

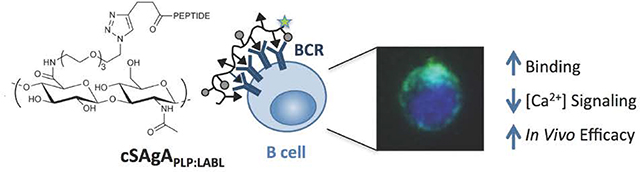
Abstract
A pressing need exists for antigen-specific immunotherapies (ASIT) that induce selective tolerance in autoimmune disease while avoiding deleterious global immunosuppression. Multivalent soluble antigen arrays (SAgAPLP:LABL), consisting of a hyaluronic acid (HA) linear polymer backbone co-grafted with multiple copies of autoantigen (PLP) and cell adhesion inhibitor (LABL) peptides, are designed to induce tolerance to a specific multiple sclerosis (MS) autoantigen. Previous studies established that hydrolyzable SAgAPLP:LABL, employing a degradable linker to codeliver PLP and LABL, was therapeutic in experimental autoimmune encephalomyelitis (EAE) in vivo and exhibited antigen-specific binding with B cells, targeted the B cell receptor (BCR), and dampened BCR-mediated signaling in vitro. Our results pointed to sustained BCR engagement as the SAgAPLP:LABL therapeutic mechanism, so we developed a new version of the SAgA molecule using non-hydrolyzable conjugation chemistry, hypothesizing it would enhance and maintain the molecule’s action at the cell surface to improve efficacy. ‘Click SAgA’ (cSAgAPLP:LABL) uses hydrolytically stable covalent conjugation chemistry (Copper-catalyzed Azide-Alkyne Cycloaddition (CuAAC)) rather than a hydrolyzable oxime bond to attach PLP and LABL to HA. We explored cSAgAPLP:LABL B cell engagement and modulation of BCR-mediated signaling in vitro through flow cytometry binding and calcium flux signaling assays. Indeed, cSAgAPLP:LABL exhibited higher avidity B cell binding and greater dampening of BCR-mediated signaling than hydrolyzable SAgAPLP:LABL. Furthermore, c SAgAPLP:LABL exhibited significantly enhanced in vivo efficacy compared to hydrolyzable SAgAPLP:LABL, achieving equivalent efficacy at one quarter of the dose. These results indicate that non-hydrolyzable conjugation increased the avidity of cSAgAPLP:LABL to drive in vivo efficacy through modulated BCR-mediated signaling.
Keywords: antigen-specific immunotherapy, autoimmune, conjugation, multivalent, binding, signaling
Graphical Abstract
INTRODUCTION
Autoimmune diseases such as multiple sclerosis (MS) are typified by a breakdown of healthy immune regulation and subsequent misrecognition of self for non-self.1–2 The autoimmune breakdown in MS is largely propagated by autoreactive T and/or B cell clonal expansion and attack against myelin sheath autoantigens, leading to demyelination and neurodegeneration.3–6 Activation of naïve T cells against autoantigen requires two signals from an antigen presenting cell (APC): (1) primary antigenic signal delivered through the major histocompatibility complex (MHC) on the APC to the T cell receptor (TCR) on the T cell, and (2) secondary costimulatory signal (i.e., CD80/CD86) delivered to the cognate receptor (i.e., CD28) on the T cell.7–15 B cells, as professional APCs that possess antigen specificity and immunological memory, play a particularly pivotal role in immune regulation.16–18 Indeed, loss of B cell tolerance has been implicated in numerous autoimmune diseases.19–21 Autoimmune therapies targeting B cells have been successful in treating MS (i.e., rituximab), but general B cell depletion or inactivation may induce global immunosuppression, trigger adverse side effects, and suffer from limited efficacy.22–24 Development of an antigen-specific immunotherapy (ASIT) that targets and silences autoreactive B cells in a selective manner would address an important need for safer and more effective treatment.25–27
Modulation of B cells in a direct, antigen-specific manner requires targeting of the B cell receptor (BCR).18 Antigen binding to the BCR can trigger receptor clustering and antigen-specific B cell activation.28–32 However, continuous antigen binding and occupation of the BCR in the absence of secondary costimulatory signals results in B cell anergy, or a state of antigen unresponsiveness, that is marked by reduced calcium signaling27,33–35 Induction of B cell anergy can have a two-fold therapeutic effect by inducing (1) an effector B cell population that is not responsive to autoantigen and (2) B cells with reduced APC capacity due to downregulation of costimulatory signals CD80 and CD86.15, 25–26, 36 Thus, a promising avenue for modulating the immune response in an antigen-specific manner is to induce these B cell phenotypes through BCR engagement.
Multivalent linear polymers are especially adept at engaging cell surface receptors such as the BCR, where high avidity multivalent antigen is aided by the molecule’s conformational flexibility to allow for greater interaction with the cell surface to bind and cluster receptors.28, 30, 37–43 For example, the spacing and orientation of ligands on a linear polymer may adapt to the contour and dynamic receptor spacing of the cell surface, whereas spherical or globular particles (i.e., dendrimers, nanoparticles, liposomes) are inherently more rigid with relatively fixed spacing and orientation of ligands. As such, multivalent linear polymers may be particularly suited for B cell and BCR targeted therapies. We previously reported on multivalent soluble antigen arrays (SAgAPLP:LABL) consisting of a linear hyaluronic acid (HA) polymer co-grafted with myelin autoantigen peptide (proteolipid protein peptide, PLP139–151) and intercellular adhesion molecule-1 (ICAM-1) inhibitor peptide (LABL) derived from leukocyte function associated antigen-1 (LFA-1).44–50 In this molecule, PLP acts as the primary antigenic signal to drive antigen-specific B cell binding, while LABL enhances cellular engagement by targeting ICAM-1, exploiting the ICAM-1/LFA-1 interaction that promotes and sustains intercellular adhesion.44, 51–54 Our previous studies established that in vivo treatment with SAgAPLP:LABL significantly alleviated experimental autoimmune encephalomyelitis (EAE), a murine model of relapsing-remitting multiple sclerosis.46–50 Importantly, multivalent presentation of both PLP and LABL on a polymer carrier was necessary for therapeutic efficacy,45, 47, 49 and presentation on soluble linear HA was more effective than presentation on insoluble PLGA nanoparticles.48
The SAgAPLP:LABL molecule studied up to this point employed a degradable linker to codeliver PLP and LABL. ‘Hydrolyzable SAgAPLP:LABL exhibited antigen-specific binding with B cells by targeting the BCR, remained on the cell surface for an extended period of time, and dampened BCR-mediated signaling in vitro.44 Our results pointed to sustained BCR engagement as the molecule’s therapeutic mechanism, so we hypothesized that using non-hydrolyzable conjugation chemistry to develop a non-degradable SAgA would enhance and maintain the molecule’s action at the cell surface to improve efficacy. Here, we have developed a new version of the SAgA molecule, termed ‘click SAgA’ (cSAgAPLP:LABL), using a hydrolytically stable covalent conjugation chemistry rather than hydrolyzable grafting of multivalent PLP139–151 and LABL peptides to HA using a hydrolyzable oxime bond. We explored whether this non-hydrolyzable conjugation chemistry improved B cell engagement and modulation of BCR-mediated signaling, and if in vivo efficacy was correspondingly improved.
B cell binding, signaling, and therapeutic efficacy in EAE were compared between SAgAPLP:LABL (oxime conjugation chemistry) and cSAgAPLP:LABL (‘click’ conjugation chemistry) through a combination of in vitro and in vivo studies. Binding avidity was evaluated in immortalized human Raji B cells as a model APC system using flow cytometry binding assays developed previously.44 Modulation of BCR-mediated signaling was assessed using flow cytometry calcium flux assays. Engagement and organization of BCR on the cell surface was observed through real-time fluorescence microscopy. Lastly, in vivo efficacy was compared across various doses in the EAE model.
MATERIALS AND METHODS
Materials
Hyaluronic acid (HA) sodium salt (MW 16 kDa) was purchased from Lifecore Biomedical (Chaska, MN). 11 -azido-3,6,9-trioxaundecan-1-amine (NH2-PEG3-N3), N-hydroxysuccinimide, N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), 2-(N-morpholino)ethane-sulfonic acid sodium salt (MES), tris(3-hydroxypropyltriazolylmethyl)amine, and sodium ascorbate (NaAsc) were purchased from Sigma-Aldrich (St. Louis, MO) and used as received without further purification. Copper(II) sulfate pentahydrate (CuSO4 · 5H2O) was purchased from Acros Organics (Geel, Belgium). Alkyne-functionalized peptides bearing an N-terminal 4-pentynoic acid (homopropargyl, hp) modification, hpPLP139–151 (hp-HSLGKWLGHPDKF-OH) and hpLABL (hp-ITDGEATDSG-OH), were originally synthesized in our laboratory via solid phase peptide synthesis. Larger quantities of both hpPLP139–151 and hpLABL peptides were obtained from Biomatik USA, LLC (Wilmington, DE). Unmodified PLP (NH2-HSLGKWLGHPDKL-OH) peptide was purchased from PolyPeptide Laboratories (San Diego, CA). Incomplete Freund’s adjuvant (IFA) and killed Mycobacterium tuberculosis strain H37RA were purchased from Difco (Sparks, MD). Pertussis toxin was purchased from List Biological Laboratories (Campbell, CA). Fluo-4 AM calcium indicator were purchased from Thermo Fisher Scientific (Waltham, MA). Immortalized human Raji B cells were purchased from American Type Culture Collection (ATCC, Manassas, VA). AffiniPure F(ab’)2 fragment goat anti-human IgM and AlexaFluor® 647 AffiniPure F(ab’)2 fragment goat anti-human IgM were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA). All other chemicals and reagents were analytical grade and used as received.
Synthesis and Labeling of Soluble Antigen Arrays
Soluble antigen arrays (SAgAs) and fluorescein isothiocyanate (FITC)-labeled SAgAs (fSAgAs) were synthesized and characterized as previously reported.44, 47 Aminooxy peptides AoPLP and/or AoLABL were grafted to HA using oxime conjugation chemistry to synthesize HAPLP (HA and AoPLP), HALABL (HA and AoLABL), and SAgAPLP:LABL (HA, AoPLP, and AoLABL). Peptide conjugation was determined through gradient reverse-phase analytical high-performance liquid chromatography (RP-HPLC) following cleavage of peptides in 0.1N HCl. Relative FITC fluorescence of labeled samples was determined spectrofluorometrically.
Synthesis of Penn Green-Alk (Scheme 1A)
Scheme 1.

Synthesis of cSAgA components and molecules: (A) Alkyne-functionalized Pennsylvania Green. (B) Structures of homopropargyl-modified peptides. (C) ‘Click’ soluble antigen array (cSAgA).
Synthesis of 4-(2,7-difluoro-6-hydroxy-3-oxo-3H-xanthen-9-yl)-3-methyl-N-(prop-2-yn-1-yl)benzamide (Penn Green-Alk) was adapted from Meng et al.55 To a mixture of 2,5-dioxopyrrolidin-1-yl 4-(2,7-difluoro-6-hydroxy-3-oxo-3H-xanthen-9-y1)-3-methylbenzoate (55.3 μmol) in DMF (0.5 mL), propargylamine (61.2 μmol) in 0.5 mL H3BO3 buffer (pH = 8.5, 50 mM) was added and stirred at room temperature for 6 hours. The reaction mixture was frozen and lyophilized to give the crude product as an orange liquid. The crude product was dissolved in DMSO and purified by preparative RP-HPLC (Waters XBridge C18, 5 μm, 10×250 mm, linear gradient from 5–95% MeCN (+ 0.05% TFA) in H2O (+ 0.05% TFA) over 30 minutes, detection at 280 nm) to give the final product (22.5 mg, 84.7%) as an orange-yellow solid; 1H NMR (400 MHz, DMSO-d6) δ 7.99 (s, 1H), 7.93–7.87 (m, 1H), 7.40 (d, J = 7.9 Hz, 1H), 6.85 (br s, 2H), 6.63 (d, J= 11.2 Hz, 2H), 4.11 (dd, J = 5.6, 2.5 Hz, 2H), 3.17 (t, J = 2.5 Hz, 1H), 2.08 (s, 3H); HRMS (TOF ESI+) expected [M+Na]+: 442.0867, found: 442.0870.
Synthesis of HA-N3 (Scheme 1C)
Synthesis of FLA-N3 was adapted from Hu et al and Di Meo et al.56–57 Sodium hyaluronate (93.9 μmol, 16 kDa average MW) was added to a 250 mL round bottom flask with stir bar, followed by 100 mL of 50 mM MES buffer (pH = 4.0). The mixture was stirred until in solution (~15 minutes) before EDC (23.1 mmol) was added neat, then N-hydroxysuccinimide (18.8 mmol) added neat. The mixture was stirred for 5 minutes before H2N-PEG3-N3 (4.51 mmol) in 20 mL MES buffer was added. The solution was then stirred for 24 hours at room temperature before being dialyzed in 6–8 kDa cutoff dialysis tubing against 4.5 L of 1.0 M NaCl solution for 24 hours, then 4.5 L of deionized water (4 ×12 hours). The volume in the bag was then transferred to vials, frozen, and lyophilized to yield a white powder (1.61 g, 95.0%).
Synthesis and Labeling of Click Soluble Antigen Arrays (Scheme 1C)
HA-N3 (2 μmol) was added as a 50 μM solution in deionized H2O to a 250 mL round bottom flask with stir bar. Each component peptide (40 μmol) was then added as a ~3 mM solution in deionized H2O, followed by a premixed solution of THPTA (70 μmol) and CuSO4 · 5H2O (14 μmol) in deionized H2O. In the case with fluorescently-labeled variants, a 2.4 mM Penn Green-Alk solution in DMF (2.0 equivalents relative to HA-N3) was also added. The solution was allowed to stir for 1–2 minutes before a 100 μL aliquot was removed for HPLC analysis. NaAsc (300 μmol) was then added to the reaction mixture as a 100 mM solution in deionized H2O. The reaction was allowed to proceed under varying conditions depending on the starting components and desired valency (cHAPLP: 18 hours at 37°C; cHALABL and cSAgAPLP:LABL: 18 hours at 50°C; fcHA and fcHAPLP: 24 hours at 37°C; fcHALABL and fcSAgAPLP:LABL: 24 hours at 50°C). Additional 100 μL aliquots were removed throughout the course of the reaction to determine the extent of conjugation. Once the target conjugation values were achieved, the reaction solution was transferred to 6–8 kDa dialysis tubing and dialyzed against 4.5 L of 1.0 M NaCl (3×8 hours), then 4.5 L of deionized H2O (5×8 hours). The volume in the bag was then transferred to vials, frozen, and lyophilized.
Analytical Characterization of Click Soluble Antigen Arrays
FTIR spectra were collected on a Bruker Tensor 27 FTIR spectrometer equipped with an Attenuated Total Reflectance (ATR) cell, analyzing purified samples at ambient temperature in the solid state, and collecting a total of 32 scans per sample. NMR spectra were collected on a Bruker Avance AVIII 500 MHz: spectrometer equipped with a dual carbon/proton cryoprobe (unless otherwise noted), and all samples were dissolved in 650 μL of D2O for analysis. MestReNova 11.0 was used for NMR data analysis. The amide methyl resonance (δ ~ 1.90–2.05 ppm) of all 1H NMR spectra was normalized to an integration of 3.0, and the sum of all other signals in the range of δ ~ 1.0–4.0 ppm was used to ratiometrically determine the number of azide functionalization sites during FLA-N3 synthesis.
RP-HPLC and SEC analysis were conducted using a Waters Alliance HPLC system equipped with either a diode array detector or dual wavelength UV/Vis detector. For the quantitative determination of peptide conjugation by RP-HPLC, the following equation was used:
| Equation 1 |
where Ncon = number of conjugated peptides per backbone, npep = moles of peptide used in reaction, nHA = moles of HA-N3 used in reaction, Vpre = total reaction volume before NaAsc is added, Vsam = volume of “pre-NaAsc” sample removed from reaction mixture, PAt = measured peak area of peptide at time t, PAstart = measured peak area of free peptide before NaAsc is added to the reaction. General chromatographic conditions employed a Waters XBridge C4, 3.5 μm, 300 Å stationary phase under ion pairing (0.05% TFA in H2O and MeCN) mobile phase conditions, utilizing a linear elution gradient (5–60%) with detection at 214 nm.
Cell Culture
Raji B cells (human B lymphocytes, ATCC) were cultured in RPMI-1640 supplemented with L-glutamine, 10% fetal bovine serum (FBS), and 1% penicillin/streptomycin (P/S) at 37°C and 5% CO2. Cell assays were consistently performed after cells reached confluency (~2 weeks) and following no more than 8–10 passages, per ATCC guidelines.
Flow Cytometry Binding Assay
Association binding studies were performed by flow cytometry (MoFlo XDP Cell Sorter, Beckman Coulter Inc., Brea, CA), as previously reported.44 Cell nuclei were stained with Hoechst and propidium iodide (PI) was used as a dead cell indicator; data acquisition was triggered off the Hoechst signal. Cell samples were warmed to 37°C for 2 minutes prior to the flow cytometry run. Fluorescence was excited using 488, 405, and 640 nm lasers and was collected using 529/28, 457/40, and 670/30 nm bandpass emission filters.
To observe maximum steady state binding, cells were mixed with the treatment to achieve a final concentration of 1×106 cells/mL immediately before injecting on the flow cytometer. Sample concentration was determined from preliminary saturation studies.44 Samples were added at an equimolar PLP dose (353 μM PLP for fHAPLP and fSAgAPLP:LABL, or 353 μM LABL for fHALABL) and fHA was dosed at 39 μM (the HA molar equivalent to a 353 μM PLP dose of fSAgAPLP:LABL) to mimic the dosing scheme from in vivo studies. The sample was allowed to run for 5 minutes to ensure that maximum steady state was established, which occurs after approximately 3–4 minutes.
Flow cytometry binding data was first gated to remove doublets, dead cells, and debris using Kaluza Flow Analysis software (Beckman Coulter, Inc., Brea, CA) (Supplementary Figure 1). Additional data processing was performed using KNIME software (Konstanz Information Miner, KNIME, Zurich, Switzerland). Nonlinear regression and additional statistical analysis was performed using GraphPad Prism (GraphPad Software, Inc., La Jolla, CA).
Calcium Flux Signaling Assay
Raji B cells were loaded with 5 μM Fluo-4 AM for 30 minutes at room temperature in PBS, then kept on ice in BBSS (Hanks Balanced Salt Solution) containing 1.3 mM Ca2+ and 0.9 mM Mg2+ before analysis. Cells were run through a BD FACSFusion cytometer and fluorescence was monitored in the 530/30 nm channel. After baseline quantification for ~30 seconds, crosslinking goat anti-human IgM (Jackson ImmunoResearch) was added at a final concentration of 20 μg/mL to stimulate the cells. This concentration was determined from a preliminary study where a range of anti-human IgM concentrations (5–40 μg/mL) was evaluated and 20 μg/mL achieved greatest stimulation of Raji B cells. Changes in Fluo-4 fluorescence were measured for 1 minute to establish an anti-IgM stimulated baseline, followed by addition of (c)SAgA treatment (dosed at 353 μM PLP, same concentration used in binding studies) to determine the effect on IgM-stimulated signaling. Data was acquired for an additional 3 minutes until steady state was established. To measure inhibition of anti-IgM stimulation, (c)SAgAPLP:LABL was added to cells prior to anti-IgM stimulation. KNIME was used to process and plot the kinetic data, while Kaluza and GraphPad Prism were used for the remaining analysis.
Fluorescence Microscopy
Live cell imaging of fcSAgA binding and surface IgM clustering was observed under fluorescence microscopy (Olympus IX81 Inverted Epifluorescence Microscope) using the same concentrations from flow cytometry association binding experiments. CellASIC ONIX M04S Microfluidics Switching Plates and Microfluidics Platform (EMD Millipore, Billerica, MA) were utilized for controlled perfusion of fluorescent samples and media with cells during real-time imaging. Raji B cells were stained with Hoechst and mixed with AlexaFluor® 647 goat anti-human IgM (Jackson ImmunoResearch) at 20 μg/mL to stimulate the cells and label surface IgM, then loaded into the imaging chamber. fcSAgA was perfused into the chamber for 10 minutes (3 psi for 5 minutes, 0.25 psi for 5 minutes) to allow binding with cells, followed by gentle media perfusion (0.25 psi for 5 minutes) to rinse unbound fcSAgA, followed by immediate image capture. Images were processed using Slidebook 5.5 (Intelligent Imaging Innovations, Inc., Denver, CO) and quantitative BCR clustering analysis was performed using ImageJ (Supplementary Figure 2). Otsu thresholding was applied to the IgM channel to identify IgM capping, where cells with a single localized area of IgM fluorescence above a brightness threshold of 65 were considered positive for capping (Supplementary Figure 2B). This method was applied to 100–250 IgM-positive stained cells per sample to determine % IgM capping. IgM pixel intensity was plotted for individual cells (n=10 per sample) as a function of diameter to generate profile plots relative to normalized diameter (d/D) and to determine the maximum IgM pixel intensity per cell (Supplementary Figure 2C).
Preclinical EAE Study in Mice
In vivo studies were carried out with 4–6 week old SJL/J (H-2) female mice purchased from Envigo Laboratories (Indianapolis, IN). Mice were housed under specified, pathogen-free conditions at the University of Kansas and all experiments were approved by the University’s Institutional Animal Care and Use Committee. Complete Freund’s adjuvant (CFA) was made by combining IFA and killed M. tuberculosis strain H37RA at a final concentration of 4 mg/mL. Animals were induced with experimental autoimmune encephalomyelitis (EAE), the PLP-specific mouse model of relapsing-remitting MS, on day 0 of the study. Immunization was accomplished using a 0.2 mL emulsion containing 200 μg PLP139–151 peptide, plus equal volumes of PBS and CFA. The emulsion was administered subcutaneously (s.c.) as a total of four 50 μL injections, located above each shoulder and each hind flank. Pertussis toxin (100 ng in 100 μL) was injected intraperitoneally on day 0 and day 2 post-immunization.
Treatments were administered on days 4, 7, and 10 as 100 μL subcutaneous injections at the nape of the neck (n=3–6 mice per treatment group), with the exception of one group in the dosing study that received treatments on days 4 and 7 only. Samples were administered at a dose equivalent to 50, 133, or 200 nmol PLP per 100 μL (0.5, 1.33, or 2 mM PLP, respectively). This three-day dosing schedule and dose of 200 nmol PLP were found to be efficacious in a previous SAgAPLP:LABL study.49 Disease progression was evaluated by a single observer using the following clinical score system: 0, no clinical disease symptoms; 1, weakness or limpness of the tail; 2, weakness or partial paralysis of one or two hind limbs (paraparesis); 3, full paralysis of both hind limbs (paraplegia); 4, paraplegia plus weakness or paralysis of forelimbs; 5, moribund (at which point mice were euthanized). In addition to animal scoring, body weight measurements were performed daily for the 26-day duration of the EAE study.
Statistical Analysis
GraphPad Prism was used to perform statistical analysis including sigmoidal nonlinear regression, ordinary one-way or two-way analysis of variance (ANOVA), and unpaired t-test. ANOVA was followed by Tukey’s or Sidak’s post-hoc test, where appropriate. The threshold for statistical significance was set to p<0.05.
RESULTS AND DISCUSSION
Structural Design of Click Soluble Antigen Arrays
Multivalent soluble antigen arrays (SAgAPLP:LABL) consist of a 16 kDa HA linear polymer conjugated with approximately 10 PLP and 10 LABL peptides. The molecule was rationally designed based on studies by Dintzis et al that suggested multivalent linear polymers with a valency of 10–20 antigens and MW<100 kDa could induce a tolerogenic immune response,42, 58–59 combined with studies by Siahaan et al that showed linking PLP and LABL peptides in a bifunctional molecule was therapeutic in EAE. 53–54, 60–64 Previous SAgAPLP:LABL molecules studied in our research group employed a hydrolyzable linker chemistry to conjugate both PLP139–151 and LABL peptides to HA, and have been shown to significantly suppress disease severity in EAE.44–50 This approach was built upon our earlier two-signal hypothesis that SAgAPLP:LABL inhibited autoimmune activation via the immunological synapse by promoting antigen processing without the necessary secondary signal, a mechanism that would necessitate antigen uptake, processing, and presentation. However, our recent in vitro studies instead pointed to a therapeutic mechanism in which SAgAPLP:LABL acted through sustained BCR engagement, targeting BCR signaling while exhibiting prolonged residence on the cell surface.44 Whereas our early hypothesis motivated a degradable SAgA molecule to allow for antigen uptake and processing, these results motivated the development of a non-degradable SAgA molecule to enhance and maintain the molecule’s surface activity. Thus, we synthesized ‘click’ cSAgA variants that exploit a non-cleavable linker chemistry to evaluate whether possible release of PLP139–151 and LABL influenced the efficacy of the molecule. cSAgA multivalent arrays utilize the Copper-catalyzed Azide-Alkyne Cycloaddition (CuAAC) reaction as a stable attachment chemistry, which carries significant literature precedence with respect to application65–66 and optimization67–68. The versatility of the CuAAC reaction was a major consideration in the implementation of an alternative conjugation chemistry, as the wide range of available reaction conditions can enable improved control over valency.
Analytical Characterization of Click Soluble Antigen Arrays
Characterization was completed using a variety of qualitative and quantitative analytical techniques. Initial azide-functionalization was confirmed by FTIR spectroscopy, showing the presence of a characteristic azide stretching band after synthesis of HA-N3, which disappeared following utilization of the azide moiety during conjugation (Figure 1A). Quantitation by 1H NMR proved challenging due to signal broadening of the increasingly heterogeneous polymeric systems, and the high molecular weight of the multivalent arrays led to decreasing sensitivity with increasing conjugation, eventually limited by sample solubility. For an analysis of azide functionalization, integration ranges were used to account for signal overlap between the polymer backbone resonances and those on the linker. To assess the validity of this approach, two additional batches of FLA-N3 were prepared using a reduced number of molar equivalents of linker relative to the starting HA, resulting in a reduced number of azide functionalized sites (Supplementary Table 1). It should be noted, however, that manual integration was required to perform this analysis, and coupled with the broad resonances observed from the polymeric system, this technique is considered to be a semi-quantitative approach to justify subsequent peptide conjugation. Additional NMR studies in the solid-state or observation of alternative nuclei may aid in the quantitative nature of the analytical methodology. 1H/13C Heteronuclear Single Quantum Coherence (HSQC) NMR spectroscopy was used qualitatively to confirm the existence of resonances present in both peptide samples, which carried over to the final dialyzed products (Figure 2). Importantly, these experiments also showed the disappearance of the terminal alkyne resonance from the linker on each peptide along with the concomitant appearance of a broadened aromatic resonance not present in any individual component, corresponding to the new triazole ring.
Figure 1.

Analytical characterization data for cSAgA compounds: (A) FTIR spectra collected after functionalization and conjugation. (B) Representative HPLC chromatograms used to quantify the number of conjugated peptides as a function of reaction temperature.
Figure 2.

Qualitative confirmation of conjugation by 2D HSQC NMR where a 1H spectrum is shown on the x-axis and a 1H-decoupled 13C spectrum is shown on the y-axis.
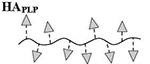
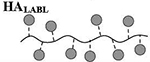
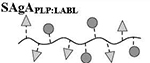
Quantitative analysis of peptide conjugation efficiency was conducted via RP-HPLC by measuring the decrease in peak area of the free alkyne-containing peptide(s) throughout the course of the reaction. The CuAAC conjugation chemistry requires an active Cu1+ catalyst for the reaction to proceed, which is generated in situ through addition of the reducing agent NaAsc to an inactive Cu2+ in solution. Prior to this final NaAsc addition step, an aliquot of the reaction mixture was removed for HPLC analysis to establish a baseline response correlating to the molar excess of peptide used in the reaction. Subsequent to the addition of NaAsc, any decrease in peak area of free alkyne-containing peptide was attributed to conjugation (Figure 1B). Standard curves for both peptides were linear to 110% of the nominal concentrations used in the reaction mixture (0.81 mM for hpPLP139–151 and 0.71 mM for hpLABL), exhibiting R2 values >0.99 upon linear regression analysis. Additional control experiments showed both hpPLP and hpLABL displayed <5% degradation (0.8% and 4.5%, respectively) at 37°C in H2O over 20 hours in the absence of all other reaction components, indicating a minimal impact of peptide degradation on the accuracy of the analytical methodology. Further, the final dialyzed products showed no evidence of peptide release after 18 hours at room temperature in either pH ~ 2.4, 5.0, or 7.0 buffers, confirming the basis behind the non-hydrolyzable linker design of the cSAgA molecular platform (Supplementary Figure 3). An abbreviated reaction optimization study showed that conjugation ratios could be significantly influenced by buffer, temperature, reactant concentrations, and molar excess of free peptide. From these observations, reaction conditions were identified that achieved a desired peptide valency. Quantitative peptide conjugation of representative test articles is provided in Table 1, showing that target conjugation efficiencies of approximately 25% per peptide (relative to theoretically available disaccharide monomers) were achieved in cHAPLP (10 PLP139–151), cHALABL (12 LABL), and cSAgAPLP:LABL (11 PLP139–151, 9 LABL).
Table 1.
Peptide molar conjugation of hydrolyzable and click conjugates, as determined by RP-HPLC.a
| Sample | Approx. MW (kDa)b | Average Molar Ratio per Polymerc | % Molar Conjugation | ||
|---|---|---|---|---|---|
| PLP:HA | LABL:HA | PLP | LABL | ||
 |
30.4 | 9 | 0 | 21 | 0 |
 |
26.0 | 0 | 10 | 0 | 24 |
 |
46.3 | 10 | 13 | 24 | 31 |
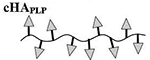 |
41.2 | 10 | 0 | 24 | 0 |
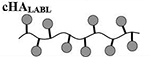 |
37.2 | 0 | 12 | 0 | 28 |
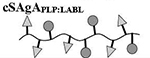 |
52.1 | 11 | 9 | 26 | 21 |
Results are an average of triplicate injections from a single batch preparation. In the molecule schematics, dotted lines represent hydrolyzable oxime linker chemistry while solid lines represent non-hydrolyzable ‘click’ linker chemistry.
Calculated from RP-HPLC data. MW, molecular weight.
HA, hyaluronic acid; PLP, proteolipid protein peptide; LABL, inhibitor peptide derived from leukocyte function associated antigen-1
Size exclusion chromatography (SEC) was primarily used to verify the success of dialysis, showing no evidence of free peptide or other reaction components in any purified cSAgA samples (Supplementary Figure 4). HA samples of varying molecular weight were used as standards to compare against purified reaction products. HA-N3 exhibited a slight decrease in retention time compared to 16 kDa HA, indicating an increase in molecular weight following azide functionalization. Retention time increased following peptide conjugation; however, this was likely a reflection of the molecule’s altered physicochemical properties and increased secondary interactions with the stationary phase rather than a reflection of molecular size. Given that hpPLP139–151 eluted after the retention time observed for salts, the increased retention of peptide-conjugated samples was likely due to secondary interactions between peptide in solution and the stationary phase overcoming the primary interactions that drive SEC separation. This secondary interaction was evident throughout method development regardless of mobile phase pH, salt concentration, and stationary phase composition.
Analytical Characterization of Soluble Antigen Arrays
SAgAs and fSAgAs were analyzed by RP-HPLC to determine molecular weight and peptide conjugation, as described previously.47 Quantitative peptide conjugation of representative test articles is provided in Table 1, showing target conjugation efficiencies of approximately 25% per peptide were achieved in HAPLP(9 PLP), HALABL (10 LABL), and SAgAPLP:LABL (10 PLP, 13 LABL).
Flow Cytometry Binding Assay
A flow cytometry binding assay was used to compare the relative binding avidities of hydrolyzable fSAgA and click-conjugated fcSAgA with Raji B cells. Binding kinetics were observed during association between the fluorescently labeled polymer arrays and Raji B cells until maximum steady state (max. SS) was reached, illustrated in Figure 4A. It was previously observed that fSAgAPLP:LABL, co-grafted with both PLP139–151 and LABL, exhibited greater binding with Raji B cells than the polymer alone (fHA) or the homopolymers grafted with only one signal (fHAPLP or fHALABL).44 A similar trend was observed with click-conjugated arrays: fcSAgAPLP:LABL exhibited the highest amount of binding, followed by fcHAPLP, while fcHA exhibited the lowest amount of binding (Figure 3A). Comparison of the maximum SS indicated that fcSAgAPLP:LABL binding was significantly greater than that of fcHAPLP, fcHALABL, and fcHA, while fcHAPLP binding was significantly greater than that of fcHALABL and fcHA (Figure 3B). Thus, multivalent PLP139–151 and LABL appear to have a cooperative effect on avidity. We previously reported that SAgAPLP:LABL exhibited PLP-specific binding and BCR targeting, implying that PLP may enhance B cell avidity by providing specific affinity for the BCR.44 Meanwhile, LABL, derived from LFA-1 and specific for ICAM-1, may enhance B cell avidity by promoting cell adhesion through the LFA-1/ICAM-1 interaction.7, 11–13, 51–54, 69 [CAM-1 and LFA-1 expression are upregulated on B cells during surface BCR engagement to promote intercellular adhesion, as the ICAM-1/LFA-1 interaction is critical for B cell:T cell conjugate formation during signaling. 70 This may explain why multivalent LABL exerted a cooperative effect on binding avidity when presented alongside PLP in fcSAgAPLP:LABL but a minimal effect when presented alone in fcHALABL.
Figure 4.

Comparing SAgAPLP:LABL and cSAgAPLP:LABL binding and IgM-stimulated (BCR-mediated) calcium flux signaling in Raji B cells through flow cytometry assays: (A) Binding kinetics and (B) maximum steady state (max. SS) binding with fSAgAPLP:LABL, fcSAgAPLP:LABL, and fcHA. (C) Calcium flux inhibition: Fluo-4 loaded cells were first pretreated with vehicle (HBSS), SAgAPLP:LABL, or cSAgAPLP:LABL, then stimulated with anti-IgM (αIgM, black arrow) to evaluate signaling inhibition. (D) Relative IgM signaling stimulation following pretreatment; baseline-adjusted values determined from mean steady state values. (E) Calcium flux reduction: Fluo-4 loaded cells were first stimulated with αIgM at ~30 s (black arrow), then treated with vehicle (HBSS), SAgAPLP:LABL, or cSAgAPLP:LABL after ~60 s (black arrow) to evaluate signaling reduction. (F) Percent reduction from IgM-stimulated baseline following sample addition, determined from mean steady state values. Statistical significance was determined by ANOVA followed by Tukey’s post hoc test with p<0.05 and n=3 (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). Calcium flux kinetics in (C) and (D) show median Fluo-4 fluorescence values. Robust curve fitting in (A) was performed using sigmoidal nonlinear regression. Calcium flux data was pooled from three independent experiments.
Figure 3.

Binding of fcHA, fcHALABL, fcHAPLP, and fcSAgAPLP:LABL with Raji B cells determined by flow cytometry: (A) Binding kinetics showing association through steady state. (B) Relative binding at maximum steady state (max. SS). (C) Comparison of max. SS binding with hydrolyzable versus click-conjugated arrays. Statistical significance determined by ANOVA followed by Tukey’s (B) or Sidak’s (C) post hoc test with p<0.05 and n=3 (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). Robust curve fitting in (A) was performed using sigmoidal nonlinear regression.
Comparison of click-conjugated versus hydrolyzable compound binding revealed that both fcSAgAPLP:LABL and fcHAPLP, exhibited significantly enhanced binding compared to their hydrolyzable counterparts, fSAgAPLP:LABL and fHAPLP, respectively (Figure 3C). Differences in kinetics and maximum SS binding between fcSAgAPLP:LABL, fSAgAPLP:LABL, and fHA illustrate how avidity was altered when multivalent peptide was conjugated to HA in a hydrolyzable versus non-hydrolyzable manner (Figure 4AB). While both methods of multivalent modification resulted in significantly increased binding compared to the polymer alone, click-conjugated fcSAgAPLP:LABL. exhibited significantly greater maximum SS binding (p<0.001) than hydrolyzable SAgAPLP:LABL. Thus, multivalent co-presentation of PLP and LABL through non-hydrolyzable modification increased the avidity of the polymer array more than hydrolyzable modification. This result is supportive of literature stating that multivalent antigens exhibit superior binding avidity, higher ‘effective concentration’, and an enhanced ability to engage cell receptors compared to monovalent (or in this case, hydrolyzable) antigen.27, 38–39, 41, 71
Calcium Flux Signaling Flow Cytometry Assay
Flow cytometry calcium flux assays were used to compare the ability of SAgA and cSAgA molecules to modulate BCR-mediated signaling in Raji B cells. Signaling modulation was evaluated in Fluo-4 loaded Raji B cells prior to stimulation (Figure 4CD) and after stimulation (Figure 4EF) with crosslinking αIgM. The relative signal increase from resting baseline (Figure 4D) or reduction from stimulated baseline (Figure 4F) was determined using mean Fluo-4 fluorescence values at steady state.
We reported previously that SAgAPLP:LABL was capable of both inhibiting and reducing IgM-stimulated signaling.44 Here, pre-treatment with SAgAPLP:LABL prior to addition of αIgM significantly inhibited IgM-stimulated calcium signaling compared to the vehicle (p<0.05) (Figure 4CD). However, pre-treatment with cSAgAPLP:LABL significantly inhibited IgM-stimulated calcium signaling to a greater extent (p<0.01), largely preventing even the initial spike in calcium flux observed with SAgAPLP:LABL after αIgM addition. Similarly, addition of cSAgAPLP:LABL after αIgM stimulation caused a greater reduction in calcium signaling than SAgAPLP:LABL (p<0.01) (Figure 4EF). cSAgAPLP:LABL reduced signaling by ~60% while SAgAPLP:LABL reduced signaling by ~40% relative to the vehicle control. These results indicated that click-conjugated cSAgAPLP:LABL was significantly more effective at dampening BCR-mediated signaling – both through inhibition and reduction – compared to its hydrolyzable counterpart.
Previously, we reported a significant reduction in signaling from addition of SAgAPLP:LABL, HAPLP, and HALABL, but negligible change in signaling from addition of vehicle (HBSS) or HA44 The click conjugates exhibited a similar trend: cSAgAPLP:LABL caused the greatest reduction in signaling while cHA caused the smallest reduction (p<0.05) (Figure 5A). However, while cSAgAPLP:LABL caused a greater reduction than hydrolyzable SAgAPLP:LABL, there was not a significant difference between cHAPLP vs. HAPLP and cHALABL vs. HALABL (Figure 5B). It is also interesting to note that cHA (HA-N3) caused a greater reduction than unmodified HA, which may be due to the presence of azide groups on the HA backbone leading to a greater degree of nonspecific binding.
Figure 5.

Reduction in IgM-stimulated (BCR-mediated) calcium flux signaling in Fluo-4 loaded Raji B cells determined by flow cytometry: (A) Percent reduction from αIgM-stimulated baseline following addition of cHA, cHALABL, cHAPLP, or cSAgAPLP:LABL, determined from mean steady state values. (B) Comparison of reduction in αIgM-stimulated signaling from hydrolyzable versus click-conjugated arrays. Data was pooled from three independent experiments. Statistical significance was determined by ANOVA followed by Tukey’s post hoc test (A) or unpaired t-test (B) with p<0.05 and n=3 (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).
Fluorescence Microscopy
Fluorescence microscopy was performed using a microfluidics platform that enabled real time observation of binding and BCR clustering on the cell surface. Previously, we observed that fSAgAPLP:LABL binding induced mature receptor clustering in Raji B cells while the polymer alone did not, and concluded that LABL may contribute to the SAgAPLP:LABL molecule’s ability to cluster receptors.44 Here, we observed receptor clustering following binding and also labeled IgM to monitor BCR organization on the cell surface. Negative control cells treated with media exhibited diffuse BCR staining (Figure 6A), while cells treated with fcHALABL (Figure 6C), fcHAPLP (Figure 6D), and fcSAgAPLP:LABL (Figure 6E) exhibited BCR capping. BCR capping occurs when BCR clusters coalesce to form a single aggregate (i.e., one area of high intensity IgM fluorescence).72 In contrast, cells treated with unmodified fcHA polymer exhibited BCR microclustering, which occurs when BCR clusters fail to coalesce into a single aggregate (i.e., multiple areas of moderate intensity IgM fluorescence) (Figure 6B). BCR capping on individual cells was quantified by plotting IgM pixel intensity relative to normalized cell diameter (Figure 7A), revealing that fcHALABL, fcHAPLP, and fcSAgAPLP:LABL induced clusters with significantly higher IgM intensity than the vehicle or fcHA (Figure 7B). Additionally, fcHALABL, fcHAPLP, and fcSAgAPLP:LABL induced IgM capping in a significantly higher fraction of cells than the vehicle or fcHA (Figure 7C). fcHALABL and fcSAgAPLP:LABL appeared particularly adept at inducing a high degree of IgM capping in a large fraction of cells. These trends echoed our previous observations that polymer modified with multivalent PLP139–151 and LABL, but not unmodified HA, induced mature receptor clustering.44 PLP may promote clustering due to its antigen-specific affinity for BCR. LABL may promote clustering due to its affinity for ICAM-1, which (along with LFA-1) plays an integral role in the organization of supramolecular activation clusters (SMACs) during BCR signaling by forming a peripheral ring (pSMAC) around the central BCR cluster (cSMAC). 7, 52, 73–74
Figure 6.

Fluorescence microscopy showing binding and BCR clustering in Raji B cells following perfusion of (A) vehicle, (B) fcHA, (C) fcHALABL, (D) fcHAPLP, and (E) fcSAgAPLP:LABL Cell nuclei were stained with Hoechst (violet – Panel 1) and surface IgM was stained with AlexaFluor® 647-conjugated αIgM (blue – Panel 3). Penn Green-labeled polymer arrays are shown binding to the cell surface (green – Panel 2). In contrast to the diffuse IgM fluorescence in (A), highly localized punctate IgM fluorescence in (C), (D), and (E) indicates BCR clustering and capping in cells treated with fcHALABL, fcHAPLP, and fcSAgAPLP:LABL. Capping occurs when BCR clusters coalesce to form a single aggregate (i.e., one area of high intensity IgM fluorescence). Captured using the M04S plate and CellASIC Onyx Microfluidics platform on an Olympus IX81 inverted Epifluorescence microscope. Magnification: 60X air. Scale bar equals 10 μm.
Figure 7.

Quantification of BCR clustering in Raji B cells following perfusion of vehicle, fcHA, fcHALABL, cHAPLP, or fcSAgAPLP:LABL in the microfluidics plate. (A) Representative cell profile plots of IgM pixel intensity relative to normalized cell diameter, d/D. (B) Maximum IgM pixel intensity per cell determined from profile plots of individual cells (n=40). (C) Percent of cells positive for IgM capping (fully coalesced IgM clustering) determined using otsu thresholding (n=100–250 cells per sample). Statistical significance was determined by ANOVA followed by Tukey’s post hoc test with p<0.05 (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). Mean+ SD shown. Images were captured using the M04S plate and CellASIC Onyx Microfluidics platform on an Olympus IX81 inverted Epifluorescence microscope and analyzed in ImageJ.
It has been suggested that quantitative differences in the degree of BCR clustering and crosslinking may drive qualitative differences in BCR signaling. 75 In general, our observations of BCR clustering corroborated our calcium flux results, as cHAPLP, cHALABL, and in particular cSAgAPLP:LABL reduced BCR-mediated signaling to a greater extent than cHA. Combined with previous evidence supporting BCR as a target for SAgAPLP:LABL binding,44 these results suggested that cSAgAPLP:LABL engagement and subsequent clustering of the BCR may dampen signaling. Our observations are consistent with reports that continuous BCR engagement and clustering are a mechanism for inducing B cell anergy that is accompanied by reduced calcium flux signaling.34–35
Preclinical EAE Studies
Therapeutic efficacy of SAgAPLP:LABL and cSAgAPLP:LABL was evaluated in EAE mice induced with PLP139–151 to model the relapsing-remitting form of MS. Disease symptoms emerged on day 10–12 with peak of disease occurring on day 13–15 before progressing to remission around day 20–25. Efficacy was measured by clinical score, weight change, and clinical score area under the curve (AUC) relative to the PBS control. AUC representation of clinical data has been reported as an informative secondary measure for overall extent of disease because it provides a cumulative measure not weighted by the scaling or time course of disease.76 Disease incidence and mortality rate are provided as supplemental information (Supplementary Figure 5). Statistical differences were determined by comparing treated groups with the negative PBS control.
A three-day dosing schedule with a dose equivalent to 200 nmol PLP139–151 administered on days 4, 7, and 10 was found to be efficacious in previous SAgAPLP:LABL studies.45–50 This dose and schedule were mirrored in a preliminary in vivo study with cSAgAPLP:LABL. It is important to note that shortly after the third administration on day 10, five out of six mice that received cSAgAPLP:LABL died from apparent anaphylaxis. This result may be due to a greater effective concentration of PLP antigen being delivered to immune cells with the click-conjugated platform over the acid-labile platform. As is the case with allergen tolerization therapy, where an offending allergen is administered in gradually increasing doses over time to desensitize the allergic immune response, care must be taken to achieve an effective cumulative dose that induces tolerance without activating a severe hypersensitive response.77–79 Our observation reflects the greater potency of cSAgAPLP:LABL compared to SAgAPLP:LABL, such that a lower dose was required to tip the immune response towards severe hypersensitivity. Therefore, a combination of lower doses was investigated in a small-scale dosing study (Figure 8). To determine whether the total cumulative dose or the number of injections caused the negative response, a group was included with the same dose per injection (200 nmol PLP139–151) but only administered on two days (days 4, 7). In another group, an equivalent cumulative dose was administered over three days (133 nmol PLP139–151 on days 4, 7, 10). A final group was included with a low dose of 50 nmol PLP139–151, administered on all three days. All dosing groups significantly alleviated disease compared to the PBS control according to clinical disease score (Figure 8A) and clinical score AUC (Figure 8C). The cSAgAPLP:LABL dose of 50 nmol PLP139–151 caused a significant reduction in clinical score on the greatest number of days (days 12–18) and exhibited the greatest reduction in clinical score AUC compared to PBS (p<0.001). Therefore, a cSAgAPLP:LABL dose of 50 nmol PLP139–151 was selected for studies going forward.
Figure 8.

Clinical EAE dosing study with cSAgAPLP:LABL: cSAgAPLP:LABL was administered on days 4 and 7 at a dose equivalent to 200 nmol PLP and on days 4, 7, and 10 at a dose equivalent to 50 or 133 nmol PLP. Therapeutic efficacy evaluated by comparing (A) clinical disease score, (B) percent weight change, and (C) clinical score area under the curve (AUC). Statistical significance (compared to the negative PBS control) was determined by ANOVA followed by Tukey’s post hoc test with p<0.05 and n=3 (*p<0.05, **p<0.01, #/***p<0.001, ##/****p<0.0001).
Next, in vivo efficacy of click-conjugated cSAgAPLP:LABL was compared to hydrolyzable SAgAPLP:LABL (Figure 9). At the original therapeutic dose equivalent to 200 nmol PLP139–151, SAgAPLP:LABL significantly reduced clinical score on days 11–20 (Figure 9A) and significantly reduced total disease score AUC compared to PBS (p<0.0001) (Figure 9E). At only a quarter of the dose, cSAgAPLP:LABL (50 nmol PLP139–151) significantly reduced total clinical score AUC to an equivalent extent as SAgAPLP:LABL at 200 nmol PLP139–151 (Figure 9E). Furthermore, cSAgAPLP:LABL (50 nmol PLP139–151) significantly reduced clinical score on days 10–17 to a greater extent than SAgAPLP:LABL at the 200 nmol dose (Figure 9B). In contrast, the 50 nmol dose of SAgAPLP:LABL significantly reduced clinical score only on days 11 and 14 (Figure 9B), and reduced clinical score AUC to a significantly lesser extent (p<0.001) than cSAgAPLP:LABL (50 nmol PLP139–151) (Figure 9E). While SAgAPLP:LABL (200 nmol PLP139–151) significantly alleviated weight loss on days 11–22 (Figure 9C), cSAgAPLP:LABL (50 nmol PLP139–151) significantly alleviated weight loss over a larger portion of the study, on days 11–25 (Figure 9D). In contrast, SAgAPLP:LABL (50 nmol PLP139–151) did not alleviate weight loss on any day of the study (Figure 9D).
Figure 9.

Comparing SAgAPLP:LABL and cSAgAPLP:LABL therapeutic efficacy in EAE: (A) SAgAPLP:LABL (200 nmol PLP dose) clinical scores (n=6), (B) cSAgAPLP:LABL versus SAgAPLP:LABL (50 nmol PLP dose) clinical scores (n=5), (C) SAgAPLP:LABL (200 nmol PLP dose) weight change, (D) cSAgAPLP:LABL versus SAgAPLP:LABL (50 nmol PLP dose) weight change, and (E) clinical score area under the curve (AUC) relative to PBS. Statistical significance was determined by ANOVA followed by Dunnet’s (A-D) or Tukey’s (E) post hoc test with p<0.05 (*p<0.05, **p<0.01, #/***p<0.001, ##/****p<0.0001).
Lastly, cSAgAPLP:LABL (50 nmol PLP139–151) reduced incidence of disease to a greater extent than SAgAPLP:LABL at either dose (Supplementary Figure 5). During peak of disease, 100% of the mice treated with PBS or SAgAPLP:LABL (50 nmol PLP139–151) and over 75% of the mice treated with SAgAPLP:LABL (200 nmol PLP139–151) exhibited disease symptoms. In contrast, less than 25% of the mice treated with cSAgAPLP:LABL (50 nmol PLP139–151) exhibited disease symptoms during peak of disease. Therefore, when considering multiple measures of efficacy, cSAgAPLP:LABL achieved equivalent or greater in vivo efficacy as SAgAPLP:LABL at one quarter of the antigen dose. We conclude that non-hydrolyzable click conjugation rendered greater therapeutic efficacy than hydrolyzable modification.
CONCLUSIONS
Click-conjugated multivalent soluble antigen arrays were developed and evaluated in vitro and in vivo as therapeutic agents in a murine model of MS. Hydrolyzable SAgAPLP:LABL which we have studied extensively and shown to significantly suppress EAE,44–50 employed a degradable linker to co-deliver antigen (PLP) and cell adhesion inhibitor (LABL) peptides. This approach was built upon our earlier two-signal hypothesis that SAgAPLP:LABL inhibited autoimmune activation via the immunological synapse, a mechanism that would necessitate antigen uptake, processing, and presentation and motivated the design of a degradable SAgA molecule. Recent in vitro studies, however, pointed instead to a therapeutic mechanism whereby SAgAPLP:LABL acted through sustained BCR engagement, targeting BCR signaling while exhibiting prolonged residence on the cell surface.44 These results motivated the development of a non-degradable SAgA molecule to enhance and maintain the molecule’s surface activity, which we hypothesized would improve therapeutic efficacy. cSAgAPLP:LABL was developed as a modified version of the SAgAPLP:LABL molecule with multiple PLP139–151 and LABL peptides conjugated to HA using non-hydrolyzable linker chemistry (Copper-catalyzed Azide-Alkyne Cycloaddition (CuAAC)).
Building upon previous work, these studies sought to establish therapeutic efficacy of cSAgAPLP:LABL in vivo while identifying a potential therapeutic mechanism by evaluating binding avidity and signaling modulation in vitro. Click-conjugated cSAgAPLP:LABL exhibited greatly enhanced binding in B cells compared to hydrolyzable SAgAPLP:LABL, indicating that non-hydrolyzable multivalent ligand increased the avidity of the molecule. Furthermore, cSAgAPLP:LABL exhibited greater capacity for reducing and inhibiting BCR-mediated signaling as compared to SAgAPLP:LABL Imaging revealed that c SAgAPLP:LABL binding caused BCR clustering, another marker indicative of BCR engagement and signaling modulation. Our in vitro observations pointed to B cell anergy, induced by continuous BCR engagement and clustering and accompanied by reduced calcium flux signaling, as a likely cSAgAPLP:LABL therapeutic cellular mechanism. Lastly, cSAgAPLP:LABL exhibited enhanced in vivo efficacy against EAE, achieving equivalent therapeutic efficacy as SAgAPLP:LABL at one quarter of the dose. Taken together, these results indicated that non-hydrolyzable conjugation increased the avidity of cSAgAPLP:LABL, driving in vivo efficacy through modulated BCR-mediated signaling. The click-conjugated c SAgAPLP:LABL molecule shows promising potential for ASIT for the improved treatment of autoimmune disease.
Supplementary Material
ACKNOWLEDGEMENTS
We gratefully acknowledge support from the National Institutes of Health Graduate Training Program in Dynamic Aspects of Chemical Biology Grant (T32 GM008545) from the National Institutes of General Medical Sciences (C.J.P. and M.L.), the Madison and Lila Self Graduate Fellowship at the University of Kansas (B.L.H.), and the Howard Rytting pre-doctoral fellowship from the Department of Pharmaceutical Chemistry at the University of Kansas (C.J.P.). We would like to thank Dr. Blake Peterson for graciously donating the Pennsylvania Green parent compound. We thank Laura Northrup and Sharadvi Thati for their assistance with animal studies. Additionally, we thank the Macromolecule and Vaccine Stabilization Center, KU NMR Lab, Microscopy and Analytical Imaging Core Lab, and the Kansas Vaccine Institute at the University of Kansas for their collaboration and instrument use.
Footnotes
SUPPORTING INFORMATION AVAILABLE
Supplementary Table 1 chronicles azide functionalization of HA across various batches, as measured by 1H NMR. Flow cytometry histograms show the gating protocol used to remove dead cells and debris from analysis in Kaluza Flow Analysis software (Supplementary Figure 1). Supplementary Figure 2 illustrates the image quantification methods used to generate BCR clustering data shown in Figure 7. RP-HPLC chromatograms from a room temperature solution stability study with cHAPLP at pH 2.4 show negligible degradation and release of PLP over time (Supplementary Figure 3). SEC chromatograms show purified reaction products compared to HA of various molecular weights (Supplementary Figure 4). Incidence of disease and mortality plots provide additional measures of in vivo efficacy (Supplementary Figure 5).
CONFLICT OF INTEREST
The authors declare no competing financial interests.
REFERENCES
- 1.Gonsette R, Self-tolerance in multiple sclerosis. Acta Neurol. Belg 2012, 112 (2), 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carson MJ; Doose JM; Melchior B; Schmid CD; Ploix CC, CNS immune privilege: hiding in plain sight. Immunol. Rev 2006, 213 (1), 48–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fletcher JM; Lalor S; Sweeney C; Tubridy N; Mills K, T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin. Exp. Immunol 2010, 162 (1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amor S; Puentes F; Baker D; Van Der Valk P, Inflammation in neurodegenerative diseases. Immunology 2010, 129 (2), 154–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herz J; Zipp F; Siffrin V, Neurodegeneration in autoimmune CNS inflammation. Exp. Neurol 2010, 225(1), 9–17. [DOI] [PubMed] [Google Scholar]
- 6.Lassmann H, Mechanisms of inflammation induced tissue injury in multiple sclerosis. J. Neurol. Sci 2008, 274 (1), 45–47. [DOI] [PubMed] [Google Scholar]
- 7.Grakoui A; Bromley SK; Sumen C; Davis MM; Shaw AS; Allen PM; Dustin ML, The immunological synapse: a molecular machine controlling T cell activation. Science 1999, 285 (5425), 221–227. [DOI] [PubMed] [Google Scholar]
- 8.Iezzi G; Karjalainen K; Lanzavecchia A, The duration of antigenic stimulation determines the fate of naive and effector T cells. Immunity 1998, 8 (1), 89–95. [DOI] [PubMed] [Google Scholar]
- 9.Baxter AG; Hodgkin PD, Activation rules: the two-signal theories of immune activation. Nat. Rev. Immunol 2002, 2 (6), 439–446. [DOI] [PubMed] [Google Scholar]
- 10.Bromley SK; Iaboni A; Davis SJ; Whitty A; Green JM; Shaw AS; Weiss A; Dustin ML, The immunological synapse and CD28-CD80 interactions. Nat. Immunol 2001, 2 (12), 1159–1166. [DOI] [PubMed] [Google Scholar]
- 11.Chen L; Flies DB, Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol 2013, 13 (4), 227–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frauwirth KA; Thompson CB, Activation and inhibition of lymphocytes by costimulation. J. Clin. Invest 2002, 109 (109 (3)), 295–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jun JE; Goodnow CC, Scaffolding of antigen receptors for immunogenic versus tolerogenic signaling. Nat. Immunol 2003, 4 (11), 1057–1064. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Q; Vignali DA, Co-stimulatory and co-inhibitory pathways in autoimmunity. Immunity 2016, 44 (5), 1034–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mueller DL; Jenkins MK; Schwartz RH, Clonal expansion versus functional clonal inactivation: a costimulatory signalling pathway determines the outcome of T cell antigen receptor occupancy. Annu. Rev. Immunol 1989, 7(1), 445–480. [DOI] [PubMed] [Google Scholar]
- 16.Rock KL; Benacerraf B; Abbas AK, Antigen presentation by hapten-specific B lymphocytes. I. Role of surface immunoglobulin receptors. J. Exp. Med 1984, 160 (4), 1102–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Constant SL, B lymphocytes as antigen-presenting cells for CD4+ T cell priming in vivo. J. Immunol 1999, 162 (10), 5695–5703. [PubMed] [Google Scholar]
- 18.Kontos S; Grimm AJ; Hubbell JA, Engineering antigen-specific immunological tolerance. Curr. Opin. Immunol 2015, 35, 80–88. [DOI] [PubMed] [Google Scholar]
- 19.Kinzel S; Weber MS, B Cell-Directed Therapeutics in Multiple Sclerosis: Rationale and Clinical Evidence. CNS drugs 2016, 1–12. [DOI] [PubMed] [Google Scholar]
- 20.Jackson SW; Kolhatkar NS; Rawlings DJ, B cells take the front seat: dysregulated B cell signals orchestrate loss of tolerance and autoantibody production. Curr. Opin. Immunol 2015, 33, 70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khan WN; Wright JA; Kleiman E; Boucher JC; Castro I; Clark ES, B-lymphocyte tolerance and effector function in immunity and autoimmunity. Immunol. Res 2013, 57(1–3), 335–353. [DOI] [PubMed] [Google Scholar]
- 22.Boster A; Ankeny DP; Racke MK, The potential role of B cell-targeted therapies in multiple sclerosis. Drugs 2010, 70 (18), 2343–2356. [DOI] [PubMed] [Google Scholar]
- 23.Oh S; Cudrici C; Ito T; Rus H, B-cells and humoral immunity in multiple sclerosis. Implications for therapy. Immunol. Res 2008, 40 (3), 224–234. [DOI] [PubMed] [Google Scholar]
- 24.Bates D, Treatment effects of immunomodulatory therapies at different stages of multiple sclerosis in short-term trials. Neurology 2011, 76 (1 Supplement 1), S14–S25. [DOI] [PubMed] [Google Scholar]
- 25.Feldmann M; Steinman L, Design of effective immunotherapy for human autoimmunity. Nature 2005, 435 (7042), 612. [DOI] [PubMed] [Google Scholar]
- 26.Miller SD; Turley DM; Podojil JR, Antigen-specific tolerance strategies for the prevention and treatment of autoimmune disease. Nat. Rev. Immunol 2007, 7 (9), 665. [DOI] [PubMed] [Google Scholar]
- 27.Jones DS, Multivalent compounds for antigen-specific B cell tolerance and treatment of autoimmune diseases. Curr. Med. Chem 2005, 12 (16), 1887–1904. [DOI] [PubMed] [Google Scholar]
- 28.Ketchum C; Miller H; Song W; Upadhyaya A, Ligand mobility regulates B cell receptor clustering and signaling activation. Biophys. J 2014, 106 (1), 26–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Batista FD; Iber D; Neuberger MS, B cells acquire antigen from target cells after synapse formation. Nature 2001, 411 (6836), 489–494. [DOI] [PubMed] [Google Scholar]
- 30.Puffer EB; Pontrello JK; Hollenbeck JJ; Kink JA; Kiessling LL, Activating B cell signaling with defined multivalent ligands. ACS Chem. Biol 2007, 2 (4), 252–262. [DOI] [PubMed] [Google Scholar]
- 31.Tolar P; Sohn HW; Pierce SK, The initiation of antigen-induced B cell antigen receptor signaling viewed in living cells by fluorescence resonance energy transfer. Nat. Immunol 2005, 6 (11), 1168–1176. [DOI] [PubMed] [Google Scholar]
- 32.Harwood NE; Batista FD, Early events in B cell activation. Annu. Rev. Immunol 2009, 28, 185–210. [DOI] [PubMed] [Google Scholar]
- 33.Getahun A; O’Neill SK; Cambier JC, Establishing anergy as a bona fide in vivo mechanism ofB cell tolerance. J. Immunol 2009, 183 (9), 5439–5441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gauld SB; Benschop RJ; Merrell KT; Cambier JC, Maintenance of B cell anergy requires constant antigen receptor occupancy and signaling. Nat. Immunol 2005, 6(11), 1160–1167. [DOI] [PubMed] [Google Scholar]
- 35.Cambier JC; Gauld SB; Merrell KT; Vilen BJ, B-cell anergy: from transgenic models to naturally occurring anergic B cells? Nat. Rev. Immunol 2007, 7 (8), 633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mueller DL, Mechanisms maintaining peripheral tolerance. Nat. Immunol 2010, 11 (1), 21–27. [DOI] [PubMed] [Google Scholar]
- 37.Cairo CW; Gestwicki JE; Kanai M; Kiessling LL, Control of multivalent interactions by binding epitope density. J. Am. Chem. Soc 2002, 124 (8), 1615–1619. [DOI] [PubMed] [Google Scholar]
- 38.Gestwicki JE; Cairo CW; Strong LE; Oetjen KA; Kiessling LL, Influencing receptor-ligand binding mechanisms with multivalent ligand architecture. J. Am. Chem. Soc. 2002, 124 (50), 14922–14933. [DOI] [PubMed] [Google Scholar]
- 39.Kiessling LL; Gestwicki JE; Strong LE, Synthetic multivalent ligands in the exploration of cell-surface interactions. Curr. Opin. Chem. Biol 2000, 4 (6), 696–703. [DOI] [PubMed] [Google Scholar]
- 40.Hartwell BL; Antunez L; Sullivan BP; Thati S; Sestak JO; Berkland C, Multivalent Nanomaterials: Learning from Vaccines and Progressing to Antigen - Specific Immunotherapies. J. Pharm. Sci 2015, 104 (2), 346–361. [DOI] [PubMed] [Google Scholar]
- 41.Krishnamurthy VM; Estroff LA; Whitesides GM, Multivalency in ligand design. Fragment-based approaches in drug discovery, 2006, 34, 11–53. [Google Scholar]
- 42.Dintzis H; Dintzis R; Vogelstein B, Molecular determinants of immunogenicity: the immunon model of immune response. Proc. Natl. Acad. Sci. U. S. A 1976, 73 (10), 3671–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kiessling LL; Gestwicki JE; Strong LE, Synthetic multivalent ligands as probes of signal transduction. Angew. Chem., Int. Ed 2006, 45 (15), 2348–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hartwell BL; Martinez-Becerra FJ; Chen J; Shinogle H; Samowski M; Moore DS; Berkland C, Antigen-Specific Binding of Multivalent Soluble Antigen Arrays Induces Receptor Clustering and Impedes B Cell Receptor Mediated Signaling. Biomacromolecules 2016, 17 (3), 710–722. [DOI] [PubMed] [Google Scholar]
- 45.Hartwell BL; Smalter Hall A; Swafford D; Sullivan BP; Garza A; Sestak JO; Northrup L; Berkland C, Molecular Dynamics of Multivalent Soluble Antigen Arrays Support a Two-Signal Co-delivery Mechanism in the Treatment of Experimental Autoimmune Encephalomyelitis. Mol. Pharmaceutics 2016, 13 (2), 330–343. [DOI] [PubMed] [Google Scholar]
- 46.Northrup L; Sestak JO; Sullivan BP; Thati S; Hartwell BL; Siahaan TJ; Vines CM; Berkland C, Co-delivery of autoantigen and b7 pathway modulators suppresses experimental autoimmune encephalomyelitis. AAPS J. 2014,16 (6), 1204–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sestak J; Mullins M; Northrup L; Thati S; Forrest ML; Siahaan TJ; Berkland C, Single-step grafting of aminooxy-peptides to hyaluronan: a simple approach to multifunctional therapeutics for experimental autoimmune encephalomyelitis. J. Controlled Release 2013, 168 (3), 334–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sestak JO; Fakhari A; Badawi AH; Siahaan TJ; Berkland C, Structure, size, and solubility of antigen arrays determines efficacy in experimental autoimmune encephalomyelitis. AAPS J. 2014, 16(6), 1185–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sestak JO; Sullivan BP; Thati S; Northrup L; Hartwell B; Antunez L; Forrest ML; Vines CM; Siahaan TJ; Berkland C, Codelivery of antigen and an immune cell adhesion inhibitor is necessary for efficacy of soluble antigen arrays in experimental autoimmune encephalomyelitis. Mol. Ther. Methods Clin. Dev 2014, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thati S; Kuehl C; Hartwell B; Sestak J; Siahaan T; Forrest ML; Berkland C, Routes of administration and dose optimization of soluble antigen arrays in mice with experimental autoimmune encephalomyelitis. J. Pharm. Sci 2015, 104 (2), 714–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anderson ME; Siahaan TJ, Targeting ICAM-l/LFA-1 interaction for controlling autoimmune diseases: designing peptide and small molecule inhibitors. Peptides 2003, 24 (3), 487–501. [DOI] [PubMed] [Google Scholar]
- 52.Carrasco YR; Fleire SJ; Cameron T; Dustin ML; Batista FD, LFA-l/ICAM-1 interaction lowers the threshold of B cell activation by facilitating B cell adhesion and synapse formation. Immunity 2004, 20 (5), 589–599. [DOI] [PubMed] [Google Scholar]
- 53.Tibbetts SA; Jois DS; Siahaan TJ; Benedict SH; Chan MA, Linear and cyclic LFA-1 and ICAM-1 peptides inhibit T cell adhesion and function. Peptides 2000, 21 (8), 1161–1167. [DOI] [PubMed] [Google Scholar]
- 54.Badawi A; Kiptoo P; Siahaan T, Immune Tolerance Induction against Experimental Autoimmune Encephalomyelitis (EAE) Using A New PLP-B7AP Conjugate that Simultaneously Targets B7/CD28 Costimulatory Signal and TCR/MHC-II Signal. J Mult Scler 2014, 1 (131), 2376–0389.1000131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meng Q; Yu M; Zhang H; Ren J; Huang D, Synthesis and application of N-hydroxysuccinimidyl rhodamine B ester as an amine-reactive fluorescent probe. Dyes and Pigments 2007, 73 (2), 254–260. [Google Scholar]
- 56.Hu X; Li D; Zhou F; Gao C, Biological hydrogel synthesized from hyaluronic acid, gelatin and chondroitin sulfate by click chemistry. Acta Biomater. 2011, 7 (4), 1618–1626. [DOI] [PubMed] [Google Scholar]
- 57.Di Meo C; Panza L; Campo F; Capitani D; Mannina L; Banzato A; Rondina M; Rosato A; Crescenzi V, Novel Types of Carborane - Carrier Hyaluronan Derivatives via “ Click Chemistry”. Macromol. Biosci. 2008, 8 (7), 670–681. [DOI] [PubMed] [Google Scholar]
- 58.Dintzis R; Middleton M; Dintzis H, Studies on the immunogenicity and tolerogenicity of T-independent antigens. J. Immunol 1983, 131 (5), 2196–2203. [PubMed] [Google Scholar]
- 59.Dintzis RZ; Vogelstein B; Dintzis HM, Specific cellular stimulation in the primary immune response: experimental test of a quantized model. Proc. Natl. Acad. Sci. U. S. A 1982, 79 (3), 884–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Badawi AH; Kiptoo P; Wang W-T; Choi I-Y; Lee P; Vines CM; Siahaan TJ, Suppression of EAE and prevention of blood-brain barrier breakdown after vaccination with novel bifunctional peptide inhibitor. Neuropharmacology 2012, 62 (4), 1874–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Badawi AH; Siahaan TJ, Suppression of MOG-and PLP-induced experimental autoimmune encephalomyelitis using a novel multivalent bifunctional peptide inhibitor. J. Neuroimmunol 2013, 263 (1), 20–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kiptoo P; Buyuktimkin B; Badawi A; Stewart J; Ridwan R; Siahaan T, Controlling immune response and demyelination using highly potent bifunctional peptide inhibitors in the suppression of experimental autoimmune encephalomyelitis. Clin. Exp. Immunol 2013, 172 (1), 23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kobayashi N; Kobayashi H; Gu L; Malefyt T; Siahaan TJ, Antigen-specific suppression of experimental autoimmune encephalomyelitis by a novel bifunctional peptide inhibitor. J. Pharmacol. Exp. Ther 2007, 322 (2), 879–886. [DOI] [PubMed] [Google Scholar]
- 64.Manikwar P; Tejo BA; Shinogle H; Moore DS; Zimmerman T; Blanco F; Siahaan TJ, Utilization of I-domain of LFA-1 to Target Drug and Marker Molecules to Leukocytes. Theranostics 2011, 1, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tang W; Becker ML, “Click” reactions: a versatile toolbox for the synthesis of peptide-conjugates. Chem. Soc. Rev 2014, 43 (20), 7013–7039. [DOI] [PubMed] [Google Scholar]
- 66.Sokolova NV; Nenajdenko VG, Recent advances in the Cu (i)-catalyzed azide–alkyne cycloaddition: focus on functionally substituted azides and alkynes. RSC Adv. 2013, 3 (37), 16212–16242. [Google Scholar]
- 67.Presolski SI; Hong VP; Finn M, Copper - Catalyzed Azide-Alkyne Click Chemistry for Bioconjugation. Curr. Protoc. Chem. Biol 2011, 153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Presolski SI; Hong V; Cho S-H; Finn M, Tailored ligand acceleration of the Cu-catalyzed azide– alkyne cycloaddition reaction: practical and mechanistic implications. J. Am. Chem. Soc 2010, 132 (41), 14570–14576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Delon I; Germain RN, Information transfer at the immunological synapse. Curr. Biol 2000, 10 (24), R923–R933. [DOI] [PubMed] [Google Scholar]
- 70.Dang LH; Rock KL, Stimulation of B lymphocytes through surface Ig receptors induces LFA-1 and ICAM-1-dependent adhesion. J. Immunol 1991, 146 (10), 3273–3279. [PubMed] [Google Scholar]
- 71.Symer DE; Dintzis RZ; Diamond DJ; Dintzis HM, Inhibition or activation of human T cell receptor transfectants is controlled by defined, soluble antigen arrays. J. Exp. Med 1992, 176 (5), 1421–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pierce SK; Liu W, The tipping points in the initiation of B cell signalling: how small changes make big differences. Nat. Rev. Immunol 2010, 10 (11), 767–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Penninger JM; Crabtree GR, The actin cytoskeleton and lymphocyte activation. Cell 1999, 96 (1), 9–12. [DOI] [PubMed] [Google Scholar]
- 74.Monks CR; Freiberg BA; Kupfer H; Sciaky N; Kupfer A, Three-dimensional segregation of supramolecular activation clusters in T cells. Nature 1998, 395 (6697), 82–86. [DOI] [PubMed] [Google Scholar]
- 75.Hartley SB; Crosbie J, Elimination from peripheral lymphoid tissues of self-reactive B lymphocytes recognizing membrane-bound antigens. Nature 1991, 353 (6346), 765. [DOI] [PubMed] [Google Scholar]
- 76.Fleming KK; Bovaird JA; Mosier MC; Emerson MR; LeVine SM; Marquis JG, Statistical analysis of data from studies on experimental autoimmune encephalomyelitis. J. Neuroimmunol 2005, 170 (1), 71–84. [DOI] [PubMed] [Google Scholar]
- 77.Akdis M; Akdis CA, Mechanisms of allergen-specific immunotherapy: multiple suppressor factors at work in immune tolerance to allergens. Journal of Allergy and Clinical Immunology 2014, 133 (3), 621–631. [DOI] [PubMed] [Google Scholar]
- 78.Burks AW; Calderon MA; Casale T; Cox L; Demoly P; Jutel M; Nelson H; Akdis CA, Update on allergy immunotherapy: American academy of allergy, asthma & immunology/European academy of allergy and clinical immunology/PRACTALL consensus report. Journal of Allergy and Clinical Immunology 2013, 131 (5), 1288–1296. e3. [DOI] [PubMed] [Google Scholar]
- 79.Sabatos-Peyton CA; Verhagen J; Wraith DC, Antigen-specific immunotherapy of autoimmune and allergic diseases. Curr. Opin. Immunol 2010, 22 (5), 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.