to facilitate gene/protein list analysis. Bioinformatics. 2012;28(13):1805–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bindea G, Mlecnik B, Hackl H, et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25(8):1091–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shen S, Park JW, Lu ZX, et al. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc Natl Acad Sci U S A. 2014;111(51):E5593–5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Franz M, Rodriguez H, Lopes C, et al. GeneMANIA update 2018. Nucleic Acids Res. 2018;46(W1):W60–w64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Y, Chen K, Sloan SA, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34(36):11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Herbeck YE, Gulevich RG. Neuropeptides as facilitators of domestication. Cell Tissue Res. 2019;375(1):295–307. [DOI] [PubMed] [Google Scholar]
- 54.Buttner AP. Neurobiological underpinnings of dogs’ human-like social competence: How interactions between stress response systems and oxytocin mediate dogs’ social skills. Neurosci Biobehav Rev. 2016;71:198–214. [DOI] [PubMed] [Google Scholar]
- 55.Nagasawa M, Mitsui S, En S, et al. Social evolution. Oxytocin-gaze positive loop and the coevolution of human-dog bonds. Science. 2015;348(6232):333–336. [DOI] [PubMed] [Google Scholar]
- 56.Persson ME, Trottier AJ, Belteky J, Roth LSV, Jensen P. Intranasal oxytocin and a polymorphism in the oxytocin receptor gene are associated with human-directed social behavior in golden retriever dogs. Horm Behav. 2017;95:85–93. [DOI] [PubMed] [Google Scholar]
- 57.Wang J, Vasaikar S, Shi Z, Greer M, Zhang B. WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017;45(W1):W130–w137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. https://cran.r-project.org/web/packages/WGCNA/index.html.
- 59.Michel V, Yuan Z, Ramsubir S, Bakovic M. Choline transport for phospholipid synthesis. Exper Biol Med. 2006;231(5):490–504. [DOI] [PubMed] [Google Scholar]
- 60.Blusztajn JK, Mellott TJ. Choline nutrition programs brain development via DNA and histone methylation. Central Nervous System Agents Medicinal Chem. 2012;12(2):82–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ferreira-Vieira TH, Guimaraes IM, Silva FR, Ribeiro FM. Alzheimer’s disease: Targeting the cholinergic system. Curr Neuropharmacol. 2016;14(1):101–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lin D, Boyle MP, Dollar P, et al. Functional identification of an aggression locus in the mouse hypothalamus. Nature. 2011;470(7333):221–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Takahashi A, Miczek KA. Neurogenetics of aggressive behavior: studies in rodents. Curr Top Behav Neurosci. 2014;17:3–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reichardt LF, Tomaselli KJ. Extracellular matrix molecules and their receptors: functions in neural development. Annu Rev Neurosci. 1991;14:531–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110(6):673–687. [DOI] [PubMed] [Google Scholar]
- 66.Einheber S, Schnapp LM, Salzer JL, Cappiello ZB, Milner TA. Regional and ultrastructural distribution of the alpha 8 integrin subunit in developing and adult rat brain suggests a role in synaptic function. J Comp Neurol. 1996;370(1):105–134. [DOI] [PubMed] [Google Scholar]
- 67.McCarty JH, Lacy-Hulbert A, Charest A, et al. Selective ablation of alphav integrins in the central nervous system leads to cerebral hemorrhage, seizures, axonal degeneration and premature death. Development. 2005;132(1):165–176. [DOI] [PubMed] [Google Scholar]
- 68.Rehberg K, Kliche S, Madencioglu DA, et al. The serine/threonine kinase Ndr2 controls integrin trafficking and integrin-dependent neurite growth. J Neurosci. 2014;34(15):5342–5354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kerrisk ME, Cingolani LA, Koleske AJ. ECM receptors in neuronal structure, synaptic plasticity, and behavior. Prog Brain Res. 2014;214:101–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chan CS, Chen H, Bradley A, Dragatsis I, Rosenmund C, Davis RL. alpha8-integrins are required for hippocampal long-term potentiation but not for hippocampal-dependent learning. Genes Brain Behavior. 2010;9(4):402–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang Z, Shimazu K, Woo NH, et al. Distinct roles of the beta 1-class integrins at the developing and the mature hippocampal excitatory synapse. J Neurosci. 2006;26(43):11208–11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu X, Reddy DS. Integrins as receptor targets for neurological disorders. Pharmacol Ther. 2012;134(1):68–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Supriyanto I, Watanabe Y, Mouri K, et al. A missense mutation in the ITGA8 gene, a cell adhesion molecule gene, is associated with schizophrenia in Japanese female patients. Prog Neuropsychopharmacol Biol Psychiatry. 2013;40:347–352. [DOI] [PubMed] [Google Scholar]
- 74.Lesniak W, Slomnicki LP, Filipek A. S100A6 - new facts and features. Biochem Biophys Res Commun. 2009;390(4):1087–1092. [DOI] [PubMed] [Google Scholar]
- 75.Donato R, Cannon BR, Sorci G, et al. Functions of S100 proteins. Curr Mol Med. 2013;13(1):24–57. [PMC free article] [PubMed] [Google Scholar]
- 76.Jurewicz E, Goral A, Filipek A. S100A6 is secreted from Wharton’s jelly mesenchymal stem cells and interacts with integrin beta1. Int J Biochem Cell Biol. 2014;55:298–303. [DOI] [PubMed] [Google Scholar]
- 77.Bartkowska K, Swiatek I, Aniszewska A, et al. Stress-dependent changes in the CacyBP/SIP interacting protein S100A6 in the mouse brain. PLoS One. 2017;12(1):e0169760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Baker K, Beales PL. Making sense of cilia in disease: the human ciliopathies. Am J Med Genet C Semin Med Genet. 2009;151C(4):281–295. [DOI] [PubMed] [Google Scholar]
- 79.Rezabkova L, Kraatz SH, Akhmanova A, Steinmetz MO, Kammerer RA. Biophysical and structural characterization of the centriolar protein Cep104 interaction network. J Biol Chem. 2016;291(35):18496–18504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Srour M, Hamdan FF, McKnight D, et al. Joubert Syndrome in French Canadians and identification of mutations in CEP104. Am J Hum Genet. 2015;97(5):744–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Heydeck W, Zeng H, Liu A. Planar cell polarity effector gene Fuzzy regulates cilia formation and Hedgehog signal transduction in mouse. Dev Dyn. 2009;238(12):3035–3042. [DOI] [PubMed] [Google Scholar]
- 82.Zhang Z, Wlodarczyk BJ, Niederreither K, et al. Fuz regulates craniofacial development through tissue specific responses to signaling factors. PLoS One. 2011;6(9):e24608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Seo JH, Zilber Y, Babayeva S, et al. Mutations in the planar cell polarity gene, Fuzzy, are associated with neural tube defects in humans. Hum Mol Genet. 2011;20(22):4324–4333. [DOI] [PubMed] [Google Scholar]
- 84.Cohen M, Varki A. The sialome--far more than the sum of its parts. OMICS. 2010;14(4):455–464. [DOI] [PubMed] [Google Scholar]
- 85.Wang B, Brand-Miller J. The role and potential of sialic acid in human nutrition. Eur J Clin Nutr. 2003;57(11):1351–1369. [DOI] [PubMed] [Google Scholar]
- 86.Lo TH, Silveira PA, Fromm PD, et al. Characterization of the expression and function of the C-type lectin receptor CD302 in mice and humans reveals a role in dendritic cell migration. J Immunol. 2016;197(3):885–898. [DOI] [PubMed] [Google Scholar]
- 87.Zhang Y, Qin C, Yang L, Lu R, Zhao X, Nie G. A comparative genomics study of carbohydrate/glucose metabolic genes: from fish to mammals. BMC Genomics. 2018;19(1):246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Albert FW, Shchepina O, Winter C, et al. Phenotypic differences in behavior, physiology and neurochemistry between rats selected for tameness and for defensive aggression towards humans. Horm Behav. 2008;53(3):413–421. [DOI] [PubMed] [Google Scholar]
- 89.Li-Byarlay H, Rittschof CC, Massey JH, Pittendrigh BR, Robinson GE. Socially responsive effects of brain oxidative metabolism on aggression. Proc Natl Acad Sci U S A. 2014;111(34):12533–12537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chandrasekaran S, Rittschof CC, Djukovic D, et al. Aggression is associated with aerobic glycolysis in the honey bee brain(1). Genes Brain Behavior. 2015;14(2):158–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pozo M, Claret M. Hypothalamic control of systemic glucose homeostasis: the pancreas connection. Trends Endocrinol Metab 2018;29(8):581–594. [DOI] [PubMed] [Google Scholar]
- 92.Wan L, Lou J, Xia Y, et al. hPrimpol1/CCDC111 is a human DNA primase-polymerase required for the maintenance of genome integrity. EMBO Reports. 2013;14(12):1104–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Guilliam TA, Brissett NC, Ehlinger A, et al. Molecular basis for PrimPol recruitment to replication forks by RPA. Nature Commun. 2017;8:15222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lee SH, Kim CH. DNA-dependent protein kinase complex: a multifunctional protein in DNA repair and damage checkpoint. Mol Cells. 2002;13(2):159–166. [PubMed] [Google Scholar]
- 95.Traini M, Quinn CM, Sandoval C, et al. Sphingomyelin phosphodiesterase acid-like 3A (SMPDL3A) is a novel nucleotide phosphodiesterase regulated by cholesterol in human macrophages. J Biol Chem. 2014;289(47):32895–32913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ozato K, Shin DM, Chang TH, Morse HC 3rd. TRIM family proteins and their emerging roles in innate immunity. Nat Rev Immunol. 2008;8(11):849–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fan W, Liu T, Li X, et al. TRIM52: A nuclear TRIM protein that positively regulates the nuclear factor-kappa B signaling pathway. Mol Immunol. 2017;82:114–122. [DOI] [PubMed] [Google Scholar]
- 98.Cruciat CM, Niehrs C. Secreted and transmembrane wnt inhibitors and activators. Cold Spring Harb Perspect Biol. 2013;5(3):a015081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Krupnik VE, Sharp JD, Jiang C, et al. Functional and structural diversity of the human Dickkopf gene family. Gene. 1999;238(2):301–313. [DOI] [PubMed] [Google Scholar]
- 100.Sibbe M, Jarowyj J. Region-specific expression of Dickkopf-like1 in the adult brain. Abbreviated title: Dkkl1 in the adult brain. Neurosci Lett. 2013;535:84–89. [DOI] [PubMed] [Google Scholar]
- 101.Veeck J, Dahl E. Targeting the Wnt pathway in cancer: the emerging role of Dickkopf-3. Biochim Biophys Acta. 2012;1825(1):18–28. [DOI] [PubMed] [Google Scholar]
- 102.Bruggink KA, Kuiperij HB, Gloerich J, et al. Dickkopf-related protein 3 is a potential Abeta-associated protein in Alzheimer’s Disease. J Neurochem. 2015;134(6):1152–1162. [DOI] [PubMed] [Google Scholar]
- 103.Ikeuchi T, de Vega S, Forcinito P, et al. Extracellular protein fibulin-7 and its C-terminal fragment have in vivo antiangiogenic activity. Sci Rep. 2018;8(1):17654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.de Vega S, Iwamoto T, Nakamura T, et al. TM14 is a new member of the fibulin family (fibulin-7) that interacts with extracellular matrix molecules and is active for cell binding. J Biol Chem. 2007;282(42):30878–30888. [DOI] [PubMed] [Google Scholar]
- 105.de Vega S, Kondo A, Suzuki M, et al. Fibulin-7 is overexpressed in glioblastomas and modulates glioblastoma neovascularization through interaction with angiopoietin-1. Int J Cancer. 2019. [DOI] [PubMed] [Google Scholar]
- 106.Charrin S, Le Naour F, Oualid M, et al. The major CD9 and CD81 molecular partner. Identification and characterization of the complexes. J Biol Chem. 2001;276(17):14329–14337. [DOI] [PubMed] [Google Scholar]
- 107.Orlicky DJ, Nordeen SK. Cloning, sequencing and proposed structure for a prostaglandin F2 alpha receptor regulatory protein. Prostaglandins Leukot Essent Fatty Acids. 1996;55(4):261–268. [DOI] [PubMed] [Google Scholar]
- 108.Stipp CS, Orlicky D, Hemler ME. FPRP, a major, highly stoichiometric, highly specific CD81- and CD9-associated protein. J Biol Chem. 2001;276(7):4853–4862. [DOI] [PubMed] [Google Scholar]
- 109.Boucheix C, Rubinstein E. Tetraspanins. Cell Mol Life Sci. 2001;58(9):1189–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Levy S, Shoham T. The tetraspanin web modulates immune-signalling complexes. Nat Rev Immunol. 2005;5(2):136–148. [DOI] [PubMed] [Google Scholar]
- 111.Chambrion C, Le Naour F. The tetraspanins CD9 and CD81 regulate CD9P1-induced effects on cell migration. PLoS One. 2010;5(6):e11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rocha-Perugini V, Sanchez-Madrid F, Martinez Del Hoyo G. Function and dynamics of tetraspanins during antigen recognition and immunological synapse formation. Front Immunol. 2015;6:653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Orlicky DJ. Negative regulatory activity of a prostaglandin F2 alpha receptor associated protein (FPRP). Prostaglandins Leukot Essent Fatty Acids. 1996;54(4):247–259. [DOI] [PubMed] [Google Scholar]
- 114.Liu M, Shi X, Bi Y, et al. SHCBP1L, a conserved protein in mammals, is predominantly expressed in male germ cells and maintains spindle stability during meiosis in testis. Mol Hum Reprod. 2014;20(6):463–475. [DOI] [PubMed] [Google Scholar]
- 115.Bernstein KA, Granneman S, Lee AV, Manickam S, Baserga SJ. Comprehensive mutational analysis of yeast DEXD/H box RNA helicases involved in large ribosomal subunit biogenesis. Mol Cell Biol. 2006;26(4):1195–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schmid M, Jensen TH. The exosome: a multipurpose RNA-decay machine. Trends Biochem Sci. 2008;33(10):501–510. [DOI] [PubMed] [Google Scholar]
- 117.Kopplin LJ, Igo RP Jr., Wang Y, et al. Genome-wide association identifies SKIV2L and MYRIP as protective factors for age-related macular degeneration. Genes Immun. 2010;11(8):609–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Filiano AJ, Gadani SP, Kipnis J. Interactions of innate and adaptive immunity in brain development and function. Brain Res. 2015;1617:18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hale MW, Spencer SJ, Conti B, et al. Diet, behavior and immunity across the lifespan. Neurosci Biobehav Rev. 2015;58:46–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fakhoury M Role of immunity and inflammation in the pathophysiology of neurodegenerative diseases. Neurodegener Dis. 2015;15(2):63–69. [DOI] [PubMed] [Google Scholar]
- 121.Hare B, Plyusnina I, Ignacio N, et al. Social cognitive evolution in captive foxes is a correlated by-product of experimental domestication. Curr Biol. 2005;15(3):226–230. [DOI] [PubMed] [Google Scholar]
- 122.Albert FW, Somel M, Carneiro M, et al. A comparison of brain gene expression levels in domesticated and wild animals. PLoS Genet. 2012;8(9):e1002962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Long K, Mao K, Che T, et al. Transcriptome differences in frontal cortex between wild boar and domesticated pig. Animal Sci J. 2018;89(6):848–857. [DOI] [PubMed] [Google Scholar]
- 124.Yang X, Zhang H, Shang J, et al. Comparative analysis of the blood transcriptomes between wolves and dogs. Anim Genet. 2018;49(4):291–302. [DOI] [PubMed] [Google Scholar]
- 125.Belteky J, Agnvall B, Jensen P. Gene expression of behaviorally relevant genes in the cerebral hemisphere changes after selection for tameness in red junglefowl. PLoS One. 2017;12(5):e0177004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Freedman AH, Schweizer RM, Ortega-Del Vecchyo D, et al. Demographically-based evaluation of genomic regions under selection in domestic dogs. PLoS Genet. 2016;12(3):e1005851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Brusini I, Carneiro M, Wang C, et al. Changes in brain architecture are consistent with altered fear processing in domestic rabbits. Proc Natl Acad Sci USA. 2018;115(28):7380–7385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Polster A, Dittmer PJ, Perni S, Bichraoui H, Sather WA, Beam KG. Stac Proteins suppress Ca(2+)-dependent inactivation of neuronal l-type Ca(2+) channels. J Neurosci. 2018;38(43):9215–9227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Crosetto N, Bienko M, van Oudenaarden A. Spatially resolved transcriptomics and beyond. Nature Rev Genet. 2015;16(1):57–66. [DOI] [PubMed] [Google Scholar]
- 130.Khan AM, Grant AH, Martinez A, et al. Mapping molecular datasets back to the brain regions they are extracted from: remembering the native countries of hypothalamic expatriates and refugees. Adv Neurobiol. 2018;21:101–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bentosela M, Wynne CD, D’Orazio M, Elgier A, Udell MA. Sociability and gazing toward humans in dogs and wolves: Simple behaviors with broad implications. J Exp Anal Behav. 2016;105(1):68–75. [DOI] [PubMed] [Google Scholar]
- 132.Hare B, Brown M, Williamson C, Tomasello M. The domestication of social cognition in dogs. Science. 2002;298(5598):1634–1636. [DOI] [PubMed] [Google Scholar]
- 133.Hare B, Tomasello M. Human-like social skills in dogs? Trends Cogn Sci. 2005;9(9):439–444. [DOI] [PubMed] [Google Scholar]
- 134.Persson ME, Roth LS, Johnsson M, Wright D, Jensen P. Human-directed social behaviour in dogs shows significant heritability. Genes Brain Behavior. 2015;14(4):337–344. [DOI] [PubMed] [Google Scholar]
- 135.Rossano F, Nitzschner M, Tomasello M. Domestic dogs and puppies can use human voice direction referentially. Proc Biol Sci. 2014;281(1785):20133201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Werhahn G, Viranyi Z, Barrera G, Sommese A, Range F. Wolves (Canis lupus) and dogs (Canis familiaris) differ in following human gaze into distant space but respond similar to their packmates’ gaze. J Comp Psychol. 2016;130(3):288–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. http://heatmapper.ca/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Sequence of primers used for qPCR analyses.
Table S2. Sequence reads and mapping rate of samples.
Table S3. Hypothalamus DEGs that are DE in one or more of the three other brain regions: PFC 31, BFB 31, and anterior pituitary 26.
Table S4. qPCR expression for other hypothalamic genes that did not differ between aggressive vs. tame foxes. Average ± SEM for 2−ΔΔCt values.
Figure S2. Anatomical location (red) and connectivity (blue) of the basal forebrain (BFB) dissection samples. Connectivity includes the olfactory tubercule, piriform lobe, stria terminalis, amygdala, medial temporal lobe, and lateral temporal cortex (ectosylvian and suprasylvian gyri).
Figure S3. Anatomical location (red) and connectivity (blue) of the medial prefrontal cortex (PFC) dissection samples. Connectivity includes the olfactory tubercule, stria terminalis, caudate, cingulum bundle, and medial temporal lobe.
Figure S1. Anatomical location (red) and connectivity (blue) of the hypothalamus (HYP) dissection samples. Connectivity includes the olfactory tubercule, piriform lobe, amygdala, brainstem, medial prefrontal cortex, and medial temporal lobe.
Figure S4. Expression pattern of three groups of DEG in the different brain regions. A) Expression pattern of DEG within PFC (cortex) in the three brain regions, cortex, forebrain, and hypothalamus. B) Expression pattern of DEG within BFB (forebrain) within the three brain regions. C) Expression pattern of DEG within hypothalamus within the three brain regions.
Figure S6. qPCR analysis for AVP and OXT. No differences in gene expression were detected for these genes between tame and aggressive foxes.
Figure S5. Expression pattern of hypothalamic DEG (between tame and aggressive foxes) within the hypothalamus for the three brain regions. A) Expression pattern of hypothalamic DEG in the PFC (cortex). B) Expression pattern of hypothalamic DEG in the BFB (forebrain). C) Expression pattern of hypothalamic DEG in the hypothalamus. The count data included a meta data information specifying which gene was either up-regulated or downregulated in HYP.
Figure S9. Gene interactions as determined by the GeneMANIA app 51 in Cytoscape 48,for the pale turquoise and midnight blue modules. A) Pale Turquoise. B) Midnight blue. Extensive interactions for genes within both of these modules is evident. Legends for the colors of the nodes and edges are detailed in the figure.
Figure S8. Diagram showing BPHL as a hub gene with 135 genes within the midnight blue module connecting to this central gene. This figure though only shows 70 genes that have corresponding gene symbols of the 135 genes based on Ensembl IDs.
Figure S7. Identification and characterization of weighted gene co-expression network analyses (WGCNA) and clustering dendrogram of genes. The clustering dendrogram is based on variation in topological overlap and assigned module colors are shown above and to the side of the heatmap.
Figure S10. Gene interactions as determined by the GeneMANIA app 51 in Cytoscape 48, for the dark turquoise and the dark orange modules. A) Dark Turquoise. B) Dark Orange. Extensive interactions for genes within both of these modules is evident. Legends for the colors of the nodes and edges are detailed in the figure.
Figure S11. Functional enrichment pathway analyses for the dark turquoise and dark orange modules, as determined by the ClueGo App 47 in Cytoscape 48. A) For the dark turquoise module, the cGMP-PKG signaling pathway was the primary enriched pathway. B) In the dark orange module, regulation of gene expression, cellular response to cytokine stimulus, and cell surface receptor signaling pathway were enriched in the dark orange module.
Supplementary Information Appendix S1: Imaging studies for fox brain samples
Supplementary Information Appendix S3. Behavioral scores for aggressive and tame foxes.
Supplementary Information Appendix S4. rMATS splice form analysis.
Supplementary Information Appendix S5. Gene module membership for those that are significantly correlated with tameness vs. aggressiveness.
Supplementary Information Appendix S6. Distribution of the other 69 DEG within the various color modules. The two modules that contained most of the DEG were Grey (n = 34 DEG), which is a module generally made up of genes that do not fit into any of the modules, and Turquoise (n = 18).
Supplementary Information Appendix S7. DEG and other genes within the Turquoise module. Highlighted genes within this module are DEG.
Supplementary Information Appendix S8. GO terms associated with genes in modules that are significantly correlated with tameness vs. aggressiveness.
Supplementary Information Appendix S2. Differentially expressed genes (DEGs) and all other genes.